ERWINAZE- asparaginase injection, powder, lyophilized, for solution
ERWINAZE by
Drug Labeling and Warnings
ERWINAZE by is a Prescription medication manufactured, distributed, or labeled by Jazz Pharmaceuticals, Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use ERWINAZE® safely and effectively. See full prescribing information for ERWINAZE.
ERWINAZE (asparaginase Erwinia chrysanthemi) for injection, for intramuscular or intravenous use
Initial U.S. Approval: 2011INDICATIONS AND USAGE
ERWINAZE is an asparagine specific enzyme indicated as a component of a multi-agent chemotherapeutic regimen for the treatment of patients with acute lymphoblastic leukemia (ALL) who have developed hypersensitivity to E. coli-derived asparaginase. (1)
DOSAGE AND ADMINISTRATION
- To substitute for a dose of pegaspargase: The recommended dose for each planned dose of pegaspargase is 25,000 International Units/m2 administered intramuscularly or intravenously 3 times a week (Monday/Wednesday/Friday) for 6 doses. (2.1)
- To substitute for a dose of native E. coli asparaginase: The recommended dose is 25,000 International Units/m2 administered intramuscularly or intravenously for each scheduled dose of native E. coli asparaginase. (2.1)
DOSAGE FORMS AND STRENGTHS
For injection: 10,000 International Units as a lyophilized powder in single-dose vial for reconstitution. (3)
CONTRAINDICATIONS
- ERWINAZE is contraindicated in patients with a history of:
- Serious hypersensitivity reactions to ERWINAZE, including anaphylaxis (4)
- Serious pancreatitis with prior L-asparaginase therapy (4)
- Serious thrombosis with prior L-asparaginase therapy (4)
- Serious hemorrhagic events with prior L-asparaginase therapy (4)
WARNINGS AND PRECAUTIONS
- Hypersensitivity Reactions: Monitor for signs or symptoms. Discontinue ERWINAZE for serious reaction. (5.1)
- Pancreatitis: Monitor for symptoms. Discontinue if pancreatitis occurs. (5.2)
- Glucose Intolerance: Monitor and manage medically. (5.3)
- Thrombosis: Discontinue for severe or life-threatening thrombosis. Provide anticoagulation therapy as indicated. (5.4)
- Hemorrhage: Discontinue for severe or life-threatening hemorrhage. (5.4)
ADVERSE REACTIONS
Most common adverse reactions (incidence ≥ 1%) are: systemic hypersensitivity, hyperglycemia, transaminases abnormal, fever, pancreatitis, local reactions, vomiting, nausea, thrombosis, hyperbilirubinemia, abdominal pain/discomfort, and diarrhea.
To report SUSPECTED ADVERSE REACTIONS, contact Jazz Pharmaceuticals, Inc. at 1-800-520-5568 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
USE IN SPECIFIC POPULATIONS
Lactation: Breastfeeding not recommended. (8.2)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 12/2019
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
2.2 Preparation and Handling Instructions
2.3 Administration Instructions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity Reactions
5.2 Pancreatitis
5.3 Glucose Intolerance
5.4 Thrombosis and Hemorrhage
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Immunogenicity
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
To substitute for a dose of pegaspargase:
The recommended dose for each planned dose of pegaspargase is 25,000 International Units/m2 administered intramuscularly or intravenously three times a week (Monday/Wednesday/Friday) for six doses.
To substitute for a dose of native E. coli asparaginase:
The recommended dose is 25,000 International Units/m2 administered intramuscularly or intravenously for each scheduled dose of native E. coli asparaginase within a treatment.
When administering ERWINAZE intravenously, consider monitoring nadir (pre-dose) serum asparaginase activity (NSAA) levels and switching to intramuscular administration if desired NSAA levels are not achieved [see Clinical Pharmacology (12.3)].
2.2 Preparation and Handling Instructions
- Visually inspect the ERWINAZE powder for foreign particulate matter and discoloration prior to reconstitution. Discard vial if present.
