FULPHILA- pegfilgrastim injection
Fulphila by
Drug Labeling and Warnings
Fulphila by is a Prescription medication manufactured, distributed, or labeled by Mylan Institutional LLC. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use FULPHILA safely and effectively. See full prescribing information for FULPHILA.
FULPHILA® (pegfilgrastim-jmdb) injection, for subcutaneous use
Initial U.S. Approval: 2018
FULPHILA® (pegfilgrastim-jmdb) is biosimilar* to NEULASTA® (pegfilgrastim). (1)
RECENT MAJOR CHANGES
INDICATIONS AND USAGE
Fulphila is a leukocyte growth factor indicated to
- Decrease the incidence of infection, as manifested by febrile neutropenia, in patients with non-myeloid malignancies receiving myelosuppressive anti-cancer drugs associated with a clinically significant incidence of febrile neutropenia. (1.1)
Limitations of Use
Fulphila is not indicated for the mobilization of peripheral blood progenitor cells for hematopoietic stem cell transplantation.
DOSAGE AND ADMINISTRATION
- Patients with cancer receiving myelosuppressive chemotherapy
DOSAGE FORMS AND STRENGTHS
- Injection: 6 mg/0.6 mL solution in a single-dose prefilled syringe for manual use only. (3)
CONTRAINDICATIONS
Patients with a history of serious allergic reactions to human granulocyte colony-stimulating factors such as pegfilgrastim products or filgrastim products. (4)
WARNINGS AND PRECAUTIONS
- Fatal splenic rupture: Evaluate patients who report left upper abdominal or shoulder pain for an enlarged spleen or splenic rupture. (5.1)
- Acute respiratory distress syndrome (ARDS): Evaluate patients who develop fever, lung infiltrates, or respiratory distress. Discontinue Fulphila in patients with ARDS. (5.2)
- Serious allergic reactions, including anaphylaxis: Permanently discontinue Fulphila in patients with serious allergic reactions. (5.3)
- Fatal sickle cell crises: Have occurred. (5.4)
- Glomerulonephritis: Evaluate and consider dose-reduction or interruption of Fulphila if causality is likely. (5.5)
ADVERSE REACTIONS
Most common adverse reactions (≥ 5% difference in incidence compared to placebo) are bone pain and pain in extremity. (6.1)
*Biosimilar means that the biological product is approved based on data demonstrating that it is highly similar to an FDA-approved biological product, known as a reference product, and that there are no clinically meaningful differences between the biosimilar product and the reference product.
Biosimilarity of Fulphila has been demonstrated for the condition(s) of use (e.g. indication(s), dosing regimen(s)), strength(s), dosage form(s), and route(s) of administration described in its Full Prescribing Information.
To report SUSPECTED ADVERSE REACTIONS, contact Mylan at 1-877-446-3679 (1-877-4-INFO-RX) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 5/2019
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Patients with Cancer Receiving Myelosuppressive Chemotherapy
2 DOSAGE AND ADMINISTRATION
2.1 Patients with Cancer Receiving Myelosuppressive Chemotherapy
2.2 Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Splenic Rupture
5.2 Acute Respiratory Distress Syndrome
5.3 Serious Allergic Reactions
5.4 Use in Patients with Sickle Cell Disorders
5.5 Glomerulonephritis
5.6 Leukocytosis
5.7 Capillary Leak Syndrome
5.8 Potential for Tumor Growth Stimulatory Effects on Malignant Cells
5.9 Aortitis
5.10 Nuclear Imaging
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Immunogenicity
6.3 Postmarketing Experience
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Patients with Cancer Receiving Myelosuppressive Chemotherapy
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
1.1 Patients with Cancer Receiving Myelosuppressive Chemotherapy
Fulphila is indicated to decrease the incidence of infection, as manifested by febrile neutropenia, in patients with non-myeloid malignancies receiving myelosuppressive anti-cancer drugs associated with a clinically significant incidence of febrile neutropenia [see Clinical Studies (14.1)].
-
2 DOSAGE AND ADMINISTRATION
2.1 Patients with Cancer Receiving Myelosuppressive Chemotherapy
The recommended dosage of Fulphila is a single subcutaneous injection of 6 mg administered once per chemotherapy cycle. For dosing in pediatric patients weighing less than 45 kg, refer to Table 1. Do not administer Fulphila between 14 days before and 24 hours after administration of cytotoxic chemotherapy.
2.2 Administration
Fulphila is administered subcutaneously via a single-dose prefilled syringe for manual use.
