FERAHEME- ferumoxytol injection
Feraheme by
Drug Labeling and Warnings
Feraheme by is a Prescription medication manufactured, distributed, or labeled by AMAG Pharmaceuticals, Inc., Baxter Pharmaceutical Solutions, Patheon Manufacturing Services LLC. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use Feraheme safely and effectively. See full prescribing information for Feraheme.
Feraheme® (ferumoxytol injection), for intravenous use
Initial U.S. Approval: 2009WARNING: RISK FOR SERIOUS HYPERSENSITIVITY/ANAPHYLAXIS REACTIONS
See full prescribing information for complete boxed warning.Fatal and serious hypersensitivity reactions including anaphylaxis have occurred in patients receiving Feraheme. Initial symptoms may include hypotension, syncope, unresponsiveness, cardiac/cardiorespiratory arrest.
- Only administer Feraheme as an intravenous infusion over at least 15 minutes and only when personnel and therapies are immediately available for the treatment of anaphylaxis and other hypersensitivity reactions. (5.1)
- Observe for signs or symptoms of hypersensitivity reactions during and for at least 30 minutes following Feraheme infusion including monitoring of blood pressure and pulse during and after Feraheme administration. (5.1)
- Hypersensitivity reactions have occurred in patients in whom a previous Feraheme dose was tolerated. (5.1)
RECENT MAJOR CHANGES
INDICATIONS AND USAGE
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
Injection: 510 mg iron per 17 mL (30 mg per mL) in single-dose vials. (3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- Greater risk of anaphylaxis in patients with multiple drug allergies. (5.1)
- Hypotension: Feraheme may cause hypotension. Monitor for signs and symptoms of hypotension following each administration of Feraheme. (5.2)
- Iron Overload: Regularly monitor hematologic responses during Feraheme therapy. Do not administer Feraheme to patients with iron overload. (5.3)
- Magnetic Resonance Imaging Test Interference: Feraheme can alter magnetic resonance imaging (MRI) studies. (5.4)
ADVERSE REACTIONS
The most common adverse reactions (≥ 2%) are diarrhea, headache, nausea, dizziness, hypotension, constipation, and peripheral edema. (6.1)
To report SUSPECTED ADVERSE REACTIONS with Feraheme, contact AMAG Pharmaceuticals, Inc. at 1-877-411-2510, or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 2/2018
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: RISK FOR SERIOUS HYPERSENSITIVITY/ANAPHYLAXIS REACTIONS
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Serious Hypersensitivity Reactions
5.2 Hypotension
5.3 Iron Overload
5.4 Magnetic Resonance (MR) Imaging Test Interference
6 ADVERSE REACTIONS
6.1 Clinical Trial Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Iron Deficiency Anemia in Patients Who Are Intolerant to Oral Iron or Have Had Unsatisfactory Response to Oral Iron
14.2 Iron Deficiency Anemia in Patients with Chronic Kidney Disease
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Stability and Storage
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: RISK FOR SERIOUS HYPERSENSITIVITY/ANAPHYLAXIS REACTIONS
Fatal and serious hypersensitivity reactions including anaphylaxis have occurred in patients receiving Feraheme. Initial symptoms may include hypotension, syncope, unresponsiveness, cardiac/cardiorespiratory arrest.
- Only administer Feraheme as an intravenous infusion over at least 15 minutes and only when personnel and therapies are immediately available for the treatment of anaphylaxis and other hypersensitivity reactions. [see Warnings and Precautions (5.1)].
- Observe for signs or symptoms of hypersensitivity reactions during and for at least 30 minutes following Feraheme infusion including monitoring of blood pressure and pulse during and after Feraheme administration [see Warnings and Precautions (5.1)].
- Hypersensitivity reactions have occurred in patients in whom a previous Feraheme dose was tolerated [see Warnings and Precautions (5.1)].
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
The recommended dose of Feraheme is an initial 510 mg dose followed by a second 510 mg dose 3 to 8 days later. Administer Feraheme as an intravenous infusion in 50-200 mL 0.9% Sodium Chloride Injection, USP or 5% Dextrose Injection, USP over at least 15 minutes. Administer while the patient is in a reclined or semi-reclined position.
Feraheme does not contain antimicrobial preservatives. Discard unused portion. Feraheme, when added to intravenous infusion bags containing either 0.9% Sodium Chloride Injection, USP (normal saline), or 5% Dextrose Injection, USP, at concentrations of 2-8 mg elemental iron per mL, should be used immediately but may be stored at controlled room temperature (25°C ± 2°C) for up to 4 hours or refrigerated (2-8° C) for up to 48 hours.
The dosage is expressed in terms of mg of elemental iron, with each mL of Feraheme containing 30 mg of elemental iron. Evaluate the hematologic response (hemoglobin, ferritin, iron and transferrin saturation) at least one month following the second Feraheme infusion. The recommended Feraheme dose may be readministered to patients with persistent or recurrent iron deficiency anemia.
For patients receiving hemodialysis, administer Feraheme once the blood pressure is stable and the patient has completed at least one hour of hemodialysis. Monitor for signs and symptoms of hypotension following each Feraheme infusion.
Allow at least 30 minutes between administration of Feraheme and administration of other medications that could potentially cause serious hypersensitivity reactions and/or hypotension, such as chemotherapeutic agents or monoclonal antibodies.
