HALAVEN- eribulin mesylate injection
Halaven by
Drug Labeling and Warnings
Halaven by is a Prescription medication manufactured, distributed, or labeled by Eisai Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use HALAVEN safely and effectively. See full prescribing information for HALAVEN.
HALAVEN® (eribulin mesylate) injection, for intravenous use
Initial U.S. Approval: 2010
INDICATIONS AND USAGE
HALAVEN is a microtubule inhibitor indicated for the treatment of patients with:
- Metastatic breast cancer who have previously received at least two chemotherapeutic regimens for the treatment of metastatic disease. Prior therapy should have included an anthracycline and a taxane in either the adjuvant or metastatic setting. (1.1)
- Unresectable or metastatic liposarcoma who have received a prior anthracycline-containing regimen. (1.2)
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
Injection: 1 mg per 2 mL (0.5 mg per mL) (3)
CONTRAINDICATIONS
None (4)
WARNINGS AND PRECAUTIONS
-
Neutropenia: Monitor peripheral blood cell counts and adjust dose as appropriate. (5.1)
-
Peripheral Neuropathy: Monitor for signs of neuropathy. Manage with dose delay and adjustment. (5.2)
-
Embryo-Fetal Toxicity: Can cause fetal harm. Advise females of reproductive potential of the potential risk to the fetus and to use effective contraception. (5.3, 8.1, 8.3)
- QT Prolongation: Monitor for prolonged QT intervals in patients with congestive heart failure, bradyarrhythmias, drugs known to prolong the QT interval, and electrolyte abnormalities. Avoid in patients with congenital long QT syndrome. (5.4)
ADVERSE REACTIONS
The most common adverse reactions (≥25%) in metastatic breast cancer were neutropenia, anemia, asthenia/fatigue, alopecia, peripheral neuropathy, nausea, and constipation. (6.1)
The most common adverse reactions (≥25%) in liposarcoma and leiomyosarcoma were fatigue, nausea, alopecia, constipation, peripheral neuropathy, abdominal pain, and pyrexia. The most common (≥5%) Grade 3-4 laboratory abnormalities in liposarcoma and leiomyosarcoma were neutropenia, hypokalemia, and hypocalcemia. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Eisai Inc. at (1-877-873-4724) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatchUSE IN SPECIFIC POPULATIONS
- Lactation: Do not breastfeed. (8.2)
- Hepatic Impairment: A lower starting dose is recommended for patients with mild (Child-Pugh A) and moderate (Child-Pugh B) hepatic impairment. Patients with severe hepatic impairment (Child-Pugh C) were not studied. (8.6)
- Renal Impairment: A lower starting dose is recommended for patients with moderate (CLcr 30-49 mL/min) or severe (CLcr 15-29 mL/min) renal impairment. (8.7)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 12/2017
- Metastatic breast cancer who have previously received at least two chemotherapeutic regimens for the treatment of metastatic disease. Prior therapy should have included an anthracycline and a taxane in either the adjuvant or metastatic setting. (1.1)
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Metastatic Breast Cancer
1.2 Liposarcoma
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dose
2.2 Dose Modification
2.3 Instructions for Preparation and Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Neutropenia
5.2 Peripheral Neuropathy
5.3 Embryo-Fetal Toxicity
5.4 QT Prolongation
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Effects of Other Drugs on HALAVEN
7.2 Effects of HALAVEN on Other Drugs
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
8.7 Renal Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Metastatic Breast Cancer
14.2 Liposarcoma
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
1.1 Metastatic Breast Cancer
HALAVEN is indicated for the treatment of patients with metastatic breast cancer who have previously received at least two chemotherapeutic regimens for the treatment of metastatic disease. Prior therapy should have included an anthracycline and a taxane in either the adjuvant or metastatic setting [see Clinical Studies (14.1)].
1.2 Liposarcoma
HALAVEN is indicated for the treatment of patients with unresectable or metastatic liposarcoma who have received a prior anthracycline-containing regimen [see Clinical Studies (14.2)].
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dose
The recommended dose of HALAVEN is 1.4 mg/m2 administered intravenously over 2 to 5 minutes on Days 1 and 8 of a 21-day cycle.
The recommended dose of HALAVEN in patients with mild hepatic impairment (Child-Pugh A) is 1.1 mg/m2 administered intravenously over 2 to 5 minutes on Days 1 and 8 of a 21-day cycle [see Use in Specific Populations (8.6)].
The recommended dose of HALAVEN in patients with moderate hepatic impairment (Child-Pugh B) is 0.7 mg/m2 administered intravenously over 2 to 5 minutes on Days 1 and 8 of a 21-day cycle [see Use in Specific Populations (8.6)].
The recommended dose of HALAVEN in patients with moderate or severe renal impairment (creatinine clearance (CLcr) 15-49 mL/min) is 1.1 mg/m2 administered intravenously over 2 to 5 minutes on Days 1 and 8 of a 21-day cycle [see Use in Specific Populations (8.7)].
2.2 Dose Modification
Assess for peripheral neuropathy and obtain complete blood cell counts prior to each dose.
Recommended dose delays
- Do not administer HALAVEN on Day 1 or Day 8 for any of the following:
- ANC < 1,000/mm3
- Platelets < 75,000/mm3
- Grade 3 or 4 non-hematological toxicities.
- The Day 8 dose may be delayed for a maximum of 1 week.
- If toxicities do not resolve or improve to ≤ Grade 2 severity by Day 15, omit the dose.
- If toxicities resolve or improve to ≤ Grade 2 severity by Day 15, administer HALAVEN at a reduced dose and initiate the next cycle no sooner than 2 weeks later.
Recommended dose reductions
- If a dose has been delayed for toxicity and toxicities have recovered to Grade 2 severity or less, resume HALAVEN at a reduced dose as set out in Table 1.
- Do not re-escalate HALAVEN dose after it has been reduced.
Table 1: Recommended Dose Reductions Event Description Recommended HALAVEN
DosePermanently reduce the 1.4 mg/m2 HALAVEN dose for any of the
following:1.1 mg/m2 ANC <500/mm3 for >7 days ANC <1,000 /mm3 with fever or infection Platelets <25,000/mm3 Platelets <50,000/mm3 requiring transfusion Non-hematologic Grade 3 or 4 toxicities Omission or delay of Day 8 HALAVEN dose in previous cycle for toxicity Occurrence of any event requiring permanent dose reduction while receiving 1.1 mg/m2 0.7 mg/m2 Occurrence of any event requiring permanent dose reduction while receiving 0.7 mg/m2 Discontinue HALAVEN ANC = absolute neutrophil count.
Toxicities graded in accordance with National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events
(CTCAE) version 3.0.2.3 Instructions for Preparation and Administration
Aseptically withdraw the required amount of HALAVEN from the single-use vial and administer undiluted or diluted in 100 mL of 0.9% Sodium Chloride Injection, USP.
