VORANIGO- vorasidenib tablet, film coated
VORANIGO by
Drug Labeling and Warnings
VORANIGO by is a Prescription medication manufactured, distributed, or labeled by Servier Pharmaceuticals LLC. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use VORANIGO safely and effectively. See full prescribing information for VORANIGO.
VORANIGO® (vorasidenib) tablets, for oral use
Initial U.S. Approval: 2024RECENT MAJOR CHANGES
INDICATIONS AND USAGE
VORANIGO is an isocitrate dehydrogenase-1 (IDH1) and isocitrate dehydrogenase-2 (IDH2) inhibitor indicated for the treatment of adult and pediatric patients 12 years and older with Grade 2 astrocytoma or oligodendroglioma with a susceptible IDH1 or IDH2 mutation, as detected by an FDA-approved test, following surgery including biopsy, sub-total resection, or gross total resection. (1)
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
Tablets: 10 mg and 40 mg. (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
- Hepatotoxicity: Monitor liver function tests every 2 weeks during the first 2 months of treatment, then monthly for the first 2 years of treatment, and as clinically indicated. Withhold, reduce the dose or discontinue VORANIGO based on severity. (2.3, 5.1)
- Embryo-Fetal Toxicity: VORANIGO can cause fetal harm. Advise patients of the potential risk to a fetus and to use effective nonhormonal contraception. (5.2, 8.1, 8.3)
ADVERSE REACTIONS
The most common (≥15%) adverse reactions include fatigue, headache, COVID-19, musculoskeletal pain, diarrhea, nausea, and seizure.
Grade 3 or 4 (≥2%) laboratory abnormalities were ALT increased, AST increased, GGT increased, and neutrophils decreased. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Servier Pharmaceuticals at 1-800-807-6124 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- CYP1A2 Inhibitors: Avoid concomitant use of strong and moderate CYP1A2 inhibitors. (7.1)
- CYP1A2 Inducers: Avoid concomitant use of moderate CYP1A2 inducers and smoking tobacco. (7.1)
- Certain CYP3A Substrates: Avoid concomitant use with CYP3A substrates, where a minimal concentration change can reduce efficacy. (7.2)
- Hormonal Contraception: If concomitant use cannot be avoided, use with nonhormonal contraception methods. (7.2)
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 4/2025
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Evaluation Before Initiating VORANIGO
2.2 Patient Selection
2.3 Recommended Dosage and Administration
2.4 Dosage Modifications, Management and Monitoring for Adverse Reactions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hepatotoxicity
5.2 Embryo-Fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on VORANIGO
7.2 Effect of VORANIGO on Other Drugs
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and Pharmacology
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
VORANIGO is indicated for the treatment of adult and pediatric patients 12 years and older with Grade 2 astrocytoma or oligodendroglioma with a susceptible isocitrate dehydrogenase-1 (IDH1) or isocitrate dehydrogenase-2 (IDH2) mutation, as detected by an FDA-approved test, following surgery including biopsy, sub-total resection, or gross total resection [see Dosage and Administration (2.1), Clinical Pharmacology (12.1) and Clinical Studies (14)].
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Evaluation Before Initiating VORANIGO
Before initiating VORANIGO, evaluate blood chemistry and liver laboratory tests [see Warnings and Precautions (5.1) and Adverse Reactions (6.1)].
2.2 Patient Selection
Select patients with Grade 2 astrocytoma or oligodendroglioma for treatment with VORANIGO based on the presence of IDH1 or IDH2 mutations in tumor specimens [see Clinical Studies (14)].
Information on FDA-approved tests for detection of IDH1 or IDH2 mutations in Grade 2 astrocytoma or oligodendroglioma for selecting patients for treatment with VORANIGO is available at: https://www.fda.gov/CompanionDiagnostics.
2.3 Recommended Dosage and Administration
Recommended Dosage
Adult Patients
The recommended dosage of VORANIGO in adult patients is 40 mg orally once daily until disease progression or unacceptable toxicity.
Pediatric Patients 12 Years and Older
The recommended dosage of VORANIGO in pediatric patients 12 years and older is based on body weight:
- Patients weighing ≥40 kg: 40 mg orally once daily
- Patients weighing <40 kg: 20 mg orally once daily
Continue treatment with VORANIGO until disease progression or unacceptable toxicity.
Administration
Swallow VORANIGO tablets whole with water with or without food [see Clinical Pharmacology (12.3)]. Do not split, crush or chew tablets.
2.4 Dosage Modifications, Management and Monitoring for Adverse Reactions
The recommended VORANIGO dosage reductions for adverse reactions are provided in Table 1.
Table 1: Recommended VORANIGO Dosage Reductions for Adverse Reactions Dosage Reduction Recommended Dose and Schedule Adult patients and Pediatric patients 12 years and older weighing at least 40 kg First 20 mg once daily Second 10 mg once daily Pediatric patients 12 years and older weighing less than 40 kg First 10 mg once daily Permanently discontinue VORANIGO in patients unable to tolerate 10 mg once daily. The recommended management for adverse reactions and VORANIGO dosage modifications for adverse reactions are provided in Table 2.
Table 2: Recommended VORANIGO Dosage Modifications and Management for Adverse Reactions Adverse Reaction Severity* Management and Dosage Modifications Abbreviations: ALT = Alanine aminotransferase; AST = Aspartate aminotransferase; ULN = Upper limit of normal - * Adverse reactions graded by the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI-CTCAE) version 5.0.
