PRECEDEX- dexmedetomidine hydrochloride injection, solution
Precedex by
Drug Labeling and Warnings
Precedex by is a Prescription medication manufactured, distributed, or labeled by Hospira, Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use PRECEDEX safely and effectively. See full prescribing information for PRECEDEX.
PRECEDEX™ (dexmedetomidine hydrochloride) in sodium chloride injection, for intravenous use
Initial U.S. Approval: 1999RECENT MAJOR CHANGES
INDICATIONS AND USAGE
PRECEDEX is a alpha2-adrenergic receptor agonist indicated for:
- Sedation of initially intubated and mechanically ventilated adult patients during treatment in an intensive care setting. Administer PRECEDEX by continuous infusion not to exceed 24 hours. (1.1)
- Sedation of non-intubated adult patients prior to and/or during surgical and other procedures. (1.2)
- Sedation of non-intubated pediatric patients aged 1 month to less than 18 years prior to and during non-invasive procedures. (1.2)
DOSAGE AND ADMINISTRATION
- Individualize and titrate PRECEDEX dosing to desired clinical effect. (2.1)
- Administer PRECEDEX using a controlled infusion device. (2.1)
- The 80 mcg/20 mL single-dose vial, and 200 mcg/50 mL, 400 mcg/100 mL, and 1,000 mcg/250 mL single-dose bottles do not require further dilution prior to administration. (2.4)
- For Adult Intensive Care Unit Sedation: Initiate at one mcg/kg over 10 minutes, followed by a maintenance infusion of 0.2 to 0.7 mcg/kg/hour. (2.2)
- For Adult Procedural Sedation: Initiate at one mcg/kg over 10 minutes, followed by a maintenance infusion initiated at 0.6 mcg/kg/hour and titrated to achieve desired clinical effect with doses ranging from 0.2 to 1 mcg/kg/hour. (2.2)
- For Sedation of Pediatric Patients During Non-invasive Procedures: Patients 1 month to less than 2 years old initiate at 1.5 mcg/kg over 10 minutes followed by a maintenance infusion of 1.5 mcg/kg/hour and titrated to achieve desired clinical effect with dosage ranging from 0.5 to 1.5 mcg/kg/hour; patients 2 to less than 18 years old initiate at 2.0 mcg/kg over 10 minutes followed by a maintenance infusion of 1.5 mcg/kg/hour and titrated to achieve desired clinical effect with dosage ranging from 0.5 to 1.5 mcg/kg/hour. (2.2)
- Alternative Doses: Recommended for patients over 65 years of age and awake fiberoptic intubation patients. (2.2)
DOSAGE FORMS AND STRENGTHS
- PRECEDEX in 0.9% Sodium Chloride Injection, 80 mcg/20 mL (4 mcg/mL) in a single-dose vial. Ready to use. (3)
- PRECEDEX in 0.9% Sodium Chloride Injection, 200 mcg/50 mL, 400 mcg/100 mL in single-dose glass bottles. Ready to use. (3)
- PRECEDEX in 0.9% Sodium Chloride Injection, 1,000 mcg/250 mL (4 mcg/mL) in a glass bottle. Ready to use. (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
- Monitoring: Continuously monitor patients while receiving PRECEDEX. (5.1)
- Bradycardia and Sinus Arrest: Have occurred in young healthy volunteers with high vagal tone or with different routes of administration, e.g., rapid intravenous or bolus administration. (5.2)
- Hypotension and Bradycardia: May necessitate medical intervention. May be more pronounced in patients with hypovolemia, diabetes mellitus, or chronic hypertension, and in the elderly. Use with caution in patients with advanced heart block or severe ventricular dysfunction. (5.2)
- Co-administration with Other Vasodilators or Negative Chronotropic Agents: Use with caution due to additive pharmacodynamic effects. (5.2)
- Transient Hypertension: Observed primarily during the loading dose. Consider reduction in loading infusion rate. (5.3)
- Arousability: Patients can become aroused/alert with stimulation; this alone should not be considered as lack of efficacy. (5.4)
- Tolerance and Tachyphylaxis: Prolonged exposure to dexmedetomidine beyond 24 hours may be associated with tolerance and tachyphylaxis and a dose-related increase in adverse events. (5.6)
ADVERSE REACTIONS
- The most common adverse reactions (incidence >2%) in adults are hypotension, bradycardia, and dry mouth. (6.1)
- The most common adverse reactions (incidence >5%) in pediatric patients aged 1 month to less than 17 years are bradypnea, bradycardia, hypertension, and hypotension. (6.1)
- Adverse reactions in adults, associated with infusions >24 hours in duration include ARDS, respiratory failure, and agitation. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Hospira, Inc. at 1-800-441-4100, or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
Anesthetics, Sedatives, Hypnotics, Opioids: Enhancement of pharmacodynamic effects. Reduction in dosage of PRECEDEX or the concomitant medication may be required. (7.1)
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 12/2022
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Intensive Care Unit Sedation
1.2 Procedural Sedation
2 DOSAGE AND ADMINISTRATION
2.1 Administration Instructions
2.2 Recommended Dosage
2.3 Dosage Adjustment
2.4 Preparation of Solution
2.5 Administration with Other Fluids
2.6 Compatibility with Natural Rubber
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Drug Administration
5.2 Hypotension, Bradycardia, and Sinus Arrest
5.3 Transient Hypertension
5.4 Arousability
5.5 Withdrawal
5.6 Tolerance and Tachyphylaxis
5.7 Hyperthermia or Pyrexia
5.8 Hepatic Impairment
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Anesthetics, Sedatives, Hypnotics, Opioids
7.2 Neuromuscular Blockers
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
9 DRUG ABUSE AND DEPENDENCE
9.1 Controlled Substance
9.3 Dependence
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
14.1 Intensive Care Unit Sedation
14.2 Procedural Sedation
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
1.1 Intensive Care Unit Sedation
PRECEDEX is indicated for sedation of initially intubated and mechanically ventilated adult patients during treatment in an intensive care setting. PRECEDEX should be administered by continuous infusion not to exceed 24 hours.
PRECEDEX has been continuously infused in mechanically ventilated adult patients prior to extubation, during extubation, and post-extubation. It is not necessary to discontinue PRECEDEX prior to extubation.
-
2 DOSAGE AND ADMINISTRATION
2.1 Administration Instructions
- PRECEDEX dosing should be individualized and titrated to desired clinical response.
- PRECEDEX is not indicated for infusions lasting longer than 24 hours.
- PRECEDEX should be administered using a controlled infusion device.
2.2 Recommended Dosage
Table 1: Recommended Dosage in Adult Patients INDICATION DOSAGE AND ADMINISTRATION Initiation of Intensive Care Unit Sedation
For adult patients: a loading infusion of one mcg/kg over 10 minutes.
For adult patients being converted from alternate sedative therapy: a loading dose may not be required.
For patients over 65 years of age: Consider a dose reduction [see Use in Specific Populations (8.5)].
For adult patients with impaired hepatic function: Consider a dose reduction [see Use in Specific Populations (8.6), Clinical Pharmacology (12.3)].
Maintenance of Intensive Care Unit Sedation
For adult patients: a maintenance infusion of 0.2 to 0.7 mcg/kg/hour. The rate of the maintenance infusion should be adjusted to achieve the desired level of sedation.
For patients over 65 years of age: Consider a dose reduction [see Use in Specific Populations (8.5)].
For adult patients with impaired hepatic function: Consider a dose reduction [see Use in Specific Populations (8.6), Clinical Pharmacology (12.3)]
Initiation of Procedural Sedation
For adult patients: a loading infusion of one mcg/kg over 10 minutes. For less invasive procedures such as ophthalmic surgery, a loading infusion of 0.5 mcg/kg given over 10 minutes may be suitable.
For awake fiberoptic intubation in adult patients: a loading infusion of one mcg/kg over 10 minutes.
For patients over 65 years of age: a loading infusion of 0.5 mcg/kg over 10 minutes [see Use in Specific Populations (8.5)].
For adult patients with impaired hepatic function: Consider a dose reduction [see Use in Specific Populations (8.6), Clinical Pharmacology (12.3)].
Maintenance of Procedural Sedation
For adult patients: the maintenance infusion is generally initiated at 0.6 mcg/kg/hour and titrated to achieve desired clinical effect with doses ranging from 0.2 to 1 mcg/kg/hour. Adjust the rate of the maintenance infusion to achieve the targeted level of sedation.
For awake fiberoptic intubation in adult patients: a maintenance infusion of 0.7 mcg/kg/hour is recommended until the endotracheal tube is secured.
For patients over 65 years of age: Consider a dose reduction [see Use in Specific Populations (8.5)].
For adult patients with impaired hepatic function: Consider a dose reduction [see Use in Specific Populations (8.6), Clinical Pharmacology (12.3)].
Table 2: Recommended Dosage in Pediatric Patients INDICATION
DOSAGE AND ADMINISTRATION
Initiation of Sedation During Non‑invasive Procedures
For pediatric patients:
- 1 month to less than 2 years: a loading infusion of 1.5 mcg/kg over 10 minutes.