Do not dilute in or administer through an intravenous line containing solutions with dextrose. Do not administer in the same intravenous line concurrent with the other medicinal products.
Store undiluted HALAVEN in the syringe for up to 4 hours at room temperature or for up to 24 hours under refrigeration (40°F or 4°C). Store diluted solutions of HALAVEN for up to 4 hours at room temperature or up to 24 hours under refrigeration.
Discard unused portions of the vial.
- Do not administer HALAVEN on Day 1 or Day 8 for any of the following:
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Neutropenia
In Study 1, severe neutropenia (ANC < 500/mm3) lasting more than one week occurred in 12% (62/503) of patients with metastatic breast cancer, leading to discontinuation in <1% of patients. Febrile neutropenia (fever ≥38.5°C with Grade 3 or 4 neutropenia) occurred in 5% (23/503) of patients; two patients (0.4%) died from complications of febrile neutropenia [see Adverse Reactions (6.1)].
In Study 1, patients with alanine aminotransferase (ALT) or aspartate aminotransferase (AST) > 3 × ULN (upper limit of normal) experienced a higher incidence of Grade 4 neutropenia and febrile neutropenia than patients with normal aminotransferase levels. Patients with bilirubin > 1.5 × ULN also had a higher incidence of Grade 4 neutropenia and febrile neutropenia.
In Study 2, severe neutropenia (ANC < 500/mm3) lasting more than one week occurred in 12% (26/222) of patients with liposarcoma or leiomyosarcoma. Febrile neutropenia occurred in 0.9% of patients treated with HALAVEN and fatal neutropenic sepsis in 0.9% [see Adverse Reactions (6.1)].
Monitor complete blood counts prior to each dose; increase the frequency of monitoring in patients who develop Grade 3 or 4 cytopenias. Delay administration of HALAVEN and reduce subsequent doses in patients who experience febrile neutropenia or Grade 4 neutropenia lasting longer than 7 days [see Dosage and Administration (2.2)]. Clinical studies of HALAVEN did not include patients with baseline neutrophil counts below 1,500/mm3.
5.2 Peripheral Neuropathy
In Study 1, Grade 3 peripheral neuropathy occurred in 8% (40/503) of patients, and Grade 4 in 0.4% (2/503) of patients with metastatic breast cancer (MBC). Peripheral neuropathy was the most common toxicity leading to discontinuation of HALAVEN (5% of patients; 24/503) in Study 1. Neuropathy lasting more than one year occurred in 5% (26/503) of patients. Twenty-two percent (109/503) of patients developed a new or worsening neuropathy that had not recovered within a median follow-up duration of 269 days (range 25-662 days).
In Study 2, Grade 3 peripheral neuropathy occurred in 3.1% (7/223) of HALAVEN-treated patients. Peripheral neuropathy led to discontinuation of HALAVEN in 0.9% of patients. The median time to first occurrence of peripheral neuropathy of any severity was 5 months (range: 3.5 months to 9 months). Neuropathy lasting more than 60 days occurred in 58% (38/65) of patients. Sixty three percent (41/65) had not recovered within a median follow-up duration of 6.4 months (range: 27 days to 29 months).
Monitor patients closely for signs of peripheral motor and sensory neuropathy. Withhold HALAVEN in patients who experience Grade 3 or 4 peripheral neuropathy, until resolution to Grade 2 or less [see Dosage and Administration (2.2)].
5.3 Embryo-Fetal Toxicity
Based on findings from an animal reproduction study and its mechanism of action, HALAVEN can cause fetal harm when administered to a pregnant woman. There are no adequate and well-controlled studies of HALAVEN in pregnant women. In animal reproduction studies, eribulin mesylate caused embryo-fetal toxicity when administered to pregnant rats during organogenesis at doses below the recommended human dose. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with HALAVEN and for at least 2 weeks following the final dose. Advise males with female partners of reproductive potential to use effective contraception during treatment with HALAVEN and for 3.5 months following the final dose [see Use in Specific Populations (8.1)].
5.4 QT Prolongation
In an uncontrolled open-label ECG study in 26 patients, QT prolongation was observed on Day 8, independent of eribulin concentration, with no QT prolongation observed on Day 1. ECG monitoring is recommended if therapy is initiated in patients with congestive heart failure, bradyarrhythmias, drugs known to prolong the QT interval, including Class Ia and III antiarrhythmics, and electrolyte abnormalities. Correct hypokalemia or hypomagnesemia prior to initiating HALAVEN and monitor these electrolytes periodically during therapy. Avoid HALAVEN in patients with congenital long QT syndrome.
-
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, the adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in other clinical trials and may not reflect the rates observed in clinical practice.
The following adverse reactions are discussed in detail in other sections of the labeling:
- Neutropenia [see Warnings and Precautions (5.1)]
- Peripheral neuropathy [see Warnings and Precautions (5.2)]
- QT prolongation [see Warnings and Precautions (5.4)]
In clinical trials, HALAVEN has been administered to 1963 patients including 467 patients exposed to HALAVEN for 6 months or longer. The majority of the 1963 patients were women (92%) with a median age of 55 years (range: 17 to 85 years). The racial and ethnic distribution was White (72%), Black (4%), Asian (9%), and other (3%).
Metastatic Breast Cancer
The most common adverse reactions (≥25%) reported in patients receiving HALAVEN were neutropenia, anemia, asthenia/fatigue, alopecia, peripheral neuropathy, nausea, and constipation. The most common serious adverse reactions reported in patients receiving HALAVEN were febrile neutropenia (4%) and neutropenia (2%). The most common adverse reaction resulting in discontinuation of HALAVEN was peripheral neuropathy (5%).
The adverse reactions described in Table 2 were identified in 750 patients treated in Study 1 [see Clinical Studies (14.1)]. In Study 1, patients were randomized (2:1) to receive either HALAVEN (1.4 mg/m2 on Days 1 and 8 of a 21-day cycle) or single agent treatment chosen by their physician (control group). A total of 503 patients received HALAVEN and 247 patients in the control group received therapy consisting of chemotherapy [total 97% (anthracyclines 10%, capecitabine 18%, gemcitabine 19%, taxanes 15%, vinorelbine 25%, other chemotherapies 10%)] or hormonal therapy (3%). The median duration of exposure was 118 days for patients receiving HALAVEN and 63 days for patients receiving control therapy. Table 2 reports the most common adverse reactions occurring in at least 10% of patients in either group.
