These highlights do not include all the information needed to use XTANDI® safely and effectively. See full prescribing information for XTANDI.XTANDI® (enzalutamide) capsules, for oral use XTANDI® (enzalutamide) tablets, for oral useInitial U.S. Approval: 2012
Xtandi by
Drug Labeling and Warnings
Xtandi by is a Prescription medication manufactured, distributed, or labeled by Astellas Pharma US, Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
XTANDI- enzalutamide tablet
Astellas Pharma US, Inc.
----------
HIGHLIGHTS OF PRESCRIBING INFORMATIONThese highlights do not include all the information needed to use XTANDI® safely and effectively. See full prescribing information for XTANDI.
XTANDI® (enzalutamide) capsules, for oral use XTANDI® (enzalutamide) tablets, for oral use Initial U.S. Approval: 2012 RECENT MAJOR CHANGESDosage and Administration – Dosing Information (2.1) 10/2020 INDICATIONS AND USAGEDOSAGE AND ADMINISTRATIONXTANDI 160 mg (two 80 mg tablets or four 40 mg tablets or four 40 mg capsules) administered orally once daily. Swallow capsules or tablets whole. (2.1) Patients receiving XTANDI should also receive a gonadotropin-releasing hormone (GnRH) analog concurrently or should have had bilateral orchiectomy. (2.3) CONTRAINDICATIONSNone. (4) WARNINGS AND PRECAUTIONS
ADVERSE REACTIONSThe most common adverse reactions (≥ 10%) that occurred more frequently (≥ 2% over placebo) in the XTANDI-treated patients are asthenia/fatigue, back pain, hot flush, constipation, arthralgia, decreased appetite, diarrhea, and hypertension. (6.1) To report SUSPECTED ADVERSE REACTIONS, contact Astellas Pharma US, Inc. at 1-800-727-7003 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch. DRUG INTERACTIONS
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling. Revised: 5/2021 |
FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
XTANDI® is indicated for the treatment of patients with:
- castration-resistant prostate cancer (CRPC)
- metastatic castration-sensitive prostate cancer (mCSPC).
2 DOSAGE AND ADMINISTRATION
2.1 Dosing Information
The recommended dose of XTANDI is 160 mg (two 80 mg tablets or four 40 mg tablets or four 40 mg capsules) administered orally once daily. XTANDI can be taken with or without food [see Clinical Pharmacology (12.3)]. Swallow capsules or tablets whole. Do not chew, dissolve, or open the capsules. Do not cut, crush, or chew the tablets [see How Supplied/Storage and Handling (16)].
2.2 Dose Modifications
If a patient experiences a ≥ Grade 3 toxicity or an intolerable side effect, withhold dosing for one week or until symptoms improve to ≤ Grade 2, then resume at the same or a reduced dose (120 mg or 80 mg), if warranted [see Warnings and Precautions (5.1), (5.2)].
Concomitant Strong CYP2C8 Inhibitors
The concomitant use of strong CYP2C8 inhibitors should be avoided if possible. If patients must be co-administered a strong CYP2C8 inhibitor, reduce the XTANDI dose to 80 mg once daily. If co-administration of the strong inhibitor is discontinued, the XTANDI dose should be returned to the dose used prior to initiation of the strong CYP2C8 inhibitor [see Drug Interactions (7.1) and Clinical Pharmacology (12.3)].
Concomitant Strong CYP3A4 Inducers
The concomitant use of strong CYP3A4 inducers should be avoided if possible. If patients must be co-administered a strong CYP3A4 inducer, increase the XTANDI dose from 160 mg to 240 mg once daily. If co-administration of the strong CYP3A4 inducer is discontinued, the XTANDI dose should be returned to the dose used prior to initiation of the strong CYP3A4 inducer [see Drug Interactions (7.2) and Clinical Pharmacology (12.3)].
3 DOSAGE FORMS AND STRENGTHS
XTANDI 40 mg capsules are white to off-white oblong soft gelatin capsules imprinted in black ink with ENZ.
XTANDI 40 mg tablets are yellow, round, film-coated and debossed with E 40.
XTANDI 80 mg tablets are yellow, oval, film-coated and debossed with E 80.
5 WARNINGS AND PRECAUTIONS
5.1 Seizure
Seizure occurred in 0.5% of patients receiving XTANDI in seven randomized clinical trials. In these trials, patients with predisposing factors for seizure were generally excluded. Seizure occurred from 13 to 1776 days after initiation of XTANDI. Patients experiencing seizure were permanently discontinued from therapy, and all seizure events resolved.
In a single-arm trial designed to assess the risk of seizure in patients with pre-disposing factors for seizure, 8 of 366 (2.2%) XTANDI-treated patients experienced a seizure. Three of the 8 patients experienced a second seizure during continued treatment with XTANDI after their first seizure resolved. It is unknown whether anti-epileptic medications will prevent seizures with XTANDI. Patients in the study had one or more of the following pre-disposing factors: the use of medications that may lower the seizure threshold (~ 54%), history of traumatic brain or head injury (~ 28%), history of cerebrovascular accident or transient ischemic attack (~ 24%), and Alzheimer’s disease, meningioma, or leptomeningeal disease from prostate cancer, unexplained loss of consciousness within the last 12 months, past history of seizure, presence of a space occupying lesion of the brain, history of arteriovenous malformation, or history of brain infection (all < 5%). Approximately 17% of patients had more than one risk factor.
Advise patients of the risk of developing a seizure while receiving XTANDI and of engaging in any activity where sudden loss of consciousness could cause serious harm to themselves or others.
Permanently discontinue XTANDI in patients who develop a seizure during treatment.
5.2 Posterior Reversible Encephalopathy Syndrome (PRES)
There have been reports of posterior reversible encephalopathy syndrome (PRES) in patients receiving XTANDI [see Adverse Reactions (6.2)]. PRES is a neurological disorder which can present with rapidly evolving symptoms including seizure, headache, lethargy, confusion, blindness, and other visual and neurological disturbances, with or without associated hypertension. A diagnosis of PRES requires confirmation by brain imaging, preferably magnetic resonance imaging (MRI). Discontinue XTANDI in patients who develop PRES.
5.3 Hypersensitivity
Hypersensitivity reactions, including edema of the face (0.5%), tongue (0.1%), or lip (0.1%) have been observed with enzalutamide in seven randomized clinical trials. Pharyngeal edema has been reported in post-marketing cases. Advise patients who experience any symptoms of hypersensitivity to temporarily discontinue XTANDI and promptly seek medical care. Permanently discontinue XTANDI for serious hypersensitivity reactions.
5.4 Ischemic Heart Disease
In the combined data of four randomized, placebo-controlled clinical studies, ischemic heart disease occurred more commonly in patients on the XTANDI arm compared to patients on the placebo arm (2.9% vs 1.3%). Grade 3-4 ischemic events occurred in 1.4% of patients on the XTANDI arm compared to 0.7% on the placebo arm. Ischemic events led to death in 0.4% of patients on the XTANDI arm compared to 0.1% on the placebo arm.
Monitor for signs and symptoms of ischemic heart disease. Optimize management of cardiovascular risk factors, such as hypertension, diabetes, or dyslipidemia. Discontinue XTANDI for Grade 3-4 ischemic heart disease.
5.5 Falls and Fractures
Falls and fractures occurred in patients receiving XTANDI. Evaluate patients for fracture and fall risk. Monitor and manage patients at risk for fractures according to established treatment guidelines and consider use of bone-targeted agents.
In the combined data of four randomized, placebo-controlled clinical studies, falls occurred in 11% of patients treated with XTANDI compared to 4% of patients treated with placebo. Falls were not associated with loss of consciousness or seizure. Fractures occurred in 10% of patients treated with XTANDI and in 4% of patients treated with placebo. Grade 3-4 fractures occurred in 3% of patients treated with XTANDI and in 2% of patients treated with placebo. The median time to onset of fracture was 336 days (range: 2 to 1914 days) for patients treated with XTANDI. Routine bone density assessment and treatment of osteoporosis with bone-targeted agents were not performed in the studies.
5.6 Embryo-Fetal Toxicity
The safety and efficacy of XTANDI have not been established in females. Based on animal reproductive studies and mechanism of action, XTANDI can cause fetal harm and loss of pregnancy when administered to a pregnant female. Advise males with female partners of reproductive potential to use effective contraception during treatment with XTANDI and for 3 months after the last dose of XTANDI [see Use in Specific Populations (8.1, 8.3)].
6 ADVERSE REACTIONS
The following is discussed in more detail in other sections of the labeling:
- Seizure [see Warnings and Precautions (5.1)]
- Posterior Reversible Encephalopathy Syndrome (PRES) [see Warnings and Precautions (5.2)]
- Hypersensitivity [see Warnings and Precautions (5.3)]
- Ischemic Heart Disease [see Warnings and Precautions (5.4)]
- Falls and Fractures [see Warnings and Precautions (5.5)]
6.1 Clinical Trial Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data in WARNINGS and PRECAUTIONS reflect seven randomized, controlled trials [AFFIRM, PREVAIL, TERRAIN, PROSPER, ARCHES, Asian PREVAIL (NCT02294461), and STRIVE (NCT01664923)] that were pooled to conduct safety analyses in patients with CRPC (N=3509) or mCSPC (N= 572) treated with XTANDI. Patients received XTANDI 160 mg (N= 4081) or placebo orally once daily (N= 2472) or bicalutamide 50 mg orally once daily (N= 387). All patients continued androgen deprivation therapy (ADT). In these seven trials, the median duration of treatment was 13.8 months (range: <0.1 to 87.6) in the XTANDI group.
In four placebo-controlled trials (AFFIRM, PROSPER, PREVAIL, and ARCHES), the median duration of treatment was 14.3 months (range: <0.1 to 87.6) in the XTANDI group [see Clinical Studies (14)]. In these four trials, the most common adverse reactions (≥ 10%) that occurred more frequently (≥ 2% over placebo) in the XTANDI-treated patients were asthenia/fatigue, back pain, hot flush, constipation, arthralgia, decreased appetite, diarrhea, and hypertension.
AFFIRM (NCT00974311): XTANDI versus Placebo in Metastatic CRPC Following Chemotherapy
AFFIRM enrolled 1199 patients with metastatic CRPC who had previously received docetaxel. The median duration of treatment was 8.3 months with XTANDI and 3.0 months with placebo. During the trial, 48% of patients on the XTANDI arm and 46% of patients on the placebo arm received glucocorticoids.
