MAYZENT- siponimod tablet, film coated
MAYZENT by
Drug Labeling and Warnings
MAYZENT by is a Prescription medication manufactured, distributed, or labeled by Novartis Pharmaceuticals Corporation. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use MAYZENT safely and effectively. See full prescribing information for MAYZENT.
MAYZENT® (siponimod) tablets, for oral use
Initial U.S. Approval: 2019
INDICATIONS AND USAGE
MAYZENT is a sphingosine 1-phosphate receptor modulator indicated for the treatment of relapsing forms of multiple sclerosis (MS), to include clinically isolated syndrome, relapsing-remitting disease, and active secondary progressive disease, in adults. (1)
DOSAGE AND ADMINISTRATION
- Assessments are required prior to initiating MAYZENT (2.1)
- Titration is required for treatment initiation (2.2, 2.3)
- The recommended maintenance dosage is 2 mg (2.2)
- The recommended maintenance dosage in patients with a CYP2C9*1/*3 or *2/*3 genotype is 1 mg (2.3)
- First-dose monitoring is recommended for patients with sinus bradycardia, first- or second-degree [Mobitz type I] atrioventricular (AV) block, or a history of myocardial infarction or heart failure (2.4)
DOSAGE FORMS AND STRENGTHS
Tablets: 0.25 mg and 2 mg (3)
CONTRAINDICATIONS
- Patients with a CYP2C9*3/*3 genotype (4)
- In the last 6 months, experienced myocardial infarction, unstable angina, stroke, TIA, decompensated heart failure requiring hospitalization, or Class III/IV heart failure (4)
- Presence of Mobitz type II second-degree, third-degree AV block, or sick sinus syndrome, unless patient has a functioning pacemaker (4)
WARNINGS AND PRECAUTIONS
-
Infections: MAYZENT may increase the risk. Obtain a complete blood count (CBC) before initiating treatment. Monitor for infection during treatment. Do not start in patients with active infection. (5.1)
-
Macular Edema: An ophthalmic evaluation is recommended before starting treatment and if there is any change in vision while taking MAYZENT. Diabetes mellitus and uveitis increase the risk. (5.2)
-
Bradyarrhythmia and Atrioventricular Conduction Delays: MAYZENT may result in a transient decrease in heart rate; titration is required for treatment initiation. Consider resting heart rate with concomitant beta-blocker use; obtain cardiologist consultation before concomitant use with other drugs that decrease heart rate (5.3, 7.2, 7.3)
-
Respiratory Effects: May cause a decline in pulmonary function. Assess pulmonary function (e.g., spirometry) if clinically indicated. (5.4)
-
Liver Injury: Obtain liver enzyme results before initiation. Closely monitor patients with severe hepatic impairment. Discontinue if significant liver injury occurs. (5.5)
-
Increased Blood Pressure (BP): Monitor BP during treatment. (5.6)
-
Fetal Risk: Women of childbearing potential should use effective contraception during and for 10 days after stopping MAYZENT. (5.7)
ADVERSE REACTIONS
Most common adverse reactions (incidence greater than 10%) are headache, hypertension, and transaminase increases. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Novartis Pharmaceuticals Corporation at 1-888-669-6682 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.DRUG INTERACTIONS
-
Vaccines: Avoid live attenuated vaccines during and for up to 4 weeks after treatment with MAYZENT (7.4)
- CYP2C9 and CYP3A4 Inhibitors: Increase in siponimod exposure; concomitant use of MAYZENT with moderate CYP2C9 and moderate or strong CYP3A4 inhibitors is not recommended (7.5)
- CYP2C9 and CYP3A4 Inducers: Decrease in siponimod exposure; concomitant use of MAYZENT with moderate CYP2C9 and strong CYP3A4 inducers is not recommended (7.6)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 3/2019
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Assessments Prior to First Dose of MAYZENT
2.2 Recommended Dosage in Patients With CYP2C9 Genotypes *1/*1, *1/*2, or *2/*2
2.3 Recommended Dosage in Patients With CYP2C9 Genotypes *1/*3 or *2/*3
2.4 First Dose Monitoring in Patients With Certain Preexisting Cardiac Conditions
2.5 Reinitiation of MAYZENT After Treatment Interruption
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Infections
5.2 Macular Edema
5.3 Bradyarrhythmia and Atrioventricular Conduction Delays
5.4 Respiratory Effects
5.5 Liver Injury
5.6 Increased Blood Pressure
5.7 Fetal Risk
5.8 Posterior Reversible Encephalopathy Syndrome
5.9 Unintended Additive Immunosuppressive Effects From Prior Treatment With Immunosuppressive or Immune-Modulating Therapies
5.10 Severe Increase in Disability After Stopping MAYZENT
5.11 Immune System Effects After Stopping MAYZENT
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 Anti-Neoplastic, Immune-Modulating, or Immunosuppressive Therapies
7.2 Anti-Arrhythmic Drugs, QT Prolonging Drugs, Drugs That May Decrease Heart Rate
7.3 Beta-Blockers
7.4 Vaccination
7.5 CYP2C9 and CYP3A4 Inhibitors
7.6 CYP2C9 and CYP3A4 Inducers
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 CYP2C9 Genotype
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.5 Pharmacogenomics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage and Handling
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Assessments Prior to First Dose of MAYZENT
Before initiation of treatment with MAYZENT, assess the following:
CYP2C9 Genotype Determination
Test patients for CYP2C9 variants to determine CYP2C9 genotype [see Dosage and Administration (2.2, 2.3), Contraindications (4), and Use in Specific Populations (8.6)]. An FDA-cleared or -approved test for the detection of CYP2C9 variants to direct the use of siponimod is not currently available.
Complete Blood Count
Review results of a recent complete blood count (CBC) [see Warnings and Precautions (5.1)].
Ophthalmic Evaluation
Obtain an evaluation of the fundus, including the macula [see Warnings and Precautions (5.2)].
Cardiac Evaluation
Obtain an electrocardiogram (ECG) to determine whether preexisting conduction abnormalities are present. In patients with certain preexisting conditions, advice from a cardiologist and first-dose monitoring is recommended [see Dosage and Administration (2.4) and Warnings and Precautions (5.3)].
Determine whether patients are taking drugs that could slow heart rate or atrioventricular (AV) conduction [see Drug Interactions (7.2, 7.3)].
Current or Prior Medications
If patients are taking anti-neoplastic, immunosuppressive, or immune-modulating therapies, or if there is a history of prior use of these drugs, consider possible unintended additive immunosuppressive effects before initiating treatment with MAYZENT [see Warnings and Precautions (5.1) and Drug Interactions (7.1)].
Vaccinations
Test patients for antibodies to varicella zoster virus (VZV) before initiating MAYZENT; VZV vaccination of antibody-negative patients is recommended prior to commencing treatment with MAYZENT [see Warnings and Precautions (5.1)].
Liver Function Tests
Obtain recent (i.e., within last 6 months) transaminase and bilirubin levels [see Warnings and Precautions (5.5)].
2.2 Recommended Dosage in Patients With CYP2C9 Genotypes *1/*1, *1/*2, or *2/*2
Maintenance Dosage
After treatment titration (see Treatment Initiation), the recommended maintenance dosage of MAYZENT is 2 mg taken orally once daily starting on Day 6. Dosage adjustment is required in patients with a CYP2C9*1/*3 or *2/*3 genotype [see Dosage and Administration (2.3)].
Treatment Initiation
Initiate MAYZENT with a 5-day titration, as shown in Table 1 [see Warnings and Precautions (5.3)]. A starter pack should be used for patients who will be titrated to the 2-mg maintenance dosage [see How Supplied/Storage and Handling (16.1, 16.2)].
Table 1 Dose Titration Regimen to Reach MAYZENT 2 mg Maintenance Dosage Titration Titration Dose Titration Regimen Day 1 0.25 mg 1 x 0.25 mg Day 2 0.25 mg 1 x 0.25 mg Day 3 0.50 mg 2 x 0.25 mg Day 4 0.75 mg 3 x 0.25 mg Day 5 1.25 mg 5 x 0.25 mg If one titration dose is missed for more than 24 hours, treatment needs to be reinitiated with Day 1 of the titration regimen.
2.3 Recommended Dosage in Patients With CYP2C9 Genotypes *1/*3 or *2/*3
Maintenance Dosage
In patients with a CYP2C9*1/*3 or *2/*3 genotype, after treatment titration (see Treatment Initiation), the recommended maintenance dosage of MAYZENT is 1 mg taken orally once daily starting on Day 5.