Table 2: Adverse Reactionsa with a Per-Patient Incidence of at Least 10% in Study 1
Adverse ReactionsHALAVEN
n=503Control Group
n=247All Grades ≥ Grade 3 All Grades ≥ Grade 3 Blood and lymphatic system disordersb Neutropenia 82% 57% 53% 23% Anemia 58% 2% 55% 4% Nervous system disorders Peripheral neuropathyc 35% 8% 16% 2% Headache 19% <1% 12% <1% General disorders Asthenia/Fatigue 54% 10% 40% 11% Pyrexia 21% <1% 13% <1% Mucosal inflammation 9% 1% 10% 2% Gastrointestinal disorders Nausea 35% 1% 28% 3% Constipation 25% 1% 21% 1% Vomiting 18% 1% 18% 1% Diarrhea 18% 0 18% 0 Musculoskeletal and connective tissue disorders Arthralgia/Myalgia 22% <1% 12% 1% Back pain 16% 1% 7% 2% Bone pain 12% 2% 9% 2% Pain in extremity 11% 1% 10% 1% Metabolism and nutrition disorders Decreased weight 21% 1% 14% <1% Anorexia 20% 1% 13% 1% Respiratory, thoracic, and mediastinal disorders Dyspnea 16% 4% 13% 4% Cough 14% 0 9% 0 Skin and subcutaneous tissue disorders Alopecia 45% NAd 10% NAd Infections Urinary Tract Infection 10% 1% 5% 0 a adverse reactions were graded per National Cancer Institute Criteria for Adverse Events version 4.0.
b based upon laboratory data.
c includes peripheral neuropathy, peripheral sensorimotor neuropathy, peripheral motor neuropathy, polyneuropathy, peripheral sensory neuropathy, and paraesthesia.
d not applicable; (grading system does not specify > Grade 2 for alopecia).Cytopenias: Grade 3 neutropenia occurred in 28% (143/503) of patients who received HALAVEN in Study 1, and 29% (144/503) of patients experienced Grade 4 neutropenia. Febrile neutropenia occurred in 5% (23/503) of patients; two patients (0.4%) died from complications of febrile neutropenia. Dose reduction due to neutropenia was required in 12% (62/503) of patients and discontinuation was required in <1% of patients. The mean time to nadir was 13 days and the mean time to recovery from severe neutropenia (<500/mm3) was 8 days. Grade 3 or greater thrombocytopenia occurred in 1% (7/503) of patients. G-CSF (granulocyte colony-stimulating factor) or GM-CSF (granulocyte–macrophage colony-stimulating factor) was used in 19% of patients who received HALAVEN.
Peripheral Neuropathy: In Study 1, 17% of enrolled patients had Grade 1 peripheral neuropathy and 3% of patients had Grade 2 peripheral neuropathy at baseline. Dose reduction due to peripheral neuropathy was required by 3% (14/503) of patients who received HALAVEN. Four percent (20/503) of patients experienced peripheral motor neuropathy of any grade and 2% (8/503) of patients developed Grade 3 peripheral motor neuropathy.
Liver Function Test Abnormalities: Among patients with Grade 0 or 1 ALT levels at baseline, 18% of HALAVEN-treated patients experienced Grade 2 or greater ALT elevation. One HALAVEN-treated patient without documented liver metastases had concomitant Grade 2 elevations in bilirubin and ALT; these abnormalities resolved and did not recur with re-exposure to HALAVEN.
Less Common Adverse Reactions: The following additional adverse reactions were reported in ≥5% to <10% of the HALAVEN-treated group:
-
Eye Disorders: increased lacrimation
-
Gastrointestinal Disorders: dyspepsia, abdominal pain, stomatitis, dry mouth
-
General Disorders and Administration Site Conditions: peripheral edema
-
Infections and Infestations: upper respiratory tract infection
-
Metabolism and Nutrition Disorders: hypokalemia
-
Musculoskeletal and Connective Tissue Disorders: muscle spasms, muscular weakness
-
Nervous System Disorders: dysgeusia, dizziness
-
Psychiatric Disorders: insomnia, depression
- Skin and Subcutaneous Tissue Disorders: rash
Liposarcoma
The safety of HALAVEN was evaluated in Study 2, an open-label, randomized, multicenter, active-controlled trial, in which patients were randomized (1:1) to receive either HALAVEN 1.4 mg/m2 on Days 1 and 8 of a 21-day cycle or dacarbazine at doses of 850 mg/m2 (20%), 1000 mg/m2 (64%), or 1200 mg/m2 (16%) every 3 weeks. A total of 223 patients received HALAVEN and 221 patients received dacarbazine. Patients were required to have received at least two prior systemic chemotherapy regimens. The trial excluded patients with pre-existing ≥ Grade 3 peripheral neuropathy, known central nervous system metastasis, elevated serum bilirubin or significant chronic liver disease, history of myocardial infarction within 6 months, history of New York Heart Association Class II or IV heart failure, or cardiac arrhythmia requiring treatment. The median age of the safety population in Study 2 was 56 years (range: 24 to 83 years); 67% female; 73% White, 3% Black or African American, 8% Asian/Pacific Islander, and 15% unknown; 99% received prior anthracycline-containing regimen; and 99% received ≥ 2 prior regimens. The median duration of exposure was 2.3 months (range: 21 days to 26 months) for patients receiving HALAVEN [see Clinical Studies (14.2)].
The most common adverse reactions (≥25%) reported in patients receiving HALAVEN were fatigue, nausea, alopecia, constipation, peripheral neuropathy, abdominal pain, and pyrexia. The most common (≥5%) Grade 3-4 laboratory abnormalities reported in patients receiving HALAVEN were neutropenia, hypokalemia, and hypocalcemia. The most common serious adverse reactions reported in patients receiving HALAVEN were neutropenia (4.9%) and pyrexia (4.5%). Permanent discontinuation of HALAVEN for adverse reactions occurred in 8% of patients. The most common adverse reactions resulting in discontinuation of HALAVEN were fatigue and thrombocytopenia (0.9% each). Twenty-six percent of patients required at least one dose reduction. The most frequent adverse reactions that led to dose reduction were neutropenia (18%) and peripheral neuropathy (4.0%).
Table 3 summarizes the incidence of adverse reactions occurring in at least 10% of patients in the HALAVEN-treated arm in Study 2.
