VUMERITY- diroximel fumarate capsule
Vumerity by
Drug Labeling and Warnings
Vumerity by is a Prescription medication manufactured, distributed, or labeled by Biogen Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use VUMERITY™ safely and effectively. See full prescribing information for VUMERITY.
VUMERITY™ (diroximel fumarate) delayed-release capsules, for oral use
Initial U.S. Approval: 2013INDICATIONS AND USAGE
VUMERITY is indicated for the treatment of relapsing forms of multiple sclerosis (MS), to include clinically isolated syndrome, relapsing-remitting disease, and active secondary progressive disease, in adults. (1)
DOSAGE AND ADMINISTRATION
- Blood tests are required prior to initiation of VUMERITY (2.1)
- Starting dose: 231 mg twice a day, orally, for 7 days (2.2)
- Maintenance dose after 7 days: 462 mg (administered as two 231 mg capsules) twice a day, orally (2.2)
- Swallow VUMERITY capsules whole and intact. Do not crush, chew, or sprinkle capsule contents on food (2.3)
- Avoid administration of VUMERITY with a high-fat, high-calorie meal/snack (2.3)
- Avoid co-administration of VUMERITY with alcohol (2.3)
DOSAGE FORMS AND STRENGTHS
Delayed-release capsules: 231 mg (3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- Anaphylaxis and Angioedema: Discontinue and do not restart VUMERITY if these occur. (5.1)
- Progressive Multifocal Leukoencephalopathy (PML): Withhold VUMERITY at the first sign or symptom suggestive of PML. (5.2)
- Lymphopenia: Obtain a CBC including lymphocyte count before initiating VUMERITY, after 6 months, and every 6 to 12 months thereafter. Consider interruption of VUMERITY if lymphocyte counts <0.5 × 109/L persist for more than six months. (5.3)
- Liver Injury: Obtain serum aminotransferase, alkaline phosphatase, and total bilirubin levels before initiating VUMERITY and during treatment, as clinically indicated. Discontinue VUMERITY if clinically significant liver injury induced by VUMERITY is suspected. (5.4)
ADVERSE REACTIONS
Most common adverse reactions (incidence for dimethyl fumarate [which has the same active metabolite as VUMERITY] ≥10% and ≥2% more than placebo) were flushing, abdominal pain, diarrhea, and nausea. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Biogen at 1-800-456-2255 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 10/2019
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Blood Tests Prior to Initiation of VUMERITY
2.2 Dosing Information
2.3 Administration Instructions
2.4 Blood Tests to Assess Safety After Initiation of VUMERITY
2.5 Patients With Renal Impairment
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Anaphylaxis and Angioedema
5.2 Progressive Multifocal Leukoencephalopathy
5.3 Lymphopenia
5.4 Liver Injury
5.5 Flushing
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Concomitant Dimethyl Fumarate
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
16 HOW SUPPLIED/ STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage and Handling
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Blood Tests Prior to Initiation of VUMERITY
Obtain the following prior to treatment with VUMERITY:
2.2 Dosing Information
The starting dosage for VUMERITY is 231 mg twice a day orally. After 7 days, the dosage should be increased to the maintenance dosage of 462 mg (administered as two 231 mg capsules) twice a day orally. Temporary dosage reductions to 231 mg twice a day may be considered for individuals who do not tolerate the maintenance dosage. Within 4 weeks, the recommended dosage of 462 mg twice a day should be resumed. Discontinuation of VUMERITY should be considered for patients unable to tolerate return to the maintenance dosage. Administration of non-enteric coated aspirin (up to a dose of 325 mg) 30 minutes prior to VUMERITY dosing may reduce the incidence or severity of flushing [see Clinical Pharmacology (12.3)].
2.3 Administration Instructions
Swallow VUMERITY capsules whole and intact. Do not crush or chew, or sprinkle the capsule contents on food.
If taken with food, avoid a high-fat, high-calorie meal/snack; the meal/snack should contain no more than 700 calories and no more than 30 g fat [see Warnings and Precautions (5.5) and Clinical Pharmacology (12.3)].
Avoid co-administration of VUMERITY with alcohol [see Clinical Pharmacology (12.3)].
2.4 Blood Tests to Assess Safety After Initiation of VUMERITY
Obtain a complete blood cell count (CBC), including lymphocyte count, 6 months after initiation of VUMERITY and then every 6 to 12 months thereafter, as clinically indicated [see Warnings and Precautions (5.3)].
Obtain serum aminotransferase, alkaline phosphatase, and total bilirubin levels during treatment with VUMERITY, as clinically indicated [see Warnings and Precautions (5.4)].
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Anaphylaxis and Angioedema
VUMERITY can cause anaphylaxis and angioedema after the first dose or at any time during treatment. Signs and symptoms in patients taking dimethyl fumarate (which has the same active metabolite as VUMERITY) have included difficulty breathing, urticaria, and swelling of the throat and tongue. Patients should be instructed to discontinue VUMERITY and seek immediate medical care should they experience signs and symptoms of anaphylaxis or angioedema.
5.2 Progressive Multifocal Leukoencephalopathy
Progressive multifocal leukoencephalopathy (PML) has occurred in patients with MS treated with dimethyl fumarate (which has the same active metabolite as VUMERITY). PML is an opportunistic viral infection of the brain caused by the JC virus (JCV) that typically only occurs in patients who are immunocompromised, and that usually leads to death or severe disability. A fatal case of PML occurred in a patient who received dimethyl fumarate for 4 years while enrolled in a clinical trial. During the clinical trial, the patient experienced prolonged lymphopenia (lymphocyte counts predominantly <0.5 × 109/L for 3.5 years) while taking dimethyl fumarate [see Warnings and Precautions (5.3)]. The patient had no other identified systemic medical conditions resulting in compromised immune system function and had not previously been treated with natalizumab, which has a known association with PML. The patient was also not taking any immunosuppressive or immunomodulatory medications concomitantly.