Treatment Initiation
Initiate MAYZENT with a 4-day titration, as shown in Table 2 [see Warnings and Precautions (5.3) and Use in Specific Populations (8.6)]. Do not use the starter pack for patients who will be titrated to the 1-mg maintenance dosage.
Table 2 Dose Titration Regimen to Reach MAYZENT 1 mg Maintenance Dosage Titration Titration Dose Titration Regimen Day 1 0.25 mg 1 x 0.25 mg Day 2 0.25 mg 1 x 0.25 mg Day 3 0.50 mg 2 x 0.25 mg Day 4 0.75 mg 3 x 0.25 mg If one titration dose is missed for more than 24 hours, treatment needs to be reinitiated with Day 1 of the titration regimen.
2.4 First Dose Monitoring in Patients With Certain Preexisting Cardiac Conditions
Because initiation of MAYZENT treatment results in a decrease in heart rate (HR), first-dose 6 hour monitoring is recommended for patients with sinus bradycardia [HR less than 55 beats per minute (bpm)], first- or second-degree [Mobitz type I] AV block, or a history of myocardial infarction or heart failure [see Warnings and Precautions (5.3) and Clinical Pharmacology (12.2)].
First Dose 6-Hour Monitoring
Administer the first dose of MAYZENT in a setting where resources to appropriately manage symptomatic bradycardia are available. Monitor patients for 6 hours after the first dose for signs and symptoms of bradycardia with hourly pulse and blood pressure measurement. Obtain an ECG in these patients at the end of the Day 1 observation period.
Additional Monitoring After 6-Hour Monitoring
If any of the following abnormalities are present after 6 hours (even in the absence of symptoms), continue monitoring until the abnormality resolves:
- The heart rate 6 hours postdose is less than 45 bpm
- The heart rate 6 hours postdose is at the lowest value postdose, suggesting that the maximum pharmacodynamic effect on the heart may not have occurred
- The ECG 6 hours postdose shows new onset second-degree or higher AV block
If postdose symptomatic bradycardia, bradyarrhythmia, or conduction related symptoms occur, or if ECG 6 hours post-dose shows new onset second degree or higher AV block or QTc greater than or equal to 500 msec, initiate appropriate management, begin continuous ECG monitoring, and continue monitoring until the symptoms have resolved if no pharmacological treatment is required. If pharmacological treatment is required, continue monitoring overnight and repeat 6-hour monitoring after the second dose.
Advice from a cardiologist should be sought to determine the most appropriate monitoring strategy (which may include overnight monitoring) during treatment initiation, if treatment with MAYZENT is considered in patients:
- With some preexisting heart and cerebrovascular conditions [see Warnings and Precautions (5.3)]
- With a prolonged QTc interval before dosing or during the 6-hour observation, or at additional risk for QT prolongation, or on concurrent therapy with QT prolonging drugs with a known risk of torsades de pointes [see Warnings and Precautions (5.3) and Drug Interactions (7.2)]
- Receiving concurrent therapy with drugs that slow heart rate or AV conduction [see Drug Interactions (7.2, 7.3)]
2.5 Reinitiation of MAYZENT After Treatment Interruption
After the initial titration is complete, if MAYZENT treatment is interrupted for 4 or more consecutive daily doses, reinitiate treatment with Day 1 of the titration regimen [see Dosage and Administration (2.2, 2.3)]; also complete first-dose monitoring in patients for whom it is recommended [see Dosage and Administration (2.4)].
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
MAYZENT is contraindicated in patients who have:
- A CYP2C9*3/*3 genotype [see Use in Specific Populations (8.6) and Clinical Pharmacology (12.5)]
- In the last 6 months experienced myocardial infarction, unstable angina, stroke, TIA, decompensated heart failure requiring hospitalization, or Class III or IV heart failure
- Presence of Mobitz type II second-degree, third-degree AV block, or sick sinus syndrome, unless patient has a functioning pacemaker [see Warnings and Precautions (5.3)]
-
5 WARNINGS AND PRECAUTIONS
5.1 Infections
Risk of Infections
MAYZENT causes a dose-dependent reduction in peripheral lymphocyte count to 20%-30% of baseline values because of reversible sequestration of lymphocytes in lymphoid tissues. MAYZENT may therefore increase the risk of infections, some serious in nature [see Clinical Pharmacology (12.2)]. Life-threatening and rare fatal infections have occurred in association with MAYZENT.
In Study 1 [see Clinical Studies (14)], the overall rate of infections was comparable between the MAYZENT-treated patients and those on placebo (49.0% vs. 49.1% respectively). However, herpes zoster, herpes infection, bronchitis, sinusitis, upper respiratory infection, and fungal skin infection were more common in MAYZENT-treated patients. In Study 1, serious infections occurred at a rate of 2.9% in MAYZENT-treated patients compared to 2.5% of patients receiving placebo.
Before initiating treatment with MAYZENT, results from a recent complete blood count (i.e., within 6 months or after discontinuation of prior therapy) should be reviewed.
Initiation of treatment with MAYZENT should be delayed in patients with severe active infection until resolution. Because residual pharmacodynamic effects, such as lowering effects on peripheral lymphocyte count, may persist for up to 3-4 weeks after discontinuation of MAYZENT, vigilance for infection should be continued throughout this period [see Warnings and Precautions (5.11)].
Effective diagnostic and therapeutic strategies should be employed in patients with symptoms of infection while on therapy. Suspension of treatment with MAYZENT should be considered if a patient develops a serious infection.
Cryptococcal Infections
Cases of fatal cryptococcal meningitis (CM) and disseminated cryptococcal infections have been reported with another sphingosine 1-phosphate (S1P) receptor modulator. Rare cases of CM have also occurred with MAYZENT. Physicians should be vigilant for clinical symptoms or signs of CM. Patients with symptoms or signs consistent with a cryptococcal infection should undergo prompt diagnostic evaluation and treatment. MAYZENT treatment should be suspended until a cryptococcal infection has been excluded. If CM is diagnosed, appropriate treatment should be initiated.
Herpes Viral Infections
Cases of herpes viral infection, including one case of reactivation of VZV infection leading to varicella zoster meningitis, have been reported in the development program of MAYZENT. In Study 1, the rate of herpetic infections was 4.6% in MAYZENT-treated patients compared to 3.0% of patients receiving placebo. In Study 1, an increase in the rate of herpes zoster infections was reported in 2.5% of MAYZENT-treated patients compared to 0.7% of patients receiving placebo. Patients without a healthcare professional confirmed history of varicella (chickenpox) or without documentation of a full course of vaccination against VZV should be tested for antibodies to VZV before initiating MAYZENT (see Vaccinations below).
Progressive Multifocal Leukoencephalopathy
Progressive multifocal leukoencephalopathy (PML) is an opportunistic viral infection of the brain caused by the JC virus (JCV) that typically only occurs in patients who are immunocompromised, and that usually leads to death or severe disability. Typical symptoms associated with PML are diverse, progress over days to weeks, and include progressive weakness on one side of the body or clumsiness of limbs, disturbance of vision, and changes in thinking, memory, and orientation leading to confusion and personality changes.
No cases of PML have been reported in MAYZENT-treated patients in the development program; however, PML has been reported in patients treated with a S1P receptor modulator and other multiple sclerosis (MS) therapies and has been associated with some risk factors (e.g., immunocompromised patients, polytherapy with immunosuppressants). Physicians should be vigilant for clinical symptoms or MRI findings that may be suggestive of PML. MRI findings may be apparent before clinical signs or symptoms. If PML is suspected, treatment with MAYZENT should be suspended until PML has been excluded.
Prior and Concomitant Treatment with Anti-neoplastic, Immune-Modulating, or Immunosuppressive Therapies
Anti-neoplastic, immune-modulating, or immunosuppressive therapies (including corticosteroids) should be coadministered with caution because of the risk of additive immune system effects during such therapy [see Drug Interactions (7.1)].
Vaccinations
Patients without a healthcare professional confirmed history of chickenpox or without documentation of a full course of vaccination against VZV should be tested for antibodies to VZV before initiating MAYZENT treatment. A full course of vaccination for antibody-negative patients with varicella vaccine is recommended prior to commencing treatment with MAYZENT, following which initiation of treatment with MAYZENT should be postponed for 4 weeks to allow the full effect of vaccination to occur.
