TEFLARO- ceftaroline fosamil powder, for solution
Teflaro by
Drug Labeling and Warnings
Teflaro by is a Prescription medication manufactured, distributed, or labeled by Allergan, Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use TEFLARO safely and effectively. See full prescribing information for TEFLARO.
TEFLARO ® (ceftaroline fosamil) for injection, for intravenous use
Initial U.S. Approval: 2010
INDICATIONS AND USAGE
Teflaro® is a cephalosporin antibacterial indicated in adult and pediatric patients for the treatment of the following infection caused by designated susceptible bacteria:
- Acute bacterial skin and skin structure infections (ABSSSI) in adult and pediatric patients (at least 34 weeks gestational age and 12 days postnatal age) (1.1)
- Community-acquired bacterial pneumonia (CABP) in adult and pediatric patients 2 months of age and older (1.2)
To reduce the development of drug-resistant bacteria and maintain the effectiveness of Teflaro and other antibacterial drugs, Teflaro should be used only to treat or prevent infections that are proven or strongly suspected to be caused by bacteria (1.3).
------------------------DOSAGE AND ADMINISTRATION-----------------
Dosage of Teflaro by Indication in Adult and Pediatric Patients (2.1, 2.2)
Indication Age Range Dosage Infusion Time Duration Acute Bacterial Skin and Skin Structure Infections (ABSSSI) 18 years and older 600 mg every 12 hours 5 to 60 minutes 5 to 14 days ≥2 years to < 18 years (> 33 kg) 400 mg every 8 hours
OR
600 mg every 12 hours5 to 60 minutes 5 to 14 days ≥2 years to < 18 years (≤ 33kg) 12 mg/kg every 8 hours 5 to 60 minutes 5 to 14 days 2 months to < 2 years 8 mg/kg every 8 hours 5 to 60 minutes 5 to 14 days 0* to < 2 months 6 mg/kg every 8 hours 30 to 60 minutes 5 to 14 days *Gestational age 34 weeks and older and postnatal age 12 days and older
Indication Age Range Dosage Infusion Time Duration Community Acquired Bacterial Pneumonia (CABP) 18 years and older 600 mg every 12 hours 5 to 60 minutes 5 to 7 days ≥2 years to < 18 years (> 33 kg) 400 mg every 8 hours
OR
600 mg every 12 hours5 to 60 minutes 5 to 14 days ≥2 years to < 18 years (≤ 33kg) 12 mg/kg every 8 hours 5 to 60 minutes 5 to 14 days 2 months to < 2 years 8 mg/kg every 8 hours 5 to 60 minutes 5 to 14 days DOSAGE FORMS AND STRENGTHS
For Injection: 600 mg or 400 mg of sterile ceftaroline fosamil powder in single-dose 20 mL vials. The powder is constituted and further diluted for intravenous injection. (3)
CONTRAINDICATIONS
- Known serious hypersensitivity to ceftaroline or other members of the cephalosporin class. (4)
WARNINGS AND PRECAUTIONS
- Serious hypersensitivity (anaphylactic) reactions have been reported with beta-lactam antibacterial drugs, including Teflaro. If a hypersensitivity reaction occurs, discontinue Teflaro. (5.1)
- Clostridium difficile-associated diarrhea (CDAD) has been reported with nearly all systemic antibacterial agents, including Teflaro. Evaluate if diarrhea occurs. (5.2)
- Direct Coombs’ test seroconversion has been reported with Teflaro. If anemia develops during or after therapy, a diagnostic workup for drug-induced hemolytic anemia should be performed and consideration given to discontinuation of Teflaro. (5.3)
ADVERSE REACTIONS
The most common adverse reactions occurring in >2% of adult patients and ≥3% of pediatric patients are diarrhea, nausea, and rash. Additional adverse reactions that occurred in ≥3% of pediatric patients include vomiting and pyrexia. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Allergan at 1-800-678-1605 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.See 17 for PATIENT COUNSELING INFORMATION.
Revised: 9/2019
- Acute bacterial skin and skin structure infections (ABSSSI) in adult and pediatric patients (at least 34 weeks gestational age and 12 days postnatal age) (1.1)
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1. INDICATIONS AND USAGE
1.1 Acute Bacterial Skin and Skin Structure Infections
1.2 Community-Acquired Bacterial Pneumonia
1.3 Usage
2. DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage in Adult Patients
2.2 Recommended Dosage in Pediatric Patients
2.3 Dosage Adjustments in Patients with Renal Impairment
2.4 Preparation of Teflaro for Administration
2.5 Storage of Constituted Solutions
2.6 Drug Compatibilities
3. DOSAGE FORMS AND STRENGTHS
4. CONTRAINDICATIONS
5. WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity Reactions
5.2 Clostridium difficile-Associated Diarrhea
5.3 Direct Coombs’ Test Seroconversion
5.4 Development of Drug-Resistant Bacteria
6. ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
8. USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Patients with Renal Impairment
10. OVERDOSAGE
11. DESCRIPTION
12. CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.4 Microbiology
13. NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14. CLINICAL STUDIES
14.1 Acute Bacterial Skin and Skin Structure Infections (ABSSSI)
14.2 Community-Acquired Bacterial Pneumonia (CABP)
16. HOW SUPPLIED/STORAGE AND HANDLING
17. PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1.
INDICATIONS AND USAGE
1.1 Acute Bacterial Skin and Skin Structure Infections
Teflaro is indicated in adult and pediatric patients (at least 34 weeks gestational age and 12 days postnatal age) for the treatment of acute bacterial skin and skin structure infections (ABSSSI) caused by susceptible isolates of the following Gram-positive and Gram-negative microorganisms: Staphylococcus aureus (including methicillin-susceptible and -resistant isolates), Streptococcus pyogenes, Streptococcus agalactiae, Escherichia coli, Klebsiella pneumoniae, and Klebsiella oxytoca [see Dosage and Administration (2.2) and Use in Specific Populations (8.4)].
1.2 Community-Acquired Bacterial Pneumonia
Teflaro is indicated in adult and pediatric patients 2 months of age and older for the treatment of community-acquired bacterial pneumonia (CABP) caused by susceptible isolates of the following Gram-positive and Gram-negative microorganisms: Streptococcus pneumoniae (including cases with concurrent bacteremia), Staphylococcus aureus (methicillin-susceptible isolates only), Haemophilus influenzae, Klebsiella pneumoniae, Klebsiella oxytoca, and Escherichia coli.
1.3 Usage
To reduce the development of drug-resistant bacteria and maintain the effectiveness of Teflaro and other antibacterial drugs, Teflaro should be used to treat or prevent infections that are proven or strongly suspected to be caused by susceptible bacteria. Appropriate specimens for microbiological examination should be obtained in order to isolate and identify the causative pathogens and to determine their susceptibility to ceftaroline. When culture and susceptibility information are available, they should be considered in selecting or modifying antibacterial therapy. In the absence of such data, local epidemiology and susceptibility patterns may contribute to the empiric selection of therapy.
-
2.
DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage in Adult Patients
The recommended dosage of Teflaro is 600 mg administered every 12 hours by intravenous (IV) infusion over 5 to 60 minutes in patients ≥ 18 years of age. The duration of therapy should be guided by the severity and site of infection and the patient’s clinical and bacteriological progress.
The recommended dosage and administration by infection is described in Table 1.
Table 1: Dosage of Teflaro by Indication in Adults Indication Dosage Frequency Infusion Time
Recommended Duration of Treatment Acute Bacterial Skin and Skin Structure Infections (ABSSSI) 600 mg Every 12 hours 5 to 60 minutes 5-14 days Community-Acquired Bacterial Pneumonia (CABP) 600 mg Every 12 hours 5 to 60 minutes 5-7 days 2.2 Recommended Dosage in Pediatric Patients
The recommended dosage of Teflaro in pediatric patients is based on the age and weight of the child. The duration of therapy should be guided by the severity, site of infection and the patient’s clinical and bacteriological progress.