- Reconstitute the contents of each vial by slowly injecting 1 or 2 mL of preservative free sterile sodium chloride (0.9%) injection (USP) against the inner vial wall.
- Do not forcefully inject solution for reconstitution directly onto or into the powder. When reconstituted with 1 mL the resultant concentration is 10,000 International Units per mL. When reconstituted with 2 mL the resultant concentration is 5,000 International Units per mL.
- Dissolve contents by gentle mixing or swirling. Do not shake or invert vial.
- When reconstituted, ERWINAZE should be a clear, colorless solution. Inspect the solution after reconstitution and discard if any visible particles or protein aggregates are present.
- Calculate the dose needed and the volume needed to obtain the calculated dose.
- Withdraw the volume containing the calculated dose from the vial into a polypropylene syringe within 15 minutes of reconstitution. For intravenous use, slowly inject the reconstituted ERWINAZE into an IV infusion bag containing 100 mL of normal saline acclimatized to room temperature. Do not shake or squeeze the IV bag.
- If partial vial is used, do not save or reuse the unused drug for later administration. Discard unused portions.
- Do not freeze or refrigerate reconstituted solution and administer within 4 hours or discard [see How Supplied/Storage and Handling (16)].
2.3 Administration Instructions
Administer ERWINAZE in a setting with resuscitation equipment and other agents necessary to treat anaphylaxis.
ERWINAZE solution can be administered by intramuscular injection or by intravenous infusion.
- For intramuscular use, limit the volume of reconstituted ERWINAZE at a single injection site to 2 mL; if reconstituted dose to be administered is greater than 2 mL, use multiple injection sites.
- For intravenous use, infuse ERWINAZE in 100 mL of normal saline over 1 to 2 hours. Do not infuse other intravenous drugs through the same intravenous line while infusing ERWINAZE.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity Reactions
Grade 3 and 4 hypersensitivity reactions after the use of ERWINAZE have occurred in 5% of patients in clinical trials [see Adverse Reactions (6.1)].
Administer this product in a setting with resuscitation equipment and other agents necessary to treat anaphylaxis. If a serious hypersensitivity reaction occurs, discontinue ERWINAZE and initiate appropriate therapy.
5.2 Pancreatitis
Pancreatitis has been reported in 4% of patients in clinical trials [see Adverse Reactions (6.1)].
Evaluate patients with symptoms compatible with pancreatitis to establish a diagnosis. Discontinue ERWINAZE for severe or hemorrhagic pancreatitis manifested by abdominal pain > 72 hours and amylase elevation ≥ 2.0 x ULN. Severe pancreatitis is a contraindication to additional asparaginase administration. In the case of mild pancreatitis, hold ERWINAZE until the signs and symptoms subside and amylase levels return to normal. After resolution, treatment with ERWINAZE may be resumed.
5.3 Glucose Intolerance
Glucose intolerance has been reported in 5% of patients receiving ERWINAZE in clinical trials [see Adverse Reactions (6.1)]. In some cases glucose intolerance may be irreversible. Monitor glucose levels in patients at baseline and periodically during treatment. Administer insulin therapy as necessary in patients with hyperglycemia.
5.4 Thrombosis and Hemorrhage
Serious thrombotic events, including sagittal sinus thrombosis and pulmonary embolism have been reported with both E. coli and Erwinia-derived L-asparaginase therapy. The following coagulation proteins were decreased in the majority of patients after a 2-week course of ERWINAZE by intramuscular administration: fibrinogen, protein C activity, protein S activity, and anti-thrombin III. Discontinue ERWINAZE for a thrombotic or hemorrhagic event until symptoms resolve; after resolution, treatment with ERWINAZE may be resumed.
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are discussed in greater detail in other sections of the label:
- Hypersensitivity reactions [see Warnings and Precautions (5.1)]
- Pancreatitis [see Warnings and Precautions (5.2)]
- Glucose intolerance [see Warnings and Precautions (5.3)]
- Thrombosis and hemorrhage [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under controlled, but widely varying conditions, adverse reaction rates observed in clinical trials of ERWINAZE cannot be directly compared to rates in the clinical trials of other drugs and may not reflect the rates observed in practice.
