KEVEYIS- dichlorphenamide tablet
Keveyis by
Drug Labeling and Warnings
Keveyis by is a Prescription medication manufactured, distributed, or labeled by Strongbridge US Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use KEVEYIS® safely and effectively. See full prescribing information for KEVEYIS®.
KEVEYIS® (dichlorphenamide) tablets, for oral use
Initial U.S. Approval: 1958RECENT MAJOR CHANGES
INDICATIONS AND USAGE
KEVEYIS is an oral carbonic anhydrase inhibitor indicated for the treatment of primary hyperkalemic periodic paralysis, primary hypokalemic periodic paralysis, and related variants (1)
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
Tablets: 50 mg (3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- Hypersensitivity and Other Life-Threatening Reactions: discontinue KEVEYIS at the first appearance of skin rash or any sign of immune-mediated or idiosyncratic adverse reaction (5.1)
- Hypokalemia: baseline and periodic measurements of serum potassium are recommended; if hypokalemia develops or persists, consider reducing the dose or discontinuing KEVEYIS and correcting potassium levels (5.3)
- Metabolic acidosis: baseline and periodic measurements of serum bicarbonate are recommended; if metabolic acidosis develops or persists, consider reducing the dose or discontinuing KEVEYIS (5.4)
- Falls: consider reducing the dose or discontinuing KEVEYIS in patients who experience falls (5.5)
ADVERSE REACTIONS
Most common adverse reactions (incidence at least 10% and greater than placebo) include paresthesias, cognitive disorder, dysgeusia, and confusional state (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Strongbridge Biopharma at 1-855-324-8912, or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.DRUG INTERACTIONS
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 11/2019
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Dosage Information
2.2 Monitoring to Assess Effectiveness
2.3 Monitoring to Assess Safety
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity and Other Life-Threatening Reactions
5.2 Concomitant Use of Aspirin or Other Salicylates
5.3 Hypokalemia
5.4 Metabolic Acidosis
5.5 Falls
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Aspirin and Other Salicylates
7.2 Drugs that are Substrates of Organic Anion Transporter1 (OAT1)
7.3 Drugs that Cause Hypokalemia
7.4 Drugs that Cause Metabolic Acidosis
7.5 Drugs that are Inhibitors of OAT1 or OAT3
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Dosage Information
Initiate dosing at 50 mg by mouth once or twice daily. The dosage may be increased or decreased based on individual response, at weekly intervals (or sooner in case of adverse reaction). The minimum recommended total daily dosage is 50 mg, and the maximum recommended total daily dosage is 200 mg.
2.2 Monitoring to Assess Effectiveness
Primary hyperkalemic periodic paralysis, primary hypokalemic periodic paralysis, and related variants are a heterogeneous group of conditions, for which the response to KEVEYIS may vary. Therefore, prescribers should evaluate the patient's response to KEVEYIS after 2 months of treatment to decide whether KEVEYIS should be continued.
2.3 Monitoring to Assess Safety
Baseline and periodic measurements of serum potassium and serum bicarbonate during KEVEYIS treatment is recommended [see Warnings and Precautions (5.3, 5.4)].
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
KEVEYIS is contraindicated in the following circumstances:
- Hypersensitivity to dichlorphenamide or other sulfonamides [see Warnings and Precautions (5.1)]
- Concomitant use of KEVEYIS and high dose aspirin [see Warnings and Precautions (5.2) and Drug Interactions (7.1)]
- Severe pulmonary disease, limiting compensation to metabolic acidosis caused by KEVEYIS [see Warnings and Precautions (5.4)]
- Hepatic insufficiency: KEVEYIS may aggravate hepatic encephalopathy.
-
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity and Other Life-Threatening Reactions
Fatalities associated with the administration of sulfonamides have occurred because of adverse reactions including Stevens-Johnson syndrome, toxic epidermal necrolysis, fulminant hepatic necrosis, agranulocytosis, aplastic anemia and other blood dyscrasias. Pulmonary involvement can occur in isolation or as part of a systemic reaction.