The use of live attenuated vaccines should be avoided while patients are taking MAYZENT and for 4 weeks after stopping treatment [see Drug Interactions (7.1)].
Vaccinations may be less effective if administered during MAYZENT treatment. MAYZENT treatment discontinuation 1 week prior to and until 4 weeks after a planned vaccination is recommended.
5.2 Macular Edema
Macular edema was reported in 1.8% of MAYZENT-treated patients compared to 0.2% of patients receiving placebo. The majority of cases occurred within the first four months of therapy.
An ophthalmic evaluation of the fundus, including the macula, is recommended in all patients before starting treatment and at any time if there is any change in vision while taking MAYZENT.
Continuation of MAYZENT therapy in patients with macular edema has not been evaluated. A decision on whether or not MAYZENT should be discontinued needs to take into account the potential benefits and risks for the individual patient.
Macular Edema in Patients with a History of Uveitis or Diabetes Mellitus
Patients with a history of uveitis and patients with diabetes mellitus are at increased risk of macular edema during MAYZENT therapy. The incidence of macular edema is also increased in MS patients with a history of uveitis. In the clinical trial experience in adult patients with all doses of MAYZENT, the rate of macular edema was approximately 10% in MS patients with a history of uveitis or diabetes mellitus versus 2% in those without a history of these diseases. In addition to the examination of the fundus, including the macula, prior to treatment, MS patients with diabetes mellitus or a history of uveitis should have regular follow-up examinations.
5.3 Bradyarrhythmia and Atrioventricular Conduction Delays
Since initiation of MAYZENT treatment results in a transient decrease in heart rate and atrioventricular conduction delays, an up-titration scheme should be used to reach the maintenance dosage of MAYZENT [see Dosage and Administration (2.2, 2.3) and Clinical Pharmacology (12.2)].
MAYZENT was not studied in patients who had:
- In the last 6 months experienced myocardial infarction, unstable angina, stroke, TIA, or decompensated heart failure requiring hospitalization
- New York Heart Association Class II-IV heart failure
- Cardiac conduction or rhythm disorders, including complete left bundle branch block, sinus arrest or sino-atrial block, symptomatic bradycardia, sick sinus syndrome, Mobitz type II second degree AV-block or higher grade AV-block (either history or observed at screening), unless patient has a functioning pacemaker
- Significant QT prolongation (QTc greater than 500 msec)
- Arrhythmias requiring treatment with Class Ia or Class III anti-arrhythmic drugs [see Drug Interactions (7.2)]
Reduction in Heart Rate
After the first titration dose of MAYZENT, the heart rate decrease starts within an hour, and the Day 1 decline is maximal at approximately 3-4 hours. With continued up-titration, further heart rate decreases are seen on subsequent days, with maximal decrease from Day 1-baseline reached on Day 5-6. The highest daily post-dose decrease in absolute hourly mean heart rate is observed on Day 1, with the pulse declining on average 5-6 bpm. Post-dose declines on the following days are less pronounced. With continued dosing, heart rate starts increasing after Day 6 and reaches placebo levels within 10 days after treatment initiation.
In Study 1, bradycardia occurred in 4.4% of MAYZENT-treated patients compared to 2.9% of patients receiving placebo. Patients who experienced bradycardia were generally asymptomatic. Few patients experienced symptoms, including dizziness or fatigue, and these symptoms resolved within 24 hours without intervention [see Adverse Reactions (6.1)]. Heart rates below 40 bpm were rarely observed.
Atrioventricular Conduction Delays
Initiation of MAYZENT treatment has been associated with transient atrioventricular conduction delays that follow a similar temporal pattern as the observed decrease in heart rate during dose titration. The AV conduction delays manifested in most of the cases as first-degree AV block (prolonged PR interval on ECG), which occurred in 5.1% of MAYZENT-treated patients and in 1.9% of patients receiving placebo in Study 1. Second-degree AV blocks, usually Mobitz type I (Wenckebach), have been observed at the time of treatment initiation with MAYZENT in less than 1.7% of patients in clinical trials. The conduction abnormalities typically were transient, asymptomatic, resolved within 24 hours, rarely required treatment with atropine, and did not require discontinuation of MAYZENT treatment.
If treatment with MAYZENT is considered, advice from a cardiologist should be sought:
- In patients with significant QT prolongation (QTc greater than 500 msec)
- In patients with arrhythmias requiring treatment with Class Ia or Class III anti-arrhythmic drugs [see Drug Interactions (7.2)]
- In patients with ischemic heart disease, heart failure, history of cardiac arrest or myocardial infarction, cerebrovascular disease, and uncontrolled hypertension
- In patients with a history of second-degree Mobitz type II or higher AV block, sick-sinus syndrome, or sino-atrial heart block [see Contraindications (4)]
Treatment-Initiation Recommendations
- Obtain an ECG in all patients to determine whether preexisting conduction abnormalities are present.
- In all patients, a dose titration is recommended for initiation of MAYZENT treatment to help reduce cardiac effects [see Dosage and Administration (2.2, 2.3)].
- In patients with sinus bradycardia (HR less than 55 bpm), first- or second-degree [Mobitz type I] AV block, or a history of myocardial infarction or heart failure with onset > 6 months prior to initiation, ECG testing and first-dose monitoring is recommended [see Dosage and Administration (2.1, 2.4)].
- Since significant bradycardia may be poorly tolerated in patients with history of cardiac arrest, cerebrovascular disease, uncontrolled hypertension, or severe untreated sleep apnea, MAYZENT is not recommended in these patients. If treatment is considered, advice from a cardiologist should be sought prior to initiation of treatment in order to determine the most appropriate monitoring strategy.
- Use of MAYZENT in patients with a history of recurrent syncope or symptomatic bradycardia should be based on an overall benefit-risk assessment. If treatment is considered, advice from a cardiologist should be sought prior to initiation of treatment in order to determine the most appropriate monitoring.
- Experience with MAYZENT is limited in patients receiving concurrent therapy with drugs that decrease heart-rate (e.g., beta-blockers, calcium channel blockers - diltiazem and verapamil, and other drugs that may decrease heart rate, such as ivabradine and digoxin). Concomitant use of these drugs during MAYZENT initiation may be associated with severe bradycardia and heart block.
- For patients receiving a stable dose of a beta-blocker, the resting heart rate should be considered before introducing MAYZENT treatment. If the resting heart rate is greater than 50 bpm under chronic beta-blocker treatment, MAYZENT can be introduced. If resting heart rate is less than or equal to 50 bpm, beta-blocker treatment should be interrupted until the baseline heart-rate is greater than 50 bpm. Treatment with MAYZENT can then be initiated and treatment with a beta-blocker can be reinitiated after MAYZENT has been up-titrated to the target maintenance dosage [see Drug Interactions (7.3)].
- For patients taking other drugs that decrease heart rate, treatment with MAYZENT should generally not be initiated without consultation from a cardiologist because of the potential additive effect on heart rate [see Dosage and Administration (2.4) and Drug Interactions (7.2)].
Missed Dose During Treatment Initiation and Reinitiation of Therapy Following Interruption
If a titration dose is missed or if 4 or more consecutive daily doses are missed during maintenance treatment, reinitiate Day 1 of the dose titration and follow titration monitoring recommendations [see Dosage and Administration (2.2, 2.3)].
5.4 Respiratory Effects
Dose-dependent reductions in absolute forced expiratory volume over 1 second (FEV1) were observed in MAYZENT-treated patients as early as 3 months after treatment initiation. In a placebo-controlled trial in adult patients, the decline in absolute FEV1 from baseline compared to placebo was 88 mL [95% confidence interval (CI): 139, 37] at 2 years. The mean difference between MAYZENT-treated patients and patients receiving placebo in percent predicted FEV1 at 2 years was 2.8% (95% CI: -4.5, -1.0). There is insufficient information to determine the reversibility of the decrease in FEV1 after drug discontinuation. In Study 1, five patients discontinued MAYZENT because of decreases in pulmonary function testing. MAYZENT has been tested in MS patients with mild to moderate asthma and chronic obstructive pulmonary disease. The changes in FEV1 were similar in this subgroup compared with the overall population. Spirometric evaluation of respiratory function should be performed during therapy with MAYZENT if clinically indicated.
5.5 Liver Injury
Elevations of transaminases may occur in MAYZENT-treated patients. Recent (i.e., within last 6 months) transaminase and bilirubin levels should be reviewed before initiation of MAYZENT therapy.