Prior to use‚ remove the carton from the refrigerator and allow the Fulphila prefilled syringe to reach room temperature for a minimum of 30 minutes. Discard any prefilled syringe left at room temperature for greater than 72 hours.
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. Do not administer Fulphila if discoloration or particulates are observed.
Pediatric Patients weighing less than 45 kg
The Fulphila prefilled syringe is not designed to allow for direct administration of doses less than 0.6 mL (6 mg). The syringe does not bear graduation marks, which are necessary to accurately measure doses of Fulphila less than 0.6 mL (6 mg) for direct administration to patients. Thus, the direct administration to patients requiring dosing of less than 0.6 mL (6 mg) is not recommended due to the potential for dosing errors. Refer to Table 1.
Table 1. Dosing of Fulphila for pediatric patients weighing less than 45 kg - * For pediatric patients weighing less than 10 kg, administer 0.1 mg/kg (0.01 mL/kg) of Fulphila.
Body Weight
Fulphila Dose
Volume to Administer
Less than 10 kg*
See below*
See below*
10 to 20 kg
1.5 mg
0.15 mL
21 to 30 kg
2.5 mg
0.25 mL
31 to 44 kg
4 mg
0.4 mL
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
Fulphila is contraindicated in patients with a history of serious allergic reactions to pegfilgrastim products or filgrastim products. Reactions have included anaphylaxis [see Warnings and Precautions (5.3)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Splenic Rupture
Splenic rupture, including fatal cases, can occur following the administration of pegfilgrastim products. Evaluate for an enlarged spleen or splenic rupture in patients who report left upper abdominal or shoulder pain after receiving Fulphila.
5.2 Acute Respiratory Distress Syndrome
Acute respiratory distress syndrome (ARDS) can occur in patients receiving pegfilgrastim products. Evaluate patients who develop fever and lung infiltrates or respiratory distress after receiving Fulphila, for ARDS. Discontinue Fulphila in patients with ARDS.
5.3 Serious Allergic Reactions
Serious allergic reactions, including anaphylaxis, can occur in patients receiving pegfilgrastim products. The majority of reported events occurred upon initial exposure. Allergic reactions, including anaphylaxis, can recur within days after the discontinuation of initial anti-allergic treatment. Permanently discontinue Fulphila in patients with serious allergic reactions. Do not administer Fulphila to patients with a history of serious allergic reactions to pegfilgrastim products or filgrastim products.
5.4 Use in Patients with Sickle Cell Disorders
Severe and sometimes fatal sickle cell crises can occur in patients with sickle cell disorders receiving pegfilgrastim products. Discontinue Fulphila if sickle cell crisis occurs.
5.5 Glomerulonephritis
Glomerulonephritis has occurred in patients receiving pegfilgrastim. The diagnoses were based upon azotemia, hematuria (microscopic and macroscopic), proteinuria, and renal biopsy. Generally, events of glomerulonephritis resolved after dose reduction or discontinuation of pegfilgrastim. If glomerulonephritis is suspected, evaluate for cause. If causality is likely, consider dose-reduction or interruption of Fulphila.
5.6 Leukocytosis
White blood cell (WBC) counts of 100 x 109/L or greater have been observed in patients receiving pegfilgrastim products. Monitoring of complete blood count (CBC) during Fulphila therapy is recommended.
5.7 Capillary Leak Syndrome
Capillary leak syndrome has been reported after G-CSF administration, including pegfilgrastim, and is characterized by hypotension, hypoalbuminemia, edema and hemoconcentration. Episodes vary in frequency, severity and may be life-threatening if treatment is delayed. Patients who develop symptoms of capillary leak syndrome should be closely monitored and receive standard symptomatic treatment, which may include a need for intensive care.
5.8 Potential for Tumor Growth Stimulatory Effects on Malignant Cells
The granulocyte colony-stimulating factor (G-CSF) receptor through which pegfilgrastim products and filgrastim products act has been found on tumor cell lines. The possibility that pegfilgrastim products act as a growth factor for any tumor type, including myeloid malignancies and myelodysplasia, diseases for which pegfilgrastim products are not approved, cannot be excluded.
5.9 Aortitis
Aortitis has been reported in patients receiving pegfilgrastim. It may occur as early as the first week after start of therapy. Manifestations may include generalized signs and symptoms such as fever, abdominal pain, malaise, back pain, and increased inflammatory markers (e.g., c-reactive protein and white blood cell count). Consider aortitis in patients who develop these signs and symptoms without known etiology. Discontinue Fulphila if aortitis is suspected.
