PARICALCITOL injection, solution
Paricalcitol by
Drug Labeling and Warnings
Paricalcitol by is a Prescription medication manufactured, distributed, or labeled by Akorn, Inc., Akorn Operating Company LLC, Akorn, Inc. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
DESCRIPTION
Paricalcitol, USP, the active ingredient in Paricalcitol Injection, is a synthetically manufactured analog of calcitriol, the metabolically active form of vitamin D indicated for the prevention and treatment of secondary hyperparathyroidism associated with chronic kidney disease (CKD) Stage 5. Paricalcitol Injection, USP is available as a sterile, clear, colorless, aqueous solution for intravenous injection. Each mL contains paricalcitol, 5 mcg and the following inactive ingredients: dehydrated alcohol, 20% (v/v), propylene glycol, 30% (v/v) and water for injection.
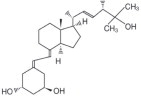
Paricalcitol is a white powder chemically designated as 19-nor-1α,3β,25-trihydroxy-9,10-secoergosta-5(Z),7(E),22(E)-triene and has the following structural formula:
Molecular formula is C27H44O3.
Molecular weight is 416.64.
-
CLINICAL PHARMACOLOGY
Secondary hyperparathyroidism is characterized by an elevation in parathyroid hormone (PTH) associated with inadequate levels of active vitamin D hormone. The source of vitamin D in the body is from synthesis in the skin and from dietary intake. Vitamin D requires two sequential hydroxylations in the liver and the kidney to bind to and to activate the vitamin D receptor (VDR). The endogenous VDR activator, calcitriol [1,25(OH)2 D3], is a hormone that binds to VDRs that are present in the parathyroid gland, intestine, kidney, and bone to maintain parathyroid function and calcium and phosphorus homeostasis, and to VDRs found in many other tissues, including prostate, endothelium and immune cells. VDR activation is essential for the proper formation and maintenance of normal bone. In the diseased kidney, the activation of vitamin D is diminished, resulting in a rise of PTH, subsequently leading to secondary hyperparathyroidism, and disturbances in the calcium and phosphorus homeostasis. The decreased levels of 1,25(OH)2 D3 and resultant elevated PTH levels, both of which often precede abnormalities in serum calcium and phosphorus, affect bone turnover rate and may result in renal osteodystrophy.
Mechanism of Action
Paricalcitol is a synthetic, biologically active vitamin D analog of calcitriol with modifications to the side chain (D2) and the A (19-nor) ring. Preclinical and in vitro studies have demonstrated that paricalcitol's biological actions are mediated through binding of the VDR, which results in the selective activation of vitamin D responsive pathways. Vitamin D and paricalcitol have been shown to reduce parathyroid hormone levels by inhibiting PTH synthesis and secretion.
Pharmacokinetics
Within two hours after administering paricalcitol injection intravenous doses ranging from 0.04 to 0.24 mcg/kg, concentrations of paricalcitol decreased rapidly; thereafter, concentrations of paricalcitol declined log-linearly. No accumulation of paricalcitol was observed with three times a week dosing.
Distribution
Paricalcitol is extensively bound to plasma proteins (≥99.8%). In healthy subjects, the steady state volume of distribution is approximately 23.8 L. The mean volume of distribution following a 0.24 mcg/kg dose of paricalcitol in CKD Stage 5 subjects requiring hemodialysis (HD) and peritoneal dialysis (PD) is between 31 and 35 L.
Metabolism
After IV administration of a 0.48 mcg/kg dose of 3H-paricalcitol, parent drug was extensively metabolized, with only about 2% of the dose eliminated unchanged in the feces and no parent drug found in the urine. Several metabolites were detected in both the urine and feces. Most of the systemic exposure was from the parent drug. Two minor metabolites, relative to paricalcitol, were detected in human plasma. One metabolite was identified as 24(R)-hydroxy paricalcitol, while the other metabolite was unidentified. The 24(R)-hydroxy paricalcitol is less active than paricalcitol in an in vivo rat model of PTH suppression.