Table 3: Adverse Reactionsa Occurring in ≥10% (all Grades) of Patients Treated on the HALAVEN arm and at a Higher Incidence than in the Dacarbazine Arm (Between Arm Difference of ≥5% for All Grades or ≥2% for Grades 3 and 4) (Study 2)b
Adverse ReactionHALAVEN
n=223Dacarbazine
n=221All Grades Grades 3-4 All Grades Grades 3-4 Nervous system disorders Peripheral Neuropathyc 29% 3.1% 8% 0.5% Headache 18% 0% 10% 0% General disorders Pyrexia 28% 0.9% 14% 0.5% Gastrointestinal disorders Constipation 32% 0.9% 26% 0.5% Abdominal paind 29% 1.8% 23% 4.1% Stomatitis 14% 0.9% 5% 0.5% Skin and subcutaneous tissue disorders Alopecia 35% NAe 2.7% NAe Infections Urinary tract infection 11% 2.2% 5% 0.5% a Adverse reactions were graded per National Cancer Institute Criteria for Adverse Events version 4.03 (NCI CTCAE v4.03).
b Safety data from one study site enrolling six patients were excluded.
c Includes peripheral neuropathy, peripheral sensorimotor neuropathy, peripheral motor neuropathy, polyneuropathy, peripheral sensory neuropathy, and paraesthesia.
d Includes abdominal pain, upper abdominal pain, lower abdominal pain, abdominal discomfort.
e Not applicable; (grading system does not specify > Grade 2 for alopecia).Other clinically important adverse reactions occurring in ≥10% of the HALAVEN-treated patients were:
-
Gastrointestinal Disorders: nausea (41%); vomiting (19%), diarrhea (17%)
-
General Disorders: asthenia/fatigue (62%); peripheral edema (12%)
-
Metabolism and Nutrition Disorders: decreased appetite (19%)
-
Musculoskeletal and Connective Tissue Disorders: arthralgia/myalgia (16%); back pain (16%)
- Respiratory Disorders: cough (18%)
Less Common Adverse Reactions: The following additional clinically important adverse reactions were reported in ≥5% to <10% of the HALAVEN-treated group:
-
Blood and Lymphatic System Disorders: thrombocytopenia
-
Eye Disorders: increased lacrimation
-
Gastrointestinal Disorders: dyspepsia
-
Metabolism and Nutrition Disorders: hyperglycemia
-
Musculoskeletal and Connective Tissue Disorders: muscle spasms, musculoskeletal pain
-
Nervous System Disorders: dizziness, dysgeusia
-
Psychiatric Disorders: insomnia, anxiety
-
Respiratory, Thoracic, and Mediastinal Disorders: oropharyngeal pain
- Vascular Disorders: hypotension
Table 4: Laboratory Abnormalities Occurring in ≥10% (all Grades) of Patients Treated on the HALAVEN arm and at a Higher Incidence than in the Dacarbazine Arm (Between Arm Difference of ≥5% for All Grades or ≥2% for Grades 3 and 4)a (Study 2)† Laboratory Abnormality Halaven Dacarbazine All Grades Grades 3 - 4 All Grades Grades 3 – 4 Hematology Anemia 70% 4.1% 52% 6% Neutropenia 63% 32% 30% 8.9% Chemistry Increased alanine aminotransferase (ALT) 43% 2.3% 28% 2.3% Increased aspartate aminotransferase (AST) 36% 0.9% 16% 0.5% Hypokalemia 30% 5.4% 14% 2.8% Hypocalcemia 28% 5% 18% 1.4% Hypophosphatemia 20% 3.2% 11% 1.4% a Each test incidence is based on the number of patients who had both baseline and at least one on-study measurement and at least 1 grade increase from baseline.Halaven group (range 221-222) and dacarbazine group (range 214-215)
† Laboratory results were graded per NCI CTCAE v4.03.6.2 Postmarketing Experience
The following adverse drug reactions have been identified during post-approval of HALAVEN. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
-
Blood and Lymphatic System Disorders: lymphopenia
-
Gastrointestinal Disorders: pancreatitis
-
Hepatobiliary Disorders: hepatotoxicity
-
Immune System Disorders: drug hypersensitivity
-
Infections and Infestations: pneumonia, sepsis/neutropenic sepsis
-
Metabolism and Nutrition Disorders: hypomagnesemia, dehydration
-
Respiratory, thoracic and mediastinal disorders: interstitial lung disease
- Skin and Subcutaneous Tissue Disorders: pruritus, Stevens-Johnson syndrome, toxic epidermal necrolysis
- Neutropenia [see Warnings and Precautions (5.1)]
-
7 DRUG INTERACTIONS
7.1 Effects of Other Drugs on HALAVEN
No drug-drug interactions are expected with CYP3A4 inhibitors, CYP3A4 inducers or P-glycoprotein (P-gp) inhibitors. Clinically meaningful differences in exposure (AUC) were not observed in patients with advanced solid tumors when HALAVEN was administered with or without ketoconazole (a strong inhibitor of CYP3A4 and a P-gp inhibitor) and when HALAVEN was administered with or without rifampin (a CYP3A4 inducer) [see Clinical Pharmacology (12.3)].
7.2 Effects of HALAVEN on Other Drugs
Eribulin does not inhibit CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP2E1 or CYP3A4 enzymes or induce CYP1A2, CYP2C9, CYP2C19 or CYP3A4 enzymes at relevant clinical concentrations. Eribulin is not expected to alter the plasma concentrations of drugs that are substrates of these enzymes [see Clinical Pharmacology (12.3)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings from an animal reproduction study and its mechanism of action, HALAVEN can cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)]. There are no available data on the use of HALAVEN during pregnancy. In an animal reproduction study, eribulin mesylate caused embryo-fetal toxicity when administered to pregnant rats during organogenesis at doses below the recommended human dose [see Data]. Advise pregnant women of the potential risk to a fetus.
The estimated background risks of major birth defects and miscarriage for the indicated populations are unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically-recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
In an embryo-fetal developmental toxicity study, pregnant rats received intravenous infusion of eribulin mesylate during organogenesis (Gestation Days 8, 10, and 12) at doses approximately 0.04, 0.13, 0.43 and 0.64 times the recommended human dose, based on body surface area. Increased abortion and severe fetal external or soft tissue malformations, including the absence of a lower jaw and tongue, or stomach and spleen, were observed at doses 0.64 times the recommended human dose of 1.4 mg/m2 based on body surface area. Increased embryo-fetal death/resorption, reduced fetal weights, and minor skeletal anomalies consistent with developmental delay were also reported at doses at or above a maternally toxic dose of approximately 0.43 times the recommended human dose.
8.2 Lactation
Risk Summary
There is no information regarding the presence of eribulin mesylate or its metabolites in human milk, the effects on the breastfed infant, or the effects on milk production. No lactation studies in animals were conducted. Because of the potential for serious adverse reactions in breastfed infants from eribulin mesylate, advise women not to breastfeed during treatment with HALAVEN and for 2 weeks after the final dose.
8.3 Females and Males of Reproductive Potential
Contraception
Females
Based on findings from an animal reproduction study and its mechanism of action, HALAVEN can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)]. Advise females of reproductive potential to use effective contraception during treatment with HALAVEN and for at least 2 weeks following the final dose.
Males
Based on its mechanism of action, advise males with female partners of reproductive potential to use effective contraception during treatment with HALAVEN and for 3.5 months following the final dose.
Infertility
Males
Based on animal data, HALAVEN may result in damage to male reproductive tissues leading to impaired fertility of unknown duration [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of HALAVEN in pediatric patients below the age of 18 years have not been established.