- 2 to less than 18 years: a loading infusion of 2 mcg/kg over 10 minutes.
Consider a reduction in dosage if clinically indicated.
Maintenance of Sedation During Non-invasive Procedures
For pediatric patients:
- 1 month to less than 18 years: the maintenance infusion is generally initiated at 1.5 mcg/kg/hour and titrated to achieve desired clinical effect with dosage ranging from 0.5 to 1.5 mcg/kg/hour.
As clinically warranted, titrate the maintenance dose to individual patient clinical response.
2.3 Dosage Adjustment
Due to possible pharmacodynamic interactions, a reduction in dosage of PRECEDEX or other concomitant anesthetics, sedatives, hypnotics or opioids may be required when co-administered [see Drug Interactions (7.1)].
Dosage reductions may need to be considered for adult patients with hepatic impairment, and geriatric patients [see Warnings and Precautions (5.8), Use in Specific Populations (8.6), Clinical Pharmacology (12.3)].
2.4 Preparation of Solution
Strict aseptic technique must always be maintained during handling of PRECEDEX.
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. Do not use if product is discolored or if precipitate matter is present.
PRECEDEX in 0.9% Sodium Chloride Injection, 80 mcg/20 mL (4 mcg/mL), 200 mcg/50 mL (4 mcg/mL), 400 mcg/100 mL (4 mcg/mL), and 1,000 mcg/250 mL (4 mcg/mL)
PRECEDEX in 0.9% Sodium Chloride Injection is supplied in glass containers containing a premixed, ready to use dexmedetomidine hydrochloride solution in 0.9% sodium chloride in water. No further dilution of these preparations is necessary.
2.5 Administration with Other Fluids
PRECEDEX infusion should not be co-administered through the same intravenous catheter with blood or plasma because physical compatibility has not been established.
PRECEDEX has been shown to be incompatible when administered with the following drugs: amphotericin B, diazepam.
PRECEDEX has been shown to be compatible when administered with the following intravenous fluids:
- 0.9% sodium chloride in water
- 5% dextrose in water
- 20% mannitol
- Lactated Ringer's solution
- 100 mg/mL magnesium sulfate solution
- 0.3% potassium chloride solution
-
3 DOSAGE FORMS AND STRENGTHS
PRECEDEX (dexmedetomidine hydrochloride) in 0.9% sodium chloride injection is a clear and colorless solution, ready to use. It is available as:
PRECEDEX 80 mcg/20 mL (4 mcg/mL) single-dose vial.
PRECEDEX 200 mcg/50 mL (4 mcg/mL) single-dose glass bottle.
PRECEDEX 400 mcg/100 mL (4 mcg/mL) single-dose glass bottle.
PRECEDEX 1,000 mcg dexmedetomidine/250 mL (4 mcg/mL) in a glass bottle. Ready to use.
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Drug Administration
PRECEDEX should be administered only by persons skilled in the management of patients in the intensive care or operating room setting. Due to the known pharmacological effects of PRECEDEX, patients should be continuously monitored while receiving PRECEDEX.
5.2 Hypotension, Bradycardia, and Sinus Arrest
Clinically significant episodes of bradycardia and sinus arrest have been reported with PRECEDEX administration in young, healthy adult volunteers with high vagal tone or with different routes of administration including rapid intravenous or bolus administration.
Reports of hypotension and bradycardia have been associated with PRECEDEX infusion. Some of these cases have resulted in fatalities. If medical intervention is required, treatment may include decreasing or stopping the infusion of PRECEDEX, increasing the rate of intravenous fluid administration, elevation of the lower extremities, and use of pressor agents. Because PRECEDEX has the potential to augment bradycardia induced by vagal stimuli, clinicians should be prepared to intervene. The intravenous administration of anticholinergic agents (e.g., glycopyrrolate, atropine) should be considered to modify vagal tone. In clinical trials, glycopyrrolate or atropine were effective in the treatment of most episodes of PRECEDEX-induced bradycardia. However, in some patients with significant cardiovascular dysfunction, more advanced resuscitative measures were required.
Caution should be exercised when administering PRECEDEX to patients with advanced heart block and/or severe ventricular dysfunction. Because PRECEDEX decreases sympathetic nervous system activity, hypotension and/or bradycardia may be expected to be more pronounced in patients with hypovolemia, diabetes mellitus, or chronic hypertension and in elderly patients.
In clinical trials where other vasodilators or negative chronotropic agents were co-administered with PRECEDEX an additive pharmacodynamic effect was not observed. Nonetheless, caution should be used when such agents are administered concomitantly with PRECEDEX.
5.3 Transient Hypertension
Transient hypertension has been observed primarily during the loading dose in association with the initial peripheral vasoconstrictive effects of PRECEDEX. Treatment of the transient hypertension has generally not been necessary, although reduction of the loading infusion rate may be desirable.
5.4 Arousability
Some patients receiving PRECEDEX have been observed to be arousable and alert when stimulated. This alone should not be considered as evidence of lack of efficacy in the absence of other clinical signs and symptoms.
5.5 Withdrawal
Intensive Care Unit Sedation
With administration up to 7 days, regardless of dose, 12 (5%) PRECEDEX adult subjects experienced at least 1 event related to withdrawal within the first 24 hours after discontinuing study drug and 7 (3%) PRECEDEX adult subjects experienced at least 1 event 24 to 48 hours after end of study drug. The most common events were nausea, vomiting, and agitation [see Adverse Reactions (6.1)].
In adult subjects, tachycardia and hypertension requiring intervention in the 48 hours following study drug discontinuation occurred at frequencies of <5%.
Procedural Sedation
In adult subjects, withdrawal symptoms were not seen after discontinuation of short-term infusions of PRECEDEX (<6 hours).
In pediatric patients, mild transient withdrawal symptoms of emergence delirium or agitation were seen after discontinuation of short‑term infusions of PRECEDEX (<2 hours) [see Adverse Reactions (6.1)].
5.6 Tolerance and Tachyphylaxis
Use of dexmedetomidine beyond 24 hours has been associated with tolerance and tachyphylaxis and a dose-related increase in adverse reactions [see Adverse Reactions (6.1)].
5.7 Hyperthermia or Pyrexia
PRECEDEX may induce hyperthermia or pyrexia, which may be resistant to traditional cooling methods, such as administration of cooled intravenous fluids and antipyretic medications. Discontinue PRECEDEX if drug-related hyperthermia or pyrexia is suspected and monitor patients until body temperature normalizes.
5.8 Hepatic Impairment
Since PRECEDEX clearance decreases with severity of hepatic impairment, dose reduction should be considered in patients with impaired hepatic function [see Dosage and Administration (2.2, 2.3)].
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Hypotension, bradycardia and sinus arrest [see Warnings and Precautions (5.2)]
- Transient hypertension [see Warnings and Precautions (5.3)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reactions rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Most common treatment-emergent adverse reactions, occurring in greater than 2% of adult patients in both Intensive Care Unit and procedural sedation studies include hypotension, bradycardia and dry mouth.
Intensive Care Unit Sedation
Adverse reaction information is derived from the continuous infusion trials of PRECEDEX for sedation in the Intensive Care Unit setting in which 1,007 adult patients received PRECEDEX. The mean total dose was 7.4 mcg/kg (range: 0.8 to 84.1), mean dose per hour was 0.5 mcg/kg/hr (range: 0.1 to 6.0) and the mean duration of infusion of 15.9 hours (range: 0.2 to 157.2). The population was between 17 to 88 years of age, 43% ≥65 years of age, 77% male and 93% Caucasian. Treatment-emergent adverse reactions occurring at an incidence of >2% are provided in Table 3. The most frequent adverse reactions were hypotension, bradycardia and dry mouth [see Warnings and Precautions (5.2)].
Table 3: Adverse Reactions with an Incidence >2%-Adult Intensive Care Unit Sedation Population <24 hours* Adverse Event All PRECEDEX
(N = 1007)
(%)Randomized PRECEDEX
(N = 798)
(%)Placebo
(N = 400)
(%)Propofol
(N = 188)
(%)- * 26 subjects in the all PRECEDEX group and 10 subjects in the randomized PRECEDEX group had exposure for greater than 24 hours.
Hypotension
25%
24%
12%
13%
Hypertension
12%
13%
19%
4%
Nausea
9%
9%
9%
11%
Bradycardia
5%
5%
3%
0
Atrial Fibrillation
4%
5%
3%
7%
Pyrexia
4%
4%
4%
4%
Dry Mouth
4%
3%
1%
1%
Vomiting
3%
3%
5%
3%
Hypovolemia
3%
3%
2%
5%
Atelectasis
3%
3%
3%
6%
Pleural Effusion
2%
2%
1%
6%
Agitation
2%
2%
3%
1%
Tachycardia
2%
2%
4%
1%
Anemia
2%
2%
2%
2%
Hyperthermia
2%
2%
3%
0
Chills
2%
2%
3%
2%
Hyperglycemia
2%
2%
2%
3%
Hypoxia
2%
2%
2%
3%
Post-procedural Hemorrhage
2%
2%
3%
4%
Pulmonary Edema
1%
1%
1%
3%
Hypocalcemia
1%
1%
0
2%
Acidosis
1%
1%
1%
2%
Urine Output Decreased
1%
1%
0
2%
Sinus Tachycardia
1%
1%
1%
2%
Ventricular Tachycardia
<1%
1%
1%
5%
Wheezing
<1%
1%
0
2%
Edema Peripheral
<1%
0
1%
2%
Adverse reaction information was also derived from the placebo-controlled, continuous infusion trials of PRECEDEX for sedation in the surgical intensive care unit setting in which 387 adult patients received PRECEDEX for less than 24 hours. The most frequently observed treatment-emergent adverse events included hypotension, hypertension, nausea, bradycardia, fever, vomiting, hypoxia, tachycardia and anemia (see Table 4).