In vitro data suggest that paricalcitol is metabolized by multiple hepatic and nonhepatic enzymes, including mitochondrial CYP24, as well as CYP3A4 and UGT1A4. The identified metabolites include the product of 24(R)-hydroxylation (present at low levels in plasma), as well as 24,26- and 24,28- dihydroxylation and direct glucuronidation.
Elimination
Paricalcitol is excreted primarily by hepatobiliary excretion. Approximately 63% of the radioactivity was eliminated in the feces and 19% was recovered in the urine in healthy subjects. In healthy subjects, the mean elimination half-life of paricalcitol is about five to seven hours over the studied dose range of 0.04 to 0.16 mcg/kg. The pharmacokinetics of paricalcitol has been studied in CKD Stage 5 subjects requiring hemodialysis (HD) and peritoneal dialysis (PD). The mean elimination half-life of paricalcitol after administration of 0.24 mcg/kg paricalcitol IV bolus dose in CKD Stage 5 HD and PD patients is 13.9 and 15.4 hours, respectively (Table 1).
Table 1 Mean ± SD Paricalcitol Pharmacokinetic Parameters in CKD Stage 5 Subjects Following Single 0.24 mcg/kg IV Bolus Dose †harmonic mean ± pseudo standard deviation, HD: hemodialysis, PD: peritoneal dialysis
CKD Stage 5-HD
(n=14)CKD Stage 5-PD
(n=8)Cmax (ng/mL) 1.680 ± 0.511 1.832 ± 0.315 AUC0-∞ (ngh/mL) 14.51 ± 4.12 16.01 ± 5.98 β (1/h) 0.050 ± 0.023 0.045 ± 0.026 t1/2 (h)† 13.9 ± 7.3 15.4 ± 10.5 CL (L/h) 1.49 ± 0.60 1.54 ± 0.95 Vdβ (L) 30.8 ± 7.5 34.9 ± 9.5 No accumulation of paricalcitol was observed with three times a week dosing which is consistent with the observed half-life.
Special Populations
Geriatric
The pharmacokinetics of paricalcitol have not been investigated in geriatric patients greater than 65 years.
Pediatrics
The pharmacokinetics of paricalcitol have not been investigated in patients less than 18 years of age.
Hepatic Impairment
The disposition of paricalcitol (0.24 mcg/kg) was compared in patients with mild (n=5) and moderate (n=5) hepatic impairment (as indicated by the Child-Pugh method) and subjects with normal hepatic function (n=10). The pharmacokinetics of unbound paricalcitol were similar across the range of hepatic function evaluated in this study. No dose adjustment is required in patients with mild and moderate hepatic impairment. The influence of severe hepatic impairment on the pharmacokinetics of paricalcitol has not been evaluated.
Renal Impairment
The pharmacokinetics of paricalcitol have been studied in CKD Stage 5 subjects requiring hemodialysis (HD) and peritoneal dialysis (PD). Hemodialysis procedure has essentially no effect on paricalcitol elimination. However, compared to healthy subjects, CKD Stage 5 subjects showed a decreased CL and increased half-life (see Pharmacokinetics-Elimination).
Drug Interactions
An in vitro study indicates that paricalcitol is not an inhibitor of CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, or CYP3A at concentrations up to 50 nM (21 ng/mL) (approximately 20-fold greater than that obtained after highest tested dose). In fresh primary cultured hepatocytes, the induction observed at paricalcitol concentrations up to 50 nM was less than two-fold for CYP2B6, CYP2C9 or CYP3A, where the positive controls rendered a six- to nineteen-fold induction. Hence, paricalcitol is not expected to inhibit or induce the clearance of drugs metabolized by these enzymes.
Drug interactions with paricalcitol injection have not been studied.
Omeprazole
The pharmacokinetic interaction between paricalcitol capsule (16 mcg) and omeprazole (40 mg; oral), a strong inhibitor of CYP2C19, was investigated in a single dose, crossover study in healthy subjects. The pharmacokinetics of paricalcitol were unaffected when omeprazole was administrated approximately 2 hours prior to the paricalcitol dose.