Hepatotoxicity
(Elevation of ALT or AST)
[see Warnings and Precautions (5.1)]Grade 1
ALT or AST increase >ULN to 3 × ULN without concurrent total bilirubin >2 × ULNContinue VORANIGO at current dose.
Monitor liver laboratory tests weekly until recovery to <Grade 1.Grade 2
ALT or AST >3 to 5 × ULN without concurrent total bilirubin >2 × ULNFirst Occurrence: Withhold VORANIGO until recovery to ≤Grade 1 or baseline. - Recovery in ≤28 days, resume VORANIGO at the same dose.
- Recovery in >28 days, resume VORANIGO at reduced dose [see Table 1].
Grade 3
ALT or AST >5 to 20 × ULN without concurrent total bilirubin >2 × ULNFirst Occurrence: Withhold VORANIGO until recovery to ≤Grade 1 or baseline. - Recovery in ≤28 days, resume VORANIGO at reduced dose [see Table 1].
- If not recovered in ≤28 days, permanently discontinue VORANIGO.
Grade 2 or 3
Any ALT or AST >3 to 20 × ULN with concurrent total bilirubin >2 × ULNFirst Occurrence: Withhold VORANIGO until recovery to ≤Grade 1 or baseline. - Resume VORANIGO at reduced dose [see Table 1].
Grade 4
Any ALT or AST >20 × ULNPermanently discontinue VORANIGO. Other Adverse Reactions
[see Adverse Reactions (6.1)]Grade 3 First Occurrence: Withhold VORANIGO until recovery to ≤Grade 1 or baseline. - Resume VORANIGO at reduced dose [see Table 1].
Grade 4 Permanently discontinue VORANIGO. -
3 DOSAGE FORMS AND STRENGTHS
Tablets:
- 10 mg: White to off-white, round film-coated tablet imprinted with "10" in black ink on one side and plain on the other side. Each tablet contains 10 mg of vorasidenib.
- 40 mg: White to off-white, oblong film-coated tablet imprinted with "40" in black ink on one side and plain on the other side. Each tablet contains 40 mg of vorasidenib.
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Hepatotoxicity
VORANIGO can cause hepatic transaminase elevations, which can lead to hepatic failure, hepatic necrosis, and autoimmune hepatitis.
In the pooled safety population [see Adverse Reactions (6.1)], 58% of patients treated with VORANIGO experienced increased ALT and 44% of patients experienced increased AST. Grade 3 or 4 increased ALT or AST occurred in 9% and 4.8% of patients respectively. Among these patients, 4.1% (10/244) had concurrent Grade 3 to 4 ALT or AST elevations. A total of 34% of patients treated with VORANIGO had increased gamma-glutamyl transferase (GGT), of these 2.2% were Grade 3 or 4. Bilirubin increases occurred in 4.8% of patients treated with VORANIGO, with 0.4% Grade 3 or 4. Nine percent of patients treated with VORANIGO had increased alkaline phosphatase, with 0.9% Grade 3 or 4.
Two patients met the laboratory criteria for Hy's Law and had concurrent elevations in ALT or AST >3 times the upper limit of normal and total bilirubin >2 times the upper limit of normal; these events were associated with cases of autoimmune hepatitis and hepatic failure. The median time to first onset of increased ALT or AST was 57 days (range: 1 to 1049).
Permanent discontinuation of VORANIGO was required for 2.9% of patients with ALT elevations, 1.6% of AST elevations, and 0.4% of GGT elevations. Dosage reductions of VORANIGO were required for 7% of patients with ALT elevations, 1.2% of AST elevations, and 0.4% of GGT elevations. Dosage interruptions were required in 14% of patients with ALT elevations, 6% of AST elevations, and 1.6% of GGT elevations.
Monitor liver laboratory tests (AST, ALT, GGT, total bilirubin and alkaline phosphatase) prior to the start of VORANIGO, every 2 weeks during the first 2 months of treatment, then monthly for the first 2 years of treatment, and as clinically indicated, with more frequent testing in patients who develop transaminase elevations.
Reduce the dose, withhold, or permanently discontinue VORANIGO based on severity [see Dosage and Administration (2.3) and Adverse Reactions (6.1)].
5.2 Embryo-Fetal Toxicity
Based on findings from animal studies, VORANIGO can cause fetal harm when administered to a pregnant woman. In animal embryo-fetal development studies, oral administration of vorasidenib to pregnant rats during the period of organogenesis caused embryo-fetal toxicities at doses ≥45 times the human exposure based on the area under the concentration-time curve (AUC) at the highest recommended dose. Oral administration of vorasidenib to pregnant rabbits during the period of organogenesis resulted in embryo-fetal toxicity at doses ≥8 times the human exposure based on the AUC at the highest recommended dose.
Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females of reproductive potential to use effective nonhormonal contraception during treatment with VORANIGO and for 3 months after the last dose, since VORANIGO can render some hormonal contraceptives ineffective [see Drug Interactions (7.2)]. Advise male patients with female partners of reproductive potential to use effective contraception during treatment with VORANIGO and for 3 months after the last dose [see Use in Specific Populations (8.1, 8.3)].