Grade 3 and higher adverse reactions were reported among 47% of XTANDI-treated patients. Discontinuations due to adverse events were reported for 16% of XTANDI-treated patients. The most common adverse reaction leading to treatment discontinuation was seizure, which occurred in 0.9% of the XTANDI-treated patients compared to none (0%) of the placebo-treated patients. Table 1 shows adverse reactions reported in AFFIRM that occurred at a ≥ 2% higher frequency in the XTANDI arm compared to the placebo arm.
| XTANDI
(N = 800) | Placebo
(N = 399) |
|||
|---|---|---|---|---|
| Grade 1-4*
(%) | Grade 3-4
(%) | Grade 1-4
(%) | Grade 3-4
(%) |
|
|
|
||||
|
General Disorders |
||||
|
Asthenic Conditions† |
51 |
9.0 |
44 |
9.3 |
|
Peripheral Edema |
15 |
1.0 |
13 |
0.8 |
|
Musculoskeletal and Connective Tissue Disorders |
||||
|
Back Pain |
26 |
5.3 |
24 |
4.0 |
|
Arthralgia |
21 |
2.5 |
17 |
1.8 |
|
Musculoskeletal Pain |
15 |
1.3 |
12 |
0.3 |
|
Muscular Weakness |
9.8 |
1.5 |
6.8 |
1.8 |
|
Musculoskeletal Stiffness |
2.6 |
0.3 |
0.3 |
0.0 |
|
Gastrointestinal Disorders |
||||
|
Diarrhea |
22 |
1.1 |
18 |
0.3 |
|
Vascular Disorders |
||||
|
Hot Flush |
20 |
0.0 |
10 |
0.0 |
|
Hypertension |
6.4 |
2.1 |
2.8 |
1.3 |
|
Nervous System Disorders |
||||
|
Headache |
12 |
0.9 |
5.5 |
0.0 |
|
Dizziness‡ |
9.5 |
0.5 |
7.5 |
0.5 |
|
Spinal Cord Compression and Cauda Equina Syndrome |
7.4 |
6.6 |
4.5 |
3.8 |
|
Paresthesia |
6.6 |
0.0 |
4.5 |
0.0 |
|
Mental Impairment Disorders§ |
4.3 |
0.3 |
1.8 |
0.0 |
|
Hypoesthesia |
4.0 |
0.3 |
1.8 |
0.0 |
|
Infections and Infestations |
||||
|
Upper Respiratory Tract Infection¶ |
11 |
0.0 |
6.5 |
0.3 |
|
Lower Respiratory Tract And Lung Infection# |
8.5 |
2.4 |
4.8 |
1.3 |
|
Psychiatric Disorders |
||||
|
Insomnia |
8.8 |
0.0 |
6.0 |
0.5 |
|
Anxiety |
6.5 |
0.3 |
4.0 |
0.0 |
|
Renal and Urinary Disorders |
||||
|
Hematuria |
6.9 |
1.8 |
4.5 |
1.0 |
|
Pollakiuria |
4.8 |
0.0 |
2.5 |
0.0 |
|
Injury, Poisoning and Procedural Complications |
||||
|
Fall |
4.6 |
0.3 |
1.3 |
0.0 |
|
Non-pathologic Fractures |
4.0 |
1.4 |
0.8 |
0.3 |
|
Skin and Subcutaneous Tissue Disorders |
||||
|
Pruritus |
3.8 |
0.0 |
1.3 |
0.0 |
|
Dry Skin |
3.5 |
0.0 |
1.3 |
0.0 |
|
Respiratory Disorders |
||||
|
Epistaxis |
3.3 |
0.1 |
1.3 |
0.3 |
PREVAIL (NCT01212991): XTANDI versus Placebo in Chemotherapy-naïve Metastatic CRPC
PREVAIL enrolled 1717 patients with metastatic CRPC who had not received prior cytotoxic chemotherapy, of whom 1715 received at least one dose of study drug. The median duration of treatment was 17.5 months with XTANDI and 4.6 months with placebo. Grade 3-4 adverse reactions were reported in 44% of XTANDI-treated patients and 37% of placebo-treated patients. Discontinuations due to adverse events were reported for 6% of XTANDI-treated patients. The most common adverse reaction leading to treatment discontinuation was fatigue/asthenia, which occurred in 1% of patients on each treatment arm. Table 2 includes adverse reactions reported in PREVAIL that occurred at a ≥ 2% higher frequency in the XTANDI arm compared to the placebo arm.
| XTANDI
(N = 871) | Placebo
(N = 844) |
|||
|---|---|---|---|---|
| Grade 1-4*
(%) | Grade 3-4
(%) | Grade 1-4
(%) | Grade 3-4
(%) |
|
|
|
||||
|
General Disorders |
||||
|
Asthenic Conditions† |
47 |
3.4 |
33 |
2.8 |
|
Peripheral Edema |
12 |
0.2 |
8.2 |
0.4 |
|
Musculoskeletal and Connective Tissue Disorders |
||||
|
Back Pain |
29 |
2.5 |
22 |
3.0 |
|
Arthralgia |
21 |
1.6 |
16 |
1.1 |
|
Gastrointestinal Disorders |
||||
|
Constipation |
23 |
0.7 |
17 |
0.4 |
|
Diarrhea |
17 |
0.3 |
14 |
0.4 |
|
Vascular Disorders |
||||
|
Hot Flush |
18 |
0.1 |
7.8 |
0.0 |
|
Hypertension |
14 |
7.2 |
4.1 |
2.3 |
|
Nervous System Disorders |
||||
|
Dizziness‡ |
11 |
0.3 |
7.1 |
0.0 |
|
Headache |
11 |
0.2 |
7.0 |
0.4 |
|
Dysgeusia |
7.6 |
0.1 |
3.7 |
0.0 |
|
Mental Impairment Disorders§ |
5.7 |
0.0 |
1.3 |
0.1 |
|
Restless Legs Syndrome |
2.1 |
0.1 |
0.4 |
0.0 |
|
Respiratory Disorders |
||||
|
Dyspnea¶ |
11 |
0.6 |
8.5 |
0.6 |
|
Infections and Infestations |
||||
|
Upper Respiratory Tract Infection# |
16 |
0.0 |
11 |
0.0 |
|
Lower Respiratory Tract And Lung InfectionÞ |
7.9 |
1.5 |
4.7 |
1.1 |
|
Psychiatric Disorders |
||||
|
Insomnia |
8.2 |
0.1 |
5.7 |
0.0 |
|
Renal and Urinary Disorders |
||||
|
Hematuria |
8.8 |
1.3 |
5.8 |
1.3 |
|
Injury, Poisoning and Procedural Complications |
||||
|
Fall |
13 |
1.6 |
5.3 |
0.7 |
|
Non-Pathological Fracture |
8.8 |
2.1 |
3.0 |
1.1 |
|
Metabolism and Nutrition Disorders |
||||
|
Decreased Appetite |
19 |
0.3 |
16 |
0.7 |
|
Investigations |
||||
|
Weight Decreased |
12 |
0.8 |
8.5 |
0.2 |
|
Reproductive System and Breast Disorders |
||||
|
Gynecomastia |
3.4 |
0.0 |
1.4 |
0.0 |
TERRAIN (NCT01288911): XTANDI versus Bicalutamide in Chemotherapy-naïve Metastatic CRPC
TERRAIN enrolled 375 patients with metastatic CRPC who had not received prior cytotoxic chemotherapy, of whom 372 received at least one dose of study drug. The median duration of treatment was 11.6 months with XTANDI and 5.8 months with bicalutamide. Discontinuations with an adverse event as the primary reason were reported for 7.6% of XTANDI-treated patients and 6.3% of bicalutamide-treated patients. The most common adverse reactions leading to treatment discontinuation were back pain and pathological fracture, which occurred in 3.8% of XTANDI-treated patients for each event and in 2.1% and 1.6% of bicalutamide-treated patients, respectively. Table 3 shows overall and common adverse reactions (≥ 10%) in XTANDI-treated patients.
| XTANDI
(N = 183) | Bicalutamide
(N = 189) |
|||
|---|---|---|---|---|
| Grade 1-4*
(%) | Grade 3-4
(%) | Grade 1-4
(%) | Grade 3-4
(%) |
|
|
|
||||
|
Overall |
94 |
39 |
94 |
38 |
|
General Disorders |
||||
|
Asthenic Conditions† |
32 |
1.6 |
23 |
1.1 |
|
Musculoskeletal and Connective Tissue Disorders |
||||
|
Back Pain |
19 |
2.7 |
18 |
1.6 |
|
Musculoskeletal Pain‡ |
16 |
1.1 |
14 |
0.5 |
|
Vascular Disorders |
||||
|
Hot Flush |
15 |
0 |
11 |
0 |
|
Hypertension |
14 |
7.1 |
7.4 |
4.2 |
|
Gastrointestinal Disorders |
||||
|
Nausea |
14 |
0 |
18 |
0 |
|
Constipation |
13 |
1.1 |
13 |
0.5 |
|
Diarrhea |
12 |
0 |
9.0 |
1.1 |
|
Infections and Infestations |
||||
|
Upper Respiratory Tract Infection§ |
12 |
0 |
6.3 |
0.5 |
|
Investigational |
||||
|
Weight Loss |
11 |
0.5 |
7.9 |
0.5 |
PROSPER (NCT02003924): XTANDI versus Placebo in Non-metastatic CRPC Patients
PROSPER enrolled 1401 patients with non-metastatic CRPC, of whom 1395 received at least one dose of study drug. Patients were randomized 2:1 and received either XTANDI at a dose of 160 mg once daily (N = 930) or placebo (N = 465). The median duration of treatment at the time of analysis was 18.4 months (range: 0.0 to 42 months) with XTANDI and 11.1 months (range: 0.0 to 43 months) with placebo.
