QUVIVIQ- daridorexant tablet, film coated
QUVIVIQ by
Drug Labeling and Warnings
QUVIVIQ by is a Prescription medication manufactured, distributed, or labeled by Idorsia Pharmaceuticals Ltd. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use QUVIVIQ safely and effectively. See full prescribing information for QUVIVIQ.
QUVIVIQ (daridorexant) tablets, for oral use, CIV
Initial U.S. Approval: 2022INDICATIONS AND USAGE
QUVIVIQ is an orexin receptor antagonist indicated for the treatment of adult patients with insomnia, characterized by difficulties with sleep onset and/or sleep maintenance. (1)
DOSAGE AND ADMINISTRATION
- The recommended dosage is 25 mg to 50 mg once per night, taken orally within 30 minutes before going to bed, with at least 7 hours remaining prior to planned awakening. (2.1)
- Time to sleep onset may be delayed if taken with or soon after a meal. (2.1)
- Hepatic Impairment: (2.3)
- Moderate hepatic impairment: Maximum recommended dosage is 25 mg no more than once per night.
- Severe hepatic impairment: Not recommended.
DOSAGE FORMS AND STRENGTHS
Tablets: 25 mg, 50 mg. (3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- CNS-Depressant Effects and Daytime Impairment: Impairs alertness and motor coordination including morning impairment. Risk increases when used with other central nervous system (CNS) depressants. For patients taking QUVIVIQ, caution against next-day driving and other activities requiring complete mental alertness. (5.1)
- Worsening of Depression/Suicidal Ideation: Worsening of depression or suicidal thinking may occur. (5.2)
- Sleep Paralysis, Hypnagogic/Hypnopompic Hallucinations, and Cataplexy-like Symptoms: May occur with use of QUVIVIQ. (5.3)
- Complex Sleep Behaviors: Behaviors including sleepwalking, sleep driving, and engaging in other activities while not fully awake may occur. Discontinue immediately if complex sleep behavior occurs. (5.4)
- Compromised Respiratory Function: Effect on respiratory function should be considered. (5.5, 8.7)
- Need to Evaluate for Co-morbid Diagnoses: Reevaluate if insomnia persists after 7 to 10 days. (5.6)
ADVERSE REACTIONS
The most common adverse reactions (reported in ≥ 5% of patients treated with QUVIVIQ and at an incidence ≥ than placebo) were headache and somnolence or fatigue. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Idorsia Pharmaceuticals Ltd at toll-free phone 1-833-400-9611 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 9/2024
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
2.2 Dosage Recommendations for Concomitant Use with CYP3A4 Inhibitors or CYP3A4 Inducers
2.3 Dosage Recommendations for Patients with Hepatic Impairment
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 CNS-Depressant Effects and Daytime Impairment
5.2 Worsening of Depression/Suicidal Ideation
5.3 Sleep Paralysis, Hypnagogic/Hypnopompic Hallucinations, and Cataplexy-like Symptoms
5.4 Complex Sleep Behaviors
5.5 Patients with Compromised Respiratory Function
5.6 Need to Evaluate for Co-morbid Diagnoses
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Post-Marketing Experience
7 DRUG INTERACTIONS
7.1 Effects of Other Drugs on QUVIVIQ
7.2 Effects of QUVIVIQ on Other Drugs
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
8.7 Patients with Compromised Respiratory Function
9 DRUG ABUSE AND DEPENDENCE
9.1 Controlled Substance
9.2 Abuse
9.3 Dependence
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
14.1 Controlled Clinical Studies
14.2 Special Safety Studies
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage and Handling
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
QUVIVIQ is indicated for the treatment of adult patients with insomnia, characterized by difficulties with sleep onset and/or sleep maintenance [see Clinical Studies (14.1)].
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
The recommended dosage range is 25 mg to 50 mg of QUVIVIQ taken orally no more than once per night within 30 minutes of going to bed (with at least 7 hours remaining prior to planned awakening).
Time to sleep onset may be delayed if taken with or soon after a meal [see Clinical Pharmacology (12.3)].
2.2 Dosage Recommendations for Concomitant Use with CYP3A4 Inhibitors or CYP3A4 Inducers
Co-administration with Strong CYP3A4 Inhibitors
Avoid concomitant use of QUVIVIQ with strong inhibitors of CYP3A4 [see Drug Interactions (7.1), Clinical Pharmacology (12.3)].
Co-administration with Moderate CYP3A4 Inhibitors
The recommended dosage of QUVIVIQ is 25 mg no more than once per night when used with moderate inhibitors of CYP3A4 [see Drug Interactions (7.1), Clinical Pharmacology (12.3)].
Co-administration with Strong or Moderate CYP3A4 Inducers
Avoid concomitant use of QUVIVIQ with strong or moderate CYP3A4 inducers [see Drug Interactions (7.1)].
2.3 Dosage Recommendations for Patients with Hepatic Impairment
The maximum recommended dosage in patients with moderate hepatic impairment (Child-Pugh score 7–9) is 25 mg of QUVIVIQ no more than once per night [see Use in Specific Populations (8.6), Clinical Pharmacology (12.3)].
QUVIVIQ is not recommended in patients with severe hepatic impairment (Child-Pugh score ≥ 10) [see Use in Specific Populations (8.6)].
-
3 DOSAGE FORMS AND STRENGTHS
QUVIVIQ (daridorexant) tablets are available as:
25 mg: light purple, arc-triangle shaped, film-coated tablet debossed with "25" on one side and "i" (Idorsia logo) on the other side, containing 25 mg daridorexant.
50 mg: light orange, arc-triangle shaped, film-coated tablet debossed with "50" on one side and "i" (Idorsia logo) on the other side, containing 50 mg daridorexant.
-
4 CONTRAINDICATIONS
QUVIVIQ is contraindicated:
- in patients with narcolepsy.
- in patients with a history of hypersensitivity to daridorexant or any components of QUVIVIQ. Angioedema with pharyngeal involvement has been reported [see Adverse Reactions (6.2)].
-
5 WARNINGS AND PRECAUTIONS
5.1 CNS-Depressant Effects and Daytime Impairment
QUVIVIQ is a central nervous system (CNS) depressant that can impair daytime wakefulness even when used as prescribed. CNS-depressant effects may persist in some patients for up to several days after discontinuing QUVIVIQ. Prescribers should advise patients about the potential for next-day somnolence.
Driving ability was impaired in some subjects taking QUVIVIQ 50 mg [see Clinical Studies (14.2)]. The risk of daytime impairment is increased if QUVIVIQ is taken with less than a full night of sleep remaining or if a higher than recommended dose is taken [see Dosage and Administration (2.1)]. If QUVIVIQ is taken in these circumstances, caution patients against driving and other activities requiring complete mental alertness.
Co-administration with other CNS depressants (e.g., benzodiazepines, opioids, tricyclic antidepressants, alcohol) increases the risk of CNS depression, which can cause daytime impairment. Dosage adjustments of QUVIVIQ and of concomitant CNS depressants may be necessary when administered together because of potentially additive effects. The use of QUVIVIQ with other drugs to treat insomnia is not recommended. Advise patients not to consume alcohol in combination with QUVIVIQ because co-administration of QUVIVIQ with alcohol resulted in additive effects on psychomotor performance [see Drug Interactions (7.1)].
Because QUVIVIQ can cause drowsiness, patients, particularly the elderly, are at a higher risk of falls.
5.2 Worsening of Depression/Suicidal Ideation
Patients with psychiatric disorders, including insomnia, are at increased risk of suicide. In primarily depressed patients treated with hypnotics, worsening of depression and suicidal thoughts and actions (including completed suicides) have been reported. As with other hypnotics, QUVIVIQ should be administered with caution in patients exhibiting symptoms of depression. Monitoring of suicide risk and protective measures may be required.