KEVEYIS should be discontinued at the first appearance of skin rash or any sign of immune-mediated or other life-threatening adverse reaction.
5.2 Concomitant Use of Aspirin or Other Salicylates
Carbonic anhydrase inhibitors, including KEVEYIS, can cause metabolic acidosis [see Warnings and Precautions (5.4)], which can increase the risk of salicylate toxicity. Anorexia, tachypnea, lethargy, and coma have been reported with concomitant use of dichlorphenamide and high-dose aspirin. Therefore, the concomitant use of KEVEYIS and high-dose aspirin is contraindicated. Patients with concomitant use of KEVEYIS and low-dose aspirin should be carefully monitored.
5.3 Hypokalemia
KEVEYIS increases potassium excretion and can cause hypokalemia. The risk of hypokalemia is greater when KEVEYIS is used in patients with conditions associated with hypokalemia (e.g., adrenocortical excess, renal tubular acidosis type 1 and 2), and in patients receiving other drugs that may cause hypokalemia [see Drug Interactions (7.3)].
Baseline and periodic measurements of serum potassium during KEVEYIS treatment is recommended.
If hypokalemia develops or persists, consideration should be given to reducing the dose or discontinuing KEVEYIS and correction of potassium levels.
5.4 Metabolic Acidosis
KEVEYIS can cause hyperchloremic non-anion gap metabolic acidosis. Concomitant use of KEVEYIS with other drugs that cause metabolic acidosis may increase the severity of acidosis. Concomitant use of KEVEYIS in compensated patients with respiratory acidosis, such as in advanced lung diseases, may lead to respiratory decompensation.
Baseline and periodic measurements of serum bicarbonate during KEVEYIS treatment are recommended.
If metabolic acidosis develops or persists, consideration should be given to reducing the dose or discontinuing KEVEYIS [see Drug Interactions (7.4)].
-
6 ADVERSE REACTIONS
The following serious adverse reactions are described elsewhere in labeling:
- Hypersensitivity and Other Life-Threatening Reactions [see Warnings and Precautions (5.1)]
- Hypokalemia [see Warnings and Precautions (5.3)]
- Metabolic Acidosis [see Warnings and Precautions (5.4)]
- Falls [see Warnings and Precautions (5.5)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
In a 9-week randomized controlled trial in adults with hyperkalemic or hypokalemic periodic paralysis (Study 1), the most common adverse reactions in patients treated with KEVEYIS, with rates greater than placebo, were paresthesia, cognitive disorder, dysgeusia, and confusional state. The mean dose of KEVEYIS was 94 mg/day in patients with hypokalemic periodic paralysis and 82 mg/day in patients with hyperkalemic periodic paralysis.
Table 1 lists the incidence of adverse reactions that occurred in ≥ 5% of patients treated with KEVEYIS and more commonly than in patients treated with placebo in Study 1.
Table 1: Adverse Reactions in Patients Treated with KEVEYIS with Incidence ≥ 5% and more common than in Patients Treated with Placebo in Study 1 Adverse Reaction KEVEYIS
N = 36
(%)Placebo
N = 29
(%)- * Cognitive disorder combined cases with the preferred terms of cognitive disorder, disturbance in attention, and mental impairment.