Ketoconazole
Although no data are available for the drug interaction between paricalcitol injection and ketoconazole, a strong inhibitor of CYP3A, the effect of multiple doses of ketoconazole administered as 200 mg BID for 5 days on the pharmacokinetics of paricalcitol capsule has been studied in healthy subjects. The Cmax of paricalcitol was minimally affected, but AUC0-∞ approximately doubled in the presence of ketoconazole. The mean half-life of paricalcitol was 17.0 hours in the presence of ketoconazole as compared to 9.8 hours, when paricalcitol was administered alone (see PRECAUTIONS).
-
CLINICAL STUDIES
In three 12-week, placebo-controlled, phase 3 studies in chronic kidney disease Stage 5 patients on dialysis, the dose of paricalcitol injection was started at 0.04 mcg/kg 3 times per week. The dose was increased by 0.04 mcg/kg every 2 weeks until intact parathyroid hormone (iPTH) levels were decreased at least 30% from baseline or a fifth escalation brought the dose to 0.24 mcg/kg, or iPTH fell to less than 100 pg/mL, or the Ca × P product was greater than 75 within any 2 week period, or serum calcium became greater than 11.5 mg/dL at any time.
Patients treated with paricalcitol injection achieved a mean iPTH reduction of 30% within 6 weeks. In these studies, there was no significant difference in the incidence of hypercalcemia or hyperphosphatemia between paricalcitol injection and placebo-treated patients. The results from these studies are as follows:
Group
(No. of Pts.)Baseline Mean (Range) Mean (SE)
Change from
Baseline to
Final EvaluationPTH (pg/mL) Paricalcitol injection (n = 40) 783 (291 to 2076) -379 (43.7) placebo (n = 38) 745 (320 to 1671) -69.6 (44.8) Alkaline
Phosphatase (U/L)Paricalcitol injection (n = 31) 150 (40 to 600) -41.5 (10.6) placebo (n = 34) 169 (56 to 911) +2.6 (10.1) Calcium (mg/dL) Paricalcitol injection (n = 40) 9.3 (7.2 to 10.4) +0.47 (0.1) placebo (n = 38) 9.1 (7.8 to 10.7) +0.02 (0.1) Phosphorous (mg/dL) Paricalcitol injection (n = 40) 5.8 (3.7 to 10.2) +0.47 (0.3) placebo (n = 38) 6.0 (2.8 to 8.8) -0.47 (0.3) Calcium x
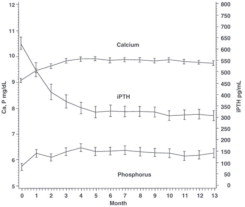
Phosphorous ProductParicalcitol injection (n = 40) 54 (32 to 106) +7.9 (2.2) placebo (n = 38) 54 (26 to 77) -3.9 (2.3) A long-term, open-label safety study of 164 CKD Stage 5 patients (mean dose of 7.5 mcg three times per week), demonstrated that mean serum Ca, P, and Ca × P remained within clinically appropriate ranges with PTH reduction (mean decrease of 319 pg/mL at 13 months).
- INDICATIONS AND USAGE
-
CONTRAINDICATIONS
Paricalcitol injection should not be given to patients with evidence of vitamin D toxicity, hypercalcemia, or hypersensitivity to any ingredient in this product (see WARNINGS).
-
WARNINGS
Acute overdose of paricalcitol injection may cause hypercalcemia, and require emergency attention (see OVERDOSAGE). During dose adjustment, serum calcium and phosphorus levels should be monitored closely (e.g., twice weekly). If clinically significant hypercalcemia develops, the dose should be reduced or interrupted. Chronic administration of paricalcitol injection may place patients at risk of hypercalcemia, elevated Ca × P product, and metastatic calcification. Chronic hypercalcemia can lead to generalized vascular calcification and other soft-tissue calcification.
Concomitant administration of high doses of calcium-containing preparations or thiazide diueretics with paricalcitol injection may increase the risk of hypercalcemia. High intake of calcium and phosphate concomitant with vitamin D compounds may lead to serum abnormalities requiring more frequent patient monitoring and individualized dose titration. Patients also should be informed about the symptoms of elevated calcium, which include feeling tired, difficulty thinking clearly, loss of appetite, nausea, vomiting, constipation, increased thirst, increased urination and weight loss.