The data presented below are based on information collected from Study 1, a single-arm, multi-center, open-label, safety and clinical pharmacology trial (intramuscular administration), the ERWINAZE Master Treatment Protocol (EMTP), an expanded access program (both intramuscular, intravenous, and other or unknown administration), and Study 2, a single-arm, multi-center, open-label, pharmacokinetic (PK) study trial of intravenous administration of ERWINAZE.
Study 1 enrolled 58 patients treated on National Cancer Institute (NCI)-sponsored cooperative group ALL protocols who were unable to continue to receive pegaspargase due to hypersensitivity reactions. Patients received 6 doses of ERWINAZE 25,000 International Units/m2 intramuscularly on a Monday, Wednesday, and Friday schedule as a replacement for each scheduled dose of pegaspargase remaining on their original treatment protocol. The Study 1 population included patients with a median age of 11 years (2 to 18 years); 59% were male, 78% were White, 10% were Black/African American, 5% were Asian, and 7% were other or unknown. A total of 35% were Hispanic or Latino. In Study 1, the number of ERWINAZE courses ranged from 1 to 9. In this study, 76% (44 of 58) completed all planned therapy. Fourteen (24%) patients stopped therapy prior to completion; seven due to allergic reactions, five due to physician or patient choice, one due to disease progression, and one due to discontinuation during frontline protocol. All other chemotherapy was continued according to the patient’s prescribed treatment regimen [see Clinical Studies (14)].
Study 2 enrolled 30 patients [29 were being treated for ALL and one for lymphoblastic lymphoma (LBL)] following allergy to native E. coli asparaginase or pegaspargase. Patients received ERWINAZE 25,000 International Unit/m2/dose, administered by intravenous infusion on a Monday, Wednesday, and Friday schedule (6 doses) as a replacement for doses remaining on their original treatment plan. The Study 2 population included patients with a median age of 7 years (1 to 17 years); 63% were male, 27% were Hispanic or Latino, 83% were White, 3% were Black/African American, 7% were Asian, and 7% were other (American Indian, Alaska Native or Indian) [see Clinical Studies (14)].The EMTP trial enrolled 1368 patients with ALL or lymphoblastic lymphoma who received ERWINAZE after developing systemic hypersensitivity to an E. coli-derived asparaginase. Of these 1368 patients, safety data were received for 940 patients with a median age of 9 years (0 to 76 years), 63% were male, 91% with leukemia, 3% with lymphoma, and 6% with unknown disease information. Patients received ERWINAZE according to several schedules, and treatment center specifications with doses that ranged from 20,000 to 25,000 International Units/m2. The route of administration was intramuscular n=852, intravenous n=29, other or unknown n=59. In the EMTP trial, the planned number of doses of ERWINAZE ranged from 3 to 48 doses. Seventy-eight percent of patients (693 of 893) were able to receive all planned doses to complete their prescribed treatment regimen.
In Study 1 and Study 2, safety information was prospectively and systematically collected. In Study 1, all Grades of adverse events were reported for the following adverse events of special interest: allergy, pancreatitis, coagulopathy (hemorrhage, thrombosis or infarct), hyperbilirubinemia, hyperglycemia, hyperlipidemia, ketoacidosis, and CNS events (hemorrhage, thrombosis or infarction, and cerebral venous thrombosis) and only Grade 3 and 4 events were reported for other adverse events. In Study 2 all adverse events of all Grades were prospectively collected. In the EMTP trial, safety data were derived from case report forms that collected adverse event information. The forms specifically requested information on occurrence of allergic reactions, thrombotic events, hemorrhagic events, hepatobiliary disorders, pancreatic disorders, and hyperglycemia.
The most common adverse reactions (incidence 1% or greater) with ERWINAZE treatment are systemic hypersensitivity, hyperglycemia, transaminases abnormal, fever, pancreatitis, local reactions, vomiting, nausea, thrombosis, hyperbilirubinemia, abdominal pain/discomfort, and diarrhea.
