ADCIRCA- tadalafil tablet
ADCIRCA by
Drug Labeling and Warnings
ADCIRCA by is a Prescription medication manufactured, distributed, or labeled by Aphena Pharma Solutions - Tennessee, LLC. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use ADCIRCA safely and effectively. See full prescribing information for ADCIRCA.
ADCIRCA (tadalafil) tablets for oral administration
Initial U.S. Approval: 2003RECENT MAJOR CHANGES
Warnings and Precautions (5.5) 05/2017
INDICATIONS AND USAGE
ADCIRCA is a phosphodiesterase 5 (PDE5) inhibitor indicated for the treatment of pulmonary arterial hypertension (PAH) (WHO Group 1) to improve exercise ability. Studies establishing effectiveness included predominately patients with NYHA Functional Class II – III symptoms and etiologies of idiopathic or heritable PAH (61%) or PAH associated with connective tissue diseases (23%). (1.1)
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
Tablets (not scored): 20 mg (3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- Cardiovascular effects: Carefully consider whether patients with certain underlying conditions (e.g., cardiovascular disease, impaired autonomic control of blood pressure, aortic stenosis) could be adversely affected by vasodilatory effects of ADCIRCA. Not recommended in patients with pulmonary veno-occlusive disease. (5.1)
- Concomitant alpha-blockers or alcohol: Note additive blood pressure-lowering effects. (5.1)
- Use with Ritonavir: Requires dosage adjustment. (2.3, 5.2)
- Other concomitant potent CYP3A inhibitors: Avoid use with ADCIRCA. (5.2)
- Potent Inducers of CYP3A: Avoid use of ADCIRCA in patients chronically taking potent inducers of CYP3A (e.g., rifampin). (5.2, 7.2)
- Effects on the eye: Patients should seek immediate medical attention if sudden loss of vision occurs, which could be a sign of non-arteritic ischemic optic neuropathy (NAION). (5.5)
- Hearing impairment: Advise patients to seek immediate medical attention if sudden decrease or loss of hearing occurs. (5.6)
- Concomitant PDE5 inhibitors: Avoid use with CIALIS or other PDE5 inhibitors. (5.7)
- Prolonged erection: Advise patients to seek emergency treatment if an erection lasts >4 hours. (5.8)
ADVERSE REACTIONS
The most common adverse reaction is headache. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Eli Lilly and Company at 1-800-545 5979 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 8/2017
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Pulmonary Arterial Hypertension
2 DOSAGE AND ADMINISTRATION
2.1 Pulmonary Arterial Hypertension
2.2 Use in Special Populations
2.3 Use with Ritonavir
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
4.1 Concomitant Organic Nitrates
4.2 Concomitant Guanylate Cyclase (GC) Stimulators
4.3 Hypersensitivity Reactions
5 WARNINGS AND PRECAUTIONS
5.1 Cardiovascular Effects
5.2 Use with Potent CYP3A Inhibitors or Inducers
5.3 Use in Renal Impairment
5.4 Use in Hepatic Impairment
5.5 Visual Loss
5.6 Hearing Impairment
5.7 Combination with Other PDE5 Inhibitors
5.8 Prolonged Erection
5.9 Effects on Bleeding
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Potential for Pharmacodynamic Interactions with ADCIRCA
7.2 Potential for Other Drugs to Affect ADCIRCA
7.3 Potential for ADCIRCA to Affect Other Drugs
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.3 Nursing Mothers
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
13.3 Reproductive Toxicology Studies
14 CLINICAL STUDIES
14.1 ADCIRCA for Pulmonary Arterial Hypertension
14.2 Long-Term Treatment of Pulmonary Arterial Hypertension
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
1.1 Pulmonary Arterial Hypertension
ADCIRCA® is indicated for the treatment of pulmonary arterial hypertension (PAH) (WHO Group 1) to improve exercise ability. Studies establishing effectiveness included predominately patients with NYHA Functional Class II – III symptoms and etiologies of idiopathic or heritable PAH (61%) or PAH associated with connective tissue diseases (23%).
-
2 DOSAGE AND ADMINISTRATION
2.1 Pulmonary Arterial Hypertension
The recommended dose of ADCIRCA is 40 mg (two 20 mg tablets) taken once daily with or without food. Dividing the dose (40 mg) over the course of the day is not recommended.
2.2 Use in Special Populations
Renal Impairment
- Mild (creatinine clearance 51 to 80 mL/min) or moderate (creatinine clearance 31 to 50 mL/min): Start dosing at 20 mg once daily. Increase to 40 mg once daily based on individual tolerability.
- Severe (creatinine clearance <30 mL/min and on hemodialysis): Avoid use of ADCIRCA because of increased tadalafil exposure (AUC), limited clinical experience, and the lack of ability to influence clearance by dialysis [see Warnings and Precautions (5.3) and Use in Specific Populations (8.6)].
Hepatic Impairment
- Mild or moderate (Child Pugh Class A or B): Because of limited clinical experience in patients with mild to moderate hepatic cirrhosis, consider a starting dose of 20 mg once per day.
- Severe (Child Pugh Class C): Patients with severe hepatic cirrhosis have not been studied. Avoid use of ADCIRCA [see Warnings and Precautions (5.4) and Use in Specific Populations (8.7)].
2.3 Use with Ritonavir
Co-administration of ADCIRCA in Patients on Ritonavir
In patients receiving ritonavir for at least one week, start ADCIRCA at 20 mg once daily. Increase to 40 mg once daily based upon individual tolerability [see Warnings and Precautions (5.2), Drug Interactions (7.2) and Clinical Pharmacology (12.3)].
Co-administration of Ritonavir in Patients on ADCIRCA
Avoid use of ADCIRCA during the initiation of ritonavir. Stop ADCIRCA at least 24 hours prior to starting ritonavir. After at least one week following the initiation of ritonavir, resume ADCIRCA at 20 mg once daily. Increase to 40 mg once daily based upon individual tolerability [see Warnings and Precautions (5.2), Drug Interactions (7.2) and Clinical Pharmacology (12.3)].
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
4.1 Concomitant Organic Nitrates
Do not use ADCIRCA in patients who are using any form of organic nitrate, either regularly or intermittently. ADCIRCA potentiates the hypotensive effect of nitrates. This potentiation is thought to result from the combined effects of nitrates and ADCIRCA on the nitric oxide/cGMP pathway [see Clinical Pharmacology (12.2)].
4.2 Concomitant Guanylate Cyclase (GC) Stimulators
Do not use ADCIRCA in patients who are using a GC stimulator, such as riociguat. ADCIRCA may potentiate the hypotensive effects of GC stimulators.
4.3 Hypersensitivity Reactions
ADCIRCA is contraindicated in patients with a known serious hypersensitivity to tadalafil (ADCIRCA or CIALIS). Hypersensitivity reactions have been reported, including Stevens-Johnson syndrome and exfoliative dermatitis [see Adverse Reactions (6.2)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Cardiovascular Effects
Discuss with patients the appropriate action to take in the event that they experience anginal chest pain requiring nitroglycerin following intake of ADCIRCA. At least 48 hours should elapse after the last dose of ADCIRCA before taking nitrates. If a patient has taken ADCIRCA within 48 hours, administer nitrates under close medical supervision with appropriate hemodynamic monitoring. Patients who experience anginal chest pain after taking ADCIRCA should seek immediate medical attention.
PDE5 inhibitors, including tadalafil, have mild systemic vasodilatory properties that may result in transient decreases in blood pressure. Prior to prescribing ADCIRCA, carefully consider whether patients with underlying cardiovascular disease could be affected adversely by such vasodilatory effects. Patients with severely impaired autonomic control of blood pressure or with left ventricular outflow obstruction, (e.g., aortic stenosis and idiopathic hypertrophic subaortic stenosis) may be particularly sensitive to the actions of vasodilators, including PDE5 inhibitors.
Pulmonary vasodilators may significantly worsen the cardiovascular status of patients with pulmonary veno-occlusive disease (PVOD). Since there are no clinical data on administration of ADCIRCA to patients with veno-occlusive disease, administration of ADCIRCA to such patients is not recommended. Should signs of pulmonary edema occur when ADCIRCA is administered, the possibility of associated PVOD should be considered.