Pediatric Patients 2 Months of Age and Older
- For pediatric patients 2 months of age and older, Teflaro is administered every 8 hours by intravenous infusion over 5 to 60 minutes.
- Teflaro dosing regimen is dependent on the type of infection (ABSSSI, CABP). See dosing table 2 below.
Table 2: Dosage of Teflaro by Indication in Pediatric Patients 2 Months of Age and Older Indication Age Range Dosage and Frequency Infusion time Recommended Duration of Treatment Acute Bacterial Skin and Skin Structure Infections (ABSSSI)
OR
Community-Acquired Bacterial Pneumonia (CABP)
2 months to < 2 years 8 mg/kg every 8 hours 5 to 60 minutes 5-14 days > 2 years to < 18 years (< 33 kg) 12 mg/kg every 8 hours > 2 years to < 18 years (> 33 kg) 400 mg every 8 hours
OR
600 mg every 12 hoursPediatric Patients Less Than 2 Months of Age
- Teflaro is administered every 8 hours by intravenous infusion over 30 to 60 minutes for patients less than 2 months of age.
- Teflaro dosing regimen is only recommended for patients with ABSSSI. See dosing Table 3 below.
- Concentrations of Teflaro in the cerebrospinal fluid have not been evaluated [see Use in Specific Populations (8.4)].
- There is no information for dosing Teflaro in infants less than 34 weeks gestational age and less than 12 days postnatal age.
Table 3: Dosage of Teflaro in Pediatric Patients less Than 2 Months of Age Indication Age Range Dosage and Frequency Infusion time Recommended Duration of Treatment Acute Bacterial Skin and Skin Structure Infections (ABSSSI)
0* to < 2 months 6 mg/kg every 8 hours 30 to 60 minutes 5-14 days *Gestational age 34 weeks and older and postnatal age 12 days and older.
2.3 Dosage Adjustments in Patients with Renal Impairment
Adults: No dosage adjustment is required in adult patients with CrCL > 50 mL/min. The dose in adult patients should be adjusted when creatinine clearance (CrCL) is < 50 mL/min as shown below (see Table 4).
Table 4: Dosage of Teflaro in Adult Patients with Renal Impairment Estimated CrCla (mL/min) Recommended Dosage Regimen for Teflaro > 50 No dosage adjustment necessary > 30 to ≤ 50 400 mg IV (over 5 to 60 minutes) every 12 hours ≥ 15 to ≤ 30 300 mg IV (over 5 to 60 minutes) every 12 hours End-stage renal disease,
including hemodialysisb200 mg IV (over 5 to 60 minutes) every 12 hoursc a Creatinine clearance (CrCl) estimated using the Cockcroft-Gault formula.
b End-stage renal disease is defined as CrCl < 15 mL/min.
c Teflaro is hemodialyzable; thus Teflaro should be administered after hemodialysis on hemodialysis days.
Pediatrics: No dosage adjustment is required in pediatric patients with CrCL > 50 mL/min/1.73 m2, estimated using the Schwartz equation. There is insufficient information to recommend a dosage regimen for pediatric patients with CrCL < 50 mL/min/1.73 m2.
2.4 Preparation of Teflaro for Administration
Constitution of Teflaro Powder for Injection
Aseptic technique must be followed in preparing the infusion solution. The contents of Teflaro vial should be constituted with 20 mL Sterile Water for Injection, USP; or 0.9% of sodium chloride injection (normal saline); or 5% of dextrose injection; or lactated ringer’s injection. Constitution time is less than 2 minutes. Mix gently to constitute and check to see that the contents have dissolved completely. The preparation of Teflaro solutions is summarized in Table 5.
Table 5: Preparation of Teflaro for Intravenous Use Dosage Strength
(mg)Volume of Diluent To Be Added
(mL)Approximate Ceftaroline fosamil Concentration
(mg/mL)Amount to Be Withdrawn 400 20 20 Adults: Total Volume
Pediatric*: Volume based on age and weight600 20 30 Adults: Total Volume
Pediatric*: Volume based on age and weight* The recommended dosage of Teflaro is based on the age and weight of the child. See Table 2
Dilution of the Constituted Solution of Teflaro
The constituted solution must be further diluted in a range between 50 mL to 250 mL before intravenous infusion into patients. Use the same diluent used for constitution of the powder for this further dilution, unless sterile water for injection was used earlier. If sterile water for injection was used earlier, then appropriate infusion solutions include: 0.9% Sodium Chloride Injection, USP (normal saline); 5% Dextrose Injection, USP; 2.5% Dextrose Injection, USP, and 0.45% Sodium Chloride Injection, USP; or Lactated Ringer’s Injection, USP.
Dilution of the Constituted Solution of Teflaro in the 50 mL Infusion Bags Only
Preparation of 600 mg of Teflaro dose in 50 mL infusion bag (for adult patients): Withdraw 20 mL of diluent from the infusion bag. Proceed to inject entire content of the Teflaro vial into the bag to provide a total volume of 50 mL. The resultant concentration is approximately 12 mg/mL.
Preparation of 400 mg of Teflaro dose in 50 mL infusion bag (for adult patients or pediatric patients weighing > 33 kg): Withdraw 20 mL of diluent from the infusion bag. Proceed to inject entire content of the Teflaro vial into the bag to provide a total volume of 50 mL. The resultant concentration is approximately 8 mg/mL.
Preparation of Teflaro dose in the infusion bag (for pediatric patients weighing ≤ 33 kg): The amount of solution withdrawn from the constituted Teflaro vial for pediatric patients weighing < 33 kg for dilution in the infusion bag will vary according to the weight and age of the child. The infusion solution concentration for administration should not exceed 12 mg/ml ceftaroline fosamil.
The color of Teflaro infusion solutions ranges from clear, light to dark yellow depending on the concentration and storage conditions. When stored as recommended, the product potency is not affected. Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit.
2.5 Storage of Constituted Solutions
Stability in Baxter® Mini-Bag Plus™: Solutions of Teflaro in concentrations ranging from 4 to 12 mg/mL in Baxter Mini-Bag Plus containers with 0.9% Sodium Chloride Injection may be stored for up to 6 hours at room temperature or for up to 24 hours at 2°C to 8°C (36°F to 46°F). Stability testing in the Baxter Mini-Bag Plus has solely been conducted on 50 mL and 100 mL containers (0.9% Sodium Chloride Injection).
Stability in Infusion Bag: Studies have shown that the constituted solution in the infusion bag should be used within 6 hours when stored at room temperature or within 24 hours when stored under refrigeration at 2 to 8º C (36 to 46º F).
- For pediatric patients 2 months of age and older, Teflaro is administered every 8 hours by intravenous infusion over 5 to 60 minutes.
-
3.
DOSAGE FORMS AND STRENGTHS
For Injection: Teflaro is supplied as 600 mg or 400 mg of sterile ceftaroline fosamil (equivalent to 668 mg and 446 mg, respectively, of ceftaroline fosamil monoacetate monohydrate) powder in single-dose, 20 mL clear glass vials. The powder is constituted and further diluted for intravenous injection.
- 4. CONTRAINDICATIONS
-
5.
WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity Reactions
Serious and occasionally fatal hypersensitivity (anaphylactic) reactions and serious skin reactions have been reported in patients receiving beta-lactam antibacterial drugs. Before therapy with Teflaro is instituted, careful inquiry about previous hypersensitivity reactions to other cephalosporins, penicillins, or carbapenems should be made. Maintain clinical supervision if this product is to be given to a penicillin- or other beta-lactam-allergic patient, because cross sensitivity among beta-lactam antibacterial agents has been clearly established.
If an allergic reaction to Teflaro occurs, discontinue Teflaro and institute appropriate treatment and supportive measures.