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Hepatotoxicity [see Warnings and Precautions (5.1)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Adverse reactions described in the WARNINGS AND PRECAUTIONS reflect exposure to VORANIGO 40 mg orally once daily until disease progression or unacceptable toxicity in the 244 patients with astrocytoma or oligodendroglioma with susceptible IDH1 or IDH2 mutation in trials AG881-C-002 (NCT02481154, n=11), AG120-881-001 (NCT03343197, n=14) and INDIGO (NCT04164901, n=167 randomized patients and n=52 crossover patients). Among the 244 patients who received VORANIGO, 78% were exposed for 6 months or longer and 44% were exposed for greater than one year. In this pooled safety population, the most common (≥15%) adverse reactions were fatigue (33%), headache (28%), COVID-19 (28%), musculoskeletal pain (24%), diarrhea (21%), nausea (20%), and seizure (16%). In this pooled safety population, the most common (≥2%) Grade 3 or 4 laboratory abnormalities were increased ALT (9%), increased AST (4.8%), increased GGT (2.2%), and decreased neutrophils (2.2%).
INDIGO
The safety of VORANIGO was evaluated in 330 patients with Grade 2 astrocytoma or oligodendroglioma with an IDH1 or IDH2 mutation who received at least one dose of either VORANIGO 40 mg daily (N=167) or placebo (N=163) in the INDIGO trial [see Clinical Studies (14)]. Patients received VORANIGO 40 mg orally once daily or placebo orally once daily until disease progression or unacceptable toxicity. Among the 167 patients who were randomized and received VORANIGO, the median duration of exposure to VORANIGO was 12.7 months (range: 1 to 30 months) with 153 patients (92%) exposed to VORANIGO for at least 6 months and 89 (53%) exposed for at least 1 year.
The demographics of patients randomized to VORANIGO were: median age 41 years (range: 21 to 71 years); 60% male, 74% White, 20% race not reported, 3% Asian, and 1.2% Black or African American; and 5% were Hispanic or Latino.
Serious adverse reactions occurred in 7% of patients who received VORANIGO. The most common serious adverse reactions occurring in ≥2% of patients who received VORANIGO includes seizure (3%).
Permanent discontinuation of VORANIGO due to an adverse reaction occurred in 3.6% of patients. Adverse reactions which resulted in permanent discontinuation of VORANIGO in ≥2% of patients included ALT increased (3%).
Dosage interruptions of VORANIGO due to an adverse reaction occurred in 30% of patients. Adverse reactions which required dose interruption in ≥5% of patients included ALT increased (14%), COVID-19 (9%), and AST increased (6%).
Dose reductions of VORANIGO due to an adverse reaction occurred in 11% of patients. Adverse reactions which required dose reduction in ≥5% of patients included ALT increased (8%).
The most common (≥15%) adverse reactions were fatigue (37%), COVID-19 (33%), musculoskeletal pain (26%), diarrhea (25%), and seizure (16%).
Grade 3 or 4 (≥2%) laboratory abnormalities were ALT increased (10%), AST increased (4.8%), GGT increased (3%) and neutrophil decreased (2.4%).
Adverse reactions and select laboratory abnormalities reported in the INDIGO trial are shown in Tables 3 and 4.
Table 3: Adverse Reactions (≥5%) in Patients with Grade 2 IDH1/2 Mutant Glioma Who Received VORANIGO Compared with Placebo in the INDIGO Trial VORANIGO
40 mg daily
(n=167)Placebo
(n=163)Adverse Reaction* All Grades (%) Grades 3 or 4 (%) All Grades (%) Grades 3 or 4 (%) - * Adverse reactions are based on NCI CTCAE v5.0.
- † Grouped term includes asthenia.
- ‡ Grouped term includes partial seizures, generalized tonic-clonic seizure, epilepsy, clonic convulsion, and simple partial seizures.
- § Grouped term includes arthralgia, back pain, non-cardiac chest pain, pain in extremity, myalgia, neck pain, musculoskeletal chest pain, arthritis, and musculoskeletal stiffness.
- ¶ Grouped term includes feces soft and frequent bowel movements.
- # Grouped term includes abdominal pain upper, abdominal discomfort, abdominal pain lower, abdominal tenderness, and epigastric discomfort.
General Disorders Fatigue† 37 0.6 36 1.2 Infections and Infestations COVID-19 33 0 29 0 Nervous System Disorders Seizure‡ 16 4.2 15 3.7 Musculoskeletal and Connective Tissue Disorders Musculoskeletal pain§ 26 0 25 1.8 Gastrointestinal Disorders Diarrhea¶ 25 0.6 17 0.6 Constipation 13 0 12 0 Abdominal pain# 13 0 12 0 Decreased appetite 9 0 3.7 0 Table 4: Select Laboratory Abnormalities (≥5%) That Worsened from Baseline in Patients with Grade 2 IDH1/2 Mutant Glioma Who Received VORANIGO in the INDIGO Trial VORANIGO
40 mg daily
N=167Placebo
N=163Parameter All Grades* (%†) Grades* 3 or 4 (%†) All Grades* (%†) Grades* 3 or 4 (%†) Abbreviations: AST = Aspartate Aminotransferase; ALT = Alanine Aminotransferase; GGT = Gamma-Glutamyl Transferase; ALP = Alkaline Phosphatase - * Based on NCI CTCAE v5.0.
- † The denominator used to calculate percentages is N, the number of subjects in the Safety Analysis Set within each treatment group.
- ‡ Includes adverse reaction term hyperglycemia.
- § Includes adverse reaction terms hypophosphatemia and blood phosphorus decreased.