PML has occurred in patients taking dimethyl fumarate in the postmarketing setting in the presence of lymphopenia (<0.8 × 109/L) persisting for more than 6 months. While the role of lymphopenia in these cases is uncertain, the majority of cases occurred in patients with lymphocyte counts <0.5×109/L.
At the first sign or symptom suggestive of PML, withhold VUMERITY and perform an appropriate diagnostic evaluation. Typical symptoms associated with PML are diverse, progress over days to weeks, and include progressive weakness on one side of the body or clumsiness of limbs, disturbance of vision, and changes in thinking, memory, and orientation leading to confusion and personality changes.
Magnetic resonance imaging (MRI) findings may be apparent before clinical signs or symptoms. Cases of PML diagnosed based on MRI findings and the detection of JCV DNA in the cerebrospinal fluid in the absence of clinical signs or symptoms specific to PML, have been reported in patients treated with other MS medications associated with PML. Many of these patients subsequently became symptomatic with PML. Therefore, monitoring with MRI for signs that may be consistent with PML may be useful, and any suspicious findings should lead to further investigation to allow for an early diagnosis of PML, if present. Lower PML-related mortality and morbidity have been reported following discontinuation of another MS medication associated with PML in patients with PML who were initially asymptomatic compared to patients with PML who had characteristic clinical signs and symptoms at diagnosis. It is not known whether these differences are due to early detection and discontinuation of MS treatment or due to differences in disease in these patients.
5.3 Lymphopenia
VUMERITY may decrease lymphocyte counts. In the MS placebo-controlled trials with dimethyl fumarate (which has the same active metabolite as VUMERITY), mean lymphocyte counts decreased by approximately 30% during the first year of treatment with dimethyl fumarate and then remained stable. Four weeks after stopping dimethyl fumarate, mean lymphocyte counts increased but did not return to baseline. Six percent (6%) of dimethyl fumarate patients and <1% of placebo patients experienced lymphocyte counts <0.5 × 109/L (lower limit of normal 0.91 × 109/L). The incidence of infections (60% vs 58%) and serious infections (2% vs 2%) was similar in patients treated with dimethyl fumarate or placebo, respectively. There was no increased incidence of serious infections observed in patients with lymphocyte counts <0.8 × 109/L or ≤0.5 × 109/L in controlled trials, although one patient in an extension study developed PML in the setting of prolonged lymphopenia (lymphocyte counts predominantly <0.5 × 109/L for 3.5 years) [see Warnings and Precautions (5.2)].
In controlled and uncontrolled clinical trials with dimethyl fumarate, 2% of patients experienced lymphocyte counts <0.5 × 109/L for at least six months, and in this group the majority of lymphocyte counts remained <0.5 × 109/L with continued therapy. Neither VUMERITY nor dimethyl fumarate have been studied in patients with preexisting low lymphocyte counts.
Obtain a complete blood count (CBC), including lymphocyte count, before initiating treatment with VUMERITY, 6 months after starting treatment, and then every 6 to 12 months thereafter, and as clinically indicated. Consider interruption of VUMERITY in patients with lymphocyte counts less than 0.5 × 109/L persisting for more than six months. Given the potential for delayed recovery of lymphocyte counts, continue to obtain lymphocyte counts until their recovery if VUMERITY is discontinued or interrupted because of lymphopenia. Consider withholding treatment from patients with serious infections until resolution. Decisions about whether or not to restart VUMERITY should be individualized based on clinical circumstances.
5.4 Liver Injury
Clinically significant cases of liver injury have been reported in patients treated with dimethyl fumarate (which has the same active metabolite as VUMERITY) in the postmarketing setting. The onset has ranged from a few days to several months after initiation of treatment with dimethyl fumarate. Signs and symptoms of liver injury, including elevation of serum aminotransferases to greater than 5-fold the upper limit of normal and elevation of total bilirubin to greater than 2-fold the upper limit of normal have been observed. These abnormalities resolved upon treatment discontinuation. Some cases required hospitalization. None of the reported cases resulted in liver failure, liver transplant, or death. However, the combination of new serum aminotransferase elevations with increased levels of bilirubin caused by drug-induced hepatocellular injury is an important predictor of serious liver injury that may lead to acute liver failure, liver transplant, or death in some patients.
Elevations of hepatic transaminases (most no greater than 3 times the upper limit of normal) were observed during controlled trials with dimethyl fumarate [see Adverse Reactions (6.1)].
Obtain serum aminotransferase, alkaline phosphatase (ALP), and total bilirubin levels prior to treatment with VUMERITY and during treatment, as clinically indicated. Discontinue VUMERITY if clinically significant liver injury induced by VUMERITY is suspected.
5.5 Flushing
VUMERITY may cause flushing (e.g., warmth, redness, itching, and/or burning sensation). In clinical trials of dimethyl fumarate (which has the same active metabolite as VUMERITY), 40% of dimethyl fumarate-treated patients experienced flushing. Flushing symptoms generally began soon after initiating dimethyl fumarate and usually improved or resolved over time. In the majority of patients who experienced flushing, it was mild or moderate in severity. Three percent (3%) of patients discontinued dimethyl fumarate for flushing and <1% had serious flushing symptoms that were not life-threatening but led to hospitalization.
Administration of VUMERITY with food may reduce the incidence of flushing [see Dosage and Administration (2.3)]. Studies with dimethyl fumarate show that administration of non-enteric coated aspirin (up to a dose of 325 mg) 30 minutes prior to dosing may reduce the incidence or severity of flushing [see Clinical Pharmacology (12.3)].
-
6 ADVERSE REACTIONS
The following important adverse reactions are described elsewhere in labeling:
- Anaphylaxis and Angioedema [see Warnings and Precautions (5.1)].
- Progressive Multifocal Leukoencephalopathy [see Warnings and Precautions Section (5.2)].
- Lymphopenia [see Warnings and Precautions (5.3)].
- Liver Injury [see Warnings and Precautions (5.4)].
- Flushing [see Warnings and Precautions (5.5)].