Inspect parenteral drug products visually for the absence of particulate matter and discoloration prior to administration.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Serious Hypersensitivity Reactions
Fatal and serious hypersensitivity reactions including anaphylaxis, presenting with cardiac/ cardiorespiratory arrest, clinically significant hypotension, syncope, or unresponsiveness have occurred in patients receiving Feraheme. Other adverse reactions potentially associated with hypersensitivity have occurred (pruritus, rash, urticaria, and wheezing). These reactions have occurred following the first dose or subsequent doses in patients in whom a previous Feraheme dose was tolerated.
Patients with a history of multiple drug allergies may have a greater risk of anaphylaxis with parenteral iron products. Carefully consider the potential risks and benefits before administering Feraheme to these patients.
Only administer Feraheme as an intravenous infusion over at least 15 minutes and only when personnel and therapies are immediately available for the treatment of anaphylaxis and other hypersensitivity reactions. Closely observe patients for signs and symptoms of hypersensitivity including monitoring of blood pressure and pulse during and after Feraheme administration for at least 30 minutes and until clinically stable following completion of each infusion [see Adverse Reactions (6.2)].
In a clinical study in patients with IDA, regardless of etiology, hypersensitivity reactions were reported in 0.4% (4/997) of subjects receiving Feraheme administered as intravenous infusion over at least 15 minutes. These included one patient with severe hypersensitivity reaction and three patients with moderate hypersensitivity reactions.
In clinical studies predominantly in patients with IDA and CKD, serious hypersensitivity reactions were reported in 0.2% (4/1,806) of subjects receiving Feraheme (administered as a rapid intravenous injection – prior method of administration no longer approved). Other adverse reactions potentially associated with hypersensitivity (e.g., pruritus, rash, urticaria or wheezing) were reported in 3.5% (63/1,806) of these subjects.
In the post-marketing experience, fatal and serious anaphylactic type reactions presenting with cardiac/ cardiorespiratory arrest, clinically significant hypotension, syncope, and unresponsiveness have been reported. Elderly patients with multiple or serious co-morbidities who experience hypersensitivity reactions and/or hypotension following administration of Feraheme may have more severe outcomes [see Boxed Warning, Adverse Reactions (6.2) and Use in Specific Populations (8.5)].
5.2 Hypotension
Feraheme may cause clinically significant hypotension.
In a clinical study with Feraheme in patients with IDA, regardless of etiology, moderate hypotension was reported in 0.2% (2/997) of subjects receiving Feraheme administered as intravenous infusion over at least 15 minutes.
In clinical studies in patients with IDA and CKD, hypotension was reported in 1.9% (35/1,806) of subjects, including three patients with serious hypotensive reactions, who had received Feraheme as a rapid intravenous injection (prior method of administration no longer approved).
Hypotension has also been reported in the post-marketing experience [see Adverse Reactions (6.2)]. Monitor patients for signs and symptoms of hypotension following each Feraheme administration [see Dosage and Administration (2) and Warnings and Precautions (5.1)].
5.3 Iron Overload
Excessive therapy with parenteral iron can lead to excess storage of iron with the possibility of iatrogenic hemosiderosis. Regularly monitor the hematologic response during parenteral iron therapy [see Dosage and Administration (2)]. Do not administer Feraheme to patients with iron overload.
In the 24 hours following administration of Feraheme, laboratory assays may overestimate serum iron and transferrin bound iron by also measuring the iron in the Feraheme complex.
5.4 Magnetic Resonance (MR) Imaging Test Interference
Administration of Feraheme may transiently affect the diagnostic ability of MR imaging. Conduct anticipated MR imaging studies prior to the administration of Feraheme. Alteration of MR imaging studies may persist for up to 3 months following the last Feraheme dose. If MR imaging is required within 3 months after Feraheme administration, use T1- or proton density-weighted MR pulse sequences to minimize the Feraheme effects; MR imaging using T2-weighted pulse sequences should not be performed earlier than 4 weeks after the administration of Feraheme. Maximum alteration of vascular MR imaging is anticipated to be evident for 1 – 2 days following Feraheme administration [see Clinical Pharmacology (12.3)].
Feraheme will not interfere with X-ray, computed tomography (CT), positron emission tomography (PET), single photon emission computed tomography (SPECT), ultrasound or nuclear medicine imaging.
-
6 ADVERSE REACTIONS
The following serious adverse reactions are described elsewhere in the labeling:
- Serious Hypersensitivity Reactions [see Warnings and Precautions (5.1)]
- Hypotension [see Warnings and Precautions (5.2)]
- Iron Overload [see Warnings and Precautions (5.3)]
- Magnetic Resonance (MR) Imaging Test Interference [see Warnings and Precautions (5.4)]
6.1 Clinical Trial Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
In clinical studies, 3,968 subjects were exposed to Feraheme. Of these subjects 31% were male and the median age was 54 years (range of 18 to 96 years).
The data described below reflect exposure to Feraheme in 997 patients exposed to a 1.02 g course of ferumoxytol administered as two 510 mg intravenous (IV) doses: 992 subjects (99.5%) received at least 1 complete dose of ferumoxytol and 946 subjects (94.9%) received 2 complete doses. The mean cumulative IV Iron exposure was 993.80 ±119.085 mg.