Chemistry Increased ALT 59 10 25 0 Increased AST 46 4.8 20 0 Increased Creatinine 11 0.6 7 0 Decreased Calcium 10 0 7 0 Increased Glucose‡ 10 0 4.3 0 Increased GGT 38 3 10 1.8 Decreased Phosphate§ 8 0.6 4.9 0 Increased Potassium 23 0.6 20 0 Increased ALP 10 1.2 7 0.6 Hematology Increased Hemoglobin 13 0 3.1 0 Decreased Lymphocytes 11 1.8 8 0.6 Decreased Leukocytes 13 0.6 12 0.6 Decreased Neutrophils 14 2.4 12 1.8 Decreased Platelets 12 0 4.3 0 -
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on VORANIGO
Table 5: Effect of Other Drugs on VORANIGO Strong and Moderate CYP1A2 Inhibitors Clinical Impact - Concomitant use of VORANIGO with a strong or moderate CYP1A2 inhibitor may increase vorasidenib plasma concentrations [see Clinical Pharmacology (12.3)], which may increase the risk of adverse reactions.
Prevention or Management - Avoid concomitant use of VORANIGO with strong and moderate CYP1A2 inhibitors.
- If concomitant use of moderate CYP1A2 inhibitors cannot be avoided, monitor for increased adverse reactions and modify the dosage for adverse reactions as recommended [see Dosage and Administration (2.4)].
Moderate CYP1A2 Inducers Clinical Impact - Concomitant use of VORANIGO with moderate CYP1A2 inducers and smoking tobacco may decrease vorasidenib plasma concentrations [see Clinical Pharmacology (12.3)], which may reduce the anti-tumor activity of VORANIGO.
Prevention or Management - Avoid concomitant use of VORANIGO with moderate CYP1A2 inducers and smoking tobacco.
7.2 Effect of VORANIGO on Other Drugs
Table 6: Effect of VORANIGO on Other Drugs Certain CYP3A Substrates Clinical Impact - Concomitant use of VORANIGO with CYP3A substrates may decrease plasma concentrations of CYP3A substrates.
Prevention or Management - Avoid concomitant use of VORANIGO with CYP3A substrates, where a minimal concentration change may lead to reduced therapeutic effect.
Hormonal Contraception Clinical Impact - Concomitant use of VORANIGO may decrease the concentrations of hormonal contraceptives, which may lead to contraception failure and/or an increase in breakthrough bleeding.
Prevention or Management - If concomitant use cannot be avoided, use with nonhormonal contraception methods.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings from animal studies and its mechanism of action [see Clinical Pharmacology (12.1)], VORANIGO can cause fetal harm when administered to a pregnant woman. There are no available data on VORANIGO use in pregnant women to inform a drug-associated risk. In animal embryo-fetal development studies, oral administration of vorasidenib to pregnant rats and rabbits during the period of organogenesis caused embryo-fetal toxicity at ≥8 times the human exposure based on the AUC at the highest recommended dose (see Data). Advise pregnant women of the potential risk to the fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
In an embryo-fetal development study, vorasidenib was administered to pregnant rats via oral gavage at dose levels of 10, 25, and 75 mg/kg/day during the period of organogenesis (gestation days 6 to 17). Embryo-fetal toxicity (higher incidence of early resorptions, and visceral malformations of kidney and testes) occurred in rats at the maternally toxic dose of 75 mg/kg/day (approximately 170 times the human exposure based on the AUC at the highest recommended dose). Malformation of heart occurred in a rat at a dose of 25 mg/kg (approximately 97 times the human exposure based on the AUC at the highest recommended dose). Dose-related delayed ossification of bones and short ribs associated with decreased fetal body weights was observed at 10 and 25 mg/kg/day in the absence of maternal toxicity and at 75 mg/kg/day. The dose of 10 mg/kg/day is ≥45 times the human exposure based on the AUC at the highest recommended dose.
In an embryo-fetal development study, oral administration of vorasidenib to pregnant rabbits at dose levels of 2, 6, and 18 mg/kg/day during the period of organogenesis (gestation days 6 to 19) resulted in maternal toxicity at all doses (≥1.5 times the human exposure based on the AUC at the highest recommended dose) and caused higher incidence of late resorptions at 18 mg/kg/day as well as decreased fetal weights and delayed ossification at doses ≥6 mg/kg/day (≥8 times the human exposure based on the AUC at the highest recommended dose).
8.2 Lactation
Risk Summary
There are no data on the presence of vorasidenib or its metabolites in human milk, their effects on the breastfed child, or on milk production. Because of the potential for adverse reactions in breastfed children from VORANIGO, advise women not to breastfeed during treatment with VORANIGO and for 2 months after the last dose.
8.3 Females and Males of Reproductive Potential
Based on animal embryo-fetal toxicity studies, VORANIGO can cause fetal harm when administered to pregnant women [see Use in Specific Populations (8.1)].
Pregnancy Testing
Verify pregnancy status in females of reproductive potential prior to starting VORANIGO [see Use in Specific Populations (8.1)].
Contraception
Females
Advise females of reproductive potential to use effective nonhormonal contraception during treatment with VORANIGO and for 3 months after the last dose. VORANIGO can render some hormonal contraceptives ineffective [see Drug Interactions (7.2)].