In Study 1, elevations in transaminases and bilirubin were observed in 10.1% of MAYZENT-treated patients compared to 3.7% of patients receiving placebo, mainly because of transaminase [alanine aminotransferase/aspartate aminotransferase/gamma-glutamyltransferase (ALT/AST/GGT)] elevations.
In Study 1, ALT or AST increased to three and five times the upper limit of normal (ULN) in 5.6% and 1.4% of MAYZENT-treated patients, respectively, compared to 1.5% and 0.5% of patients receiving placebo, respectively. ALT or AST increased eight and ten times ULN in MAYZENT-treated patients (0.5% and 0.2%, respectively) compared to no patients receiving placebo. The majority of elevations occurred within 6 months of starting treatment. ALT levels returned to normal within approximately 1 month after discontinuation of MAYZENT. In clinical trials, MAYZENT was discontinued if the elevation exceeded a 3-fold increase and the patient showed symptoms related to hepatic dysfunction.
Patients who develop symptoms suggestive of hepatic dysfunction, such as unexplained nausea, vomiting, abdominal pain, fatigue, anorexia, rash with eosinophilia, or jaundice and/or dark urine during treatment, should have liver enzymes checked. MAYZENT should be discontinued if significant liver injury is confirmed.
Although there are no data to establish that patients with preexisting liver disease are at increased risk to develop elevated liver function test values when taking MAYZENT, caution should be exercised when using MAYZENT in patients with a history of significant liver disease.
5.6 Increased Blood Pressure
In Study 1, MAYZENT-treated patients had an average increase over placebo of approximately 3 mmHg in systolic pressure and 1.2 mmHg in diastolic pressure, which was first detected after approximately 1 month of treatment initiation and persisted with continued treatment. Hypertension was reported as an adverse reaction in 12.5% of MAYZENT-treated patients and in 9.2% of patients receiving placebo. Blood pressure should be monitored during treatment with MAYZENT and managed appropriately.
5.7 Fetal Risk
Based on animal studies, MAYZENT may cause fetal harm [see Use in Specific Populations (8.1)]. Because it takes approximately 10 days to eliminate MAYZENT from the body, women of childbearing potential should use effective contraception to avoid pregnancy during and for 10 days after stopping MAYZENT treatment.
5.8 Posterior Reversible Encephalopathy Syndrome
Rare cases of posterior reversible encephalopathy syndrome (PRES) have been reported in patients receiving a sphingosine 1-phosphate (S1P) receptor modulator. Such events have not been reported for MAYZENT-treated patients in the development program. However, should a MAYZENT-treated patient develop any unexpected neurological or psychiatric symptoms/signs (e.g., cognitive deficits, behavioral changes, cortical visual disturbances, or any other neurological cortical symptoms/signs), any symptom/sign suggestive of an increase of intracranial pressure, or accelerated neurological deterioration, the physician should promptly schedule a complete physical and neurological examination and should consider a MRI. Symptoms of PRES are usually reversible but may evolve into ischemic stroke or cerebral hemorrhage. Delay in diagnosis and treatment may lead to permanent neurological sequelae. If PRES is suspected, MAYZENT should be discontinued.
5.9 Unintended Additive Immunosuppressive Effects From Prior Treatment With Immunosuppressive or Immune-Modulating Therapies
When switching from drugs with prolonged immune effects, the half-life and mode of action of these drugs must be considered to avoid unintended additive immunosuppressive effects while at the same time minimizing risk of disease reactivation, when initiating MAYZENT.
Initiating treatment with MAYZENT after treatment with alemtuzumab is not recommended [see Drug Interactions (7.1)].
5.10 Severe Increase in Disability After Stopping MAYZENT
Severe exacerbation of disease, including disease rebound, has been rarely reported after discontinuation of a S1P receptor modulator. The possibility of severe exacerbation of disease should be considered after stopping MAYZENT treatment. Patients should be observed for a severe increase in disability upon MAYZENT discontinuation and appropriate treatment should be instituted, as required.
5.11 Immune System Effects After Stopping MAYZENT
After stopping MAYZENT therapy, siponimod remains in the blood for up to 10 days. Starting other therapies during this interval will result in concomitant exposure to siponimod.
Lymphocyte counts returned to the normal range in 90% of patients within 10 days of stopping therapy [see Clinical Pharmacology (12.2)]. However, residual pharmacodynamics effects, such as lowering effects on peripheral lymphocyte count, may persist for up to 3-4 weeks after the last dose. Use of immunosuppressants within this period may lead to an additive effect on the immune system, and therefore caution should be applied 3-4 weeks after the last dose of MAYZENT [see Drug Interactions (7.1)].
-
6 ADVERSE REACTIONS
The following serious adverse reactions are described elsewhere in labeling:
- Infections [see Warnings and Precautions (5.1)]
- Macular Edema [see Warnings and Precautions (5.2)]
- Bradyarrhytmia and Atrioventricular (AV) Conduction Delays [see Warnings and Precautions (5.3)]
- Respiratory Effects [see Warnings and Precautions (5.4)]
- Liver Injury [see Warnings and Precautions (5.5)]
- Increased Blood Pressure [see Warnings and Precautions (5.6)]
- Fetal Risk [see Warnings and Precautions (5.7)]
- Posterior Reversible Encephalopathy Syndrome [see Warnings and Precautions (5.8)]
- Unintended Additive Immunosuppressive Effects From Prior Treatment With Immunosuppressive or Immune-Modulating Therapies [see Warnings and Precautions (5.9)]
- Severe Increase in Disability After Stopping MAYZENT [see Warnings and Precautions (5.10)]
- Immune System Effects After Stopping MAYZENT [see Warnings and Precautions (5.11)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reactions rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
A total of 1737 MS patients have received MAYZENT at doses of at least 2 mg daily. These patients were included in Study 1 [see Clinical Studies (14)] and in a Phase 2 placebo-controlled study in patients with MS. In Study 1, 67% of MAYZENT-treated patients completed the double-blind part of the study, compared to 59.0% of patients receiving placebo. Adverse events led to discontinuation of treatment in 8.5% of MAYZENT-treated patients, compared to 5.1% of patients receiving placebo. The most common adverse reactions (incidence at least 10%) in MAYZENT-treated patients in Study 1 were headache, hypertension, and transaminase increases.
Table 3 lists adverse reactions that occurred in at least 5% of MAYZENT-treated patients and at a rate at least 1% higher than in patients receiving placebo.
Table 3 Adverse Reactions Reported in Study 1 (Occurring in at Least 5% of MAYZENT-Treated Patients and at a Rate at Least 1% Higher Than in Patients Receiving Placebo) Terms were combined as follows:
aheadache, tension headache, sinus headache, cervicogenic headache, drug withdrawal headache, and procedural headache.
bhypertension, blood pressure increased, blood pressure systolic increased, essential hypertension, blood pressure diastolic increased.
calanine aminotransferase increased, gamma-glutamyltransferase increased, hepatic enzyme increased, aspartate aminotransferase increased,
blood alkaline phosphatase increased, liver function test increased, hepatic function abnormal, liver function test abnormal, transaminases increased.
dedema peripheral, joint swelling, fluid retention, swelling face.
ebradycardia, sinus bradycardia, heart rate decreased.
fpain in extremity and limb discomfort.Adverse Reaction MAYZENT 2 mg
(N = 1099)
%Placebo
(N = 546)
%Headachea 15 14 Hypertensionb 13 9 Transaminase increasedc 11 3 Falls 11 10 Edema peripherald 8 4 Nausea 7 4 Dizziness 7 5 Diarrhea 6 4 Bradycardiae 6 3 Pain in extremityf 6 4 The following adverse reactions have occurred in less than 5% of MAYZENT-treated patients but at a rate at least 1% higher than in patients receiving placebo: herpes zoster, lymphopenia, seizure, tremor, macular edema, AV block (1st and 2nd degree), asthenia, and pulmonary function test decreased [see Warnings and Precautions (5.1, 5.2, 5.3, 5.4)].
Seizures
In Study 1, cases of seizures were reported in 1.7% of MAYZENT-treated patients, compared to 0.4% in patients receiving placebo. It is not known whether these events were related to the effects of MS, to MAYZENT, or to a combination of both.