Nervous system disorders Paresthesia 44 14 Cognitive disorder* 14 7 Dysgeusia 14 0 Confusional state 11 0 Headache 8 7 Hypoesthesia 8 0 Lethargy 8 0 Dizziness 6 0 Gastrointestinal disorders Diarrhea 6 3 Nausea 6 0 General disorders and administration site conditions Fatigue 8 0 Malaise 6 0 Investigations Weight decreased 6 0 Musculoskeletal and connective tissue disorders Muscle spasms 8 0 Arthralgia 6 3 Muscle twitching 6 0 Respiratory Dyspnea 6 0 Pharyngolaryngeal pain 6 0 Skin Rash 8 0 Pruritus 6 0 6.2 Postmarketing Experience
Adverse reactions have been identified during postapproval use of dichlorphenamide. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
The following are adverse reactions which have been reported during postapproval use of dichlorphenamide and were serious or are not reported in the previous section of labeling [see Clinical Trials Experience (6.1)]: amnesia, cardiac failure, condition aggravated, convulsion, hallucination, nephrolithiasis, pancytopenia, psychotic disorder, renal tubular necrosis, stupor, syncope, tremor.
-
7 DRUG INTERACTIONS
7.1 Aspirin and Other Salicylates
Carbonic anhydrase inhibitors, including KEVEYIS, can cause metabolic acidosis [see Warnings and Precautions (5.2, 5.4)], which can increase the risk of salicylate toxicity. Anorexia, tachypnea, lethargy, and coma have been reported with concomitant use of dichlorphenamide and high-dose aspirin. Therefore, concomitant use of KEVEYIS and high-dose aspirin is contraindicated. Patients with concomitant use of KEVEYIS and low-dose aspirin should be carefully monitored [see Contraindications (4) and Warnings and Precautions (5.2)].
7.2 Drugs that are Substrates of Organic Anion Transporter1 (OAT1)
In vitro, dichlorphenamide is an inhibitor of OAT1 transporters. The concomitant administration of KEVEYIS may increase the plasma exposures of OAT1 substrates. Use of KEVEYIS with drugs that are sensitive to OAT1 inhibition (e.g., methotrexate, famotidine, oseltamivir) is not recommended [see Clinical Pharmacology (12.3)].
7.3 Drugs that Cause Hypokalemia
The risk of hypokalemia is greater with coadministration of KEVEYIS and other drugs that can cause hypokalemia (e.g., loop diuretics, thiazide diuretics, laxatives, antifungals, penicillins, and theophylline) [see Warnings and Precautions (5.3)].
7.4 Drugs that Cause Metabolic Acidosis
Coadministration of KEVEYIS and other drugs that can cause metabolic acidosis may increase the severity of the acidosis [see Warnings and Precautions (5.4)].
7.5 Drugs that are Inhibitors of OAT1 or OAT3
An in vitro transporter study indicated that dichlorphenamide is a substrate of human transporters OAT1 and OAT3 [see Clinical Pharmacology (12.3)]. Therefore, signs of dichlorphenamide toxicity should be monitored when administered with OAT1 or OAT3 inhibitors.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no adequate data on the developmental risk associated with the use of KEVEYIS in pregnant women. A no-effect dose has not been established. Dichlorphenamide was teratogenic when administered orally to pregnant rats.The background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4%, and 15% to 20%, respectively.
Clinical Considerations
Fetal/Neonatal adverse reactions
KEVEYIS treatment can cause metabolic acidosis [see Warnings and Precautions (5.4)]. The effect of dichlorphenamide-induced metabolic acidosis has not been studied in pregnancy; however, metabolic acidosis in pregnancy (due to other causes) can cause decreased fetal growth, decreased fetal oxygenation, and fetal death, and may affect the fetus’ ability to tolerate labor. Pregnant patients should be monitored for metabolic acidosis and treated as in the nonpregnant state. Newborns of mothers treated with KEVEYIS should be monitored for metabolic acidosis because of possible occurrence of transient metabolic acidosis following birth.Labor or Delivery
Although the effect of KEVEYIS on labor and delivery in humans has not been established, the development of dichlorphenamide-induced metabolic acidosis in the mother and/or in the fetus might affect the fetus’ ability to tolerate labor.Data
Animal Data
Teratogenic effects (fetal limb reduction defects) were reported following oral administration of dichlorphenamide to pregnant rats during organogenesis at 350 mg/kg, or 17 times the maximum recommended human dose (200 mg/day) on a body surface area (mg/m2) basis. A no-effect dose for adverse effects on embryofetal development has not been established.8.2 Lactation
Risk Summary
There are no data on the presence of dichlorphenamide in human milk, the effects on the breastfed infant, or the effects on milk production.The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for KEVEYIS and any potential adverse effects on the breastfed infant from KEVEYIS or from the underlying maternal condition.