5.2 Clostridium difficile-Associated Diarrhea
Clostridium difficile-associated diarrhea (CDAD) has been reported for nearly all systemic antibacterial agents, including Teflaro, and may range in severity from mild diarrhea to fatal colitis.
Treatment with antibacterial agents alters the normal flora of the colon and may permit overgrowth of C. difficile.
C. difficile produces toxins A and B which contribute to the development of CDAD. Hypertoxin-producing strains of C. difficile cause increased morbidity and mortality, as these infections can be refractory to antimicrobial therapy and may require colectomy. CDAD must be considered in all patients who present with diarrhea following antibiotic use. Careful medical history is necessary because CDAD has been reported to occur more than 2 months after the administration of antibacterial agents.
If CDAD is suspected or confirmed, antibacterials not directed against C. difficile should be discontinued, if possible. Appropriate fluid and electrolyte management, protein supplementation, antibiotic treatment of C. difficile, and surgical evaluation should be instituted as clinically indicated [see Adverse Reactions (6.1)].
5.3 Direct Coombs’ Test Seroconversion
Seroconversion from a negative to a positive direct Coombs’ test result occurred in 120/1114 (10.8%) of adult patients receiving Teflaro and 49/1116 (4.4%) of patients receiving comparator drugs in the four pooled adult Phase 3 trials.
In the pooled adult Phase 3 CABP trials, 51/520 (9.8%) of Teflaro-treated patients compared to 24/534 (4.5%) of ceftriaxone-treated patients seroconverted from a negative to a positive direct Coombs’ test result. No adverse reactions representing hemolytic anemia were reported in any treatment group.
Seroconversion from a negative to a positive direct Coombs’ test result occurred in 42/234 (17.9%) of children receiving Teflaro and 3/93 (3.2%) of patients receiving comparator drugs in the three pooled pediatric trials. No adverse reactions representing hemolytic anemia were reported in any treatment group.
If anemia develops during or after treatment with Teflaro, drug-induced hemolytic anemia should be considered. Diagnostic studies including a direct Coombs’ test, should be performed. If drug-induced hemolytic anemia is suspected, discontinuation of Teflaro should be considered and supportive care should be administered to the patient (i.e. transfusion) if clinically indicated.
-
6.
ADVERSE REACTIONS
The following serious adverse reactions are described in greater detail in the Warnings and Precautions section
- Hypersensitivity Reactions [see Warnings and Precautions (5.1)]
-
Clostridium difficile-Associated diarrhea [see Warnings and Precautions (5.2)]
- Direct Coombs’ Test Seroconversion [see Warnings and Precautions (5.3)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in clinical trials of a drug cannot be compared directly to rates from clinical trials of another drug and may not reflect rates observed in practice.
Adult Patients
Teflaro was evaluated in four controlled comparative Phase 3 clinical trials (two in ABSSSI and two in CABP) which included 1300 adult patients treated with Teflaro (600 mg administered by IV over 1 hour every 12h) and 1297 patients treated with comparator (vancomycin plus aztreonam or ceftriaxone) for a treatment period up to 21 days. The median age of patients treated with Teflaro was 54 years, ranging between 18 and 99 years old. Patients treated with Teflaro were predominantly male (63%) and Caucasian (82%).
Serious Adverse Reactions and Adverse Reactions Leading to Discontinuation
In the four pooled adult Phase 3 clinical trials, serious adverse reactions (SARs) occurred in 98/1300 (7.5%) of patients receiving Teflaro and 100/1297 (7.7%) of patients receiving comparator drugs. Treatment discontinuation due to adverse reactions occurred in 35/1300 (2.7%) of patients receiving Teflaro and 48/1297 (3.7%) of patients receiving comparator drugs with the most common adverse reactions leading to discontinuation being hypersensitivity for both treatment groups at a rate of 0.3% in the Teflaro group and 0.5% in comparator group.
Most Common Adverse Reactions
No adverse reactions occurred in greater than 5% of adult patients receiving Teflaro. The most common adverse reactions occurring in > 2% of patients receiving Teflaro in the pooled adult phase 3 clinical trials were diarrhea, nausea, and rash.
Table 6 lists adverse reactions occurring in ≥ 2% of patients receiving Teflaro in the pooled adult Phase 3 clinical trials.
Table 6: Adverse Reactions Occurring in ≥ 2% of Patients Receiving Teflaro in the Pooled Adult Phase 3 Clinical Trials Adverse Reactions Pooled Phase 3 Clinical Trials
(four trials, two in ABSSSI and two in CABP)Teflaro
(N=1300)Pooled Comparatorsa
(N=1297)Gastrointestinal Disorders Diarrhea 5 % 3 % Nausea 4 % 4 % Constipation 2 % 2 % Vomiting 2 % 2 % Laboratory Investigations Increased transaminases 2% 3 % Metabolism and Nutrition disorders Hypokalemia 2 % 3 % Skin and Subcutaneous Tissue Disorders Rash 3% 2% Vascular Disorders Phlebitis 2% 1% a Comparators included vancomycin 1 gram IV every 12h plus aztreonam 1 gram IV every 12h in the Phase 3 ABSSSI trials, and ceftriaxone 1 gram IV every 24h in the Phase 3 CABP trials.
Other Adverse Reactions Observed During Clinical Trials of Teflaro
Following is a list of additional adverse reactions reported by the 1740 adult patients who received Teflaro in any clinical trial with incidences less than 2%.
Blood and lymphatic system disorders - Anemia, Eosinophilia, Neutropenia, Thrombocytopenia
Cardiac disorders - Bradycardia, Palpitations
Gastrointestinal disorders - Abdominal pain
General disorders and administration site conditions - Pyrexia
Hepatobiliary disorders - Hepatitis
Immune system disorders - Hypersensitivity, Anaphylaxis
Infections and infestations - Clostridium difficile colitis
Metabolism and nutrition disorders - Hyperglycemia, Hyperkalemia
Nervous system disorders - Dizziness, Convulsion
Renal and urinary disorders - Renal failure
Skin and subcutaneous tissue disorders - Urticaria
Pediatric Patients
Teflaro was evaluated in three clinical trials (one in ABSSSI and two in CABP) which included 257 pediatric patients 2 months to < 18 years of age treated with Teflaro, and 102 patients treated with comparator agents for a treatment period up to 21 days. In two trials, one in ABSSSI and one in CABP, the dose was selected to result in exposures comparable to adult exposure with 600 mg administered by IV infusion every 12h. In an additional pediatric trial in complicated CABP the dose was higher. The median age of pediatric patients treated with Teflaro was 5 years, ranging from 2 months to < 18 years of age. Patients treated with Teflaro were predominantly male (55%) and Caucasian (92%).
A single study enrolled 11 pediatric patients with a gestational age of ≥34 weeks and a postnatal age of 12 days to less than 2 months of age. The safety findings were similar to those observed in adult and pediatric patients 2 months of age and older.
Serious Adverse Reactions and Adverse Reactions Leading to Discontinuation
In the three pooled pediatric clinical trials, SARs occurred in 10/257 (4%) of patients receiving Teflaro and 3/102 (3%) of patients receiving comparator drugs. Treatment discontinuation due to adverse reactions occurred in 10/257 (3.9%) of patients receiving Teflaro and 2/102 (2%) of patients receiving comparator drugs with the most common adverse reaction leading to discontinuation being rash in 2/257 (0.8%) of patients treated with Teflaro.
Most Common Adverse Reactions
No adverse reactions occurred in greater than 8% of pediatric patients receiving Teflaro. The most common adverse reactions occurring in ≥ 3% of patients receiving Teflaro in the pooled pediatric clinical trials were diarrhea, nausea, vomiting, pyrexia and rash.
Table 7 lists adverse reactions occurring in ≥ 3% of patients receiving Teflaro in the pooled pediatric clinical trials.