Respiratory Effects
Dose-dependent reductions in forced expiratory volume over 1 second (FEV1) were observed in patients treated with MAYZENT [see Warnings and Precautions (5.4)].
Vascular Events
Vascular events, including ischemic strokes, pulmonary embolisms, and myocardial infarctions, were reported in 3.0% of MAYZENT-treated patients compared to 2.6% of patients receiving placebo. Some of these events were fatal. Physicians and patients should remain alert for the development of vascular events throughout treatment, even in the absence of previous vascular symptoms. Patients should be informed about the symptoms of cardiac or cerebral ischemia caused by vascular events and the steps to take if they occur.
Malignancies
Malignancies such as malignant melanoma in situ and seminoma were reported in MAYZENT-treated patients in Study 1. An increased risk of cutaneous malignancies has been reported in association with another S1P modulator.
-
7 DRUG INTERACTIONS
7.1 Anti-Neoplastic, Immune-Modulating, or Immunosuppressive Therapies
MAYZENT has not been studied in combination with anti-neoplastic, immune-modulating, or immunosuppressive therapies. Caution should be used during concomitant administration because of the risk of additive immune effects during such therapy and in the weeks following administration [see Warnings and Precautions (5.1)].
When switching from drugs with prolonged immune effects, the half-life and mode of action of these drugs must be considered in order to avoid unintended additive immunosuppressive effects [see Warnings and Precautions (5.9)].
Because of the characteristics and duration of alemtuzumab immune suppressive effects, initiating treatment with MAYZENT after alemtuzumab is not recommended.
MAYZENT can generally be started immediately after discontinuation of beta interferon or glatiramer acetate.
7.2 Anti-Arrhythmic Drugs, QT Prolonging Drugs, Drugs That May Decrease Heart Rate
MAYZENT has not been studied in patients taking QT prolonging drugs.
Class Ia (e.g., quinidine, procainamide) and Class III (e.g., amiodarone, sotalol) anti-arrhythmic drugs have been associated with cases of Torsades de Pointes in patients with bradycardia. If treatment with MAYZENT is considered, advice from a cardiologist should be sought.
Because of the potential additive effects on heart rate, treatment with MAYZENT should generally not be initiated in patients who are concurrently treated with QT prolonging drugs with known arrhythmogenic properties, heart rate lowering calcium channel blockers (e.g., verapamil, diltiazem), or other drugs that may decrease heart rate (e.g., ivabradine, digoxin) [see Warnings and Precautions (5.3) and Drug Interactions (7.3)]. If treatment with MAYZENT is considered, advice from a cardiologist should be sought regarding the switch to non-heart-rate lowering drugs or appropriate monitoring for treatment initiation.
7.3 Beta-Blockers
Caution should be applied when MAYZENT is initiated in patients receiving treatment with a beta-blocker because of the additive effects on lowering heart rate; temporary interruption of the beta-blocker treatment may be needed prior to initiation of MAYZENT [see Warnings and Precautions (5.3)]. Beta-blocker treatment can be initiated in patients receiving stable doses of MAYZENT [see Clinical Pharmacology (12.2)].
7.4 Vaccination
During and for up to one month after discontinuation of treatment with MAYZENT, vaccinations may be less effective; therefore MAYZENT treatment should be paused 1 week prior and for 4 weeks after vaccination [see Warnings and Precautions (5.1)].
The use of live attenuated vaccines may carry the risk of infection and should therefore be avoided during MAYZENT treatment and for up to 4 weeks after discontinuation of treatment with MAYZENT [see Warnings and Precautions (5.1)].
7.5 CYP2C9 and CYP3A4 Inhibitors
Because of a significant increase in exposure to siponimod, concomitant use of MAYZENT and drugs that cause moderate CYP2C9 and moderate or strong CYP3A4 inhibition is not recommended. This concomitant drug regimen can consist of a moderate CYP2C9/CYP3A4 dual inhibitor (e.g., fluconazole) or a moderate CYP2C9 inhibitor in combination with a separate - moderate or strong CYP3A4 inhibitor.
Caution should be exercised for concomitant use of MAYZENT with moderate CYP2C9 inhibitors.
7.6 CYP2C9 and CYP3A4 Inducers
Because of a significant decrease in siponimod exposure, concomitant use of MAYZENT and drugs that cause moderate CYP2C9 and strong CYP3A4 induction is not recommended for all patients. This concomitant drug regimen can consist of moderate CYP2C9/strong CYP3A4 dual inducer (e.g., rifampin or carbamazepine) or a moderate CYP2C9 inducer in combination with a separate strong CYP3A4 inducer.
Caution should be exercised for concomitant use of MAYZENT with moderate CYP2C9 inducers.
Concomitant use of MAYZENT and moderate (e.g., modafinil, efavirenz) or strong CYP3A4 inducers is not recommended for patients with CYP2C9*1/*3 and*2/*3 genotype [see Clinical Pharmacology (12.3)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no adequate data on the developmental risk associated with the use of MAYZENT in pregnant women. Based on animal data and its mechanism of action, MAYZENT can cause fetal harm when administered to a pregnant woman (see Data). Reproductive and developmental studies in pregnant rats and rabbits have demonstrated MAYZENT-induced embryotoxicity and fetotoxicity in rats and rabbits and teratogenicity in rats. Increased incidences of post-implantation loss and fetal abnormalities (external, urogenital and skeletal) in rat and of embryo-fetal deaths, abortions and fetal variations (skeletal and visceral) in rabbit were observed following prenatal exposure to siponimod starting at a dose 2 times the exposure in humans at the highest recommended dose of 2 mg/day.
In the US general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2%-4% and 15%-20%, respectively. The background risk of major birth defects and miscarriage for the indicated population is unknown.
Data
Animal Data
When siponimod (0, 1, 5, or 40 mg/kg) was orally administered to pregnant rats during the period of organogenesis, post implantation loss and fetal malformations (visceral and skeletal) were increased at the lowest dose tested, the only dose with fetuses available for evaluation. A no-effect dose for adverse effects on embryo-fetal development in rats was not identified. Plasma exposure AUC at the lowest dose tested was approximately 18 times that in humans at the recommended human dose (RHD) of 2 mg/day.
When siponimod (0, 0.1, 1, or 5 mg/kg) was orally administered to pregnant rabbits during the period of organogenesis, embryolethality and increased incidences of fetal skeletal variations were observed at all but the lowest dose tested. Plasma exposure (AUC) at the no-effect dose (0.1 mg/kg) for adverse effects on embryo-fetal development in rabbits is less that than in humans at the RHD.
When siponimod (0, 0.05, 0.15, or 0.5 mg/kg) was orally administered to female rats throughout pregnancy and lactation, increased mortality, decreased body weight, and delayed sexual maturation were observed in the offspring at all but the lowest dose tested. An increase in malformations was observed at all doses. A no-effect dose for adverse effects on pre- and postnatal development in rats was not identified. The lowest dose tested (0.05 mg/kg) is less than the RHD, on a mg/m2 basis.
8.2 Lactation
Risk Summary
There are no data on the presence of siponimod in human milk, the effects of MAYZENT on the breastfed infant, or the effects of the drug on milk production. A study in lactating rats has shown excretion of siponimod and/or its metabolites in milk. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for MAYZENT and any potential adverse effects on the breastfed infant from MAYZENT or from the underlying maternal condition.
8.3 Females and Males of Reproductive Potential
Contraception
Females
Before initiation of MAYZENT treatment, women of childbearing potential should be counselled on the potential for a serious risk to the fetus and the need for effective contraception during treatment with MAYZENT [see Use in Specific Populations (8.1)]. Since it takes approximately 10 days to eliminate the compound from the body after stopping treatment, the potential risk to the fetus may persist and women should use effective contraception during this period [see Warnings and Precautions (5.7)].
8.5 Geriatric Use
Clinical studies of MAYZENT did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
8.6 CYP2C9 Genotype
Before initiation of treatment with MAYZENT, test patients to determine CYP2C9 genotype. MAYZENT is contraindicated in patients homozygous for CYP2C9*3 (i.e., CYP2C9*3/*3 genotype), which is approximately 0.4%-0.5% of Caucasians and less in others, because of substantially elevated siponimod plasma levels. MAYZENT dosage adjustment is recommended in patients with CYP2C9*1/*3 or *2/*3 genotype because of an increase in exposure to siponimod [see Dosage and Administration (2.3) and Clinical Pharmacology (12.5)].