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are discussed in greater detail in other sections of the labeling:
- Splenic Rupture [See Warnings and Precautions (5.1)]
- Acute Respiratory Distress Syndrome [See Warnings and Precautions (5.2)]
- Serious Allergic Reactions [See Warnings and Precautions (5.3)]
- Use in Patients with Sickle Cell Disorders [See Warnings and Precautions (5.4)]
- Glomerulonephritis [See Warnings and Precautions (5.5)]
- Leukocytosis [See Warnings and Precautions (5.6)]
- Capillary Leak Syndrome [See Warnings and Precautions (5.7)]
- Potential for Tumor Growth Stimulatory Effects on Malignant Cells [See Warnings and Precautions (5.8)]
- Aortitis [see Warnings and Precautions (5.9)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
Pegfilgrastim clinical trials safety data are based upon 932 patients receiving pegfilgrastim in seven randomized clinical trials. The population was 21 to 88 years of age and 92% female. The ethnicity was 75% Caucasian, 18% Hispanic, 5% Black, and 1% Asian. Patients with breast (n = 823), lung and thoracic tumors (n = 53) and lymphoma (n = 56) received pegfilgrastim after nonmyeloablative cytotoxic chemotherapy. Most patients received a single 100 mcg/kg (n = 259) or a single 6 mg (n = 546) dose per chemotherapy cycle over 4 cycles.
The following adverse reaction data in Table 2 are from a randomized, double-blind, placebo-controlled study in patients with metastatic or non-metastatic breast cancer receiving docetaxel 100 mg/m2 every 21 days (Study 3). A total of 928 patients were randomized to receive either 6 mg pegfilgrastim (n = 467) or placebo (n = 461). The patients were 21 to 88 years of age and 99% female. The ethnicity was 66% Caucasian, 31% Hispanic, 2% Black, and < 1% Asian, Native American, or other.
The most common adverse reactions occurring in ≥ 5% of patients and with a between-group difference of ≥ 5% higher in the pegfilgrastim arm in placebo-controlled clinical trials are bone pain and pain in extremity.
Table 2. Adverse Reactions with ≥ 5% Higher Incidence in Pegfilgrastim Patients Compared to Placebo in Study 3 Body System
Adverse Reaction
Placebo
(N = 461)
Pegfilgrastim 6 mg SC on Day 2
(N = 467)
Musculoskeletal and connective tissue disorders
Bone pain
26%
31%
Pain in extremity
4%
9%
6.2 Immunogenicity
As with all therapeutic proteins, there is a potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors, including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies in the studies described below with the incidence of antibodies in other studies or to other pegfilgrastim products may be misleading.
Binding antibodies to pegfilgrastim were detected using a BIAcore assay. The approximate limit of detection for this assay is 500 ng/mL. Pre-existing binding antibodies were detected in approximately 6% (51/849) of patients with metastatic breast cancer. Four of 521 pegfilgrastim-treated subjects who were negative at baseline developed binding antibodies to pegfilgrastim following treatment. None of these 4 patients had evidence of neutralizing antibodies detected using a cell-based bioassay.
6.3 Postmarketing Experience
The following adverse reactions have been identified during post approval use of pegfilgrastim products. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- Splenic rupture and splenomegaly (enlarged spleen) [see Warnings and Precautions (5.1)]
- Acute respiratory distress syndrome (ARDS) [see Warnings and Precautions (5.2)]
- Allergic reactions/hypersensitivity, including anaphylaxis, skin rash, and urticaria, generalized erythema, and flushing [see Warnings and Precautions (5.3)]
- Sickle cell crisis [see Warnings and Precautions (5.4)]
- Glomerulonephritis [see Warnings and Precautions (5.5)]
- Leukocytosis [see Warnings and Precautions (5.6)]
- Capillary Leak Syndrome [see Warnings and Precautions (5.7)]
- Injection site reactions
- Sweet’s syndrome, (acute febrile neutrophilic dermatosis), cutaneous vasculitis
- Aortitis [see Warnings and Precautions (5.9)]
- Alveolar hemorrhage
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Although available data with Fulphila or pegfilgrastim product use in pregnant women are insufficient to establish whether there is a drug associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes, there are available data from published studies in pregnant women exposed to filgrastim products. These studies have not established an association of filgrastim product use during pregnancy with major birth defects, miscarriage or adverse maternal or fetal outcomes.
In animal studies, no evidence of reproductive/developmental toxicity occurred in the offspring of pregnant rats that received cumulative doses of pegfilgrastim approximately 10 times the recommended human dose (based on body surface area). In pregnant rabbits, increased embryolethality and spontaneous abortions occurred at 4 times the maximum recommended human dose simultaneously with signs of maternal toxicity (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Data
Human Data
Retrospective studies indicate that exposure to pegfilgrastim is without significant adverse effect on fetal outcomes and neutropenia. Preterm deliveries have been reported in some patients.
