SIGNIFOR- pasireotide injection
Signifor by
Drug Labeling and Warnings
Signifor by is a Prescription medication manufactured, distributed, or labeled by Novartis Pharmaceuticals Corporation. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use SIGNIFOR safely and effectively. See full prescribing information for SIGNIFOR.
SIGNIFOR® (pasireotide) injection, for subcutaneous use
Initial U.S. Approval: 2012
INDICATIONS AND USAGE
SIGNIFOR is a somatostatin analog indicated for the treatment of adult patients with Cushing’s disease for whom pituitary surgery is not an option or has not been curative (1)
DOSAGE AND ADMINISTRATION
- Recommended initial dosage is either 0.6 mg or 0.9 mg by subcutaneous injection twice a day; recommended dosage range is 0.3 mg to 0.9 mg twice a day (2.1)
- Titrate dosage based on treatment response [clinically meaningful reduction in 24-hour urinary free cortisol (UFC) and/or improvements in signs and symptoms of disease] and tolerability (2.1)
- Testing Prior to Dosing: fasting plasma glucose (FPG), hemoglobin A1c (HbA1c), liver tests, electrocardiogram (ECG), gallbladder ultrasound, and serum potassium and magnesium levels (2.2)
- Patients With Hepatic Impairment:
DOSAGE FORMS AND STRENGTHS
Injection: 0.3 mg/mL, 0.6 mg/mL, and 0.9 mg/mL in a single-dose ampule (3)
CONTRAINDICATIONS
None (4)
WARNINGS AND PRECAUTIONS
- Hypocortisolism: Decreases in circulating levels of cortisol may occur resulting in biochemical and/or clinical hypocortisolism. SIGNIFOR dose reduction or interruption and/or adding a low-dose short-term glucocorticoid may be necessary (5.1)
- Hyperglycemia and Diabetes (occurs with initiation): Intensive glucose monitoring is recommended and may require initiation or adjustment of anti-diabetic treatment per standard of care (5.2)
- Bradycardia and QT Prolongation: Use with caution in at-risk patients; ECG testing prior to dosing and on treatment (5.3, 7.1)
- Liver Test Elevations: Evaluate liver tests prior to and during treatment (5.4)
- Cholelithiasis and Complications of Cholelithiasis: Monitor periodically. Discontinue if complications of cholelithiasis are suspected (5.5)
ADVERSE REACTIONS
Most common adverse reactions occurring in ≥ 20% of patients are diarrhea, nausea, hyperglycemia, cholelithiasis, headache, abdominal pain, fatigue, and diabetes mellitus (6)
To report SUSPECTED ADVERSE REACTIONS, contact Novartis Pharmaceuticals Corporation at 1-888-669-6682 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.DRUG INTERACTIONS
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 1/2020
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Cushing’s Disease
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage Range
2.2 Recommendations Prior to Initiation of SIGNIFOR
2.3 Dosage in Patients With Hepatic Impairment
2.4 Important Administration Instructions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hypocortisolism
5.2 Hyperglycemia and Diabetes
5.3 Bradycardia and QT Prolongation
5.4 Liver Test Elevations
5.5 Cholelithiasis and Complications of Cholelithiasis
5.6 Monitoring for Deficiency of Pituitary Hormones
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Effects of Other Drugs on SIGNIFOR
7.2 Effects of SIGNIFOR on Other Drugs
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
8.7 Renal Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2
DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage Range
The recommended dosage range of SIGNIFOR is 0.3 mg to 0.9 mg by subcutaneous injection twice a day. The recommended initial dose is either 0.6 mg or 0.9 mg twice a day. Titrate dose based on response and tolerability.
Patients should be evaluated for a treatment response [clinically meaningful reduction in 24-hour urinary free cortisol (UFC) levels and/or improvement in signs or symptoms of the disease] and should continue receiving therapy with SIGNIFOR as long as benefit is derived [see Clinical Studies (14)]. Maximum UFC reduction is typically seen by two months of treatment [see Clinical Studies (14)]. For patients who are started on 0.6 mg twice a day, a dosage increase to 0.9 mg twice a day may be considered based on the response to the treatment, as long as the 0.6 mg dosage is well tolerated by the patient.
Management of suspected adverse reactions may require temporary dose reduction of SIGNIFOR. Dose reduction by 0.3 mg decrements per injection is suggested.
2.2 Recommendations Prior to Initiation of SIGNIFOR
Prior to the start of SIGNIFOR, patients should have baseline levels of the following:
- fasting plasma glucose (FPG) [see Warnings and Precautions (5.2)]
- hemoglobin A1c (HbA1c) [see Warnings and Precautions (5.2)]
- liver tests [see Warnings and Precautions (5.4)]
- serum potassium and magnesium levels [see Warnings and Precautions (5.3)]
Patients should also have a baseline electrocardiogram (ECG) and gallbladder ultrasound [see Warnings and Precautions (5.3, 5.5)].
Treatment of patients with poorly controlled diabetes mellitus should be intensively optimized with anti-diabetic therapy prior to starting SIGNIFOR [see Warnings and Precautions (5.2)].
2.3 Dosage in Patients With Hepatic Impairment
For patients with moderate hepatic impairment (Child-Pugh B), the recommended initial dosage is 0.3 mg twice a day and the maximum dosage is 0.6 mg twice a day. Avoid the use of SIGNIFOR in patients with severe hepatic impairment (Child-Pugh C) [see Use in Specific Populations (8.6)].
2.4 Important Administration Instructions
Instruct patients to:
- Refer to the FDA-approved patient labeling (Instructions for Use) for detailed administration instructions.
- Prior to injection, visually inspect the product for particulate matter and discoloration. Do not use if particulates and/or discoloration are observed.
- Avoid injection in sites showing signs of inflammation or irritation.
- Prior to injection, gently pinch the skin at the injection site and hold the needle/syringe at an angle of approximately 45 degrees.
- Administer SIGNIFOR subcutaneously by self-injection into the top of the thigh or the abdomen.
- Avoid multiple subcutaneous injections at the same site within short periods of time. Use of the same injection site for 2 consecutive injections is not recommended.
- If a dose of SIGNIFOR is missed, the next injection should be administered at the scheduled time. Do not double doses to make up for a missed dose.
- fasting plasma glucose (FPG) [see Warnings and Precautions (5.2)]
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5
WARNINGS AND PRECAUTIONS
5.1 Hypocortisolism
Treatment with SIGNIFOR leads to suppression of adrenocorticotropic hormone (ACTH) secretion in Cushing’s disease. Suppression of ACTH may lead to a decrease in circulating levels of cortisol and potentially hypocortisolism.
Monitor and instruct patients on the signs and symptoms associated with hypocortisolism (e.g., weakness, fatigue, anorexia, nausea, vomiting, hypotension, hyponatremia, or hypoglycemia). If hypocortisolism occurs, consider temporary dose reduction or interruption of treatment with SIGNIFOR, as well as temporary, exogenous glucocorticoid replacement therapy.
5.2 Hyperglycemia and Diabetes
Blood glucose elevations have been seen in healthy volunteers and patients treated with SIGNIFOR. In the clinical study [see Clinical Studies (14)], patients developed pre-diabetes and diabetes. Nearly all patients in the study, including those with normal glucose status at baseline, pre-diabetes, and diabetes, developed worsening glycemia in the first two weeks of treatment. Cushing’s disease patients with poor glycemic control (HbA1c > 8%) may be at a higher risk of developing severe hyperglycemia and associated complications, e.g., ketoacidosis.
Assess the patient’s glycemic status prior to starting treatment with SIGNIFOR. In patients with uncontrolled diabetes mellitus, optimize anti-diabetic therapy prior to SIGNIFOR initiation. Glycemic monitoring should be done every week for the first two to three months and periodically thereafter, as well as over the first two to four weeks after any dose increase. If hyperglycemia develops, initiate or adjust anti-diabetic treatment per standard of care. If uncontrolled hyperglycemia persists despite appropriate treatment, reduce the dose or discontinue SIGNIFOR and perform glycemic monitoring according to clinical practice. Patients who were initiated on anti-diabetic treatment as a result of SIGNIFOR require closer monitoring after discontinuation of SIGNIFOR, especially if the anti-diabetic therapy has a risk of causing hypoglycemia.
5.3 Bradycardia and QT Prolongation
Bradycardia
Bradycardia has been reported with the use of SIGNIFOR [see Adverse Reactions (6)]. Patients with cardiac disease and/or risk factors for bradycardia, such as history of clinically significant bradycardia, high-grade heart block, or concomitant use of drugs associated with bradycardia, should be carefully monitored. Dose adjustments of beta-blockers, calcium channel blockers, or correction of electrolyte disturbances may be necessary.