8.5 Geriatric Use
Study 1 did not include sufficient numbers of subjects with metastatic breast cancer aged 65 years and older to determine whether they respond differently from younger subjects. Of the 827 subjects who received the recommended dose and schedule of HALAVEN in clinical studies with advanced breast cancer, 15% (121/827) were 65 and older, and 2% (17/827) patients were 75 and older. No overall differences in safety were observed between these subjects and younger subjects.
Clinical studies of HALAVEN did not include a sufficient number of subjects in Study 2 aged 65 years and older to determine whether they respond differently from younger subjects.
8.6 Hepatic Impairment
Administration of HALAVEN at a dose of 1.1 mg/m2 to patients with mild hepatic impairment and 0.7 mg/m2 to patients with moderate hepatic impairment resulted in similar exposure to eribulin as a dose of 1.4 mg/m2 to patients with normal hepatic function. Therefore, a lower starting dose of 1.1 mg/m2 is recommended for patients with mild hepatic impairment (Child-Pugh A) and of 0.7 mg/m2 is recommended for patients with moderate hepatic impairment (Child-Pugh B). HALAVEN was not studied in patients with severe hepatic impairment (Child-Pugh C) [see Dosage and Administration (2.1), Clinical Pharmacology (12.3)].
8.7 Renal Impairment
For patients with moderate or severe renal impairment (CLcr 15-49 mL/min), reduce the starting dose to 1.1 mg/m2 [see Dosage and Administration (2.1), Clinical Pharmacology (12.3)].
- 10 OVERDOSAGE
-
11 DESCRIPTION
HALAVEN contains eribulin mesylate, a microtubule dynamics inhibitor. Eribulin mesylate is a synthetic analogue of halichondrin B, a product isolated from the marine sponge Halichondria okadai. The chemical name for eribulin mesylate is 11,15:18,21:24,28-Triepoxy-7,9-ethano-12,15-methano-9H,15H-furo[3,2-i]furo[2',3':5,6]pyrano[4,3-b][1,4]dioxacyclopentacosin-5(4H)-one, 2-[(2S)-3-amino-2-hydroxypropyl]hexacosahydro-3-methoxy-26-methyl-20,27-bis(methylene)-, (2R,3R,3aS,7R,8aS,9S,10aR,11S,12R,13aR,13bS,15S,18S,21S,24S,26R,28R,29aS)-, methanesulfonate (salt). It has a molecular weight of 826.0 (729.9 for free base). The empirical formula is C40H59NO11CH4O3S. Eribulin mesylate has the following structural formula:
![a product isolated from the marine sponge Halichondria okadai. The chemical name for eribulin mesylate is 11,15:18,21:24,28-Triepoxy-7,9-ethano-12,15-methano-9H,15H-furo[3,2-i]furo[2',3':5,6]pyrano[4,3-b][1,4]dioxacyclopentacosin-5(4H)-one, 2-[(2S)-3-amino-2-hydroxypropyl]hexacosahydro-3-methoxy-26-methyl-20,27-bis(methylene)-, (2R,3R,3aS,7R,8aS,9S,10aR,11S,12R,13aR,13bS,15S,18S,21S,24S,26R,28R,29aS)-, methanesulfonate (salt). It has a molecular weight of 826.0 (729.9 for free base). The empirical formula is C40H59NO11CH4O3S. Eribulin mesylate has the following structural formula:](https://fda.report/DailyMed/31ce4750-ded5-4a0b-95e9-f229fa6bc822/halaven-01.jpg)
HALAVEN is a clear, colorless, sterile solution for intravenous administration. Each vial contains 1 mg of eribulin mesylate as a 0.5 mg/mL solution in ethanol: water (5:95).
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Eribulin inhibits the growth phase of microtubules without affecting the shortening phase and sequesters tubulin into nonproductive aggregates. Eribulin exerts its effects via a tubulin-based antimitotic mechanism leading to G2/M cell-cycle block, disruption of mitotic spindles, and, ultimately, apoptotic cell death after prolonged mitotic blockage.
In addition, eribulin treatment of human breast cancer cells caused changes in morphology and gene expression as well as decreased migration and invasiveness in vitro. In mouse xenograft models of human breast cancer, eribulin treatment was associated with increased vascular perfusion and permeability in the tumor cores, resulting in reduced tumor hypoxia, and changes in the expression of genes in tumor specimens associated with a change in phenotype.
12.2 Pharmacodynamics
Cardiac Electrophysiology
The effect of HALAVEN on the QTc interval was assessed in an open-label, uncontrolled, multicenter, single-arm dedicated QT trial. A total of 26 patients with solid tumors received 1.4 mg/m2 of HALAVEN on Days 1 and 8 of a 21-day cycle. A delayed QTc prolongation was observed on Day 8, with no prolongation observed on Day 1. The maximum mean QTcF change from baseline (95% upper confidence interval) was 11.4 (19.5) ms.
12.3 Pharmacokinetics
The pharmacokinetics (PK) of eribulin is linear with a mean elimination half-life of approximately 40 hours, a mean volume of distribution of 43 L/m2 to 114 L/m2 and mean clearance of 1.16 L/hr/m2 to 2.42 L/hr/m2 over the dose range of 0.25 mg/m2 to 4.0 mg/m2. The human plasma protein binding of eribulin at concentrations of 100 ng/mL to 1,000 ng/mL ranges from 49% to 65%. Eribulin exposure after multiple dosing is comparable to that following a single dose. No accumulation of eribulin is observed with weekly administration.
Elimination
Metabolism
Unchanged eribulin was the major circulating species in plasma following administration of 14C-eribulin to patients. Metabolite concentrations represented <0.6% of parent compound, confirming that there are no major human metabolites of eribulin. Cytochrome P450 3A4 (CYP3A4) negligibly metabolizes eribulin in vitro.
Excretion
Eribulin is eliminated primarily in feces unchanged. After administration of 14C-eribulin to patients, approximately 82% of the dose was eliminated in feces and 9% in urine. Unchanged eribulin accounted for approximately 88% and 91% of total eribulin in feces and urine, respectively.
Specific Populations
Age, Sex, and Race/Ethnicity: Based on a population pharmacokinetic analysis with data collected from 340 patients, sex, race, and age do not have a clinically meaningful effect on the exposure of eribulin.
Hepatic Impairment
In a study evaluating the effect of hepatic impairment on the PK of eribulin, eribulin exposures increased by 1.8-fold in patients with mild hepatic impairment (Child-Pugh A; n=7) and by 2.5-fold in patients with moderate (Child-Pugh B; n=5) hepatic impairment as compared to patients with normal hepatic function (n=6). Administration of HALAVEN at a dose of 1.1 mg/m2 to patients with mild hepatic impairment and 0.7 mg/m2 to patients with moderate hepatic impairment resulted in similar exposure to eribulin at a dose of 1.4 mg/m2 to patients with normal hepatic function [see Dosage and Administration (2.1), Use in Specific Populations (8.6)].