The safety of Feraheme was studied in a randomized, multicenter, double-blind clinical trial in patients with IDA (IDA Trial 3), [see Clinical Studies (14.1)]. In this trial, patients were randomized to two intravenous infusions of 510 mg (1.02 g) of Feraheme (n=997), or two intravenous infusions of 750 mg (1.500 g) of ferric carboxymaltose (FCM) (n=1000). Both intravenous irons were infused over a period of at least 15 minutes. Most patients received their second infusion of Feraheme and FCM 7(+1) days after Dose 1.
The mean (SD) age of the study population (N=1997) was 55.2 (17.16) years. The majority of patients were female (76.1%), white (71.4%) and non-Hispanic (81.8%). The mean (SD) hemoglobin at baseline for all patients was 10.4 (1.5) g/dl.
Serious adverse events were reported in 3.6% (71/1997) of ferumoxytol- and FCM- treated patients. The most common (≥2 subjects) serious AEs reported in Feraheme-treated patients were syncope, gastroenteritis, seizure, pneumonia, hemorrhagic anemia, and acute kidney injury. In FCM-treated patients the most common (≥2 subjects) serious AEs were syncope, cardiac failure congestive, angina pectoris, and atrial fibrillation.
Adverse reactions related to Feraheme and reported by ≥1% of Feraheme-treated patients in IDA Trial 3 are listed in Table 1.
Table 1: Adverse Reactions to Feraheme Reported in ≥1% of IDA Patients in IDA Trial 3 Adverse Reactions Feraheme
2 x 510 mg
(N = 997)
%Ferric Carboxymaltose
2 x 750 mg
(N = 1000)
%Headache 3.4 3.1 Nausea 1.8 3.4 Dizziness 1.5 1.6 Fatigue 1.5 1.2 Diarrhea 1 0.8 Back Pain 1 0.4 In IDA Trial 3, adverse reactions leading to treatment discontinuation and occurring in ≥ 2 Feraheme-treated patients included arthralgia (0.3%), dyspnea (0.3%), flushing (0.2%), chest discomfort (0.2%), chest pain (0.2%), nausea (0.2%), back pain (0.2%), dizziness (0.2%) and headache (0.2%).
Across two clinical trials in patients with IDA (IDA Trial 1 and 2), [see Clinical Studies (14.1)], patients were randomized to: two injections (rapid intravenous injection - prior method of administration no longer approved) of 510 mg of Feraheme (n=1,014), placebo (n=200), or five injections/infusions of 200 mg of iron sucrose (n=199). Most patients received their second Feraheme injection 3 to 8 days after the first injection. Adverse reactions related to Feraheme and reported by ≥ 1% of Feraheme-treated patients in these trials were similar to those seen in Trial 3.
In Trials 1 and 2, adverse reactions leading to treatment discontinuation and occurring in ≥ 2 Feraheme-treated patients included hypersensitivity (0.6%), hypotension (0.3%), and rash (0.2%).
In addition, a total of 634 subjects enrolled in and completed participation in a Phase 3 open label extension study. Of these, 337 subjects met IDA treatment criteria and received Feraheme. Adverse reactions following this repeat Feraheme dosing were generally similar in type and frequency to those observed after the first two intravenous injections.
Across three randomized clinical trials in patients with IDA and CKD (CKD Trials 1, 2, and 3), [see Clinical Studies (14.2)], a total of 605 patients were exposed to two injections of 510 mg of Feraheme and a total of 280 patients were exposed to 200 mg/day of oral iron for 21 days. Most patients received their second Feraheme injection 3 to 8 days after the first injection.
Adverse reactions related to Feraheme and reported by ≥ 1% of Feraheme-treated patients in the CKD randomized clinical trials are listed in Table 2 . Diarrhea (4%), constipation (2.1%) and hypertension (1%) have also been reported in Feraheme-treated patients.
Table 2: Adverse Reactions to Feraheme Reported in ≥1% of Patients with IDA and CKD Trials 1, 2 and 3 Adverse Reactions Feraheme
2 x 510 mg
(n = 605)
%Oral Iron
(n = 280)
%Nausea 3.1 7.5 Dizziness 2.6 1.8 Hypotension 2.5 0.4 Peripheral Edema 2 3.2 Headache 1.8 2.1 Edema 1.5 1.4 Vomiting 1.5 5 Abdominal Pain 1.3 1.4 Chest Pain 1.3 0.7 Cough 1.3 1.4 Pruritus 1.2 0.4 Pyrexia 1 0.7 Back Pain 1 0 Muscle Spasms 1 1.4 Dyspnea 1 1.1 Rash 1 0.4 In these clinical trials in patients with IDA and CKD, adverse reactions leading to treatment discontinuation and occurring in ≥ 2 Feraheme-treated patients included hypotension (0.4%), chest pain (0.3%), and dizziness (0.3%).
Following completion of the controlled phase of the trials, 69 patients received two additional 510 mg intravenous injections of Feraheme (for a total cumulative dose of 2.04 g). Adverse reactions following this repeat Feraheme dosing were similar in character and frequency to those observed following the first two intravenous injections.