There is a lack of data on safety and efficacy in the following groups who were specifically excluded from the PAH clinical trials:
- Patients with clinically significant aortic and mitral valve disease
- Patients with pericardial constriction
- Patients with restrictive or congestive cardiomyopathy
- Patients with significant left ventricular dysfunction
- Patients with life-threatening arrhythmias
- Patients with symptomatic coronary artery disease
- Patients with hypotension (<90/50 mm Hg) or uncontrolled hypertension
Use with Alpha Blockers and Antihypertensives
PDE5 inhibitors, including ADCIRCA, and alpha–adrenergic blocking agents are vasodilators with blood pressure–lowering effects. When vasodilators are used in combination, an additive effect on blood pressure may be anticipated. In some patients, concomitant use of these two drug classes can lower blood pressure significantly [see Drug Interactions (7.1) and Clinical Pharmacology (12.2)], which may lead to symptomatic hypotension (e.g., fainting). Safety of combined use of PDE5 inhibitors and alpha blockers may be affected by other variables, including intravascular volume depletion and use of other antihypertensive drugs [see Drug Interactions (7.1)].
5.2 Use with Potent CYP3A Inhibitors or Inducers
Co-administration of ADCIRCA in Patients on Ritonavir
In patients receiving ritonavir for at least one week, start ADCIRCA at 20 mg once daily. Increase to 40 mg once daily based upon individual tolerability [see Dosage and Administration (2.3), Drug Interactions (7.2) and Clinical Pharmacology (12.3)].
Co-administration of Ritonavir in Patients on ADCIRCA
Avoid use of ADCIRCA during the initiation of ritonavir. Stop ADCIRCA at least 24 hours prior to starting ritonavir. After at least one week following the initiation of ritonavir, resume ADCIRCA at 20 mg once daily. Increase to 40 mg once daily based upon individual tolerability [see Dosage and Administration (2.3), Drug Interactions (7.2) and Clinical Pharmacology (12.3)].
5.3 Use in Renal Impairment
5.4 Use in Hepatic Impairment
5.5 Visual Loss
Physicians should advise patients to seek immediate medical attention in the event of a sudden loss of vision in one or both eyes. Such an event may be a sign of non–arteritic anterior ischemic optic neuropathy (NAION), a cause of decreased vision, including permanent loss of vision, that has been reported postmarketing in temporal association with the use of all PDE5 inhibitors. Most, but not all, of these patients had underlying anatomic or vascular risk factors for development of NAION, including but not necessarily limited to: low cup to disc ratio (“crowded disc”), age over 50, diabetes, hypertension, coronary artery disease, hyperlipidemia, and smoking. Based on published literature, the annual incidence of NAION is 2.5-11.8 cases per 100,000 in males aged ≥50 in the general population. An observational case-crossover study evaluated the risk of NAION when PDE5 inhibitor use, as a class, typical of erectile dysfunction treatment, occurred immediately before NAION onset (within 5 half-lives), compared to PDE5 inhibitor use in a prior time period. The results suggest an approximate 2-fold increase in the risk of NAION, with a risk estimate of 2.15 (95% CI 1.06, 4.34). A similar study reported a consistent result, with a risk estimate of 2.27 (95% CI 0.99, 5.20). Other risk factors for NAION, such as the presence of “crowded” optic disc, may have contributed to the occurrence of NAION in these studies.
Neither the rare postmarketing reports, nor the association of PDE5 inhibitor use and NAION in the observational studies, substantiate a causal relationship between PDE5 inhibitor use and NAION [see Adverse Reactions (6.2)].
Physicians should also discuss with patients the increased risk of NAION in individuals who have already experienced NAION in one eye, including whether such individuals could be adversely affected by use of vasodilators such as PDE5 inhibitors.
Patients with known hereditary degenerative retinal disorders, including retinitis pigmentosa, were not included in the clinical trials, and use in these patients is not recommended.
5.6 Hearing Impairment
Physicians should advise patients to seek immediate medical attention in the event of sudden decrease or loss of hearing. These events, which may be accompanied by tinnitus and dizziness, have been reported in temporal association to the intake of PDE5 inhibitors, including ADCIRCA. It is not possible to determine whether these events are related directly to the use of PDE5 inhibitors or to other factors [see Adverse Reactions (6.2)].
5.7 Combination with Other PDE5 Inhibitors
Tadalafil is also marketed as CIALIS. The safety and efficacy of taking ADCIRCA together with CIALIS or other PDE5 inhibitors have not been studied. Inform patients taking ADCIRCA not to take CIALIS or other PDE5 inhibitors.
5.8 Prolonged Erection
There have been rare reports of prolonged erections greater than 4 hours and priapism (painful erections greater than 6 hours in duration) for this class of compounds. Priapism, if not treated promptly, can result in irreversible damage to the erectile tissue. Patients who have an erection lasting greater than 4 hours, whether painful or not, should seek emergency medical attention.
ADCIRCA should be used with caution in patients who have conditions that might predispose them to priapism (such as sickle cell anemia, multiple myeloma, or leukemia), or in patients with anatomical deformation of the penis (such as angulation, cavernosal fibrosis, or Peyronie's disease).
5.9 Effects on Bleeding
PDE5 is found in platelets. When administered in combination with aspirin, tadalafil 20 mg did not prolong bleeding time, relative to aspirin alone. ADCIRCA has not been administered to patients with bleeding disorders or significant active peptic ulceration. Although ADCIRCA has not been shown to increase bleeding times in healthy subjects, use in patients with bleeding disorders or significant active peptic ulceration should be based upon a careful risk-benefit assessment.
-
6 ADVERSE REACTIONS
The following serious adverse reactions are discussed elsewhere in the labeling:
- Hypotension [see Warnings and Precautions (5.1)]
- Visual Loss [see Warnings and Precautions (5.5) and Patient Counseling Information (17)]
- Hearing loss [see Warnings and Precautions (5.6)]
- Priapism [see Warnings and Precautions (5.8)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Tadalafil was administered to 398 patients with PAH during clinical trials worldwide. In trials of ADCIRCA, a total of 311 and 251 subjects have been treated for at least 182 days and 360 days, respectively. The overall rates of discontinuation because of an adverse event (AE) in the placebo-controlled trial were 9% for ADCIRCA 40 mg and 15% for placebo. The rates of discontinuation because of AEs, other than those related to worsening of PAH, in patients treated with ADCIRCA 40 mg was 4% compared to 5% in placebo-treated patients.
In the placebo-controlled study, the most common AEs were generally transient and mild to moderate in intensity. Table 1 presents treatment-emergent adverse events reported by ≥9% of patients in the ADCIRCA 40 mg group and occurring more frequently than with placebo.
Table 1: Treatment-Emergent Adverse Events Reported by ≥9% of Patients in ADCIRCA and More Frequent than Placebo by 2% EVENT Placebo (%)
(N=82)ADCIRCA 20 mg (%)
(N=82)ADCIRCA 40 mg (%)
(N=79)Headache 15 32 42 Myalgia 4 9 14 Nasopharyngitis 7 2 13 Flushing 2 6 13 Respiratory Tract Infection (Upper and Lower) 6 7 13 Pain in Extremity 2 5 11 Nausea 6 10 11 Back Pain 6 12 10 Dyspepsia 2 13 10 Nasal Congestion (Including sinus congestion) 1 0 9 6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of tadalafil. These events have been chosen for inclusion either because of their seriousness, reporting frequency, lack of clear alternative causation, or a combination of these factors. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to estimate reliably their frequency or establish a causal relationship to drug exposure. The list does not include adverse events that are reported from clinical trials and that are listed elsewhere in this section.
Cardiovascular and cerebrovascular — Serious cardiovascular events, including myocardial infarction, sudden cardiac death, stroke, chest pain, palpitations, and tachycardia, have been reported postmarketing in temporal association with the use of tadalafil. Most, but not all, of these patients had preexisting cardiovascular risk factors. Many of these events were reported to occur during or shortly after sexual activity, and a few were reported to occur shortly after the use of tadalafil without sexual activity. Others were reported to have occurred hours to days after the use of tadalafil and sexual activity. It is not possible to determine whether these events are related directly to tadalafil, to sexual activity, to the patient's underlying cardiovascular disease, to a combination of these factors, or to other factors [see Warnings and Precautions (5.1)].
Body as a whole — Hypersensitivity reactions including urticaria, Stevens–Johnson syndrome, and exfoliative dermatitis
Nervous — Migraine, seizure and seizure recurrence, and transient global amnesia
Ophthalmologic — Visual field defect, retinal vein occlusion, retinal artery occlusion, and NAION [see Warnings and Precautions (5.5) and Patient Counseling Information (17)].