Prescription-based doses of vitamin D and its derivatives should be withheld during paricalcitol injection treatment to avoid hypercalcemia.
Aluminum-containing preparations (e.g., antacids, phosphate binders) should not be administered chronically with paricalcitol injection, as increased blood levels of aluminum and aluminum bone toxicity may occur.
-
PRECAUTIONS
General
Digitalis toxicity is potentiated by hypercalcemia of any cause, so caution should be applied when digitalis compounds are prescribed concomitantly with paricalcitol injection. A dynamic bone lesion may develop if PTH levels are suppressed to abnormal levels.
Information for Patients
The patient should be instructed that, to ensure effectiveness of paricalcitol injection therapy, it is important to adhere to a dietary regimen of calcium supplementation and phosphorus restriction. Appropriate types of phosphate-binding compounds may be needed to control serum phosphorus levels in patients with chronic kidney disease (CKD) Stage 5, but excessive use of aluminum containing compounds should be avoided (see WARNINGS). Patients should also be carefully informed about the symptoms of elevated calcium (see WARNINGS).
Laboratory Tests
During the initial phase of medication, serum calcium and phosphorus should be determined frequently (e.g., twice weekly). Once dosage has been established, serum calcium and phosphorus should be measured at least monthly. Measurements of serum or plasma PTH are recommended every 3 months. During dose adjustment of paricalcitol injection, laboratory tests may be required more frequently.
Drug Interactions
Specific interaction studies were not performed with paricalcitol injection. Paricalcitol is not expected to inhibit the clearance of drugs metabolized by cytochrome P450 enzymes CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, or CYP3A nor induce the clearance of drug metabolized by CYP2B6, CYP2C9 or CYP3A.
A multiple dose drug-drug interaction study with ketoconazole and paricalcitol capsule demonstrated that ketoconazole approximately doubled paricalcitol AUC0-∞ (see CLINICAL PHARMACOLOGY). Since paricalcitol is partially metabolized by CYP3A and ketoconazole is known to be a strong inhibitor of cytochrome P450 3A enzyme, care should be taken while paricalcitol is co-administered with ketoconazole and other strong P450 3A inhibitors including the following drugs but not limited to: atazanavir, clarithromycin, indinavir, itraconazole, nefazodone, nelfinavir, ritonavir, saquinavir, telithromycin or voriconazole.
Digitalis toxicity is potentiated by hypercalcemia of any cause, so caution should be applied when digitalis compounds are prescribed concomitantly with paricalcitol injection.
Carcinogenesis, Mutagenesis, Impairment of Fertility
In a 104-week carcinogenicity study in CD-1 mice, an increased incidence of uterine leiomyoma and leiomyosarcoma was observed at subcutaneous doses of 1, 3, 10 mcg/kg (2 to 15 times the AUC at a human dose of 14 mcg, equivalent to 0.24 mcg/kg based on AUC). The incidence rate of uterine leiomyoma was significantly different than the control group at the highest dose of 10 mcg/kg.
In a 104-week carcinogenicity study in rats, there was an increased incidence of benign adrenal pheochromocytoma at subcutaneous doses of 0.15, 0.5, 1.5 mcg/kg (< 1 to 7 times the exposure following a human dose of 14 mcg, equivalent to 0.24 mcg/kg based on AUC). The increased incidence of pheochromocytomas in rats may be related to the alteration of calcium homeostasis by paricalcitol.
Paricalcitol did not exhibit genetic toxicity in vitro with or without metabolic activation in the microbial mutagenesis assay (Ames Assay), mouse lymphoma mutagenesis assay (L5178Y), or a human lymphocyte cell chromosomal aberration assay. There was also no evidence of genetic toxicity in an in vivo mouse micronucleus assay. Paricalcitol injection had no effect on fertility (male or female) in rats at intravenous doses up to 20 mcg/kg/dose [equivalent to 13 times the highest recommended human dose (0.24 mcg/kg) based on surface area, mg/m2].