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
The data described in the following sections were obtained using dimethyl fumarate delayed-release capsules, which has the same active metabolite as VUMERITY.
Adverse Reactions in Placebo-Controlled Trials with Dimethyl Fumarate
In the two well-controlled studies demonstrating effectiveness, 1529 patients received dimethyl fumarate with an overall exposure of 2244 person-years [see Clinical Studies (14)].
The adverse reactions presented in Table 1 below are based on safety information from 769 patients treated with dimethyl fumarate 240 mg twice a day and 771 placebo-treated patients. The most common adverse reactions (incidence ≥10% and ≥2% more than placebo) for dimethyl fumarate were flushing, abdominal pain, diarrhea, and nausea.
Table 1: Adverse Reactions in Study 1 and 2 Reported for Dimethyl Fumarate at ≥2% Higher Incidence than Placebo Adverse Reactions Dimethyl Fumarate
240 mg Twice Daily
(N=769)
%Placebo
(N=771)
%Flushing 40 6 Abdominal pain 18 10 Diarrhea 14 11 Nausea 12 9 Vomiting 9 5 Pruritus 8 4 Rash 8 3 Albumin urine present 6 4 Erythema 5 1 Dyspepsia 5 3 Aspartate aminotransferase increased 4 2 Lymphopenia 2 <1 Gastrointestinal
Dimethyl fumarate caused GI events (e.g., nausea, vomiting, diarrhea, abdominal pain, and dyspepsia). The incidence of GI events was higher early in the course of treatment (primarily in month 1) and usually decreased over time in patients treated with dimethyl fumarate compared with placebo. Four percent (4%) of patients treated with dimethyl fumarate and less than 1% of placebo patients discontinued due to gastrointestinal events. The incidence of serious GI events was 1% in patients treated with dimethyl fumarate.
Hepatic Transaminases
An increased incidence of elevations of hepatic transaminases in patients treated with dimethyl fumarate was seen primarily during the first six months of treatment, and most patients with elevations had levels <3 times the upper limit of normal (ULN) during controlled trials. Elevations of alanine aminotransferase and aspartate aminotransferase to ≥3 times the ULN occurred in a small number of patients treated with both dimethyl fumarate and placebo and were balanced between groups. There were no elevations in transaminases ≥3 times the ULN with concomitant elevations in total bilirubin >2 times the ULN. Discontinuations due to elevated hepatic transaminases were <1% and were similar in patients treated with dimethyl fumarate or placebo.
Eosinophilia
A transient increase in mean eosinophil counts was seen during the first 2 months of therapy.
Adverse Reactions in Clinical Studies with VUMERITY
In clinical studies assessing safety in patients with RRMS, approximately 700 patients were treated with VUMERITY and approximately 490 patients received more than 1 year of treatment with VUMERITY. The adverse reaction profile of VUMERITY was consistent with the experience in the placebo-controlled clinical trials with dimethyl fumarate.
6.2 Postmarketing Experience
The following adverse reaction has been identified during post approval use of dimethyl fumarate. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Liver function abnormalities (elevations in transaminases ≥3 times ULN with concomitant elevations in total bilirubin >2 times ULN) have been reported following dimethyl fumarate administration in post marketing experience [see Warnings and Precautions (5.4)].
-
7 DRUG INTERACTIONS
7.1 Concomitant Dimethyl Fumarate
VUMERITY is contraindicated in patients currently taking dimethyl fumarate, which is also metabolized to monomethyl fumarate. VUMERITY may be initiated the day following discontinuation of dimethyl fumarate [see Contraindications (4)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no adequate data on the developmental risk associated with the use of VUMERITY or dimethyl fumarate (which has the same active metabolite as VUMERITY) in pregnant women. In animal studies, administration of diroximel fumarate during pregnancy or throughout pregnancy and lactation resulted in adverse effects on embryofetal and offspring development (increased incidences of skeletal abnormalities, increased mortality, decreased body weights, neurobehavioral impairment) at clinically relevant drug exposures [see Data].
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively. The background risk of major birth defects and miscarriage for the indicated population is unknown.
Data
Animal Data
Oral administration of diroximel fumarate (0, 40, 100, or 400 mg/kg/day) to pregnant rats throughout organogenesis resulted in a decrease in fetal body weight and an increase in fetal skeletal variations at the highest dose tested, which was associated with maternal toxicity. Plasma exposures (AUC) for MMF and HES (the major circulating drug-related compound in humans) at the no-effect dose (100 mg/kg/day) for adverse effects on embryofetal development were approximately 2 times those in humans at the recommended human dose (RHD) of 924 mg/day.
Oral administration of diroximel fumarate (0, 50, 150, or 350 mg/kg/day) to pregnant rabbits throughout organogenesis resulted in an increase in fetal skeletal malformations at the mid and high doses and reduced fetal body weight and increases in embryofetal death and fetal skeletal variations at the highest dose tested. The high dose was associated with maternal toxicity. Plasma exposures (AUC) for MMF and HES at the no-effect dose (50 mg/kg/day) for adverse effects on embryofetal development were similar to (MMF) or less than (HES) those in humans at the RHD.
Oral administration of diroximel fumarate (0, 40, 100, or 400 mg/kg/day) to rats throughout gestation and lactation resulted in reduced weight, which persisted into adulthood, and adverse effects on neurobehavioral function in offspring at the highest dose tested. Plasma exposures (AUC) for MMF and HES at the no-effect dose for adverse effects on postnatal development (100 mg/kg/day) were approximately 3 times (MMF) or similar to (HES) those in humans at the RHD.
8.2 Lactation
Risk Summary
There are no data on the presence of diroximel fumarate or metabolites (MMF, HES) in human milk. The effects on the breastfed infant and on milk production are unknown.
The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for VUMERITY and any potential adverse effects on the breastfed infant from the drug or from the underlying maternal condition.
8.5 Geriatric Use
Clinical studies of dimethyl fumarate and VUMERITY did not include sufficient numbers of patients aged 65 years and over to determine whether they respond differently from younger patients.