5.3 Sleep Paralysis, Hypnagogic/Hypnopompic Hallucinations, and Cataplexy-like Symptoms
Sleep paralysis, an inability to move or speak for up to several minutes during sleep-wake transitions, and hypnagogic/hypnopompic hallucinations, including vivid and disturbing perceptions, can occur with the use of QUVIVIQ [see Adverse Reactions (6.1)]. Prescribers should explain the nature of these events to patients when prescribing QUVIVIQ.
Symptoms similar to mild cataplexy have been reported with orexin receptor antagonists. Such symptoms can include periods of leg weakness lasting from seconds to a few minutes, can occur either at night or during the day, and may not be associated with an identified triggering event (e.g., laughter or surprise).
5.4 Complex Sleep Behaviors
Complex sleep behaviors, including sleepwalking, sleep driving, and engaging in other activities while not fully awake (e.g., preparing and eating food, making phone calls, having sex), have been reported to occur with the use of hypnotics, including orexin receptor antagonists such as QUVIVIQ. These events can occur in hypnotic-naïve as well as in hypnotic-experienced persons. Patients usually do not remember these events. Complex sleep behaviors may occur following the first or any subsequent use of hypnotics, such as QUVIVIQ, with or without the concomitant use of alcohol and other CNS depressants [see Drug Interactions (7.1)]. Discontinue QUVIVIQ immediately if a patient experiences a complex sleep behavior.
5.5 Patients with Compromised Respiratory Function
QUVIVIQ has been studied in mild to severe OSA not using CPAP, and in patients with moderate COPD. The effects of QUVIVIQ on respiratory function should be considered if prescribed to patients with compromised respiratory function. QUVIVIQ has not been studied in patients with mild or severe COPD [see Use in Specific Populations (8.7)].
5.6 Need to Evaluate for Co-morbid Diagnoses
Because sleep disturbances may be the presenting manifestation of a medical and/or psychiatric disorder, treatment of insomnia should be initiated only after careful evaluation of the patient. The failure of insomnia to remit after 7 to 10 days of treatment may indicate the presence of a primary psychiatric and/or medical illness that should be evaluated. Worsening of insomnia or the emergence of new cognitive or behavioral abnormalities may be the result of an unrecognized underlying psychiatric or medical disorder and can emerge during the course of treatment with sleep-promoting drugs such as QUVIVIQ.
-
6 ADVERSE REACTIONS
The following are discussed in detail in other sections of the labeling:
- CNS-Depressant Effects and Daytime Impairment [see Warnings and Precautions (5.1)]
- Worsening of Depression/Suicidal Ideation [see Warnings and Precautions (5.2)]
- Sleep Paralysis, Hypnagogic/Hypnopompic Hallucinations, and Cataplexy-like Symptoms [see Warnings and Precautions (5.3)]
- Complex Sleep Behaviors [see Warnings and Precautions (5.4)]
- Patients with Compromised Respiratory Function [see Warnings and Precautions (5.5)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in clinical trials of a drug cannot be directly compared to rates in clinical trials of another drug and may not reflect the rates observed in practice.
The safety of QUVIVIQ was evaluated in three placebo-controlled clinical studies (two 3-month studies of identical design [Study 1 and Study 2], and a 9-month extension study [Study 3]). Study 1 evaluated 50 mg and 25 mg doses of QUVIVIQ, while Study 2 evaluated a 25 mg dose and a 10 mg dose of QUVIVIQ. The 10 mg dose is not an approved dose. A total of 1232 patients (including approximately 40% elderly patients [≥ 65 years old]), received QUVIVIQ 50 mg (N = 308); 25 mg (N = 618); or 10 mg (an unapproved dose) (N = 306). A total of 576 patients were treated with QUVIVIQ for at least 6 months and 331 for at least 12 months.
Most Common Adverse Reactions
The most common reported adverse reaction (in at least 5% of patients and greater than placebo) during double-blind treatment in Study 1 was headache.
Table 1 shows adverse reactions that occurred in at least 2% of patients treated with QUVIVIQ and more frequently than in patients who received placebo in Study 1.
Table 1 Adverse Reactions Reported in ≥ 2% of QUVIVIQ-treated Patients and Greater than in Placebo-treated Patients in a 3-Month Placebo-Controlled Study (Study 1) QUVIVIQ QUVIVIQ Placebo 25 mg 50 mg (N=310) (N=308) (N=309) % % % - * The following terms were combined:
Headache includes: headache, tension headache, migraine, migraine with aura, head discomfort
Somnolence or fatigue includes: somnolence, sedation, fatigue, hypersomnia, lethargy
Dizziness includes: dizziness, vertigo, labyrinthitis
Nausea includes: nausea, vomiting, procedural nauseaNervous System Disorders Headache* 6 7 5 Somnolence or fatigue* 6 5 4 Dizziness* 2 3 2 Gastro-intestinal disorders Nausea* 0 3 2 Other Adverse Reactions Observed During Clinical Trials (Study 1 and Study 2)
Other adverse reactions of < 2% frequency but greater than placebo are shown below. The following do not include adverse reactions 1) for which a drug cause was remote, 2) that were so general as to be uninformative, or 3) that were not considered to have clinically significant implications.
- Sleep paralysis was reported in 0.5% and 0.3% of patients receiving QUVIVIQ 25 mg and 50 mg, respectively, compared to no reports for placebo.
- Hypnagogic and hypnopompic hallucinations were reported in 0.6% of patients receiving QUVIVIQ 25 mg compared to no cases with QUVIVIQ 50 mg or placebo.
6.2 Post-Marketing Experience
The following adverse reactions have been identified during post-approval use of QUVIVIQ. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Psychiatric disorders: Nightmares or abnormal dreams
Immune system disorders: Hypersensitivity (including angioedema, rash, urticaria)
-
7 DRUG INTERACTIONS
7.1 Effects of Other Drugs on QUVIVIQ
Table 2 describes clinically significant drug interactions where the concomitant use of other drugs affects QUVIVIQ.
Table 2 Effects of Other Drugs on QUVIVIQ Strong or Moderate CYP3A4 Inhibitors Clinical Implications: Concomitant use with a strong or moderate CYP3A4 inhibitor increases exposure to daridorexant [see Clinical Pharmacology (12.3)], which may increase the risk of QUVIVIQ adverse reactions. Prevention or Management: The recommended dose of QUVIVIQ is 25 mg when used with a moderate CYP3A4 inhibitor [see Dosage and Administration (2.2)].
Concomitant use of QUVIVIQ with a strong inhibitor of CYP3A4 is not recommended [see Dosage and Administration (2.2)].Strong and Moderate CYP3A4 Inducers Clinical Implications: Concomitant use with a strong or moderate CYP3A4 inducer decreases exposure to daridorexant [see Clinical Pharmacology (12.3)], which may reduce the efficacy of QUVIVIQ. Prevention or Management: Concomitant use of QUVIVIQ with a strong or moderate inducer of CYP3A4 is not recommended [see Dosage and Administration (2.2)]. Alcohol and Other CNS Depressants Clinical Implications: Concomitant use of alcohol or other CNS depressants with QUVIVIQ may lead to additive impairment of psychomotor performance and risk of CNS depression [see Clinical Pharmacology (12.2)]. Prevention or Management: Avoid alcohol consumption with QUVIVIQ [see Warnings and Precautions (5.1)].