Otologic — Cases of sudden decrease or loss of hearing have been reported postmarketing in temporal association with the use of PDE5 inhibitors, including tadalafil. In some of the cases, medical conditions and other factors were reported that may have also played a role in the otologic adverse events. In many cases, medical follow-up information was limited. It is not possible to determine whether these reported events are related directly to the use of tadalafil, to the patient's underlying risk factors for hearing loss, a combination of these factors, or to other factors [see Warnings and Precautions (5.6) and Patient Counseling Information (17)].
Urogenital — Priapism [see Warnings and Precautions (5.8)].
-
7 DRUG INTERACTIONS
7.1 Potential for Pharmacodynamic Interactions with ADCIRCA
Nitrates
Do not use ADCIRCA in patients who are using any form of organic nitrate [see Contraindications (4.1)]. In clinical pharmacology studies ADCIRCA potentiated the hypotensive effect of nitrates [see Clinical Pharmacology (12.2)]. In a patient who has taken ADCIRCA, where nitrate administration is deemed medically necessary in a life–threatening situation, at least 48 hours should elapse after the last dose of ADCIRCA before nitrate administration is considered. In such circumstances, nitrates should still only be administered under close medical supervision with appropriate hemodynamic monitoring.
Alpha-Blockers
PDE5 inhibitors, including ADCIRCA, and alpha–adrenergic blocking agents are both vasodilators with blood-pressure-lowering effects. When vasodilators are used in combination, an additive effect on blood pressure may be anticipated. Clinical pharmacology studies have been conducted with coadministration of tadalafil with doxazosin, alfuzosin or tamsulosin [see Warnings and Precautions (5.1) and Clinical Pharmacology (12.2)].
Antihypertensives
PDE5 inhibitors, including ADCIRCA, are mild systemic vasodilators. Clinical pharmacology studies were conducted to assess the effect of tadalafil on the potentiation of the blood–pressure–lowering effects of selected antihypertensive medications (amlodipine, angiotensin II receptor blockers, bendroflumethiazide, enalapril, and metoprolol). Small reductions in blood pressure occurred following coadministration of tadalafil with these agents compared with placebo [see Warnings and Precautions (5.1) and Clinical Pharmacology (12.2)].
Alcohol
Both alcohol and tadalafil, a PDE5 inhibitor, act as mild vasodilators. When mild vasodilators are taken in combination, blood pressure–lowering effects of each individual compound may be increased. Substantial consumption of alcohol (e.g., 5 units or greater) in combination with ADCIRCA can increase the potential for orthostatic signs and symptoms, including increase in heart rate, decrease in standing blood pressure, dizziness, and headache. Tadalafil (10 mg or 20 mg) did not affect alcohol plasma concentrations and alcohol did not affect tadalafil plasma concentrations. [See Warnings and Precautions (5.1) and Clinical Pharmacology (12.2)].
7.2 Potential for Other Drugs to Affect ADCIRCA
Ritonavir
Ritonavir initially inhibits and later induces CYP3A, the enzyme involved in the metabolism of tadalafil. At steady state of ritonavir (about 1 week), the exposure to tadalafil is similar as in the absence of ritonavir [see Dosage and Administration (2.3), Warnings and Precautions (5.2), and Clinical Pharmacology (12.3)].
7.3 Potential for ADCIRCA to Affect Other Drugs
Cytochrome P450 Substrates
Tadalafil is not expected to cause clinically significant inhibition or induction of the clearance of drugs metabolized by cytochrome P450 (CYP) isoforms (e.g., theophylline, warfarin, midazolam, lovastatin, bosentan) [see Clinical Pharmacology (12.3)].
Aspirin
Tadalafil (10 mg and 20 mg once daily) does not potentiate the increase in bleeding time caused by aspirin [see Clinical Pharmacology (12.3)].
P-glycoprotein (e.g., digoxin)
Coadministration of tadalafil (40 mg once daily) for 10 days did not significantly alter digoxin pharmacokinetics in healthy subjects [see Clinical Pharmacology (12.3)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Category B
Animal reproduction studies in rats and mice revealed no evidence of fetal harm. There are, however, no adequate and well-controlled studies of tadalafil in pregnant women. Because animal reproduction studies are not always predictive of human response, tadalafil should be used during pregnancy only if clearly needed.
Non–teratogenic effects
Animal reproduction studies showed no evidence of teratogenicity, embryotoxicity, or fetotoxicity when tadalafil was given to pregnant rats or mice at unbound tadalafil exposures up to 7 times the maximum recommended human dose (MRHD) of 40 mg/day during organogenesis. In one of two perinatal/postnatal developmental studies in rats, postnatal pup survival decreased following maternal exposure to unbound tadalafil concentrations greater than 5 times the MRHD based on AUC. Signs of maternal toxicity occurred at doses greater than 8 times the MRHD based on AUC. Surviving offspring had normal development and reproductive performance [see Nonclinical Toxicology (13.3)].
8.3 Nursing Mothers
It is not known whether tadalafil is excreted into human milk. While tadalafil or some metabolite of tadalafil was excreted into rat milk, drug levels in animal breast milk may not accurately predict levels of drug in human breast milk. Because many drugs are excreted in human milk, caution should be exercised when ADCIRCA is administered to a nursing woman.
8.4 Pediatric Use
Safety and effectiveness of ADCIRCA in pediatric patients have not been established.
8.5 Geriatric Use
Of the total number of subjects in the clinical study of tadalafil for pulmonary arterial hypertension, 28 percent were 65 and over, while 8 percent were 75 and over. No overall differences in safety were observed between subjects over 65 years of age compared to younger subjects or those over 75 years of age. No dose adjustment is warranted based on age alone; however, a greater sensitivity to medications in some older individuals should be considered. [See Dosage and Administration (2.2) and Clinical Pharmacology (12.3)].
8.6 Renal Impairment
For patients with mild or moderate renal impairment, start ADCIRCA at 20 mg once daily. Increase the dose to 40 mg once daily based upon individual tolerability [see Dosage and Administration (2.2), Warnings and Precautions (5.3) and Clinical Pharmacology (12.3)].
In patients with severe renal impairment, avoid use of ADCIRCA because of increased tadalafil exposure (AUC), limited clinical experience, and the lack of ability to influence clearance by dialysis [see Warnings and Precautions (5.3) and Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
Because of limited clinical experience in patients with mild to moderate hepatic cirrhosis (Child-Pugh Class A or B), consider a starting dose of ADCIRCA 20 mg once daily. Patients with severe hepatic cirrhosis (Child-Pugh Class C) have not been studied, thus avoid use of ADCIRCA in such patients [see Dosage and Administration (2.2), Warnings and Precautions (5.4) and Clinical Pharmacology (12.3)].
-
10 OVERDOSAGE
Single doses up to 500 mg have been given to healthy male subjects, and multiple daily doses up to 100 mg have been given to male patients with erectile dysfunction. Adverse reactions were similar to those seen at lower doses. Doses greater than 40 mg have not been studied in patients with pulmonary arterial hypertension. In cases of overdose, standard supportive measures should be adopted as needed. Hemodialysis contributes negligibly to tadalafil elimination.
-
11 DESCRIPTION
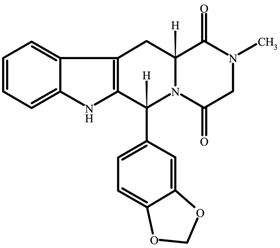
ADCIRCA (tadalafil), an oral treatment for pulmonary arterial hypertension, is a selective inhibitor of cyclic guanosine monophosphate (cGMP)–specific phosphodiesterase type 5 (PDE5). Tadalafil has the empirical formula C22H19N3O4 representing a molecular weight of 389.41. The structural formula is:
The chemical designation is pyrazino[1´,2´:1,6]pyrido[3,4–b]indole-1,4-dione, 6-(1,3-benzodioxol-5-yl)-2,3,6,7,12,12a-hexahydro-2-methyl-, (6R,12aR)-. It is a crystalline solid that is practically insoluble in water and very slightly soluble in ethanol.