QT Prolongation
SIGNIFOR is associated with QT prolongation. In two thorough QT studies with SIGNIFOR, QT prolongation occurred at therapeutic and supra-therapeutic doses. SIGNIFOR should be used with caution in patients who are at significant risk of developing prolongation of QTc, such as those:
- with congenital long QT prolongation.
- with uncontrolled or significant cardiac disease, including recent myocardial infarction, congestive heart failure, unstable angina, or clinically significant bradycardia.
- on anti-arrhythmic therapy or other substances that are known to lead to QT prolongation.
- with hypokalemia and/or hypomagnesemia.
A baseline ECG is recommended prior to initiating therapy with SIGNIFOR and monitoring for an effect on the QTc interval is advisable. Hypokalemia and hypomagnesemia must be corrected prior to SIGNIFOR administration and should be monitored periodically during therapy.
5.4 Liver Test Elevations
In the Phase III trial, 5% of patients had an alanine aminotransferase (ALT) or aspartate aminotransferase (AST) level greater than 3 times the upper limit of normal (ULN). In the entire clinical development program of SIGNIFOR, there were 4 cases of concurrent elevations in ALT greater than 3 x ULN and bilirubin greater than 2 x ULN: one patient with Cushing’s disease and 3 healthy volunteers [see Adverse Reactions (6)]. In these cases, total bilirubin elevations were seen either concomitantly or preceding the transaminase elevation.
Monitoring of liver tests should be done after 1- to 2 weeks on treatment, then monthly for 3 months, and every 6 months thereafter. If ALT is normal at baseline and elevations of ALT of 3-5 times the ULN are observed on treatment, repeat the test within a week or within 48 hours if exceeding 5 times ULN. If ALT is abnormal at baseline and elevations of ALT of 3 to 5 times the baseline values are observed on treatment, repeat the test within a week or sooner if exceeding 5 times ULN. Tests should be done in a laboratory that can provide same-day results. If the values are confirmed or rising, interrupt SIGNIFOR treatment and investigate for probable cause of the findings, which may or may not be SIGNIFOR-related. Serial measures of ALT, AST, alkaline phosphatase, and total bilirubin, should be done weekly, or more frequently, if any value exceeds 5 times the baseline value in case of abnormal baselines, or 5 times the ULN in case of normal baselines. If resolution of abnormalities to normal or near normal occurs, resuming treatment with SIGNIFOR may be done cautiously, with close observation, and only if some other likely cause has been found.
5.5 Cholelithiasis and Complications of Cholelithiasis
Cholelithiasis has been frequently reported in clinical studies with SIGNIFOR [see Adverse Reactions (6)]. There have been postmarketing reports of cholelithiasis (gallstones) resulting in complications, including cholecystitis or cholangitis and requiring cholecystectomy in patients taking SIGNIFOR. Ultrasonic examination of the gallbladder before, and periodically during SIGNIFOR therapy is recommended. If complications of cholelithiasis are suspected, discontinue SIGNIFOR and treat appropriately.
5.6 Monitoring for Deficiency of Pituitary Hormones
As the pharmacological activity of SIGNIFOR mimics that of somatostatin, inhibition of pituitary hormones, other than ACTH, may occur. Monitoring of pituitary function [e.g., thyroid-stimulating harmone (TSH)/free T4, GH/IGF-1] should occur prior to initiation of therapy with SIGNIFOR and periodically during treatment should be considered as clinically appropriate. Patients who have undergone transsphenoidal surgery and pituitary irradiation are particularly at increased risk for deficiency of pituitary hormones.
-
6
ADVERSE REACTIONS
Clinically significant adverse reactions that appear in other sections of the labeling include:
- Hypocortisolism [see Warnings and Precautions (5.1)]
- Hyperglycemia and Diabetes [see Warnings and Precautions (5.2)]
- Bradycardia and QT prolongation [see Warnings and Precautions (5.3)]
- Liver Test Elevations [see Warnings and Precautions (5.4)]
- Cholelithiasis and Complications of Cholelithiasis [see Warnings and Precautions (5.5)]
- Pituitary Hormone Deficiency [see Warnings and Precautions (5.6)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in clinical trials of a drug cannot be directly compared to rates in clinical trials of another drug and may not reflect the rates observed in practice.
A total of 162 Cushing’s disease patients were exposed to SIGNIFOR in the Phase III study [see Clinical Studies (14)]. At study entry, patients were randomized to receive twice a day doses of either 0.6 mg or 0.9 mg of SIGNIFOR given subcutaneously. The mean age of patients was approximately 40 years old with a predominance of female patients (78%). The majority of the patients had persistent or recurrent Cushing’s disease (83%) and few patients (≤ 5%) in either treatment group had received previous pituitary irradiation. The median exposure to the treatment was 10.4 months (0.03-37.8) with 68% of patients having at least 6-months exposure.
In the Phase III trial, adverse reactions were reported in 98% of patients. The most common adverse reactions (frequency ≥ 20% in either group) were diarrhea, nausea, hyperglycemia, cholelithiasis, headache, abdominal pain, fatigue, and diabetes mellitus. There were no deaths during the study. Serious adverse events were reported in 25% of patients. Adverse events leading to study discontinuation were reported in 17% of patients.
Adverse reactions with an overall frequency higher than 5% are presented in Table 1 by randomized dose group and overall. Adverse reactions are ranked by frequency, with the most frequent reactions listed first.
Table 1 - Adverse Reactions [n (%)] With an Overall Frequency of More Than 5% in the Combined Dose Group in the Phase III Study in Cushing’s Disease Patients SIGNIFOR
0.6 mg twice a day
N = 82SIGNIFOR
0.9 mg twice a day
N = 80Overall
N = 162Diarrhea 48 (59) 46 (58) 94 (58) Nausea 38 (46) 46 (58) 84 (52) Hyperglycemia 31 (38) 34 (43) 65 (40) Cholelithiasis 25 (30) 24 (30) 49 (30) Headache 23 (28) 23 (29) 46 (28) Abdominal pain 19 (23) 20 (25) 39 (24) Fatigue 12 (15) 19(24) 31 (19) Diabetes mellitus 13 (16) 16 (20) 29 (18) Injection-site reactions 14 (17) 14 (18) 28 (17) Nasopharyngitis 10 (12) 11 (14) 21 (13) Alopecia 10 (12) 10 (13) 20 (12) Asthenia 13 (16) 5 (6) 18 (11) Glycosylated hemoglobin increased 10 (12) 8 (10) 18 (11) Alanine aminotransferase increased 11 (13) 6 (8) 17 (10) Gamma-glutamyl transferase increased 10 (12) 7 (9) 17 (10) Edema peripheral 9 (11) 8 (10) 17 (10) Abdominal pain upper 10 (12) 6 (8) 16 (10) Decreased appetite 7 (9) 9 (11) 16 (10) Hypercholesterolemia 7 (9) 9 (11) 16 (10) Hypertension 8 (10) 8 (10) 16 (10) Dizziness 8 (10) 7 (9) 15 (9) Hypoglycemia 12 (15) 3 (4) 15 (9) Type 2 diabetes mellitus 10 (12) 5 (6) 15 (9) Anxiety 5 (6) 9 (11) 14 (9) Influenza 9 (11) 5 (6) 14 (9) Insomnia 3 (4) 11 (14) 14 (9) Myalgia 10 (12) 4 (5) 14 (9) Arthralgia 5 (6) 8 (10) 13 (8) Pruritus 6 (7) 7 (9) 13 (8) Lipase increased 7 (9) 5 (6) 12 (7) Constipation 7 (9) 4 (5) 11 (7) Hypotension 5 (6) 6 (8) 11 (7) Vomiting 3 (4) 8 (10) 11 (7) Back pain 4 (5) 6 (8) 10 (6) Dry skin 5 (6) 5 (6) 10 (6) Electrocardiogram QT prolonged 5 (6) 5 (6) 10 (6) Hypokalemia 6 (7) 4 (5) 10 (6) Pain in extremity 6 (7) 4 (5) 10 (6) Sinus bradycardia 8 (10) 2 (3) 10 (6) Vertigo 4 (5) 6 (8) 10 (6) Abdominal distension 4 (5) 5 (6) 9 (6) Adrenal insufficiency 4 (5) 5 (6) 9 (6) Aspartate aminotransferase increased 6 (7) 3 (4) 9 (6) Blood glucose increased 6 (7) 3 (4) 9 (6) Other notable adverse reactions which occurred with a frequency less than 5% were: anemia (4%), blood amylase increased (2%), and prothrombin time prolonged (2%).
Gastrointestinal Disorders
Gastrointestinal disorders, predominantly diarrhea, nausea, abdominal pain, and vomiting were reported frequently in the Phase III trial (see Table 1). These events began to develop primarily during the first month of treatment with SIGNIFOR and required no intervention.