8.4 Pediatric Use
Safety and effectiveness of KEVEYIS in pediatric patients have not been established.
8.5 Geriatric Use
The risk of falls and of metabolic acidosis are greater in elderly patients [see Warnings and Precautions (5.4, 5.5)].
-
10 OVERDOSAGE
Symptoms of overdosage or toxicity may include drowsiness, anorexia, nausea, vomiting, dizziness, ataxia, tremor, and tinnitus.
In the event of overdosage, induce emesis or perform gastric lavage. The electrolyte disturbances most likely to be encountered from overdosage are hypokalemia and hyperchloremic metabolic acidosis.
-
11 DESCRIPTION
KEVEYIS tablets contain dichlorphenamide, an oral carbonic anhydrase inhibitor. Dichlorphenamide, a dichlorinated benzenedisulfonamide, is known chemically as 4, 5–dichloro-1,3-benzenedisulfonamide.
Its empirical formula is C6H6Cl2N2O4S2 and its structural formula is:
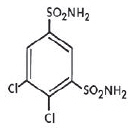
Dichlorphenamide USP is a white or practically white, crystalline compound with a molecular weight of 305.16. It is very slightly soluble in water but soluble in dilute solutions of sodium carbonate and sodium hydroxide. Dilute alkaline solutions of dichlorphenamide are stable at room temperature.
KEVEYIS (dichlorphenamide) tablets are supplied as tablets, for oral administration, each containing 50 mg dichlorphenamide. Inactive ingredients are lactose monohydrate, magnesium stearate and pregelatinized starch.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Dichlorphenamide is a carbonic anhydrase inhibitor. However, the precise mechanism by which dichlorphenamide exerts its therapeutic effects in patients with primary periodic paralysis is unknown.
12.2 Pharmacodynamics
KEVEYIS can cause metabolic acidosis, which can increase the risk of salicylate toxicity with coadministration [see Warnings and Precautions (5.2)]. KEVEYIS-induced metabolic acidosis can also increase in severity with coadministration of other drugs that cause metabolic acidosis [see Warnings and Precautions (5.4)].
12.3 Pharmacokinetics
After single-dose administration in healthy subjects in fasted state, dichlorphenamide Cmax and AUC increased in a dose-proportional manner within the range of 25 mg to 400 mg (2 times the maximum recommended dose). The steady-state is expected to be achieved within 10 days of twice-daily dosing.
Absorption
The median time to reach maximum concentration (Tmax) of dichlorphenamide was about 1.5 to 3 hours postdose after both single and multiple dose administrations.Distribution
The plasma protein binding of dichlorphenamide is approximately 88%.Elimination
Following a single-dose administration, mean terminal half-life was in the range of 32 to 66 hours.Metabolism
Dichlorphenamide is not a substrate for CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 and CYP3A4 isoforms when tested in vitro.Drug Interaction Studies
In vitro Assessment of Drug Interactions
Drug-Metabolizing Enzyme Inhibition
Dichlorphenamide is not an inhibitor for CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6, or 3A4 enzymes when tested in vitro.Drug-Metabolizing Enzyme Induction
Dichlorphenamide is not an inducer for CYP1A2, 2B6, or 3A4 enzymes when tested in vitro.In vitro Assessment of Transporter-Drug Interactions
Dichlorphenamide is neither a substrate nor inhibitor for p-gp, BCRP, OATP1B1, OATP1B3, OAT2, OAT4, OCT1, OCT2, MATE1, or MATE2-K when tested in vitro.Dichlorphenamide is not an inhibitor of OAT3, but is an inhibitor of OAT1 based on in vitro studies [see Drug Interactions (7.4)].