Use with caution in patients receiving CNS depressants. Consider dose adjustment of QUVIVIQ and/or the CNS depressant(s) if used concomitantly [see Warnings and Precautions (5.1)].7.2 Effects of QUVIVIQ on Other Drugs
Table 3 describes clinically significant drug interactions where the concomitant use of QUVIVIQ affects other drugs.
Table 3 Effects of QUVIVIQ on Other Drugs CYP3A4 Substrates Clinical Implications: Concomitant use of QUVIVIQ with CYP3A4 substrates increases the exposure to CYP3A4 substrate [see Clinical Pharmacology (12.3)]. Prevention or Management: Use with caution in patients receiving CYP3A4 substrates with narrow therapeutic index. P-gp Substrates Clinical Implications: Concomitant use of QUVIVIQ with P-gp substrates increases the exposure to P-gp substrate [see Clinical Pharmacology (12.3)]. Prevention or Management: Use with caution in patients receiving P-gp substrates with a narrow therapeutic index. -
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Exposure Registry
There will be a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to QUVIVIQ during pregnancy. Pregnant women exposed to QUVIVIQ and healthcare providers are encouraged to call Idorsia Pharmaceuticals Ltd at 1-833-400-9611.
Risk Summary
There are no available data on QUVIVIQ use in pregnant women to evaluate for drug-associated risks of major birth defects, miscarriage, or other adverse maternal or fetal outcomes. In animal reproduction studies, oral administration of daridorexant to pregnant rats and rabbits during the period of organogenesis did not cause fetal toxicity or malformation at doses up to 8 and 10 times the maximum recommended human dose (MRHD) of 50 mg, respectively, based on AUC. Oral administration of daridorexant to pregnant and lactating rats did not cause any maternal or developmental toxicity at doses up to 9 times the MRHD, based on AUC (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
Daridorexant was administered orally to pregnant rats during the period of organogenesis at doses of 30, 100, and 300 mg/kg/day, which are approximately 1, 3, and 8 times the MRHD of 50 mg, respectively, based on AUC. Daridorexant did not cause any maternal or embryofetal toxicities or fetal malformation at doses up to 300 mg/kg/day. The NOAEL for maternal and fetal toxicity is 300 mg/kg/day, which is approximately 8 times the MRHD of 50 mg, based on AUC.
Daridorexant was administered orally to pregnant rabbits during the period of organogenesis at doses of 30, 60, and 120 mg/kg/day, which are approximately 3, 4, and 10 times the MRHD of 50 mg, respectively, based on AUC. Daridorexant did not cause any fetal toxicity or malformation at doses up to 120 mg/kg/day. Daridorexant caused maternal toxicities of decreased weight gain and food consumption at the dose of 120 mg/kg/day. The NOAELs for maternal and fetal toxicity are 60 and 120 mg/kg/day, respectively, which are approximately 4 and 10 times the MRHD of 50 mg, respectively, based on AUC.
Daridorexant was administered orally to pregnant rats during gestation and lactation at doses of 50, 100, and 300 mg/kg/day, which are approximately 1, 3, and 9 times the MRHD of 50 mg, respectively, based on AUC. Daridorexant did not cause any maternal or developmental toxicities at doses up to 300 mg/kg/day. The NOAEL for maternal and developmental toxicity is 300 mg/kg/day, which is approximately 9 times the MRHD of 50 mg, based on AUC.
8.2 Lactation
Risk Summary
Daridorexant is present in human breast milk in low amounts. In a clinical lactation study, daridorexant was detected in human milk at a mean daily infant dose of 0.0016 mg/kg, with a relative infant dose of <1% the maternal weight-adjusted dose (see Data). There are no data on the effects of daridorexant on the breastfed infant or its effects on milk production.
Infants exposed to QUVIVIQ through breastmilk should be monitored for excessive sedation. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for QUVIVIQ and any potential adverse effects on the breastfed infant from QUVIVIQ or from the underlying maternal condition.
Data
A lactation study in 10 healthy adult lactating women evaluated the concentrations of daridorexant in plasma and breast milk following a single dose of 50 mg daridorexant. The calculated mean daily infant dose is 0.0016 mg/kg based on an assumed infant body weight of 6 kg, and the relative infant dose is 0.22% of the maternal weight-adjusted dose. Approximately 87% of the total amount of daridorexant present in milk was detected by 24 hours after administration. The amount of daridorexant present in breast milk at steady state is expected to be similar to that after a single dose.
8.4 Pediatric Use
The safety and effectiveness of QUVIVIQ have not been established in pediatric patients.
8.5 Geriatric Use
No dose adjustment is required in patients over the age of 65 years.
Of the total number of subjects in the clinical studies of QUVIVIQ with insomnia (N = 1854), approximately 39% (N = 727) were ≥ 65 years and 5.9% (N = 110) were ≥ 75 years. The likelihood of somnolence and fatigue increased with patient age.
Because QUVIVIQ can increase somnolence and drowsiness, patients, particularly the elderly, are at higher risk of falls [see Warnings and Precautions (5.1)].
8.6 Hepatic Impairment
QUVIVIQ has not been studied in patients with severe hepatic impairment (Child-Pugh score ≥ 10). Use in this population is not recommended [see Clinical Pharmacology (12.3)].
Reduce the dose of QUVIVIQ in patients with moderate hepatic impairment (Child-Pugh score 7–9) [see Dosage and Administration (2.3)]. Moderate hepatic impairment may increase daridorexant systemic exposure to a clinically relevant extent [see Clinical Pharmacology (12.3)], which may increase the frequency or severity of adverse reactions.
8.7 Patients with Compromised Respiratory Function
Obstructive Sleep Apnea
The respiratory depressant effect of QUVIVIQ was evaluated after one night and after five consecutive nights of treatment with QUVIVIQ 50 mg in a randomized, placebo-controlled, two-period crossover study in 25 patients with mild to moderate OSA (apnea-hypopnea index [AHI] 5 to 30 events per hour of sleep) not using CPAP. Following once-daily dosing of 50 mg in the evening, the mean treatment difference (daridorexant – placebo) on Day 5 for AHI was 0.74 (90% CI, -1.43 to 2.92).
QUVIVIQ was also evaluated after 5 consecutive nights of treatment with QUVIVIQ 50 mg in a randomized, placebo-controlled, double-blind, two-period crossover study in 16 patients with severe OSA (apnea-hypopnea index ≥ 30 events per hour of sleep; studied range 30.8–82.2 events per hour) not using CPAP. Following once daily dosing of 50 mg in the evening, the mean treatment difference (daridorexant – placebo) on Day 5 for AHI was -3.74 (90% CI, -11.71 to 4.23).
Due to study limitations, including the short duration of the studies, clinically meaningful respiratory effects of QUVIVIQ in OSA cannot be excluded, including for long-term treatment [see Warnings and Precautions (5.5)].
Chronic Obstructive Pulmonary Disease
The respiratory depressant effect of QUVIVIQ was evaluated after one night and after five consecutive nights of treatment with QUVIVIQ 50 mg in a randomized, placebo-controlled, two-period crossover study in 25 patients with moderate COPD (FEV1/FVC ratio ≤ 70% and 40% ≤ FEV1 < 80% of predicted). Following once-daily dosing of 50 mg in the evening, the mean SpO2 treatment difference (daridorexant – placebo) on Day 5 was 0.18% (90% CI, -0.21 to 0.57).
QUVIVIQ has not been studied in patients with mild (FEV1 ≥ 80% predicted) or severe COPD (FEV1 < 40% of predicted).
Clinically meaningful respiratory effects of QUVIVIQ in patients with compromised respiratory function cannot be excluded [see Warnings and Precautions (5.5)].