Hyperglycemia and Diabetes
Hyperglycemia-related terms were reported frequently in the Phase III trial. For all patients, these terms included: hyperglycemia (40%), diabetes mellitus (18%), increased HbA1c (11%), type 2 diabetes mellitus (9%). In general, increases in FPG and HbA1c were seen soon after initiation of SIGNIFOR and were sustained during the treatment period. In the SIGNIFOR 0.6 mg group, mean FPG levels increased from 98.6 mg/dL at baseline to 125.1 mg/dL at Month 6. In the SIGNIFOR 0.9 mg group, mean fasting FPG levels increased from 97.0 mg/dL at baseline to 128.0 mg/dL at Month 6. In the SIGNIFOR 0.6 mg group, HbA1c increased from 5.8% at baseline to 7.2% at Month 6. In the SIGNIFOR 0.9 mg group, HbA1c increased from 5.8% at baseline to 7.3% at Month 6 [see Warnings and Precautions (5.2)].
At one-month follow-up visits, following discontinuation of SIGNIFOR, mean FPG and HbA1c levels decreased but remained above baseline values. Long-term follow-up data are not available.
Elevated Liver Tests
In the Phase III trial, there were transient mean elevations in aminotransferase values in patients treated with SIGNIFOR. Mean values returned to baseline levels by Month 4 of treatment. The elevations were not associated with clinical symptoms of hepatic disease.
In the clinical development program of SIGNIFOR, there were 4 patients with concurrent elevations in ALT greater than 3 x ULN and bilirubin greater than 2 x ULN: one patient with Cushing’s disease and 3 healthy volunteers [see Warnings and Precautions (5.4)]. In all 4 cases, the elevations were noted within the first 10 days of treatment. In all of these cases, total bilirubin elevations were seen either concomitantly or preceding the transaminase elevation. The patient with Cushing’s disease developed jaundice. All 4 cases had resolution of the laboratory abnormalities with discontinuation of SIGNIFOR.
Hypocortisolism
Cases of hypocortisolism were reported in the Phase III study in Cushing’s disease patients [see Adverse Reactions (6), Clinical Studies (14)]. The majority of cases were manageable by reducing the dose of SIGNIFOR and/or adding low-dose, short-term glucocorticoid therapy [see Warnings and Precautions (5.1)].
Injection-Site Reactions
Injection-site reactions were reported in 17% of patients enrolled in the Phase III trial in Cushing’s disease. The events were most frequently reported as local pain, erythema, hematoma, hemorrhage, and pruritus. These events resolved spontaneously and required no intervention.
Thyroid Function
Hypothyroidism, with the use of SIGNIFOR, was reported for seven patients participating in the Phase III study in Cushing’s disease. All seven patients presented with a TSH close to or below the lower limit at study entry, which precludes establishing a conclusive relationship between the adverse event and the use of SIGNIFOR.
Other Abnormal Laboratory Findings
Asymptomatic and reversible elevations in lipase and amylase were observed in patients receiving SIGNIFOR in clinical studies. Pancreatitis is a potential adverse reaction associated with the use of somatostatin analogs due to the association between cholelithiasis and acute pancreatitis.
For hemoglobin levels, mean decreases that remained within normal range were observed. Also, post-baseline elevations in prothrombin time (PT) and partial thromboplastin time (PTT) were noted in 33% and 47% of patients, respectively. The PT and PTT elevations were minimal.
These laboratory findings are of unclear clinical significance.
6.2 Postmarketing Experience
Additional adverse reactions have been identified during postapproval use of SIGNIFOR. Because these reactions are reported voluntarily from a population of uncertain size, it is generally not possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- Cholelithiasis resulting in complications, including cholecystitis and cholangitis, which have sometimes required cholecystectomy
- Hypocortisolism [see Warnings and Precautions (5.1)]
-
7
DRUG INTERACTIONS
7.1 Effects of Other Drugs on SIGNIFOR
Drugs That Prolong QT
Coadministration of drugs that prolong the QT interval with SIGNIFOR may have additive effects on the prolongation of the QT interval. Caution is required when coadministering SIGNIFOR with drugs that may prolong the QT interval [see Warnings and Precautions (5.3)].
7.2 Effects of SIGNIFOR on Other Drugs
Cyclosporine
Concomitant administration of cyclosporine with pasireotide may decrease the relative bioavailability of cyclosporine and, therefore, dose adjustment of cyclosporine to maintain therapeutic levels may be necessary.
Bromocriptine
Coadministration of somatostatin analogues with bromocriptine may increase the blood levels of bromocriptine. Dose reduction of bromocriptine may be necessary.
-
8
USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
The limited data with SIGNIFOR in pregnant women are insufficient to inform a drug-associated risk for major birth defects and miscarriage. In embryo-fetal development studies in rabbits, findings indicating developmental delay were observed with subcutaneous administration of pasireotide during organogenesis at doses less than the exposure in humans at the highest recommended dose; maternal toxicity was not observed at this dose (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2%-4% and 15%-20%, respectively.
Data
Animal Data
In embryo-fetal development studies in rats given 1, 5, and 10 mg/kg/day subcutaneously throughout organogenesis, maternal toxicity was observed at all doses, including the lowest dose tested which had exposures 4 times higher than that at the maximum therapeutic dose based on area under the curve (AUC) comparisons across species. An increased incidence of early/total resorptions and malrotated limbs was observed in rats at 10 mg/kg/day. At 10 mg/kg/day in rats, the maternal systemic exposure (AUC) was 42179 ng*hr/mL, approximately 144 times the exposure in humans at the highest recommended dose of 900 mcg SIGNIFOR administered as a subcutaneous injection twice a day.
In embryo-fetal development studies in rabbits given 0.05, 1, and 5 mg/kg/day subcutaneously through organogenesis, maternal toxicity was observed at 1 mg/kg/day, at a maternal systemic exposure (AUC) of 1906 ng*hr/mL, approximately 7 times higher than the maximum human therapeutic exposure. An increased incidence of unossified forepaw phalanx, indicative of a developmental retardation, was observed in rabbits at 0.05 mg/kg/day, with maternal systemic exposures less than the systemic exposure in humans at the highest recommended dose.
In pre- and post-natal developmental studies in rats given subcutaneous doses of 2, 5, and 10 mg/kg/day during gestation through lactation and weaning, maternal toxicity was observed at all doses, including the lowest dose (12 times higher than the maximum therapeutic dose based on surface area comparisons across species). Retardation of physiological growth, attributed to GH inhibition was observed at 2 mg/kg/day during a pre- and post-natal study in rats. After weaning, body weight gains in the rat pups (F1 generation) exposed to pasireotide were comparable to controls, showing reversibility of this developmental delay.
8.2 Lactation
Risk Summary
There is no information available on the presence of SIGNIFOR in human milk, the effects of the drug on the breastfed infant, or the effects of the drug on milk production. Studies show that pasireotide administered subcutaneously passes into the milk of lactating rats; however, due to species-specific differences in lactation physiology, animal data may not reliably predict drug levels in human milk (see Data). The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for SIGNIFOR and any potential adverse effects on the breastfed child from SIGNIFOR or from the underlying maternal condition.
Data
Available data in animals have shown excretion of pasireotide in milk. After a single 1 mg/kg [14C]-pasireotide subcutaneous dose to lactating rats, the transfer of radioactivity into milk was observed. The overall milk:plasma (M/P) exposure ratio of total radioactivity was 0.28, based on AUC0-∞ values.
8.3 Females and Males of Reproductive Potential
Discuss the potential for unintended pregnancy with premenopausal women as the therapeutic benefits of a reduction or normalization of serum cortisol levels in female patients with Cushing’s disease treated with pasireotide may lead to improved fertility.
8.4 Pediatric Use
Safety and effectiveness of SIGNIFOR have not been established in pediatric patients.
8.5 Geriatric Use
Clinical studies of SIGNIFOR did not include sufficient numbers of patients aged 65 and over to determine whether they respond differently from younger patients. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and concomitant disease or other drug therapy [see Clinical Pharmacology (12.3)].
8.6 Hepatic Impairment
Dose adjustment is not required in patients with mild impaired hepatic function (Child-Pugh A), but is required for patients with moderately impaired hepatic function (Child-Pugh B) [see Dosage and Administration (2.3), Clinical Pharmacology (12.3)]. Avoid the use of SIGNIFOR in patients with severe hepatic impairment (Child-Pugh C).
-
10
OVERDOSAGE
No cases of overdosage have been reported in patients with Cushing’s disease receiving SIGNIFOR subcutaneously. Doses up to 2.1 mg twice a day have been used in healthy volunteers with adverse reactions of diarrhea being observed at a high frequency.
In the event of overdosage, it is recommended that appropriate supportive treatment be initiated, as dictated by the patient’s clinical status, until resolution of the symptoms.
Up-to-date information about the treatment of overdose can be obtained from a certified Regional Poison Center. Contact Poison Control (1-800-222-1222) for latest recommendations.