Overall, 32 patients (3.4%) receiving XTANDI died from adverse events. The reasons for death with ≥ 2 patients included coronary artery disorders (n = 7), sudden death (n = 2), cardiac arrhythmias (n = 2), general physical health deterioration (n = 2), stroke (n = 2), and secondary malignancy (n = 5; one each of acute myeloid leukemia, brain neoplasm, mesothelioma, small cell lung cancer, and malignant neoplasm of unknown primary site). Three patients (0.6%) receiving placebo died from adverse events of cardiac arrest (n = 1), left ventricular failure (n = 1), and pancreatic carcinoma (n = 1). Grade 3 or higher adverse reactions were reported among 31% of XTANDI-treated patients and 23% of placebo-treated patients. Discontinuations with an adverse event as the primary reason were reported for 9.4% of XTANDI-treated patients and 6.0% of placebo-treated patients. Of these, the most common adverse event leading to treatment discontinuation was fatigue, which occurred in 1.6% of the XTANDI-treated patients compared to none of the placebo-treated patients. Table 4 shows adverse reactions reported in PROSPER that occurred at a ≥ 2% higher frequency in the XTANDI arm than in the placebo arm.
| XTANDI
(N = 930) | Placebo
(N = 465) |
|||
|---|---|---|---|---|
| Grade 1-4*
(%) | Grade 3-4
(%) | Grade 1-4
(%) | Grade 3-4
(%) |
|
|
|
||||
|
Metabolism and Nutrition Disorders |
||||
|
Decreased Appetite |
9.6 |
0.2 |
3.9 |
0.2 |
|
Nervous System Disorders |
||||
|
Dizziness† |
12 |
0.5 |
5.2 |
0 |
|
Headache |
9.1 |
0.2 |
4.5 |
0 |
|
Cognitive and Attention Disorders‡ |
4.6 |
0.1 |
1.5 |
0 |
|
Vascular Disorders |
||||
|
Hot Flush |
13 |
0.1 |
7.7 |
0 |
|
Hypertension |
12 |
4.6 |
5.2 |
2.2 |
|
Gastrointestinal Disorders |
||||
|
Nausea |
11 |
0.3 |
8.6 |
0 |
|
Constipation |
9.1 |
0.2 |
6.9 |
0.4 |
|
General Disorders and Administration Site Conditions |
||||
|
Asthenic Conditions§ |
40 |
4.0 |
20 |
0.9 |
|
Investigations |
||||
|
Weight Decreased |
5.9 |
0.2 |
1.5 |
0 |
|
Injury, Poisoning and Procedural Complications |
||||
|
Fall |
11 |
1.3 |
4.1 |
0.6 |
|
Fractures¶ |
9.8 |
2.0 |
4.9 |
1.7 |
|
Psychiatric Disorders |
||||
|
Anxiety |
2.8 |
0.2 |
0.4 |
0 |
ARCHES (NCT02677896): XTANDI versus Placebo in Metastatic CSPC Patients
ARCHES randomized 1150 patients with mCSPC, of whom 1146 received at least one dose of study drug. All patients received either a gonadotropin-releasing hormone (GnRH) analogue concurrently or had bilateral orchiectomy. Patients received either XTANDI at a dose of 160 mg once daily (N=572) or placebo (N=574). The median duration of treatment was 12.8 months (range: 0.2 to 26.6 months) with XTANDI and 11.6 months (range: 0.2 to 24.6 months) with placebo.
Overall, 10 patients (1.7%) receiving XTANDI died from adverse events. The reasons for death in ≥ 2 patients included heart disease (n=3), sepsis (n=2) and pulmonary embolism (n=2). Eight patients (1.4%) receiving placebo died from adverse events. The reasons for death in ≥ 2 patients included heart disease (n=2) and sudden death (n=2). Grade 3 or higher adverse events were reported in 24% of patients treated with XTANDI. Permanent discontinuation due to adverse events as the primary reason was reported in 4.9% of XTANDI-treated patients and 3.7% of placebo-treated patients. The most common adverse events resulting in permanent discontinuation in XTANDI-treated patients were alanine aminotransferase increased, aspartate aminotransferase elevation, and seizure, each in 0.3%. The most common adverse events leading to permanent discontinuation in placebo-treated patients were arthralgia, and fatigue, each in 0.3%.
Dose reductions due to an adverse reaction occurred in 4.4% of patients who received XTANDI. Fatigue/asthenia was the most frequent adverse reaction requiring dose reduction in 2.1% of XTANDI-treated patients and 0.7% of placebo-treated patients.
Table 5 shows adverse reactions reported in ARCHES that occurred at a ≥ 2% higher frequency in the XTANDI arm than in the placebo arm.
| XTANDI
(N = 572) | Placebo
(N = 574) |
|||
|---|---|---|---|---|
| Grade 1-4*
(%) | Grade 3-4
(%) | Grade 1-4
(%) | Grade 3-4
(%) |
|
|
|
||||
|
Metabolism and Nutrition Disorders |
||||
|
4.9 |
0.2 |
2.6 |
0 |
|
Nervous System Disorders |
||||
|
4.5 |
0.7 |
2.1 |
0 |
|
2.4 |
0 |
0.3 |
0 |
|
Vascular Disorders |
||||
|
27 |
0.3 |
22 |
0 |
|
8.0 |
3.3 |
5.6 |
1.7 |
|
General Disorders and Administration Site Conditions |
||||
|
24 |
1.7 |
20 |
1.6 |
|
Musculoskeletal and Connective Tissue Disorders |
||||
|
6.3 |
0.2 |
4.0 |
0.2 |
|
Injury, Poisoning and Procedural Complications |
||||
|
6.5 |
1.0 |
4.2 |
1.0 |
Laboratory Abnormalities
Table 6 shows laboratory abnormalities that occurred in ≥ 5% of patients, and more frequently (> 2%) in the XTANDI arm compared to placebo in the pooled, randomized, placebo-controlled studies.
| XTANDI
(N = 3173) | Placebo
(N = 2282) |
|||
|---|---|---|---|---|
| Grade 1-4
(%) | Grade 3-4
(%) | Grade 1-4
(%) | Grade 3-4
(%) |
|
|
Hematology |
||||
|
20 |
0.9 |
17 |
0.4 |
|
17 |
0.4 |
9.8 |
0.2 |
|
Chemistry |
||||
|
83 |
3.2 |
75 |
3.1 |
|
16 |
0.1 |
13 |
0 |
|
13 |
1.4 |
8.6 |
1.5 |
|
6.8 |
0.1 |
4.5 |
0 |
Hypertension
In the combined data from four randomized placebo-controlled clinical trials, hypertension was reported in 12% of patients receiving XTANDI and 5% of patients receiving placebo. Medical history of hypertension was balanced between arms. Hypertension led to study discontinuation in < 1% of patients in each arm.
6.2 Post-Marketing Experience
The following additional adverse reactions have been identified during post-approval use of XTANDI. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate the frequency or establish a causal relationship to drug exposure.
Gastrointestinal Disorders: vomiting
Immune System Disorders: hypersensitivity (edema of the face, tongue, lip, or pharynx)
Neurological Disorders: posterior reversible encephalopathy syndrome (PRES)
Skin and Subcutaneous Tissue Disorders: rash, severe cutaneous adverse reactions (including Stevens-Johnson syndrome (SJS), erythema multiforme, toxic epidermal necrolysis (TEN), drug reaction with eosinophilia and systemic symptoms (DRESS) and acute generalized exanthematous pustulosis (AGEP))
7 DRUG INTERACTIONS
7.1 Drugs that Inhibit CYP2C8
Co-administration of a strong CYP2C8 inhibitor (gemfibrozil) increased the composite area under the plasma concentration-time curve (AUC) of enzalutamide plus N-desmethyl enzalutamide by 2.2-fold. Co‑administration of XTANDI with strong CYP2C8 inhibitors should be avoided if possible. If co-administration of XTANDI with a strong CYP2C8 inhibitor cannot be avoided, reduce the dose of XTANDI [see Dosage and Administration (2.2) and Clinical Pharmacology (12.3)].
7.2 Drugs that Induce CYP3A4
Co-administration of rifampin (strong CYP3A4 inducer and moderate CYP2C8 inducer) decreased the composite AUC of enzalutamide plus N-desmethyl enzalutamide by 37%. Co-administration of strong CYP3A4 inducers (e.g., carbamazepine, phenobarbital, phenytoin, rifabutin, rifampin, rifapentine) with XTANDI should be avoided if possible. St John’s wort may decrease enzalutamide exposure and should be avoided. If co-administration of a strong CYP3A4 inducer with XTANDI cannot be avoided, increase the dose of XTANDI [see Dosage and Administration (2.2) and Clinical Pharmacology (12.3)].
7.3 Effect of XTANDI on Drug Metabolizing Enzymes
Enzalutamide is a strong CYP3A4 inducer and a moderate CYP2C9 and CYP2C19 inducer in humans. At steady-state, XTANDI reduced the plasma exposure to midazolam (CYP3A4 substrate), warfarin (CYP2C9 substrate), and omeprazole (CYP2C19 substrate). Concomitant use of XTANDI with narrow therapeutic index drugs that are metabolized by CYP3A4 (e.g., alfentanil, cyclosporine, dihydroergotamine, ergotamine, fentanyl, pimozide, quinidine, sirolimus and tacrolimus), CYP2C9 (e.g., phenytoin, warfarin) and CYP2C19 (e.g., S-mephenytoin, clopidogrel) should be avoided, as enzalutamide may decrease their exposure. If co-administration with warfarin cannot be avoided, conduct additional INR monitoring [see Clinical Pharmacology (12.3)].
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
The safety and efficacy of XTANDI have not been established in females. Based on animal reproductive studies and mechanism of action, XTANDI can cause fetal harm and loss of pregnancy. There are no human data on the use of XTANDI in pregnant females. In animal reproduction studies, oral administration of enzalutamide in pregnant mice during organogenesis caused adverse developmental effects at doses lower than the maximum recommended human dose (see Data).
Data
Animal Data
In an embryo-fetal developmental toxicity study in mice, enzalutamide caused developmental toxicity when administered at oral doses of 10 or 30 mg/kg/day throughout the period of organogenesis (gestational days 6-15). Findings included embryo-fetal lethality (increased post-implantation loss and resorptions) and decreased anogenital distance at ≥ 10 mg/kg/day, and cleft palate and absent palatine bone at 30 mg/kg/day. Doses of 30 mg/kg/day caused maternal toxicity. The doses tested in mice (1, 10 and 30 mg/kg/day) resulted in systemic exposures (AUC) approximately 0.04, 0.4 and 1.1 times, respectively, the exposures in patients. Enzalutamide did not cause developmental toxicity in rabbits when administered throughout the period of organogenesis (gestational days 6-18) at dose levels up to 10 mg/kg/day (approximately 0.4 times the exposures in patients based on AUC).
In a pharmacokinetic study in pregnant rats with a single oral 30 mg/kg enzalutamide administration on gestation day 14, enzalutamide and/or its metabolites were present in the fetus at a Cmax that was approximately 0.3 times the concentration found in maternal plasma and occurred 4 hours after administration.