Dichlorphenamide is a substrate for transporters OAT1 and OAT3 based on in vitro studies [see Drug Interactions (7.2)].
In Vivo Drug Interactions
The use of dichlorphenamide in combination with high-dose aspirin is contraindicated as it may lead to salicylate toxicity. The mechanism(s) of this interaction is not known.Specific Populations
Geriatrics
The pharmacokinetics of dichlorphenamide in the elderly has not been determined. - 13 NONCLINICAL TOXICOLOGY
-
14 CLINICAL STUDIES
The efficacy of KEVEYIS was evaluated in two clinical studies, Study 1 and Study 2.
Study 1
Study 1 was a 9-week, double blind, placebo-controlled multi-center study. Study 1 consisted of two substudies: a substudy in patients with hypokalemic periodic paralysis (n=44), and a substudy in patients with hyperkalemic periodic paralysis (n=21). The primary efficacy endpoint in both substudies was the average number of self-reported attacks of muscle weakness per week over the final 8 weeks of the trial. Withdrawal from the study for acute severe worsening (increase in attack frequency or severity) was also assessed as an endpoint.
In Study 1, the dose of KEVEYIS was 50 mg b.i.d. for treatment-naïve patients. Patients already on dichlorphenamide prior to the study continued on the same dose while on KEVEYIS during the study. In patients taking acetazolamide prior to the study, the dose of KEVEYIS was set at 20% of the acetazolamide dose. Dose reduction for tolerability was permitted.
Hypokalemic Periodic Paralysis Substudy of Study 1
In the hypokalemic periodic paralysis substudy, median age of patients was 45 years and 73% of patients were male. Patients treated with KEVEYIS (n=24) had 2.2 fewer attacks per week than patients (n=20) treated with placebo (p=0.02). None of the patients randomized to KEVEYIS reached the endpoint of withdrawal from the study for acute worsening, vs. five patients randomized to placebo. The mean dose of KEVEYIS at Week 9 was 94 mg/day.
Hyperkalemic Periodic Paralysis Substudy of Study 1
In the Hyperkalemic Periodic Paralysis substudy, median age of patients was 43 years and 43% of patients were male. During the double-blind treatment period, patients treated with KEVEYIS (n=12) had 3.9 fewer attacks per week than patients (n=9) treated with placebo (p=0.08). None of the patients randomized to KEVEYIS reached the endpoint of withdrawal from the study for acute worsening, vs. two patients randomized to placebo. The mean dose of KEVEYIS at Week 9 was 82 mg/day.
Study 2
Study 2 was a 35-week, double blind, placebo-controlled, multi-center, two-period crossover study. Study 2 also consisted of two substudies: a substudy in patients with hypokalemic periodic paralysis (n=42), and a substudy in patients with hyperkalemic periodic paralysis (n=31), including patients with Paramyotonia Congenita. The primary endpoint in the hypokalemic periodic paralysis substudy was the incidence of acute intolerable worsening (based on attack frequency or severity) necessitating withdrawal. The primary endpoint in the hyperkalemic periodic paralysis substudy was the average number of self-reported attacks of muscle weakness per week. Dosing was determined similarly to Study 1.
Hypokalemic Periodic Paralysis Substudy of Study 2
The hypokalemic periodic paralysis substudy included patients with a mean age of 38 years; 79% of patients were male. Acute intolerable worsening was observed in 2 patients on KEVEYIS vs. 11 patients on placebo (p=0.02). The mean dose of KEVEYIS at the end of the study was 96 mg/day.