The incidence of non-hematologic, non-infectious, adverse events (all Grades) in Study 1, Study 2, and the EMTP trial is provided in Table 1.
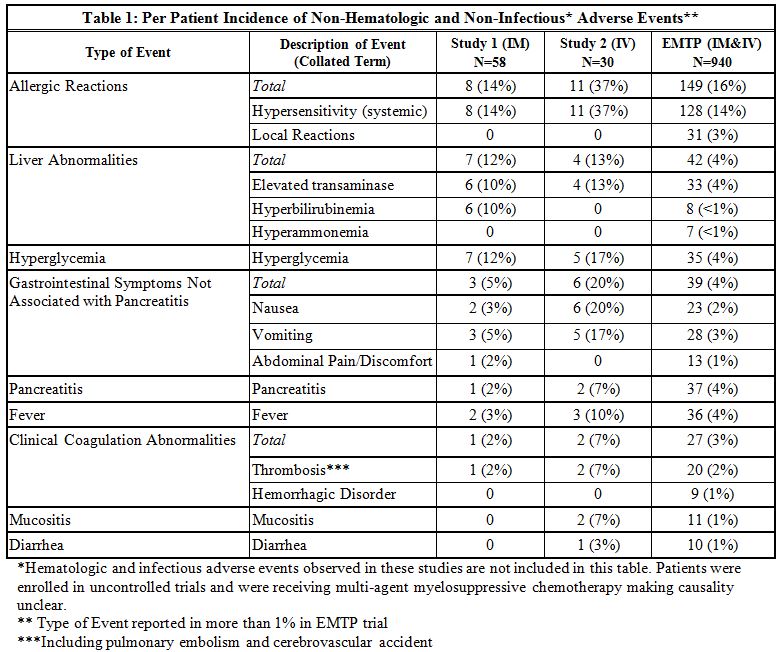
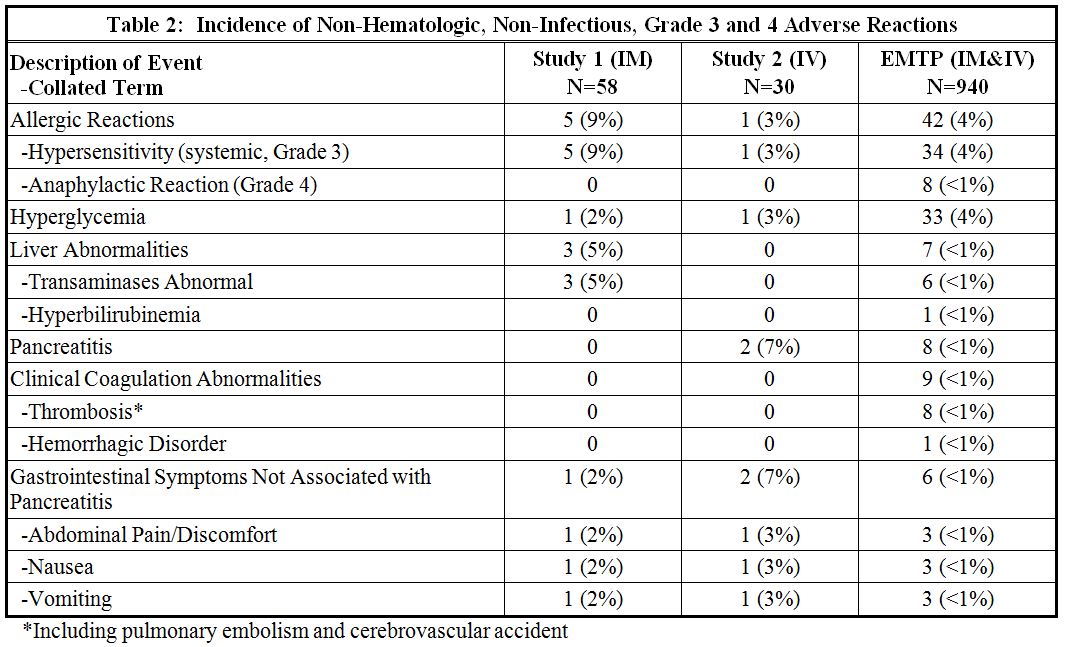
The incidence of Grade 3 or greater non-hematologic, non-infectious adverse reactions occurring with ERWINAZE in Study 1, Study 2 and EMTP trial is provided in Table 2.6.2 Immunogenicity
As with all therapeutic proteins, there is potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies in other studies or to other asparaginase Erwinia chrysanthemi products may be misleading.