Pregnancy
Category C
Paricalcitol has been shown to cause minimal decreases in fetal viability (5%) when administered daily to rabbits at a dose 0.5 times the 0.24 mcg/kg human dose (based on surface area, mg/m2) and when administered to rats at a dose 2 times the 0.24 mcg/kg human dose (based on plasma levels of exposure). At the highest dose tested (20 mcg/kg 3 times per week in rats, 13 times the 0.24 mcg/kg human dose based on surface area), there was a significant increase of the mortality of newborn rats at doses that were maternally toxic (hypercalcemia). No other effects on offspring development were observed. Paricalcitol was not teratogenic at the doses tested.
There are no adequate and well-controlled studies in pregnant women. Paricalcitol injection should be used during pregnancy only if the potential benefit to the mother justifies the potential risk to the fetus.
Nursing Mothers
Studies in rats have shown that paricalcitol is present in the milk. It is not known whether paricalcitol is excreted in human milk. In the nursing patient, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
Pediatric Use
The safety and effectiveness of paricalcitol injection were examined in a 12-week randomized, double-blind, placebo-controlled study of 29 pediatric patients, aged 5 to 19 years, with end-stage renal disease on hemodialysis and nearly all had received some form of vitamin D prior to the study. Seventy-six percent of the patients were male, 52% were Caucasian and 45% were African-American. The initial dose of paricalcitol injection was 0.04 mcg/kg 3 times per week based on baseline iPTH level of less than 500 pg/mL, or 0.08 mcg/kg 3 times a week, based on baseline iPTH level of ≥ 500 pg/mL, respectively. The dose of paricalcitol injection was adjusted in 0.04 mcg/kg increments based on the levels of serum iPTH, calcium and Ca x P. The mean baseline levels of iPTH were 841 pg/mL for the 15 paricalcitol injection-treated patients and 740 pg/mL for the 14 placebo-treated subjects. The mean dose of paricalcitol injection administered was 4.6 mcg (range: 0.8 mcg to 9.6 mcg). Ten of the 15 (67%) paricalcitol injection-treated patients and 2 of the 14 (14%) placebo-treated patients completed the trial. Ten of the placebo patients (71%) were discontinued due to excessive elevations in iPTH levels as defined by 2 consecutive iPTH levels > 700 pg/mL and greater than baseline after 4 weeks of treatment.
In the primary efficacy analysis, 9 of 15 (60%) subjects in the paricalcitol injection group had 2 consecutive 30% decreases from baseline iPTH compared with 3 of 14 (21%) patients in the placebo group (95% CI for the difference between groups –1%, 63%). Twenty-three percent of paricalcitol injection vs. 31% of placebo patients had at least one serum calcium level > 10.3 mg/dL, and 40% vs. 14% of paricalcitol injection vs. placebo subjects had at least one Ca x P ion product > 72 (mg/dL)2 . The overall percentage of serum calcium measurements > 10.3 mg/dL was 7% in the paricalcitol injection group and 7% in the placebo group; the overall percentage of patients with Ca x P product > 72 (mg/dL)2 was 8% in the paricalcitol injection group and 7% in the placebo group. No subjects in either the paricalcitol injection group or placebo group developed hypercalcemia (defined as at least one calcium value > 11.2 mg/dL) during the study.
-
ADVERSE REACTIONS
Paricalcitol injection has been evaluated for safety in clinical studies in 609 CKD Stage 5 patients. In four, placebo-controlled, double-blind, multicenter studies, discontinuation of therapy due to any adverse event occurred in 6.5% of 62 patients treated with paricalcitol injection (dosage titrated as tolerated, see CLINICAL PHARMACOLOGY - Clinical Studies) and 2.0% of 51 patients treated with placebo for 1 to 3 months. Adverse events occurring in the paricalcitol injection group at a frequency of 2% or greater and with an incidence greater than that in the placebo group, regardless of causality, are presented in the following table:
Adverse Event Incidence Rates for All Treated Patients In All Placebo-Controlled Studies Adverse Event Paricalcitol injection (n=62)
%Placebo (n=51)
%Overall 71 78 Cardiac Disorders Palpitations 3.2 0.0 Gastrointestinal Disorders Dry Mouth 3.2 2.0 Gastrointestinal Hemorrhage 4.8 2.0 Nausea 12.9 7.8 Vomiting 8.1 5.9 General Disorders and Administration Site Conditions Chills 4.8 2.0 Edema 6.5 0.0 Malaise 3.2 0.0 Pyrexia 4.8 2.0 Infections and Infestations Influenza 4.8 3.9 Pneumonia 4.8 0.0 Sepsis 4.8 2.0 Musculoskeletal and Connective Tissue Disorders Arthralgia 4.8 3.9 A patient who reported the same medical term more than once was counted only once for that medical term.