-
9 DRUG ABUSE AND DEPENDENCE
9.2 Abuse
Drug abuse is the intentional, non-therapeutic use of a drug, even once, for its desirable psychological or physiological effects. The abuse potential of daridorexant was evaluated in preclinical models, recreational sedative drug users, and insomnia subjects.
In a human abuse potential study conducted in 63 recreational sedative drug users, the effect of single-dose administration of QUVIVIQ [50 mg, 100 mg (two times the maximum recommended dose), and 150 mg (three times the maximum recommended dose)], zolpidem (30 mg), suvorexant (150 mg), and placebo on subjective rating of "drug liking" was evaluated. At the dose of 50 mg, QUVIVIQ showed significantly lower "drug liking" ratings than zolpidem (30 mg) and suvorexant (150 mg), but significantly higher than placebo. At doses of 100 mg (two times the maximum recommended dose) and 150 mg (three times the maximum recommended dose), QUVIVIQ showed similar "drug liking" ratings to zolpidem (30 mg) and suvorexant (150 mg).
In placebo-controlled Phase 3 clinical studies in which 1232 subjects with insomnia were treated with QUVIVIQ for up to 12 months, there were no reports indicative of abuse liability. Because individuals with a history of abuse of or addiction to alcohol or other drugs may be at increased risk for abuse of or addiction to QUVIVIQ, follow such patients carefully.
9.3 Dependence
Physical dependence is a state that develops as a result of physiological adaptation in response to repeated drug use, manifested by withdrawal signs and symptoms upon abrupt treatment discontinuation or a significant dose reduction of a drug.
In animal studies and clinical trials evaluating physical dependence, chronic administration of daridorexant did not produce withdrawal signs or symptoms upon drug discontinuation. This suggests that daridorexant does not produce physical dependence.
-
10 OVERDOSAGE
There is limited clinical experience with QUVIVIQ overdose. In clinical pharmacology studies, healthy subjects were administered single doses of up to 200 mg (4 times the maximum recommended dose) of QUVIVIQ. The following adverse reactions were observed: somnolence, muscle weakness, cataplexy-like symptoms, sleep paralysis, disturbance in attention, fatigue, headache, and constipation.
There is no specific antidote to an overdosage of QUVIVIQ. In the event of an overdose, general symptomatic and supportive medical care, along with immediate gastric lavage where appropriate, should be provided and patients should be carefully monitored. Dialysis is unlikely to be effective as daridorexant is highly protein bound. Consider contacting the Poison Help Line (1-800-222-1222) or a medical toxicologist for additional overdosage management recommendations.
-
11 DESCRIPTION
QUVIVIQ contains daridorexant, an orexin receptor antagonist, present as daridorexant hydrochloride salt. The chemical name of daridorexant hydrochloride is (S)-(2-(5-chloro-4-methyl-1H-benzo[d]imidazol-2-yl)-2-methylpyrrolidin-1-yl)(5-methoxy-2-(2H-1,2,3-triazol-2-yl)phenyl)methanone hydrochloride. The molecular formula is C23H23N6O2Cl * HCl. The molecular weight is 487.38 g/mol.
The structural formula is:
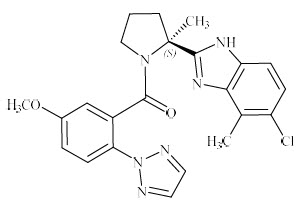
Daridorexant hydrochloride is a white to light yellowish powder that is very slightly soluble in water.
QUVIVIQ tablets are intended for oral administration. Each film-coated tablet contains daridorexant 25 mg or 50 mg, equivalent to 27 mg or 54 mg of daridorexant hydrochloride, respectively. The inactive ingredients are croscarmellose sodium, magnesium stearate, mannitol, microcrystalline cellulose, povidone, and silicon dioxide.
In addition, the film coating contains the following inactive ingredients: glycerin, hypromellose, iron oxide black, iron oxide red, microcrystalline cellulose, talc, titanium dioxide, and, in the 50 mg tablet only, iron oxide yellow.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
The mechanism of action of daridorexant in the treatment of insomnia is presumed to be through antagonism of orexin receptors. The orexin neuropeptide signaling system plays a role in wakefulness. Blocking the binding of wake-promoting neuropeptides orexin A and orexin B to receptors OX1R and OX2R is thought to suppress wake drive.
12.2 Pharmacodynamics
Daridorexant binds to and inhibits the orexin receptors OX1R and OX2R (Ki = 0.47 and 0.93 nM, respectively).
Cardiac Electrophysiology
At a dose 4 times the maximum recommended dose, QUVIVIQ does not prolong the QTc interval to any clinically relevant extent.
Alcohol
Co-administration of a single 50 mg dose of QUVIVIQ with alcohol at a blood level of 0.6 g/L led to additive effects on impairment of psychomotor performance (postural stability and alertness). Daridorexant did not affect alcohol concentrations and alcohol did not affect daridorexant concentrations [see Warnings and Precautions (5.1, 5.4), Drug Interactions (7.1)].
12.3 Pharmacokinetics
Daridorexant plasma exposure is dose proportional from 25 mg to 50 mg. The daridorexant pharmacokinetic profile is similar following multiple-dose and single-dose administration with no accumulation.
Absorption
Daridorexant reaches peak plasma concentrations within 1–2 hours (Tmax). Daridorexant has an absolute bioavailability of 62%.
Distribution
Daridorexant has a volume of distribution of 31 L. Daridorexant is 99.7% bound to plasma proteins. The blood to plasma ratio is 0.64.
Elimination
The terminal half-life of daridorexant is approximately 8 hours.
Specific Populations
Age, sex, race (White, Black, Asian), body size, and mild to severe renal impairment (Cockcroft-Gault < 30 mL/min, not on dialysis) did not have a clinically significant effect on the pharmacokinetics of daridorexant. The effect of severe hepatic impairment (Child-Pugh score ≥ 10) on the pharmacokinetics of daridorexant has not been studied.
Effects of hepatic impairment and renal impairment on the exposure to daridorexant are summarized in Figure 1.
Figure 1 Effects of hepatic impairment and renal impairment on daridorexant PK
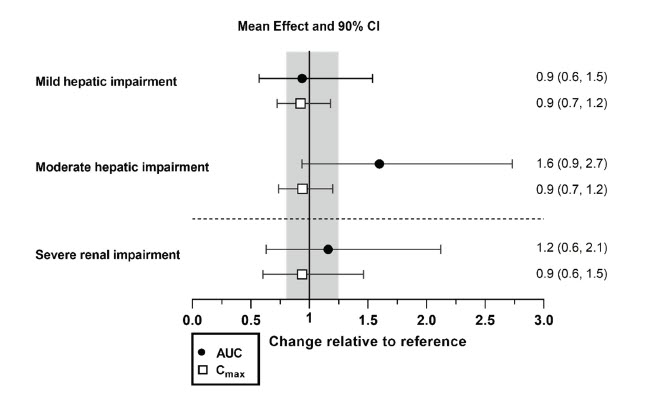
Daridorexant dose: 25 mg. Data are GMRs and 90% CIs. Hepatic impairment PK variables are based on the unbound fraction of daridorexant. Reference = matched healthy subjects. AUC = area under the plasma concentration-time curve from zero to infinity; CI = confidence interval; Cmax = maximum plasma concentration; GMR = geometric mean ratio; PK = pharmacokinetics.
Drug Interaction Studies
The effects of other compounds on the exposure to daridorexant are summarized in Figure 2. The effects of daridorexant on the exposure to other compounds are summarized in Figure 3.