In a study with ERWINAZE treatment by intramuscular administration (Study 1), 6 of 56 (11%) patients treated with ERWINAZE developed antibodies to ERWINAZE. Of these 6 anti-drug antibody (ADA) positive patients, one experienced a hypersensitivity reaction during Study 1 (2%, 1 of 56). None of these 6 patients had neutralizing antibodies.
In a study with ERWINAZE treatment by intravenous administration (Study 2), 4 of 30 (13.3%) patients treated with ERWINAZE developed anti-ERWINAZE antibodies. Of these 4 patients who developed anti-ERWINAZE antibodies, 3 experienced hypersensitivity reactions (10%, 3 of 30) during the study. None of these 4 patients had neutralizing antibodies.
The presence of ADA to ERWINAZE is associated with a higher risk of hypersensitivity reactions in patients who received ERWINAZE through intravenous infusion compared to intramuscular administration of ERWINAZE. -
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings from animal reproduction studies, ERWINAZE can cause fetal harm when administered to a pregnant woman. In animal reproduction studies, intramuscular administration of asparaginase Erwinia chrysanthemi to pregnant rats and rabbits during organogenesis at doses approximately 0.005-0.5 times the maximum recommended human dose resulted in structural abnormalities and embryo-fetal mortality (see Data). There are no available data on ERWINAZE use in pregnant women to evaluate the drug-associated risk of major birth defects, miscarriage or adverse maternal or fetal outcomes. Advise pregnant women of the potential risk to the fetus.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. Adverse outcomes in pregnancy occur regardless of the health of the mother or the use of medications. In the U.S. general population, the estimated background risks of major birth defects and miscarriage in clinically recognized pregnancies are 2 to 4% and 15 to 20%, respectively.
Data
Animal Data
In embryofetal development studies, asparaginase Erwinia chrysanthemi was administered intramuscularly every other day during the period of organogenesis to pregnant rats (at 3000, 6000, or 12000 IU/m2) and rabbits (at 120, 300, or 480 IU/m2). In rats given 12000 IU/m2 (approximately 0.5 times the maximum recommended human dose), maternal toxicity of decreased body weight gain was observed, as well as a fetal finding of increased incidence of partially undescended thymic tissue.
In rabbits, maternal toxicity consisting of decreased body weight was observed at 480 IU/m2 (approximately 0.02 times the maximum recommended human dose). Increased post-implantation loss, a decrease in the number of live fetuses, and gross abnormalities (e.g., absent kidney, absent accessory lung lobe, additional subclavian artery, and delayed ossification) were observed at doses of ≥120 IU/m2 (approximately 0.005 times the maximum recommended human dose).
8.2 Lactation
Risk Summary
There are no data on the presence of asparaginase Erwinia chrysanthemi in human or animal milk, the effects on the breastfed child, or the effects on milk production. Because of the potential for serious adverse reactions in the breastfed child, advise patients that breastfeeding is not recommended during treatment with ERWINAZE, and for 3 months after the last dose.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Pregnancy testing is recommended for females of reproductive potential before starting ERWINAZE treatment.
Contraception
Females
ERWINAZE can cause embryo-fetal harm when administered to pregnant women [see Use in Specific Populations (8.1)]. Advise females of reproductive potential to use effective contraception during treatment with ERWINAZE and for 3 months after the final dose. Since an indirect interaction between oral contraceptives and ERWINAZE cannot be ruled out, a method of contraception other than oral contraceptives should be used in women of childbearing potential.