8.2 Lactation
Risk Summary
The safety and efficacy of XTANDI have not been established in females. There is no information available on the presence of XTANDI in human milk, the effects of the drug on the breastfed infant, or the effects of the drug on milk production. Enzalutamide and/or its metabolites were present in milk of lactating rats (see Data).
Data
Following a single oral administration in lactating rats on postnatal day 14, enzalutamide and/or its metabolites were present in milk at a Cmax that was 4 times higher than concentrations in the plasma and occurred 4 hours after administration.
8.3 Females and Males of Reproductive Potential
Contraception
Males
Based on findings in animal reproduction studies, advise male patients with female partners of reproductive potential to use effective contraception during treatment and for 3 months after the last dose of XTANDI [see Use in Specific Populations (8.1)].
Infertility
Males
Based on animal studies, XTANDI may impair fertility in males of reproductive potential [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
Safety and effectiveness of XTANDI in pediatric patients have not been established.
8.5 Geriatric Use
Of 4081 patients who received XTANDI in seven randomized, controlled clinical trials, 78% were 65 and over, while 35% were 75 and over. No overall differences in safety or effectiveness were observed between these patients and younger patients. Other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
8.6 Patients with Renal Impairment
A dedicated renal impairment trial for XTANDI has not been conducted. Based on the population pharmacokinetic analysis using data from clinical trials in patients with metastatic CRPC and healthy volunteers, no significant difference in enzalutamide clearance was observed in patients with pre-existing mild to moderate renal impairment (30 mL/min ≤ creatinine clearance [CrCL] ≤ 89 mL/min) compared to patients and volunteers with baseline normal renal function (CrCL ≥ 90 mL/min). No initial dosage adjustment is necessary for patients with mild to moderate renal impairment. Severe renal impairment (CrCL < 30 mL/min) and end-stage renal disease have not been assessed [see Clinical Pharmacology (12.3)].
8.7 Patients with Hepatic Impairment
Dedicated hepatic impairment trials compared the composite systemic exposure of enzalutamide plus N-desmethyl enzalutamide in volunteers with baseline mild, moderate, or severe hepatic impairment (Child-Pugh Class A, B, or C, respectively) versus healthy controls with normal hepatic function. The composite AUC of enzalutamide plus N-desmethyl enzalutamide was similar in volunteers with mild, moderate, or severe baseline hepatic impairment compared to volunteers with normal hepatic function. No initial dosage adjustment is necessary for patients with baseline mild, moderate, or severe hepatic impairment [see Clinical Pharmacology (12.3)].
10 OVERDOSAGE
In the event of an overdose, stop treatment with XTANDI and initiate general supportive measures taking into consideration the half-life of 5.8 days. In a dose escalation study, no seizures were reported at < 240 mg daily, whereas 3 seizures were reported, 1 each at 360 mg, 480 mg, and 600 mg daily. Patients may be at increased risk of seizure following an overdose.
11 DESCRIPTION
Enzalutamide is an androgen receptor inhibitor. The chemical name is 4-{3-[4-cyano-3-(trifluoromethyl)phenyl]-5,5-dimethyl-4-oxo-2-sulfanylideneimidazolidin-1-yl}-2-fluoro-N-methylbenzamide.
The molecular weight is 464.44 and molecular formula is C21H16F4N4O2S. The structural formula is:
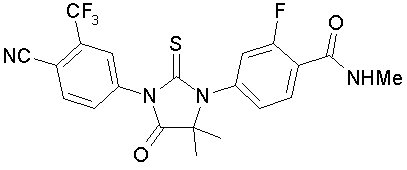
Enzalutamide is a white crystalline non-hygroscopic solid. It is practically insoluble in water.
XTANDI is available as liquid-filled soft gelatin capsules for oral administration. Each capsule contains 40 mg of enzalutamide as a solution in caprylocaproyl polyoxylglycerides. The inactive ingredients are caprylocaproyl polyoxylglycerides, butylated hydroxyanisole, butylated hydroxytoluene, gelatin, sorbitol sorbitan solution, glycerin, purified water, titanium dioxide, and black iron oxide.
XTANDI is also available as film-coated tablets for oral administration. Each tablet contains 40 mg or 80 mg of enzalutamide. The inactive ingredients are hypromellose acetate succinate, microcrystalline cellulose, colloidal silicon dioxide, croscarmellose sodium, and magnesium stearate. The tablet film-coat contains hypromellose, talc, polyethylene glycol, titanium dioxide, and ferric oxide.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Enzalutamide is an androgen receptor inhibitor that acts on different steps in the androgen receptor signaling pathway. Enzalutamide has been shown to competitively inhibit androgen binding to androgen receptors; and consequently, inhibits nuclear translocation of androgen receptors and their interaction with DNA. A major metabolite, N‑desmethyl enzalutamide, exhibited similar in vitro activity to enzalutamide. Enzalutamide decreased proliferation and induced cell death of prostate cancer cells in vitro, and decreased tumor volume in a mouse prostate cancer xenograft model.
12.2 Pharmacodynamics
Cardiac Electrophysiology
The effect of enzalutamide 160 mg/day at steady-state on the QTc interval was evaluated in 796 patients with metastatic CRPC. No large difference (i.e., greater than 20 ms) was observed between the mean QT interval change from baseline in patients treated with XTANDI and that in patients treated with placebo, based on the Fridericia correction method. However, small increases in the mean QTc interval (i.e., less than 10 ms) due to enzalutamide cannot be excluded due to limitations of the study design.
12.3 Pharmacokinetics
The pharmacokinetics of enzalutamide and its major active metabolite (N-desmethyl enzalutamide) were evaluated in patients with metastatic CRPC and healthy male volunteers. The plasma enzalutamide pharmacokinetics are adequately described by a linear two-compartment model with first-order absorption.
Absorption
Following oral administration of XTANDI capsules (160 mg daily) in patients with metastatic CRPC, the median time to reach maximum plasma enzalutamide concentrations (Cmax) is 1 hour (range 0.5 to 3 hours). At steady-state, the plasma mean Cmax values for enzalutamide and N-desmethyl enzalutamide are 16.6 μg/mL (23% CV) and 12.7 μg/mL (30% CV), respectively, and the plasma mean predose trough values are 11.4 μg/mL (26% CV) and 13.0 μg/mL (30% CV), respectively. Following a single dose administration of 160 mg enzalutamide in healthy male volunteers, enzalutamide extent of absorption (AUC) was comparable between XTANDI tablet and XTANDI capsule, but the mean Cmax was 10%-28% lower than that of XTANDI capsules. The steady-state pharmacokinetic profiles (AUC and Cmax) of enzalutamide and N-desmethyl enzalutamide are similar for XTANDI tablet and XTANDI capsule.
With the daily dosing regimen, enzalutamide steady-state is achieved by Day 28, and enzalutamide accumulates approximately 8.3-fold relative to a single dose. Daily fluctuations in enzalutamide plasma concentrations are low (mean peak-to-trough ratio of 1.25). At steady-state, enzalutamide showed approximately dose proportional pharmacokinetics over the daily dose range of 30 to 360 mg.
A single 160 mg oral dose of XTANDI was administered to healthy volunteers with a high-fat meal or in the fasted condition. A high-fat meal did not alter the AUC to enzalutamide or N-desmethyl enzalutamide. The results are summarized in Figure 1.
Distribution and Protein Binding
The mean apparent volume of distribution (V/F) of enzalutamide in patients after a single oral dose is 110 L (29% CV).
Enzalutamide is 97% to 98% bound to plasma proteins, primarily albumin. N-desmethyl enzalutamide is 95% bound to plasma proteins. In vitro, there was no protein binding displacement between enzalutamide and other highly protein bound drugs (warfarin, ibuprofen, and salicylic acid) at clinically relevant concentrations.
Metabolism
Following single oral administration of 14C-enzalutamide 160 mg, plasma samples were analyzed for enzalutamide and its metabolites up to 77 days post dose. Enzalutamide, N-desmethyl enzalutamide, and a major inactive carboxylic acid metabolite accounted for 88% of the 14C-radioactivity in plasma, representing 30%, 49%, and 10%, respectively, of the total 14C-AUC0-inf.
In vitro, human CYP2C8 and CYP3A4 are responsible for the metabolism of enzalutamide. Based on in vivo and in vitro data, CYP2C8 is primarily responsible for the formation of the active metabolite (N-desmethyl enzalutamide). In vitro data suggest that carboxylesterase 1 metabolizes N-desmethyl enzalutamide and enzalutamide to the inactive carboxylic acid metabolite.
In vitro, N-desmethyl enzalutamide is not a substrate of human CYP1A1, CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C18, CYP2C19, CYP2D6, CYP2E1 and CYP3A4/5.
Elimination
Enzalutamide is primarily eliminated by hepatic metabolism. Following single oral administration of 14C-enzalutamide 160 mg, 85% of the radioactivity is recovered by 77 days post dose: 71% is recovered in urine (including only trace amounts of enzalutamide and N-desmethyl enzalutamide), and 14% is recovered in feces (0.4% of dose as unchanged enzalutamide and 1% as N-desmethyl enzalutamide).
The mean apparent clearance (CL/F) of enzalutamide in patients after a single oral dose is 0.56 L/h (range 0.33 to 1.02 L/h).
The mean terminal half-life (t1/2) for enzalutamide in patients after a single oral dose is 5.8 days (range 2.8 to 10.2 days). Following a single 160 mg oral dose of enzalutamide in healthy volunteers, the mean terminal t1/2 for N-desmethyl enzalutamide is approximately 7.8 to 8.6 days.
Pharmacokinetics in Special Populations
Renal Impairment:
A population pharmacokinetic analysis (based on pre-existing renal function) was carried out with data from 59 healthy male volunteers and 926 patients with metastatic CRPC enrolled in clinical trials, including 512 with normal renal function (CrCL ≥ 90 mL/min), 332 with mild renal impairment (CrCL 60 to < 90 mL/min), 88 with moderate renal impairment (CrCL 30 to < 60 mL/min), and 1 with severe renal impairment (CrCL < 30 mL/min). The apparent clearance of enzalutamide was similar in patients with pre-existing mild and moderate renal impairment (CrCL 30 to < 90 mL/min) compared to patients and volunteers with normal renal function. The potential effect of severe renal impairment or end-stage renal disease on enzalutamide pharmacokinetics cannot be determined as clinical and pharmacokinetic data are available from only one patient [see Use in Specific Populations (8.6)].