Table 7: Adverse Reactions Occurring in ≥ 3% of Patients Receiving Teflaro in the Pooled Pediatric Clinical Trials Adverse Reactions Pooled Pediatric Clinical Trials
(three trials, one in ABSSSI and two in CABP)Teflaro
(N=257)Pooled Comparatorsa
(N=102)Gastrointestinal Disorders Diarrhea 8 % 10 % Nausea 3 % 1 % Vomiting 5 % 12 % General and Administrative Site disorders Pyrexia 3% 2 % Skin and Subcutaneous Tissue Disorders Rash 7% 4% a Comparators included vancomycin or cefazolin with or without aztreonam in the ABSSSI trial and ceftriaxone alone or ceftriaxone plus vancomycin in the CABP trials
Following is a list of additional adverse reactions reported by the 257 patients who received Teflaro in the pediatric clinical trials with incidences less than 3%.
Investigations – Alanine aminotransferase increased, Aspartate aminotransferase increased
Nervous system disorders – Headache
Skin and subcutaneous tissue disorders- Pruritus
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of Teflaro in adult patients. Because these adverse reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Blood and lymphatic system disorders: Agranulocytosis, leukopenia, eosinophilic pneumonia.
- Hypersensitivity Reactions [see Warnings and Precautions (5.1)]
-
8.
USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no adequate studies with Teflaro in pregnant women that informed any drug associated risks. The background risk of major birth defects and miscarriage for the indicated population is unknown. The background risk of major birth defects is 2-4% and of miscarriage is 15-20% of clinically recognized pregnancies within the general population.
In developmental toxicity studies conducted in animals, no malformations or other adverse developmental effects were observed in offspring of rats exposed to Teflaro at up to 4 times the maximum recommended human dose (MRHD) during the period of organogenesis through lactation. In rabbits exposed to Teflaro during organogenesis at levels approximately equal to the MRHD, no drug-induced fetal malformations were observed despite maternal toxicity.
Data
Animal Data
Developmental toxicity studies performed with ceftaroline fosamil in rats at IV doses up to 300 mg/kg demonstrated no maternal toxicity and no effects on the fetus. A separate toxicokinetic study showed that ceftaroline exposure in rats (based on AUC) at this dose level was approximately 4 times the exposure in humans given 600 mg every 12 hours. There were no drug-induced malformations in the offspring of rabbits given IV doses of 25, 50, and 100 mg/kg, despite maternal toxicity. Signs of maternal toxicity appeared secondary to the sensitivity of the rabbit gastrointestinal system to broad-spectrum antibacterials and included changes in fecal output in all groups and dose-related reductions in body weight gain and food consumption at > 50 mg/kg; these were associated with an increase in spontaneous abortion at 50 and 100 mg/kg. The highest dose was also associated with maternal moribundity and mortality. An increased incidence of a common rabbit skeletal variation, angulated hyoid alae, was also observed at the maternally toxic doses of 50 and 100 mg/kg. A separate toxicokinetic study showed that ceftaroline exposure in rabbits (based on AUC) was approximately 0.4 times the exposure in humans given 600 mg every 12 hours at 25 mg/kg and 0.7 times the human exposure at 50 mg/kg.
Ceftaroline fosamil did not affect the postnatal development or reproductive performance of the offspring of rats given IV doses up to 450 mg/kg/day. Results from a toxicokinetic study conducted in pregnant rats with doses up to 300 mg/kg suggest that exposure was ≥ 4 times the exposure in humans given 600 mg every 12 hours.
8.2 Lactation
Risk Summary
No data is available regarding the presence of ceftaroline in human milk, the effects of ceftaroline on breastfed infants, or the effects on milk production.
The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for Teflaro and any potential adverse effects on the breastfed child from Teflaro or from the underlying maternal condition.
8.4 Pediatric Use
The safety and effectiveness of Teflaro in the treatment of ABSSSI have been established in pediatric patients (at least 34 weeks gestational age and 12 days postnatal age).
The safety and effectiveness of Teflaro in the treatment of CABP have been established in the age groups 2 months to less than 18 years old.
Use of Teflaro in these age groups is supported by evidence from adequate and well-controlled studies of Teflaro in adults with additional pharmacokinetic and safety data in pediatric patients 2 months of age and older with ABSSSI or CABP [see Clinical Studies (14.1 and 14.2)]. Use of Teflaro in pediatric patients less than 2 months of age was supported by pharmacokinetic and safety data in 11 infants at least 34 weeks gestational age and 12 days postnatal age. In these infants, concentrations of Teflaro in the cerebrospinal fluid were not evaluated [see Adverse Reactions (6.1), Clinical Pharmacology (12.3) and Clinical Studies (14.2)].
Results from the clinical studies in pediatric patients show that Teflaro demonstrated a safety profile that was comparable with treatment of ABSSSI and CABP in adults at the clinical dosages studied.
Safety and effectiveness of Teflaro in pediatric patients less than 34 weeks gestational age and less than 12 days postnatal age for the treatment of ABSSSI have not been established.
Safety and effectiveness of Teflaro in pediatric patients below the age of 2 months for the treatment of CABP have not been established as no data are available.
8.5 Geriatric Use
Of the 1300 adult patients treated with Teflaro in the Phase 3 ABSSSI and CABP trials, 397 (30.5%) were ≥ 65 years of age. The clinical cure rates in the Teflaro group (Clinically Evaluable [CE] Population) were similar in patients ≥ 65 years of age compared with patients < 65 years of age in both the ABSSSI and CABP trials.
The adverse reaction profiles in patients ≥ 65 years of age and in patients < 65 years of age were similar. The percentage of patients in the Teflaro group who had at least one adverse reaction was 52.4% in patients ≥ 65 years of age and 42.8% in patients < 65 years of age for the two indications combined.
Ceftaroline is excreted primarily by the kidney, and the risk of adverse reactions may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection in this age group and it may be useful to monitor renal function. Elderly subjects had greater ceftaroline exposure relative to non-elderly subjects when administered the same single dose of Teflaro. However, higher exposure in elderly subjects was mainly attributed to age-related changes in renal function. Dosage adjustment for elderly patients should be based on renal function [see Dosage and Administration (2.2) and Clinical Pharmacology (12.3)].
8.6 Patients with Renal Impairment
Dosage adjustment is required in adult patients with moderate (CrCl > 30 to ≤ 50 mL/min) or severe (CrCl ≥ 15 to ≤ 30 mL/min) renal impairment and in patients with end-stage renal disease (ESRD – defined as CrCl < 15 mL/min), including patients on hemodialysis (HD). There is insufficient information to recommend a dosage regimen for pediatric patients with CrCl < 50 ml/min [see Dosage and Administration (2.2) and Clinical Pharmacology (12.3)].
-
10.
OVERDOSAGE
In the event of overdose, Teflaro should be discontinued and general supportive treatment given.
Ceftaroline can be removed by hemodialysis. In subjects with ESRD administered 400 mg of Teflaro, the mean total recovery of ceftaroline in the dialysate following a 4-hour hemodialysis session started 4 hours after dosing was 76.5 mg (21.6% of the dose). However, no information is available on the use of hemodialysis to treat overdosage [see Clinical Pharmacology (12.3)].
-
11.
DESCRIPTION
Teflaro is a sterile, semi-synthetic, prodrug of the cephalosporin antibacterial class of beta-lactams (β-lactams). Chemically, the prodrug, ceftaroline fosamil monoacetate monohydrate is (6R,7R)-7-{(2Z)-2-(ethoxyimino)-2-[5-(phosphonoamino)-1,2,4-thiadiazol-3-yl]acetamido}-3-{[4-(1-methylpyridin-1-ium-4-yl)-1,3-thiazol-2-yl]sulfanyl}-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylate monoacetate monohydrate. Its molecular weight is 762.75. The empirical formula is C22H21N8O8PS4.C2H4O2.H2O.