-
11
DESCRIPTION
SIGNIFOR (pasireotide) injection is prepared as a sterile solution of pasireotide diaspartate in a tartaric acid buffer for administration by subcutaneous injection. SIGNIFOR is a somatostatin analog. Pasireotide diaspartate, chemically known as (2-Aminoethyl) carbamic acid (2R,5S,8S,11S,14R,17S,19aS)-11-(4-aminobutyl)-5-benzyl-8-(4-benzyloxybenzyl)-14-(1H-indol-3-ylmethyl)-4,7,10,13,16,19-hexaoxo-17-phenyloctadecahydro-3a,6,9,12,15,18-hexaazacyclopentacyclooctadecen-2-yl ester, di[(S)-2-aminosuccinic acid] salt, is a cyclohexapeptide with pharmacologic properties mimicking those of the natural hormone somatostatin.
The molecular formula of pasireotide diaspartate is C58H66N10O9 2 C4H7NO4 and the molecular weight is 1313.41 g/mol. The structural formula is:
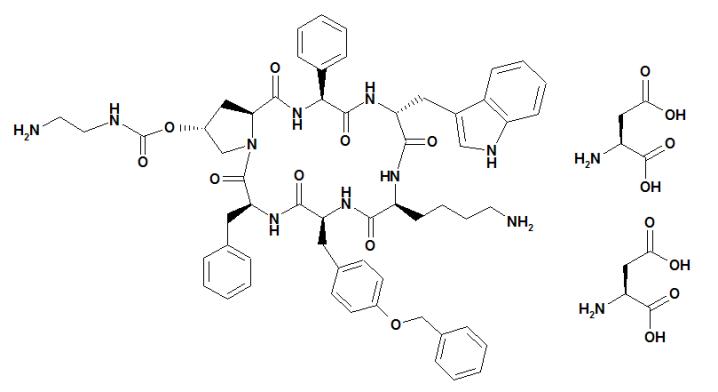
SIGNIFOR is supplied as a sterile solution in a single-dose, 1 mL colorless glass ampule containing pasireotide in 0.3 mg/mL, 0.6 mg/mL, or 0.9 mg/mL strengths for subcutaneous injection.
Each glass ampule contains:
*corresponds to 0.3/0.6/0.9 mg pasireotide base.
Note: Each ampule contains an overfill of 0.1 mL to allow accurate administration of 1 mL from the ampule.0.3 mg 0.6 mg 0.9 mg Pasireotide diaspartate 0.3762* 0.7524* 1.1286* Mannitol 49.50 49.50 49.50 Tartaric acid 1.501 1.501 1.501 Sodium hydroxide ad pH 4.2 ad pH 4.2 ad pH 4.2 Water for injection ad 1 mL ad 1 mL ad 1 mL -
12
CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
SIGNIFOR is an injectable cyclohexapeptide somatostatin analogue. Pasireotide exerts its pharmacological activity via binding to somatostatin receptors (SSTRs). Five human somatostatin receptor subtypes are known: SSTR 1, 2, 3, 4, and 5. These receptor subtypes are expressed in different tissues under normal physiological conditions. Corticotroph tumor cells from Cushing’s disease patients frequently over-express SSTR5 whereas the other receptor subtypes are often not expressed or are expressed at lower levels. Pasireotide binds and activates the SSTRs resulting in inhibition of ACTH secretion, which leads to decreased cortisol secretion.
The binding affinities of endogenous somatostatin and pasireotide are shown in Table 2.
Table 2 - Binding Affinities of Somatostatin (SRIF-14) and Pasireotide to the Five Human Somatostatin Receptor Subtypes (SSTR1-5) Results are the mean ± SEM of IC50 values expressed as nmol/L. Compound SSTR1 SSTR2 SSTR3 SSTR4 SSTR5 Somatostatin (SRIF-14) 0.93 ± 0.12 0.15 ± 0.02 0.56 ± 0.17 1.5 ± 0.4 0.29 ± 0.04 Pasireotide 9.3 ± 0.1 1.0 ± 0.1 1.5 ± 0.3 > 100 0.16 ± 0.01 12.2 Pharmacodynamics
Glucose Metabolism
In a randomized, double-blind mechanism study conducted in healthy volunteers, the development of hyperglycemia with pasireotide at doses of 0.6 mg twice a day and 0.9 mg twice a day was related to significant decreases in insulin secretion as well as incretin hormones (i.e., glucagon-like peptide-1 [GLP-1] and glucose-dependent insulinotropic polypeptide [GIP]) [see Warnings and Precautions (5.2), Adverse Reactions (6.1)].
Cardiac Electrophysiology
QTcI interval was evaluated in a randomized, blinded, crossover study in healthy subjects investigating pasireotide doses of 0.6 mg twice a day and 1.95 mg twice a day. The maximum mean (95% upper confidence bound) placebo-subtracted QTcI change from baseline was 12.7 (14.7) ms and 16.6 (18.6) ms, respectively. Both pasireotide doses decreased heart rate, with a maximum mean (95% lower confidence bound) placebo-subtracted change from baseline of -10.9 (-11.9) beats per minute (bpm) observed at 1.5 hours for pasireotide 0.6 mg twice a day, and -15.2 (-16.5) bpm at 0.5 hours for pasireotide 1.95 mg twice a day. The supra-therapeutic dose (1.95 mg twice a day) produced mean steady-state Cmax values 3.3-fold the mean Cmax for the 0.6 mg twice a day dose in the study.
12.3 Pharmacokinetics
In healthy volunteers, pasireotide demonstrates approximately linear pharmacokinetics (PK) for a dose range from 0.0025 to 1.5 mg. In Cushing’s disease patients, pasireotide demonstrates linear dose-exposure relationship in a dose range from 0.3 mg to 1.2 mg.
Absorption and Distribution
In healthy volunteers, pasireotide peak plasma concentration is reached within Tmax 0.25-0.5 hour. Cmax and AUC are dose-proportional following administration of single and multiple doses.
No studies have been conducted to evaluate the absolute bioavailability of pasireotide in humans. Food effect is unlikely to occur since SIGNIFOR is administered via a parenteral route.
In healthy volunteers, pasireotide is widely distributed with large apparent volume of distribution (Vz/F > 100 L). Distribution between blood and plasma is concentration independent and shows that pasireotide is primarily located in the plasma (91%). Plasma protein binding is moderate (88%) and independent of concentration.
Pasireotide has low passive permeability and is likely to be a substrate of P-glycoprotein (P-gp), but the impact of P-gp on ADME (absorption, distribution, metabolism, excretion) of pasireotide is expected to be low. In clinical testing in healthy volunteers, P-gp inhibition (e.g., verapamil) did not affect the rate or extent of pasireotide availability. Pasireotide is not a substrate of efflux transporter BCRP (breast cancer resistance protein), influx transporter OCT1 (organic cation transporter 1), or influx transporters OATP (organic anion-transporting polypeptide) 1B1, 1B3, or 2B1.
Metabolism and Excretion
Pasireotide was shown to be metabolically stable in human liver and kidney microsomes systems. In healthy volunteers, pasireotide in its unchanged form is the predominant form found in plasma, urine, and feces. Somatropin may increase CYP450 enzymes and, therefore, suppression of growth hormone secretion by somatostatin analogs, including pasireotide may decrease the metabolic clearance of compounds metabolized by CYP450 enzymes.
Pasireotide is eliminated mainly via hepatic clearance (biliary excretion) with a small contribution of the renal route. In a human ADME study 55.9 ± 6.63% of the radioactivity dose was recovered over the first 10 days post dosing, including 48.3 ± 8.16% of the radioactivity in feces and 7.63 ± 2.03% in urine.
The clearance (CL/F) of pasireotide in healthy volunteers and Cushing’s disease patients is ~7.6 L/h and ~3.8 L/h, respectively.
Steady-State Pharmacokinetics
Following multiple subcutaneous doses, pasireotide demonstrates linear pharmacokinetics in the dose range of 0.05 to 0.6 mg once a day in healthy volunteers, and 0.3 mg to 1.2 mg twice a day in Cushing’s disease patients. Based on the accumulation ratios of AUC, the calculated effective half-life (t1/2,eff) in healthy volunteers was approximately 12 hours (on average between 10 and 13 hours for 0.05, 0.2 and 0.6 mg once a day doses).
Specific Populations
Population PK analyses of SIGNIFOR indicate that race, body weight, age, and gender do not have a clinically relevant influence on PK parameters. No dose adjustment is required for demographics.