Animal Data
Pregnant rabbits were dosed with pegfilgrastim subcutaneously every other day during the period of organogenesis. At cumulative doses ranging from the approximate human dose to approximately 4 times the recommended human dose (based on body surface area), the treated rabbits exhibited decreased maternal food consumption, maternal weight loss, as well as reduced fetal body weights and delayed ossification of the fetal skull; however, no structural anomalies were observed in the offspring from either study. Increased incidences of post-implantation losses and spontaneous abortions (more than half the pregnancies) were observed at cumulative doses approximately 4 times the recommended human dose, which were not seen when pregnant rabbits were exposed to the recommended human dose.
Three studies were conducted in pregnant rats dosed with pegfilgrastim at cumulative doses up to approximately 10 times the recommended human dose at the following stages of gestation: during the period of organogenesis, from mating through the first half of pregnancy, and from the first trimester through delivery and lactation. No evidence of fetal loss or structural malformations was observed in any study. Cumulative doses equivalent to approximately 3 and 10 times the recommended human dose resulted in transient evidence of wavy ribs in fetuses of treated mothers (detected at the end of gestation but no longer present in pups evaluated at the end of lactation).
8.2 Lactation
Risk Summary
There are no data on the presence of pegfilgrastim products in human milk, the effects on the breastfed child, or the effects on milk production. Other filgrastim products are secreted poorly into breast milk, and filgrastim products are not absorbed orally by neonates. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for Fulphila and any potential adverse effects on the breastfed child from Fulphila or from the underlying maternal condition.
8.4 Pediatric Use
The safety and effectiveness of pegfilgrastim have been established in pediatric patients. No overall differences in safety were identified between adult and pediatric patients based on postmarketing surveillance and review of the scientific literature.
Use of pegfilgrastim in pediatric patients for chemotherapy-induced neutropenia is based on adequate and well-controlled studies in adults with additional pharmacokinetic and safety data in pediatric patients with sarcoma [see Clinical Pharmacology (12.3) and Clinical Studies (14.1)].
-
10 OVERDOSAGE
Overdosage of pegfilgrastim products may result in leukocytosis and bone pain. Events of edema, dyspnea, and pleural effusion have been reported in a single patient who administered pegfilgrastim on 8 consecutive days in error. In the event of overdose, the patient should be monitored for adverse reactions [see Adverse Reactions (6)].
-
11 DESCRIPTION
Pegfilgrastim-jmdb is a covalent conjugate of recombinant methionyl human G-CSF and monomethoxypolyethylene glycol. Recombinant methionyl human G-CSF is a water-soluble 175 amino acid protein with a molecular weight of approximately 19 kilodaltons (kD). Recombinant methionyl human G-CSF is obtained from the bacterial fermentation of a strain of E coli transformed with a genetically engineered plasmid containing the human G-CSF gene. To produce pegfilgrastim-jmdb a 20 kD monomethoxypolyethylene glycol molecule is covalently bound to the N-terminal methionyl residue of recombinant methionyl human G-CSF. The average molecular weight of pegfilgrastim-jmdb is approximately 39 kD.
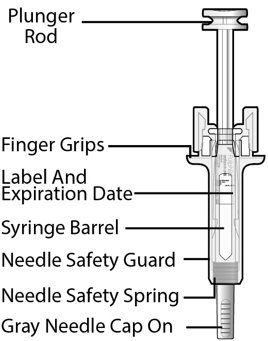
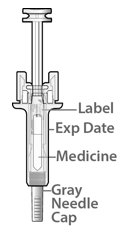
Fulphila (pegfilgrastim-jmdb) injection is intended for subcutaneous use only and is supplied in a single-dose prefilled syringe with a 29 gauge needle, with UltraSafe Passive Plus™ Needle Guard. The prefilled syringe does not bear graduation marks and is designed to deliver the entire contents of the syringe (6 mg/0.6 mL).
The delivered 0.6 mL dose from the prefilled syringe contains 6 mg pegfilgrastim-jmdb (based on protein mass only) in a sterile, clear, colorless, preservative-free solution (pH 4.0) containing acetate (0.7 mg), D-sorbitol (30 mg), polysorbate 20 (0.024 mg) and sodium (0.01 mg) in Water for Injection, USP.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Pegfilgrastim products are colony-stimulating factors that act on hematopoietic cells by binding to specific cell surface receptors, thereby stimulating proliferation, differentiation, commitment, and end cell functional activation.