Effect of Other Compounds on QUVIVIQ
Figure 2 Effect of co-administered compounds on the PK of daridorexant
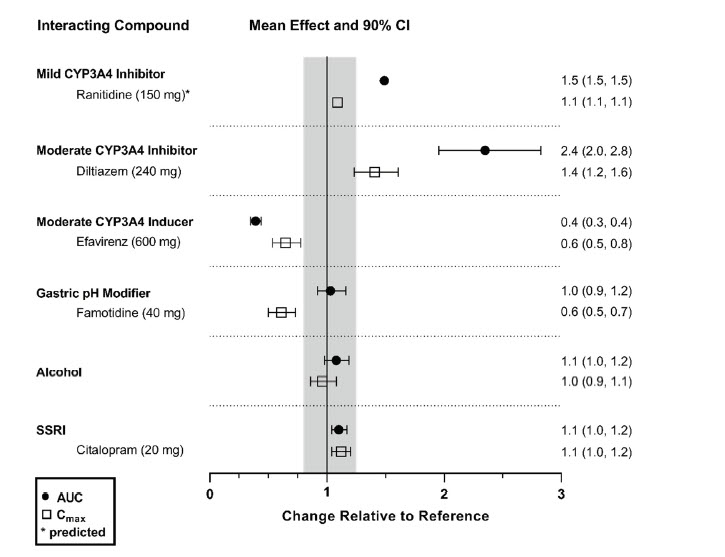
Daridorexant 50 mg was administered with interacting drugs, except with diltiazem (25 mg daridorexant). Interacting drugs were administered in multiple-dose fashion, except famotidine (single dose) and alcohol (5 h infusion at 0.6 g/L). Based on PBPK analysis: Concomitant use of itraconazole (a strong CYP3A4 inhibitor) increased daridorexant AUC by more than 400%. Concomitant use of rifampin (a strong CYP3A4 inducer) decreased daridorexant AUC by more than 50%. Data are GMRs and 90% CIs. Some 90% CIs are too narrow to be shown. AUC = area under the plasma concentration-time curve; CI = confidence interval; Cmax = maximum plasma concentration; GMR = geometric mean ratio; SSRI = selective serotonin reuptake inhibitor.
Effect of QUVIVIQ on Other Compounds
Figure 3 Effect of daridorexant on the PK of other compounds
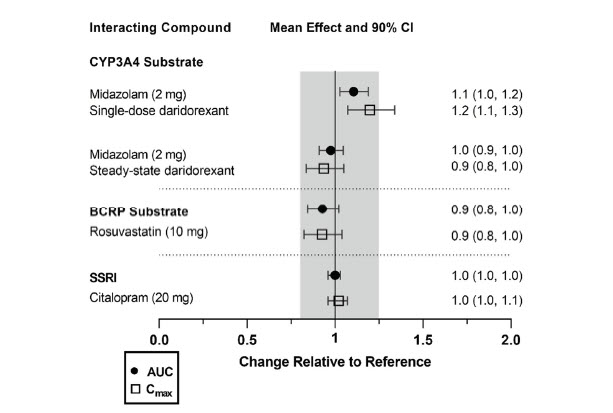
AUC = area under the plasma concentration-time curve; BCRP = breast cancer resistance protein; CI = confidence interval; Cmax = maximum plasma concentration; CP = cytochrome P450 enzyme; P-gp = P-glycoprotein; SSRI = selective serotonin reuptake inhibitor.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Daridorexant did not increase the incidence of tumors in rats treated for 2 years at oral doses of 15, 50, and 150 mg/kg/day. The high dose of 150 mg/kg/day is approximately 4 times the MRHD of 50 mg, based on AUC. Daridorexant did not increase the incidence of tumors in Tg.rasH2 mice treated for 26 weeks at oral doses of 100, 300, and 1000 mg/kg/day in males or 100, 200, and 1000 mg/kg/day in females.
Mutagenesis
Daridorexant was not mutagenic in the in vitro bacterial reverse mutation (Ames) assay or in the in vitro mammalian chromosome aberration assay in human lymphocytes and was not clastogenic in the in vivo rat micronucleus assay.
Impairment of Fertility
Daridorexant was orally administered to female rats at doses of 30, 100, and 300 mg/kg/day prior to and throughout mating and continuing to gestation Day 6. These doses are approximately 0.5, 3, and 9 times the MRHD of 50 mg, based on AUC. Daridorexant increased pre-implantation loss and decreased implantation sites without affecting mating or fertility at 300 mg/kg/day. The NOAEL for female fertility is 100 mg/kg/day, which is approximately 3 times the MRHD of 50 mg, based on AUC.
Daridorexant did not affect fertility when orally administered to male rats at doses of 50, 150, and 450 mg/kg/day prior to and throughout mating. These doses are approximately 1, 3, and 7 times the MRHD of 50 mg, based on AUC.
13.2 Animal Toxicology and/or Pharmacology
In dogs, daily oral administration of daridorexant at ≥ 30 mg/kg/day resulted in behavior characteristic of cataplexy when presented with positive stimulation. The no-observed-effect level (NOEL) for cataplexy is 20 mg/kg/day, which is approximately 3 times the MRHD of 50 mg, based on Cmax and AUC.
-
14 CLINICAL STUDIES
14.1 Controlled Clinical Studies
The efficacy of QUVIVIQ was evaluated in two multicenter, randomized, double-blind, placebo-controlled, parallel-group studies, Study 1 (NCT03545191) and Study 2 (NCT03575104).
A total of 1854 patients with Diagnostic and Statistical Manual of Mental Disorders, 5th edition (DSM-5®) insomnia were randomized to receive QUVIVIQ or placebo once daily, in the evening, for 3 months. Study 1 randomized 930 subjects to QUVIVIQ 50 mg (N = 310), 25 mg (N = 310) or placebo (N = 310). Study 2 randomized 924 subjects to QUVIVIQ 25 mg (N = 309), 10 mg (N = 307), or placebo (N = 308). The 10 mg dose is not an approved dose.
At the end of the 3-month treatment period, both studies included a 7-day placebo run-out period, after which patients could enter a 9-month, double-blind, placebo-controlled extension study (Study 3, NCT03679884).
In Study 1, patients had a mean age of 55.4 years (range 18 to 88 years), with 39.1% of subjects ≥ 65 years of age, including 5.8% ≥ 75 years of age. Patients were identified as female or male and by US census-based racial and ethnic categories. The percentages of patients in the respective categories were: female sex (67.1%), White (90%), Black or African American (8%), Asian (1.0%), or Other race (< 1%).
In Study 2, patients had a mean age of 56.7 years (range 19 to 85 years), with 39.3% of subjects ≥ 65 years of age, including 6.1% ≥ 75 years of age. Patients were identified as female or male and by US census-based racial and ethnic categories. The percentages of patients in the respective categories were: female sex (69.0%), White (88%), Black or African American (8%), Asian (4%), or Other race (< 1%).
Primary efficacy endpoints for both studies were the change from baseline to Month 1 and Month 3 in Latency to Persistent Sleep (LPS) and Wake After Sleep Onset (WASO), measured objectively by polysomnography in a sleep laboratory. LPS is a measure of sleep induction and WASO is a measure of sleep maintenance.
Secondary endpoint included in the statistical testing hierarchy with Type 1 error control was patient-reported Total Sleep Time (sTST), evaluated every morning at home using a validated Sleep Diary Questionnaire (SDQ).
In Study 1, doses of 25 and 50 mg QUVIVIQ showed a statistically significant improvement vs placebo on polysomnography (LPS, WASO) and self-reported total sleep (sTST), at Month 1 and Month 3 (Table 4).
In Study 2, QUVIVIQ 25 mg showed a statistically significant improvement vs placebo on WASO and sTST at Month 1 and Month 3 (Table 5). QUVIVIQ 10 mg did not show a statistically significant improvement on LPS, WASO, or sTST at Month 1 or Month 3.