Hyperkalemic Periodic Paralysis Substudy of Study 2
The hyperkalemic periodic paralysis substudy included patients with a mean age of 37 years; and 79% of patients were male. Patients treated had 2.3 fewer attacks per week on KEVEYIS than on placebo (p=0.006). The mean dose of KEVEYIS at the end of the study was 73 mg/day.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
Each KEVEYIS (dichlorphenamide) tablet, 50 mg is round, white, scored on one side, engraved with "D" above the score and "50" below the score. The other side is plain.
KEVEYIS (dichlorphenamide) tablets are supplied as follows:
Bottles of 100......................................NDC: 71090-001-01 -
17 PATIENT COUNSELING INFORMATION
Worsening of Symptoms
Advise patients to notify their physician if they experience acute worsening of symptoms of periodic paralysis.Hypersensitivity and Other Life-Threatening Reactions
Inform patients that hypersensitivity and immune mediated reactions can occur with KEVEYIS, and could be fatal. Advise patients to discontinue KEVEYIS and notify their healthcare provider immediately if they develop a rash or signs and symptoms of anaphylaxis or other life-threatening reactions [see Warnings and Precautions (5.1)].Drug Interactions
Instruct patients to notify their healthcare provider of all of the drugs and over-the-counter medications that they take and to not take aspirin or other salicylates without first discussing with their healthcare provider [see Drug Interactions (7.1, 7.2, 7.3, 7.4)].Metabolic Acidosis
Instruct patients to contact their healthcare provider immediately if they develop possible manifestations of metabolic acidosis (e.g., fast breathing, fatigue/tiredness, loss of appetite, or irregular heart beat or palpitations) [see Warnings and Precautions (5.4)].Falls
Inform patients that KEVEYIS can increase their risk of falls [see Warnings and Precautions (5.5)].Cognitive Impairment
Advise patients to notify their healthcare provider if they experience symptoms of cognitive impairment including confusion and memory lapse [see Adverse Reactions (6.1)].Driving and Operating Machinery
KEVEYIS may cause drowsiness/fatigue in some patients [see Adverse Reactions (6.1)]. Caution patients on the potential for impaired ability to drive and operate machinery. -
SPL UNCLASSIFIED SECTION
Manufactured by: Taro Pharmaceutical Industries Ltd., Haifa Bay, Israel 2624761
Distributed by: Strongbridge US Inc., Trevose, PA 19053KEVEYIS® is a registered trademark licensed exclusively in the US to Strongbridge Biopharma plc.
STRONGBRIDGE BIOPHARMA® is a registered trademark of the Strongbridge Biopharma plc. companies, which includes Strongbridge Dublin Limited and Strongbridge U.S. Inc.
21061-0117-0
STRONGBRIDGE BIOPHARMA
-
PRINCIPAL DISPLAY PANEL - 50 mg Tablet Bottle Label
NDC: 71090-001-01
100 TabletsKeveyis®
(dichlorphenamide)
Tablets 50 mgKeep this and all medications out
of the reach of children.
Rx only
-
INGREDIENTS AND APPEARANCE
KEVEYIS
dichlorphenamide tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 71090-001 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength Dichlorphenamide (UNII: VVJ6673MHY) (Dichlorphenamide - UNII:VVJ6673MHY) Dichlorphenamide 50 mg Inactive Ingredients Ingredient Name Strength lactose monohydrate (UNII: EWQ57Q8I5X) magnesium stearate (UNII: 70097M6I30) starch, corn (UNII: O8232NY3SJ) Product Characteristics Color WHITE Score 2 pieces Shape ROUND Size 6mm Flavor Imprint Code D;50 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 71090-001-01 100 in 1 BOTTLE; Type 0: Not a Combination Product 02/21/2017 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA011366 08/07/2015 Labeler - Strongbridge US Inc. (080121041)
Trademark Results [Keveyis]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 KEVEYIS 86724075 5034655 Live/Registered |
TARO PHARMACEUTICALS U.S.A., INC. 2015-08-13 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.