Hepatic Impairment:
The plasma pharmacokinetics of enzalutamide and N-desmethyl enzalutamide were examined in volunteers with normal hepatic function (N = 22) and with pre-existing mild (N = 8, Child-Pugh Class A) moderate (N = 8, Child-Pugh Class B), or severe (N = 8, Child-Pugh Class C) hepatic impairment. XTANDI was administered as a single 160 mg dose. The composite AUC of enzalutamide plus N-desmethyl enzalutamide was similar in volunteers with mild, moderate, or severe baseline hepatic impairment compared to volunteers with normal hepatic function. The results are summarized in Figure 1 [see Use in Specific Populations (8.7)].
Body Weight and Age:
Population pharmacokinetic analyses showed that weight (range: 46 to 163 kg) and age (range: 41 to 92 yr) do not have a clinically meaningful influence on the exposure to enzalutamide.
Gender:
The effect of gender on the pharmacokinetics of enzalutamide has not been evaluated.
Race:
The majority of XTANDI-treated patients in the randomized clinical trials were Caucasian (81%). Based on pharmacokinetic data from studies in Japanese and Chinese patients with prostate cancer, there were no clinically relevant differences in exposure among the populations. There are insufficient data to evaluate potential differences in the pharmacokinetics of enzalutamide in other races.
Drug Interactions
Effect of Other Drugs on XTANDI:
In a drug-drug interaction trial in healthy volunteers, a single 160 mg oral dose of XTANDI was administered alone or after multiple oral doses of gemfibrozil (strong CYP2C8 inhibitor). Gemfibrozil increased the AUC0‑inf of enzalutamide plus N-desmethyl enzalutamide by 2.2-fold with minimal effect on Cmax. The results are summarized in Figure 1 [see Dosage and Administration (2.2) and Drug Interactions (7.1)].
In a drug-drug interaction trial in healthy volunteers, a single 160 mg oral dose of XTANDI was administered alone or after multiple oral doses of rifampin (strong CYP3A4 and moderate CYP2C8 inducer). Rifampin decreased the AUC0‑inf of enzalutamide plus N-desmethyl enzalutamide by 37% with no effect on Cmax. The results are summarized in Figure 1[see Dosage and Administration (2.2) and Drug Interactions (7.2)].
In a drug-drug interaction trial in healthy volunteers, a single 160 mg oral dose of XTANDI was administered alone or after multiple oral doses of itraconazole (strong CYP3A4 inhibitor). Itraconazole increased the AUC0‑inf of enzalutamide plus N-desmethyl enzalutamide by 1.3-fold with no effect on Cmax. The results are summarized in Figure 1.
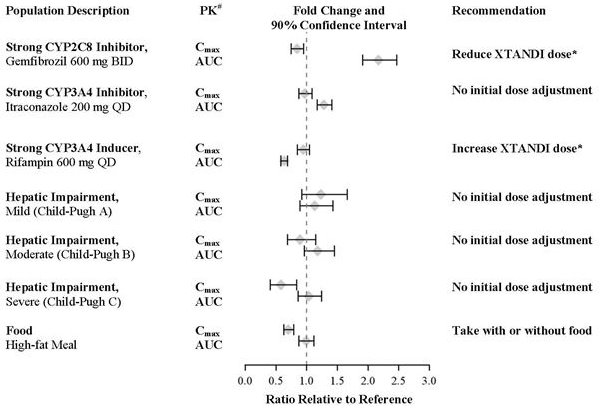
# PK parameters (Cmax and AUC0-inf) are for enzalutamide plus N-desmethyl enzalutamide, except in the food-effect trial, where they are for enzalutamide alone.
* See Dosage and Administration (2.2).
Effect of XTANDI on Other Drugs:
In an in vivo phenotypic cocktail drug-drug interaction trial in patients with metastatic CRPC, a single oral dose of the CYP probe substrate cocktail (for CYP2C8, CYP2C9, CYP2C19, and CYP3A4) was administered before and concomitantly with XTANDI (following at least 55 days of dosing at 160 mg daily). The results are summarized in
Figure 2. Results showed that in vivo, at steady-state, XTANDI is a strong CYP3A4 inducer and a moderate CYP2C9 and CYP2C19 inducer [see Drug Interactions (7.3)]. XTANDI did not cause clinically meaningful changes in exposure to the CYP2C8 substrate.
In an in vivo phenotypic cocktail drug-drug interaction trial in patients with CRPC, a single oral dose of the CYP probe substrate cocktail for CYP1A2 and CYP2D6 was administered before and concomitantly with XTANDI (following at least 49 days of dosing at 160 mg daily). The results are summarized in Figure 2. Results showed that in vivo, at steady-state, XTANDI did not cause clinically meaningful changes in exposure to the CYP1A2 or CYP2D6 substrates.
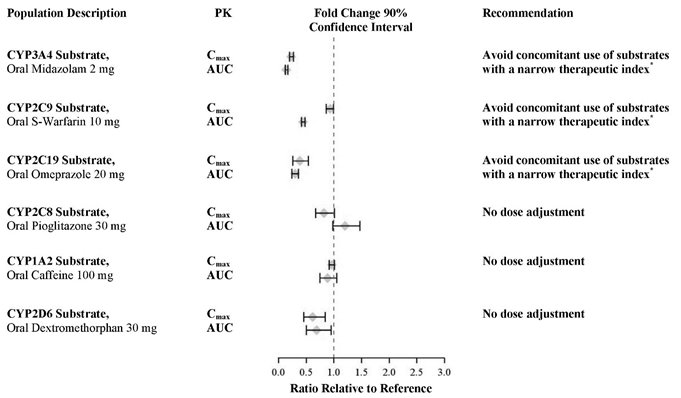
*See Drug Interactions (7.3).
In vitro, enzalutamide, N-desmethyl enzalutamide, and the major inactive carboxylic acid metabolite caused direct inhibition of multiple CYP enzymes including CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4/5; however, subsequent clinical data showed that XTANDI is an inducer of CYP2C9, CYP2C19, and CYP3A4 and had no clinically meaningful effect on CYP2C8 (see Figure 2). In vitro, enzalutamide caused time-dependent inhibition of CYP1A2.
In vitro studies showed that enzalutamide induces CYP2B6 and CYP3A4 and does not induce CYP1A2 at therapeutically relevant concentrations.
In vitro, enzalutamide, N-desmethyl enzalutamide, and the major inactive carboxylic acid metabolite are not substrates for human P-glycoprotein. In vitro, enzalutamide and N-desmethyl enzalutamide are inhibitors of human P-glycoprotein, while the major inactive carboxylic acid metabolite is not.
In vitro, enzalutamide and N-desmethyl enzalutamide do not appear to be substrates of human breast cancer resistance protein (BCRP); however, enzalutamide and N-desmethyl enzalutamide are inhibitors of human BCRP at clinically relevant concentrations.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
A two-year carcinogenicity study was conducted in male and female rats at oral enzalutamide doses of 10, 30, and 100 mg/kg/day. Enzalutamide increased the incidence of benign Leydig cell tumors in the testes at all dose levels tested (≥ 0.3 times the human exposure based on AUC) and combined incidence of urothelial papilloma and carcinoma in the urinary bladder in male rats at 100 mg/kg/day (1.4 times the human exposure based on AUC). The findings in the testes are considered to be related to the pharmacological activity of enzalutamide. Rats are regarded as more sensitive than humans to developing interstitial cell tumors in the testes. Administration of enzalutamide to male and female rasH2 transgenic mice by oral gavage daily for 26 weeks did not result in increased incidence of neoplasms at doses up to 20 mg/kg/day.
Enzalutamide did not induce mutations in the bacterial reverse mutation (Ames) assay and was not genotoxic in either the in vitro mouse lymphoma thymidine kinase (Tk) gene mutation assay or the in vivo mouse micronucleus assay.
Based on nonclinical findings in repeat-dose toxicology studies, which were consistent with the pharmacological activity of enzalutamide, male fertility may be impaired by treatment with XTANDI. In a 26-week study in rats, atrophy of the prostate and seminal vesicles was observed at ≥ 30 mg/kg/day (equal to the human exposure based on AUC). In 4-, 13-, and 39-week studies in dogs, hypospermatogenesis and atrophy of the prostate and epididymides were observed at ≥ 4 mg/kg/day (0.3 times the human exposure based on AUC).
14 CLINICAL STUDIES
The efficacy of XTANDI in patients with CRPC (N = 4692) or mCSPC (N = 1150) was demonstrated in five randomized, multicenter clinical trials. All patients received concomitant GnRH therapy or had prior bilateral orchiectomy. Patients were allowed, but not required, to continue or initiate glucocorticoids.
AFFIRM (NCT00974311): XTANDI versus Placebo in Metastatic CRPC Following Chemotherapy
In AFFIRM, a total of 1199 patients who had received prior docetaxel-based chemotherapy were randomized 2:1 to receive either XTANDI orally at a dose of 160 mg once daily (N = 800) or placebo orally once daily (N = 399). Study treatment continued until disease progression (evidence of radiographic progression, a skeletal-related event, or clinical progression), initiation of new systemic antineoplastic treatment, unacceptable toxicity, or withdrawal. Patients with a previous history of seizure, taking medicines known to decrease the seizure threshold, or with other risk factors for seizure were not eligible [see Warnings and Precautions (5.1)].
The following patient demographics and baseline disease characteristics were balanced between the treatment arms. The median age was 69 years (range 41-92) and the racial distribution was 92.7% Caucasian, 3.9% Black, 1.1% Asian, and 2.1% Other. Ninety-two percent of patients had an ECOG performance status score of 0-1 and 28% had a mean Brief Pain Inventory score of ≥ 4. Ninety-one percent of patients had metastases in bone and 23% had visceral involvement in the lung and/or liver. Fifty-nine percent of patients had radiographic evidence of disease progression and 41% had PSA-only progression on study entry. All patients had received prior docetaxel-based therapy and 24% had received two cytotoxic chemotherapy regimens. During the trial, 48% of patients on the XTANDI arm and 46% of patients on the placebo arm received glucocorticoids.