6.2 Postmarketing Experience
Because adverse reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
The following serious adverse reactions have been reported from the post-marketing experience with Feraheme: fatal, life-threatening, and serious anaphylactic-type reactions, cardiac/cardiorespiratory arrest, clinically significant hypotension, syncope, unresponsiveness, loss of consciousness, tachycardia/rhythm abnormalities, angioedema, ischemic myocardial events, congestive heart failure, pulse absent, and cyanosis. These adverse reactions have usually occurred within 30 minutes after the administration of Feraheme. Reactions have occurred following the first dose or subsequent doses of Feraheme.
- 7 DRUG INTERACTIONS
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Limited available data with ferumoxytol use in pregnant women are insufficient to inform a drug associated risk of adverse developmental outcomes. There are risks to the mother and fetus associated with untreated iron deficiency anemia (IDA) in pregnancy (see Clinical Considerations). In animal studies, administration of ferumoxytol to pregnant rabbits during organogenesis caused adverse developmental outcomes including fetal malformations and decreased fetal weights at maternally toxic doses of 6 times the estimated human daily dose.
The estimated background risk of major birth defects and miscarriage for the indicated populations is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defect and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Disease-associated maternal and/or embryo/fetal risk
Untreated iron deficiency anemia (IDA) in pregnancy is associated with adverse maternal outcomes such as post-partum anemia. Adverse pregnancy outcomes associated with IDA include increased risk for preterm delivery and low birth weight.
Data
Animal Data
Administration of ferumoxytol during organogenesis, at doses of 31.6 mg Fe/kg/day in rats and 16.5 mg Fe/kg/day in rabbits, did not result in maternal or fetal effects. These doses are approximately 2 times the estimated human daily dose based on body surface area. In rats, administration of ferumoxytol during organogenesis at a maternally toxic dose of 100 mg Fe/kg/day, approximately 6 times the estimated human daily dose based on body surface area, caused a decrease in fetal weights. In rabbits, administration of ferumoxytol during organogenesis at a maternally toxic dose of 45 mg Fe/kg/day, approximately 6 times the estimated human daily dose based on body surface area, was associated with external and soft tissue fetal malformations and decreased fetal weights.
8.2 Lactation
Risk Summary
There are no data on the presence of ferumoxytol in human milk, the effects on the breastfed child, or the effects on milk production. Ferumoxytol has been detected in the milk of lactating rats. However, due to species-specific differences in lactation physiology, the clinical relevance of these data are not clear. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for Feraheme and any potential adverse effects on the breastfed child from Feraheme or from the underlying maternal condition.
8.4 Pediatric Use
The safety and effectiveness of Feraheme in pediatric patients (less than 18 years old) have not been established.
8.5 Geriatric Use
In controlled clinical trials, 833 patients ≥ 65 years of age were treated with Feraheme. No overall differences in safety and efficacy were observed between older and younger patients in these trials, but greater sensitivity of older individuals cannot be ruled out. In general, dose administration to an elderly patient should be cautious, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy. Elderly patients with multiple or serious co-morbidities who experience hypersensitivity reactions and/or hypotension following administration of Feraheme may have more severe outcomes. The potential risks and benefits of Feraheme administration should be carefully considered in these patients [see Dosage and Administration (2), Warnings and Precautions (5.1), and Clinical Studies (14)].
-
10 OVERDOSAGE
Limited data are available regarding overdosage of Feraheme in humans.
Excessive dosages of Feraheme may lead to accumulation of iron in storage sites potentially leading to hemosiderosis. Do not administer Feraheme to patients with iron overload [Warnings and Precautions (5.3)].
Feraheme is not removed by hemodialysis. -
11 DESCRIPTION
Feraheme is an iron replacement product containing ferumoxytol for intravenous infusion. Ferumoxytol is a non-stoichiometric magnetite (superparamagnetic iron oxide) coated with polyglucose sorbitol carboxymethylether. The overall colloidal particle size is 17-31 nm in diameter. The chemical formula of Feraheme is Fe5874O8752-C11719H18682O9933Na414 with an apparent molecular weight of 750 kDa.
Feraheme Injection is a sterile aqueous colloidal product that is formulated with mannitol. It is a black to reddish brown liquid, and is provided in single-dose vials containing 510 mg of elemental iron. Each mL of the sterile colloidal solution of Feraheme Injection contains 30 mg of elemental iron, 30 mg polyglucose sorbitol carboxymethylether, and 44 mg of mannitol. The formulation is isotonic with an osmolality of 270-330 mOsm/kg. The product contains no preservatives, and has a pH of 6 to 8.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Feraheme consists of a superparamagnetic iron oxide that is coated with a carbohydrate shell, which helps to isolate the bioactive iron from plasma components until the iron-carbohydrate complex enters the reticuloendothelial system macrophages of the liver, spleen and bone marrow. The iron is released from the iron-carbohydrate complex within vesicles in the macrophages. Iron then either enters the intracellular storage iron pool (e.g., ferritin) or is transferred to plasma transferrin for transport to erythroid precursor cells for incorporation into hemoglobin.