8.6 Renal Impairment
No dosage adjustment is necessary in patients with mild renal impairment. Because of an increase in the exposure of a major metabolite [2-hydroxyethyl succinimide (HES)], use of VUMERITY is not recommended in patients with moderate or severe renal impairment [see Clinical Pharmacology (12.3)].
-
11 DESCRIPTION
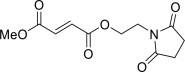
VUMERITY contains diroximel fumarate. The chemical name of diroximel fumarate is 2-Butenedioic acid (2E)-, 1-[2-(2,5-dioxo-1-pyrrolidinyl)ethyl] 4-methyl ester, which has a molecular formula of C11H13NO6 and molecular weight of 255.22. Diroximel fumarate has the following structure:
Diroximel fumarate is a white to off-white powder that is slightly soluble in water.
VUMERITY is provided as delayed-release capsules for oral administration. Each capsule contains 231 mg of diroximel fumarate and the following inactive ingredients: crospovidone, colloidal silicon dioxide, magnesium stearate (non-bovine), methacrylic acid and ethyl acrylate copolymer, microcrystalline cellulose, talc, and triethyl citrate.
The capsule shell contains carrageenan, hypromellose, potassium chloride, and titanium dioxide. It is printed with black ink that contains iron oxide, potassium hydroxide, propylene glycol, and shellac.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
The mechanism by which diroximel fumarate exerts its therapeutic effect in multiple sclerosis is unknown. MMF, the active metabolite of diroximel fumarate, has been shown to activate the nuclear factor (erythroid-derived 2)-like 2 (Nrf2) pathway in vitro and in vivo in animals and humans. The Nrf2 pathway is involved in the cellular response to oxidative stress. MMF has been identified as a nicotinic acid receptor agonist in vitro.
12.3 Pharmacokinetics
After oral administration of VUMERITY, diroximel fumarate undergoes rapid presystemic hydrolysis by esterases and is converted to its active metabolite, monomethyl fumarate (MMF). Diroximel fumarate is not quantifiable in plasma following oral administration of VUMERITY. Therefore, all pharmacokinetic analyses related to VUMERITY were performed with plasma MMF concentrations. Pharmacokinetic data were obtained in subjects with relapsing forms of multiple sclerosis (MS) and healthy volunteers.
Absorption
Following oral administration of VUMERITY, the median tmax of MMF is 2.5 to 3 hours. The peak plasma concentration (Cmax) and overall exposure (AUC) increased dose proportionally in the recommended daily dose range (462 mg to 924 mg). Following administration of VUMERITY 462 mg in patients with MS, the mean Cmax of MMF was 2.11 mg/L. The mean steady state AUC of MMF was estimated to be 8.32 mg.hr/L following twice daily dosing in patients with MS.
Effect of Food
In an open-label, randomized, balanced, crossover study, co-administration of VUMERITY with a high-fat, high-calorie meal (900-1000 calories, 50% of calories from fat) did not affect the AUC of MMF, but resulted in an approximately 44% reduction in Cmax compared to fasted state [see Dosage and Administration (2.3)]. The MMF Cmax with low-fat, low-calorie (350 to 400 calories, 10 to 15 g fat) and medium-fat, medium-calorie (650 to 700 calories, 25 to 30 g fat) meals was reduced by approximately 12% and 25%, respectively.
Relative to fasted state, the tmax of MMF was delayed from 2.5 hours (fasted state) to 4.5 hours (low-fat, low-calorie meal or a medium-fat, medium-calorie meal) and 7.0 hours (high-fat, high-calorie meal) in the fed state. There was no impact of low, medium, or high-fat meals on the AUC of MMF after administration of VUMERITY.
Distribution
The apparent volume of distribution for MMF is between 72 L and 83 L in healthy subjects after administration of VUMERITY. Human plasma protein binding of MMF is 27-45% and independent of concentration.
Elimination
Metabolism
In humans, diroximel fumarate is extensively metabolized by esterases, which are ubiquitous in the gastrointestinal tract, blood, and tissues, to the major active metabolite, MMF, before it reaches the systemic circulation. Further metabolism of MMF occurs through the tricarboxylic acid (TCA) cycle, with no involvement of the cytochrome P450 (CYP) system. Fumaric and citric acid, and glucose are the major metabolites of MMF in plasma.
Esterase metabolism of diroximel fumarate also produces 2-hydroxyethyl succinimide (HES), an inactive major metabolite.
Excretion
MMF is mainly eliminated as carbon dioxide in the expired air with only trace amounts (less than 0.3% of the total dose) recovered in urine.
The terminal half-life of MMF is approximately 1 hour, and accumulation of MMF does not occur with multiple doses of VUMERITY.
HES is mainly eliminated in urine (58-63% of the dose was excreted as HES in urine).
Specific Populations
Age (18-79 years), sex, and race (White, African American, and Asian) did not have clinically meaningful effects on the pharmacokinetics of MMF after administration of VUMERITY. The effect of hepatic impairment or severe renal impairment (CrCl <30 mL/min, Cockcroft-Gault) requiring hemodialysis on MMF pharmacokinetics is unknown.
Patients with Renal Impairment
A single-dose clinical study investigating the effect of renal impairment on the pharmacokinetics of diroximel fumarate and its metabolites MMF and HES was conducted. The study included cohorts with mild, moderate, and severe renal impairment and a healthy cohort (8 subjects per cohort) and found no clinically relevant changes in MMF exposure. However, HES exposure increased by 1.3, 1.8, and 2.7-fold with mild, moderate, and severe renal impairment, respectively, compared to the healthy cohort [see Use in Specific Populations (8.6)]. There are no data available on long-term use of VUMERITY in patients with moderate or severe renal impairment.
Drug Interaction Studies
Diroximel fumarate metabolism does not involve CYP enzymes, therefore, no clinically meaningful interactions are expected when administered with CYP inhibitors or inducers.
In vitro studies found diroximel fumarate and its metabolites did not inhibit CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4/5 enzymes in human liver microsomes or induce CYP1A2, 2B6, or 3A4/5 in cultured human hepatocytes.