Hepatic Impairment
In a clinical study with a single subcutaneous dose of 600 mcg pasireotide in subjects with impaired hepatic function (Child-Pugh A, B, and C), subjects with moderate and severe hepatic impairment (Child-Pugh B and C) showed significantly higher exposures than subjects with normal hepatic function. Upon comparison with the control group, AUCinf was increased by 12%, 56%, and 42%; and Cmax increased by 3%, 46%, and 33%, respectively, in the mild, moderate, and severe hepatic impairment groups [see Use in Specific Populations (8.6), Dosage and Administration (2.3)].
Pediatric Patients
No studies have been performed in pediatric patients [see Use in Specific Populations (8.4)].
Geriatric Patients
No clinical pharmacology studies have been performed in geriatric patients.
Renal Impairment
Renal clearance has a minor contribution to the elimination of pasireotide in humans. In a clinical study with a single subcutaneous dose of 900 mcg pasireotide, in subjects with impaired renal function, renal impairment of mild, moderate, or severe degree or end stage renal failure, did not have a significant impact on the pharmacokinetics of pasireotide [see Use in Specific Populations (8.7)].
Drug Interaction Studies
There was no significant drug interaction between pasireotide and metformin, nateglinide or liraglutide.
-
13
NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
A life-time carcinogenicity study was conducted in rats and transgenic mice. Rats were given daily subcutaneous doses of pasireotide at 0.01, 0.05, 0.3 mg/kg/day for 104 weeks. There were no drug-related tumors in rats at exposures up to 7-fold higher than the maximum recommended clinical exposure at the 1.8 mg/day dose. Mice were given subcutaneous doses of pasireotide at 0.5, 1.0, 2.5 mg/kg/day for 26 weeks and did not identify any carcinogenic potential.
Mutagenesis
Pasireotide was not genotoxic in a battery of in vitro assays (Ames mutation test in Salmonella and Escherichia coli and mutation test in human peripheral lymphocytes). Pasireotide was not genotoxic in an in vivo rat bone marrow nucleus test.
Impairment of Fertility
Subcutaneous dosing at 0.1 mg/kg/day before mating and continuing into gestation in rats at exposures less than the human clinical exposure based on body surface area comparisons across species resulted in statistically significant increased implantation loss and decreased viable fetuses, corpora lutea, and implantation sites. Abnormal cycles or acyclicity were observed at 1 mg/kg/day (5-fold higher than the maximum therapeutic exposure based on surface area, comparisons across species).
-
14
CLINICAL STUDIES
A Phase III, multicenter, randomized study was conducted to evaluate the safety and efficacy of two dose levels of SIGNIFOR over a 6-month treatment period in Cushing’s disease patients with persistent or recurrent disease despite pituitary surgery or de novo patients for whom surgery was not indicated or who had refused surgery.
Patients with a baseline 24-hour urine free cortisol (UFC) >1.5 x ULN were randomized to receive a SIGNIFOR dosage of either 0.6 mg subcutaneous twice a day or 0.9 mg subcutaneous twice a day. After 3 months of treatment, patients with a mean 24-hour UFC ≤ 2.0 x ULN and below or equal to their baseline values continued blinded treatment at the randomized dose until Month 6. Patients who did not meet these criteria were unblinded and the dose was increased by 0.3 mg twice a day. After the initial 6 months in the study, patients entered an additional 6-month open-label treatment period. The dosage could be reduced by 0.3 mg twice a day at any time during the study for intolerability.
A total of 162 patients were enrolled in this study. The majority of patients were female (78%) and had persistent or recurrent Cushing’s disease despite pituitary surgery (83%) with a mean age of 40 years. A few patients (4%) in either treatment group received previous pituitary irradiation. The median value of the baseline 24-hour UFC for all patients was 565 nmol/24 hours (normal range 30 to 145 nmol/24 hours). About two-thirds of all randomized patients completed 6 months of treatment.
The primary efficacy endpoint was the proportion of patients who achieved normalization of mean 24-hour UFC levels after 6 months of treatment and did not dose increase during this period.
24-Hour Urinary Free Cortisol Results
At Month 6, the percentages of responders for the primary endpoint were 15% and 26% in the 0.6 mg twice a day and 0.9 mg twice a day groups, respectively (Table 3). The percentages of patients with mUFC ≤ ULN or ≥ 50% reduction from baseline, a less stringent endpoint than the primary endpoint, were 34% in the 0.6 mg twice a day and 41% in the 0.9 mg twice a day groups. Dose increases appeared to have minimal effect on 24-hour UFC response. Mean and median percentage changes in UFC from baseline are presented in Table 3.
Table 3 – 24-Hour Urinary Free Cortisol Study Results at Month 6 in Patients With Cushing’s Disease SIGNIFOR
0.6 mg twice a day
N = 82SIGNIFOR
0.9 mg twice a day
N = 80UFC Responders
n/N
% (95% CI)12/82
15% (7%, 22%)21/80
26% (17%, 36%)UFC Levels (nmol/24 hr)
Baseline
Mean (SD)
MedianN = 78
868 (764)
704N = 72
750 (930)
470% Change from baseline
Mean (95% CI)
Median
-22% (-44%, +1%)
-47%
-42% (-50%, -33%)
-46%SIGNIFOR resulted in a decrease in the mean 24-hour UFC after 1 month of treatment (Figure 1). For patients (n = 78) who stayed in the trial, similar UFC lowering was observed at Month 12.
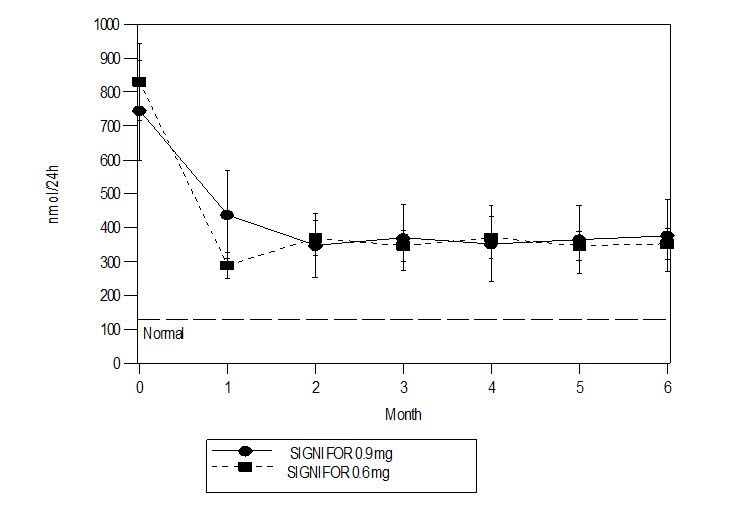
Figure 1 – Mean (± SE) Urinary Free Cortisol (nmol/24h) at Time Points up to Month 6 by Randomized Dose Group
Note: Only patients who completed 6 months of treatment are included in this analysis (n = 110). The reference line is the ULN for UFC, which is 145 nmol/24hour; +/-Standard errors are displayed.
Other Endpoints
Decreases from baseline for blood pressure were observed at Month 6, including patients who did not receive any anti-hypertensive medication. However, due to the fact that the study allowed initiation of anti-hypertensive medication and dose increases in patients already receiving such medications, the individual contribution of SIGNIFOR or of anti-hypertensive medication adjustments cannot be clearly established.
The mean decreases from baseline at Month 6 for weight, body mass index, and waist circumference were 4.4 kg, 1.6 kg/m2 and 2.6 cm, respectively. Individual patients showed varying degrees of improvement in Cushing's disease manifestations, but because of the variability in response and the absence of a control group in this trial, it is uncertain whether these changes could be ascribed to the effects of SIGNIFOR.
-
16
HOW SUPPLIED/STORAGE AND HANDLING
SIGNIFOR is supplied as a single dose, colorless glass ampule packaged in a box of 60 ampules, arranged in 10 packs of 6 ampules each. The following packaging configurations are available.
0.3 mg/1 mL pasireotide (as diaspartate)
Box of 60 ampules NDC# 0078-0633-20
0.6 mg/1 mL pasireotide (as diaspartate)
Box of 60 ampules NDC# 0078-0634-20
0.9 mg/1 mL pasireotide (as diaspartate)
Box of 60 ampules NDC# 0078-0635-20
Store at 25°C (77°F); excursions permitted to 15°C-30°C (59°F-86°F), protect from light.
-
17
PATIENT COUNSELING INFORMATION
See FDA approved patient labeling (Medication Guide and Instructions for Use).
Counsel patients on the following possible significant adverse reactions:
- Hypocortisolism [see Warnings and Precautions (5.1)]
- Hyperglycemia and Diabetes [see Warnings and Precautions (5.2)]
- Bradycardia and QT Prolongation [see Warnings and Precautions (5.3)]
- Liver Test elevations [see Warnings and Precautions (5.4)]
- Cholelithiasis and Complications of Cholelithiasis: Advise patients to contact their healthcare provider if they experience signs or symptoms of gallstones (cholelithiasis) or complications of gallstones (e.g., cholecystitis or cholangitis) [see Warnings and Precautions (5.5)]
- Pituitary Hormone Deficiency [see Warnings and Precautions (5.6)]
- Pregnancy: Inform female patients that treatment with SIGNIFOR may result in unintended pregnancy [see Use in Specific Populations (8.3)]
Instruct the patients on the proper use of SIGNIFOR, including instructions to:
- Carefully review the Medication Guide.