12.2 Pharmacodynamics
Animal data and clinical data in humans suggest a correlation between pegfilgrastim products exposure and the duration of severe neutropenia as a predictor of efficacy. Selection of the dosing regimen of Fulphila is based on reducing the duration of severe neutropenia.
12.3 Pharmacokinetics
The pharmacokinetics of pegfilgrastim was studied in 379 patients with cancer. The pharmacokinetics of pegfilgrastim was nonlinear, and clearance decreased with increases in dose. Neutrophil receptor binding is an important component of the clearance of pegfilgrastim, and serum clearance is directly related to the number of neutrophils. In addition to numbers of neutrophils, body weight appeared to be a factor. Patients with higher body weights experienced higher systemic exposure to pegfilgrastim after receiving a dose normalized for body weight. A large variability in the pharmacokinetics of pegfilgrastim was observed. The half-life of pegfilgrastim ranged from 15 to 80 hours after subcutaneous injection.
Specific Populations
No gender-related differences were observed in the pharmacokinetics of pegfilgrastim, and no differences were observed in the pharmacokinetics of geriatric patients (≥ 65 years of age) compared with younger patients (< 65 years of age) [see Use in Specific Populations (8.5)].
Patients with Renal Impairment
In a study of 30 subjects with varying degrees of renal dysfunction, including end stage renal disease, renal dysfunction had no effect on the pharmacokinetics of pegfilgrastim.
Pediatric Patients with Cancer Receiving Myelosuppressive Chemotherapy
The pharmacokinetics and safety of pegfilgrastim were studied in 37 pediatric patients with sarcoma in Study 4 [see Clinical Studies 14.1]. The mean (± standard deviation [SD]) systemic exposure (AUC0-inf) of pegfilgrastim after subcutaneous administration at 100 mcg/kg was 47.9 (± 22.5) mcg·hr/mL in the youngest age group (0 to 5 years, n = 11), 22.0 (± 13.1) mcg·hr/mL in the (6 to 11 years age group (n = 10), and 29.3 (± 23.2) mcg·hr/mL in the 12 to 21 years age group (n = 13). The terminal elimination half-lives of the corresponding age groups were 30.1 (± 38.2) hours, 20.2 (± 11.3) hours, and 21.2 (± 16.0) hours, respectively.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No carcinogenicity or mutagenesis studies have been performed with pegfilgrastim products.
Pegfilgrastim did not affect reproductive performance or fertility in male or female rats at cumulative weekly doses approximately 6 to 9 times higher than the recommended human dose (based on body surface area).
-
14 CLINICAL STUDIES
14.1 Patients with Cancer Receiving Myelosuppressive Chemotherapy
Pegfilgrastim was evaluated in three randomized, double-blind, controlled studies. Studies 1 and 2 were active-controlled studies that employed doxorubicin 60 mg/m2 and docetaxel 75 mg/m2 administered every 21 days for up to 4 cycles for the treatment of metastatic breast cancer. Study 1 investigated the utility of a fixed dose of pegfilgrastim. Study 2 employed a weight-adjusted dose. In the absence of growth factor support, similar chemotherapy regimens have been reported to result in a 100% incidence of severe neutropenia (ANC < 0.5 x 109/L) with a mean duration of 5 to 7 days and a 30% to 40% incidence of febrile neutropenia. Based on the correlation between the duration of severe neutropenia and the incidence of febrile neutropenia found in studies with filgrastim, duration of severe neutropenia was chosen as the primary endpoint in both studies, and the efficacy of pegfilgrastim was demonstrated by establishing comparability to filgrastim-treated patients in the mean days of severe neutropenia.
In Study 1, 157 patients were randomized to receive a single subcutaneous injection of pegfilgrastim (6 mg) on day 2 of each chemotherapy cycle or daily subcutaneous filgrastim (5 mcg/kg/day) beginning on day 2 of each chemotherapy cycle. In Study 2, 310 patients were randomized to receive a single subcutaneous injection of pegfilgrastim (100 mcg/kg) on day 2 or daily subcutaneous filgrastim (5 mcg/kg/day) beginning on day 2 of each chemotherapy cycle.
Both studies met the major efficacy outcome measure of demonstrating that the mean days of severe neutropenia of pegfilgrastim-treated patients did not exceed that of filgrastim-treated patients by more than 1 day in cycle 1 of chemotherapy. The mean days of cycle 1 severe neutropenia in Study 1 were 1.8 days in the pegfilgrastim arm compared to 1.6 days in the filgrastim arm [difference in means 0.2 (95% CI -0.2, 0.6)] and in Study 2 were 1.7 days in the pegfilgrastim arm compared to 1.6 days in the filgrastim arm [difference in means 0.1 (95% CI -0.2, 0.4)].