Safety parameters (changes in mean Ca, P, Ca × P) in an open-label safety study up to 13 months in duration support the long-term safety of paricalcitol injection in this patient population (see CLINICAL STUDIES).
Other Adverse Reactions Observed During Clinical Evaluation of Paricalcitol Injection
The following adverse reactions, with a causal relationship to paricalcitol injection, occurred in <2% of the paricalcitol injection treated patients in the above double-blind, placebo-controlled clinical trial data set. In addition, the following also includes adverse reactions reported in paricalcitol injection-treated patients who participated in other studies (non placebo-controlled), including double-blind, active-controlled and open-label studies:
Blood and Lymphatic System Disorders:
Anemia, lymphadenopathy
Cardiac Disorders:
Arrhythmia, atrial flutter, cardiac arrest
Ear and Labyrinth Disorders:
Ear discomfort
Endocrine Disorders:
Hyperparathyroidism, hypoparathyroidism
Eye Disorders:
Conjunctivitis, glaucoma, ocular hyperemia
Gastrointestinal Disorders:
Abdominal discomfort, constipation, diarrhea, dysphagia, gastritis, intestinal ischemia, rectal hemorrhage
General Disorders and Administration Site Conditions:
Asthenia, chest discomfort, chest pain, condition aggravated, edema peripheral, fatigue, feeling abnormal, gait disturbance, injection site extravasation, injection site pain, pain, swelling, thirst
Infections and Infestations:
Nasopharyngitis, upper respiratory tract infection, vaginal infection
Investigations:
Aspartate aminotransferase increased, bleeding time prolonged, heart rate irregular, laboratory test abnormal, weight decreased
Metabolism and Nutrition Disorders:
Decreased appetite, hypercalcemia, hyperkalemia, hyperphosphatemia, hypocalcemia
Musculoskeletal and Connective Tissue Disorders:
Joint stiffness, muscle twitching, myalgia
Neoplasms Benign, Malignant and Unspecified:
Breast cancer
Nervous System Disorders:
Cerebrovascular accident, dizziness, dysgeusia, headache, hypoesthesia, myoclonus, paresthesia, syncope, unresponsive to stimuli
Psychiatric Disorders:
Agitation, confusional state, delirium, insomnia, nervousness, restlessness
Reproductive System and Breast Disorders:
Breast pain, erectile dysfunction
Respiratory, Thoracic and Mediastinal Disorders:
Cough, dyspnea, orthopnea, pulmonary edema, wheezing
Skin and Subcutaneous Tissue Disorders:
Alopecia, blister, hirsutism, night sweats, rash pruritic, pruritus, skin burning sensation
Vascular Disorders:
Hypertension, hypotension
-
OVERDOSAGE
Overdosage of paricalcitol injection may lead to hypercalcemia, hypercalciuria, hyperphosphatemia, and over suppression of PTH. (see WARNINGS).
Treatment of Overdosage and Hypercalcemia
The treatment of acute overdosage should consist of general supportive measures. Serial serum electrolyte determinations (especially calcium), rate of urinary calcium excretion, and assessment of electrocardiographic abnormalities due to hypercalcemia should be obtained. Such monitoring is critical in patients receiving digitalis. Discontinuation of supplemental calcium and institution of a low calcium diet are also indicated in acute overdosage.