Diroximel fumarate did not inhibit P-gp in vivo. The major circulating inactive metabolite of diroximel fumarate, HES, did not inhibit P-gp and was neither a substrate nor an inhibitor of BCRP, MATE1, MATE2-K, OAT1, OAT3, or OCT2.
Aspirin, when administered approximately 30 minutes before dimethyl fumarate, did not alter the pharmacokinetics of MMF.
Oral Contraceptives
In a study conducted with dimethyl fumarate, no relevant effects of MMF on oral contraceptive exposure were observed when administered with a combined oral contraceptive (norelgestromin and ethinyl estradiol). No interaction studies have been performed with oral contraceptives containing other progestogens.
Alcohol
Administration of VUMERITY at the same time with 5% v/v and 40% v/v ethanol did not alter total MMF exposure relative to administration with water, demonstrating that the coingestion of ethanol does not induce dose dumping. The mean peak plasma MMF concentration for diroximel fumarate was decreased by 9% and 21%, when co-administered with 240 mL of 5% v/v and 40% v/v of ethanol, respectively [see Dosage and Administration (2.3)].
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Oral administration of diroximel fumarate (0, 0, 30, 100, 300 or 1000 [females only] mg/kg/day) for 26 weeks to Tg.rasH2 mice resulted in no drug-related tumors.
Oral administration of diroximel fumarate (0, 0, 15, 50, or 150 mg/kg/day) to male and female rats resulted in an increase in tumors (Leydig cell adenomas of the testes) in males at the highest dose tested. At the higher dose (50 mg/kg/day) not associated with drug-related tumors, plasma exposures for MMF and HES (the major circulating drug-related compound in humans) were similar to (MMF) and less than (HES) those in humans at the recommended human dose (RHD) of 924 mg/day.
Mutagenesis
Diroximel fumarate was negative in the in vitro bacterial reverse mutation and in vivo rat micronucleus and comet assays. Diroximel fumarate was clastogenic (in the absence and presence of metabolic activation) in the in vitro chromosomal aberration assay in human peripheral blood lymphocytes.
Impairment of Fertility
No adverse effects on fertility were observed following oral administration of diroximel fumarate to male rats (0, 40, 120, or 400 mg/kg/day) prior to and during mating with untreated females and, in a separate study, to female rats (0, 40, 120, or 450 mg/kg/day) prior to and during mating with untreated males and continuing to Gestation Day (GD) 7. At the highest doses tested, plasma exposures (AUC) for MMF were approximately 7-9 times that in humans at the RHD. Plasma levels of HES were not quantitated.
13.2 Animal Toxicology and/or Pharmacology
Kidney toxicity, including tubular changes (degeneration, necrosis, regeneration, hypertrophy) and/or interstitial fibrosis, were observed following oral administration of diroximel fumarate in rats and monkeys. In the chronic toxicology study in rats (0, 50, 100, or 300 mg/kg/day), adverse renal findings occurred at all doses tested. Plasma exposures (AUC) at the low dose (50 mg/kg/day) were similar to (MMF) or less than (HES) those in humans at the RHD. In the chronic toxicology study in monkeys (0, 15, 50, or 150 mg/kg/day), adverse renal findings occurred at all but the lowest dose tested (15 mg/kg/day), which was associated with plasma MMF and HES exposures (AUC) less than those in humans at the RHD.
-
14 CLINICAL STUDIES
The efficacy of VUMERITY is based upon bioavailability studies in patients with relapsing forms of multiple sclerosis and healthy subjects comparing oral dimethyl fumarate delayed-release capsules to VUMERITY delayed-release capsules [see Clinical Pharmacology (12.3)].
The clinical studies described below were conducted using dimethyl fumarate.
The efficacy and safety of dimethyl fumarate were demonstrated in two studies (Studies 1 and 2) that evaluated dimethyl fumarate taken either twice or three times a day in patients with relapsing-remitting multiple sclerosis (RRMS). The starting dose for dimethyl fumarate was 120 mg twice or three times a day for the first 7 days, followed by an increase to 240 mg twice or three times a day. Both studies included patients who had experienced at least 1 relapse over the year preceding the trial or had a brain Magnetic Resonance Imaging (MRI) scan demonstrating at least one gadolinium-enhancing (Gd+) lesion within 6 weeks of randomization. The Expanded Disability Status Scale (EDSS) was also assessed and patients could have scores ranging from 0 to 5. Neurological evaluations were performed at baseline, every 3 months, and at the time of suspected relapse. MRI evaluations were performed at baseline, month 6, and year 1 and 2 in a subset of patients (44% in Study 1 and 48% in Study 2).
Study 1: Placebo-Controlled Trial in RRMS
Study 1 was a 2-year randomized, double-blind, placebo-controlled study in 1234 patients with RRMS. The primary endpoint was the proportion of patients relapsed at 2 years. Additional endpoints at 2 years included the number of new or newly enlarging T2 hyperintense lesions, number of new T1 hypointense lesions, number of Gd+ lesions, annualized relapse rate (ARR), and time to confirmed disability progression. Confirmed disability progression was defined as at least a 1 point increase from baseline EDSS (1.5 point increase for patients with baseline EDSS of 0) sustained for 12 weeks.
Patients were randomized to receive dimethyl fumarate 240 mg twice a day (n=410), dimethyl fumarate 240 mg three times a day (n=416), or placebo (n=408) for up to 2 years. The median age was 39 years, median time since diagnosis was 4 years, and median EDSS score at baseline was 2. The median time on study drug for all treatment arms was 96 weeks. The percentages of patients who completed 96 weeks on study drug per treatment group were 69% for patients assigned to dimethyl fumarate 240 mg twice a day, 69% for patients assigned to dimethyl fumarate 240 mg three times a day, and 65% for patients assigned to placebo groups.