Table 4: Treatment-Emergent Adverse Events Occurring in >1% of All Dexmedetomidine-Treated Adult Patients in the Randomized Placebo-Controlled Continuous Infusion <24 Hours ICU Sedation Studies Adverse Event Randomized Dexmedetomidine
(N = 387)Placebo
(N = 379)Hypotension
28%
13%
Hypertension
16%
18%
Nausea
11%
9%
Bradycardia
7%
3%
Fever
5%
4%
Vomiting
4%
6%
Atrial Fibrillation
4%
3%
Hypoxia
4%
4%
Tachycardia
3%
5%
Hemorrhage
3%
4%
Anemia
3%
2%
Dry Mouth
3%
1%
Rigors
2%
3%
Agitation
2%
3%
Hyperpyrexia
2%
3%
Pain
2%
2%
Hyperglycemia
2%
2%
Acidosis
2%
2%
Pleural Effusion
2%
1%
Oliguria
2%
<1%
Thirst
2%
<1%
In a controlled clinical trial, PRECEDEX was compared to midazolam for ICU sedation exceeding 24 hours duration in adult patients. Key treatment emergent adverse events occurring in dexmedetomidine or midazolam treated adult patients in the randomized active comparator continuous infusion long-term intensive care unit sedation study are provided in Table 5. The number (%) of adult subjects who had a dose-related increase in treatment-emergent adverse events by maintenance adjusted dose rate range in the PRECEDEX group is provided in Table 6.
Table 5: Key Treatment-Emergent Adverse Events Occurring in Dexmedetomidine- or Midazolam-Treated Adult Patients in the Randomized Active Comparator Continuous Infusion Long-Term Intensive Care Unit Sedation Study Adverse Event Dexmedetomidine
(N = 244)Midazolam
(N = 122)- * Hypotension was defined in absolute terms as Systolic blood pressure of <80 mmHg or Diastolic blood pressure of <50 mmHg or in relative terms as ≤30% lower than pre-study drug infusion value.
- † Bradycardia was defined in absolute terms as <40 bpm or in relative terms as ≤30% lower than pre-study drug infusion value.
- ‡ Hypertension was defined in absolute terms as Systolic blood pressure >180 mmHg or Diastolic blood pressure of >100 mmHg or in relative terms as ≥30% higher than pre-study drug infusion value.
- § Tachycardia was defined in absolute terms as >120 bpm or in relative terms as ≥30% greater than pre-study drug infusion value.
- ¶ Includes any type of hypertension.
Hypotension*
56%
56%
Hypotension Requiring Intervention
28%
27%
Bradycardia†
42%
19%
Bradycardia Requiring Intervention
5%
1%
Systolic Hypertension‡
28%
42%
Tachycardia§
25%
44%
Tachycardia Requiring Intervention
10%
10%
Diastolic Hypertension‡
12%
15%
Hypertension‡
11%
15%
Hypertension Requiring Intervention¶
19%
30%
Hypokalemia
9%
13%
Pyrexia
7%
2%
Agitation
7%
6%
Hyperglycemia
7%
2%
Constipation
6%
6%
Hypoglycemia
5%
6%
Respiratory Failure
5%
3%
Renal Failure Acute
2%
1%
Acute Respiratory Distress Syndrome
2%
1%
Generalized Edema
2%
6%
Hypomagnesemia
1%
7%
The following adverse events occurred between 2 and 5% for PRECEDEX and Midazolam, respectively: renal failure acute (2.5%, 0.8%), acute respiratory distress syndrome (2.5%, 0.8%), and respiratory failure (4.5%, 3.3%).
Table 6: Number (%) of Adult Subjects Who Had a Dose-Related Increase in Treatment Emergent Adverse Events by Maintenance Adjusted Dose Rate Range in the PRECEDEX Group PRECEDEX (mcg/kg/hr) Adverse Event ≤0.7*
(N = 95)>0.7 to ≤1.1*
(N = 78)>1.1*
(N = 71)- * Average maintenance dose over the entire study drug administration.
Constipation
6%
5%
14%
Agitation
5%
8%
14%
Anxiety
5%
5%
9%
Edema Peripheral
3%
5%
7%
Atrial Fibrillation
2%
4%
9%
Respiratory Failure
2%
6%
10%
Acute Respiratory Distress Syndrome
1%
3%
9%
Adult Procedural Sedation
Adverse reaction information is derived from the two trials for adult procedural sedation [see Clinical Studies (14.2)] in which 318 adult patients received PRECEDEX. The mean total dose was 1.6 mcg/kg (range: 0.5 to 6.7), mean dose per hour was 1.3 mcg/kg/hr (range: 0.3 to 6.1) and the mean duration of infusion of 1.5 hours (range: 0.1 to 6.2). The population was between 18 to 93 years of age, ASA I–IV, 30% ≥65 years of age, 52% male and 61% Caucasian.
Treatment-emergent adverse reactions occurring in adults at an incidence of >2% are provided in Table 7. The most frequent adverse reactions were hypotension, bradycardia, and dry mouth [see Warnings and Precautions (5.2)]. Pre-specified criteria for the vital signs to be reported as adverse reactions are footnoted below the table. The decrease in respiratory rate and hypoxia was similar between PRECEDEX and comparator groups in both studies.
Table 7: Adverse Reactions with an Incidence >2%—Adult Procedural Sedation Population Adverse Event PRECEDEX
(N = 318)
(%)Placebo
(N = 113)
(%)- * Hypotension was defined in absolute and relative terms as Systolic blood pressure of <80 mmHg or ≤30% lower than pre-study drug infusion value, or Diastolic blood pressure of <50 mmHg.
- † Respiratory depression was defined in absolute and relative terms as respiratory rate (RR) <8 beats per minute or >25% decrease from baseline.
- ‡ Bradycardia was defined in absolute and relative terms as <40 beats per minute or ≤30% lower than pre-study drug infusion value. Subjects in Study 2 were pretreated with glycopyrrolate 0.1 mg intravenously before receiving study drug [see Clinical Studies (14.2)].
- § Hypertension was defined in absolute and relative terms as Systolic blood pressure >180 mmHg or ≥30% higher than pre-study drug infusion value or Diastolic blood pressure of >100 mmHg.
- ¶ Tachycardia was defined in absolute and relative terms as >120 beats per minute or ≥30% greater than pre-study drug infusion value.
- # Hypoxia was defined in absolute and relative terms as SpO2 <90% or 10% decrease from baseline.
Hypotension*
54%
30%
Respiratory Depression†
37%
32%
Bradycardia‡
14%
4%
Hypertension§
13%
24%
Tachycardia¶
5%
17%
Nausea
3%
2%
Dry Mouth
3%
1%
Hypoxia#
2%
3%
Bradypnea
2%
4%
Pediatric Sedation for Magnetic Resonance Imaging
Adverse reaction information is derived from a trial for sedation of pediatric procedural during a non‑invasive procedure [see Clinical Studies (14.2)] in which 122 pediatric patients aged 1 month to less than 17 years undergoing magnetic resonance imaging (MRI) scans received PRECEDEX. In pediatric patients 1 month to less than 2 years old, the median total dose for the PRECEDEX low, middle, and high dose treatment groups was 8.30, 18.90, and 22.75 mcg, respectively. The median duration of treatment ranged from 52.5 to 69 minutes across treatment groups. In pediatric patients 2 to less than 17 years old, the median total dose for the PRECEDEX low, middle, and high dose treatment groups was 21.30, 43.90, and 80.25 mcg, respectively. The median duration of treatment ranged from 56.5 to 66 minutes across treatment groups.
All-causality treatment-emergent adverse reactions occurring in the combined age group of pediatric patients during the procedure at an incidence of >5% are provided in Table 8. The most frequent treatment-emergent adverse events were bradypnea, bradycardia, hypertension, and hypotension [see Warnings and Precautions (5.2, 5.3)]. In the combined age group and in each age group, increased incidence in bradycardia and hypertension was observed with increasing PRECEDEX dose. Mild transient withdrawal symptoms of emergence delirium or agitation occurred in 3 of 122 patients after discontinuation of PRECEDEX infusion [see Warnings and Precautions (5.5)]. All reported treatment‑emergent adverse reactions were mild to moderate in severity and the majority resolved without medical intervention. No subject in the study required airway intervention, including a jaw thrust or insertion of a nasal or oral airway. A similar profile was observed in the pediatric patients 1 month to less than 2 years old and in pediatric patients 2 to less than 17 years old. Pre‑specified criteria for the vital signs to be reported as adverse events are footnoted below the table.