A secondary endpoint in both studies was days of severe neutropenia in cycles 2 through 4 with results similar to those for cycle 1.
Study 3 was a randomized, double-blind, placebo-controlled study that employed docetaxel 100 mg/m2 administered every 21 days for up to 4 cycles for the treatment of metastatic or non-metastatic breast cancer. In this study, 928 patients were randomized to receive a single subcutaneous injection of pegfilgrastim (6 mg) or placebo on day 2 of each chemotherapy cycle. Study 3 met the major trial outcome measure of demonstrating that the incidence of febrile neutropenia (defined as temperature ≥ 38.2°C and ANC ≤ 0.5 x109/L) was lower for pegfilgrastim-treated patients as compared to placebo-treated patients (1% versus 17%, respectively, p < 0.001). The incidence of hospitalizations (1% versus 14%) and IV anti-infective use (2% versus 10%) for the treatment of febrile neutropenia was also lower in the pegfilgrastim-treated patients compared to the placebo-treated patients.
Study 4 was a multicenter, randomized, open-label study to evaluate the efficacy, safety, and pharmacokinetics [see Clinical Pharmacology (12.3)] of pegfilgrastim in pediatric and young adult patients with sarcoma. Patients with sarcoma receiving chemotherapy age 0 to 21 years were eligible. Patients were randomized to receive subcutaneous pegfilgrastim as a single-dose of 100 mcg/kg (n = 37) or subcutaneous filgrastim at a dose 5 mcg/kg/day (n = 6) following myelosuppressive chemotherapy. Recovery of neutrophil counts was similar in the pegfilgrastim and filgrastim groups. The most common adverse reaction reported was bone pain.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
Fulphila single-dose prefilled syringe for manual use
Fulphila (pegfilgrastim-jmdb) Injection is a clear, colorless solution supplied in a prefilled single-dose syringe for manual use containing 6 mg pegfilgrastim-jmdb, supplied with a 29 gauge, 1/2-inch needle with an UltraSafe Passive Plus™ Needle Guard.
Fulphila is provided in a dispensing pack containing one sterile 6 mg/0.6 mL prefilled syringe.
NDC: 67457-833-06
Fulphila prefilled syringe does not bear graduation marks and is intended only to deliver the entire contents of the syringe (6 mg/0.6 mL) for direct administration. Use of the prefilled syringe is not recommended for direct administration for pediatric patients weighing less than 45 kg who require doses that are less than the full contents of the syringe.
Store refrigerated between 2° to 8°C (36° to 46°F) in the carton to protect from light or physical damage. Do not shake. Discard syringes stored at room temperature for more than 72 hours. Avoid freezing; if frozen, thaw in the refrigerator before administration. Discard syringe if frozen more than once.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information and Instructions for Use).
Advise patients of the following risks and potential risks with Fulphila:
- Splenic rupture and splenomegaly [see Warnings and Precautions (5.1)]
- Acute Respiratory Distress Syndrome [see Warnings and Precautions (5.2)]
- Serious allergic reactions [see Warnings and Precautions (5.3)]
- Sickle cell crisis [see Warnings and Precautions (5.4)]
- Glomerulonephritis [see Warnings and Precautions (5.5)]
- Capillary Leak Syndrome [see Warnings and Precautions (5.7)]
- Aortitis [see Warnings and Precautions (5.9)]
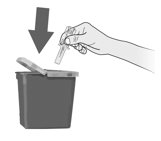
Instruct patients who self-administer Fulphila using the single-dose prefilled syringe of the:
- Importance of following the Instructions for Use.
- Dangers of reusing syringes.
- Importance of following local requirements for proper disposal of used syringes.
Manufactured by:
Mylan GmbH
Turmstrasse 24
6312 Steinhausen, SwitzerlandU.S. License No. 2062
-
Patient Information
Fulphila® (FULL-fil-ah)
(pegfilgrastim-jmdb)
Injection
Single-Dose Prefilled SyringeWhat is Fulphila?
Fulphila is a man-made form of granulocyte colony-stimulating factor (G-CSF). G-CSF is a substance produced by the body. It stimulates the growth of neutrophils, a type of white blood cell important in the body’s fight against infection.
Do not take Fulphila if you have had a serious allergic reaction to pegfilgrastim products or filgrastim products.
Before you receive Fulphila, tell your healthcare provider about all of your medical conditions, including if you:
- have a sickle cell disorder.