Infertility
Based on findings in animals, VORANIGO may impair fertility in females and males of reproductive potential. The effects on female and male fertility were not reversible in rats [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of VORANIGO have been established in pediatric patients aged 12 years and older for the treatment of Grade 2 IDH1- or IDH2-mutant astrocytoma or oligodendroglioma. Use of VORANIGO for this indication in this age group is supported by evidence from an adequate and well-controlled study of VORANIGO in adult and pediatric patients with additional population pharmacokinetic data demonstrating that age had no clinically meaningful effect on the pharmacokinetics of vorasidenib. In addition, the course of IDH1- or IDH2-mutant astrocytoma or oligodendroglioma is sufficiently similar between adults and pediatric patients to allow extrapolation of pharmacokinetic data in adults to pediatric patients [see Adverse Reactions (6.1), Clinical Pharmacology (12.3) and Clinical Studies (14)].
The exposure of vorasidenib in pediatric patients 12 years and older is predicted to be within range of exposure observed in adults at the recommended dosages [see Clinical Pharmacology (12.3)].
The safety and effectiveness of VORANIGO have not been established in pediatric patients younger than 12 years of age for any indication.
8.5 Geriatric Use
Of the 167 patients who were randomized and received VORANIGO 40 mg once daily in the INDIGO trial, 1.2% (2 patients) were 65 years or older. Clinical studies of VORANIGO did not include sufficient numbers of patients aged ≥65 to determine whether they respond differently from younger subjects.
8.6 Renal Impairment
No dosage adjustment is recommended for patients with creatinine clearance (CLcr) >40 mL/min.
The pharmacokinetics and safety of vorasidenib in patients with CLcr ≤40 mL/min or renal impairment requiring dialysis have not been studied [see Clinical Pharmacology (12.3)]. For patients with CLcr ≤40 mL/min or who require dialysis, monitor for increased adverse reactions and modify the dosage for adverse reactions as recommended [see Dosage and Administration (2.4), Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
No dosage adjustment is recommended for patients with mild or moderate (Child-Pugh Class A or B) hepatic impairment [see Clinical Pharmacology (12.3)].
The pharmacokinetics and safety of vorasidenib in patients with severe hepatic impairment (Child-Pugh Class C) have not been studied. For patients with severe hepatic impairment, monitor for increased adverse reactions and modify the dosage for adverse reactions as recommended [see Dosage and Administration (2.4), Clinical Pharmacology (12.3)].
-
11 DESCRIPTION
VORANIGO tablets contain vorasidenib, an isocitrate dehydrogenase-1 (IDH1) and isocitrate dehydrogenase-2 (IDH2) inhibitor. Vorasidenib is present as the hemicitric acid hemihydrate co-crystal. The chemical name of the co-crystal is 6-(6-chloropyridin-2-yl)-N2,N4-bis[(2R)-1,1,1-trifluoropropan-2-yl]-1,3,5-triazine-2,4-diamine, 2-hydroxypropane-1,2,3-tricarboxylic acid, hydrate (2:1:1). It has the following chemical structure:
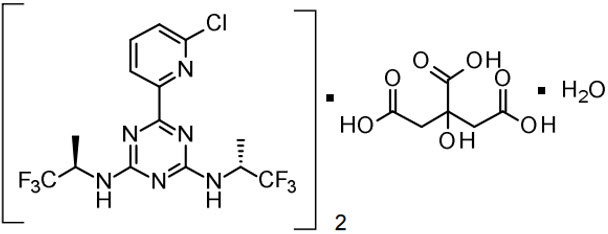
The molecular formula is C14H13ClF6N6 ½ C6H8O7 ½ H2O and the molecular weight is 519.8 g/mol. Vorasidenib hemicitric acid hemihydrate is a white to off-white solid practically insoluble in aqueous solutions between pH 1.2 to 6.8.
VORANIGO is available as a 10 mg and 40 mg strength film-coated tablet for oral administration. The strengths reflect the amount of active ingredient vorasidenib in each tablet. Each tablet core contains the following inactive ingredients: croscarmellose sodium, magnesium stearate, microcrystalline cellulose, silicified microcrystalline cellulose and sodium lauryl sulfate. The tablet coating includes hypromellose, lactose monohydrate, macrogol and titanium dioxide. Each tablet is printed with black ink that contains black iron oxide, hypromellose and propylene glycol.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Vorasidenib is a small molecule inhibitor that targets isocitrate dehydrogenase-1 and 2 (IDH1 and IDH2) enzymes. In vitro, vorasidenib inhibited the IDH1 wild type and mutant variants, including R132H and the IDH2 wild type and mutant variants. In cell-based and in vivo tumor models expressing IDH1 or IDH2 mutated proteins, vorasidenib decreased production of 2-hydroxyglutarate (2-HG) and partially restored cellular differentiation.
12.2 Pharmacodynamics
Exposure-Response Relationships
Vorasidenib decreases 2-HG tumor concentrations in patients with IDH1 or IDH2 mutated glioma. Relative to tumors from patients in the untreated group, the posterior median percentage reduction (95% credible interval) in tumor 2-HG was 64% (22%, 88%) to 93% (76%, 98%) in tumors from patients who received vorasidenib at exposures that were 0.3 to 0.8 times the exposure observed with the highest recommended dosage.
The exposure-response relationship and time course of pharmacodynamic response for the safety and effectiveness of vorasidenib have not been fully characterized.
Cardiac Electrophysiology
When the recommended dose of VORANIGO is administered 1 hour prior to a meal, a mean increase in the QTc interval >20 msec is unlikely. There is insufficient information to characterize the QTc effects of VORANIGO at higher vorasidenib concentrations, e.g., when administered with a meal or when co-administered with a moderate CYP1A2 inhibitor [see Clinical Pharmacology (12.3)].