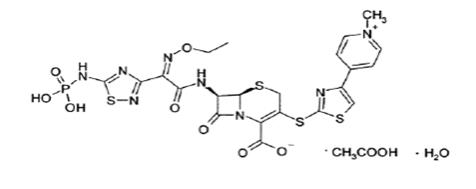
Figure 1: Chemical structure of ceftaroline fosamil
Teflaro vials contain either 600 mg or 400 mg of anhydrous ceftaroline fosamil (equivalent to 668 mg and 446 mg, respectively, of ceftaroline fosamil monoacetate monohydrate). The powder for injection is formulated from ceftaroline fosamil monoacetate monohydrate, a pale yellowish-white to light yellow sterile powder. All references to ceftaroline activity are expressed in terms of the prodrug, ceftaroline fosamil. The powder is constituted for IV injection [see Dosage and Administration (2.3)].
Each vial of Teflaro contains ceftaroline fosamil and L-arginine, which results in a constituted solution at pH 4.8 to 6.5.
-
12.
CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Ceftaroline is a cephalosporin antibacterial drug [see Microbiology (12.4)].
12.2 Pharmacodynamics
As with other beta-lactam antimicrobial agents, the time that unbound plasma concentration of ceftaroline exceeds the minimum inhibitory concentration (MIC) of the infecting organism has been shown to best correlate with efficacy in a neutropenic murine thigh infection model with S. aureus and S. pneumoniae.
Exposure-response analysis of Phase 2/3 ABSSSI trials supports the recommended dosage regimen of Teflaro 600 mg every 12 hours by IV infusion over 1 hour. For Phase 3 CABP trials, an exposure-response relationship could not be identified due to the limited range of ceftaroline exposures in the majority of patients.
Cardiac Electrophysiology
In a randomized, positive- and placebo-controlled crossover thorough QTc study, 54 healthy subjects were each administered a single dose of Teflaro 1500 mg, placebo, and a positive control by IV infusion over 1 hour. At the 1500 mg dose of Teflaro, no significant effect on QTc interval was detected at peak plasma concentration or at any other time.
12.3 Pharmacokinetics
The mean pharmacokinetic parameters of ceftaroline in healthy adults (n=6) with normal renal function after single and multiple 1-hour IV infusions of 600 mg ceftaroline fosamil administered every 12 hours are summarized in Table 8. Pharmacokinetic parameters were similar for single and multiple dose administration.
Table 8: Mean (Standard Deviation) Pharmacokinetic Parameters of Ceftaroline IV in Healthy Adults Parameter Single 600 mg Dose Administered as a 1-Hour Infusion (n=6) Multiple 600 mg Doses Administered Every 12 Hours as 1-Hour Infusions for 14 Days (n=6) Cmax (mcg/mL) 19.0 (0.71) 21.3 (4.10) Tmax (h)a 1.00 (0.92-1.25) 0.92 (0.92-1.08) AUC (mcgh/mL) b 56.8 (9.31) 56.3 (8.90) T1/2 (h) 1.60 (0.38) 2.66 (0.40) CL (L/h) 9.58 (1.85) 9.60 (1.40) a Reported as median (range)
b AUC0-∞, for single-dose administration; AUC0-tau, for multiple-dose administration; Cmax, maximum observed concentration; Tmax, time of Cmax; AUC0-∞, area under concentration-time curve from time 0 to infinity; AUC0-tau, area under concentration-time curve over dosing interval (0-12 hours); T1/2, terminal elimination half-life; CL, plasma clearanceThe Cmax and AUC of ceftaroline increase approximately in proportion to dose within the single dose range of 50 to 1000 mg. No appreciable accumulation of ceftaroline is observed following multiple IV infusions of 600 mg administered every 12 hours for up to 14 days in healthy adults with normal renal function.
The systemic exposure (AUC), T1/2, and clearance of ceftaroline were similar following administration of 600 mg ceftaroline fosamil in a volume of 50 mL to healthy subjects every 8 hours for 5 days as a 5-minute or 60-minute infusion, and the Tmax of ceftaroline occurred about 5 minutes after the end of the ceftaroline fosamil infusion for both infusion durations. The mean (SD) Cmax of ceftaroline was 32.5 (4.82) mcg/mL for the 5-minute infusion duration (n=11) and 17.4 (3.87) mcg/mL for the 60-minute infusion duration (n=12).
Distribution
The average binding of ceftaroline to human plasma proteins is approximately 20% and decreases slightly with increasing concentrations over 1-50 mcg/mL (14.5-28.0%). The median (range) steady-state volume of distribution of ceftaroline in healthy adult males (n=6) following a single 600 mg IV dose of radiolabeled ceftaroline fosamil was 20.3 L (18.3-21.6 L), similar to extracellular fluid volume.
Elimination
Metabolism
Ceftaroline fosamil is the water-soluble prodrug of the bioactive ceftaroline. Ceftaroline fosamil is converted into bioactive ceftaroline in plasma by a phosphatase enzyme and concentrations of the prodrug are measurable in plasma primarily during IV infusion. Hydrolysis of the beta-lactam ring of ceftaroline occurs to form the microbiologically inactive, open-ring metabolite ceftaroline M-1. The mean (SD) plasma ceftaroline M-1 to ceftaroline AUC0-∞ ratio following a single 600 mg IV infusion of ceftaroline fosamil in healthy adults (n=6) with normal renal function is 28% (3.1%).
When incubated with pooled human liver microsomes, ceftaroline was metabolically stable (< 12% metabolic turnover), indicating that ceftaroline is not a substrate for hepatic CYP450 enzymes.
Excretion
Ceftaroline and its metabolites are primarily eliminated by the kidneys. Following administration of a single 600 mg IV dose of radiolabeled ceftaroline fosamil to healthy male adults (n=6), approximately 88% of radioactivity was recovered in urine and 6% in feces within 48 hours. Of the radioactivity recovered in urine approximately 64% was excreted as ceftaroline and approximately 2% as ceftaroline M-1. The mean (SD) renal clearance of ceftaroline was 5.56 (0.20) L/h, suggesting that ceftaroline is predominantly eliminated by glomerular filtration.
Specific Populations
Patients with Renal Impairment
Following administration of a single 600 mg IV dose of Teflaro, the geometric mean AUC0-∞ of ceftaroline in subjects with mild (CrCl > 50 to ≤ 80 mL/min, n=6) or moderate (CrCl > 30 to ≤ 50 mL/min, n=6) renal impairment was 19% and 52% higher, respectively, compared to healthy subjects with normal renal function (CrCl > 80 mL/min, n=6). Following administration of a single 400 mg IV dose of Teflaro, the geometric mean AUC0-∞ of ceftaroline in subjects with severe (CrCl ≥ 15 to ≤30 mL/min, n=6) renal impairment was 115% higher compared to healthy subjects with normal renal function (CrCl > 80 mL/min, n=6). Dosage adjustment is recommended in patients with moderate and severe renal impairment [see Dosage and Administration (2.2)].
A single 400 mg dose of Teflaro was administered to subjects with ESRD (n=6) either 4 hours prior to or 1 hour after hemodialysis (HD). The geometric mean ceftaroline AUC0-∞ following the post-HD infusion was 167% higher compared to healthy subjects with normal renal function (CrCl > 80 mL/min, n=6). The mean recovery of ceftaroline in the dialysate following a 4-hour HD session was 76.5 mg, or 21.6% of the administered dose. Dosage adjustment is recommended in patients with ESRD (defined as CrCL < 15 mL/min), including patients on HD [see Dosage and Administration (2.2)].
Patients with Hepatic Impairment
The pharmacokinetics of ceftaroline in patients with hepatic impairment have not been established. As ceftaroline does not appear to undergo significant hepatic metabolism, the systemic clearance of ceftaroline is not expected to be significantly affected by hepatic impairment.