Table 8: Treatment-Emergent Adverse Events with Incidence >5%—Pediatric Patients During Non-invasive Procedure N = Number of pediatric patients evaluable for adverse events. - * Bradypnea was defined as respiratory rate <1st centile of the age adjusted normal range.
- † Bradycardia was defined as a decrease in HR of 30% from baseline or absolute HR ≤1st centile of the age adjusted normal range.
- ‡ For pediatric patients 1 month to less than 1 year old, hypertension was defined as supine systolic blood pressure ≥104 mm/Hg and/or diastolic blood pressure ≥56 mmHg measurements. For pediatric patients 1 to less than 17 years old: hypertension was defined as supine systolic blood pressure and/or diastolic blood pressure measurements ≥95th percentile for gender, age, and height.
- § Hypotension was defined as a decrease in systolic blood pressure ≥30% from baseline.
- ¶ Hypoxia was defined as oxygen saturation <90% for any duration.
PRECEDEX
Low Dose
(N = 42)
PRECEDEX
Middle Dose
(N = 42)
PRECEDEX
High Dose
(N = 38)
Total
(N = 122)
Number (%) of Pediatric Patients
n (%)
n (%)
n (%)
n (%)
Adverse Event
Bradypnea*
33 (79)
27 (64)
22 (58)
82 (67)
Bradycardia†
24 (57)
24 (57)
27 (71)
75 (62)
Hypertension‡
11 (26)
17 (41)
18 (47)
46 (38)
Hypotension§
13 (31)
11 (26)
6 (16)
30 (25)
Hypoxia¶
6 (14)
3 (7)
1 (3)
10 (8)
Diastolic Hypertension‡
3 (7)
3 (7)
4 (11)
10 (8)
Systolic Hypertension‡
1 (2)
5 (12)
3 (8)
9 (7)
Tachycardia
3 (7)
1 (2)
1 (3)
5 (4)
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of PRECEDEX. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Hypotension and bradycardia were the most common adverse reactions associated with the use of PRECEDEX during post-approval use of the drug.
Table 9: Adverse Reactions Experienced During Post-Approval Use of PRECEDEX System Organ Class Preferred Term Blood and Lymphatic System Disorders
Anemia
Cardiac Disorders
Arrhythmia, atrial fibrillation, atrioventricular block, bradycardia, cardiac arrest, cardiac disorder, extrasystoles, myocardial infarction, supraventricular tachycardia, tachycardia, ventricular arrhythmia, ventricular tachycardia
Eye Disorders
Photopsia, visual impairment
Gastrointestinal Disorders
Abdominal pain, diarrhea, nausea, vomiting
General Disorders and Administration Site Conditions
Chills, hyperpyrexia, pain, pyrexia, thirst
Hepatobiliary Disorders
Hepatic function abnormal, hyperbilirubinemia
Investigations
Alanine aminotransferase increased, aspartate aminotransferase increased, blood alkaline phosphatase increased, blood urea increased, electrocardiogram T wave inversion, gammaglutamyltransferase increased, electrocardiogram QT prolonged
Metabolism and Nutrition Disorders
Acidosis, hyperkalemia, hypoglycemia, hypovolemia, hypernatremia
Nervous System Disorders
Convulsion, dizziness, headache, neuralgia, neuritis, speech disorder
Psychiatric Disorders
Agitation, confusional state, delirium, hallucination, illusion
Renal and Urinary Disorders
Oliguria, polyuria
Respiratory, Thoracic and Mediastinal Disorders
Apnea, bronchospasm, dyspnea, hypercapnia, hypoventilation, hypoxia, pulmonary congestion, respiratory acidosis
Skin and Subcutaneous Tissue Disorders
Hyperhidrosis, pruritus, rash, urticaria
Surgical and Medical Procedures
Light anesthesia
Vascular Disorders
Blood pressure fluctuation, hemorrhage, hypertension, hypotension
-
7 DRUG INTERACTIONS
7.1 Anesthetics, Sedatives, Hypnotics, Opioids
Co-administration of PRECEDEX with anesthetics, sedatives, hypnotics, and opioids is likely to lead to an enhancement of effects. Specific studies have confirmed these effects with sevoflurane, isoflurane, propofol, alfentanil, and midazolam. No pharmacokinetic interactions between PRECEDEX and isoflurane, propofol, alfentanil and midazolam have been demonstrated. However, due to possible pharmacodynamic interactions, when co-administered with PRECEDEX, a reduction in dosage of PRECEDEX or the concomitant anesthetic, sedative, hypnotic or opioid may be required.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Available data from published randomized controlled trials and case reports over several decades of use with intravenously administered dexmedetomidine during pregnancy have not identified a drug-associated risk of major birth defects and miscarriage; however, the reported exposures occurred after the first trimester. Most of the available data are based on studies with exposures that occurred at the time of caesarean section delivery, and these studies have not identified an adverse effect on maternal outcomes or infant Apgar scores. Available data indicate that dexmedetomidine crosses the placenta.
In animal reproduction studies, fetal toxicity that lower fetal viability and reduced live fetuses occurred with subcutaneous administration of dexmedetomidine to pregnant rats during organogenesis at doses 1.8 times the maximum recommended human dose (MRHD) of 17.8 mcg/kg/day.
Developmental toxicity (low pup weights and adult offspring weights, decreased F1 grip strength, increased early implantation loss and decreased viability of second-generation offspring) occurred when pregnant rats were subcutaneously administered dexmedetomidine at doses less than the clinical dose from late pregnancy through lactation and weaning (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2–4% and 15–20%, respectively.
Data
Animal Data
Increased post-implantation losses and reduced live fetuses in the presence of maternal toxicity (i.e. decreased body weight) were noted in a rat embryo-fetal development study in which pregnant dams were administered subcutaneous doses of dexmedetomidine 200 mcg/kg/day (equivalent to 1.8 times the intravenous MRHD of 17.8 mcg/kg/day based on body surface area [BSA]) during the period of organogenesis (Gestation Day [GD] 6 to 15). No malformations were reported.
No malformations or embryo-fetal toxicity were noted in a rabbit embryo-fetal development study in which pregnant does were administered dexmedetomidine intravenously at doses of up to 96 mcg/kg/day (approximately half the human exposure at the MRHD based on AUC) during the period of organogenesis (GD 6 to 18).
Reduced pup and adult offspring birth weights, and grip strength were reported in a rat developmental toxicology study in which pregnant females were administered dexmedetomidine subcutaneously at doses of 8 mcg/kg/day (0.07 times the MRHD based on BSA) during late pregnancy through lactation and weaning (GD 16 to postnatal day [PND] 25). Decreased viability of second generation offspring and an increase in early implantation loss along with delayed motor development occurred in the 32 mcg/kg/day group (equivalent to less than the clinical dose based on BSA) when first generation offspring were allowed to mate. This study limited dosing to hard palate closure (GD 15 to 18) through weaning instead of dosing from implantation (GD 6 to 7) to weaning (PND 21).
In a study in the pregnant rat, placental transfer of dexmedetomidine was observed when radiolabeled dexmedetomidine was administered subcutaneously.
8.2 Lactation
Risk Summary
Available published literature reports the presence of dexmedetomidine in human milk following intravenous administration (see Data). There is no information regarding the effects of dexmedetomidine on the breastfed infant or the effects on milk production. Advise women to monitor the breastfed infant for irritability. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for PRECEDEX and any potential adverse effects on the breastfed infant from PRECEDEX or from the underlying condition.
In two published clinical studies, a total of 14 women were given intravenous dexmedetomidine 6 mcg/kg/hour for 10 minutes after delivery followed by continuous infusion of 0.2–0.7 mcg/kg/hour. Breast milk and maternal blood samples were collected at 0, 6, 12, and 24 hours after discontinuation of dexmedetomidine. Plasma and milk dexmedetomidine concentrations were detectable up to 6 hours in most subjects, up to 12 hours in one subject and undetectable in all at 24 hours. The milk-to-plasma ratio from single paired maternal milk and plasma concentrations at each time point ranged from 0.53 to 0.95. The relative infant dose was estimated to range from 0.02 to 0.098%.
8.4 Pediatric Use
Sedation for Non-Invasive Procedures
The safety and efficacy of PRECEDEX have been established in pediatric patients 1 month to less than 18 years of age for sedation during non-invasive procedures. Use in this age group is based on one randomized double-blind, dose-ranging safety and efficacy trial in non-intubated pediatric patients 1 month to less than 17 years of age who required sedation prior to undergoing MRI scans [see Clinical Studies (14.2)]. An increase in frequency of bradypnea, bradycardia, hypertension and hypotension was observed in pediatric patients treated with PRECEDEX [see Adverse Reactions (6.1)]. The overall safety profile of PRECEDEX in pediatric patients was consistent with the known safety profile in adults [see Adverse Reactions (6.1)].
The safety and effectiveness of PRECEDEX have not been established in pediatric patients less than 1 month of age.