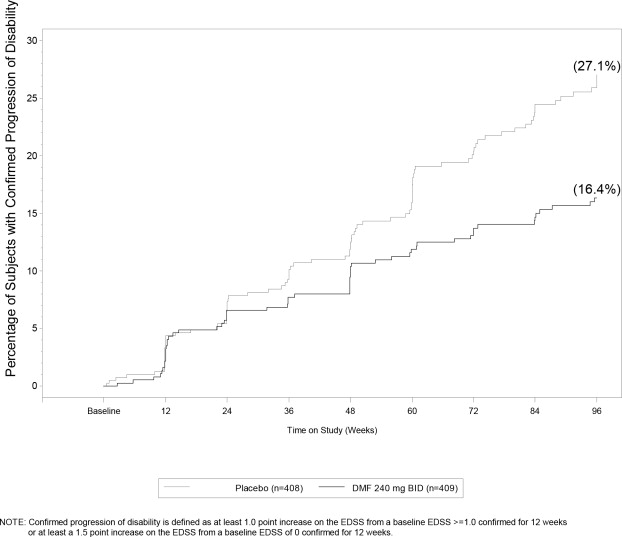
Dimethyl fumarate had a statistically significant effect on all of the endpoints described above and the 240 mg three times daily dose showed no additional benefit over the dimethyl fumarate 240 mg twice daily dose. The results for this study (240 mg twice a day vs. placebo) are shown in Table 2 and Figure 1.
Table 2: Clinical and MRI Results of Study 1 Dimethyl Fumarate 240 mg BID Placebo P-value Clinical Endpoints N=410 N=408 Proportion relapsing (primary endpoint) 27% 46% <0.0001 Relative risk reduction 49% Annualized relapse rate 0.172 0.364 <0.0001 Relative reduction 53% Proportion with disability progression 16% 27% 0.0050 Relative risk reduction 38% MRI Endpoints N=152 N=165 Mean number of new or newly enlarging T2 lesions over 2 years 2.6 17 <0.0001 Percentage of subjects with no new or newly enlarging lesions 45% 27% Number of Gd+ lesions at 2 years Mean (median) 0.1 (0) 1.8 (0) Percentage of subjects with 0 lesions 93% 62% 1 lesion 5% 10% 2 lesions <1% 8% 3 to 4 lesions 0 9% 5 or more lesions <1% 11% Relative odds reduction (percentage) 90% <0.0001 Mean number of new T1 hypointense lesions over 2 years 1.5 5.6 <0.0001 Figure 1: Time to 12-Week Confirmed Progression of Disability (Study 1)
Study 2: Placebo-Controlled Trial in RRMS
Study 2 was a 2-year multicenter, randomized, double-blind, placebo-controlled study that also included an open-label comparator arm in patients with RRMS. The primary endpoint was the annualized relapse rate at 2 years. Additional endpoints at 2 years included the number of new or newly enlarging T2 hyperintense lesions, number of T1 hypointense lesions, number of Gd+ lesions, proportion of patients relapsed, and time to confirmed disability progression as defined in Study 1.
Patients were randomized to receive dimethyl fumarate 240 mg twice a day (n=359), dimethyl fumarate 240 mg three times a day (n=345), an open-label comparator (n=350), or placebo (n=363) for up to 2 years. The median age was 37 years, median time since diagnosis was 3 years, and median EDSS score at baseline was 2.5. The median time on study drug for all treatment arms was 96 weeks. The percentages of patients who completed 96 weeks on study drug per treatment group were 70% for patients assigned to dimethyl fumarate 240 mg twice a day, 72% for patients assigned to dimethyl fumarate 240 mg three times a day, and 64% for patients assigned to placebo groups.
Dimethyl fumarate had a statistically significant effect on the relapse and MRI endpoints described above. There was no statistically significant effect on disability progression. The dimethyl fumarate 240 mg three times daily dose resulted in no additional benefit over the dimethyl fumarate 240 mg twice daily dose. The results for this study (240 mg twice a day vs. placebo) are shown in Table 3.
Table 3: Clinical and MRI Results of Study 2 Dimethyl Fumarate 240 mg BID Placebo P-value Clinical Endpoints N=359 N=363 Annualized relapse rate 0.224 0.401 <0.0001 Relative reduction 44% Proportion relapsing 29% 41% 0.0020 Relative risk reduction 34% Proportion with disability progression 13% 17% 0.25 Relative risk reduction 21% MRI Endpoints N=147 N=144 Mean number of new or newly enlarging T2 lesions over 2 years 5.1 17.4 <0.0001 Percentage of subjects with no new or newly enlarging lesions 27% 12% Number of Gd+ lesions at 2 years Mean (median) 0.5 (0.0) 2.0 (0.0) Percentage of subjects with 0 lesions 80% 61% 1 lesion 11% 17% 2 lesions 3% 6% 3 to 4 lesions 3% 2% 5 or more lesions 3% 14% Relative odds reduction (percentage) 74% <0.0001 Mean number of new T1 hypointense lesions over 2 years 3.0 7.0 <0.0001 -
16 HOW SUPPLIED/ STORAGE AND HANDLING
16.1 How Supplied
VUMERITY is available as delayed-release capsules for oral administration, containing 231 mg of diroximel fumarate. The 231 mg capsules have a white cap and a white body, printed with “DRF 231 mg” in black ink on the body. VUMERITY is available as follows:
30-day Starter dose bottle (bottle of 106 capsules), NDC: 64406-020-01.
30-day Maintenance dose bottle (bottle of 120 capsules), NDC: 64406-020-03.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Dosage and Administration
Inform patients that they will be provided a starter dose bottle: one capsule twice a day for the first 7 days and then two capsules twice a day thereafter. Advise patients to take VUMERITY as instructed. Inform patients to swallow VUMERITY capsules whole and intact. Inform patients to not crush, chew, or sprinkle capsule contents on food. Inform patients that they should avoid a high-fat, high-calorie meal/snack at the time they take VUMERITY. If taken with food, the meal/snack should contain no more than 700 calories and no more than 30 g fat. Advise patients to avoid co-administration of VUMERITY with alcohol [see Dosage and Administration (2.2)].