Geriatric Patients
Following administration of a single 600 mg IV dose of Teflaro to healthy elderly subjects (≥ 65 years of age, n=16), the geometric mean AUC0-∞ of ceftaroline was ~33% higher compared to healthy young adult subjects (18-45 years of age, n=16). The difference in AUC0-∞ was mainly attributable to age-related changes in renal function. Dosage adjustment for Teflaro in elderly patients should be based on renal function [see Dosage and Administration (2.2)].
Pediatric Patients
The pharmacokinetics of ceftaroline were evaluated in adolescent patients (ages 12 to 17, n=7) with normal renal function following administration of a single 8 mg/kg IV dose of Teflaro (or 600 mg for subjects weighing > 75 kg). The mean plasma clearance and terminal phase volume of distribution for ceftaroline in adolescent subjects were similar to healthy adults (n=6) with normal renal function in a separate study following administration of a single 600 mg IV dose. However, the mean Cmax and AUC0-∞ for ceftaroline in adolescent subjects who received a single 8 mg/kg dose were 10% and 23% less than in healthy adult subjects who received a single 600 mg IV dose. The population pharmacokinetic analyses demonstrated that the pharmacokinetics of ceftaroline in pediatric patients from 2 months to < 18 years of age were similar to those in adult patients after accounting for weight and maturational changes. No clinically significant differences in ceftaroline AUC were predicted in patients from 12 days to 2 months postnatal age and with ≥34 weeks of gestational age compared to adults and pediatric patients 2 months of age and older when given the approved recommended dosage for each patient population. [see Adverse Reactions (6), Use in Specific Populations (8.4) and Clinical Studies (14)].
Gender
Following administration of a single 600 mg IV dose of Teflaro to healthy elderly males (n=10) and females (n=6) and healthy young adult males (n=6) and females (n=10), the mean Cmax and AUC0-∞ for ceftaroline were similar between males and females, although there was a trend for higher Cmax (17%) and AUC0-∞ (6-15%) in female subjects. Population pharmacokinetic analysis did not identify any significant differences in ceftaroline AUC0-tau based on gender in Phase 2/3 patients with ABSSSI or CABP. No dose adjustment is recommended based on gender.
Race
A population pharmacokinetic analysis was performed to evaluate the impact of race on the pharmacokinetics of ceftaroline using data from Phase 2/3 adult ABSSSI and CABP trials. No significant differences in ceftaroline AUC0-tau was observed across White (n=35), Hispanic (n=34), and Black (n=17) race groups for ABSSSI patients. Patients enrolled in CABP trials were predominantly categorized as White (n=115); thus there were too few patients of other races to draw any conclusions. No dosage adjustment is recommended based on race.
Drug Interactions Studies
No clinical drug-drug interaction studies have been conducted with Teflaro. There is minimal potential for drug-drug interactions between Teflaro and CYP450 substrates, inhibitors, or inducers; drugs known to undergo active renal secretion; and drugs that may alter renal blood flow.
In vitro studies in human liver microsomes indicate that ceftaroline does not inhibit the major cytochrome P450 isoenzymes CYP1A1, CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1 and CYP3A4. In vitro studies in human hepatocytes also demonstrate that ceftaroline and its inactive open-ring metabolite are not inducers of CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, or CYP3A4/5. Therefore, Teflaro is not expected to inhibit or induce the clearance of drugs that are metabolized by these metabolic pathways in a clinically relevant manner.
Population pharmacokinetic analysis did not identify any clinically relevant differences in ceftaroline exposure (Cmax and AUC0-tau) in Phase 2/3 patients with ABSSSI or CABP who were taking concomitant medications that are known inhibitors, inducers, or substrates of the cytochrome P450 system; anionic or cationic drugs known to undergo active renal secretion; and vasodilator or vasoconstrictor drugs that may alter renal blood flow.
12.4 Microbiology
Mechanism of Action
Ceftaroline is a cephalosporin antibacterial drug with in vitro activity against Gram-positive and -negative bacteria. The bactericidal action of ceftaroline is mediated through binding to essential penicillin-binding proteins (PBPs). Ceftaroline is bactericidal against S. aureus due to its affinity for PBP2a and against Streptococcus pneumoniae due to its affinity for PBP2x.
Resistance
Ceftaroline is not active against Gram-negative bacteria producing extended spectrum beta-lactamases (ESBLs) from the TEM, SHV or CTX-M families, serine carbapenemases (such as KPC), class B metallo-beta-lactamases, or class C (AmpC cephalosporinases). Although cross-resistance may occur, some isolates resistant to other cephalosporins may be susceptible to ceftaroline.
Interaction with Other Antimicrobials
In vitro studies have not demonstrated any antagonism between ceftaroline or other commonly used antibacterial agents (e.g., vancomycin, linezolid, daptomycin, levofloxacin, azithromycin, amikacin, aztreonam, tigecycline, and meropenem).
Antimicrobial Activity
Ceftaroline has been shown to be active against most of the following bacteria, both in vitro and in clinical infections [see Indications and Usage (1)].
Skin Infections
Gram-positive Bacteria
Staphylococcus aureus (including methicillin-susceptible and -resistant isolates)
Streptococcus pyogenes
Streptococcus agalactiae
Gram-negative Bacteria
Escherichia coli
Klebsiella pneumoniae
Klebsiella oxytoca
Community-Acquired Bacterial Pneumonia (CABP)
Gram-positive Bacteria
Streptococcus pneumoniae
Staphylococcus aureus (methicillin-susceptible isolates only)
Gram-negative Bacteria
Haemophilus influenzae
Klebsiella pneumoniae
Klebsiella oxytoca
Escherichia coli
The following in vitro data are available, but their clinical significance is unknown. At least 90 percent of the following bacteria exhibit an in vitro minimum inhibitory concentration (MIC) less than or equal to the susceptible breakpoint for ceftaroline against isolates of similar genus or organism group. However, the efficacy of ceftaroline in treating clinical infections due to these bacteria has not been established in adequate and well-controlled clinical trials.
Gram-positive Bacteria
Streptococcus dysgalactiae
Gram-negative Bacteria
Citrobacter koseri
Citrobacter freundii
Enterobacter cloacae
Enterobacter aerogenes
Moraxella catarrhalis
Morganella morganii
Proteus mirabilis
Haemophilus parainfluenzae
Susceptibility Test Methods
For specific information regarding susceptibility test interpretive criteria and associated test methods and quality control standards recognized by FDA for this drug, please see: https://www.fda.gov/STICm
-
13.
NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term carcinogenicity studies have not been conducted with ceftaroline. Ceftaroline fosamil did not show evidence of mutagenic activity in in vitro tests that included a bacterial reverse mutation assay and the mouse lymphoma assay. Ceftaroline was not mutagenic in an in vitro mammalian cell assay. In vivo, ceftaroline fosamil did not induce unscheduled DNA synthesis in rat hepatocytes and did not induce the formation of micronucleated erythrocytes in mouse or rat bone marrow. Both ceftaroline fosamil and ceftaroline were clastogenic in the absence of metabolic activation in an in vitro chromosomal aberration assays, but not in the presence of metabolic activation.
IV injection of ceftaroline fosamil had no adverse effects on fertility of male and female rats given up to 450 mg/kg. This is approximately 4-fold higher than the maximum recommended human dose based on body surface area.
-
14.
CLINICAL STUDIES
14.1 Acute Bacterial Skin and Skin Structure Infections (ABSSSI)
Adult Patients
A total of 1396 adults with clinically documented complicated skin and skin structure infection were enrolled in two identical randomized, multi-center, multinational, double-blind, non-inferiority trials (Trials 1 and 2) comparing Teflaro (600 mg administered IV over 1 hour every 12 hours) to vancomycin plus aztreonam (1 g vancomycin administered IV over 1 hour followed by 1 g aztreonam administered IV over 1 hour every 12 hours). Treatment duration was 5 to 14 days. A switch to oral therapy was not allowed. The Modified Intent-to-Treat (MITT) population included all patients who received any amount of study drug according to their randomized treatment group. The Clinically Evaluable (CE) population included patients in the MITT population who demonstrated sufficient adherence to the protocol.