- Do not reuse unused portions of SIGNIFOR ampules and properly dispose of the ampules after use.
- Avoid multiple injections at or near the same site within short periods of time.
For instructions on the use of SIGNIFOR glass ampules, refer to the Medication Guide that follows.
**Trademark of Thomson Healthcare, Inc.
Distributed by:
Novartis Pharmaceuticals Corporation
East Hanover, NJ 07936T2020-01
- Hypocortisolism [see Warnings and Precautions (5.1)]
-
MEDICATION GUIDE
This Medication Guide has been approved by the U.S. Food and Drug Administration. Revised: January 2020 Medication Guide
SIGNIFOR® [sig-na-for]
(pasireotide)
InjectionRead this Medication Guide before you start using SIGNIFOR and each time you get a refill. There may be new information. This information does not take the place of talking to your doctor about your medical condition or your treatment.
What is the most important information I should know about SIGNIFOR?
SIGNIFOR can cause serious side effects, including:
-
Low cortisol levels in your blood (hypocortisolism). Tell your doctor right away if you have any signs and symptoms of hypocortisolism. Signs and symptoms of hypocortisolism may include:
- weakness
- fatigue
- loss of appetite
- nausea
- vomiting
- low blood pressure
- low level of sodium in your blood
- low blood sugar
If you get hypocortisolism while taking SIGNIFOR, your doctor may change your dose or ask you to stop taking it. - weakness
-
High blood sugar (hyperglycemia). Your doctor should check your blood sugar level before you start taking SIGNIFOR and while you take it. Signs and symptoms of hyperglycemia may include:
- excessive thirst
- high urine output
- increased appetite with weight loss
- tiredness
If you get hyperglycemia while taking SIGNIFOR, your doctor may give you another medicine to take to lower your blood sugar. Your doctor may also change your dose of SIGNIFOR or ask you to stop taking it. - excessive thirst
What is SIGNIFOR?
SIGNIFOR is a prescription medicine used to treat Cushing’s disease in adults who cannot have surgery or have failed surgery.
It is not known if SIGNIFOR is safe and effective in children.
What should I tell my doctor before using SIGNIFOR?
Before you take SIGNIFOR, tell your doctor if you:
- have or have had high blood sugar (hyperglycemia)
- have diabetes
- have or have had heart problems
- have a history of low levels of potassium or magnesium in your blood
- have or have had liver problems
- have or have had gallstones
- have any other medical conditions
- are pregnant or plan to become pregnant. SIGNIFOR may harm your unborn baby.
- are breastfeeding or plan to breastfeed. It is not known if SIGNIFOR passes into your breast milk. You and your doctor should decide if you will take SIGNIFOR or breastfeed. You should not do both.
Tell your doctor about all the medicines you take, including prescription and non-prescription medicines, vitamins, and herbal supplements.
Taking SIGNIFOR with certain other medicines can affect each other and cause side effects. Especially tell your doctor if you take:
- medicines to control your heart beat (anti-arrhythmics)
- medicines that can affect the electrical system of your heart (QT prolongation)
- medicines to control your blood pressure (such as beta-blockers or calcium channel blockers)
- medicines to control the electrolyte (such as potassium or magnesium) levels in your blood
- cyclosporine (Gengraf®, Neoral®, Restasis®, Sandimmune®)
- bromocriptine (Cycloset®, Parlodel®)
Ask your doctor for a list of these medicines, if you are not sure.
Know the medicines you take. Keep a list of them to show to your doctor and pharmacist when you get a new medicine.
How should I use SIGNIFOR?
- Read the “Instructions for Use” at the end of this Medication Guide for information about the right way to use SIGNIFOR.
- Use SIGNIFOR exactly as your doctor tells you to.
- Your doctor may change your dose if needed.
- Before you use SIGNIFOR for the first time, your doctor should do a blood test to check your blood sugar levels and your liver tests.
- Before you use SIGNIFOR for the first time, your doctor should do a test to check your heart (electrocardiogram) and your gallbladder (ultrasound).
- SIGNIFOR should be clear and colorless. Before you inject your dose, check to make sure that SIGNIFOR is clear and colorless, and does not have any clumps or particles in it.
- SIGNIFOR is given as an injection into the fat just under your skin (subcutaneous injection).
-
Do not inject SIGNIFOR into skin that is red or irritated.
- The recommended injection sites for SIGNIFOR are the top of your thigh or stomach area (abdomen).
- Change (rotate) your injection site with each dose. Do not inject SIGNIFOR into the exact same spot for each injection.
- Your doctor should show you how to prepare and give your dose of SIGNIFOR before you use it for the first time.
- You should not inject SIGNIFOR until your doctor has shown you how to use it the right way.
If you take too much SIGNIFOR, tell your doctor right away.
What are the possible side effects of SIGNIFOR?
SIGNIFOR may cause serious side effects, including:
-
See “What is the most important information I should know about SIGNIFOR?”
-
slow heart rate (bradycardia). SIGNIFOR can cause your heart to beat slower, which may cause you to feel weak, dizzy or even faint. People who have, or have had, heart problems are at higher risk for bradycardia.
-
problems with the electrical system of your heart (QT interval prolongation), which can put you at risk for abnormal heart beats, dizziness, and fainting spells that can be very serious. Call your doctor right away if you experience such spells.
-
elevation of your liver tests. Your doctor should do blood tests to monitor your liver tests while you use SIGNIFOR.
-
gallstones (cholelithiasis) and complications that can happen if you have gallstones. Gallstones are a serious but common side effect of SIGNIFOR. Possible complications of gallstones include inflammation and infection of the gallbladder. Your doctor should do a test (ultrasound) to check for gallstones before you start using SIGNIFOR and while you use it.
Tell your healthcare provider if you get any of these symptoms.- sudden pain in your upper right stomach area (abdomen)
- yellowing of your skin and whites of your eyes
- nausea
- sudden pain in your right shoulder or between your shoulder blades
- fever with chills
-
slow heart rate (bradycardia). SIGNIFOR can cause your heart to beat slower, which may cause you to feel weak, dizzy or even faint. People who have, or have had, heart problems are at higher risk for bradycardia.
The most common side effects of SIGNIFOR include:
- diarrhea
- nausea
- high blood sugar
- headache
- abdominal pain
- fatigue
- diabetes mellitus
- injection-site reactions
- common cold
- hair loss
- weakness
- fluid retention
- Abnormal blood test result for glycosylated hemoglobin (the level of glycosylated hemoglobin indicates the average blood sugar level over the previous months)
Tell your doctor if you have any side effect that bothers you or that does not go away.
These are not all the possible side effects of SIGNIFOR. For more information, ask your doctor or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1–800–FDA–1088.
How should I store SIGNIFOR?
- Store SIGNIFOR at 68°F to 77°F (20°C to 25°C).
- Keep SIGNIFOR out of the light.
Keep SIGNIFOR and all medicines out of the reach of children.
General information about the safe and effective use of SIGNIFOR.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use SIGNIFOR for a condition for which it was not prescribed. Do not give SIGNIFOR to other people, even if they have the same symptoms that you have. It may harm them.
This Medication Guide summarizes the most important information about SIGNIFOR. If you would like more information, talk to your doctor. You can ask your pharmacist or doctor for information about SIGNIFOR that is written for health professionals.
For more information go to www.SIGNIFOR.com or call 1-877-503-3377.
What are the ingredients in SIGNIFOR?
Active ingredient: Pasireotide
Inactive ingredients: Mannitol, sodium hydroxide, tartaric acid, and water for injection.
T2020-02
-
Low cortisol levels in your blood (hypocortisolism). Tell your doctor right away if you have any signs and symptoms of hypocortisolism. Signs and symptoms of hypocortisolism may include:
-
INSTRUCTIONS FOR USE
Instructions for Use
SIGNIFOR® [sig-na-for]
(pasireotide)
Injection
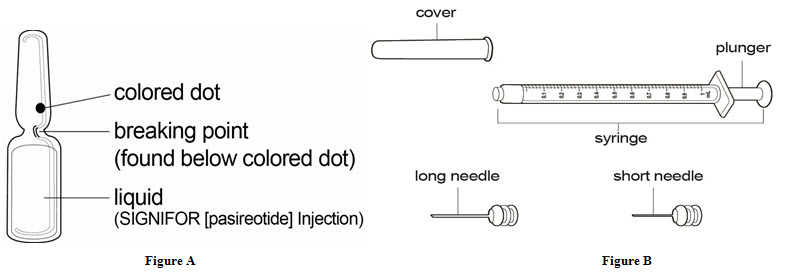
Supplies you will need to give your SIGNIFOR injection:
- 1 SIGNIFOR ampule (See Figure A)
- 1 sterile syringe (See Figure B)
- 1 long sterile needle (See Figure B)
- This needle is used to draw up your SIGNIFOR from the ampule. You should only use this needle if your doctor or nurse tells you to.