12.2 Pharmacodynamics
Cardiac Electrophysiology
In a randomized, positive- and placebo-controlled, parallel-group study, healthy subjects received a supratherapeutic regimen of Feraheme (1.02 g given as two 510 mg doses within 24 hours), placebo or a single dose of 400 mg moxifloxacin (positive control). Results demonstrated no effect of Feraheme on QT interval durations. No clinically meaningful effect of Feraheme on heart rate was observed.12.3 Pharmacokinetics
The pharmacokinetic (PK) behavior of Feraheme has been examined in healthy subjects and in patients with CKD stage 5D on hemodialysis. Feraheme exhibited dose-dependent, capacity-limited elimination from plasma with a half-life of approximately 15 hours in humans. The clearance (CL) was decreased by increasing the dose of Feraheme. Volume of distribution (Vd) was consistent with plasma volume, and the mean maximum observed plasma concentration (Cmax) and terminal half-life (t1/2) values increased with dose. The estimated values of CL and Vd following two 510 mg doses of Feraheme administered intravenously within 24 hours were 69.1 mL/hr and 3.16 L, respectively. The Cmax and time of maximum concentration (tmax) were 206 mcg/mL and 0.32 hr, respectively. Rate of infusion had no influence on Feraheme PK parameters. No gender differences in Feraheme PK parameters were observed. Feraheme is not removed by hemodialysis.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Ferumoxytol was not tested for carcinogenic effects. In standard genotoxicity tests, ferumoxytol showed no evidence of mutagenic activity in an in vitro Ames test or clastogenic activity in either an in vitro chromosomal aberration assay or an in vivo micronucleus assay.
No adverse effects on fertility or general reproductive performance were noted in animal studies. Ferumoxytol had no effect on male or female fertility or general reproductive function in rats. In a pre and postnatal development study in rats, intravenous administration of ferumoxytol from gestation day 6 until lactation day 20 at doses up to 60 mg/kg/day (approximately 3 times the daily human dose based on body surface area comparisons assuming a 60-kg person) had no effect on maternal delivery or numbers of liveborn offspring. Male offspring (F1) of pregnant rats (F0) administered ferumoxytol at a dose of 60 mg/kg/day had delayed sexual maturation and decreased reproductive competence. Female offspring (F1) of pregnant rats (F0) administered ferumoxytol at doses of 30 mg/kg/day or 60 mg/kg/day had delayed sexual maturation and decreased reproductive competence. Doses of 30 mg/kg/day and 60 mg/kg/day are approximately 2 and 3 times the daily human dose based on body surface area comparisons assuming a 60-kg person, respectively.
-
14 CLINICAL STUDIES
14.1 Iron Deficiency Anemia in Patients Who Are Intolerant to Oral Iron or Have Had Unsatisfactory Response to Oral Iron
IDA-301 Trial (referred to as IDA Trial 1) (NCT 01114139), IDA-302 Trial (referred to as IDA Trial 2) (NCT 01114204) and IDA-304 Trial (referred to as IDA Trial 3) (NCT 02694978)
The safety and efficacy of Feraheme in patients with iron deficiency anemia, regardless of etiology and a history of unsatisfactory oral iron therapy or in whom oral iron could not be used, were assessed in two randomized, controlled clinical trials (IDA Trial 1 and 2) with Feraheme administered as a rapid intravenous injection (prior method of administration no longer approved). In IDA Trial 1, patients were randomized to treatment with Feraheme or placebo. In IDA Trial 2, patients were randomized to treatment with Feraheme or iron sucrose. Feraheme (510 mg) and placebo were administered as two intravenous single dose injections over 3-8 days, and iron sucrose (200 mg) was administered as 5 intravenous injections or infusions over a period of 14 days.
In IDA Trial 1, the mean age of patients was 45 years (range, 18 to 91); 89% were female; 56% were Caucasian, 25% were Black, 16% were Asian, and 3% were other races.
In IDA Trial 2, the mean age of patients was 48 years (range, 18 to 89); 83% were female; 84% were Caucasian, 11% were Asian, 1% were Black, and 4% were other races.
Table 3 shows changes from baseline to Week 5 in hemoglobin and transferrin saturation in IDA Trial 1 and 2.
Table 3: Changes from Baseline to Week 5 in Hemoglobin (Hgb) and Transferrin Saturation in IDA Trial 1 and 2 (Intent to Treat Population) * p≤0.001 for main efficacy endpoint ENDPOINT IDA Trial 1 IDA Trial 2 Feraheme
N = 608Placebo
N = 200Feraheme
N = 406Iron Sucrose
N = 199Baseline Hgb
mean (SD), g/dL8.9 (0.9) 8.8 (0.9) 8.9 (0.9) 8.8 (1.0) Proportion of patients with
Hgb Increase of ≥2.0 g/dL at
any time from Baseline to
Week 5, %81.1 5.5 84.0 81.4 Treatment Difference
(%, 95% CI)75.6*
(71.2, 80.0)2.6
(-3.9, 9.1)Mean change in Hgb from
Baseline to Week 5
mean (SD), g/dL2.6 (1.5) 0.1 (0.9) 2.9 (1.6) 2.7 (1.3) Proportion of patients with
Hgb ≥12 g/dL at any time
from Baseline to Week 5, %50.5 3.0 66.7 48.2 Baseline TSAT
mean (SD) , %7.0 (12.9) 5.4 (4.9) 6.1 (9.9) 5.5 (10.3) Mean change in TSAT from
Baseline to Week 5
mean (SD), %11.4 (15.1) 0.4 (5.8) 15.7 (16.8) 11.9 (14.4) In IDA Trial 1, fatigue-related symptoms and impacts were assessed using a patient reported outcome instrument, FACIT-Fatigue (score range from 0 to 52 with higher scores indicating less fatigue). After 5 weeks, Feraheme-treated patients reported greater improvement from baseline in the fatigue score (+11.7 ± 11.73 points) than did patients in the placebo arm (+6.8 ± 9.51 points) with a treatment difference of 4.9 (95% CI: 3.08-6.71) points.