To evaluate the treatment effect of ceftaroline, an analysis was conducted in 797 patients with ABSSSI (such as deep/extensive cellulitis or a wound infection [surgical or traumatic]) for whom the treatment effect of antibacterials may be supported by historical evidence. This analysis evaluated responder rates based on achieving both cessation of lesion spread and absence of fever on Study Day 3 in the following subgroup of patients:
Patients with lesion size ≥ 75 cm2 and having one of the following infection types:
- Major abscess with ≥ 5 cm of surrounding erythema
- Wound infection
- Deep/extensive cellulitis
The results of this analysis are shown in Table 9.
Table 9: Clinical Responders at Study Day 3 from Two Adult Phase 3 ABSSSI Trials Teflaro
n/N (%)Vancomycin/
Aztreonam
n/N (%)Treatment Difference
(2-sided 95% CI)ABSSSI Trial 1 148/200 (74.0) 135/209 (64.6) 9.4 (0.4, 18.2) ABSSSI Trial 2 148/200 (74.0) 128/188 (68.1) 5.9 (-3.1, 14.9) The protocol-specified analyses included clinical cure rates at the Test of Cure (TOC) (visit 8 to 15 days after the end of therapy) in the co-primary CE and MITT populations (Table 10) and clinical cure rates at TOC by pathogen in the Microbiologically Evaluable (ME) population (Table 11). However, there are insufficient historical data to establish the magnitude of drug effect for antibacterial drugs compared with placebo at a TOC time point. Therefore, comparisons of Teflaro to vancomycin plus aztreonam based on clinical response rates at TOC cannot be utilized to establish non-inferiority.
Table 10: Clinical Cure Rates at TOC from Two Adult Phase 3 ABSSSI Trials Teflaro
n/N (%)Vancomycin/
Aztreonam
n/N (%)Treatment Difference
(2-sided 95% CI)Trial 1 CE 288/316 (91.1) 280/300 (93.3) -2.2 (-6.6, 2.1) MITT 304/351 (86.6) 297/347 (85.6) 1.0 (-4.2, 6.2) Trial 2 CE 271/294 (92.2) 269/292 (92.1) 0.1 (-4.4., 4.5) MITT 291/342 (85.1) 289/338 (85.5) -0.4 (-5.8, 5.0) Table 11: Clinical Cure Rates at TOC by Pathogen from Two Adult Integrated Phase 3 ABSSSI Trials Teflaro
n/N (%)Vancomycin/Aztreonam
n/N (%)Gram-positive:
MSSA (methicillin-susceptible)
MRSA (methicillin-resistant)212/228 (93.0%)
142/152 (93.4%)225/238 (94.5%)
115/122 (94.3%)Streptococcus pyogenes 56/56 (100%) 56/58 (96.6%) Streptococcus agalactiae 21/22 (95.5%) 18/18 (100%) Gram-negative:
Escherichia coli20/21 (95.2%) 19/21 (90.5%) Klebsiella pneumoniae 17/18 (94.4%) 13/14 (92.9%) Klebsiella oxytoca 10/12 (83.3%) 6/6 (100%) Of the 693 patients in the MITT population in the Teflaro arm in the two ABSSSI trials, 20 patients had baseline S. aureus bacteremia (nine MRSA and eleven MSSA). Thirteen of these twenty patients (65%) were clinical responders for ABSSSI at Study Day 3 and 18/20 (90%) were considered clinical success for ABSSSI at TOC.
Pediatric Patients
The ABSSSI pediatric trial was a randomized, parallel-group, active controlled trial in pediatric patients 2 months to < 18 years of age.
A total of 163 children from 2 months to < 18 years of age with clinically documented ABSSSI were enrolled in a randomized, multi-center, multinational, parallel group, active controlled trial comparing Teflaro to vancomycin or cefazolin (each with optional aztreonam). Treatment duration was 5 to 14 days. A switch to oral therapy with either cephalexin, clindamycin, or linezolid after Study Day 3 was allowed. The Modified Intent-to-Treat (MITT) population included all patients who received any amount of study drug according to their randomized treatment group.
The primary objective was to evaluate the safety and tolerability of Teflaro. The study was not powered for comparative inferential efficacy analysis, and no efficacy endpoint was identified as primary.
To evaluate the treatment effect of Teflaro, an analysis was conducted in 159 patients with ABSSSI in the MITT population. This analysis evaluated responder rates based on achieving both cessation of lesion spread and absence of fever on Study Day 3.
The clinical response at Study Day 3 was 80.4% (86/107) for the ceftaroline group and 75.0% (39/52) for the comparator group, with a treatment difference of 5.4% (95% CI of –7.8, 20.3).
Clinical cure rates at test of cure visit (8 to 15 days after the end of therapy) for the ABSSSI pediatric trial were 94.4% (101/107) for Teflaro and 86.5% (45/52) for the comparator, with a treatment difference of 7.9 (95% CI –1.2, 20.2). Indeterminate outcomes occurred at rates of 5.6% (6/107) for the ceftaroline group and 11.5% (6/52) for the comparator group, and rates of clinical failure were 0% (0/107) for the ceftaroline group and 1.9% (1/52) for the comparator group.
The safety and effectiveness of Teflaro were evaluated in a single study that enrolled 11 pediatric patients with a gestational age of ≥34 weeks and a postnatal age of 12 days to less than 2 months of age with known or suspected infections. The majority of patients (8 of 11) received 6 mg/kg Teflaro every 8 hours as an intravenous (IV) infusion over 60 minutes.
14.2 Community-Acquired Bacterial Pneumonia (CABP)
Adult Patients
A total of 1231 adults with a diagnosis of CABP were enrolled in two randomized, multi-center, multinational, double-blind, non-inferiority trials (Trials 1 and 2) comparing Teflaro (600 mg administered IV over 1 hour every 12 hours) with ceftriaxone (1 g ceftriaxone administered IV over 30 minutes every 24 hours). In both treatment groups of CABP Trial 1, two doses of oral clarithromycin (500 mg every 12 hours), were administered as adjunctive therapy starting on Study Day 1. No adjunctive macrolide therapy was used in CABP Trial 2. Patients with known or suspected MRSA were excluded from both trials. Patients with new or progressive pulmonary infiltrate(s) on chest radiography and signs and symptoms consistent with CABP with the need for hospitalization and IV therapy were enrolled in the trials. Treatment duration was 5 to 7 days. A switch to oral therapy was not allowed. Among all subjects who received any amount of study drug in the two CABP trials, the 30-day all-cause mortality rates were 11/609 (1.8%) for the Teflaro group vs. 12/610 (2.0%) for the ceftriaxone group, and the difference in mortality rates was not statistically significant.
To evaluate the treatment effect of ceftaroline, an analysis was conducted in CABP patients for whom the treatment effect of antibacterials may be supported by historical evidence. The analysis endpoint required subjects to meet sign and symptom criteria at Day 4 of therapy: a responder had to both (a) be in stable condition, based on temperature, heart rate, respiratory rate, blood pressure, oxygen saturation, and mental status; (b) show improvement from baseline on at least one symptom of cough, dyspnea, pleuritic chest pain, or sputum production, while not worsening on any of these four symptoms. The analysis used a microbiological intent-to-treat population (mITT population) containing only subjects with a confirmed bacterial pathogen at baseline. Results for this analysis are presented in Table 12.