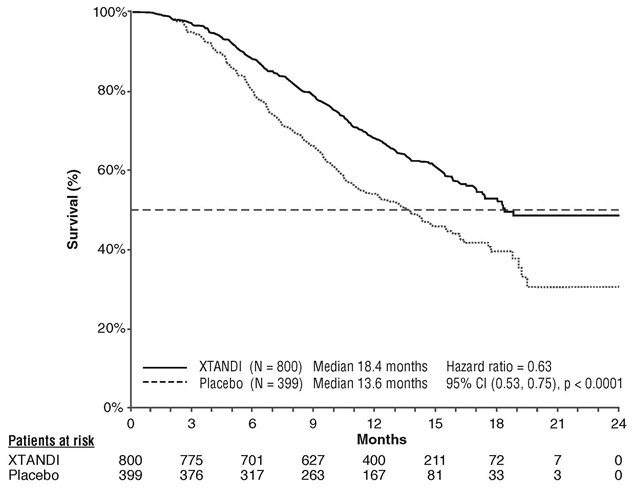
A statistically significant improvement in overall survival was demonstrated at the pre-specified interim analysis at the time of 520 deaths in patients on the XTANDI arm compared to patients on the placebo arm (Table 7 and Figure 3).
| NR = Not reached. | ||
|
|
||
|
XTANDI (N = 800) |
Placebo (N = 399) |
|
|
Number of Deaths (%) |
308 (38.5) |
212 (53.1) |
|
Median Survival, months (95% CI) |
18.4 (17.3, NR) |
13.6 (11.3, 15.8) |
|
P-value* |
p < 0.0001 |
|
|
Hazard Ratio (95% CI)† |
0.63 (0.53, 0.75) |
|
PREVAIL (NCT01212991): XTANDI versus Placebo in Chemotherapy-naïve Metastatic CRPC
In PREVAIL, 1717 chemotherapy-naïve patients were randomized 1:1 to receive either XTANDI orally at a dose of 160 mg once daily (N = 872) or placebo orally once daily (N = 845). Patients with visceral metastases, patients with a history of mild to moderate heart failure (NYHA class I or II), and patients taking medications associated with lowering the seizure threshold were allowed. Patients with a previous history of seizure or a condition that might predispose to seizure and patients with moderate or severe pain from prostate cancer were excluded. Study treatment continued until disease progression (evidence of radiographic progression, a skeletal-related event, or clinical progression) and the initiation of a cytotoxic chemotherapy or an investigational agent, unacceptable toxicity, or withdrawal. Overall survival and radiographic progression-free survival (rPFS) were assessed. Radiographic progression was assessed with the use of sequential imaging and was defined by bone scan identification of 2 or more new bone lesions with confirmation (Prostate Cancer Clinical Trials Working Group 2 criteria) and/or Response Evaluation Criteria in Solid Tumors (RECIST v 1.1) criteria for progression of soft tissue lesions. The primary analysis of rPFS utilized centrally reviewed radiographic assessment of progression.
Patient demographics and baseline disease characteristics were balanced between the treatment arms at entry. The median age was 71 years (range 42-93) and the racial distribution was 77% Caucasian, 10% Asian, 2% Black and 11% Other. The ECOG performance status score was 0 for 68% of patients, and 1 for 32% of patients. Baseline pain assessment was 0-1 (asymptomatic) in 67% of patients, and 2-3 (mildly symptomatic) in 32% of patients as defined by the Brief Pain Inventory Short Form (worst pain over past 24 hours at study entry). Fifty-four percent of patients had radiographic evidence of disease progression and 43% had PSA-only progression. Twelve percent of patients had visceral (lung and/or liver) disease involvement. During the study, 27% of patients on the XTANDI arm and 30% of patients on the placebo arm received glucocorticoids for varying reasons.
A statistically significant improvement in overall survival was demonstrated at the pre-specified interim analysis, conducted after 540 deaths in patients treated with XTANDI compared to those treated with placebo (Table 8). Forty percent of XTANDI-treated and 70% of placebo-treated patients received subsequent therapies for metastatic CRPC that may prolong overall survival. An updated survival analysis was conducted when 784 deaths were observed. The median follow-up time was 31 months. Results from this analysis were consistent with those from the pre-specified interim analysis (Table 8, Figure 4). At the updated analysis, 52% of XTANDI-treated and 81% of placebo-treated patients had received subsequent therapies that may prolong overall survival in metastatic CRPC. XTANDI was used as a subsequent therapy in 2% of XTANDI-treated patients and 29% of placebo-treated patients.
| NR = Not reached. | |||||
|
|
|||||
|
XTANDI (N = 872) |
Placebo (N = 845) |
||||
|
Pre-specified Interim Analysis* | |||||
|
Number of Deaths (%) |
241 (28) |
299 (35) |
|||
|
Median Survival, months (95% CI) |
32.4 (30.1, NR) |
30.2 (28.0, NR) |
|||
|
P-value† |
p < 0.0001 |
||||
|
Hazard Ratio (95% CI)‡ |
0.71 (0.60, 0.84) |
||||
|
Updated Survival Analysis§ | |||||
|
Number of Deaths (%) |
368 (42) |
416 (49) |
|||
|
Median Survival, months (95% CI) |
35.3 (32.2, NR) |
31.3 (28.8, 34.2) |
|||
|
Hazard Ratio (95% CI)‡ |
0.77 (0.67, 0.88) |
||||
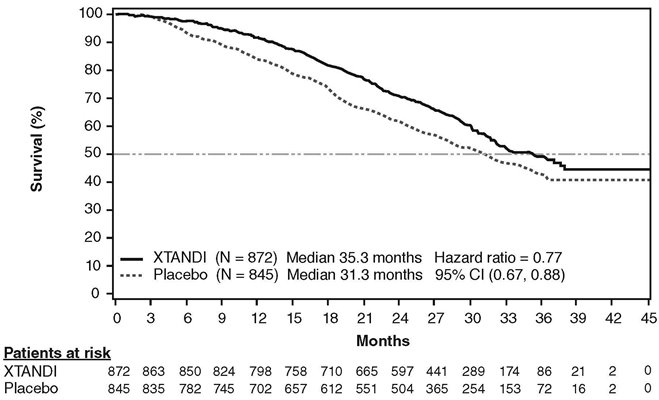
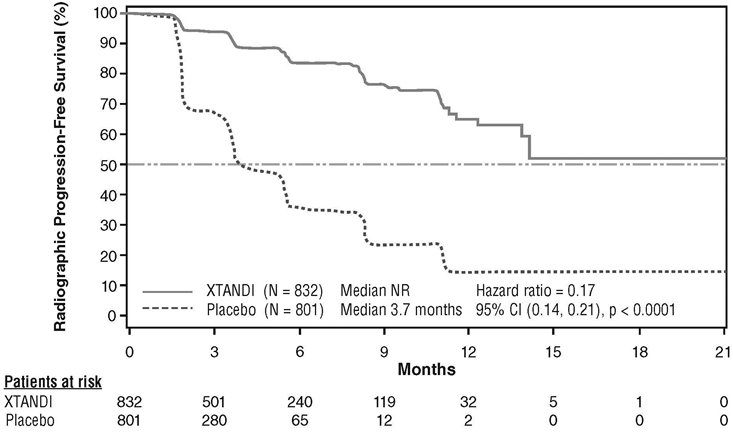
A statistically significant improvement in rPFS was demonstrated in patients treated with XTANDI compared to patients treated with placebo (Table 9, Figure 5).
| NR = Not reached. Note: As of the cutoff date for the rPFS analysis, 1633 patients had been randomized. |
||||||||
|
|
||||||||
|
XTANDI (N = 832) |
Placebo (N = 801) |
|||||||
|
Number of Progression or Deaths (%) |
118 (14) |
320 (40) |
||||||
|
Median rPFS (months) (95% CI) |
NR (13.8, NR) |
3.7 (3.6, 4.6) |
||||||
|
P-value* |
p < 0.0001 |
|||||||
|
Hazard Ratio (95% CI)† |
0.17 (0.14, 0.21) |
|||||||
Time to initiation of cytotoxic chemotherapy was prolonged after XTANDI treatment, with a median of 28.0 months for patients on the XTANDI arm versus a median of 10.8 months for patients on the placebo arm [HR = 0.35 (95% CI: 0.30, 0.40), p < 0.0001].
The median time to first skeletal‑related event was 31.1 months for patients on the XTANDI arm versus 31.3 months for patients on the placebo arm [HR = 0.72 (95% CI: 0.61, 0.84), p < 0.0001]. A skeletal‑related event was defined as radiation therapy or surgery to bone for prostate cancer, pathologic bone fracture, spinal cord compression, or change of antineoplastic therapy to treat bone pain.
TERRAIN (NCT01288911): XTANDI versus Bicalutamide in Chemotherapy-naïve Metastatic CRPC
TERRAIN was conducted in 375 chemotherapy-naïve patients who were randomized 1:1 to receive either XTANDI orally at a dose of 160 mg once daily (N = 184) or bicalutamide orally at a dose of 50 mg once daily (N = 191). Patients with a previous history of seizure or a condition that might predispose to seizure and patients with moderate to severe pain from prostate cancer were excluded. Patients could have received prior bicalutamide, but those whose disease had progressed on prior antiandrogen therapy (e.g., bicalutamide) were excluded. Study treatment continued until disease progression (evidence of radiographic progression, a skeletal-related event), the initiation of subsequent antineoplastic agent, unacceptable toxicity, or withdrawal. Radiographic disease progression was assessed by Independent Central Review (ICR) using the Prostate Cancer Clinical Trials Working Group 2 criteria and/or Response Evaluation Criteria in Solid Tumors (RECIST v 1.1) criteria for progression of soft tissue lesions. Radiographic progression-free survival (rPFS) was defined as the time from randomization to the first objective evidence of radiographic progression as assessed by ICR or death, whichever occurred first.
Patient demographics and baseline disease characteristics were balanced between the treatment arms at entry. The median age was 71 years (range 48-96) and the racial distribution was 93% Caucasian, 5% Black, 1% Asian and 1% Other. The ECOG performance status score was 0 for 74% of patients and 1 for 26% of patients. Baseline pain assessment was 0‑1 (asymptomatic) in 58% of patients, and 2‑3 (mildly symptomatic) in 36% of patients as defined by the Brief Pain Inventory Short Form Question 3 (worst pain over past 24 hours at study entry). Ninety-eight percent of patients had objective evidence of disease progression at study entry. Forty-six percent of patients had received prior treatment with bicalutamide while no patients received prior treatment with XTANDI.