- have kidney problems.
- are pregnant or plan to become pregnant. It is not known if Fulphila will harm your unborn baby.
- are breastfeeding or plan to breastfeed. It is not known if Fulphila passes into your breast milk.
Tell your healthcare provider about all of the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
How will I receive Fulphila?
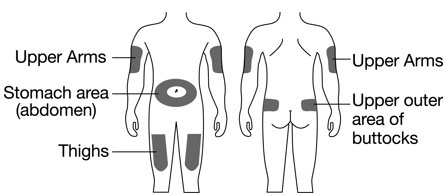
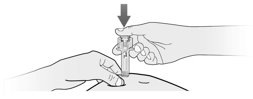
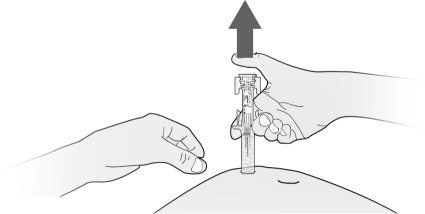
- Fulphila is given as an injection under your skin (subcutaneous injection) by a healthcare provider. If your healthcare provider decides that the subcutaneous injections can be given at home by you or your caregiver, follow the detailed “Instructions for Use” that comes with your Fulphila for information on how to prepare and inject a dose of Fulphila.
- You and your caregiver will be shown how to prepare and inject Fulphila before you use it.
- You should not inject a dose of Fulphila to children weighing less than 45 kg from a Fulphila prefilled syringe. A dose less than 0.6 mL (6 mg) cannot be accurately measured using the Fulphila prefilled syringe.
- If you are receiving Fulphila because you are also receiving chemotherapy, the last dose of Fulphila should be injected at least 14 days before and 24 hours after your dose of chemotherapy.
- If you miss a dose of Fulphila, talk to your healthcare provider about when you should give your next dose.
What are possible side effects of Fulphila?
Fulphila may cause serious side effects, including:
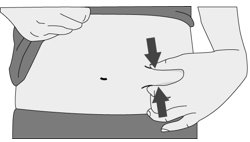
- Spleen rupture. Your spleen may become enlarged and can rupture. A ruptured spleen can cause death. Call your healthcare provider right away if you have pain in the left upper stomach area or your left shoulder.
- A serious lung problem called Acute Respiratory Distress Syndrome (ARDS). Call your healthcare provider or get emergency care right away if you have shortness of breath with or without a fever, trouble breathing, or a fast rate of breathing.
- Serious allergic reactions. Fulphila can cause serious allergic reactions. These reactions can cause a rash over your whole body, shortness of breath, wheezing, dizziness, swelling around your mouth or eyes, fast heart rate, and sweating. If you have any of these symptoms, stop using Fulphila and call your healthcare provider or get emergency medical help right away.
- Sickle cell crises. You may have a serious sickle cell crisis if you have a sickle cell disorder and receive Fulphila. Serious sickle cell crises have happened in people with sickle cell disorders receiving pegfilgrastim that has sometimes led to death. Call your healthcare provider right away if you have symptoms of sickle cell crisis such as pain or difficulty breathing.
-
Kidney injury (glomerulonephritis). Fulphila can cause kidney injury. Call your healthcare provider right away if you develop any of the following symptoms:
- swelling of your face or ankles
- blood in your urine or dark colored urine
- you urinate less than usual
- Increased white blood cell count (leukocytosis). Your healthcare provider will check your blood during treatment with Fulphila.
-
Capillary Leak Syndrome. Fulphila can cause fluid to leak from blood vessels into your body’s tissues. This condition is called “Capillary Leak Syndrome” (CLS). CLS can quickly cause you to have symptoms that may become life-threatening. Get emergency medical help right away if you develop any of the following symptoms:
- swelling or puffiness and are urinating less than usual
- trouble breathing
- swelling of your stomach-area (abdomen) and feeling of fullness
- dizziness or feeling faint
- a general feeling of tiredness
- Inflammation of the aorta (aortitis). Inflammation of the aorta (the large blood vessel which transports blood from the heart to the body) has been reported in patients who received pegfilgrastim. Symptoms may include fever, abdominal pain, feeling tired, and back pain. Call your healthcare provider if you experience these symptoms.
The most common side effects of Fulphila are pain in the bones, arms, and legs.
These are not all the possible side effects of Fulphila.
Call your health care provider for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store Fulphila?
- Store Fulphila in the refrigerator between 36°F to 46°F (2°C to 8°C).