12.3 Pharmacokinetics
Vorasidenib maximum plasma concentration (Cmax) and AUC increased approximately proportionally over the dose range of 10 to 200 mg (0.2 to 4 times the exposure of the highest approved recommended dosage) following once daily administration of single and multiple doses. At the highest approved recommended dosage, steady state mean (CV%) Cmax is 133 ng/mL (73%) and AUC is 1,988 hng/mL (95%). Steady state is achieved within 28 days of once daily dosing and the mean accumulation ratio of AUC is 4.4.
Absorption
The median (minimum, maximum) time to maximum plasma concentrations (tmax) at steady state is 2 hours (0.5 to 4 hours).
The mean absolute bioavailability of vorasidenib is 34%.
Food Effect
A high-fat and high-calorie (total 800-1,000 calories, of which 500-600 from fat) meal increased vorasidenib Cmax 3.1-fold and AUC 1.4-fold, compared to the fasting conditions.
A low-fat and low-calorie (total 400-500 calories, of which 100-125 from fat) meal increased vorasidenib Cmax 2.3-fold and AUC 1.4-fold, compared to the fasting conditions.
Distribution
The mean (CV%) volume of distribution at steady state of vorasidenib is 3,930 L (40%).
The protein binding is 97% in human plasma independent of vorasidenib concentrations in vitro.
The brain tumor-to-plasma concentration ratio is 1.6.
Elimination
The mean (CV%) steady state terminal half-life is 10 days (57%) and oral clearance is 14 L/h (56%).
Specific Populations
No clinically significant effects on the pharmacokinetics of vorasidenib were observed based on age (16 to 75 years), sex, race (White, Black or African American, Asian, American Indian/Alaskan Native, Native Hawaiian or Other Pacific Islander), ethnicity (Hispanic and non-Hispanic), body weight (43.5 to 168 kg), mild or moderate hepatic impairment (Child-Pugh Class A or B) or CLcr >40 mL/min (as Cockcroft-Gault). The pharmacokinetics of vorasidenib has not been studied in patients with severe hepatic impairment (Child-Pugh Class C), in patients with CLcr ≤40 mL/min or in patients with renal impairment who require dialysis.
Drug Interaction Studies
Clinical Studies and Model-Informed Approaches
Effect of Other Drugs on Vorasidenib
Strong and Moderate CYP1A2 Inhibitors: Concomitant use of ciprofloxacin (moderate CYP1A2 inhibitor) increased vorasidenib plasma Cmax 1.3-fold and AUC 2.5-fold.
Concomitant use with fluvoxamine (strong CYP1A2 inhibitor) is predicted to increase vorasidenib Cmax and AUC by ≥5-fold.
Effect of Vorasidenib on Other Drugs
CYP3A Substrates: Concomitant use of multiple doses of vorasidenib with CYP3A substrates is predicted to decrease the concentrations of these substrates.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been conducted with vorasidenib. Vorasidenib and its major circulating metabolite, AGI-69460, were not mutagenic in an in vitro bacterial reverse mutation (Ames) assay. Vorasidenib was not clastogenic in an in vitro human lymphocyte micronucleus assay, or an in vivo rat bone marrow micronucleus assay.
Fertility studies in animals have not been conducted with vorasidenib. In repeat-dose toxicity studies up to 13 weeks in duration with oral administration of vorasidenib in rats, adverse effects in female reproductive organs included atrophy, decreased/absent corpora lutea, increased atretic follicles, and interstitial cell vacuolation of the ovaries, atrophy, hypertrophy, and metaplasia of the uterus, hyperplasia of the cervix, atrophy, hyperplasia, and mucification of the vagina, and estrous cycle variations at doses ≥3 mg/kg/day (≥12 times the exposure based on AUC in humans at the highest recommended dose). Adverse effects in male reproductive organs in rats included tubular degeneration and atrophy of the testes, degeneration of seminiferous tubules, cellular debris in the epididymides, epithelial atrophy and inflammation in the prostate, and atrophy in the seminal vesicles at doses ≥3 mg/kg/day (≥12 times the exposure based on AUC in humans at the highest recommended dose). Findings in the male rats were not reversible. In the 4-week repeat-dose toxicity studies in monkeys, oral administration of vorasidenib led to adverse effects in male and female reproductive organs including fibrotic hypoplasia of the testes in males at doses ≥10 mg/kg/day (approximately 17 times the exposure based on AUC in humans at the highest recommended dose) and decreased uterine weights in females at doses ≥10 mg/kg/day (approximately 22 times the exposure based on AUC in humans at the highest recommended dose). Findings in male and female monkeys were reversible.
13.2 Animal Toxicology and Pharmacology
In repeat-dose toxicity studies, oral administration of vorasidenib to rats for 28 days led to ototoxicity findings of reversible neutrophil infiltration of the epithelial lining of the middle ear and Eustachian tube at doses >3 mg/kg/day (>12-times the human exposure based on the AUC at the highest recommended dose). Additional findings in a 28-day ototoxicity study included edema in the tympanic cavity at doses >30 mg/kg/day. In addition, oral administration of vorasidenib to monkeys for 13 weeks resulted in dilated cardiomyopathy and secondary congestive heart failure at a dose of 20 mg/kg/day (105 times the human exposure based on the AUC at the highest recommended dose). Skeletal muscle was a target organ in the repeat dose toxicology studies in rats and monkeys at doses ≥13 times the AUC in patients at the highest recommended dose. Findings included decreased hind limb muscle tone, abnormal gait with limited hind limb usage, and low carriage, associated with small size and atrophy of the muscle in rats and necrosis and mononuclear/mixed cell infiltrates in the muscle in monkeys.