- 1 short sterile needle (See Figure B)
- Alcohol wipes
- 1 cotton ball or gauze
- A sharps disposal container or other closeable, puncture resistant disposal container
Getting started:
Step 1: Wash your hands well with soap and water and dry them.
Step 2: Take 1 SIGNIFOR ampule out of the box.
Step 3: Look at the SIGNIFOR ampule. Check that the ampule is not cracked or broken and that the liquid medicine in the ampule is clear and colorless.
Do not use SIGNIFOR if the ampule is cracked or broken or if the liquid looks cloudy or contains particles. Take the whole box back to the pharmacy and get a new one.
Ampules should be opened just prior to administration and any unused portion discarded.
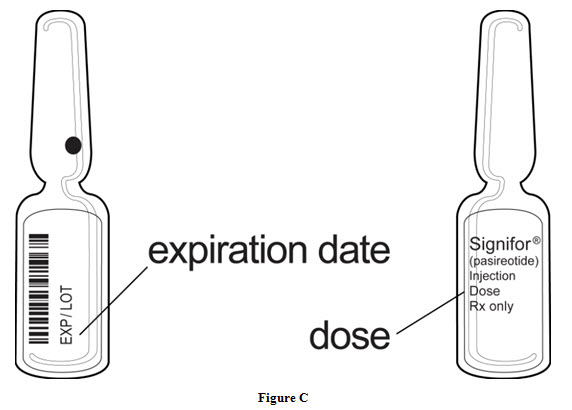
Step 4: Check the dose and expiration date printed on the ampule (See Figure C).
Preparing your SIGNIFOR dose:
Step 5: Hold the SIGNIFOR ampule at the bottom with 1 hand. With your other hand, tap the top of the SIGNIFOR ampule with your finger to make sure there is no liquid in the top of the ampule when you open it (See Figure D). 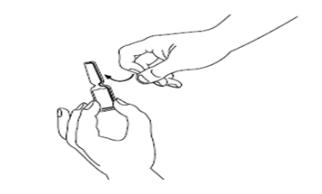
Figure DStep 6: Hold the ampule with 1 hand. With your other hand, hold the top of the ampule and pull it sideways until the top snaps off at the line marked on the ampule neck (See Figure E). - Put the ampule upright on a clean and flat surface.
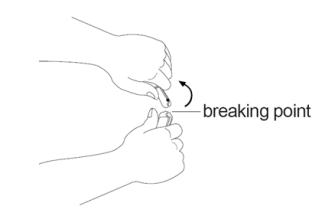
Figure EStep 7: Open the sterile needle package. Place the needle on the top of the syringe. Push down on the needle and twist it clockwise until it is tight (See Figure F). - If you have been told to use 1 long and 1 short needle, you should use the long needle for this step.
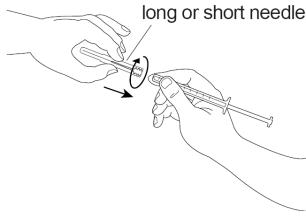
Figure FStep 8: Pull the needle cover straight off the sterile needle (See Figure G).
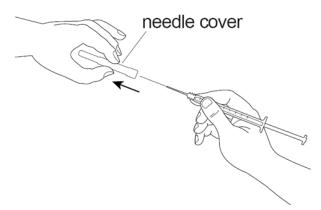
Figure GStep 9: Put the needle into the ampule making sure you do not touch the outside of the ampule and pull up on the plunger to draw up all of the SIGNIFOR liquid into the syringe (See Figure H). - If you have been told to use 1 long and 1 short needle, you should now take the long needle off of the syringe and put on the short one.
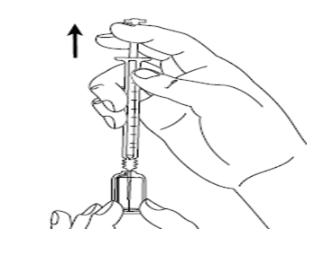
Figure HStep 10: Hold the syringe upright in 1 hand between 2 fingers with your thumb at the bottom of the plunger. With your other hand, tap the syringe with your finger to get rid of air bubbles (See Figure I).
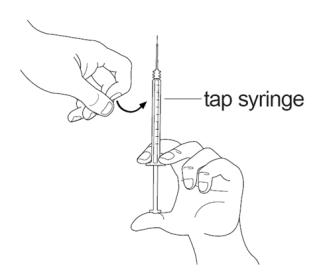
Figure IStep 11: Push up on the plunger until you see a drop of liquid on the tip of the needle (See Figure J). - Do not let the needle touch anything. You are now ready to inject your dose of SIGNIFOR.
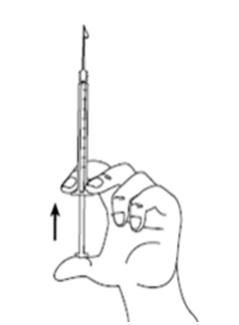
Figure JInjecting your SIGNIFOR dose:
Step 12: Choose your injection site and wipe the site with an alcohol wipe (See Figure K). Let the site dry.
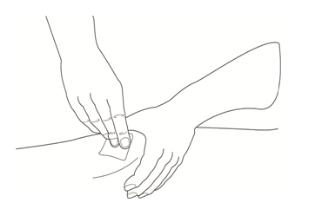
Figure KStep 13: With 1 hand, gently pinch the skin at the injection site you have chosen and wiped with an alcohol wipe. With your other hand, pick up the syringe and hold it like a pencil. Insert the needle into the pinched skin at a 45 degree angle using a quick dart like motion (See Figure L). - Do not let go of the skin.
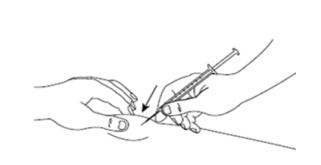
Figure L
Step 14: Gently press the plunger all the way down, until the syringe is empty (See Figure M).
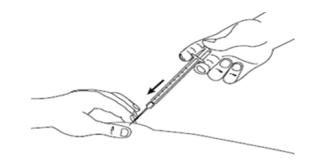
Figure MStep 15: When the syringe is empty, slowly let go of the skin and gently pull the needle out of the skin (See Figure N).
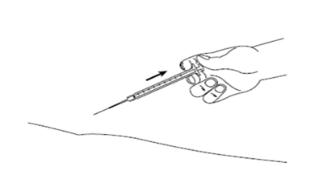
Figure NStep 16: Place a cotton ball or gauze over the injection site and press for about 5 seconds. Do not massage the injection site. - If there is bleeding, cover it with a bandage.
After injecting your SIGNIFOR dose:
- Put your used needles and syringes in a FDA-cleared sharps disposal container right away after use. Do not throw away (dispose of) loose needles and syringes in your household trash.
- If you do not have a FDA-cleared sharps disposal container, you may use a household container that is:
- made of a heavy-duty plastic,
- can be closed with a tight-fitting, puncture-resistant lid, without sharps being able to come out,
- upright and stable during use,
- leak-resistant, and
- properly labeled to warn of hazardous waste inside the container.
- made of a heavy-duty plastic,
- When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used needles and syringes. For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA’s website at: http://www.fda.gov/safesharpsdisposal.
- Do not dispose of your used sharps disposal container in your household trash unless your community guidelines permit this. Do not recycle your used sharps disposal container.
- If you do not have a FDA-cleared sharps disposal container, you may use a household container that is:
This Instructions for Use has been approved by the U.S. Food and Drug Administration. Revised: January 2019
Distributed by:
Novartis Pharmaceuticals Corporation
East Hanover, NJ 07936T2019-25
- 1 SIGNIFOR ampule (See Figure A)
-
PRINCIPAL DISPLAY PANEL
PRINCIPAL DISPLAY PANEL
Package Label – 0.3 mg/mL
Rx Only NDC: 0078-0633-20
Signifor®
(pasireotide) Injection
0.3 mg/mL
For subcutaneous use only
Dispense with enclosed Medication Guide.
60 ampules (10 packs of 6 ampules)
-
PRINCIPAL DISPLAY PANEL
PRINCIPAL DISPLAY PANEL
Package Label – 0.6 mg/mL
Rx Only NDC: 0078-0634-20
Signifor®
(pasireotide) Injection
0.6 mg/mL
For subcutaneous use only
Dispense with enclosed Medication Guide.