- Take Fulphila out of the refrigerator 30 minutes before use and allow it to reach room temperature before preparing an injection.
- Avoid freezing. If Fulphila is accidently frozen, allow the prefilled syringe to thaw in the refrigerator before injecting.
- Do not use a Fulphila prefilled syringe that has been frozen more than 1 time. Use a new Fulphila prefilled syringe.
- Keep the prefilled syringe in the original carton to protect from light or physical damage.
- Do not shake the prefilled syringe.
- Throw away (dispose of) any Fulphila that has been left at room temperature, 68°F to 77ºF (20°C to 25ºC) for more than 72 hours or frozen more than 1 time.
Keep the Fulphila prefilled syringe out of the reach of children.
General information about the safe and effective use of Fulphila.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use Fulphila for a condition for which it was not prescribed. Do not give Fulphila to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about Fulphila that is written for health professionals.
What are the ingredients in Fulphila?
Active ingredient: pegfilgrastim-jmdb
Inactive ingredients: acetate, D-sorbitol, polysorbate 20, and sodium in Water for Injection.
Manufactured by: Mylan GmbH, Turmstrasse 24, 6312 Steinhausen, Switzerland U.S. License No. 2062
Product of India. Code No.: KR/DRUGS/KTK/28D/7/2006 Distributed by: Mylan Institutional LLC, Rockford, IL 61103 U.S.A.
For more information, go to www.fulphila.com or call 1-833-695-2623.
This Patient Information has been approved by the U.S. Food and Drug Administration. Revised: 3/2019
Manufactured by:
Mylan GmbH
Turmstrasse 24
6312 Steinhausen, SwitzerlandU.S. License No. 2062
Product of India
Code No.: KR/DRUGS/KTK/28D/7/2006
Distributed by:
Mylan Institutional LLC
Rockford, IL 61103 U.S.A.Revised: 5/2019
B:PEGFIL:R3This Instructions for Use has been approved by the U.S. Food and Drug Administration.
Manufactured by:
Mylan GmbH
Turmstrasse 24
6312 Steinhausen, SwitzerlandU.S. License No. 2062
Code No.: KR/DRUGS/KTK/28D/7/2006
Revised: 3/2019
B:IFU:PEGFIL:R3BF1638/02
-
PACKAGE/LABEL PRINCIPAL DISPLAY PANEL
NDC: 67457-833-06 Rx only
Fulphila
(pegfilgrastim-jmdb)
Injection
6 mg/0.6 mLPegylated Recombinant Methionyl Human
Granulocyte Colony-Stimulating Factor
(PEG-r-metHuG-CSF) derived from E ColiFor Subcutaneous Use Only
Sterile Solution - No PreservativeOne 0.6 mL Single-Dose
Prefilled SyringeEach 0.6 mL prefilled syringe contains: 6 mg pegfilgrastim
(based on protein mass only) in a sterile, clear, colorless,
preservative-free solution (pH 4.0) containing acetate (0.7 mg),
D-sorbitol (30 mg), polysorbate 20 (0.024 mg), and sodium
(0.01 mg) in water for injection, USP.No U.S. standard of potency
Store refrigerated at 2° to 8°C (36° to 46°F) in original
carton to Protect from Light. Do Not Freeze or Shake.
Keep this and all medication out of the reach of children.Dosage: See prescribing information for dosage and
instructions for use.Manufactured by:
Mylan GmbH
Turmstrasse 24
6312 Steinhausen, Switzerland
U.S. License No. 2062Distributed by:
Mylan Institutional LLC
Rockford, IL 61103 U.S.A.
Product of IndiaMylan.com
B:833:1C:R6
Code No.: KR/DRUGS/KTK/28D/7/2006 -
INGREDIENTS AND APPEARANCE
FULPHILA
pegfilgrastim injectionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 67457-833 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength PEGFILGRASTIM (UNII: 3A58010674) (PEGFILGRASTIM - UNII:3A58010674) PEGFILGRASTIM 6 mg in 0.6 mL Inactive Ingredients Ingredient Name Strength ACETIC ACID (UNII: Q40Q9N063P) POLYSORBATE 20 (UNII: 7T1F30V5YH) SODIUM HYDROXIDE (UNII: 55X04QC32I) SORBITOL (UNII: 506T60A25R) WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 67457-833-06 1 in 1 CARTON 07/09/2018 1 0.6 mL in 1 SYRINGE; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761075 07/09/2018 Labeler - Mylan Institutional LLC (790384502)

Trademark Results [Fulphila]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 FULPHILA 86970982 5571031 Live/Registered |
Mylan Institutional Inc. 2016-04-11 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.