ADCIRCA is available as orange, film–coated, almond–shaped tablets for oral administration. Each tablet contains 20 mg of tadalafil and the following inactive ingredients: croscarmellose sodium, hydroxypropyl cellulose, hypromellose, iron oxide, lactose monohydrate, magnesium stearate, microcrystalline cellulose, sodium lauryl sulfate, talc, titanium dioxide, and triacetin.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Tadalafil is an inhibitor of phosphodiesterase type 5 (PDE5), the enzyme responsible for the degradation of cyclic guanosine monophosphate (cGMP). Pulmonary arterial hypertension is associated with impaired release of nitric oxide by the vascular endothelium and consequent reduction of cGMP concentrations in the pulmonary vascular smooth muscle. PDE5 is the predominant phosphodiesterase in the pulmonary vasculature. Inhibition of PDE5 by tadalafil increases the concentrations of cGMP resulting in relaxation of pulmonary vascular smooth muscle cells and vasodilation of the pulmonary vascular bed.
Studies in vitro have demonstrated that tadalafil is a selective inhibitor of PDE5. PDE5 is found in pulmonary vascular smooth muscle, visceral smooth muscle, corpus cavernosum, skeletal muscle, platelets, kidney, lung, cerebellum, and pancreas.
In vitro studies have shown that the effect of tadalafil is more potent on PDE5 than on other phosphodiesterases. These studies have shown that tadalafil is >10,000–fold more potent for PDE5 than for PDE1, PDE2, PDE4, and PDE7 enzymes, which are found in the heart, brain, blood vessels, liver, leukocytes, skeletal muscle, and other organs. Tadalafil is >10,000–fold more potent for PDE5 than for PDE3, an enzyme found in the heart and blood vessels. Additionally, tadalafil is 700–fold more potent for PDE5 than for PDE6, which is found in the retina and is responsible for phototransduction. Tadalafil is >9,000-fold more potent for PDE5 than for PDE8, PDE9, and PDE10. Tadalafil is 14–fold more potent for PDE5 than for PDE11A1 and 40–fold more potent for PDE5 than for PDE11A4, two of the four known forms of PDE11. PDE11 is an enzyme found in human prostate, testes, skeletal muscle and in other tissues. In vitro, tadalafil inhibits human recombinant PDE11A1 and, to a lesser degree, PDE11A4 activities at concentrations within the therapeutic range. The physiological role and clinical consequence of PDE11 inhibition in humans have not been defined.
12.2 Pharmacodynamics
Effects on Blood Pressure When Administered with Nitrates
In clinical pharmacology studies, tadalafil (5 to 20 mg) was shown to potentiate the hypotensive effect of nitrates. Do not use ADCIRCA in patients taking any form of nitrates [see Contraindications (4.1)].
A double–blind, placebo–controlled, crossover study in 150 male subjects at least 40 years of age (including subjects with diabetes mellitus and/or controlled hypertension) assessed the interaction between nitroglycerin and tadalafil. Subjects received daily doses of tadalafil 20 mg or matching placebo for 7 days and then were given a single dose of 0.4 mg sublingual nitroglycerin (NTG) at pre–specified timepoints following their last dose of tadalafil (2, 4, 8, 24, 48, 72, and 96 hours after tadalafil). A significant interaction between tadalafil and NTG was observed at each timepoint up to and including 24 hours. At 48 hours, by most hemodynamic measures, the interaction between tadalafil and NTG was not observed, although a few more tadalafil subjects compared to placebo experienced greater blood–pressure lowering effects at this timepoint. After 48 hours, the interaction was not detectable. [See Contraindications (4.1) and Warnings and Precautions (5.1)].
Effects on Blood Pressure
Tadalafil 20 mg administered to healthy male subjects produced no significant difference compared to placebo in supine systolic and diastolic blood pressure (difference in the mean maximal decrease of 1.6/0.8 mm Hg, respectively) and in standing systolic and diastolic blood pressure (difference in the mean maximal decrease of 0.2/4.6 mm Hg, respectively). In addition, there was no significant effect on heart rate.
Amlodipine — A study assessed the interaction between amlodipine (5 mg daily) and tadalafil 10 mg. There was no effect of tadalafil on amlodipine blood levels and no effect of amlodipine on tadalafil blood levels. The mean reduction in supine systolic/diastolic blood pressure because of tadalafil 10 mg in subjects taking amlodipine was 3/2 mm Hg, compared to placebo. In a similar study using tadalafil 20 mg, there were no clinically significant differences between tadalafil and placebo in subjects taking amlodipine.
Angiotensin II receptor blockers (with and without other antihypertensives) — A study assessed the interaction between angiotensin II receptor blockers and tadalafil 20 mg. Subjects in the study were taking any marketed angiotensin II receptor blocker, either alone, as a component of a combination product, or as part of a multiple antihypertensive regimen. Following dosing, ambulatory measurements of blood pressure revealed differences between tadalafil and placebo of 8/4 mm Hg in systolic/diastolic blood pressure.
Bendroflumethiazide — A study assessed the interaction between bendroflumethiazide (2.5 mg daily) and tadalafil 10 mg. Following dosing, the mean reduction in supine systolic/diastolic blood pressure because of tadalafil 10 mg in subjects taking bendroflumethiazide was 6/4 mm Hg, compared to placebo.
Enalapril — A study assessed the interaction between enalapril (10 to 20 mg daily) and tadalafil 10 mg. Following dosing, the mean reduction in supine systolic/diastolic blood pressure because of tadalafil 10 mg in subjects taking enalapril was 4/1 mm Hg, compared to placebo.
Metoprolol — A study assessed the interaction between sustained–release metoprolol (25 to 200 mg daily) and tadalafil 10 mg. Following dosing, the mean reduction in supine systolic/diastolic blood pressure because of tadalafil 10 mg in subjects taking metoprolol was 5/3 mm Hg, compared to placebo.
Effects on Blood Pressure When Administered with Alcohol
Alcohol and PDE5 inhibitors, including tadalafil, are mild systemic vasodilators. The interaction of tadalafil with alcohol was evaluated in three clinical pharmacology studies. In two of these, alcohol was administered at a dose of 0.7 g/kg, which is equivalent to approximately 6 ounces of 80–proof vodka in an 80–kg male, and tadalafil was administered at a dose of 10 mg in one study and 20 mg in another. In both these studies, all patients imbibed the entire alcohol dose within 10 minutes of starting. In one of these two studies, blood alcohol levels of 0.08% were confirmed. In these two studies, more patients had clinically significant decreases in blood pressure on the combination of tadalafil and alcohol as compared to alcohol alone. Some subjects reported postural dizziness, and orthostatic hypotension was observed in some subjects. When tadalafil 20 mg was administered with a lower dose of alcohol (0.6 g/kg, which is equivalent to approximately 4 ounces of 80–proof vodka, administered in less than 10 minutes), orthostatic hypotension was not observed, dizziness occurred with similar frequency to alcohol alone, and the hypotensive effects of alcohol were not potentiated.
Tadalafil did not affect alcohol plasma concentrations and alcohol did not affect tadalafil plasma concentrations.
Effects on Blood Pressure When Administered with Alpha-Blockers
Alpha-blockers and PDE5 inhibitors, including tadalafil, are systemic vasodilators. In subjects receiving concomitant tadalafil (20 mg single dose) and doxazosin (8 mg daily), an alpha-1 adrenergic receptor blocker, there was an augmentation of the blood pressure–lowering effect of doxazosin. This effect was still present at 12 hours postdose and had generally disappeared at 24 hours. The number of subjects with potentially clinically significant standing–blood–pressure decreases was greater for the combination.
An additional study was performed with tadalafil (20 mg single dose) and doxazosin (4 and 8 mg daily) using ambulatory blood pressure monitoring. The augmentation appeared unrelated to dosing times and resulted in a greater number of outliers for the combination than had been observed in the previous study. Both of these studies had some symptomatology associated with these blood pressure changes.
A further study was carried out with doxazosin (up to 4 mg daily) added to tadalafil (5 mg daily) and there was again an augmentation of response. In this clinical pharmacology study there were symptoms associated with the decrease in blood pressure, including syncope.
An interaction study with tadalafil (20 mg single dose) and alfuzosin, also an alpha-1 adrenergic receptor blocker, showed no clinically significant effect on blood pressure.
In two clinical pharmacology studies in healthy volunteers, tadalafil (5 mg daily, and 10 mg and 20 mg single dose) had no clinically significant effect on blood pressure changes because of tamsulosin, a selective alpha-1a adrenergic receptor blocking agent.