-
10 OVERDOSAGE
In patients with overdosage of MAYZENT, it is important to observe for signs and symptoms of bradycardia, which may include overnight monitoring. Regular measurements of pulse rate and blood pressure are required, and ECGs should be performed [see Warnings and Precautions (5.3, 5.6) and Clinical Pharmacology (12.2)].
There is no specific antidote to siponimod available. Neither dialysis nor plasma exchange would result in meaningful removal of siponimod from the body. The decrease in heart rate induced by MAYZENT can be reversed by atropine or isoprenaline.
-
11 DESCRIPTION
MAYZENT tablets contains siponimod, a sphingosine 1-phosphate receptor modulator, as 2:1 co-crystal of siponimod and fumaric acid and has the following chemical name:
1-[[4-[(1E)-1-[[[4-Cyclohexyl-3-(trifluoromethyl)phenyl]methoxy]imino]ethyl]-2-ethylphenyl]methyl]-3-azetidinecarboxylic acid (2E)-2-butenedioate (2:1). Its molecular formula is C4H4O4 2C29H35F3N2O3, and its molecular weight is 1149.29 g/mol.
Its structure is shown below:
It is a white to almost white powder.
MAYZENT is provided as 0.25 mg and 2 mg film-coated tablets for oral use. Each tablet contains 0.25 mg or 2 mg siponimod, equivalent to 0.28 mg or 2.22 mg as 2:1 co-crystal of siponimod and fumaric acid, respectively.
MAYZENT tablets contain the following inactive ingredients: colloidal silicon dioxide, crospovidone, glyceryl behenate, lactose monohydrate, microcrystalline cellulose, with a film coating containing iron oxides (black and red iron oxides for the 0.25 mg strength and red and yellow iron oxides for the 2 mg strength), lecithin (soy), polyvinyl alcohol, talc, titanium dioxide, and xanthan gum.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Siponimod is a sphingosine-1-phosphate (S1P) receptor modulator. Siponimod binds with high affinity to S1P receptors 1 and 5. Siponimod blocks the capacity of lymphocytes to egress from lymph nodes, reducing the number of lymphocytes in peripheral blood. The mechanism by which siponimod exerts therapeutic effects in multiple sclerosis is unknown, but may involve reduction of lymphocyte migration into the central nervous system.
12.2 Pharmacodynamics
Immune System
MAYZENT induces a dose-dependent reduction of the peripheral blood lymphocyte count within 6 hours of the first dose, caused by the reversible sequestration of lymphocytes in lymphoid tissues.
With continued daily dosing, the lymphocyte count continues to decrease, reaching a nadir median (90% CI) lymphocyte count of approximately 0.560 (0.271-1.08) cells/nL in a typical CYP2C9*1/*1 or *1/*2, non-Japanese patient, corresponding to 20% to 30% of baseline. Low lymphocyte counts are maintained with chronic daily dosing [see Warnings and Precautions (5.1)].
Lymphocyte counts returned to the normal range in 90% of patients within 10 days of stopping therapy. After stopping MAYZENT treatment, residual lowering effects on peripheral lymphocyte count may persist for up to 3-4 weeks after the last dose [see Warnings and Precautions (5.1)].
Heart Rate and Rhythm
MAYZENT causes a transient reduction in heart rate and atrioventricular conduction upon treatment initiation [see Warnings and Precautions (5.3)]. The maximum decline in heart rate is seen in the first 6 hours post dose. Autonomic responses of the heart, including diurnal variation of heart rate and response to exercise, are not affected by siponimod treatment.
A transient, dose-dependent decrease in heart rate was observed during the initial dosing phase of MAYZENT, which plateaued at doses greater than or equal to 5 mg, and bradyarrhythmic events (AV blocks and sinus pauses) were detected at a higher incidence under MAYZENT treatment, compared to placebo.
No second-degree AV blocks of Mobitz type II or higher degree were observed. Most AV blocks and sinus pauses occurred above the recommended dose of 2 mg, with notably higher incidence under non-titrated conditions compared to dose titration conditions [see Dosage and Administration (2.2, 2.3)].
The decrease in heart rate induced by MAYZENT can be reversed by atropine or isoprenaline.
Beta-Blockers
The negative chronotropic effect of coadministration of siponimod and propranolol was evaluated in a dedicated pharmacodynamics (PD)/safety study. The addition of propranolol on top of siponimod at steady-state had less pronounced negative chronotropic effects (less than additive effect) than the addition of siponimod to propranolol at steady state (additive HR effect) [see Drug Interactions (7.3)].
Cardiac Electrophysiology
In a thorough QT study with doses of 2 mg (recommended dose) and 10 mg (five times the recommended dose) siponimod at steady-state, siponimod treatment resulted in a prolongation of QTc , with the maximum mean (upper bound of the two-sided 90% CI) of 7.8 (9.93) ms at 2 mg dose and 7.2 (9.72) ms at 10 mg dose. There was an absence of dose- and exposure-response relationship for QTc effects with the 5-fold dose and exposures achieved by the supratherapeutic dose. No subject had absolute QTcF greater than 480 ms or ΔQTcF greater than 60 ms for siponimod treatment.
Pulmonary Function
Dose-dependent reductions in absolute forced expiratory volume over 1 second were observed in MAYZENT-treated patients and were greater than in patients taking placebo [see Warnings and Precautions (5.4)].
12.3 Pharmacokinetics
Siponimod concentration increases in an apparent dose-proportional manner after multiple once-daily doses of siponimod 0.3 mg to 20 mg. Steady-state plasma concentrations are reached after approximately 6 days of once-daily dosing, and steady-state levels are approximately 2-3-fold greater than the initial dose. An up-titration regimen is used to reach the clinical therapeutic dose of siponimod of 2 mg after 6 days, and 4 additional days of dosing are required to reach the steady-state-plasma concentrations.
Absorption
The time (Tmax) to reach maximum plasma concentrations (Cmax) after oral administration of immediate release oral dosage forms of siponimod was about 4 hours (range 3-8 hours). Siponimod absorption is extensive (greater than or equal to 70%, based on the amount of radioactivity excreted in urine and the amount of metabolites in feces extrapolated to infinity). The absolute oral bioavailability of siponimod is approximately 84%. After administration of siponimod 2 mg once-daily over 10 days, a mean Cmax of 30.4 ng/mL and mean area under plasma concentration-time curve over dosing interval (AUCtau) of 558 h*ng/mL were observed on day 10. Steady-state was reached after approximately 6 days of once-daily administration of siponimod.
Food Effect
Food intake resulted in delayed absorption (the median Tmax increased by approximately 2-3 hours). Food intake had no effect on the systemic exposure of siponimod (Cmax and AUC). Therefore, MAYZENT may be taken without regard to meals.
Distribution
Siponimod distributes to body tissues with a moderate mean volume of distribution of 124 L. Siponimod fraction found in plasma is 68% in humans. Animal studies show that siponimod readily crosses the blood-brain-barrier. Protein binding of siponimod is greater than 99.9% in healthy subjects and in hepatic and renal impaired patients.
Elimination
Metabolism
Siponimod is extensively metabolized, mainly via CYP2C9 (79.3%), followed by CYP3A4 (18.5%). The pharmacological activity of the main metabolites M3 and M17 is not expected to contribute to the clinical effect and the safety of siponimod in humans.
Excretion
An apparent systemic clearance (CL/F) of 3.11 L/h was estimated in MS patients. The apparent elimination half-life is approximately 30 hours.
Siponimod is eliminated from the systemic circulation mainly due to metabolism, and subsequent biliary/fecal excretion. Unchanged siponimod was not detected in urine.
Specific Populations
Male and Female Patients
Gender has no influence on siponimod pharmacokinetics (PK).
Racial or Ethnic Groups
The single-dose PK parameters were not different between Japanese and Caucasians healthy subjects, indicating absence of ethnic sensitivity on the PK of siponimod.
Patients with Renal Impairment
No dose adjustments are needed in patients with renal impairment. Mean siponimod half-life and Cmax (total and unbound) were comparable between subjects with severe renal impairment and healthy subjects. Unbound AUCs were only slightly increased (by 33%), compared to healthy subjects, and it is not expected to be clinically significant. The effects of end-stage renal disease or hemodialysis on the PK of siponimod has not been studied. Due to the high plasma protein binding (greater than 99.9%) of siponimod, hemodialysis is not expected to alter the total and unbound siponimod concentration and no dose adjustments are anticipated based on these considerations.