ICU Sedation
The safety and efficacy of PRECEDEX have not been established in pediatric patients for ICU sedation. One assessor-blinded trial in pediatric patients and two open label studies in neonates were conducted to assess efficacy for ICU sedation. These studies did not meet their primary efficacy endpoints and the safety data submitted were insufficient to fully characterize the safety profile of PRECEDEX for these patient populations.
8.5 Geriatric Use
Intensive Care Unit Sedation
A total of 729 patients in the clinical studies were 65 years of age and over. A total of 200 patients were 75 years of age and over. In patients greater than 65 years of age, a higher incidence of bradycardia and hypotension was observed following administration of PRECEDEX [see Warnings and Precautions (5.2)]. Therefore, a dose reduction may be considered in patients over 65 years of age [see Dosage and Administration (2.2, 2.3), Clinical Pharmacology (12.3)].
Procedural Sedation
A total of 131 patients in the clinical studies were 65 years of age and over. A total of 47 patients were 75 years of age and over. Hypotension occurred in a higher incidence in PRECEDEX-treated patients 65 years or older (72%) and 75 years or older (74%) as compared to patients <65 years (47%). A reduced loading dose of 0.5 mcg/kg given over 10 minutes is recommended and a reduction in the maintenance infusion should be considered for patients greater than 65 years of age.
8.6 Hepatic Impairment
Since PRECEDEX clearance decreases with increasing severity of hepatic impairment, dose reduction should be considered in patients with impaired hepatic function [see Dosage and Administration (2.2, 2.3), Clinical Pharmacology (12.3)].
-
9 DRUG ABUSE AND DEPENDENCE
9.3 Dependence
The dependence potential of PRECEDEX has not been studied in humans. However, since studies in rodents and primates have demonstrated that PRECEDEX exhibits pharmacologic actions similar to those of clonidine, it is possible that PRECEDEX may produce a clonidine-like withdrawal syndrome upon abrupt discontinuation [see Warnings and Precautions (5.5)].
-
10 OVERDOSAGE
The tolerability of PRECEDEX was studied in one study in which healthy adult subjects were administered doses at and above the recommended dose of 0.2 to 0.7 mcg/kg/hr. The maximum blood concentration achieved in this study was approximately 13 times the upper boundary of the therapeutic range. The most notable effects observed in two subjects who achieved the highest doses were first degree atrioventricular block and second-degree heart block. No hemodynamic compromise was noted with the atrioventricular block and the heart block resolved spontaneously within one minute.
Five adult patients received an overdose of PRECEDEX in the intensive care unit sedation studies. Two of these patients had no symptoms reported; one patient received a 2 mcg/kg loading dose over 10 minutes (twice the recommended loading dose) and one patient received a maintenance infusion of 0.8 mcg/kg/hr. Two other patients who received a 2 mcg/kg loading dose over 10 minutes, experienced bradycardia and/or hypotension. One patient who received a loading bolus dose of undiluted PRECEDEX (19.4 mcg/kg), had cardiac arrest from which he was successfully resuscitated.
-
11 DESCRIPTION
PRECEDEX (dexmedetomidine hydrochloride) in 0.9% Sodium Chloride Injection (4 mcg/mL) is a sterile, nonpyrogenic ready to use solution suitable for intravenous infusion.
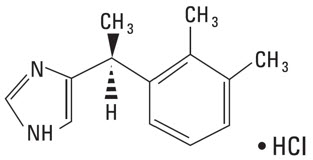
PRECEDEX contains dexmedetomidine hydrochloride as the active pharmaceutical ingredient. Dexmedetomidine hydrochloride is a central alpha2-adrenergic agonist. Dexmedetomidine hydrochloride is the S-enantiomer of medetomidine Dexmedetomidine hydrochloride chemical name is 1H-Imidazole, 4-[1-(2,3-dimethylphenyl)ethyl]-, monohydrochloride, (S). Dexmedetomidine hydrochloride has a molecular weight of 236.7 and the empirical formula is C13H16N2 ∙ HCl and the structural formula is:
PRECEDEX in 0.9% Sodium Chloride Injection is ready to be used. It is supplied as a clear, colorless, isotonic solution with a pH between 4.5 to 8.0. Each mL contains 4.72 mcg of dexmedetomidine hydrochloride (equivalent to 4 mcg or 0.004 mg of dexmedetomidine) and 9 mg sodium chloride in water for injection. The solution is preservative-free and contains no additives or chemical stabilizers.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
PRECEDEX is a relatively selective centrally acting alpha2-adrenergic agonist with sedative properties. Alpha2 selectivity is observed in animals following slow intravenous infusion of low and medium doses (10–300 mcg/kg). Both alpha1 and alpha2 activity is observed following slow intravenous infusion of high doses (≥1,000 mcg/kg) or with rapid intravenous administration.
12.2 Pharmacodynamics
In a study in healthy adult volunteers (N = 10), respiratory rate and oxygen saturation remained within normal limits and there was no evidence of respiratory depression when PRECEDEX was administered by intravenous infusion at doses within the recommended dose range (0.2–0.7 mcg/kg/hr).
12.3 Pharmacokinetics
Following intravenous administration to adults, dexmedetomidine exhibits the following pharmacokinetic parameters: a rapid distribution phase with a distribution half-life (t1/2) of approximately 6 minutes; a terminal elimination half-life (t1/2) of approximately 2 hours; and steady-state volume of distribution (Vss) of approximately 118 liters. Clearance is estimated to be approximately 39 L/h. The mean body weight associated with this clearance estimate was 72 kg.
Dexmedetomidine exhibits linear pharmacokinetics in the dosage range of 0.2 to 0.7 mcg/kg/hr when administered to adults by intravenous infusion for up to 24 hours. Table 10 shows the main pharmacokinetic parameters when PRECEDEX was infused (after appropriate loading doses) at maintenance infusion rates of 0.17 mcg/kg/hr (target plasma concentration of 0.3 ng/mL) for 12 and 24 hours, 0.33 mcg/kg/hr (target plasma concentration of 0.6 ng/mL) for 24 hours, and 0.70 mcg/kg/hr (target plasma concentration of 1.25 ng/mL) for 24 hours.
Table 10: Mean ± SD Pharmacokinetic Parameters in Adults Parameter Loading Infusion (min)/Total Infusion Duration (hrs) 10 min/12 hrs 10 min/24 hrs 10 min/24 hrs 35 min/24 hrs Dexmedetomidine Target Plasma Concentration (ng/mL) and Dose (mcg/kg/hr) 0.3/0.17 0.3/0.17 0.6/0.33 1.25/0.70 Abbreviations: t1/2 = half-life, CL = clearance, Vss = steady-state volume of distribution. - * Presented as harmonic mean and pseudo standard deviation.
- † Mean Css = Average steady-state concentration of dexmedetomidine. The mean Css was calculated based on post-dose sampling from 2.5 to 9 hours samples for 12 hour infusion and post-dose sampling from 2.5 to 18 hours for 24 hour infusions.
t1/2*, hour
1.78 ± 0.30
2.22 ± 0.59
2.23 ± 0.21
2.50 ± 0.61
CL, liter/hour
46.3 ± 8.3
43.1 ± 6.5
35.3 ± 6.8
36.5 ± 7.5
Vss, liter
88.7 ± 22.9
102.4 ± 20.3
93.6 ± 17.0
99.6 ± 17.8
Avg Css†, ng/mL
0.27 ± 0.05
0.27 ± 0.05
0.67 ± 0.10
1.37 ± 0.20
The loading doses for each of the above indicated groups were 0.5, 0.5, 1 and 2.2 mcg/kg, respectively.
Dexmedetomidine pharmacokinetic parameters in adults after PRECEDEX maintenance doses of 0.2 to 1.4 mcg/kg/hr for >24 hours were similar to the pharmacokinetic (PK) parameters after PRECEDEX maintenance dosing for <24 hours in other studies. The values for clearance (CL), volume of distribution (V), and t1/2 were 39.4 L/hr, 152 L, and 2.67 hours, respectively.
Distribution
The steady-state volume of distribution (Vss) of dexmedetomidine was approximately 118 liters. Dexmedetomidine protein binding was assessed in the plasma of normal healthy male and female subjects. The average protein binding was 94% and was constant across the different plasma concentrations tested. Protein binding was similar in males and females. The fraction of PRECEDEX that was bound to plasma proteins was significantly decreased in subjects with hepatic impairment compared to healthy subjects.
The potential for protein binding displacement of dexmedetomidine by fentanyl, ketorolac, theophylline, digoxin and lidocaine was explored in vitro, and negligible changes in the plasma protein binding of PRECEDEX were observed. The potential for protein binding displacement of phenytoin, warfarin, ibuprofen, propranolol, theophylline and digoxin by PRECEDEX was explored in vitro and none of these compounds appeared to be significantly displaced by PRECEDEX.