The efficacy of QUVIVIQ was similar across subgroups based on age, sex, race, and region.
Table 4 Primary and Secondary Efficacy Results for Change from Baseline in Sleep Onset, Sleep Maintenance, and Subjective Total Sleep Time at Month 1 and Month 3 in Patients with Insomnia (Study 1) Treatment group/dose
(N)Baseline Month 1 Month 3 Change from baseline Difference to placebo Change from baseline Difference to placebo mean (SD) mean (SD) LSM
(95%CL)LSM
(95%CL)mean (SD) LSM
(95%CL)LSM
(95%CL)CL = confidence limit; LPS = latency to persistent sleep; LSM = least squares mean; PSG = polysomnography; SD = standard deviation; sTST = subjective total sleep time; WASO = wake after sleep onset. - * doses that were statistically significantly superior (p < 0.05) to placebo after controlling for multiple comparisons.
WASO (wake after sleep onset, min): sleep maintenance, assessed by PSG 50 mg
(310)95 (38) 65 (35) -29
[-33, -25]-23*
[-28, -18]65 (39) -29
[-33, -25]-18*
[-24, -13]25 mg
(310)98 (39) 77 (42) -18
[-22, -15]-12*
[-17, -7]73 (40) -23
[-27, -19]-12*
[-17, -6]placebo
(310)103 (41) 92 (42) -6
[-10, -2]87 (43) -11
[-15, -7]LPS (latency to persistent sleep, min): sleep onset, assessed by PSG 50 mg
(310)64 (37) 34 (27) -31
[-35, -28]-11*
[-16, -7]30 (23) -35
[-38, -31]-12*
[-16, -7]25 mg
(310)67 (39) 38 (32) -28
[-32, -25]-8*
[-13, -4]36 (34) -31
[-34, -27]-8*
[-12, -3]placebo
(310)67 (40) 46 (36) -20
[-23, -17]43 (34) -23
[-26, -20]sTST (subjective total sleep time, min): patient-reported 50 mg
(310)313 (58) 358 (74) 44
[38, 49]22*
[14, 30]372 (79) 58
[51, 64]20*
[11, 29]25 mg
(310)310 (60) 345 (66) 34
[29, 40]13*
[5, 20]358 (72) 48
[41, 54]10*
[1, 19]placebo
(310)316 (53) 338 (65) 22
[16, 27]354 (73) 38
[31, 44]Table 5 Primary and Secondary Efficacy Results for Change from Baseline in Sleep Onset, Sleep Maintenance, and Subjective Total Sleep Time at Month 1 and Month 3 in Patients with Insomnia (Study 2) Treatment group/dose
(N)Baseline Month 1 Month 3 Change from baseline Difference to placebo Change from baseline Difference to placebo mean
(SD)mean
(SD)LSM
(95%CL)LSM
(95%CL)mean
(SD)LSM
(95%CL)LSM
(95%CL)CL = confidence limit; LPS = latency to persistent sleep; LSM = least squares mean; PSG = polysomnography; SD = standard deviation; sTST = subjective total sleep time; WASO = wake after sleep onset. - * doses that were statistically significantly superior (p < 0.05) to placebo after controlling for multiple comparisons.
WASO (wake after sleep onset, min): sleep maintenance, assessed by PSG 25 mg
(309)106
(49)80
(44)-24
[-28, -20]-12*
[-18, -6]80
(49)-24
[-29, -19]-10*
[-17, -4]placebo
(308)108
(49)93
(50)-13
[-17, -8]91
(47)-14
[-19, -9]LPS (latency to persistent sleep, min): sleep onset, assessed by PSG 25 mg
(309)69
(41)42
(39)-26
[-31, -22]-6
[-12, -1]39
(37)-29
[-33, -24]-9
[-15, -3]placebo
(308)72
(46)50
(40)-20
[-24, -16]49
(46)-20
[-24, -15]sTST (subjective total sleep time, min): patient-reported 25 mg
(309)308
(53)353
(67)44
[38, 49]16*
[8, 24]365
(70)56
[50, 63]19*
[10, 28]placebo
(308)308
(52)336
(63)28
[22, 33]347
(65)37
[31, 43]The effects of QUVIVIQ on LPS, WASO, and sTST were observed at Month 1 and were maintained through Month 3. The change from baseline of sTST by week in Study 1 is presented in Figure 4.
Figure 4 Change from Baseline of sTST by Week (Study 1)
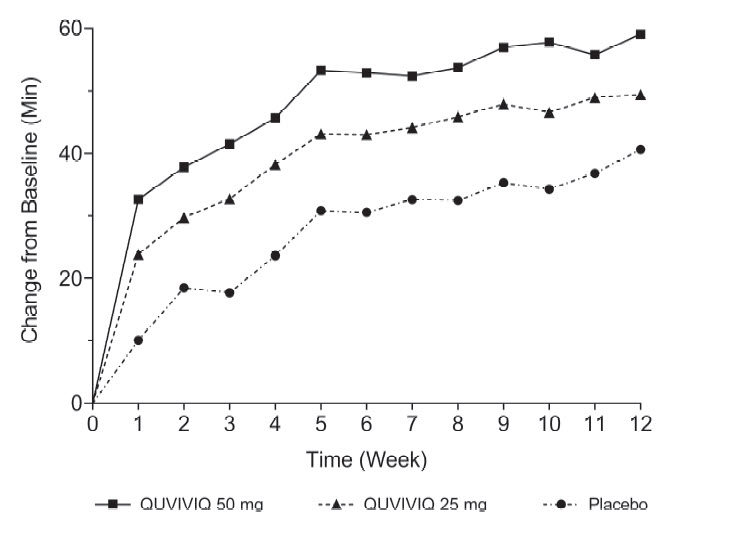
14.2 Special Safety Studies
Effects on Driving
A randomized, double-blind, placebo- and active-controlled, four-period crossover study evaluated the effects of nighttime administration of QUVIVIQ on next-morning driving performance, using a driving simulator, 9 hours after dosing in 30 healthy elderly subjects (65–79 years, median age 70 years; 15 men, 15 women) and 30 healthy adult subjects (50–64 years, median age 58 years; 15 men, 15 women). The primary driving performance outcome measure was change in Standard Deviation of Lateral Position (SDLP). Testing was conducted after one night (initial dosing) and after four consecutive nights of treatment with QUVIVIQ 50 mg and 100 mg (two times the maximum recommended daily dose). Zopiclone 7.5 mg was used as an active comparator. For both doses, QUVIVIQ caused a statistically significant impairment in next-morning driving performance in adult or elderly subjects (compared with placebo) after the first dose. Although the mean effect on driving performance was not statistically significant (compared to placebo) after 4 consecutive nights of treatment with either dose of QUVIVIQ, driving ability was impaired in some subjects taking QUVIVIQ.
Patients should be cautioned about the potential for next-morning driving impairment because there is individual variation in sensitivity to QUVIVIQ.
Withdrawal of Therapy
Withdrawal Symptoms
In controlled efficacy and safety studies, withdrawal effects were assessed by the Tyrer Benzodiazepine Withdrawal Symptom Questionnaire following discontinuation of QUVIVIQ, and by adverse event reporting during a single-blind placebo run-out period. No evidence of withdrawal symptoms was observed upon treatment discontinuation.
Loss of Treatment Effect After Discontinuation
The loss of effect from stopping treatment with QUVIVIQ was assessed during the placebo run-out period after 3 months of treatment with QUVIVIQ in Study 1 and Study 2.