Patients with Hepatic Impairment
No dose adjustments for siponimod are needed in patients with hepatic impairment. The unbound siponimod AUC parameters are 15% and 50% higher in subjects with moderate and severe hepatic impairment, respectively, in comparison with healthy subjects for the 0.25 mg single dose studied. The increased unbound siponimod AUC in subjects with moderate and severe hepatic impairment is not expected to be clinically significant. The mean half-life of siponimod was unchanged in hepatic impairment.
Drug Interaction Studies
Siponimod (and Metabolites M3, M17) as a Causative Agent of Interaction
In vitro investigations indicated that siponimod and its major systemic metabolites M3 and M17 do not show any clinically relevant drug-drug interaction potential at the therapeutic dose of 2 mg once-daily for all investigated CYP enzymes and transporters.
Siponimod as an Object of Interaction
CYP2C9 is polymorphic and the genotype influences the fractional contributions of the two oxidative metabolism pathways to overall elimination. Physiologically based PK modeling indicates a differential CYP2C9 genotype-dependent inhibition and induction of CYP3A4 pathways. With decreased CYP2C9 metabolic activity in the respective genotypes, a larger effect of the CYP3A4 perpetrators on siponimod exposure is anticipated.
Coadministration of Siponimod with CYP2C9 and CYP3A4 Inhibitors
The coadministration of fluconazole (moderate CYP2C9 and CYP3A4 dual inhibitor) 200 mg daily at steady-state and a single dose of siponimod 4 mg in CYP2C9*1/*1 healthy volunteers led to a 2-fold increase in the AUC of siponimod. Mean siponimod terminal half-life was increased by 50%. Fluconazole led to a 2- to 4-fold increase in the AUCtau,ss of siponimod across different CYP2C9 genotypes, according to in silico evaluation [see Drug Interactions (7.5)].
Coadministration of Siponimod with CYP2C9 and CYP3A4 Inducers
The coadministration of siponimod 2 mg daily in the presence of 600 mg daily doses of rifampin (strong CYP3A4 and moderate CYP2C9 dual inducer) decreased siponimod AUCtau,ss and Cmax,ss by 57% and 45%, respectively in CY2C9*1/*1 subjects. Rifampin and efavirenz (moderate CYP3A4 inducer) reduced the AUCtau,ss of siponimod by up to 78% and up to 52%, respectively, across CYP2C9 genotypes, according to in silico evaluation [see Drug Interactions (7.6)].
Oral Contraceptives
The effects of coadministration of siponimod 2 mg and 4 mg (twice the recommended dosage) once daily with a monophasic oral contraceptive (OC) containing 30 mcg ethinyl estradiol and 150 mcg levonorgestrel were assessed in 24 healthy female subjects (18 to 40 years of age; CYP2C9*1/*1 genotype). There were no clinically relevant effects on the PK or PD of the OC. No interaction studies have been performed with OCs containing other progestagens; however, an effect of siponimod on their exposure is not expected.
12.5 Pharmacogenomics
The CYP2C9 genotype has a significant impact on siponimod metabolism. After a single dose of 0.25 mg siponimod, AUCinf and AUClast was approximately 2- and 4-fold higher in subjects with the CYP2C9*2/*3 and CYP2C9*3/*3 genotypes, respectively, while there was only a minor increase of Cmax by 21% and 16%, respectively, compared to extensive metabolizers (CYP2C9*1/*1). Mean half-life is prolonged in CYP2C9*2/*3 and CYP2C9*3/*3 carriers (51 hours and 126 hours, respectively).
An apparent systemic clearance (CL/F) of about 3.11 L/h was estimated in CYP2C9 extensive metabolizer (CYP2C9*1/*1 and CYP2C9*1/*2) MS patients after multiple oral administrations of siponimod. Cl/F is 2.5, 1.9, 1.6, and 0.9 L/h in subjects with the CYP2C9*2/*2, CYP2C9*1/*3, CYP2C9*2/*3, and CYP2C9*3/*3 genotypes respectively. The resultant increase in siponimod AUC was approximatively 25, 61, 91, and 285% higher in CYP2C9*2/*2, CYP2C9*1/*3, CYP2C9*2/*3, and CYP2C9*3/*3 subjects, respectively, as compared to CYP2C9*1/*1 subjects [see Dosage and Administration (2.1, 2.3) and Contraindications (4)]. As the apparent clearance estimated for CYP2C9*1*2 subjects is comparable to that of CYP2C9*1/*1 subjects, similar siponimod exposure is expected for both genotypes.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Oral carcinogenicity studies of spinomod were conducted in mice and rats. In mice administered siponimod (0, 2, 8, or 25 mg/kg/day) for up to 104 weeks, there was an increase in malignant lymphoma in females at all doses and in hemangiosarcoma and combined hemangioma and hemangiosarcoma at all doses in males and females. The lowest dose tested is approximately 5 times the recommended human dose (RHD) of 2 mg/day, on a body surface area (mg/m2) basis.
In rats, administration of siponimod (0, 10, 30, or 90 mg/kg/day in males; 0, 3, 10, or 30 mg/kg/day in females) for up to 104 weeks, there was an increase in thyroid follicular cell adenoma and combined thyroid follicular cell adenoma and carcinoma in males at the highest dose tested. These findings are considered secondary to liver enzyme induction in rats and are not considered relevant to humans. Plasma siponimod exposure (AUC) at the highest dose tested is approximately 200 times that in humans at the RHD.
Mutagenesis
Siponimod was negative in a battery of in vitro (Ames, chromosomal aberration in mammalian cells) and in vivo (micronucleus in mouse and rat) assays.
Impairment of Fertility
When siponimod was administered orally (0, 2, 20, or 200 mg/kg) to male rats (mated with untreated females) prior to and throughout the mating period, there was a dose-related increase in precoital interval at all doses. A decrease in implantation sites, an increase in preimplantation loss, and a decrease in the number of viable fetuses were observed at the highest dose tested. The higher no-effect dose for adverse effects on fertility (20 mg/kg) is approximately 100 times the RHD on a mg/m2 basis.
When siponimod was administered orally (0, 0.1, 0.3, or 1 mg/kg) to female rats (mated with untreated males) prior to and during mating, and continuing to Day 6 of gestation, no effects on fertility were observed up to the highest dose tested (1 mg/kg). Plasma siponimod exposure (AUC) at the highest dose tested is approximately 16 times that in humans at the RHD.
-
14 CLINICAL STUDIES
The efficacy of MAYZENT was demonstrated in Study 1, a randomized, double-blind, parallel-group, placebo-controlled, time-to-event study in patients with secondary progressive multiple sclerosis (SPMS) who had evidence of disability progression in the prior 2 years, no evidence of relapse in 3 months prior to study enrollment, and an Expanded Disability Status Scale (EDSS) score of 3.0-6.5 at study entry (NCT 01665144).
Patients were randomized to receive either once daily MAYZENT 2 mg or placebo, beginning with a dose titration [see Dosage and Administration (2.2)]. Evaluations were performed at screening, every 3 months during the study, and at the time of a suspected relapse. MRI evaluations were performed at screening and every 12 months.
The primary endpoint of the study was the time to 3-month confirmed disability progression (CDP), defined as at least a 1-point increase from baseline in EDSS (0.5-point increase for patients with baseline EDSS of 5.5 or higher) sustained for 3 months. A prespecified hierarchical analysis consisted of the primary endpoint and 2 secondary endpoints, the time to 3-month confirmed worsening of at least 20% from baseline on the timed 25-foot walk test and the change from baseline in T2 lesion volume. Additional endpoints included annualized relapse rate (relapses/year) and MRI measures of inflammatory disease activity.
Study duration was variable for individual patients (median study duration was 21 months, range 1 day-37 months).
Study 1 randomized 1651 patients to either MAYZENT 2 mg (N = 1105) or placebo (N = 546); 82% of MAYZENT-treated patients and 78% of placebo-treated patients completed the study. Median age was 49.0 years, 95% of patients were white, and 60% female. The median disease duration was 16.0 years, and median EDSS score at baseline was 6.0 (56% of patients had ≥ 6.0 EDSS at baseline); 36% of patients had one or more relapses in the 2 years prior to study entry; 22% of those patients with available imaging had one or more gadolinium-enhancing lesions on their baseline MRI scan; 78% of patients had been previously treated with an MS therapy.