60 ampules (10 packs of 6 ampules)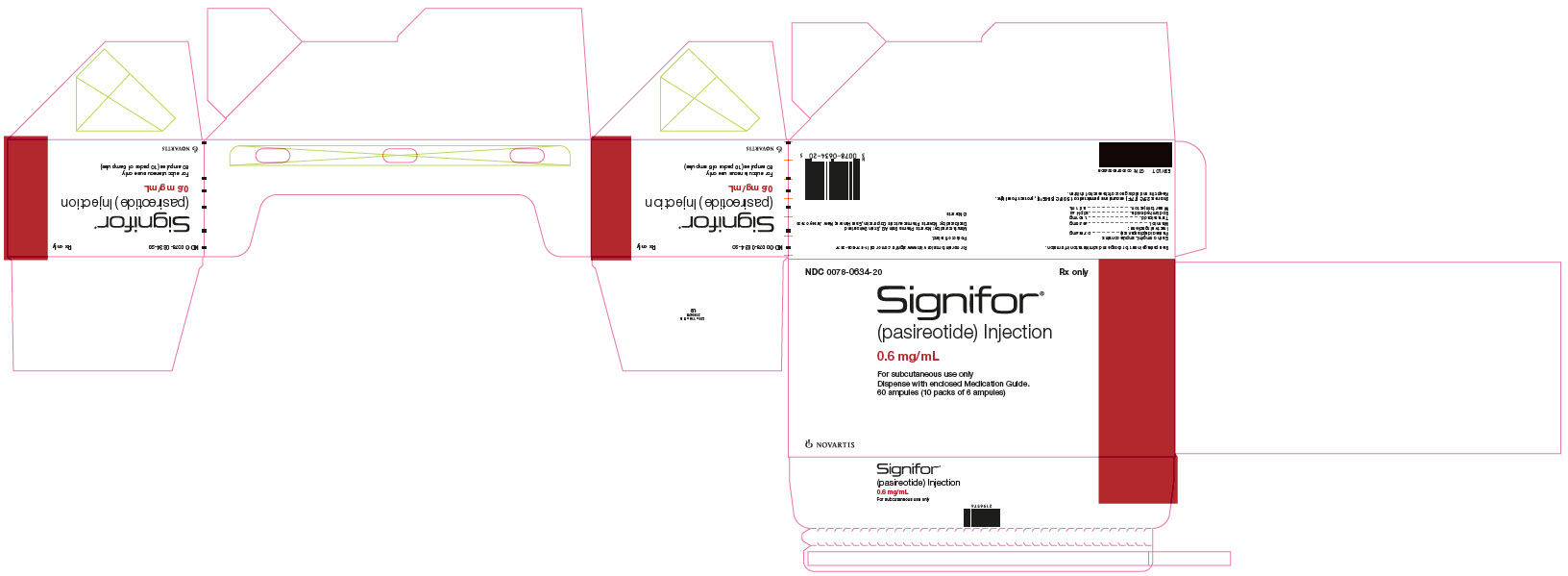
-
PRINCIPAL DISPLAY PANEL
PRINCIPAL DISPLAY PANEL
Package Label – 0.9 mg/mL
Rx Only NDC: 0078-0635-20
Signifor®
(pasireotide) Injection
0.9 mg/mL
For subcutaneous use only
Dispense with enclosed Medication Guide.
60 ampules (10 packs of 6 ampules)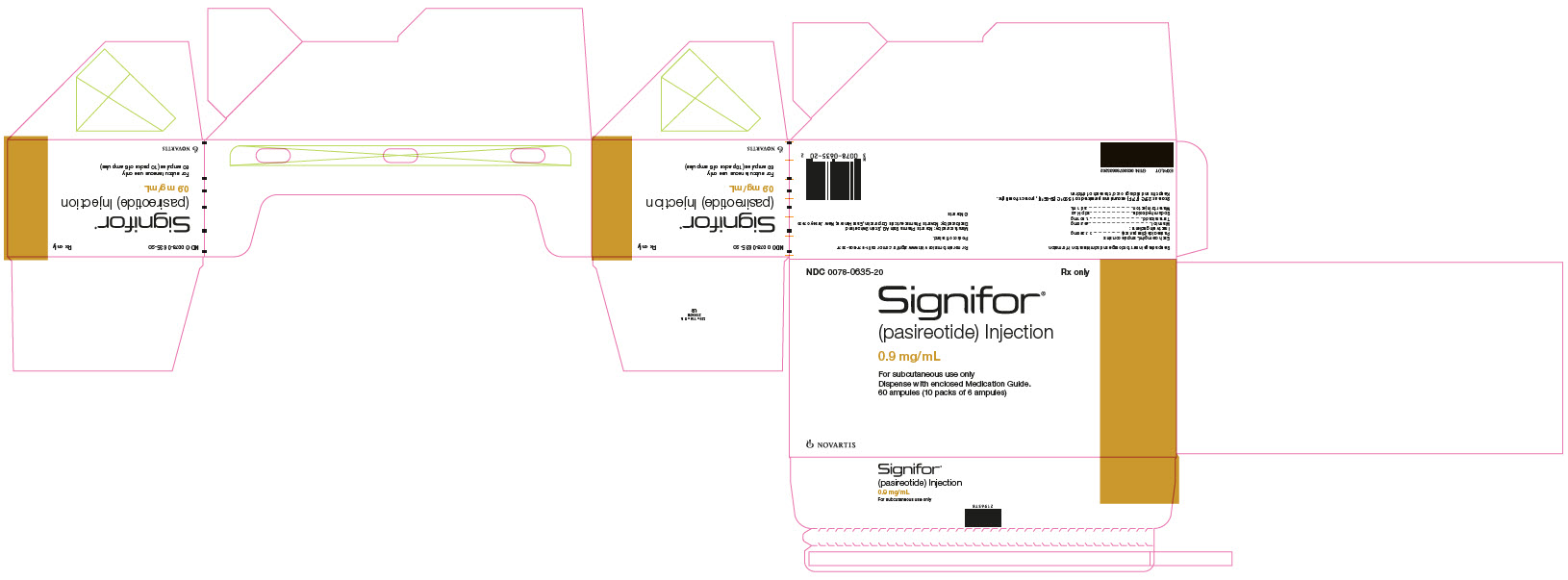
-
INGREDIENTS AND APPEARANCE
SIGNIFOR
pasireotide injectionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0078-0633 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength PASIREOTIDE (UNII: 98H1T17066) (PASIREOTIDE - UNII:98H1T17066) PASIREOTIDE 0.3 mg in 1 mL Inactive Ingredients Ingredient Name Strength MANNITOL (UNII: 3OWL53L36A) 49.50 mg in 1 mL TARTARIC ACID (UNII: W4888I119H) 1.501 mg in 1 mL SODIUM HYDROXIDE (UNII: 55X04QC32I) 4.2 mg in 1 mL WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0078-0633-20 60 in 1 BOX 12/14/2012 1 NDC: 0078-0633-06 6 in 1 PACKAGE 1 NDC: 0078-0633-61 1 mL in 1 AMPULE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA200677 12/14/2012 SIGNIFOR
pasireotide injectionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0078-0634 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength PASIREOTIDE (UNII: 98H1T17066) (PASIREOTIDE - UNII:98H1T17066) PASIREOTIDE 0.6 mg in 1 mL Inactive Ingredients Ingredient Name Strength TARTARIC ACID (UNII: W4888I119H) 1.501 mg in 1 mL SODIUM HYDROXIDE (UNII: 55X04QC32I) 4.2 mg in 1 mL WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0078-0634-20 60 in 1 BOX 12/14/2012 1 NDC: 0078-0634-06 6 in 1 PACKAGE 1 NDC: 0078-0634-61 1 mL in 1 AMPULE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA200677 12/14/2012 SIGNIFOR
pasireotide injectionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0078-0635 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength PASIREOTIDE (UNII: 98H1T17066) (PASIREOTIDE - UNII:98H1T17066) PASIREOTIDE 0.9 mg in 1 mL Inactive Ingredients Ingredient Name Strength MANNITOL (UNII: 3OWL53L36A) 49.50 mg in 1 mL TARTARIC ACID (UNII: W4888I119H) 1.501 mg in 1 mL SODIUM HYDROXIDE (UNII: 55X04QC32I) 4.2 mg in 1 mL WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0078-0635-20 60 in 1 BOX 12/14/2012 1 NDC: 0078-0635-06 6 in 1 PACKAGE 1 NDC: 0078-0635-61 1 mL in 1 AMPULE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA200677 12/14/2012 Labeler - Novartis Pharmaceuticals Corporation (002147023)
Trademark Results [Signifor]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 SIGNIFOR 79079210 3835392 Live/Registered |
Novartis AG 2009-11-24 |
 SIGNIFOR 78458907 3001565 Dead/Cancelled |
Novartis AG 2004-07-29 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.