Anaphylaxis and Angioedema
Advise patients to discontinue VUMERITY and seek medical care if they develop signs and symptoms of anaphylaxis or angioedema [see Warnings and Precautions (5.1)].
Progressive Multifocal Leukoencephalopathy
Inform patients that progressive multifocal leukoencephalopathy (PML) has occured in patients who received dimethyl fumarate, and therefore may occur with VUMERITY. Inform the patient that PML is characterized by a progression of deficits and usually leads to death or severe disability over weeks or months. Inform the patient of the importance of contacting their healthcare provider if they develop any symptoms suggestive of PML. Inform the patient that typical symptoms associated with PML are diverse, progress over days to weeks, and include progressive weakness on one side of the body or clumsiness of limbs, disturbance of vision, and changes in thinking, memory, and orientation leading to confusion and personality changes [see Warnings and Precautions (5.2)].
Lymphocyte Counts
Inform patients that VUMERITY may decrease lymphocyte counts. A blood test should be obtained before they start therapy. Blood tests are also recommended after 6 months of treatment, every 6 to 12 months thereafter, and as clinically indicated [see Warnings and Precautions (5.3) and Adverse Reactions (6.1)].
Liver Injury
Inform patients that VUMERITY may cause liver injury. Instruct patients treated with VUMERITY to report promptly to their healthcare provider any symptoms that may indicate liver injury, including fatigue, anorexia, right upper abdominal discomfort, dark urine, or jaundice. A blood test should be obtained before patients start therapy and during treatment, as clinically indicated [see Warnings and Precautions (5.4)].
Flushing and Gastrointestinal (GI) Reactions
Flushing and GI reactions (abdominal pain, diarrhea, and nausea) are the most common reactions, especially at the initiation of therapy, and may decrease over time. Advise patients to contact their healthcare provider if they experience persistent and/or severe flushing or GI reactions. Advise patients experiencing flushing that taking VUMERITY with food (avoid high-fat, high-calorie meal or snack) or taking a non-enteric coated aspirin prior to taking VUMERITY may help [see Dosage and Administration (2.2) and Adverse Reactions (6.1)].
Pregnancy
Instruct patients that if they are pregnant or plan to become pregnant while taking VUMERITY they should inform their healthcare provider [see Use in Specific Populations (8.1)].
54499-01
Manufactured for:
Biogen Inc.
Cambridge, MA 02142The VUMERITY trademark is used by Biogen under an exclusive license.
© Biogen 2019 -
PATIENT PACKAGE INSERT
This Patient Information has been approved by the U.S. Food and Drug Administration
Revised: 10/2019
Patient Information
VUMERITY (vue mer' i tee)
(diroximel fumarate) delayed-release capsulesWhat is VUMERITY?
- VUMERITY is a prescription medicine used to treat people with relapsing forms of multiple sclerosis (MS), to include clinically isolated syndrome, relapsing-remitting disease, and active secondary progressive disease in adults.
- It is not known if VUMERITY is safe and effective in children.
Do not take VUMERITY if you:
- have had an allergic reaction (such as welts, hives, swelling of the face, lips, mouth or tongue, or difficulty breathing) to diroximel fumarate, dimethyl fumarate, or any of the ingredients in VUMERITY. See “What are the ingredients in VUMERITY?” below for a complete list of ingredients.
- are taking dimethyl fumarate.
Before taking and while you take VUMERITY, tell your doctor about all of your medical conditions, including if you:
- have liver problems.
- have kidney problems.
- have or have had low white blood cell counts or an infection.
- are pregnant or plan to become pregnant. It is not known if VUMERITY will harm your unborn baby.
- are breastfeeding or plan to breastfeed. It is not known if VUMERITY passes into your breast milk. Talk to your healthcare provider about the best way to feed your baby while using VUMERITY.
Tell your doctor about all the medicines you take including prescription and over-the-counter medicines, vitamins, and herbal supplements. How should I take VUMERITY?
- Take VUMERITY exactly as your doctor tells you to take it.
- The recommended starting dose on days 1 to 7 is one capsule by mouth 2 times a day. After 7 days, the recommended dose is 2 capsules by mouth 2 times a day.
- If taken with food, avoid taking VUMERITY with a high-fat, high-calorie meal or snack.
- Your meal or snack should contain no more than 700 calories and no more than 30 g of fat.
- Swallow VUMERITY whole. Do not crush, chew, or sprinkle capsule contents on food.
- If you take too much VUMERITY, call your doctor or go to the nearest hospital emergency room right away.
What should I avoid while taking VUMERITY?
- Do not drink alcohol at the time you take a VUMERITY dose.
What are the possible side effects of VUMERITY?
VUMERITY may cause serious side effects including:
- allergic reaction (such as welts, hives, swelling of the face, lips, mouth or tongue, or difficulty breathing). Stop taking VUMERITY and get emergency medical help right away if you get any of these symptoms.
-
PML (progressive multifocal leukoencephalopathy) a rare brain infection that usually leads to death or severe disability over a period of weeks or months. Tell your doctor right away if you get any of these symptoms of PML:
- weakness on one side of the body that gets worse
- vision problems
- confusion
- clumsiness in your arms or legs
- changes in thinking and memory
- personality changes
- decreases in your white blood cell count. Your doctor should do a blood test to check your white blood cell count before you start treatment with VUMERITY and while you are on therapy. You should have blood tests after 6 months of treatment and every 6 to 12 months after that.
-
liver problems. Your doctor should do blood tests to check your liver function before you start taking VUMERITY and during treatment if needed. Tell your doctor right away if you get any of these symptoms of a liver problem during treatment.