Elimination
Metabolism
Dexmedetomidine undergoes almost complete biotransformation with very little unchanged dexmedetomidine excreted in urine and feces. Biotransformation involves both direct glucuronidation as well as cytochrome P450 mediated metabolism. The major metabolic pathways of dexmedetomidine are: direct N-glucuronidation to inactive metabolites; aliphatic hydroxylation (mediated primarily by CYP2A6 with a minor role of CYP1A2, CYP2E1, CYP2D6 and CYP2C19) of dexmedetomidine to generate 3-hydroxy-dexmedetomidine, the glucuronide of 3-hydroxy-dexmedetomidine, and 3-carboxy-dexmedetomidine; and N-methylation of dexmedetomidine to generate 3-hydroxy N-methyl-dexmedetomidine, 3-carboxy N-methyl-dexmedetomidine, and dexmedetomidine-N-methyl O-glucuronide.
Excretion
The terminal elimination half-life (t1/2) of dexmedetomidine is approximately 2 hours and clearance is estimated to be approximately 39 L/h. A mass balance study demonstrated that after nine days an average of 95% of the radioactivity, following intravenous administration of radiolabeled dexmedetomidine, was recovered in the urine and 4% in the feces. No unchanged dexmedetomidine was detected in the urine. Approximately 85% of the radioactivity recovered in the urine was excreted within 24 hours after the infusion. Fractionation of the radioactivity excreted in urine demonstrated that products of N-glucuronidation accounted for approximately 34% of the cumulative urinary excretion. In addition, aliphatic hydroxylation of parent drug to form 3-hydroxy-dexmedetomidine, the glucuronide of 3-hydroxy-dexmedetomidine, and 3-carboxylic acid-dexmedetomidine together represented approximately 14% of the dose in urine. N-methylation of dexmedetomidine to form 3-hydroxy N-methyl dexmedetomidine, 3-carboxy N-methyl dexmedetomidine, and N-methyl O-glucuronide dexmedetomidine accounted for approximately 18% of the dose in urine. The N-Methyl metabolite itself was a minor circulating component and was undetected in urine. Approximately 28% of the urinary metabolites have not been identified.
Specific Populations
Geriatric Patients
The pharmacokinetic profile of PRECEDEX was not altered by age. There were no differences in the pharmacokinetics of PRECEDEX in young (18–40 years), middle age (41–65 years), and elderly (>65 years) subjects.
Pediatric Patients
A population PK analysis was conducted with data collected from mechanically ventilated pediatric patients less than 17 years of age from 4 clinical studies. A linear 2-compartment model was found to best characterize the pooled dexmedetomidine concentration data. Mean PK parameters for each age group are summarized in Table 11.
Table 11: Geometric Mean Point Estimates and 95% Confidence Intervals of PK Parameters by Pediatric Age Group Abbreviations: CL = plasma clearance, Vc = volume of the central compartment. Age Group
N
CL (L/h)
Vc (L)
1 to less than 6 months
14
6.94 (5.46, 8.81)
4.34 (3.25, 5.81)
6 to less than 12 months
15
8.15 (7.01, 9.47)
7.29 (5.57, 9.53)
12 to less than 24 months
13
10.76 (9.09, 12.74)
7.35 (5.59, 9.67)
2 to less than 6 years
26
15.89 (14.00, 18.04)
13.78 (10.66, 17.83)
6 to less than 17 years
28
24.45 (19.34, 30.92)
24.47 (17.06, 35.10)
Patients with Hepatic Impairment
In adult subjects with varying degrees of hepatic impairment (Child-Pugh Class A, B, or C), clearance values for PRECEDEX were lower than in healthy subjects. The mean clearance values for patients with mild, moderate, and severe hepatic impairment were 74%, 64% and 53% of those observed in the normal healthy adult subjects, respectively. Mean clearances for free drug were 59%, 51% and 32% of those observed in the normal healthy adult subjects, respectively.
Although PRECEDEX is dosed to effect, it may be necessary to consider dose reduction in subjects with hepatic impairment [see Dosage and Administration (2.2), Warnings and Precautions (5.8)].
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Mutagenesis
Dexmedetomidine was not mutagenic in vitro, in either the bacterial reverse mutation assay (E. coli and Salmonella typhimurium) or the mammalian cell forward mutation assay (mouse lymphoma). Dexmedetomidine was clastogenic in the in vitro human lymphocyte chromosome aberration test with, but not without, rat S9 metabolic activation. In contrast, dexmedetomidine was not clastogenic in the in vitro human lymphocyte chromosome aberration test with or without human S9 metabolic activation. Although dexmedetomidine was clastogenic in an in vivo mouse micronucleus test in NMRI mice, there was no evidence of clastogenicity in CD-1 mice.
Impairment of Fertility
Fertility in male or female rats was not affected after daily subcutaneous injections of dexmedetomidine at doses up to 54 mcg/kg (less than the maximum recommended human intravenous dose on a mcg/m2 basis) administered from 10 weeks prior to mating in males, and 3 weeks prior to mating and during mating in females.
13.2 Animal Toxicology and/or Pharmacology
There were no differences in the adrenocorticotropic hormone (ACTH)-stimulated cortisol response in dogs following a single dose of dexmedetomidine compared to saline control. However, after continuous subcutaneous infusions of dexmedetomidine at 3 mcg/kg/hr and 10 mcg/kg/hr for one week in dogs (exposures estimated to be within the clinical range), the ACTH-stimulated cortisol response was diminished by approximately 27% and 40%, respectively, compared to saline-treated control animals indicating a dose-dependent adrenal suppression.
-
14 CLINICAL STUDIES
The safety and efficacy of PRECEDEX has been evaluated in four randomized, double-blind, placebo-controlled multicenter clinical trials in 1,185 adult patients.
14.1 Intensive Care Unit Sedation
Two randomized, double-blind, parallel-group, placebo-controlled multicenter clinical trials included 754 adult patients being treated in a surgical intensive care unit. All patients were initially intubated and received mechanical ventilation. These trials evaluated the sedative properties of PRECEDEX by comparing the amount of rescue medication (midazolam in one trial and propofol in the second) required to achieve a specified level of sedation (using the standardized Ramsay Sedation Scale) between PRECEDEX and placebo from onset of treatment to extubation or to a total treatment duration of 24 hours. The Ramsay Level of Sedation Scale is displayed in Table 12.
Table 12: Ramsay Level of Sedation Scale Clinical Score Level of Sedation Achieved 6
Asleep, no response
5
Asleep, sluggish response to light glabellar tap or loud auditory stimulus
4
Asleep, but with brisk response to light glabellar tap or loud auditory stimulus
3
Patient responds to commands
2
Patient cooperative, oriented, and tranquil
1
Patient anxious, agitated, or restless
In the first study, 175 adult patients were randomized to receive placebo and 178 to receive PRECEDEX by intravenous infusion at a dose of 0.4 mcg/kg/hr (with allowed adjustment between 0.2 and 0.7 mcg/kg/hr) following an initial loading infusion of one mcg/kg intravenous over 10 minutes. The study drug infusion rate was adjusted to maintain a Ramsay sedation score of ≥3. Patients were allowed to receive "rescue" midazolam as needed to augment the study drug infusion. In addition, morphine sulfate was administered for pain as needed. The primary outcome measure for this study was the total amount of rescue medication (midazolam) needed to maintain sedation as specified while intubated. Patients randomized to placebo received significantly more midazolam than patients randomized to PRECEDEX (see Table 13).
A second prospective primary analysis assessed the sedative effects of PRECEDEX by comparing the percentage of adult patients who achieved a Ramsay sedation score of ≥3 during intubation without the use of additional rescue medication. A significantly greater percentage of adult patients in the PRECEDEX group maintained a Ramsay sedation score of ≥3 without receiving any midazolam rescue compared to the placebo group (see Table 13).
Table 13: Midazolam Use as Rescue Medication During Intubation (ITT) Study One Placebo
(N = 175)PRECEDEX
(N = 178)p-value ITT (intent-to-treat) population includes all randomized patients. - * ANOVA model with treatment center.
- † Chi-square.
Mean Total Dose (mg) of Midazolam
19 mg
5 mg
0.0011*
Standard deviation
53 mg
19 mg
Categorized Midazolam Use
0 mg
43 (25%)
108 (61%)
<0.001†
0–4 mg
34 (19%)
36 (20%)
>4 mg
98 (56%)
34 (19%)
A prospective secondary analysis assessed the dose of morphine sulfate administered to adult patients in the PRECEDEX and placebo groups. On average, PRECEDEX -treated patients received less morphine sulfate for pain than placebo-treated patients (0.47 versus 0.83 mg/h). In addition, 44% (79 of 178 patients) of PRECEDEX patients received no morphine sulfate for pain versus 19% (33 of 175 patients) in the placebo group.
In a second study, 198 adult patients were randomized to receive placebo and 203 to receive PRECEDEX by intravenous infusion at a dose of 0.4 mcg/kg/hr (with allowed adjustment between 0.2 and 0.7 mcg/kg/hr) following an initial loading infusion of one mcg/kg intravenous over 10 minutes. The study drug infusion was adjusted to maintain a Ramsay sedation score of ≥3. Patients were allowed to receive "rescue" propofol as needed to augment the study drug infusion. In addition, morphine sulfate was administered as needed for pain. The primary outcome measure for this study was the total amount of rescue medication (propofol) needed to maintain sedation as specified while intubated.