After treatment discontinuation, in Study 1, patients previously treated with QUVIVIQ 50 mg experienced mean increases of 25 minutes in WASO, 16 minutes in LPS during the next night of sleep, and a mean decrease in sTST of 14 minutes per night over the next week, as compared to the last assessment on treatment. Following QUVIVIQ 25 mg discontinuation, a similar pattern was observed with mean increases in WASO of 19 minutes, 15 minutes in LPS, and a mean decrease in sTST of 7 minutes. Similar changes were observed with the 25 mg dose in Study 2. In both studies, patients who were on placebo and continued on placebo in the run-out period experienced minimal changes in WASO, LPS, or sTST.
Middle of the Night Safety
The effect of QUVIVIQ on middle of the night safety was evaluated in a randomized, placebo-controlled three-period crossover study in 18 healthy adult (< 65 years) and 18 healthy elderly (≥ 65 years) subjects. The ability to awaken in response to a sound stimulus, postural stability, and memory were assessed following a scheduled awakening 4 hours after administration of 25 or 50 mg QUVIVIQ, or placebo.
The ability to awaken to sound in the middle of the night was assessed using an audiometer that delivered 1000 Hz tones starting at 35 dB and increasing to 100 dB to determine the Auditory Awakening Threshold (AAT). There was no significant difference between QUVIVIQ (25 mg or 50 mg) and placebo in AAT.
Postural stability was measured by assessing body sway using a body sway meter approximately 5 minutes after awakening. Evening dosing of QUVIVIQ 25 mg and 50 mg resulted in respective increases in mean (95% confidence limit) cumulative body sway over 2 minutes of 37 mm [2 mm, 71 mm] and 66 mm [31 mm, 100 mm] at 4 hours post-dose compared to placebo.
A visual verbal learning test (VVLT) of immediate and delayed recall was administered to assess memory after middle of the night awakening (4 hours post-dose) in subjects receiving QUVIVIQ 25 mg or 50 mg. QUVIVIQ was associated with worse performance on the VVLT compared to placebo.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
QUVIVIQ tablets are available as:
- 25 mg, light purple, arc-triangle shaped film-coated tablets debossed with "25" on one side, and "i" on the other side.
NDC: 80491-7825-3, bottle of 30 with child-resistant closure - 50 mg: light orange, arc-triangle shaped film-coated tablets debossed with "50" on one side, and "i" on the other side.
NDC: 80491-7850-3, bottle of 30 with child-resistant closure
- 25 mg, light purple, arc-triangle shaped film-coated tablets debossed with "25" on one side, and "i" on the other side.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Administration Instructions
Advise patients to take QUVIVIQ only in the evening within 30 minutes before going to bed and only if they can stay in bed for a full night (at least 7 hours) before being active again [see Dosage and Administration (2.1)].
Advise patients that the effect of QUVIVIQ may be delayed if taken with or soon after a meal [see Dosage and Administration (2.1), Clinical Pharmacology (12.3)].
CNS-Depressant Effects and Daytime Impairment
Advise patients that QUVIVIQ can impair daytime wakefulness even when used as prescribed. The risk of daytime impairment is increased if QUVIVIQ is taken with less than a full night of sleep remaining or if a higher than recommended dose is taken. If QUVIVIQ is taken in these circumstances, caution patients against driving and other activities requiring complete mental alertness. Advise patients that increased drowsiness may increase the risk of falls in some patients [see Warnings and Precautions (5.1), Clinical Studies (14.2)].
Worsening of Depression/Suicidal Ideation
Tell patients to report any worsening of depression or suicidal thoughts immediately [see Warnings and Precautions (5.2)].
Sleep Paralysis, Hypnagogic/Hypnopompic Hallucinations, and Cataplexy-Like Symptoms
Advise patients and their families that QUVIVIQ may cause sleep paralysis, which is an inability to move or speak for several minutes during sleep-wake transitions and hypnagogic/hypnopompic hallucinations, including vivid and disturbing perceptions. Symptoms similar to mild cataplexy have occurred with orexin receptor antagonists [see Warnings and Precautions (5.3)].
Complex Sleep Behaviors
Instruct patients and their families that hypnotics may cause complex sleep behaviors, including sleepwalking, sleep driving, preparing and eating food, making phone calls, or having sex while not being fully awake. Tell patients to discontinue QUVIVIQ and notify their healthcare provider immediately if they develop any of these symptoms [see Warnings and Precautions (5.4)].
Concomitant Medications
Ask patients about alcohol consumption, medicines they are taking, and drugs they may be taking without a prescription. Advise patients to avoid consuming alcohol with QUVIVIQ [see Warnings and Precautions (5.1), Drug Interactions (7.1), Clinical Pharmacology (12.2)].
Pregnancy
Advise patients that there is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to QUVIVIQ during pregnancy [see Use in Specific Populations (8.1)].
Tolerance, Abuse, and Dependence
Tell patients not to increase the dose of QUVIVIQ on their own, and to inform you if they believe the drug "does not work" [see Drug Abuse and Dependence (9)].
- SPL UNCLASSIFIED SECTION
-
MEDICATION GUIDE
MEDICATION GUIDE
QUVIVIQ® (cue-VIH-vick)
(daridorexant)
tablets, for oral use, CIVThis Medication Guide has been approved by the U.S. Food and Drug Administration.
IDRSQUMG09302024Revised: 9/2024 What is the most important information I should know about QUVIVIQ?
QUVIVIQ may cause serious side effects, including:-
Decreased awareness and alertness. The morning after you take QUVIVIQ, your ability to drive safely and think clearly may be decreased. You may also have sleepiness during the day. Sleepiness may increase your risk for falls.
- Do not take more QUVIVIQ than prescribed.
- Do not take QUVIVIQ unless you are able to stay in bed for a full night (at least 7 hours) before you must be active again.
- Take QUVIVIQ in the evening within 30 minutes before going to bed.
What is QUVIVIQ? - QUVIVIQ is a prescription medicine for adults who have trouble falling asleep or staying asleep (insomnia).
- It is not known if QUVIVIQ is safe and effective for use in children.
Who should not take QUVIVIQ?
Do not take QUVIVIQ:- If you fall asleep often at unexpected times (narcolepsy).
- If you are allergic to daridorexant, or any ingredients in QUVIVIQ. See the end of this Medication Guide for a complete list of ingredients in QUVIVIQ.
Before taking QUVIVIQ, tell your healthcare provider about all of your medical conditions, including if you: - have a history of depression, mental illness, or suicidal thoughts or actions
- have a history of drug or alcohol abuse or addiction
- have a history of a sudden onset of muscle weakness (cataplexy)
- have a history of daytime sleepiness
- have lung or breathing problems, including sleep apnea
- have liver problems
- are pregnant or plan to become pregnant. It is not known if QUVIVIQ can harm your unborn baby.
- Pregnancy Registry: There is a pregnancy registry for women who are exposed to QUVIVIQ during pregnancy. The purpose of this registry is to collect information about the health of you and your baby. For more information or to participate in the registry, call 1-833-400-9611. Talk with your healthcare provider about the risk to your unborn baby if you take QUVIVIQ during pregnancy.
- are breastfeeding or plan to breastfeed. QUVIVIQ passes into your breast milk. Talk to your healthcare provider about the best way to feed your baby during treatment with QUVIVIQ.
Taking QUVIVIQ with certain medicines can cause serious side effects. QUVIVIQ may affect the way other medicines work and other medicines may affect the way QUVIVIQ works.
Do not take QUVIVIQ with other medicines that can make you sleepy unless your healthcare provider tells you to.
Know the medicines you take. Keep a list of them to show your healthcare provider and pharmacist when you get a new medicine.How should I take QUVIVIQ? - Take QUVIVIQ exactly as your healthcare provider tells you to take it.