- severe tiredness
- loss of appetite
- pain on the right side of your stomach
- have dark or brown (tea color) urine
- yellowing of your skin or the white part of your eyes
The most common side effects of VUMERITY include:
- flushing, redness, itching, or rash
- nausea, vomiting, diarrhea, stomach pain, or indigestion
- Flushing and stomach problems are the most common reactions, especially at the start of therapy, and may decrease over time. Taking VUMERITY with food (avoid high-fat, high-calorie meal or snack) may help reduce flushing. Call your doctor if you have any of these symptoms and they bother you or do not go away. Ask your doctor if taking aspirin before taking VUMERITY may reduce flushing.
These are not all the possible side effects of VUMERITY. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
For more information go to fda.reportHow should I store VUMERITY?
- Store VUMERITY at room temperature between 68°F to 77°F (20°C to 25°C).
- Keep VUMERITY and all medicines out of the reach of children.
General Information about the safe and effective use of VUMERITY
Medicines are sometimes prescribed for purposes other than those listed in this Patient Information. Do not use VUMERITY for a condition for which it was not prescribed. Do not give VUMERITY to other people, even if they have the same symptoms that you have. It may harm them.
If you would like more information, talk to your doctor or pharmacist. You can ask your pharmacist or doctor for information about VUMERITY that is written for healthcare professionals.What are the ingredients in VUMERITY?
Active ingredient: diroximel fumarate
Inactive ingredients: crospovidone, colloidal silicon dioxide, magnesium stearate (non-bovine), methacrylic acid and ethyl acrylate copolymer, microcrystalline cellulose, talc, and triethyl citrate. Capsule Shell: carrageenan, hypromellose, potassium chloride, and titanium dioxide. Capsule Shell Ink: iron oxide, potassium hydroxide, propylene glycol, and shellac.
Manufactured for: Biogen Inc., Cambridge, MA 02142, www.VUMERITY.com or call 1-800-456-2255 -
PRINCIPAL DISPLAY PANEL
Principal Display Panel – 106 Capsule Carton Label
106
CapsulesRX Only
NDC: 64406-020-01
VUMERITY™
(diroximel fumarate)
delayed-release capsules231 mg
starter
dose bottleSwallow capsule whole.
Manufactured for:
Biogen Inc.
Cambridge, MA 02142
Product of the
United KingdomBiogen®
-
PRINCIPAL DISPLAY PANEL
Principal Display Panel – 106 Capsule Bottle Label
106
CapsulesRX Only
NDC: 64406-020-01
VUMERITY™
(diroximel fumarate)
delayed-release capsules231 mg
starter
dose bottleSwallow capsule whole.
Each capsule contains 231 mg
of diroximel fumarate.Information and support:
1-800-456-2255
VUMERITY.com
54477-01
Biogen®
106
Capsules -
PRINCIPAL DISPLAY PANEL
Principal Display Panel – 120 Capsule Carton Label
120
CapsulesRX Only
NDC: 64406-020-03
VUMERITY™
(diroximel fumarate)
delayed-release capsules231 mg
maintenance
dose bottleSwallow capsule whole.
Manufactured for:
Biogen Inc.
Cambridge, MA 02142
Product of the
United Kingdom.Biogen®
-
PRINCIPAL DISPLAY PANEL
Principal Display Panel – 120 Capsule Bottle Label
120
CapsulesRX Only
NDC: 64406-020-03
VUMERITY™
(diroximel fumarate)
delayed-release capsules231 mg
maintenance
dose bottleSwallow capsule whole.
Each capsule contains 231 mg
of diroximel fumarate.Information and support:
1-800-456-2255
VUMERITY.com
54479-01
Biogen®
120
Capsules -
PRINCIPAL DISPLAY PANEL
Principal Display Panel – 106 Capsule Sample Carton Label
106
CapsulesRX Only
NDC: 64406-020-05
VUMERITY™
(diroximel fumarate)
delayed-release capsules231 mg
Swallow capsule whole.
Sample Not for Sale
Manufactured for:
Biogen Inc.
Cambridge, MA 02142
Product of the
United Kingdom.Biogen®
-
PRINCIPAL DISPLAY PANEL
Principal Display Panel – 106 Capsule Sample Bottle Label
106
CapsulesRX Only
NDC: 64406-020-05
VUMERITY™
(diroximel fumarate)
delayed-release capsules231 mg
Swallow capsule whole.
Sample Not for Sale
Each capsule contains 231 mg
of diroximel fumarate.Information and support:
1-800-456-2255
VUMERITY.com
54492-01
Biogen®
106
Capsules -
INGREDIENTS AND APPEARANCE
VUMERITY
diroximel fumarate capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 64406-020 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength diroximel fumarate (UNII: K0N0Z40J3W) (monomethyl fumarate - UNII:45IUB1PX8R) diroximel fumarate 231 mg Inactive Ingredients Ingredient Name Strength methacrylic acid - ethyl acrylate copolymer (1:1) type a (UNII: NX76LV5T8J) crospovidone (UNII: 2S7830E561) microcrystalline cellulose (UNII: OP1R32D61U) silicon dioxide (UNII: ETJ7Z6XBU4) triethyl citrate (UNII: 8Z96QXD6UM) talc (UNII: 7SEV7J4R1U) magnesium stearate (UNII: 70097M6I30) hypromellose, unspecified (UNII: 3NXW29V3WO) titanium dioxide (UNII: 15FIX9V2JP) potassium chloride (UNII: 660YQ98I10) carrageenan (UNII: 5C69YCD2YJ) Product Characteristics Color white (white) Score no score Shape CAPSULE (CAPSULE) Size 22mm Flavor Imprint Code DRF;231;mg Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 64406-020-01 1 in 1 CARTON 10/29/2019 1 106 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 2 NDC: 64406-020-03 1 in 1 CARTON 10/29/2019 2 120 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 3 NDC: 64406-020-05 1 in 1 CARTON 10/29/2019 3 106 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA211855 10/29/2019 Labeler - Biogen Inc. (121376230)






Trademark Results [Vumerity]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 VUMERITY 90777844 not registered Live/Pending |
Biogen MA Inc. 2021-06-16 |
 VUMERITY 87476946 5908620 Live/Registered |
Alkermes Pharma Ireland Limited 2017-06-06 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.