8.4 Pediatric Use
The safety and effectiveness of ERWINAZE have been established in pediatric patients ages1 year and older as a component of a multi-agent chemotherapeutic regimen for the treatment of patients with acute lymphoblastic leukemia (ALL) who have developed hypersensitivity to E. coli-derived asparaginase and the information on this use is discussed throughout the labeling.
-
11 DESCRIPTION
ERWINAZE (asparaginase Erwinia chrysanthemi) contains an asparagine specific enzyme derived from Erwinia chrysanthemi. L-asparaginase is a tetrameric enzyme consisting of four identical subunits, each having a molecular weight of about 35 kDa. The activity of ERWINAZE is expressed in terms of International Units.
ERWINAZE is supplied as a sterile, lyophilized, white powder in vials. Each vial contains 10,000 International Units of asparaginase Erwinia chrysanthemi, and the following inactive ingredients: glucose monohydrate (5.0 mg), sodium chloride (0.5 mg). -
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Asparaginase Erwinia chrysanthemi catalyzes the deamidation of asparagine to aspartic acid and ammonia, resulting in a reduction in circulating levels of asparagine. The mechanism of action of ERWINAZE is thought to be based on the inability of leukemic cells to synthesize asparagine due to lack of asparagine synthetase activity, resulting in cytotoxicity specific for leukemic cells that depend on an exogenous source of amino acid asparagine for their protein metabolism and survival.
12.3 Pharmacokinetics
Based on a population PK model, the mean (%CV) half-life of intravenous ERWINAZE was 7.51 (23.9%) hours in contrast to a mean (%CV) half-life of 15.6 (20%) hours reported for intramuscular ERWINAZE. These differences in PK between intravenous and intramuscular ERWINAZE are reflected in the proportion of patients with 2-day and 3-day nadir serum asparaginase activity (NSAA) levels of asparaginase Erwinia chrysanthemi ≥ 0.1 or 0.4 IU/mL [see Clinical Studies (14)].
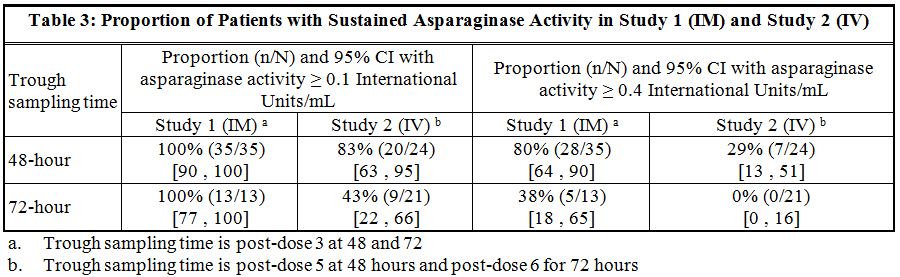
Following administration of ERWINAZE 25,000 International Units/m2 intramuscularly to 48 ALL patients aged ≥ 2 years to ≤ 18 years in Study 1 on a Monday, Wednesday, and Friday schedule for 6 doses, 100% of patients who completed Course 1 achieved NSAA levels ≥ 0.1 International Units/mL at either 48 hours (n=35) or 72 hours (n=13) post dose 3. Eighty percent (28/35) of those evaluated at 48 hours and 38% (5/13) evaluated at 72 hours had nadir serum asparaginase activity levels ≥ 0.4 International Units/mL [see Clinical Studies (14)].
Following intravenous administration of ERWINAZE 25,000 International Units/m2 to 24 evaluable patients (aged ≥ 1 year to ≤ 17 years) in Study 2 on a Monday, Wednesday, and Friday schedule, 83% (20/24) and 43% (9/21) of patients who completed Course 1 achieved NSAA levels ≥ 0.1 International Units/mL at 48 hours post-dose 5 and 72 hours post dose 6, respectively. Twenty-nine percent (7/24) of those evaluated at 48 hours and no patients (0/21) evaluated at 72 hours had nadir serum asparaginase activity levels ≥ 0.4 International Units/mL [see Clinical Studies (14)].Drug Interaction Studies
No formal drug interaction studies between ERWINAZE and other drugs have been performed
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No long-term carcinogenicity studies in animals have been performed with asparaginase Erwinia chrysanthemi. No studies that assess the mutagenic potential of asparaginase Erwinia chrysanthemi have been conducted.