An improvement in rPFS was demonstrated in patients treated with XTANDI compared to patients treated with bicalutamide (Table 10, Figure 6).
| XTANDI
(N = 184) | Bicalutamide
(N = 191) |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| NR = Not reached. | |||||||||||
|
|
|||||||||||
|
Number of Progression or Deaths (%) |
72 (39) |
74 (39) |
|||||||||
|
Median rPFS (months) (95% CI) |
19.5 (11.8, NR) |
13.4 (8.2, 16.4) |
|||||||||
|
Hazard Ratio (95% CI)* |
0.60 (0.43, 0.83) |
||||||||||
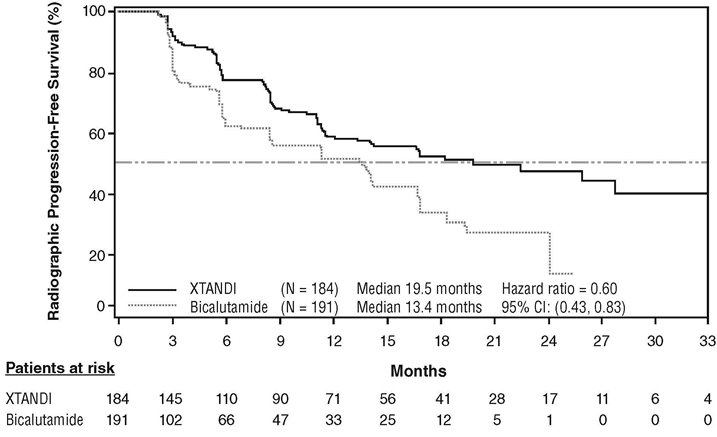
PROSPER (NCT02003924): XTANDI versus Placebo in Non-metastatic CRPC
PROSPER enrolled 1401 patients with non-metastatic CRPC who were randomized 2:1 to receive either XTANDI orally at a dose of 160 mg once daily (N = 933) or placebo orally once daily (N = 468). All patients in the PROSPER trial received a gonadotropin-releasing hormone (GnRH) analog or had a prior bilateral orchiectomy. Patients were stratified by Prostate Specific Antigen (PSA) Doubling Time (PSADT) and the use of bone-targeting agents. Patients were required to have a PSA doubling time ≤ 10 months, PSA ≥ 2 ng/mL, and confirmation of non-metastatic disease by blinded independent central review (BICR). PSA results were blinded and were not used for treatment discontinuation. Patients randomized to either arm discontinued treatment for radiographic disease progression confirmed by BICR, initiation of new treatment, unacceptable toxicity, or withdrawal.
The following patient demographics and baseline characteristics were balanced between the two treatment arms. The median age at randomization was 74 years (range 50-95) and 23% were 80 years of age or older. The racial distribution was 71% Caucasian, 16% Asian, and 2% Black. A majority of patients had a Gleason score of 7 or higher (77%). The median PSADT was 3.7 months. Fifty-four percent (54%) of patients received prior treatment for prostate cancer with either surgery or radiation. Sixty-three percent (63%) of patients received prior treatment with an anti-androgen; 56% of patients received bicalutamide and 11% of patients received flutamide. All patients had an Eastern Cooperative Oncology Group Performance Status (ECOG PS) score of 0 or 1 at study entry.
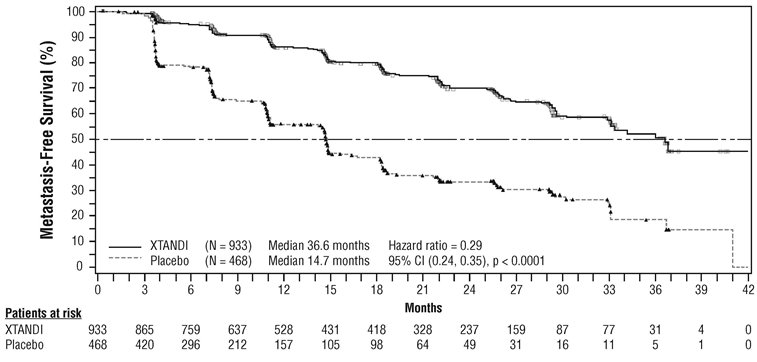
The major efficacy outcome of the study was metastasis-free survival (MFS), defined as the time from randomization to whichever of the following occurred first 1) loco-regional and/or distant radiographic progression per BICR or 2) death up to 112 days after treatment discontinuation without evidence of radiographic progression. A statistically significant improvement in MFS and OS was demonstrated in patients randomized to receive XTANDI compared with patients randomized to receive placebo. Consistent MFS results were observed when considering only distant radiographic progression events or deaths regardless of the cut-off date. Consistent MFS results were also observed in pre-specified and stratified patient sub-groups of PSADT (< 6 months or ≥ 6 months) and use of a prior bone-targeting agent (yes or no). The efficacy results from PROSPER are summarized in Table 11, Figure 7 and Figure 8.
| NR = Not reached. | |||||||||||||||
|
|
|||||||||||||||
|
XTANDI
|
Placebo
|
||||||||||||||
|
Metastasis-free survival |
|||||||||||||||
|
Number of Events (%) |
219 (23.5) |
228 (48.7) |
|||||||||||||
|
Median, months (95% CI)* |
36.6 (33.1, NR) |
14.7 (14.2, 15.0) |
|||||||||||||
|
Hazard Ratio (95% CI)† |
0.29 (0.24, 0.35) |
||||||||||||||
|
P-value† |
p < 0.0001 |
||||||||||||||
|
Overall survival‡ |
|||||||||||||||
|
Number of Events (%) |
288 (30.9) |
178 (38.0) |
|||||||||||||
|
Median, months (95% CI)* |
67.0 (64.0, NR) |
56.3 (54.4, 63.0) |
|||||||||||||
|
Hazard Ratio (95% CI)† |
0.73 (0.61, 0.88) |
||||||||||||||
|
P-value† |
p = 0.0011 |
||||||||||||||
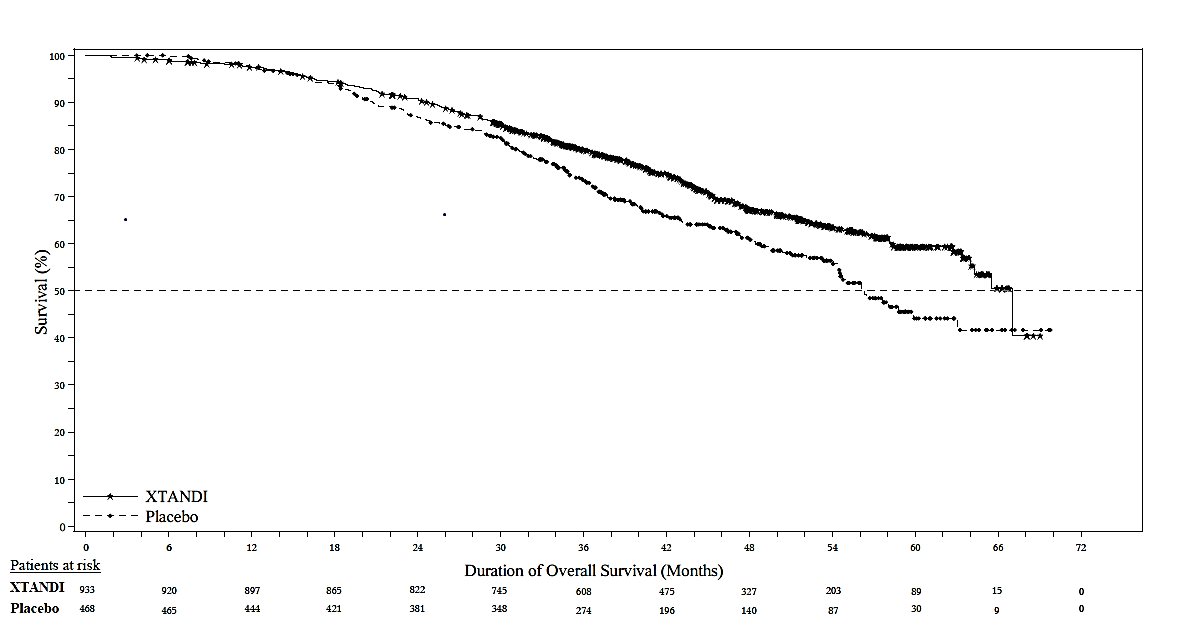
The primary efficacy outcome was also supported by a statistically significant delay in time to first use of new antineoplastic therapy (TTA) for patients in the XTANDI arm compared to those in the placebo arm. The median TTA was 39.6 months for patients on XTANDI and was 17.7 months for patients on placebo (HR = 0.21; 95% CI: [0.17, 0.26], p < 0.0001).
ARCHES (NCT02677896): XTANDI versus Placebo in Metastatic CSPC
ARCHES enrolled 1150 patients with mCSPC who were randomized 1:1 to receive XTANDI orally at a dose of 160 mg once daily (N=574) or placebo orally once daily (N=576). All patients in the trial received a GnRH analog or had a prior bilateral orchiectomy. Patients were stratified by volume of disease (low vs high) and prior docetaxel therapy for prostate cancer (no prior docetaxel, 1-5 cycles, or 6 prior cycles). High volume of disease is defined as metastases involving the viscera or, in the absence of visceral lesions, there must be 4 or more bone lesions, at least 1 of which must be in a bony structure beyond the vertebral column and pelvic bone. Treatment with concurrent docetaxel was not allowed. Patients continued treatment until radiographic disease progression, initiation of new treatment, unacceptable toxicity, or withdrawal.
The following patient demographics and baseline characteristics were balanced between the two treatment arms. The median age at randomization was 70 years (range: 42-92) and 30% were 75 years of age or older. The racial distribution was 81% Caucasian, 14% Asian, and 1% Black. Sixty-six percent (66%) of patients had a Gleason score of ≥ 8. Thirty-seven percent (37%) of patients had a low volume of disease and 63% of patients had a high volume of disease. Eighty-two percent (82%) of patients had no prior docetaxel treatment; 2% of patients had 1 to 5 cycles of docetaxel and 16% of patients had 6 prior cycles of docetaxel treatment. Twelve percent (12%) of patients received concomitant bone-targeted agents (bisphosphonates or RANKL inhibitors) which included both prostate and non-prostate cancer indications. The Eastern Cooperative Oncology Group Performance Status (ECOG PS) score was 0 for 78% of patients and 1 for 22% of patients at study entry.
The major efficacy outcome measure was radiographic progression-free survival (rPFS) based on blinded independent central review (BICR). Radiographic progression-free survival was defined as the time from randomization to radiographic disease progression at any time or death within 24 weeks after study drug discontinuation. Radiographic disease progression was defined by identification of 2 or more new bone lesions on a bone scan with confirmation (Prostate Cancer Working Group 2 criteria) and/or progression in soft tissue disease. Time to new antineoplastic therapy was an additional efficacy endpoint.