Effects on Cardiac Electrophysiology
The effect of a single 100 mg dose of tadalafil (2.5 times the recommended dose) on the QT interval was evaluated at the time of peak tadalafil concentration in a randomized, double–blinded, placebo, and active–controlled (intravenous ibutilide) crossover study in 90 healthy males aged 18 to 53 years. The mean change in QTc (Fridericia QT correction) for tadalafil, relative to placebo, was 3.5 milliseconds (two–sided 90% CI=1.9, 5.1). The mean change in QTc (Individual QT correction) for tadalafil, relative to placebo, was 2.8 milliseconds (two–sided 90% CI=1.2, 4.4). In this study, the mean increase in heart rate associated with a 100 mg dose of tadalafil compared to placebo was 3.1 beats per minute.
Effects on Exercise Stress Testing
The effects of tadalafil on cardiac function, hemodynamics, and exercise tolerance were investigated in a single clinical pharmacology study. In this blinded crossover trial, 23 subjects with stable coronary artery disease and evidence of exercise–induced cardiac ischemia were enrolled. The primary endpoint was time to cardiac ischemia. The mean difference in total exercise time was 3 seconds (tadalafil 10 mg minus placebo), which represented no clinically meaningful difference. Further statistical analysis demonstrated that tadalafil was similar to placebo with respect to time to ischemia. Of note, in this study, in some subjects who received tadalafil followed by sublingual nitroglycerin in the post–exercise period, clinically significant reductions in blood pressure were observed, consistent with the augmentation by tadalafil of the blood–pressure–lowering effects of nitrates.
Effects on Vision
Single oral doses of PDE inhibitors have demonstrated transient dose-related impairment of color discrimination (blue/green), using the Farnsworth–Munsell 100–hue test, with peak effects near the time of peak plasma levels. This finding is consistent with the inhibition of PDE6, which is involved in phototransduction in the retina. In a study to assess the effects of a single dose of tadalafil 40 mg on vision (N=59), no effects were observed on visual acuity, intraocular pressure, or pupillometry. Across all clinical studies with tadalafil, reports of changes in color vision were rare (<0.1% of patients).
Effects on Sperm Characteristics
Three studies were conducted in men to assess the potential effect on sperm characteristics of tadalafil 10 mg (one 6-month study) and 20 mg (one 6-month and one 9-month study) administered daily. There were no adverse effects on sperm morphology or sperm motility in any of the three studies. In the study of 10 mg tadalafil for 6 months and the study of 20 mg tadalafil for 9 months, results showed a decrease in mean sperm concentrations relative to placebo, although these differences were not clinically meaningful. This effect was not seen in the study of 20 mg tadalafil taken for 6 months. In addition there was no adverse effect on mean concentrations of reproductive hormones, testosterone, luteinizing hormone or follicle stimulating hormone with either 10 or 20 mg of tadalafil compared to placebo.
Dose-Response Relationship
Dose-response relationships, between 20 mg and 40 mg, were not observed for 6-minute walk distance or pulmonary vascular resistance (PVR) in subjects with PAH in the placebo-controlled study. Median change from baseline in 6-minute walk distance was 32 meters and 35 meters at 16 weeks in subjects receiving 20 mg and 40 mg daily, respectively. Mean change from baseline PVR was -254 dynes*sec*cm-5 and -209 dynes*sec*cm-5 at 16 weeks in patients receiving 20 mg and 40 mg daily, respectively.
12.3 Pharmacokinetics
Over a dose range of 2.5 to 20 mg, tadalafil exposure (AUC) increases proportionally with dose in healthy subjects. In PAH patients administered between 20 and 40 mg of tadalafil, an approximately 1.5-fold greater AUC was observed indicating a less than proportional increase in exposure over the entire dose range of 2.5 to 40 mg. During tadalafil 20 and 40 mg once daily dosing, steady-state plasma concentrations were attained within 5 days, and exposure was approximately 1.3-fold higher than after a single dose.
Absorption — After single oral-dose administration, the maximum observed plasma concentration (Cmax) of tadalafil is achieved between 2 and 8 hours (median time of 4 hours). Absolute bioavailability of tadalafil following oral dosing has not been determined.
The rate and extent of absorption of tadalafil are not influenced by food; thus ADCIRCA may be taken with or without food.
Distribution — The mean apparent volume of distribution following oral administration is approximately 77 L, indicating that tadalafil is distributed into tissues. At therapeutic concentrations, 94% of tadalafil in plasma is bound to proteins.
Metabolism — Tadalafil is predominantly metabolized by CYP3A to a catechol metabolite. The catechol metabolite undergoes extensive methylation and glucuronidation to form the methylcatechol and methylcatechol glucuronide conjugate, respectively. The major circulating metabolite is the methylcatechol glucuronide. Methylcatechol concentrations are less than 10% of glucuronide concentrations. In vitro data suggests that metabolites are not expected to be pharmacologically active at observed metabolite concentrations.
Elimination — Following 40 mg, the mean oral clearance for tadalafil is 3.4 L/hr and the mean terminal half-life is 15 hours in healthy subjects. In patients with pulmonary hypertension not receiving concomitant bosentan, the mean oral clearance for tadalafil is 1.6 L/hr, and the mean terminal half-life is 35 hours. Tadalafil is excreted predominantly as metabolites, mainly in the feces (approximately 61% of the dose) and to a lesser extent in the urine (approximately 36% of the dose).
Population pharmacokinetics — In patients with pulmonary hypertension not receiving concomitant bosentan, the average tadalafil exposure at steady-state following 40 mg was 26% higher when compared to those of healthy volunteers. The results suggest a lower clearance of tadalafil in patients with pulmonary hypertension compared to healthy volunteers.
Geriatric patients
In healthy male elderly subjects (65 years or over) after a 10 mg dose, a lower oral clearance of tadalafil, resulting in 25% higher exposure (AUC) with no effect on Cmax was observed relative to that in healthy subjects 19 to 45 years of age.
Renal impairment
In clinical pharmacology studies using single-dose tadalafil (5 to 10 mg), tadalafil exposure (AUC) doubled in subjects with mild (creatinine clearance 51 to 80 mL/min) or moderate (creatinine clearance 31 to 50 mL/min) renal impairment. In subjects with end-stage renal disease on hemodialysis, there was a two-fold increase in Cmax and 2.7- to 4.1-fold increase in AUC following single-dose administration of 10 or 20 mg tadalafil, respectively. Exposure to total methylcatechol (unconjugated plus glucuronide) was 2- to 4-fold higher in subjects with renal impairment, compared to those with normal renal function. Hemodialysis (performed between 24 and 30 hours post-dose) contributed negligibly to tadalafil or metabolite elimination [see Dosage and Administration (2.2) and Warnings and Precautions (5.3)].
Hepatic impairment
In clinical pharmacology studies, tadalafil exposure (AUC) in subjects with mild or moderate hepatic impairment (Child-Pugh Class A or B) was comparable to exposure in healthy subjects when a dose of 10 mg was administered. There are no available data for doses higher than 10 mg of tadalafil in patients with hepatic impairment. Insufficient data are available for subjects with severe hepatic impairment (Child-Pugh Class C) [see Dosage and Administration (2.2) and Warnings and Precautions (5.4)].
Patients with diabetes mellitus
In male patients with diabetes mellitus after a 10 mg tadalafil dose, exposure (AUC) was reduced approximately 19% and Cmax was 5% lower than that observed in healthy subjects. No dose adjustment is warranted.
Race
Pharmacokinetic studies have included subjects from different ethnic groups, and no differences in the typical exposure to tadalafil have been identified. No dose adjustment is warranted.
Gender
In healthy female and male subjects following single and multiple-doses of tadalafil, no clinically relevant differences in exposure (AUC and Cmax) were observed. No dose adjustment is warranted.
Drug interaction studies
Tadalafil is a substrate of and predominantly metabolized by CYP3A. Drugs that inhibit CYP3A can increase tadalafil exposure.