In a fertility and early embryonic development study in rats, asparaginase Erwinia chrysanthemi had no effect on male or female fertility when administered intramuscularly at doses of up to 12000 IU/m2 (approximately 0.50 times the maximum recommended human dose) every other day for a total of 35 doses. Findings in males included decreased sperm count at doses of more than 3000 IU/m2 (approximately 0.12 times the maximum recommended human dose).
-
14 CLINICAL STUDIES
The efficacy of ERWINAZE for the treatment of patients with acute lymphoblastic leukemia (ALL) who have developed hypersensitivity to E. Coli-derived asparaginase as a component of a multi-agent chemotherapeutic regimen was established in Study 1, a single-arm, multi-center, open-label, safety and clinical pharmacology trial. Additional safety data were obtained in the ERWINAZE Master Treatment Protocol (EMTP), an expanded access program [see Adverse Reactions (6)]. Study 1 enrolled patients treated on National Cancer Institute (NCI)-sponsored cooperative group ALL protocols who were unable to continue to receive pegaspargase due to hypersensitivity reactions. The main outcome measure was determination of the proportion of patients who achieved a serum trough asparaginase level greater than or equal to 0.1 International Units/mL. Serum trough asparaginase activity ≥ 0.1 International Units/mL has been demonstrated to correlate with asparagine depletion (asparagine < 0.4 mcg/mL or 3 µM) and to serum levels that predict clinical efficacy. Patients received ERWINAZE 25,000 International Units/m2 intramuscularly for two weeks (total 6 doses) as a replacement for each scheduled dose of pegaspargase remaining on their original treatment protocol.
Fifty-eight patients were enrolled in Study 1, of these 48 were evaluable for the main outcome measure based on availability of pharmacokinetic samples in Course 1. The median age was 11 years (2 to 18 years); 59% were male, 78% were White, 10% were Black/African American, 5% were Asian, and 7% were other or unknown. A total of 35% were Hispanic or Latino.
Study 1 met its main outcome measure of demonstrating that greater than 50% of the patients achieved the pre-specified trough asparaginase activity level of ≥ 0.1 International Units/mL at 48 or 72 hours following the third dose. Results for the main outcome measure and for an exploratory analysis using a higher cut-off (trough serum asparaginase activity levels ≥ 0.4 International Units/mL are presented in Table 3 [see Clinical Pharmacology (12.3)].
The safety and efficacy of intravenous administration were determined in Study 2 by characterizing the PK of a 25,000 International Units/m2 ERWINAZE dose given 3 days per week on a Monday, Wednesday, and Friday schedule for up to 30 weeks. This open-label, single-arm, multicenter PK study enrolled 30 patients. The main outcome measure was determination of the proportion of patients with 2-day NSAA levels (48-hour levels taken after the fifth dose) ≥ 0.1 International Unit/mL in the first 2 weeks of ERWINAZE treatment.
Of the thirty patients enrolled, 24 were evaluable for the main outcome measure based on the pharmacokinetic samples in Course 1. The median age was 7 years (1-17 years), 63% were male, 27% were Hispanic or Latino, 83% were White, 3% were Black/African American, 7% were Asian, and 7% were other (American Indian, Alaska Native, or Indian).
In Study 2, serum asparaginase activity of asparaginase Erwinia chrysanthemi was determined in 24 evaluable patients (aged ≥ 1 year to ≤17 years) following intravenous administration of ERWINAZE 25,000 International Units/m2. Five minutes after the 60-minute infusion in Course 1, the mean asparaginase activity level was 12.65 ± 3.16 International Unit/mL post-dose 1 and 12.11 ± 3.11 International Unit/mL post dose 4. The main study objective was met with an asparaginase activity level of ≥ 0.1 International Units/mL 48 hours after the fifth dose observed in 83% of patients. The 72-hour post dose 6 asparaginase activity level of ≥ 0.1 International Unit/mL was the secondary endpoint, with 43% of patients achieving this endpoint. Results are presented in Table 3 [see Clinical Pharmacology (12.3)].