Table 12: Response Rates at Study Day 4 (72-96 hours) from Two Adult Phase 3 CABP Trials Teflaro
n/N (%)Ceftriaxone
n/N (%)Treatment Difference
(2-sided 95% CI)CABP Trial 1 48/69 (69.6%) 42/72 (58.3%) 11.2 (-4.6,26.5) CABP Trial 2 58/84 (69.0%) 51/83 (61.4%) 7.6 (-6.8,21.8) The protocol-specified analyses included clinical cure rates at the TOC (8 to 15 days after the end of therapy) in the co-primary Modified Intent-to-Treat Efficacy (MITTE) and CE populations (Table 13) and clinical cure rates at TOC by pathogen in the Microbiologically Evaluable (ME) population (Table 14). However, there are insufficient historical data to establish the magnitude of drug effect for antibacterials drugs compared with placebo at a TOC time point. Therefore, comparisons of Teflaro to ceftriaxone based on clinical response rates at TOC cannot be utilized to establish non-inferiority. Neither trial established that Teflaro was statistically superior to ceftriaxone in terms of clinical response rates. The MITTE population included all patients who received any amount of study drug according to their randomized treatment group and were in PORT (Pneumonia Outcomes Research Team) Risk Class III or IV. The CE population included patients in the MITTE population who demonstrated sufficient adherence to the protocol.
Table 13: Clinical Cure Rates at TOC from Two Adult Phase 3 CABP Trials Teflaro
n/N (%)Ceftriaxone
n/N (%)Treatment Difference
(2-sided 95% CI)CABP Trial 1 CE 194/224 (86.6%) 183/234 (78.2%) 8.4 (1.4, 15.4) MITTE 244/291 (83.8%) 233/300 (77.7%) 6.2 (-0.2, 12.6) CABP Trial 2 CE 191/232 (82.3%) 165/214 (77.1%) 5.2 (-2.2, 12.8) MITTE 231/284 (81.3%) 203/269 (75.5%) 5.9 (-1.0, 12.8) Table 14: Clinical Cure Rates at TOC by Pathogen from Two Adult Integrated Phase 3 CABP Trials Teflaro
n/N (%)Ceftriaxone
n/N (%)Gram-positive:
Streptococcus pneumoniae54/63 (85.7%) 41/59 (69.5%) Staphylococcus aureus
(methicillin-susceptible isolates only)18/25 (72.0%) 14/25 (56.0%) Gram-negative:
Haemophilus influenzae15/18 (83.3%) 17/20 (85.0%) Klebsiella pneumoniae 12/12 (100%) 10/12 (83.3%) Klebsiella oxytoca 5/6 (83.3%) 7/8 (87.5%) Escherichia coli 10/12 (83.3%) 9/12 (75.0%) Pediatric Patients
The CABP pediatric trial was a randomized, parallel-group, active controlled trial in pediatric patients 2 months to < 18 years of age.
A total of 161 children with a diagnosis of CABP were enrolled in a randomized, multi-center, multinational, active controlled trial comparing Teflaro with ceftriaxone. Patients with new or progressive pulmonary infiltrate(s) on chest radiography and signs and symptoms consistent with CABP including acute onset or worsening symptoms of cough, tachypnea, sputum production, grunting, chest pain, cyanosis, or increased work of breathing with the need for hospitalization and IV therapy were enrolled in the trial. Treatment duration was 5 to 14 days. A switch to oral therapy with amoxicillin clavulanate was allowed on Study Day 4.
The primary objective was to evaluate the safety and tolerability of Teflaro. The study was not powered for comparative inferential efficacy analysis, and no efficacy endpoint was identified as primary.
To evaluate the treatment effect of Teflaro, an analysis was conducted in 143 patients with CABP in the MITT population. This analysis evaluated responder rates at Study Day 4 based on achieving improvement in at least 2 out of 7 symptoms (cough, dyspnea, chest pain, sputum production, chills, feeling of warmth / feverish and exercise intolerance or lethargy) and have worsening in none of these symptoms.
The clinical response at Study Day 4 was 69.2% (74/107) for Teflaro and 66.7% (24/36) for the comparator, with a treatment difference of 2.5% (95% CI of –13.9, 20.9).
Clinical cure rates at test of cure were 87.9% (94/107) for Teflaro and 88.9% (32/36) for the comparator, with a treatment difference of -1.0 (95% CI –11.5, 14.1).
- Major abscess with ≥ 5 cm of surrounding erythema
-
16.
HOW SUPPLIED/STORAGE AND HANDLING
Teflaro (ceftaroline fosamil) for injection is supplied in single-dose, clear glass vials containing:
- 600 mg - individual vial (NDC: 0456-0600-01) and carton containing 10 vials (NDC: 0456-0600-10)
- 400 mg - individual vial (NDC: 0456-0400-01) and carton containing 10 vials (NDC: 0456-0400-10)
Teflaro vials (unreconstituted) should be stored at 25ºC (77ºF); excursions permitted to 15-30ºC (59-86ºF) [see USP Controlled Room Temperature].
- 600 mg - individual vial (NDC: 0456-0600-01) and carton containing 10 vials (NDC: 0456-0600-10)
-
17.
PATIENT COUNSELING INFORMATION
- Advise patients that allergic reactions, including serious allergic reactions, could occur and that serious reactions require immediate treatment. They should inform their healthcare provider about any previous hypersensitivity reactions to Teflaro, other beta-lactams (including cephalosporins) or other allergens.
- Patients should be counseled that antibacterial drugs including Teflaro should only be used to treat bacterial infections. They do not treat viral infections (e.g., the common cold). When Teflaro is prescribed to treat a bacterial infection, patients should be told that although it is common to feel better early in the course of therapy, the medication should be taken exactly as directed. Skipping doses or not completing the full course of therapy may (1) decrease the effectiveness of the immediate treatment and (2) increase the likelihood that bacteria will develop resistance and will not be treatable by Teflaro or other antibacterial drugs in the future.
- Advise patients that diarrhea is a common problem caused by antibacterial drugs, including Teflaro, and usually resolves when the drug is discontinued. Sometimes, frequent watery or bloody diarrhea may occur and may be a sign of a more serious intestinal infection. If severe watery or bloody diarrhea develops, patients should contact their healthcare provider.
Distributed by:
Allergan USA, Inc.
Madison, NJ 07940Teflaro® is a registered trademark of Allergan Sales, LLC. All other trademarks are the property of their respective owners.
Made in Italy
© 2019 Allergan. All rights reserved.
v2.0USPI0600
- Advise patients that allergic reactions, including serious allergic reactions, could occur and that serious reactions require immediate treatment. They should inform their healthcare provider about any previous hypersensitivity reactions to Teflaro, other beta-lactams (including cephalosporins) or other allergens.
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
TEFLARO
ceftaroline fosamil powder, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0456-0400 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength CEFTAROLINE FOSAMIL (UNII: 7P6FQA5D21) (CEFTAROLINE - UNII:H36Z0FHR8K) CEFTAROLINE FOSAMIL 400 mg in 20 mL Inactive Ingredients Ingredient Name Strength ARGININE (UNII: 94ZLA3W45F) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0456-0400-10 10 in 1 CARTON 10/29/2010 1 NDC: 0456-0400-01 20 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA200327 10/29/2010 TEFLARO
ceftaroline fosamil powder, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0456-0600 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength CEFTAROLINE FOSAMIL (UNII: 7P6FQA5D21) (CEFTAROLINE - UNII:H36Z0FHR8K) CEFTAROLINE FOSAMIL 600 mg in 20 mL Inactive Ingredients Ingredient Name Strength ARGININE (UNII: 94ZLA3W45F) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0456-0600-10 10 in 1 CARTON 10/29/2010 1 NDC: 0456-0600-01 20 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA200327 10/29/2010 Labeler - Allergan, Inc. (144796497)
Trademark Results [Teflaro]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 TEFLARO 85073011 3949340 Live/Registered |
ALLERGAN SALES, LLC 2010-06-28 |
 TEFLARO 85018295 3935814 Live/Registered |
ALLERGAN SALES, LLC 2010-04-20 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.