Ritonavir
Ritonavir (500 mg or 600 mg twice daily at steady state), an inhibitor of CYP3A, CYP2C9, CYP2C19, and CYP2D6, increased tadalafil 20–mg single-dose exposure (AUC) by 32% with a 30% reduction in Cmax, relative to the values for tadalafil 20 mg alone. Ritonavir (200 mg twice daily), increased tadalafil 20–mg single-dose exposure (AUC) by 124% with no change in Cmax, relative to the values for tadalafil 20 mg alone. Ritonavir inhibits and induces CYP3A, the enzyme involved in the metabolism of tadalafil, in a time-dependent manner. The results suggest the initial inhibitory effect of ritonavir on CYP3A may be mitigated by a more slowly evolving induction effect so that after about 1 week of ritonavir twice daily, the exposure of tadalafil is similar in the presence of and absence of ritonavir [see Dosage and Administration (2.3), Warnings and Precautions (5.2), and Drug Interactions (7.2)]. Although specific interactions have not been studied, other HIV protease inhibitors would likely increase tadalafil exposure.
Other Cytochrome P450 inhibitors
CYP3A (e.g., ketoconazole) — Ketoconazole (400 mg daily), a selective and potent inhibitor of CYP3A, increased tadalafil 20 mg single-dose exposure (AUC) by 312% and Cmax by 22%, relative to the values for tadalafil 20 mg alone. Ketoconazole (200 mg daily) increased tadalafil 10–mg single-dose exposure (AUC) by 107% and Cmax by 15%, relative to the values for tadalafil 10 mg alone.
Although specific interactions have not been studied, other CYP3A inhibitors, such as erythromycin, itraconazole, and grapefruit juice, would likely increase tadalafil exposure.
Cytochrome P450 inducers
CYP3A (e.g., rifampin, bosentan) — Rifampin (600 mg daily), a CYP3A inducer, reduced tadalafil 10 mg single–dose exposure (AUC) by 88% and Cmax by 46%, relative to the values for tadalafil 10 mg alone.
Bosentan (125 mg twice daily), a substrate of CYP2C9 and CYP3A and a moderate inducer of CYP3A, CYP2C9 and possibly CYP2C19, reduced tadalafil (40 mg once per day) systemic exposure by 42% and Cmax by 27% following multiple-dose co-administration.
Although specific interactions have not been studied, other CYP3A inducers, such as carbamazepine, phenytoin, and phenobarbital, would likely decrease tadalafil exposure.
Cytochrome P450 substrates — Tadalafil is not expected to cause clinically significant inhibition or induction of the clearance of drugs metabolized by cytochrome P450 (CYP) isoforms.
CYP1A2 (e.g., theophylline) — Tadalafil (10 mg once per day) had no significant effect on the pharmacokinetics of theophylline. When tadalafil was administered to subjects taking theophylline, a small augmentation (3 beats per minute) of the increase in heart rate associated with theophylline was observed.
CYP2C9 (e.g., warfarin) — Tadalafil (10 mg and 20 mg once per day) had no significant effect on exposure (AUC) to S–warfarin or R–warfarin, nor did tadalafil affect changes in prothrombin time induced by warfarin.
CYP3A (e.g., midazolam, lovastatin or bosentan) — Tadalafil (10 mg and 20 mg once per day) had no significant effect on exposure (AUC) to midazolam or lovastatin. Tadalafil (40 mg once per day) had no clinically significant effect on exposure (AUC and Cmax) of bosentan, a substrate of CYP2C9 and CYP3A, or its metabolites.
Aspirin — Tadalafil (10 mg and 20 mg once per day) did not potentiate the increase in bleeding time caused by aspirin.
P-glycoprotein (e.g., digoxin) — Coadministration of tadalafil (40 mg once per day) for 10 days did not have a significant effect on the steady-state pharmacokinetics of digoxin (0.25 mg/day) in healthy subjects.
Combined oral contraceptives — At steady-state, tadalafil (40 mg once per day) increased ethinyl estradiol exposure (AUC) by 26% and Cmax by 70% relative to oral contraceptive administered with placebo. There was no significant effect of tadalafil on levonorgestrel.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis — Tadalafil was not carcinogenic to rats or mice when administered daily for 2 years at doses up to 400 mg/kg/day. Systemic drug exposures, as measured by AUC of unbound tadalafil, were approximately 5–fold for mice, and 7– and 14–fold for male and female rats, respectively, the exposures at the maximum recommended human dose (MRHD) of 40 mg.
Mutagenesis — Tadalafil was not mutagenic in the in vitro bacterial Ames assays or the forward mutation test in mouse lymphoma cells. Tadalafil was not clastogenic in the in vitro chromosomal aberration test in human lymphocytes or the in vivo rat micronucleus assays.
Impairment of Fertility — There were no effects on fertility, reproductive performance or reproductive organ morphology in male or female rats given oral doses of tadalafil up to 400 mg/kg/day, a dose producing AUCs for unbound tadalafil of 6–fold for males or 17–fold for females the exposures at the MRHD of 40 mg. In beagle dogs given tadalafil daily for 3 to 12 months, there was treatment–related non–reversible degeneration and atrophy of the seminiferous tubular epithelium in the testes in 20–100% of the dogs that resulted in a decrease in spermatogenesis in 40–75% of the dogs at doses of ≥10 mg/kg/day. Systemic exposure (based on AUC) at no–observed–adverse-effect–level (NOAEL) (10 mg/kg/day) for unbound tadalafil was similar to that expected in humans at the MRHD of 40 mg.
There were no treatment–related testicular findings in rats or mice treated with doses up to 400 mg/kg/day for 2 years.
13.2 Animal Toxicology and/or Pharmacology
Animal studies showed vascular inflammation in tadalafil–treated mice, rats, and dogs. In mice and rats, lymphoid necrosis and hemorrhage were seen in the spleen, thymus, and mesenteric lymph nodes at unbound tadalafil exposure of 1– to 17–fold the human exposure (AUCs) at the MRHD of 40 mg. In dogs, an increased incidence of disseminated arteritis was observed in 1– and 6-month studies at unbound tadalafil exposure of 0.5– to 38–fold the human exposure (AUC) at the MRHD of 40 mg. In a 12–month dog study, no disseminated arteritis was observed, but 2 dogs exhibited marked decreases in white blood cells (neutrophils) and moderate decreases in platelets with inflammatory signs at unbound tadalafil exposures of approximately 4– to 10–fold the human exposure at the MRHD of 40 mg. The abnormal blood–cell findings were reversible within 2 weeks upon removal of the drug.
13.3 Reproductive Toxicology Studies
Reproduction studies have been performed in rats and mice at exposures up to 17 times the MRHD of 40 mg and have revealed no evidence of impaired fertility or harm to the fetus because of tadalafil. In addition, there was no evidence of teratogenicity, embryotoxicity, or fetotoxicity when tadalafil was given to pregnant rats or mice at exposures up to 7 times the MRHD during the period of major organ development.
In a rat prenatal and postnatal development study at doses of 60, 200, and 1000 mg/kg, a reduction in postnatal survival of pups was observed. The no-observed-effect-level (NOEL) for maternal toxicity was 200 mg/kg/day and for developmental toxicity was 30 mg/kg/day. This gives approximately 8- and 5-fold exposure multiples, respectively, of the human AUC for the MRHD of 40 mg. Tadalafil and/or its metabolites cross the placenta, resulting in fetal exposure in rats.
Tadalafil and/or its metabolites were secreted into the milk in lactating rats at concentrations approximately 2.4–fold greater than found in the plasma.
-
14 CLINICAL STUDIES
14.1 ADCIRCA for Pulmonary Arterial Hypertension
A randomized, double-blind, 16 week placebo-controlled study was conducted in 405 patients with pulmonary arterial hypertension, defined as a resting mean pulmonary artery pressure (mPAP) ≥25 mm Hg, pulmonary capillary wedge pressure (PCWP) ≤15 mm Hg, and pulmonary vascular resistance (PVR) ≥3 Wood units via right heart catheterization. Allowed background therapy included bosentan (maintenance dosing up to 125 mg twice daily) and chronic anticoagulation. The use of prostacyclin or analogue, L–arginine, phosphodiesterase inhibitor, or other chronic PAH medications were not permitted.
Subjects were randomly assigned to 1 of 5 treatment groups (tadalafil 2.5, 10, 20, 40 mg, or placebo) in a 1:1:1:1:1 ratio. Subjects had to be at least 12 years of age and had a diagnosis of PAH that was idiopathic, heritable, related to connective tissue disease, anorexigen use, human immunodeficiency virus (HIV) infection, associated with an atrial-septal defect, or associated with surgical repair of a congenital systemic-to-pulmonary shunt of least 1 year in duration (for example, ventricular septal defect, patent ductus arteriosus). Patients with a history of left-sided heart disease, severe renal insufficiency, or pulmonary hypertension related to conditions other than specified in the inclusion criteria were not eligible for enrollment.