-
14 CLINICAL STUDIES
The efficacy of VORANIGO was evaluated in the INDIGO trial (Study AG881-C-004), a randomized, multicenter, double-blind, placebo-controlled study of 331 patients (NCT04164901). Eligible patients were required to have IDH1- or IDH2-mutant Grade 2 astrocytoma or oligodendroglioma with prior surgery including biopsy, sub-total resection, or gross total resection. Patients were required to have measurable, non-enhancing disease; patients with centrally confirmed minimal, non-nodular, non-measurable enhancement were eligible. Patients who received prior anti-cancer treatment, including chemotherapy or radiation therapy were excluded. Patients were randomized to receive either VORANIGO 40 mg orally once daily or placebo orally once daily until disease progression or unacceptable toxicity. IDH1 or IDH2 mutation status was prospectively determined by the Life Technologies Corporation Oncomine Dx Target Test.
Randomization was stratified by local 1p19q status (co-deleted or not co-deleted) and baseline tumor size (diameter ≥2 cm or <2 cm). Patients who were randomized to placebo were allowed to cross over to receive VORANIGO after documented radiographic disease progression. Tumor assessments were performed every 12 weeks.
A total of 331 patients were randomized, 168 to the VORANIGO arm and 163 to the placebo arm. The median age was 40 years (range: 16 to 71); 57% were male; 78% were White, 4% were Asian, 1% were Black or African American and 16% had race not reported; 78% were not Hispanic or Latino; 52% oligodendroglioma and 48% astrocytoma; 79% had one prior surgery and 21% had ≥2 prior surgeries. In the VORANIGO arm, 14% of patients had biopsy, 48% had sub-total resection and 51% had gross-total resection. The majority of IDH1 mutations consisted of R132H (87%). The other alleles were reported as follows: R132C (5%), R132G (3%), R132L (1%), and R132S (1%). IDH2 mutations consisted of R172K (2%) and R172G (1%).
The major efficacy outcome was progression-free survival (PFS) as evaluated by a blinded independent review committee (BIRC) per modified Response Assessment in Neuro-Oncology for Low Grade Glioma (RANO-LGG) criteria.
Efficacy results are summarized in Table 7 and Figure 1.
Table 7: Efficacy Results for the INDIGO Trial (Study AG881-C-004) Efficacy Parameter VORANIGO
40 mg daily
(n=168)Placebo
(n=163)CI = Confidence Interval - * Stratified Cox proportional hazard model, stratified by 1p19q status and baseline tumor size.
- † Based on one-sided stratified log-rank test compared to the pre-specified α of 0.000359 (one-sided).
Progression-Free Survival (PFS) Number of Events, n (%) Progressive disease 47 (28) 88 (54) Death 0 0 Hazard ratio (95% CI)* 0.39 (0.27, 0.56) p-value† <0.0001 Figure 1: Kaplan-Meier Curve for Progression-Free Survival per BIRC for the INDIGO Trial
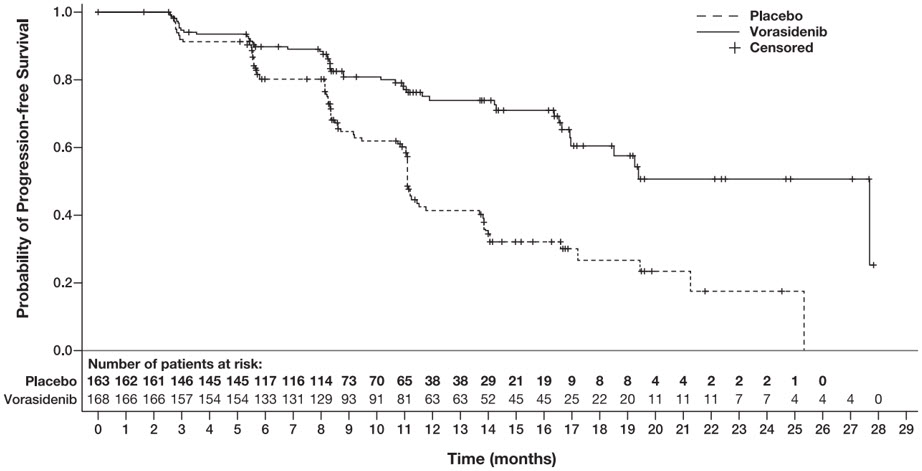
The major efficacy analyses are supported by a prospectively defined key secondary outcome measure time to next intervention (defined as the time from randomization to the initiation of first subsequent anticancer therapy or death due to any cause). The median time to next intervention was not reached for patients in the VORANIGO arm and 17.8 months for patients in the placebo arm (HR=0.26; 95% CI: [0.15, 0.43], p <0.0001).
-
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
VORANIGO® (vorasidenib) tablets are supplied in two strengths:
10 mg tablets: White to off-white, round film-coated tablet imprinted with "10" in black ink on one side and plain on the other side.
- Each carton contains one 30-count bottle of 10 mg tablets with desiccant canister(s) and child-resistant cap (NDC: 72694-879-10)
40 mg tablets: White to off-white, oblong film-coated tablet imprinted with "40" in black ink on one side and plain on the other side.
- Each carton contains one 30-count bottle of 40 mg tablets with desiccant canister(s) and child-resistant cap (NDC: 72694-728-40)
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Hepatotoxicity
Inform patients of the risk of hepatotoxicity and to promptly report any signs or symptoms of hepatotoxicity to their healthcare provider [see Warnings and Precautions (5.1)].