The safety of Feraheme in IDA patients with a history of unsatisfactory oral iron therapy or in whom oral iron could not be used was also assessed in another randomized, multicenter, double-blind safety clinical trial (IDA Trial 3). Patients were randomized in a 1:1 ratio to either two infusions of 510 mg (1.020 g) of Feraheme (n=997) or two infusions of 750 mg (1.500 g) of ferric carboxymaltose (FCM) (n=1000). Both IV irons were infused over a period of at least 15 minutes. Most patients received their second infusion of Feraheme or FCM 7(+1) days after the first infusion. This study included patients with any etiology of IDA including CKD excluding dialysis-dependent CKD.
In IDA Trial 3, the mean age of patients was 55 years (range, 18 to 96); 76% were female; 71% were Caucasian, 24% were Black, 3% were Asian, and 2% were other races.
The study met the primary endpoint to demonstrate non-inferiority to FCM with respect to the percentage of patients who experienced moderate-to-severe hypersensitivity reactions (including anaphylaxis) or moderate-to-severe hypotension (Feraheme: 0.6%; FCM: 0.7%; treatment difference: -0.1%; exact 95% confidence interval: -0.91% to +0.70%).
Table 4 shows the mean increase from baseline to week 5 in hemoglobin (Hgb) per treatment (Feraheme 2 x 510 mg; FCM 2 x 750 mg) and per gram of iron administered (Feraheme 1.020 g; FCM 1.500 g) in IDA Trial 3.
Table 4: Summary of Hemoglobin (Hgb) Changes per Treatment and per Gram of Iron Administered From Baseline to Week 5 (Intent to Treat Population) in IDA Trial 3 aAdjusted for difference in baseline Hgb ENDPOINT Feraheme
2 x 510mg
N = 997Ferric Carboxymaltose (FCM)
2 x 750mg
N = 1000Baseline Hgb
mean (SD); g/dL10.42 (1.48) 10.39 (1.46) Mean change in Hgb from
Baseline to Week 5 per Gram
of Iron Administered
mean (SD); g/dL1.35 (1.35) 1.10 (1.05) Treatment Difference Per
Gram of Irona (%, 95%CI)0.26
(0.17, 0.36)Mean change in Hgb from
Baseline to Week 5
mean (SD); g/dL1.38 (1.35) 1.63 (1.54) Treatment Differencea
(%, 95% CI)-0.24
(-0.35, -0.13)In IDA Trial 3, the incidence of severe hypophosphatemia (defined by blood phosphorous of <0.6 mmol/L at week 2) in the patients receiving Feraheme (0.4% of patients) was less than those receiving FCM (38.7% of patients).
14.2 Iron Deficiency Anemia in Patients with Chronic Kidney Disease
Trial 62745-7 (referred to as CKD Trial 1) (NCT 00255437), Trial 62745-6 (referred to as CKD Trial 2) (NCT 00255424), and Trial 62745-5 (referred to as CKD Trial 3) (NCT 00233597).
The safety and efficacy of Feraheme for the episodic treatment of iron deficiency anemia in patients with CKD were assessed in three randomized, open-label, controlled clinical trials (CKD Trial 1, 2 and 3) where Feraheme was administered as a rapid intravenous injection (prior method of administration - no longer approved). These trials also included an uncontrolled, follow-up phase in which patients with persistent iron deficiency anemia could receive two additional 510 mg intravenous injections of Feraheme. The major efficacy results from the controlled phase of each study are shown in Table 5.
In all three trials, patients with CKD and iron deficiency anemia were randomized to treatment with Feraheme or oral iron. Feraheme was administered as two 510 mg undiluted intravenous injections and oral iron (ferrous fumarate) was administered as a total daily dose of 200 mg elemental iron daily for 21 days. The major trial outcomes assessed the change in hemoglobin from baseline to Day 35. CKD Trial 1 and 2 enrolled patients with non-dialysis dependent CKD and CKD Trial 3 enrolled patients who were undergoing hemodialysis.
In CKD Trial 1, the mean age of patients was 66 years (range, 23 to 95); 60% were female; 65% were Caucasian, 32% were Black, and 2% were other races. In the Feraheme and oral iron groups, 42% and 44% of patients, respectively, were receiving erythropoiesis stimulating agents (ESAs) at baseline.
In CKD Trial 2, the mean age of patients was 65 years (range, 31 to 96); 61% were female; 58% were Caucasian, 35% were Black, and 7% were other races. In the Feraheme and oral iron groups, 36% and 43% of patients, respectively, were receiving ESAs at baseline.
In CKD Trial 3, the mean age of patients was 60 years (range, 24 to 87); 43% were female; 34% were Caucasian, 59% were Black, and 7% were other races. All patients were receiving ESAs.
Table 5 shows the Baseline and mean change to Day 35 in hemoglobin (Hgb, g/dL), transferrin saturation (TSAT, %) and ferritin (ng/mL) in each treatment group for Trial 1, 2, and 3.