Adult patients randomized to placebo received significantly more propofol than adult patients randomized to PRECEDEX (see Table 14).
A significantly greater percentage of adult patients in the PRECEDEX group compared to the placebo group maintained a Ramsay sedation score of ≥3 without receiving any propofol rescue (see Table 14).
Table 14: Propofol Use as Rescue Medication During Intubation (ITT) Study Two Placebo
(N = 198)PRECEDEX
(N = 203)p-value - * ANOVA model with treatment center.
- † Chi-square.
Mean Total Dose (mg) of Propofol
513 mg
72 mg
<0.0001*
Standard deviation
782 mg
249 mg
Categorized Propofol Use
0 mg
47 (24%)
122 (60%)
<0.001†
0–50 mg
30 (15%)
43 (21%)
>50 mg
121 (61%)
38 (19%)
A prospective secondary analysis assessed the dose of morphine sulfate administered to adult patients in the PRECEDEX and placebo groups. On average, PRECEDEX -treated patients received less morphine sulfate for pain than placebo-treated patients (0.43 versus 0.89 mg/h). In addition, 41% (83 of 203 patients) of PRECEDEX patients received no morphine sulfate for pain versus 15% (30 of 198 patients) in the placebo group.
In a controlled clinical trial, PRECEDEX was compared to midazolam for ICU sedation exceeding 24 hours duration. PRECEDEX was not shown to be superior to midazolam for the primary efficacy endpoint, the percent of time patients were adequately sedated (81% versus 81%). In addition, administration of PRECEDEX for longer than 24 hours was associated with tolerance, tachyphylaxis, and a dose-related increase in adverse events [see Adverse Reactions (6.1)].
14.2 Procedural Sedation
Adult Patients
The safety and efficacy of PRECEDEX for sedation of non-intubated adult patients prior to and/or during surgical and other procedures was evaluated in two randomized, double-blind, placebo-controlled multicenter clinical trials. Study 1 evaluated the sedative properties of PRECEDEX in adult patients having a variety of elective surgeries/procedures performed under monitored anesthesia care. Study 2 evaluated Precedex in adult patients undergoing awake fiberoptic intubation prior to a surgical or diagnostic procedure.
In Study 1, the sedative properties of PRECEDEX were evaluated by comparing the percent of adult patients not requiring rescue midazolam to achieve a specified level of sedation using the standardized Observer's Assessment of Alertness/Sedation Scale (see Table 15).
Table 15: Observer's Assessment of Alertness/Sedation Assessment Categories Responsiveness Speech Facial Expression Eyes Composite Score Responds readily to name spoken in normal tone
Normal
Normal
Clear, no ptosis
5 (alert)
Lethargic response to name spoken in normal tone
Mild slowing or thickening
Mild relaxation
Glazed or mild ptosis (less than half the eye)
4
Responds only after name is called loudly and/or repeatedly
Slurring or prominent slowing
Marked relaxation
(slack jaw)Glazed and marked ptosis (half the eye or more)
3
Responds only after mild prodding or shaking
Few recognizable words
–
–
2
Does not respond to mild prodding or shaking
–
–
–
1 (deep sleep)
Adult patients were randomized to receive a loading infusion of either PRECEDEX 1 mcg/kg, PRECEDEX 0.5 mcg/kg, or placebo (normal saline) given over 10 minutes and followed by a maintenance infusion started at 0.6 mcg/kg/hr. The maintenance infusion of study drug could be titrated from 0.2 mcg/kg/hr to 1 mcg/kg/hr to achieve the targeted sedation score (Observer's Assessment of Alertness/Sedation Scale ≤4). Adult patients were allowed to receive rescue midazolam as needed to achieve and/or maintain an Observer's Assessment of Alertness/Sedation Scale ≤4. After achieving the desired level of sedation, a local or regional anesthetic block was performed. Demographic characteristics were similar between the PRECEDEX and comparator groups. Efficacy results showed that Precedex was more effective than the comparator group when used to sedate non-intubated patients requiring monitored anesthesia care during surgical and other procedures (see Table 15).
In Study 2, the sedative properties of PRECEDEX were evaluated by comparing the percent of adult patients requiring rescue midazolam to achieve or maintain a specified level of sedation using the Ramsay Sedation Scale score ≥2 (see Table 12). Adult patients were randomized to receive a loading infusion of PRECEDEX 1 mcg/kg or placebo (normal saline) given over 10 minutes and followed by a fixed maintenance infusion of 0.7 mcg/kg/hr. After achieving the desired level of sedation, topicalization of the airway occurred. Adult patients were allowed to receive rescue midazolam as needed to achieve and/or maintain a Ramsay Sedation Scale ≥2. Demographic characteristics were similar between the PRECEDEX and comparator groups. For efficacy results see Table 16.
Table 16: Key Efficacy Results of Adult Procedural Sedation Studies Study Loading Infusion Treatment Arm Number of Patients Enrolled* % Not Requiring Midazolam Rescue Confidence† Interval on the Difference vs. Placebo Mean (SD) Total Dose (mg) of Rescue Midazolam Required Confidence† Intervals of the Mean Rescue Dose - * Based on ITT population defined as all randomized and treated patients.
- † Normal approximation to the binomial with continuity correction.
Study 1
Dexmedetomidine 0.5 mcg/kg
134
40
37 (27, 48)
1.4 (1.7)
-2.7
(-3.4, -2.0)Dexmedetomidine 1 mcg/kg
129
54
51 (40, 62)
0.9 (1.5)
-3.1
(-3.8, -2.5)Placebo
63
3
–
4.1 (3.0)
–
Study 2
Dexmedetomidine 1 mcg/kg
55
53
39 (20, 57)
1.1 (1.5)
-1.8
(-2.7, -0.9)Placebo
50
14
–
2.9 (3.0)
–
Pediatric Patients
The safety and efficacy of PRECEDEX for sedation of non-intubated pediatric patients aged 1 month to less than 17 years undergoing MRI scans was evaluated in one randomized, double-blind, dose-ranging, dose‑controlled multicenter clinical trial utilizing 3 different PRECEDEX dosages. The sedative properties of PRECEDEX were evaluated by age group and by the percent of pediatric patients at the high dose level versus the low dose level who did not require concomitant propofol to complete the MRI scan.
A total of 122 pediatric patients were randomized to the PRECEDEX low dose group (42 of 122), the middle dose group (42 of 122) or the high dose treatment group (38 of 122). All patients received a PRECEDEX loading dose infusion over 10 minutes followed by a maintenance infusion for the duration of the MRI scan (Table 17). If an adequate level of sedation was not achieved within 5 minutes after the start of the PRECEDEX maintenance infusion, patients could receive concomitant propofol as needed based on clinical judgment to achieve and/or maintain adequate sedation.
Table 17: PRECEDEX Loading and Maintenance Doses High Dose
Middle Dose
Low Dose
Age
Loading Dose
(10 min)
Maintenance Dose
Loading Dose
(10 min)
Maintenance Dose
Loading Dose
(10 min)
Maintenance Dose
1 month to less than 2 years
1.5 mcg/kg
1.5 mcg/kg/h
1 mcg/kg
1 mcg/kg/h
0.5 mcg/kg
0.5 mcg/kg/h
2 to less than 17 years
2 mcg/kg
1.5 mcg/kg/h
1.2 mcg/kg
1 mcg/kg/h
0.5 mcg/kg
0.5 mcg/kg/h
The primary efficacy results from this pediatric procedural sedation study are summarized in Table 18. In the combined age group, the percent of pediatric patients not requiring concomitant propofol was 14.3% (6/42) in the low dose group and 63.2% (24/38) in the high dose group. The percentage of patients at the PRECEDEX high dose who completed the MRI without concomitant propofol was statistically greater than the percentage in the PRECEDEX low dose group (p<0.001).
Table 18: Percent of Pediatric Patients not Requiring Concomitant Propofol by Age Group and Overall - * Exact 95% CI of proportion of subjects not requiring propofol in each dose level.
- † p-values; CI is confidence interval of odds ratio.
Age Group
High Dose
Middle Dose
Low Dose
High Dose vs Low Dose
n (%)
95% CI*
n (%)
95% CI*
n (%)
95% CI*
Odds Ratio
95% CI†
p-value†
1 month to less than 2 years
N=59
9/18 (50.0%)
(0.26, 0.74)
2/21 (9.5%)
(0.01, 0.30)
2/20 (15.0%)
(0.03, 0.38)
0.18 (0.04, 0.82)
0.022
2 to less than 17 years
N=63
15/20 (75.0%)
(0.51, 0.91)
13/21 (61.9%)
(0.38, 0.82)
3/22 (13.6%)
(0.03, 0.35)
0.05 (0.01, 0.26)
<0.001
Overall
N=122
24/38 (63.2%)
(0.46, 0.78)
15/42 (35.7%)
(0.22, 0.52)
6/42 (14.3%)
(0.05, 0.29)
0.10 (0.03, 0.29)
<0.001
Secondarily, the sedative properties were also evaluated by examining the percent of time at a target sedation score using the Pediatric Sedation State Scale (PSSS). The PSSS is a validated 6-point scale specifically designed for evaluating pediatric patients undergoing sedation for diagnostic and therapeutic procedures. The PSSS measures the effectiveness and quality of procedural sedation in children. The target sedation level was indicated by a PSSS score of 2 (i.e., patient is quiet [asleep or awake], not moving during procedure, has no frown [or brow furrow] indicating pain or anxiety and no verbalization of any complaint).