- Do not change your dose of QUVIVIQ without first talking to your healthcare provider.
- Take QUVIVIQ one time each night, within 30 minutes before going to bed.
- Only take QUVIVIQ when you can stay in bed for a full night (at least 7 hours).
- QUVIVIQ may take longer to work if you take it with or right after a meal.
- Call your healthcare provider if your insomnia (sleep problem) worsens or is not improved within 7 to 10 days. This may mean that there is another condition causing your sleep problem.
- If you take too much QUVIVIQ, call your healthcare provider, or contact your Poison Help Line at 1-800-222-1222, or go to the nearest hospital emergency room right away.
What should I avoid while taking QUVIVIQ? - Do not drink alcohol while taking QUVIVIQ. It can increase the effects of alcohol, which can be dangerous.
- You may still feel drowsy the next day after taking QUVIVIQ.
- Do not drive, operate heavy machinery, do anything dangerous, or do other activities that require clear thinking if you have taken QUVIVIQ as prescribed but do not feel fully awake, you have taken QUVIVIQ and have had less than a full night of sleep (at least 7 hours), or if you have taken more QUVIVIQ than prescribed by your healthcare provider.
What are the possible side effects of QUVIVIQ?
QUVIVIQ may cause serious side effects, including:- See "What is the most important information I should know about QUVIVIQ?"
- Worsening depression and suicidal thoughts. Call your healthcare provider right away if you have any worsening depression or thoughts of suicide or dying.
- Temporary inability to move or talk (sleep paralysis) for up to several minutes, or hallucinations while you are going to sleep or waking up.
- Temporary weakness in your legs that can happen during the day or at night.
- Complex sleep behaviors such as sleepwalking, sleep driving, preparing and eating food, making phone calls, having sex, or doing other activities while not fully awake that you may not remember the next morning. Stop taking QUVIVIQ and call your healthcare provider right away if you experience a complex sleep behavior.
These are not all of the possible side effects of QUVIVIQ.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.How should I store QUVIVIQ? - Store QUVIVIQ at room temperature between 68°F to 77°F (20°C to 25°C).
- Keep QUVIVIQ and all medicines out of the reach of children.
General information about the safe and effective use of QUVIVIQ.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use QUVIVIQ for a condition for which it was not prescribed. Do not give QUVIVIQ to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about QUVIVIQ that is written for health professionals.What are the ingredients in QUVIVIQ?
Active ingredient: daridorexant hydrochloride
Inactive ingredients: croscarmellose sodium, magnesium stearate, mannitol, microcrystalline cellulose, povidone, and silicon dioxide. The tablet film coating contains: glycerin, hypromellose, iron oxide black, iron oxide red, microcrystalline cellulose, talc, titanium dioxide, and, in the 50 mg tablet only, iron oxide yellow.Distributed by:
Idorsia Pharmaceuticals US Inc.
One Radnor Corporate Center, Suite 101
100 Matsonford Rd
Radnor, PA 19087
For more information go to QUVIVIQ.com or call 1-833-400-9611. -
Decreased awareness and alertness. The morning after you take QUVIVIQ, your ability to drive safely and think clearly may be decreased. You may also have sleepiness during the day. Sleepiness may increase your risk for falls.
-
PRINCIPAL DISPLAY PANEL - 25 mg Tablet Bottle Carton
NDC: 80491-7825-3
QUVIVIQ®
(daridorexant) tablets25 mg
CIVRx only
For oral use
30 tablets
Dispense the accompanying
Medication Guide to each patient.idorsia

-
PRINCIPAL DISPLAY PANEL - 50 mg Tablet Bottle Carton
NDC: 80491-7850-3
QUVIVIQ®
(daridorexant) tablets50 mg
CIVRx only
For oral use
30 tablets
Dispense the accompanying
Medication Guide to each patient.idorsia

-
INGREDIENTS AND APPEARANCE
QUVIVIQ
daridorexant tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 80491-7825 Route of Administration ORAL DEA Schedule CIV Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength daridorexant (UNII: LMQ24G57E9) (daridorexant - UNII:LMQ24G57E9) daridorexant 25 mg Inactive Ingredients Ingredient Name Strength Mannitol (UNII: 3OWL53L36A) MICROCRYSTALLINE CELLULOSE 101 (UNII: 7T9FYH5QMK) Povidone K30 (UNII: U725QWY32X) WATER (UNII: 059QF0KO0R) Silicon dioxide (UNII: ETJ7Z6XBU4) Magnesium stearate (UNII: 70097M6I30) MICROCRYSTALLINE CELLULOSE 102 (UNII: PNR0YF693Y) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) GLYCERIN (UNII: PDC6A3C0OX) Talc (UNII: 7SEV7J4R1U) Titanium dioxide (UNII: 15FIX9V2JP) FERRIC OXIDE RED (UNII: 1K09F3G675) FERROSOFERRIC OXIDE (UNII: XM0M87F357) Product Characteristics Color PURPLE (Light purple) Score no score Shape TRIANGLE (arc-triangle) Size 7mm Flavor Imprint Code 25;i Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 80491-7825-3 1 in 1 CARTON 04/07/2022 1 30 in 1 BOTTLE; Type 0: Not a Combination Product 2 NDC: 80491-7825-1 1 in 1 CARTON 01/01/2122 2 7 in 1 BLISTER PACK; Type 0: Not a Combination Product 3 NDC: 80491-7825-2 1 in 1 CARTON 05/01/2023 3 7 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA214985 04/07/2022 QUVIVIQ
daridorexant tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 80491-7850 Route of Administration ORAL DEA Schedule CIV Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength daridorexant (UNII: LMQ24G57E9) (daridorexant - UNII:LMQ24G57E9) daridorexant 50 mg Inactive Ingredients Ingredient Name Strength Mannitol (UNII: 3OWL53L36A) MICROCRYSTALLINE CELLULOSE 101 (UNII: 7T9FYH5QMK) Povidone K30 (UNII: U725QWY32X) WATER (UNII: 059QF0KO0R) Silicon dioxide (UNII: ETJ7Z6XBU4) Magnesium stearate (UNII: 70097M6I30) MICROCRYSTALLINE CELLULOSE 102 (UNII: PNR0YF693Y) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) GLYCERIN (UNII: PDC6A3C0OX) Talc (UNII: 7SEV7J4R1U) Titanium dioxide (UNII: 15FIX9V2JP) FERRIC OXIDE RED (UNII: 1K09F3G675) FERROSOFERRIC OXIDE (UNII: XM0M87F357) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) Product Characteristics Color ORANGE (Light orange) Score no score Shape TRIANGLE (arc-triangle) Size 7mm Flavor Imprint Code 50;i Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 80491-7850-3 1 in 1 CARTON 04/07/2022 1 30 in 1 BOTTLE; Type 0: Not a Combination Product 2 NDC: 80491-7850-1 1 in 1 CARTON 01/01/2122 2 7 in 1 BLISTER PACK; Type 0: Not a Combination Product 3 NDC: 80491-7850-2 1 in 1 CARTON 05/01/2023 3 7 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA214985 04/07/2022 Labeler - Idorsia Pharmaceuticals Ltd (480176487)
Trademark Results [QUVIVIQ]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 QUVIVIQ 79350361 not registered Live/Pending |
Idorsia Pharmaceuticals Ltd 2022-07-06 |
 QUVIVIQ 79299719 not registered Live/Pending |
Idorsia Pharmaceuticals Ltd 2020-10-08 |
 QUVIVIQ 79274313 not registered Live/Pending |
Idorsia Pharmaceuticals Ltd. 2019-11-05 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.