Renal Impairment
In a study evaluating the effect of renal impairment on the PK of eribulin, patients with moderate (CLcr 30-49 mL/min; n=7) and severe renal impairment (CLcr 15-29 mL/min; n=6) had 1.5-fold higher eribulin dose-normalized exposures compared to that in patients with normal renal function (CLcr ≥ 80 mL/min; n=6). There were no clinically meaningful changes in patients with mild renal impairment (CLcr 50-79 mL/min; n=27) [see Dosage and Administration (2.1), Use in Specific Populations (8.7)].
Drug Interaction Studies
Effect of Strong Inhibitors or Inducers of CYP3A4 on Eribulin: The effect of a strong CYP3A4 inhibitor and a P-gp inhibitor, ketoconazole, on the PK of eribulin was studied in a crossover trial of 12 patients with advanced solid tumors. No clinically relevant PK interaction was observed when HALAVEN was administered with or without ketoconazole (the geometric mean ratio of the AUC: 0.97; 90% CI: 0.83, 1.12).
The effect of a CYP3A4 inducer, rifampin, on the PK of eribulin was studied in a crossover trial of 14 patients with advanced solid tumors. No clinically relevant PK interaction was observed when HALAVEN was administered with or without rifampin (the geometric mean ratio of the AUC: 1.10; 90 CI%: 0.91, 1.34).
Effect of Eribulin on CYP Substrates: Eribulin shows no induction potential for CYP1A, CYP2B6, CYP2C9, CYP2C19, and CYP3A in primary human hepatocytes. Eribulin inhibits CYP3A4 activity in human liver microsomes, but it is unlikely that eribulin will substantially increase the plasma levels of CYP3A4 substrates. No significant inhibition of CYP1A2, CYP2C9, CYP2C19, CYP2D6, or CYP2E1 was detected with eribulin concentrations up to 5 μM in pooled human liver microsomes. In vitro drug interaction studies indicate that eribulin does not inhibit drugs that are substrates of these enzymes and it is unlikely that eribulin will affect plasma levels of drugs that are substrates of CYP enzymes.
Effect of Transporters on Eribulin: In vitro data suggest that eribulin at clinically relevant concentrations is a substrate of P-gp, but is not a substrate of breast cancer resistance protein (BCRP), multidrug resistance proteins (MRP2, MRP4), bile salt extrusion pump (BSEP), organic anion transporting polypeptides (OATP1B1, OATP1B3), organic anion transporters (OAT1, OAT3), organic cation transporters (OCT1, OCT2), or multidrug and toxin extrusion 1 (MATE1).
Effect of Eribulin on Transporters: In vitro data suggest that eribulin at clinically relevant concentrations may inhibit P-gp, but does not inhibit BCRP, OATP1B1, OCT1, OAT1, OAT3, or MATE1.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been conducted with eribulin mesylate. Eribulin mesylate was not mutagenic in in vitro bacterial reverse mutation assays (Ames test). Eribulin mesylate was positive in mouse lymphoma mutagenesis assays, and was clastogenic in an in vivo rat bone marrow micronucleus assay.
Fertility studies have not been conducted with eribulin mesylate in humans or animals; however, nonclinical findings in repeat-dose dog and rat toxicology studies suggest that male fertility may be compromised by treatment with eribulin mesylate. Rats exhibited testicular toxicity (hypocellularity of seminiferous epithelium with hypospermia/aspermia) following dosing with eribulin mesylate at or above 0.43 times the recommended human dose (based on body surface area) given once weekly for 3 weeks, or at or above 0.21 times the recommended human dose (based on body surface area) given once weekly for 3 out of 5 weeks, repeated for 6 cycles. Testicular toxicity was also observed in dogs given 0.64 times the recommended human dose (based on body surface area) weekly for 3 out of 5 weeks, repeated for 6 cycles.
-
14 CLINICAL STUDIES
14.1 Metastatic Breast Cancer
Study 1 was an open-label, randomized, multicenter trial of 762 patients with metastatic breast cancer who received at least two chemotherapeutic regimens for the treatment of metastatic disease and experienced disease progression within 6 months of their last chemotherapeutic regimen. Patients were required to receive prior anthracycline- and taxane- based chemotherapy for adjuvant or metastatic disease. Patients were randomized (2:1) to receive HALAVEN (n=508) or a single agent therapy selected prior to randomization (control arm, n=254). Randomization was stratified by geographic region, HER2/neu status, and prior capecitabine exposure. HALAVEN was administered at a dose of 1.4 mg/m2 on Days 1 and 8 of a 21-day cycle. HALAVEN-treated patients received a median of 5 cycles (range: 1 to 23 cycles) of therapy. Control arm therapy consisted of 97% chemotherapy (26% vinorelbine, 18% gemcitabine, 18% capecitabine, 16% taxane, 9% anthracycline, 10% other chemotherapy), and 3% hormonal therapy. The main efficacy outcome was overall survival.
Patient demographic and baseline characteristics were comparable between the treatment arms. The median age was 55 (range: 27 to 85 years) and 92% were White. Sixty-four percent of patients were enrolled in North America/Western Europe/Australia, 25% in Eastern Europe/Russia, and 11% in Latin America/South Africa. Ninety-one percent of patients had a baseline ECOG performance status of 0 or 1. Tumor prognostic characteristics, including estrogen receptor status (positive: 67%, negative: 28%), progesterone receptor status (positive: 49%, negative: 39%), HER2/neu receptor status (positive: 16%, negative: 74%), triple negative status (ER-, PR-, HER2/neu-: 19%), presence of visceral disease (82%, including 60% liver and 38% lung) and bone disease (61%), and number of sites of metastases (greater than two: 50%), were also similar in the HALAVEN and control arms. Patients received a median of four prior chemotherapy regimens in both arms.
In Study 1, a statistically significant improvement in overall survival was observed in patients randomized to the HALAVEN arm compared to the control arm (see Table 5). An updated, unplanned survival analysis, conducted when 77% of events had been observed (see Figure 1), was consistent with the primary analysis. In patients randomized to HALAVEN, the objective response rate by the RECIST criteria was 11% (95% CI: 8.6%, 14.3%) and the median response duration was 4.2 months (95% CI: 3.8, 5.0 months).