In the PRECEDEX high dose group, pediatric patients in both the combined and individual age group were at the target sedation rating scale score (PSSS of 2) for a mean >87% of the time during the PRECEDEX maintenance infusion. In both the combined and individual age group, an increase in the percentage of time at the target sedation rating scale score (PSSS of 2) was observed with increasing PRECEDEX dosage.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
Store at 20°C to 25°C (68°F to 77°F); excursions permitted between 15°C to 30°C (59°F to 86°F). [See USP Controlled Room Temperature.]
Do not use if product is discolored or if precipitate matter is present.
PRECEDEX (dexmedetomidine hydrochloride in 0.9% Sodium Chloride) injection (4 mcg/mL) is clear and colorless. The strength is based on the dexmedetomidine base. Discard unused portion.
Unit of Sale
Concentration
NDC: 0409-3301-10
Carton of 10 single‑dose clear glass vials
80 mcg/20 mL
(4 mcg/mL)
NDC: 0409-1454-20
Tray of 20 single‑dose clear glass bottles
200 mcg/50 mL
(4 mcg/mL)
NDC: 0409-1174-10
Tray of 10 single‑dose clear glass bottles
400 mcg/100 mL
(4 mcg/mL)
NDC: 0409-2815-01
Carton containing 1 single‑dose clear glass bottle
1,000 mcg/250 mL
(4 mcg/mL)
-
17 PATIENT COUNSELING INFORMATION
PRECEDEX is indicated for short-term intravenous sedation. Dosage must be individualized and titrated to the desired clinical effect. Blood pressure, heart rate and oxygen levels will be monitored both continuously during the infusion of PRECEDEX and as clinically appropriate after discontinuation.
- When PRECEDEX is infused for more than 6 hours, patients should be informed to report nervousness, agitation, and headaches that may occur for up to 48 hours.
- Additionally, patients should be informed to report symptoms that may occur within 48 hours after the administration of PRECEDEX.
- such as: weakness, confusion, excessive sweating, weight loss, abdominal pain, salt cravings, diarrhea, constipation, dizziness or light-headedness.
- Advise breastfeeding mothers who were exposed to PRECEDEX to monitor breastfed neonates for irritability [see Use in Specific Populations (8.2)].
- SPL UNCLASSIFIED SECTION
-
PRINCIPAL DISPLAY PANEL - 50 mL Bottle Label
50 mL
Rx only
NDC: 0409-1454-01Single-dose bottle. Discard unused portion.
Precedex™
Dexmedetomidine HCl
in 0.9% Sodium Chloride
Injection200 mcg/50 mL
(4 mcg/mL)For Intravenous Use
-
PRINCIPAL DISPLAY PANEL - 50 mL Bottle Tray
20 Units X 50 mL
Rx only
NDC: 0409-1454-20
Contains 20 of NDC: 0409-1454-01Single-dose bottle. Discard unused portion.
Precedex™
Dexmedetomidine HCl
in 0.9% Sodium Chloride Injection200 mcg/50 mL
(4 mcg/mL)For Intravenous Use
novaplus™
-
PRINCIPAL DISPLAY PANEL - 20 mL Vial Label
20 mL
NDC: 0409-3301-01Single-dose vial.
Discard unused portion.Precedex™
Dexmedetomidine HCl in
0.9% Sodium Chloride Injection80 mcg/20 mL
(4 mcg/mL) -
PRINCIPAL DISPLAY PANEL - 20 mL Vial Carton
10 units X 20 mL
Rx only
NDC: 0409-3301-10
Contains 10 of NDC: 0409-3301-01Single-dose vial. Discard unused portion.
Precedex™
Dexmedetomidine HCl in 0.9% Sodium Chloride Injection80 mcg/20 mL
(4 mcg/mL)For Intravenous Use
novaplus™
-
PRINCIPAL DISPLAY PANEL - 100 mL Bottle Label
100 mL
Rx only
NDC: 0409-1174-01Single-dose bottle. Discard unused portion.
Precedex™
Dexmedetomidine HCl
in 0.9% Sodium Chloride
Injection400 mcg/100 mL
(4 mcg/mL)For Intravenous Use
-
PRINCIPAL DISPLAY PANEL - 100 mL Bottle Tray
10 Units X 100 mL
Rx only
NDC: 0409-1174-10
Contains 10 of NDC: 0409-1174-01Single-dose bottle. Discard unused portion.
Precedex™
Dexmedetomidine HCl
in 0.9% Sodium Chloride Injection400 mcg/100 mL
(4 mcg/mL)For Intravenous Use
novaplus™
-
PRINCIPAL DISPLAY PANEL - 250 mL Bottle Label
250 mL
Rx only
NDC: 0409-2815-01Single-dose bottle. Discard unused portion.
Precedex™
Dexmedetomidine HCl
in 0.9% Sodium
Chloride Injection1000 mcg/250 mL
(4 mcg/mL)For Intravenous Use
-
PRINCIPAL DISPLAY PANEL - 250 mL Bottle Carton
1 Unit X 250 mL
Rx only
NDC: 0409-2815-01Single-dose bottle.
Discard unused portion.Precedex™
Dexmedetomidine HCl
in 0.9% Sodium Chloride
Injection1000 mcg/250 mL
(4 mcg/mL)For Intravenous Use
novaplus™
-
INGREDIENTS AND APPEARANCE
PRECEDEX
dexmedetomidine hydrochloride injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0409-1454 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength DEXMEDETOMIDINE HYDROCHLORIDE (UNII: 1018WH7F9I) (DEXMEDETOMIDINE - UNII:67VB76HONO) DEXMEDETOMIDINE 4 ug in 1 mL Inactive Ingredients Ingredient Name Strength SODIUM CHLORIDE (UNII: 451W47IQ8X) 9 mg in 1 mL WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0409-1454-20 20 in 1 TRAY 03/07/2022 1 NDC: 0409-1454-01 50 mL in 1 BOTTLE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA021038 03/07/2022 PRECEDEX
dexmedetomidine hydrochloride injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0409-3301 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength DEXMEDETOMIDINE HYDROCHLORIDE (UNII: 1018WH7F9I) (DEXMEDETOMIDINE - UNII:67VB76HONO) DEXMEDETOMIDINE 4 ug in 1 mL Inactive Ingredients Ingredient Name Strength SODIUM CHLORIDE (UNII: 451W47IQ8X) 9 mg in 1 mL WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0409-3301-10 10 in 1 CARTON 03/07/2022 1 NDC: 0409-3301-01 20 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA021038 03/07/2022 PRECEDEX
dexmedetomidine hydrochloride injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0409-1174 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength DEXMEDETOMIDINE HYDROCHLORIDE (UNII: 1018WH7F9I) (DEXMEDETOMIDINE - UNII:67VB76HONO) DEXMEDETOMIDINE 4 ug in 1 mL Inactive Ingredients Ingredient Name Strength SODIUM CHLORIDE (UNII: 451W47IQ8X) 9 mg in 1 mL WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0409-1174-10 10 in 1 TRAY 03/07/2022 1 NDC: 0409-1174-01 100 mL in 1 BOTTLE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA021038 03/07/2022 PRECEDEX
dexmedetomidine hydrochloride injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0409-2815 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength DEXMEDETOMIDINE HYDROCHLORIDE (UNII: 1018WH7F9I) (DEXMEDETOMIDINE - UNII:67VB76HONO) DEXMEDETOMIDINE 4 ug in 1 mL Inactive Ingredients Ingredient Name Strength SODIUM CHLORIDE (UNII: 451W47IQ8X) 9 mg in 1 mL WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0409-2815-01 1 in 1 CARTON 01/17/2023 1 250 mL in 1 BOTTLE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA021038 01/17/2023 Labeler - Hospira, Inc. (141588017) Establishment Name Address ID/FEI Business Operations Hospira, Inc. 030606222 ANALYSIS(0409-1454, 0409-3301, 0409-1174) , MANUFACTURE(0409-1454, 0409-3301, 0409-1174) , PACK(0409-1454, 0409-3301, 0409-1174) , LABEL(0409-1454, 0409-3301, 0409-1174) Establishment Name Address ID/FEI Business Operations Hospira, Inc. 093132819 ANALYSIS(0409-1454, 0409-3301, 0409-1174) , MANUFACTURE(0409-1454, 0409-3301, 0409-1174) , PACK(0409-1454, 0409-3301, 0409-1174) , LABEL(0409-1454, 0409-3301, 0409-1174)








Trademark Results [Precedex]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 PRECEDEX 75383541 2419482 Live/Registered |
Orion Corporation 1997-11-03 |
 PRECEDEX 75197776 not registered Dead/Abandoned |
Orion Corporation 1996-11-13 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.