The mean age of all subjects was 54 years (range 14 - 90 years) with the majority of subjects being Caucasian (81%) and female (78%). PAH etiologies were predominantly idiopathic or heritable PAH (61%) and related to connective tissue disease (23%). More than half (53%) of the subjects in the study were receiving concomitant bosentan therapy. The majority of subjects had a World Health Organization (WHO) Functional Class III (65%) or II (32%). The mean baseline 6-minute walk distance (6-MWD) was 343 meters. Of the 405 subjects, 341 completed the study.
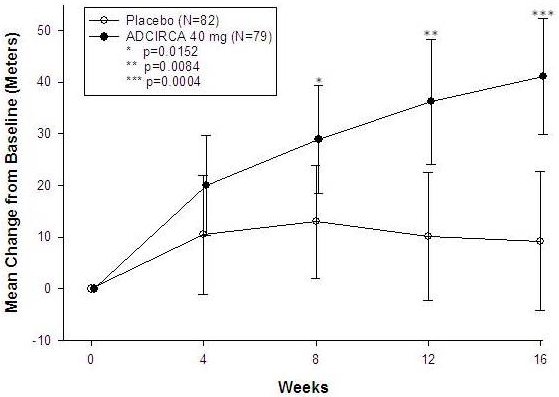
The primary efficacy endpoint was the change from baseline at week 16 in 6-MWD (see Figure 1). In the ADCIRCA 40 mg treatment group, the placebo-adjusted mean change increase in 6-MWD was 33 meters (95% C.I. 15-50 meters; p=0.0004). The improvement in 6-MWD was apparent at 8 weeks of treatment and then maintained at week 12 and week 16.
Placebo-adjusted changes in 6-MWD at 16 weeks were evaluated in subgroups (see Figure 2). In patients taking only ADCIRCA 40 mg (i.e., without concomitant bosentan), the placebo-adjusted mean change in 6-MWD was 44 meters. In patients taking ADCIRCA 40 mg and concomitant bosentan therapy, the placebo adjusted mean change in 6-MWD was 23 meters.
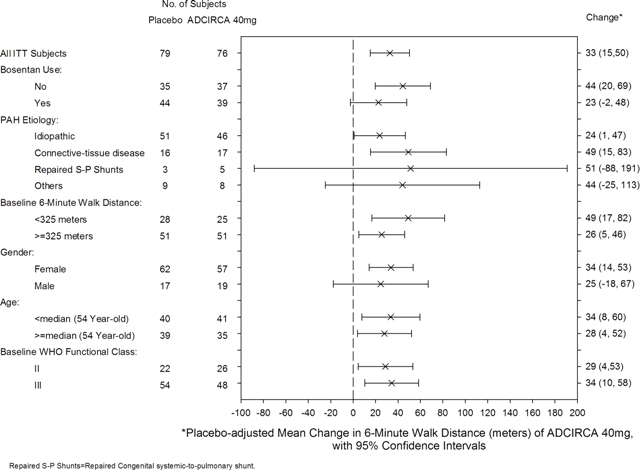
Figure 2: Placebo-adjusted Mean Change in 6-Minute Walk Distance (meters) of ADCIRCA 40 mg, with 95% Confidence Intervals
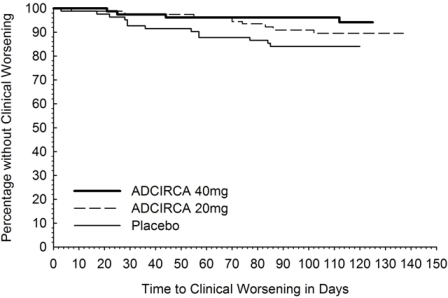
There was less clinical worsening (defined as death, lung transplantation, atrial septostomy, hospitalization because of worsening PAH, initiation of new PAH therapy [prostacyclin or analog, endothelin receptor antagonist, PDE5 inhibitor], or worsening WHO functional class) in the ADCIRCA 40 mg group compared to the placebo group and the groups that used lower doses of ADCIRCA.
Table 2: Number (percent) with Clinical Worseninga a Subjects may be counted in more than one category
ADCIRCA Placebo
N=822.5 mg
N=8210 mg
N=8020 mg
N=8240 mg
N=79Total with clinical worsening 13 (16) 10 (12) 7 (9) 8 (10) 4 (5) Death 1 0 1 0 0 Hospitalization for worsening PAH 2 2 3 0 1 New PAH therapy 0 1 0 2 1 Worsening WHO class 11 10 6 6 3 The Kaplan-Meier plot of times to clinical worsening is shown below in Figure 3.
14.2 Long-Term Treatment of Pulmonary Arterial Hypertension
Patients (N=357) from the placebo-controlled study entered a long-term extension study. Of these, 311 patients have been treated with tadalafil for at least 6 months and 182 for 1 year (median exposure 356 days; range 2 days to 415 days). The survival rate in the extension study was 96.5 per 100 patient years. Without a control group, these data must be interpreted cautiously.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
ADCIRCA (tadalafil) is supplied as follows:
20 mg orange, film–coated, almond–shaped tablets (not scored), debossed with “4467”
Bottles of 60 NDC: 66302-467-60
-
17 PATIENT COUNSELING INFORMATION
See FDA-Approved Patient Labeling (Patient Information)
- Inform patients of contraindication of ADCIRCA with any use of organic nitrates or GC stimulators.
- Inform patients that tadalafil is also marketed as CIALIS for erectile dysfunction (ED) and for the signs and symptoms of benign prostatic hyperplasia (BPH). Advise patients taking ADCIRCA not to take CIALIS or other PDE5 inhibitors.
- Advise patients to seek immediate medical attention in the event of a sudden loss of vision in one or both eyes while taking ADCIRCA. Such an event may be a sign of NAION.
- Advise patients to seek prompt medical attention in the event of sudden decrease or loss of hearing while taking ADCIRCA. These events may be accompanied by tinnitus and dizziness.
-
PATIENT PACKAGE INSERT
PATIENT INFORMATION
ADCIRCA® (Ad-sur-kuh)
(tadalafil) tablets
Read this patient information before you start taking ADCIRCA and each time you get a refill. There may be new information. This information does not take the place of talking with your healthcare provider about your medical condition or treatment.
What is the most important information I should know about ADCIRCA?
Never take ADCIRCA with any nitrate or guanylate cyclase stimulator medicines:
- Your blood pressure could drop quickly to an unsafe level
- You could get dizzy, faint and even have a heart attack or stroke.
Nitrates include:
- Medicines that treat chest pain (angina)
- Nitroglycerin in any form including tablets, patches, sprays, and ointments
- Other nitrate medicines (isosorbide mononitrate or dinitrate)
- Street drugs that are inhaled, called “poppers” (amyl nitrate, butyl nitrate or nitrite)
Guanylate cyclase stimulators include:
- Riociguat (Adempas®) a medicine that treats pulmonary arterial hypertension and chronic-thromboembolic pulmonary hypertension
Ask your healthcare provider or pharmacist if you are not sure if you take a nitrate or guanylate cyclase stimulator medicine.
What is ADCIRCA?
ADCIRCA is a prescription medicine used to treat pulmonary arterial hypertension (PAH, high blood pressure in your lungs) to improve your ability to exercise.
It is not known if ADCIRCA is safe or effective in children.
Who should not take ADCIRCA?
Do not take ADCIRCA if you
- take any medicines called nitrates.
- use recreational drugs called “poppers” like amyl nitrate, butyl nitrate or nitrite.
- take any medicines called guanylate cyclase stimulators
- are allergic to tadalafil or any other ingredient in ADCIRCA. See “What are the ingredients in ADCIRCA?” at the end of this leaflet.
See “What is the most important information I should know about ADCIRCA?”
What should I tell my healthcare provider before taking ADCIRCA?
Before taking ADCIRCA, tell your healthcare provider about all of your medical conditions, including if you:
- are allergic to ADCIRCA or Cialis or any of its ingredients. See the end of this leaflet for a complete list of ingredients in ADCIRCA.
- have pulmonary veno-occlusive disease (PVOD)
- have heart problems such as angina (chest pain), heart failure, irregular heartbeats, or have had a heart attack
- have low blood pressure or high blood pressure that is not controlled
- have had a stroke
- have liver problems
- have kidney problems or get dialysis
- have stomach ulcers
- have retinitis pigmentosa, a rare genetic eye disease
- have ever had any sudden vision loss, including any damage to your optic nerve or NAION.