Table 5: Comparison of Overall Survival in HALAVEN and Control Arm - Study 1 Overall Survival HALAVEN
(n=508)Control Arm
(n=254)Primary survival analysis Number of deaths 274 148 Median, months (95% CI) 13.1 (11.8, 14.3) 10.6 (9.3, 12.5) Hazard Ratio (95% CI)a 0.81 (0.66, 0.99) P valueb 0.041 Updated survival analysis Number of deaths 386 203 Median, months (95% CI) 13.2 (12.1, 14.4) 10.6 (9.2, 12.0) CI = confidence interval
a Based on Cox proportional hazards model stratified by geographic region, HER2 status, and prior capecitabine therapy.
b Based on a log-rank test stratified by geographic region, HER2 status, and prior capecitabine therapy.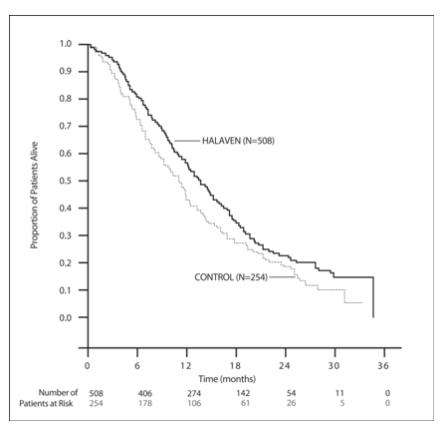
Figure 1: Updated Overall Survival Analysis for Study 1
14.2 Liposarcoma
The efficacy and safety of HALAVEN were evaluated in Study 2, an open-label, randomized (1:1), multicenter, active-controlled trial. Eligible patients were required to have unresectable, locally advanced or metastatic liposarcoma or leiomyosarcoma, at least two prior systemic chemotherapies (one of which must have included an anthracycline), and disease progression within 6 months of the most recent chemotherapy regimen. Patients were randomized to HALAVEN 1.4 mg/m2 administered intravenously on Days 1 and 8 of a 21-day cycle or to dacarbazine at a dose of 850 mg/m2, 1000 mg/m2, or 1200 mg/m2 administered intravenously every 21 days (dacarbazine dose was selected by the investigator prior to randomization). Treatment continued until disease progression or unacceptable toxicity. Randomization was stratified by histology (liposarcoma or leiomyosarcoma), number of prior therapies (2 vs. > 2), and geographic region (U.S. and Canada vs. Western Europe, Australia, and Israel vs. Eastern Europe, Latin America, and Asia). The major efficacy outcome measure was overall survival (OS). Additional efficacy outcome measures were progression-free survival (PFS) and confirmed objective response rate (ORR) as assessed by the investigator according to Response Evaluation Criteria in Solid Tumors (RECIST v1.1). Patients in the dacarbazine arm were not offered HALAVEN at the time of disease progression.
A total of 446 patients were randomized, 225 to the HALAVEN arm and 221 to the dacarbazine arm. The median age was 56 years (range: 24 to 83); 33% were male; 73% were White; 44% had ECOG performance status (PS) 0 and 53% had ECOG PS 1; 68% had leiomyosarcoma and 32% had liposarcoma; 39% were enrolled in U.S. and Canada (Region 1) and 46% were enrolled in Western Europe, Australia, and Israel (Region 2); and 47% received more than two prior systemic chemotherapies. The most common (>40%) prior systemic chemotherapies were doxorubicin (90%), ifosfamide (62%), gemcitabine (59%), trabectedin (50%), and docetaxel (48%).
Of the 143 patients with liposarcoma, the median age was 55 years (range: 32 to 83); 62% were male, 72% were White; 41% had ECOG PS of 0 and 53% had ECOG PS of 1; 35% were enrolled in Region 1 and 51% were enrolled in Region 2; and 44% received more than two prior systemic chemotherapies. The distribution of subtypes of liposarcoma, based on local histologic assessment, were 45% dedifferentiated, 37% myxoid/round cell, and 18% pleomorphic.
Study 2 demonstrated a statistically significant improvement in OS in patients randomized to HALAVEN compared with dacarbazine (see Table 6). There was no significant difference in progression-free survival in the overall population. Treatment effects of HALAVEN were limited to patients with liposarcoma based on pre-planned, exploratory subgroup analyses of OS and PFS (see Tables 6 and 7 and Figure 2). There was no evidence of efficacy of HALAVEN in patients with advanced or metastatic leiomyosarcoma in Study 2 (see Table 7).
Table 6: Efficacy Results for the Liposarcoma Stratum and All Patients* in Study 2a Liposarcoma
StratumAll Patients* Halaven
(n=71)Dacarbazine
(n=72)Halaven
(n=225)Dacarbazine
(n=221)Overall survival Deaths, n (%) 52 (73) 63 (88) 173 (77) 179 (81) Median, months
(95% CI)15.6
(10.2, 18.6)8.4
(5.2, 10.1)13.5
(11.1, 16.5)11.3
(9.5, 12.6)Hazard ratio (HR)
(95% CI)0.51
(0.35, 0.75)0.75
(0.61, 0.94)Stratified log-rank p value N/A† 0.011 Progression-free survival Events, n (%) 57 (80) 59 (82) 194 (86) 185 (84) Disease progression 53 52 180 170 Death 4 7 14 15 Median, months
(95% CI)2.9
(2.6, 4.8)1.7
(1.4, 2.6)2.6
(2.0, 2.8)2.6
(1.7, 2.7)HR
(95% CI)0.52
(0.35, 0.78)0.86
(0.69, 1.06)Objective response rate Objective response rate (%)
(95% CI)1.4
(0, 7.6)0
(0, 4.2)4.0
(1.8, 7.5)5.0
(2.5, 8.7)a Efficacy data from one study site enrolling six patients were excluded.
*All patients = liposarcoma and leiomyosarcoma.
† N/A = not applicable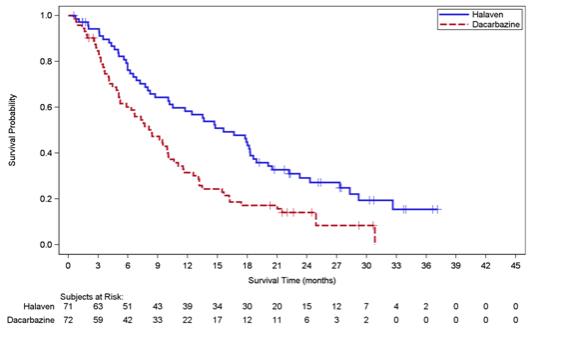
Figure 2: Kaplan-Meier Curves of Overall Survival in the Liposarcoma Stratum in Study 2
Table 7: Efficacy Results for the Leiomyosarcoma Stratum in Study 2a Leiomyosarcoma
StratumHalaven
(n=154)Dacarbazine
(n=149)Overall survival Deaths, n (%) 121 (79) 116 (78) Median, months
(95% CI)12.8
(10.3, 14.8)12.3
(11.0, 15.1)HR (95% CI) 0.90 (0.69, 1.18) Progression-free survival Events, n (%) 137 (89) 126 (85) Disease progression 127 118 Death 10 8 Median, months
(95% CI)2.2
(1.5, 2.7)2.6
(2.2, 2.9)HR (95% CI) 1.05 (0.81, 1.35) Objective response rate (%)
(95% CI)5.2
(2.3, 10)7.4
(3.7, 12.8)a Efficacy data from one study site enrolling six patients were excluded.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
NDC: 62856-389-01
Injection: 1 mg/2 mL, in a single-use vial. One vial per carton.