Embryo-Fetal Toxicity
Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females of reproductive potential to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.2) and Use in Specific Populations (8.1, 8.3)].
Advise females of reproductive potential to use effective nonhormonal contraception during treatment with VORANIGO and for 3 months after the last dose [see Use in Specific Populations (8.3)] since VORANIGO can render some hormonal contraceptives ineffective [see Drug Interactions (7.2)].
Advise males with female partners of reproductive potential to use effective contraception during treatment with VORANIGO and for 3 months after the last dose [see Use in Specific Populations (8.3)].
Lactation
Advise women not to breastfeed during treatment with VORANIGO and for 2 months after the last dose [see Use in Specific Populations (8.2)].
Infertility
Advise females and males of reproductive potential that VORANIGO may impair fertility [see Use in Specific Populations (8.3) and Nonclinical Toxicology (13.1)].
Drug Interactions
Advise patients and caregivers to inform their healthcare provider of all concomitant medications, including prescription medicines, over-the counter drugs, vitamins and herbal products [see Drug Interactions (7)].
Advise patients and caregivers to inform their healthcare provider if they currently smoke tobacco as it may affect how well VORANIGO works [see Drug Interactions (7)].
Instructions for Taking VORANIGO
Advise patients to swallow tablets whole with a glass of water, with or without food, and to not split, crush or chew VORANIGO tablets [see Dosage and Administration (2.3)].
If a patient misses a dose by less than 6 hours, instruct patients to take the missed dose right away. If a patient misses a dose by 6 or more hours, instruct patients to skip the dose for that day. Advise patients to take the next dose at the usual time [see Dosage and Administration (2.3)].
If a patient vomits a dose, instruct patients not to take another dose. Advise patients to take the next dose at the usual time [see Dosage and Administration (2.3)].
-
SPL UNCLASSIFIED SECTION
Manufactured for Servier Pharmaceuticals LLC, Boston, MA 02210
Servier and the Servier logo are trademarks of Les Laboratoires Servier.
VORANIGO® is a registered trademark of Servier Pharmaceuticals LLC, a wholly owned, indirect subsidiary of Les Laboratoires Servier.
Oncomine™ Dx Target Test is a trademark of Life Sciences Corporation, a part of Thermo Fisher Scientific Inc.
© 2024 Servier Pharmaceuticals LLC
-
PATIENT PACKAGE INSERT
-
PRINCIPAL DISPLAY PANEL - 10 mg Tablet Bottle Carton
NDC: 72694-879-10
Voranigo®
(vorasidenib)
tablets10 mg
Swallow tablets whole.
Do not split, crush, or chew the tablet.1 bottle containing 30 tablets
Rx only
-
PRINCIPAL DISPLAY PANEL - 40 mg Tablet Bottle Carton
NDC: 72694-728-40
Voranigo®
(vorasidenib)
tablets40 mg
Swallow tablets whole.
Do not split, crush, or chew the tablet.1 bottle containing 30 tablets
Rx only
-
INGREDIENTS AND APPEARANCE
VORANIGO
vorasidenib tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 72694-879 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength vorasidenib (UNII: 789Q85GA8P) (vorasidenib - UNII:789Q85GA8P) vorasidenib 10 mg Inactive Ingredients Ingredient Name Strength microcrystalline cellulose (UNII: OP1R32D61U) croscarmellose sodium (UNII: M28OL1HH48) silicon dioxide (UNII: ETJ7Z6XBU4) sodium lauryl sulfate (UNII: 368GB5141J) magnesium stearate (UNII: 70097M6I30) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) lactose monohydrate (UNII: EWQ57Q8I5X) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) titanium dioxide (UNII: 15FIX9V2JP) FERROSOFERRIC OXIDE (UNII: XM0M87F357) propylene glycol (UNII: 6DC9Q167V3) Product Characteristics Color WHITE (off-white) Score no score Shape ROUND Size 6mm Flavor Imprint Code 10 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 72694-879-10 1 in 1 CARTON 08/07/2024 1 30 in 1 BOTTLE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA218784 08/07/2024 VORANIGO
vorasidenib tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 72694-728 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength vorasidenib (UNII: 789Q85GA8P) (vorasidenib - UNII:789Q85GA8P) vorasidenib 40 mg Inactive Ingredients Ingredient Name Strength microcrystalline cellulose (UNII: OP1R32D61U) croscarmellose sodium (UNII: M28OL1HH48) silicon dioxide (UNII: ETJ7Z6XBU4) sodium lauryl sulfate (UNII: 368GB5141J) magnesium stearate (UNII: 70097M6I30) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) lactose monohydrate (UNII: EWQ57Q8I5X) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) titanium dioxide (UNII: 15FIX9V2JP) FERROSOFERRIC OXIDE (UNII: XM0M87F357) propylene glycol (UNII: 6DC9Q167V3) Product Characteristics Color WHITE (off-white) Score no score Shape OVAL (oblong) Size 15mm Flavor Imprint Code 40 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 72694-728-40 1 in 1 CARTON 08/07/2024 1 30 in 1 BOTTLE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA218784 08/07/2024 Labeler - Servier Pharmaceuticals LLC (116608503)
Trademark Results [VORANIGO]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 VORANIGO 97578483 not registered Live/Pending |
BIOFARMA 2022-09-05 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.