XTANDI demonstrated a statistically significant improvement in rPFS compared to placebo. Consistent rPFS results were observed in patients with high or low volume of disease and patients with and without prior docetaxel therapy. Overall survival (OS) data were not mature at the time of rPFS analysis (7.3% deaths in the ITT population had been reported). Efficacy results for rPFS from ARCHES are summarized in Table 12 and Figure 9.
| NR = Not reached | ||||||||||||||||||
|
|
||||||||||||||||||
|
XTANDI
|
Placebo
|
|||||||||||||||||
|
Radiographic Progression-free Survival |
||||||||||||||||||
|
Number of events (%) |
89 (15.5) |
198 (34.4) |
||||||||||||||||
|
77 (13.4) |
185 (32.1) |
||||||||||||||||
|
12 (2.1) |
13 (2.3) |
||||||||||||||||
|
Median, months (95% CI)* |
NR |
19.4 (16.6, NR) |
||||||||||||||||
|
Hazard ratio (95% CI)† |
0.39 (0.30, 0.50) |
|||||||||||||||||
|
P-value‡ |
p < 0.0001 |
|||||||||||||||||
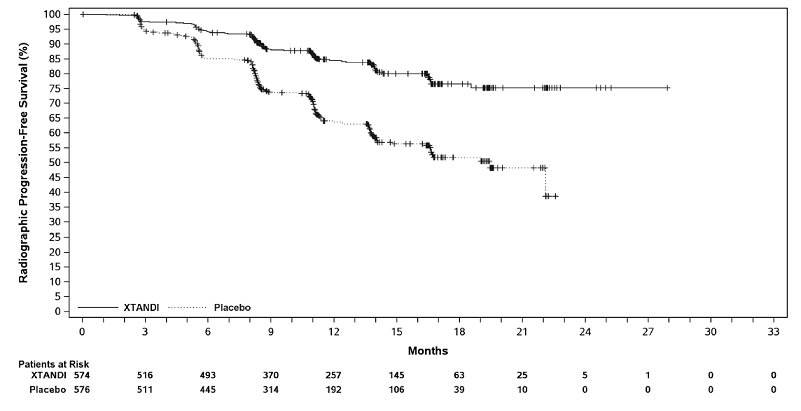
A statistically significant improvement was also reported on the XTANDI arm compared to placebo in time to initiation of a new antineoplastic therapy (HR = 0.28 [95% CI: 0.20, 0.40]; p < 0.0001).
16 HOW SUPPLIED/STORAGE AND HANDLING
XTANDI (enzalutamide) 40 mg capsules are supplied as white to off-white oblong soft gelatin capsules imprinted in black ink with ENZ and are available in the following package size:
- Bottles of 120 capsules with child resistant closures (NDC: 0469-0125-99)
XTANDI (enzalutamide) 40 mg tablets are supplied as yellow, round, film-coated tablets debossed with E 40, and are available in the following package size:
- Bottles of 120 tablets with child resistant closures (NDC: 0469-0625-99)
XTANDI (enzalutamide) 80 mg tablets are supplied as yellow, oval, film-coated tablets debossed with E 80, and are available in the following package size:
- Bottles of 60 tablets with child resistant closures (NDC: 0469-0725-60)
Recommended storage: Store XTANDI capsules and tablets at 20°C to 25°C (68°F to 77°F) in a dry place and keep the container tightly closed. Excursions permitted from 15°C to 30°C (59°F to 86°F).
Swallow capsules or tablets whole. Do not chew, dissolve or open the capsules. Do not cut, crush, or chew the tablets.
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Seizure
- Inform patients that XTANDI has been associated with an increased risk of seizure. Discuss conditions that may predispose to seizures and medications that may lower the seizure threshold. Advise patients of the risk of engaging in any activity where sudden loss of consciousness could cause serious harm to themselves or others. Inform patients to contact their healthcare provider right away if they have loss of consciousness or seizure [see Warnings and Precautions (5.1)].
Posterior Reversible Encephalopathy Syndrome (PRES)
- Inform patients to contact their healthcare provider right away if they experience rapidly worsening symptoms possibly indicative of PRES such as seizure, headache, decreased alertness, confusion, reduced eyesight, or blurred vision [see Warnings and Precautions (5.2)].
Hypersensitivity
- Inform patients that XTANDI may be associated with hypersensitivity reactions that include swelling of the face, lip, tongue, or throat [see Warnings and Precautions (5.3)]. Advise patients who experience these types of symptoms of hypersensitivity to discontinue XTANDI and promptly contact their healthcare provider.
Ischemic Heart Disease
- Inform patients that XTANDI has been associated with an increased risk of ischemic heart disease. Advise patients to seek immediate medical attention if any symptoms suggestive of a cardiovascular event occur [see Warnings and Precautions (5.4)].
Falls and Fractures
- Inform patients that XTANDI is associated with an increased incidence of dizziness/vertigo, falls, and fractures. Advise patients to report these adverse reactions to their healthcare provider [see Warnings and Precautions (5.5)].
Hypertension
- Inform patients that XTANDI is associated with an increased incidence of hypertension [see Adverse Reactions (6.1)].
Dosing and Administration
- Inform patients who have not undergone bilateral orchiectomy and are receiving GnRH therapy that they need to maintain this treatment during the course of treatment with XTANDI.
- Instruct patients to take their dose at the same time each day (once daily). XTANDI can be taken with or without food. Each capsule or tablet should be swallowed whole. Do not chew, dissolve, or open the capsules. Do not cut, crush, or chew the tablets.
- Inform patients that they should not interrupt, modify the dose, or stop XTANDI without first consulting their healthcare provider.
- Inform patients that if they miss a dose, then they should take it as soon as they remember. If they forget to take the dose for the whole day, then they should take their normal dose the next day. They should not take more than their prescribed dose per day [see Dosage and Administration (2.1)].
Embryo-Fetal Toxicity
- Inform patients that XTANDI can be harmful to a developing fetus and can cause loss of pregnancy.
- Advise male patients with female partners of reproductive potential to use effective contraception during treatment and for 3 months after the last dose of XTANDI. Advise male patients to use a condom if having sex with a pregnant woman [see Warnings and Precautions (5.6)].
Infertility
- Inform male patients that XTANDI may impair fertility [see Use in Specific Populations (8.3)].
Manufactured for and Distributed by: Astellas Pharma US, Inc., Northbrook, IL 60062
Marketed by:
Astellas Pharma US, Inc., Northbrook, IL 60062 Pfizer Inc., New York, NY 10017
280168-XTA-USA
Rx Only
© 2020 Astellas Pharma US, Inc.
XTANDI® is a registered trademark of Astellas Pharma Inc.
|
PATIENT INFORMATION XTANDI® (ex TAN dee) (enzalutamide) capsules and tablets |
|
|
What is XTANDI®? XTANDI is a prescription medicine used to treat men with prostate cancer that:
It is not known if XTANDI is safe and effective in females. It is not known if XTANDI is safe and effective in children. |
|
|
Before taking XTANDI, tell your healthcare provider about all your medical conditions, including if you:
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. XTANDI may affect the way other medicines work, and other medicines may affect how XTANDI works. You should not start or stop any medicine before you talk with the healthcare provider that prescribed XTANDI. Know the medicines you take. Keep a list of them with you to show your healthcare provider and pharmacist when you get a new medicine. |
|
|
How should I take XTANDI?
If you take too much XTANDI, call your healthcare provider or go to the nearest emergency room right away. You may have an increased risk of seizure if you take too much XTANDI. |
|
|
What are the possible side effects of XTANDI? XTANDI may cause serious side effects including:
Your healthcare provider will stop treatment with XTANDI if you have serious side effects. The most common side effects of XTANDI include: |
|
|
|
|
XTANDI may cause fertility problems in males, which may affect the ability to father children. Talk to your healthcare provider if you have concerns about fertility. These are not all the possible side effects of XTANDI. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
|
|
How should I store XTANDI?
Keep XTANDI and all medicines out of the reach of children. |
|
|
General information about the safe and effective use of XTANDI. Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use XTANDI for a condition for which it was not prescribed. Do not give XTANDI to other people, even if they have the same symptoms that you have. It may harm them. You can ask your healthcare provider or pharmacist for information about XTANDI that is written for health professionals. |
|
|
What are the ingredients in XTANDI? XTANDI capsules Active ingredient: enzalutamide Inactive ingredients: caprylocaproyl polyoxylglycerides, butylated hydroxyanisole, butylated hydroxytoluene, gelatin, sorbitol sorbitan solution, glycerin, purified water, titanium dioxide, black iron oxide XTANDI tablets Active ingredient: enzalutamide Inactive ingredients: hypromellose acetate succinate, microcrystalline cellulose, colloidal silicon dioxide, croscarmellose sodium, and magnesium stearate. The tablet film-coat contains hypromellose, talc, polyethylene glycol, titanium dioxide, and ferric oxide. Marketed by: Astellas Pharma US, Inc., Northbrook, IL 60062 Pfizer Inc., New York, NY 10017 280168-XTA-USA XTANDI® is a registered trademark of Astellas Pharma Inc. For more information go to www.Xtandi.com or call 1-800-727-7003. |
|
This Patient Information has been approved by the U.S. Food and Drug Administration. Revised: October 2020
PACKAGE/LABEL PRINCIPAL DISPLAY PANEL

NDC: 0469-0625-99
Rx Only
Xtandi®
(enzalutamide)
tablets
40 mg
Swallow tablets whole.
Do not cut, crush, or chew the tablets.
120 Tablets
Manufactured by Pfizer Pharmaceuticals LLC, for Astellas Pharma US, Inc., Northbrook, IL 60062
PAA135442
234833-XTA-USA
PACKAGE/LABEL PRINCIPAL DISPLAY PANEL

NDC: 0469-0725-60
Rx Only
Xtandi®
(enzalutamide)
tablets
80 mg
Swallow tablets whole.
Do not cut, crush, or chew the tablets.
60 Tablets
Manufactured by Pfizer Pharmaceuticals LLC, for Astellas Pharma US, Inc., Northbrook, IL 60062
PAA135444
234833-XTA-USA
| XTANDI
enzalutamide tablet |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| XTANDI
enzalutamide tablet |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Astellas Pharma US, Inc. (605764828) |
Trademark Results [Xtandi]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 XTANDI 79110148 4181564 Live/Registered |
Astellas Pharma Inc. 2011-12-27 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.