Store at 25°C (77°F); excursions permitted to 15° – 30° C (59° - 86° F). Do not freeze. Store the vials in their original cartons.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Neutropenia
Advise patients to contact their health care provider for a fever of 100.5°F or greater or other signs or symptoms of infection such as chills, cough, or burning or pain on urination [see Warnings and Precautions (5.1)].
Peripheral Neuropathy
Advise patients to inform their healthcare providers of new or worsening numbness, tingling and pain in their extremities [see Warnings and Precautions (5.2)].
Embryo-Fetal Toxicity
- Advise females of reproductive potential of the potential risk to a fetus and to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.3), Use in Specific Populations (8.1)].
- Advise females of reproductive potential to use effective contraception during treatment with HALAVEN and for at least 2 weeks after the final dose [see Use in Specific Populations (8.3)].
- Advise males with female partners of reproductive potential to use effective contraception during treatment with HALAVEN and for 3.5 months following the final dose [see Use in Specific Populations (8.3)].
Lactation
Advise women not to breastfeed during treatment with HALAVEN and for 2 weeks after the final dose [see Use in Specific Populations (8.2)].
Distributed by:
Eisai Inc.
Woodcliff Lake, NJ 07677© 2016 Eisai Inc.
- Advise females of reproductive potential of the potential risk to a fetus and to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.3), Use in Specific Populations (8.1)].
-
PATIENT PACKAGE INSERT
PATIENT INFORMATION
HALAVEN® (HAL-ih-ven)
(eribulin mesylate)
injection, for intravenous useWhat is the most important information I should know about HALAVEN?
HALAVEN can cause serious side effects, including:
-
Low white blood cell count (neutropenia). This can lead to serious infections that could lead to death. Your healthcare provider will check your blood cell counts before you receive each dose of HALAVEN and during treatment. Call your healthcare provider right away if you develop any of these symptoms of infection:
° fever (temperature above 100.5°F)
° chills
° cough
° burning or pain when you urinate
-
Numbness, tingling, or pain in your hands or feet (peripheral neuropathy). Peripheral neuropathy is common with HALAVEN and sometimes can be severe. Tell your healthcare provider if you have new or worsening symptoms of peripheral neuropathy.
- Your healthcare provider may delay, decrease your dose, or stop treatment with HALAVEN if you have side effects.
What is HALAVEN?
HALAVEN is a prescription medicine used to treat people with:
- Breast cancer
° that has spread to other parts of the body, and
° who have already received certain types of anticancer medicines after the cancer has spread
- Liposarcoma
° that cannot be treated with surgery or has spread to other parts of the body, and
° who have received treatment with a certain type of anticancer medicine
Before you receive HALAVEN, tell your healthcare provider about all of your medical conditions, including if you:
- have liver or kidney problems
- have heart problems, including a problem called congenital long QT syndrome
- have low potassium or low magnesium in your blood
- are pregnant or plan to become pregnant. HALAVEN can harm your unborn baby. Tell your healthcare provider right away if you become pregnant or think you are pregnant during treatment with HALAVEN.
° Females who are able to become pregnant should use an effective birth control during treatment with HALAVEN and for at least 2 weeks after the final dose of HALAVEN.
° Males should use an effective form of birth control when having sex with female partners who are able to become pregnant during treatment with HALAVEN and for 3 1/2 months (14 weeks) after the final dose of HALAVEN.
- are breastfeeding or plan to breastfeed. It is not known if HALAVEN passes into your breast milk. Do not breastfeed during treatment with HALAVEN and for 2 weeks after the final dose of HALAVEN.
How will I receive HALAVEN? - HALAVEN is given by intravenous (IV) injection in your vein.
- HALAVEN is given in “cycles” of treatment, with each cycle lasting 21 days.
- HALAVEN is usually given on day 1 and day 8 of a treatment cycle.
What are the possible side effects of HALAVEN?
HALAVEN may cause serious side effects, including:
- See “What is the most important information I should know about HALAVEN?”
- HALAVEN can cause changes in your heartbeat (called QT prolongation). This can cause irregular heartbeats. Your healthcare provider may do heart monitoring (electrocardiogram or ECG) or blood tests during your treatment with HALAVEN to check for heart problems.
- low white blood cell count (neutropenia)
- low red blood cell count (anemia)
- weakness or tiredness
- hair loss (alopecia)
- nausea
- constipation
- tiredness
- nausea
- hair loss (alopecia)
- constipation
- stomach pain
- fever
- low white blood cell count (neutropenia)
- decreased blood levels of potassium or calcium
These are not all the possible side effects of HALAVEN. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.General information about HALAVEN
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. You can ask your pharmacist or healthcare provider for information about HALAVEN that is written for health professionals.What are the ingredients in HALAVEN?
Active Ingredient: eribulin mesylate
Inactive Ingredients: ethanol, water
HALAVEN® is a registered trademark used by Eisai Inc. under license from Eisai R&D Management Co., Ltd.
Distributed by: Eisai Inc. Woodcliff Lake, NJ 07677
For more information, go to www.HALAVEN.com or call Eisai Inc. at 1-877-873-4724. If you would like a leaflet with larger printing, please contact Eisai Inc. at 1-877-873-4724.This Patient Information has been approved by the U.S. Food and Drug Administration. Revised: 01/2016
-
Low white blood cell count (neutropenia). This can lead to serious infections that could lead to death. Your healthcare provider will check your blood cell counts before you receive each dose of HALAVEN and during treatment. Call your healthcare provider right away if you develop any of these symptoms of infection:
-
PRINCIPAL DISPLAY PANEL
PRINCIPAL DISPLAY PANEL
NDC: 62856-389-01
Halaven™
(eribulin mesylate) Injection
1 mg/2 mL
(0.5 mg/mL)
For Intravenous Use
-
INGREDIENTS AND APPEARANCE
HALAVEN
eribulin mesylate injectionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 62856-389 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ERIBULIN MESYLATE (UNII: AV9U0660CW) (ERIBULIN - UNII:LR24G6354G) ERIBULIN MESYLATE 0.5 mg in 1 mL Inactive Ingredients Ingredient Name Strength ALCOHOL (UNII: 3K9958V90M) WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 62856-389-01 1 in 1 CARTON 11/15/2010 1 2 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA201532 11/15/2010 Labeler - Eisai Inc. (831600833)
Trademark Results [Halaven]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 HALAVEN 77886054 not registered Dead/Abandoned |
Eisai R&D Management Co., Ltd. 2009-12-04 |
 HALAVEN 77097283 3962019 Live/Registered |
Eisai R&D Management Co., Ltd. 2007-02-01 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.