-
16 HOW SUPPLIED/STORAGE AND HANDLING
ERWINAZE is a sterile, white lyophilized powder supplied in a clear 3 mL glass vial. Each carton of ERWINAZE (NDC: 57902-249-05) contains 5 vials. Each single vial (NDC: 57902-249-01) contains 10,000 International Units asparaginase Erwinia chrysanthemi.
Store unused or unopened vials and cartons at 36°F to 46°F (2°C to 8°C). Protect from light. Do not use ERWINAZE after the expiration date on the vial.
-
17 PATIENT COUNSELING INFORMATION
- Hypersensitivity
- Inform patients of the risk of allergic reactions, including anaphylaxis. Instruct patients on the symptoms of allergic reactions and to seek medical advice immediately if they experience such symptoms [see Warnings and Precautions (5.1)].
- Pancreatitis
- Instruct patients on the risk of pancreatitis and to seek medical advice immediately if they experience abdominal pain [see Warnings and Precautions (5.2)].
- Glucose Intolerance
- Instruct patients on the risk of hyperglycemia and glucose intolerance. Advise patients to seek medical advice if they experience excessive thirst or any increase in the volume or frequency of urination [see Warnings and Precautions (5.3)].
- Thrombosis
- Instruct patients on the risk of thrombosis and hemorrhage and to seek medical advice immediately if they experience headache, arm or leg swelling, shortness of breath, and chest pain [see Warnings and Precautions (5.4)].
- Pregnancy and Lactation
- Advise female patients of reproductive potential to use effective contraceptive methods while receiving ERWINAZE and for at least 3 months after the last dose. Advise patients to notify their healthcare provider immediately in the event of a pregnancy or if pregnancy is suspected during ERWINAZE treatment [see Use in Specific Populations (8.3)]. Advise lactating women not to breastfeed during treatment with ERWINAZE and for at least 3 months after the last dose [see Use in Specific Populations (8.2)].
-
PRINCIPAL DISPLAY PANEL
PRINCIPAL DISPLAY PANEL
5 Vials NDC: 57902-249-05
asparaginase Erwinia chrysanthemi
Erwinaze®
For injection, Intramuscular or Intravenous Use
10,000 International Units per Vial
Rx Only Single Use Vial. Discard unused portion. -
INGREDIENTS AND APPEARANCE
ERWINAZE
asparaginase injection, powder, lyophilized, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 57902-249 Route of Administration INTRAMUSCULAR, INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ASPARAGINASE ERWINIA CHRYSANTHEMI (UNII: D733ET3F9O) (ASPARAGINASE - UNII:G4FQ3CKY5R) ASPARAGINASE ERWINIA CHRYSANTHEMI 10000 [iU] in 1 mL Inactive Ingredients Ingredient Name Strength DEXTROSE MONOHYDRATE (UNII: LX22YL083G) 5 mg in 1 mL SODIUM CHLORIDE (UNII: 451W47IQ8X) 0.5 mg in 1 mL Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 57902-249-05 5 in 1 CARTON 11/18/2011 1 NDC: 57902-249-01 1 mL in 1 VIAL, GLASS; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125359 11/18/2011 Labeler - Jazz Pharmaceuticals, Inc. (135926363) Registrant - Jazz Pharmaceuticals, Inc. (135926363)
Trademark Results [ERWINAZE]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 ERWINAZE 90081574 not registered Live/Pending |
Porton Biopharma Limited 2020-07-29 |
 ERWINAZE 85245951 4147569 Live/Registered |
PORTON BIOPHARMA LIMITED 2011-02-18 |
 ERWINAZE 85031446 not registered Dead/Abandoned |
EUSA Pharma (Europe) Limited 2010-05-06 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.