- have ever had hearing problems such as ringing in the ears, dizziness, or loss of hearing
- have a deformed penis shape or Peyronie's disease
- have had an erection that lasted more than 4 hours
- have blood cell problems such as sickle cell anemia, multiple myeloma, or leukemia
- are pregnant or planning to become pregnant. It is not known if ADCIRCA will harm your unborn baby. Talk to your healthcare provider if you are pregnant or plan to become pregnant.
- are breastfeeding or plan to breast feed. It is not known if ADCIRCA passes into your breast milk. You and your healthcare provider should decide if you will take ADCIRCA or breastfeed. You should not do both.
Tell your healthcare provider about all the medicines you take, including prescription and non-prescription medicines, vitamins, and herbal supplements. ADCIRCA and other medicines may affect each other.
Especially tell your healthcare provider if you take any of these medicines*:
- nitrates or guanylate cyclase stimulators (see “What is the most important information I should know about ADCIRCA?”)
- anti-hypertensives, used to treat high blood pressure. Your blood pressure could suddenly drop. You could get dizzy or faint.
- alpha blockers, used to treat prostate disease and high blood pressure. Your blood pressure could suddenly drop. You could get dizzy or faint.
- protease inhibitors, used to treat HIV infection, such as ritonavir (Norvir®, Kaletra®)
- ketoconazole (Extina®, Xolegel®, Ketozole®, Nizoral A-D®, Nizoral®) itraconazole (Sporanox®)
- erythromycin (several brand names exist. Please consult your healthcare provider to determine if you are taking this medicine)
- rifampin (Rifadin®, Rifamate®, Rifater®, Rimactane®)
- bosentan (Tracleer®)
- phenobarbital, phenytoin (Dilantin®), carbamazepine (Tegretol®)
- CIALIS® or other medicines or treatments for erectile dysfunction (impotence).
- ADCIRCA is also marketed as CIALIS for the treatment of male erectile dysfunction (ED, impotence) and for the signs and symptoms of benign prostatic hyperplasia (BPH, enlarged prostate). Do not take both ADCIRCA and CIALIS. Do not take ADCIRCA and other medicines or treatments for erectile dysfunction.
Ask your healthcare provider or pharmacist for a list of these medicines, if you are not sure.
Know the medicines you take. Keep a list of them and show it to your healthcare provider and pharmacist when you get a new medicine.
How should I take ADCIRCA?
- Take ADCIRCA exactly as your healthcare provider tells you.
- Take ADCIRCA tablets at the same time every day. You should take both tablets at the same time, one after the other, every day. Do not split your dose.
- ADCIRCA can be taken with or without food.
- Do not change your dose or stop taking ADCIRCA without speaking to your healthcare provider.
- If you take too much ADCIRCA, call your healthcare provider or go to an emergency department right away.
What should I avoid while taking ADCIRCA?
Do not have more than 4 alcohol-containing drinks in a short period of time while you take ADCIRCA. Drinking too much alcohol can lower your blood pressure. You could get dizzy or faint.
What are the possible side effects of ADCIRCA?
The following side effects were reported rarely in patients taking tadalafil:
- Decreased eyesight or loss of vision in one or both eyes (NAION). If you notice a sudden decrease or loss of vision in one or both eyes, contact a healthcare provider right away.
- Sudden decrease or loss of hearing, sometimes with ringing in the ears and dizziness. If you notice a sudden decrease or loss of hearing, contact a healthcare provider right away.
- In men, an erection that lasts more than 4 hours (with or without pain). Talk to your healthcare provider or go to the emergency department right away. An erection that lasts more than 4 hours must be treated as soon as possible or you can have lasting damage to your penis, including the inability to have erections.
See “What is the most important information I should know about ADCIRCA?”
The most common side effects with ADCIRCA include:
- headache
- muscle pain
- getting red or hot in the face (flushing)
- nausea
- pain in the arms, legs, or back
- upset stomach
- stuffy or congested nose
Tell your healthcare provider about any side effect that bothers you or does not go away.
These are not all the possible side effects of ADCIRCA. For more information, ask your healthcare provider or pharmacist. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How Should I Store ADCIRCA?
Store ADCIRCA at room temperature between 59° and 86°F (15° and 30°C).
Keep ADCIRCA and all medicines out of the reach of children.
General Information about the safe and effective use of ADCIRCA
Medicines are sometimes prescribed for conditions that are not mentioned in patient information leaflets. Do not use ADCIRCA for a condition for which it was not prescribed. Do not give ADCIRCA to other people, even if they have the same symptoms you have. It may harm them.
This patient information leaflet summarizes the most important information about ADCIRCA. If you would like more information, talk with your healthcare provider. You can ask your healthcare provider or pharmacist for information about ADCIRCA that is written for healthcare professionals. For more information, go to www.ADCIRCA.com or call 1-800-LILLYRX (1-800-545-5979).
What Are The Ingredients In ADCIRCA?
Active Ingredient: tadalafil
Inactive Ingredients: croscarmellose sodium, hydroxypropyl cellulose, hypromellose, iron oxide, lactose monohydrate, magnesium stearate, microcrystalline cellulose, sodium lauryl sulfate, talc, titanium dioxide, and triacetin.
Rx only
ADCIRCA® (tadalafil) is a trademark of Eli Lilly and Company.
*The brands listed are trademarks of their respective owners and are not trademarks of Eli Lilly and Company. The makers of these brands are not affiliated with and do not endorse Eli Lilly and Company or its products.
Revised 08/2017
Manufactured by: Eli Lilly and Company, Indianapolis, IN 46285, USA
Marketed by: United Therapeutics Corporation
Copyright © 2009, 2017, Eli Lilly and Company. All rights reserved.
ADC-0003-PPI-20170801
-
Repackaging Information
Please reference the How Supplied section listed above for a description of individual tablets. This drug product has been received by Aphena Pharma - TN in a manufacturer or distributor packaged configuration and repackaged in full compliance with all applicable cGMP regulations. The package configurations available from Aphena are listed below:
Count 20mg 12 43353-070-12 Store between 20°-25°C (68°-77°F). See USP Controlled Room Temperature. Dispense in a tight light-resistant container as defined by USP. Keep this and all drugs out of the reach of children.
Repackaged by:

Cookeville, TN 38506
20171017JH - PRINCIPAL DISPLAY PANEL - 20mg
-
INGREDIENTS AND APPEARANCE
ADCIRCA
tadalafil tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 43353-070(NDC:66302-467) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength Tadalafil (UNII: 742SXX0ICT) (tadalafil - UNII:742SXX0ICT) Tadalafil 20 mg Inactive Ingredients Ingredient Name Strength Croscarmellose sodium (UNII: M28OL1HH48) Hydroxypropyl cellulose, Unspecified (UNII: 9XZ8H6N6OH) Hypromellose, unspecified (UNII: 3NXW29V3WO) Microcrystalline cellulose (UNII: OP1R32D61U) Lactose monohydrate (UNII: EWQ57Q8I5X) Magnesium stearate (UNII: 70097M6I30) Sodium lauryl sulfate (UNII: 368GB5141J) Talc (UNII: 7SEV7J4R1U) Titanium dioxide (UNII: 15FIX9V2JP) Triacetin (UNII: XHX3C3X673) Ferric oxide yellow (UNII: EX438O2MRT) Ferric oxide red (UNII: 1K09F3G675) Product Characteristics Color ORANGE (ORANGE) Score no score Shape OVAL (ALMOND) Size 12mm Flavor Imprint Code 4467 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 43353-070-12 12 in 1 BOTTLE; Type 0: Not a Combination Product 06/22/2017 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA022332 05/22/2009 Labeler - Aphena Pharma Solutions - Tennessee, LLC (128385585) Establishment Name Address ID/FEI Business Operations Aphena Pharma Solutions - Tennessee, LLC 128385585 REPACK(43353-070)

Trademark Results [ADCIRCA]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 ADCIRCA 78737811 not registered Dead/Abandoned |
ELI LILLY AND COMPANY 2005-10-21 |
 ADCIRCA 77860178 3844162 Live/Registered |
Eli Lilly and Company 2009-10-29 |
 ADCIRCA 77262532 3677077 Live/Registered |
Eli Lilly and Company 2007-08-23 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.