General treatment of hypercalcemia due to overdosage consists of immediate dose reduction or suspension of paricalcitol injection therapy, institution of a low calcium diet, withdrawal of calcium supplements, patient mobilization, and attention to fluid and electrolyte imbalances. Serum calcium levels should be determined at least weekly until normocalcemia ensues. When serum calcium levels have returned to within normal limits, paricalcitol injection may be reinitiated at a lower dose. If persistent and markedly elevated serum calcium levels occur, there are a variety of therapeutic alternatives that may be considered. These include the use of drugs such as phosphates and corticosteroids as well as measures to induce diuresis. Also, one may consider dialysis against a calcium-free dialysate.
Paricalcitol injection is not significantly removed by dialysis.
-
DOSAGE AND ADMINISTRATION
The currently accepted target range for iPTH levels in CKD Stage 5 patients is no more than 1.5 to 3 times the non-uremic upper limit of normal.
The recommended initial dose of paricalcitol injection is 0.04 mcg/kg to 0.1 mcg/kg (2.8 to 7 mcg) administered as a bolus dose no more frequently than every other day at any time during dialysis.
If a satisfactory response is not observed, the dose may be increased by 2 to 4 mcg at 2- to 4-week intervals. During any dose adjustment period, serum calcium and phosphorus levels should be monitored more frequently, and if an elevated calcium level or a Ca × P product greater than 75 is noted, the drug dosage should be immediately reduced or interrupted until these parameters are normalized. Then, paricalcitol injection should be reinitiated at a lower dose. If a patient is on a calcium-based phosphate binder, the dose may be decreased or withheld, or the patient may be switched to a non-calcium-based phosphate binder. Paricalcitol injection doses may need to be decreased as the PTH levels decrease in response to therapy. Thus, incremental dosing must be individualized.
The following table is a suggested approach in dose titration:
Suggested Dosing Guidelines PTH Level Paricalcitol injection Dose The same or increasing increase decreasing by < 30% increase decreasing by > 30%, <60% maintain decreasing by > 60% decrease one and one-half to three times upper limit of normal maintain The influence of mild to moderately impaired hepatic function on paricalcitol pharmacokinetics is sufficiently small that no dosing adjustment is required.
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration whenever solution and container permit.
After initial vial use, the contents of the multi-dose vial remain stable up to seven days when stored at controlled room temperature (see HOW SUPPLIED).
-
HOW SUPPLIED
Paricalcitol Injection, USP 5 mcg/mL is supplied as follows:
NDC: 17478-033-02 2 mL vials in packages of 25
-
PRINCIPAL DISPLAY PANEL
Principal Display Panel Text for Container Label:
NDC: 17478-033-02
Paricalcitol
Injection, USP
10 mcg/2 mL
(5 mcg/mL)
For Intravenous Use
Multi-dose Vial
Rx only 2 mL
-
PRINCIPAL DISPLAY PANEL
Principal Display Panel Text for Carton Label:
NDC: 17478-033-02
Paricalcitol Injection, USP
10 mcg/2 mL
(5 mcg/mL)
For Intravenous Use
Rx only 25 x 2 mL Multi-dose Vials Akorn logo
-
INGREDIENTS AND APPEARANCE
PARICALCITOL
paricalcitol injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 17478-033 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength Paricalcitol (UNII: 6702D36OG5) (Paricalcitol - UNII:6702D36OG5) Paricalcitol 5 ug in 1 mL Inactive Ingredients Ingredient Name Strength Propylene Glycol (UNII: 6DC9Q167V3) Alcohol (UNII: 3K9958V90M) Water (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 17478-033-02 25 in 1 CARTON 10/17/2017 1 2 mL in 1 VIAL, MULTI-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA207692 10/17/2017 Labeler - Akorn, Inc. (062649876) Establishment Name Address ID/FEI Business Operations Akorn, Inc 063434679 PACK(17478-033) , LABEL(17478-033) Establishment Name Address ID/FEI Business Operations Akorn, Inc 155135783 MANUFACTURE(17478-033) , ANALYSIS(17478-033) , STERILIZE(17478-033)


© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.