Table 5: Changes from Baseline to Day 35 in Hemoglobin (Hgb), Transferrin Saturation and Ferritin (Intent to Treat Population) in CKD Trials 1, 2 and 3 * p≤0.001 for main efficacy endpoint ENDPOINT CKD Trial 1
Non-DialysisCKD Trial 2
Non-DialysisCKD Trial 3
DialysisFeraheme
N = 226Oral Iron
N = 77Feraheme
N = 228Oral Iron
N = 76Feraheme
N = 114Oral Iron
N = 116Baseline Hgb
mean (SD), g/dL9.9 (0.8) 9.9 (0.7) 10.0 (0.7) 10.0 (0.8) 10.6 (0.7) 10.7(0.6) Hgb change from Baseline at Day 35
mean (SD), g/dL1.2* (1.3) 0.5(1.0) 0.8* (1.2) 0.2 (1.0) 1.0*(1.1) 0.5 (1.1) Baseline TSAT
mean (SD), %9.8 (5.4) 10.4 (5.2) 11.3 (6.1) 10.1 (5.5) 15.7(7.2) 15.9 (6.3) TSAT change from Baseline at Day 35
mean (SD), %9.2 (9.4) 0.3 (4.7) 9.8 (9.2) 1.3 (6.4) 6.4 (12.6) 0.6 (8.3) Baseline ferritin
mean (SD), ng/mL123.7
(125.4)146.2
(136.3)146.1
(173.6)143.5
(144.9)340.5
(159.1)357.6
(171.7)Ferritin change from Baseline at Day 35
mean (SD), ng/mL300.7
(214.9)0.3
(82.0)381.7
(278.6)6.9
(60.1)233.9
(207.0)-59.2
(106.2)Following completion of the controlled phase of each of the Phase 3 trials, patients who were iron deficient and anemic could receive two additional 510 mg intravenous injections of Feraheme for a total cumulative dose of 2.04 g. Overall, 69 patients received two additional 510 mg intravenous injections of Feraheme, and on Day 35 following these additional injections, the majority of these patients (70%) experienced an increase in hemoglobin and iron parameters (TSAT and ferritin). The mean change (±SD) in hemoglobin level from the retreatment baseline for patients with an increase in hemoglobin was 0.86 (± 0.68) g/dL and was 0.5 (± 0.8) g/dL for all patients.
In a randomized, controlled clinical trial of 162 IDA patients with CKD (92 Non-Dialysis and 70 on Dialysis), mean change in hemoglobin from Baseline to Week 5 was 0.71 ±1.03 g/dL for Feraheme-treated patients and 0.61 ±0.97 g/dL for iron sucrose-treated patients.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
Feraheme is available in single-dose vials in the following package sizes (Table 6).
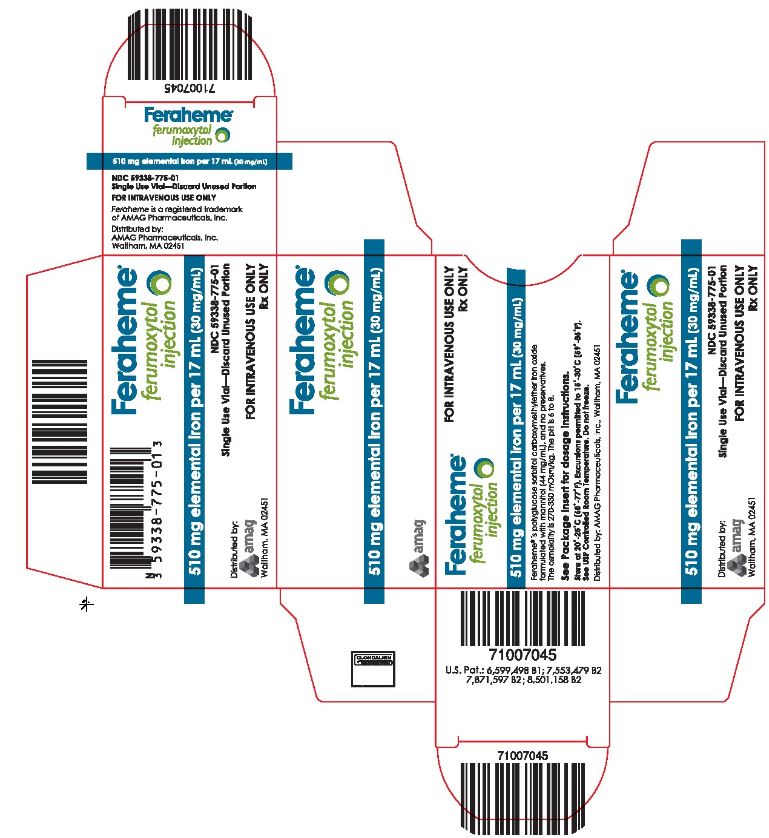
Table 6: Feraheme Packaging Description NDC Code Dose / Total volume per vial Vials / Carton NDC: 59338-775-01 510 mg/ 17 mL 1 NDC: 59338-775-10 510 mg/ 17 mL 10 -
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Prior History of Allergies to Parenteral Iron Products
Question patients regarding any prior history of allergies to parenteral iron products [see Warnings and Precautions (5.1)].
Hypersensitivity Reactions
Advise patients to immediately report any symptoms of hypersensitivity that may develop during and following Feraheme administration, such as rash, itching, dizziness, light-headedness, swelling, and breathing problems [see Warnings and Precautions (5.1)].
U.S Patents: 6,599,498 B1; 7,553,479 B2; 7,871,597 B2; 8,501,158 B2; 8,591,864 B2; 8,926,947 B2
Distributed by: AMAG Pharmaceuticals, Inc. Waltham, MA 02451
-
PATIENT PACKAGE INSERT
This Patient Information has been approved by the U.S. Food and Drug Administration. Revised: 02/2018 Patient Information
Feraheme (FER-uh-heem)
(ferumoxytol injection)What is the most important information I should know about Feraheme?
Feraheme may cause serious side effects including:
-
Serious allergic reactions that can lead to death. Serious allergic reactions have happened in people after receiving the first dose of Feraheme or after receiving additional doses in people who did not previously have an allergic reaction. If you have a history of allergies to many different medicines, you may have an increased risk of serious allergic reactions to Feraheme. Tell your healthcare provider or get medical help right away if you get any of these signs or symptoms:
- rash
- itching
- dizziness or lightheadedness
- swelling of the tongue or throat
- wheezing or trouble breathing
See “What are the possible side effects of Feraheme?” for more information about side effects.
What is Feraheme?
Feraheme is a prescription medicine used to treat iron deficiency anemia in adults who have:
- intolerance to oral iron or who have not responded well to treatment with oral iron or
- chronic kidney disease (CKD)
It is not known if Feraheme is safe and effective in children less than 18 years of age.
Who should not receive Feraheme?
Do not receive Feraheme if you:
- are allergic to Feraheme or any of the ingredients in Feraheme. See the end of this leaflet for a complete list of ingredients in Feraheme.
- have had an allergic reaction to any iron medicine given into your vein by intravenous (IV) infusion.
Before receiving Feraheme, tell your healthcare provider about all of your medical conditions, including if you:
- have allergies to many different medicines
- have iron overload
- have low blood pressure (hypotension).
- are pregnant or plan to become pregnant. It is not known if Feraheme will harm your unborn baby.
- are breastfeeding or plan to breastfeed. It is not known if Feraheme passes into your breast milk. You and your healthcare provider should decide if you will receive Feraheme or breastfeed.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
How will I receive Feraheme?
- Feraheme will be given to you into your vein by intravenous (IV) infusion over at least 15 minutes by your healthcare provider. You will receive Feraheme in 2 doses 3 to 8 days apart.
- Your healthcare provider will watch you during and for at least 30 minutes after you receive Feraheme.
What are the possible side effects of Feraheme?
Feraheme can cause serious side effects, including:
- See “What is the most important information I should know about Feraheme?”
- Low blood pressure (hypotension) is a common side effect of Feraheme and can sometimes be serious. Your healthcare provider will check you for signs and symptoms of hypotension after each Feraheme infusion.
- Iron overload. Your healthcare provider will do blood tests to check your iron levels during treatment with Feraheme.
The most common side effects of Feraheme include: diarrhea, headache, nausea, dizziness, constipation, and swelling of your legs, feet, arms, or hands.
These are not all of the possible side effects of Feraheme. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
General information about the safe and effective use of Feraheme.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. If you would like more information, talk to your healthcare provider. You can ask your pharmacist or healthcare provider for information about Feraheme that is written for health professionals.
What are the ingredients in Feraheme?
Active ingredient: ferumoxytol
Inactive ingredient: mannitol
Distributed by: AMAG Pharmaceuticals, Inc. Waltham, MA 02451
For more information, go to www.feraheme.com or call 1--877-411-2510.
-
Serious allergic reactions that can lead to death. Serious allergic reactions have happened in people after receiving the first dose of Feraheme or after receiving additional doses in people who did not previously have an allergic reaction. If you have a history of allergies to many different medicines, you may have an increased risk of serious allergic reactions to Feraheme. Tell your healthcare provider or get medical help right away if you get any of these signs or symptoms:
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
FERAHEME
ferumoxytol injectionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 59338-775 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength FERUMOXYTOL NON-STOICHIOMETRIC MAGNETITE (UNII: CLH5FT6412) (FERUMOXYTOL NON-STOICHIOMETRIC MAGNETITE - UNII:CLH5FT6412) FERUMOXYTOL NON-STOICHIOMETRIC MAGNETITE 510 mg in 17 mL Inactive Ingredients Ingredient Name Strength MANNITOL (UNII: 3OWL53L36A) 748 mg in 17 mL Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 59338-775-01 1 in 1 CARTON 07/13/2009 1 17 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product 2 NDC: 59338-775-10 10 in 1 CARTON 07/13/2009 2 17 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA022180 07/13/2009 Labeler - AMAG Pharmaceuticals, Inc. (017511155) Establishment Name Address ID/FEI Business Operations AMAG Pharmaceuticals, Inc. 017511155 MANUFACTURE(59338-775) Establishment Name Address ID/FEI Business Operations Patheon Manufacturing Services LLC 079415560 MANUFACTURE(59338-775)

Trademark Results [Feraheme]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 FERAHEME 77368571 3706009 Live/Registered |
AMAG Pharmaceuticals, Inc. 2008-01-10 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.