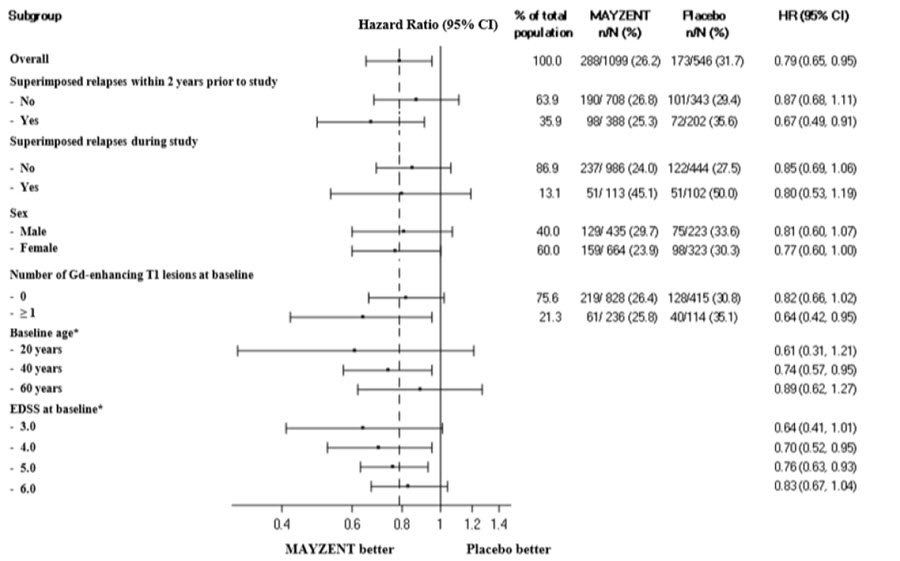
Results are presented in Table 4. MAYZENT was superior to placebo in reducing the risk of confirmed disability progression, based on a time-to-event analysis (hazard ratio 0.79, p < 0.0134; see Figure 1). MAYZENT did not significantly delay the time to 20% deterioration in the timed 25-foot walk, compared to placebo. Patients treated with MAYZENT had a 55% relative reduction in annualized relapse rate, compared to patients on placebo (nominal p-value < 0.0001). The absolute reduction in the annualized relapse rate was 0.089. Although MAYZENT had a significant effect on disability progression compared to placebo in patients with active SPMS (e.g., SPMS patients with an MS relapse in the 2 years prior to the study), the effect of MAYZENT in patients with non-active SPMS was not statistically significant (see Figure 2).
Table 4 Clinical and MRI Results From Study 1 All analyses are based on the full analysis set (FAS), which includes all randomized subjects who took at least one dose of study medication.
p-values are two-sided.
(1)Defined as an increase of 1.0 point or more from the baseline Expanded Disability Status Scale (EDSS) score for patients with baseline score of 5.5
or less, or 0.5 or more when the baseline score is greater than 5.5. Progression confirmed at 3 months. Cox proportional hazard model.
(2)Defined as the average number of confirmed relapses per year (estimated from negative binomial regression model for recurrent events).
(3)Adjusted mean averaged over Months 12 and 24.
*Statistically significant.
NS, Not statistically significant.
^Nominal p value, not corrected for multiple comparisons.MAYZENT PLACEBO Clinical Outcomes Proportion of patients with confirmed
disability progression126% 32% Relative risk reduction 21% (p = 0.0134)* Absolute risk Reduction 6% Proportion of patients with confirmed
worsening in timed 25-foot walk40% 41% p = NS Annualized relapse rate2 0.071 0.160 Relative reduction (%) 55% (p < 0.01)^ Absolute reduction 0.089 p < 0.01^ MRI Endpoints Change from baseline in T2 lesion
volume (mm3) (95% CI)3184
(54; 314)879
(712; 1047)p < 0.01^ Figure 1 Time to Confirmed Disability Progression Based on EDSS (Study 1)
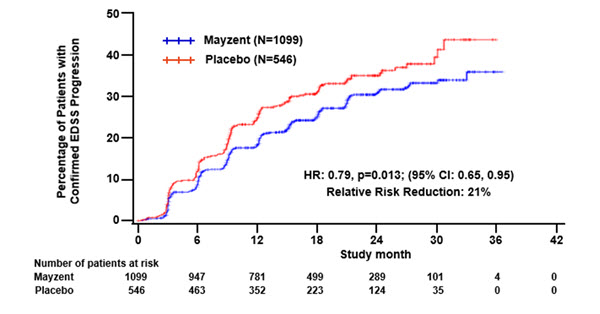
Figure 2 Time to Confirmed Disability Progression Based on EDSS (Study 1), Subgroup Analysis
* HR and 95% CI presented are model-based estimates for a range of values of age and EDSS.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
MAYZENT film-coated tablets are supplied as follows:
0.25 mg tablet: Pale red, unscored, round biconvex film-coated tablet with beveled edges, debossed with
 on one side and ‘T’ on other side.
on one side and ‘T’ on other side.Starter Pack* – blister card of twelve 0.25 mg tablets in a calendarized blister wallet...............NDC: 0078-0979-12
*This starter pack is only intended for patients who will receive the 2 mg maintenance dosage.
Bottle of 28 tablets........................................................................................................................NDC: 0078-0979-50
2 mg tablet: Pale yellow, unscored, round biconvex film-coated tablet with beveled edges, debossed with
on one side and ‘II’ on other side.Bottle of 30 tablets.........................................................................................................................NDC: 0078-0986-15
16.2 Storage and Handling
Unopened Containers
Store unopened containers of MAYZENT 0.25 mg and 2 mg film-coated tablets in a refrigerator between 2°C to 8°C (36°F to 46°F).
Opened Containers
Store opened containers of MAYZENT as follows:
Starter Pack/Blister Card
MAYZENT 0.25 mg film-coated tablets in the Starter Pack may be stored at 20°C to 25°C (68°F to 77°F) [see USP Controlled Room Temperature] for up to 1 week after opening the blister. Store in original container.
Bottles
MAYZENT 0.25 mg and 2 mg film-coated tablets in bottles may be stored at 20°C to 25°C (68°F to 77°F) [see USP Controlled Room Temperature] for up to 1 month after opening the bottles.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Tell patients not to discontinue MAYZENT without first discussing this with the prescribing physician. Advise patients to contact their physician if they accidently take more MAYZENT than prescribed.
Risk of Infections
Inform patients that they may have an increased risk of infections, some of which could be life-threatening, when taking MAYZENT, and that they should contact their physician if they develop symptoms of infection [see Warnings and Precautions (5.1)]. Advise patients that the use of some vaccines containing live virus (live attenuated vaccines) should be avoided during treatment with MAYZENT and MAYZENT should be paused 1 week prior and until 4 weeks after a planned vaccination. Recommend that patients postpone treatment with MAYZENT for at least 1 month after VZV vaccination. Inform patients that prior or concomitant use of drugs that suppress the immune system may increase the risk of infection.
Macular Edema
Advise patients that MAYZENT may cause macular edema, and that they should contact their physician if they experience any changes in their vision while taking MAYZENT [see Warnings and Precautions (5.2)]. Inform patients with diabetes mellitus or a history of uveitis that their risk of macular edema is increased.
Cardiac Effects
Advise patients that initiation of MAYZENT treatment results in transient decrease in heart rate [see Warnings and Precautions (5.3)]. Inform patients that to reduce this effect, dosage titration is required. Advise patients that dosage titration is also required if a dose is missed for more than 24 hours during the titration or if 4 or more consecutive daily maintenance doses are missed [see Dosage and Administration (2.2, 2.3, 2.5) and Warnings and Precautions (5.3)]. Inform certain patients with certain pre-existing cardiac conditions that they will need to be observed in the doctor's office or other facility for at least 6 hours after the first dose and after reinitiation if treatment is interrupted or discontinued for certain periods [see Dosage and Administration (2.4)].
Respiratory Effects
Advise patients that they should contact their physician if they experience new onset or worsening of dyspnea [see Warnings and Precautions (5.4)].
Liver Injury
Inform patients that MAYZENT may increase liver enzymes. Advise patient that they should contact their physician if they experience any unexplained nausea, vomiting, abdominal pain, fatigue, anorexia, or jaundice and/or dark urine during treatment [see Warnings and Precautions (5.5)].
Pregnancy and Fetal Risk
Inform patients that, based on animal studies MAYZENT may cause fetal harm [see Warnings and Precautions (5.7)]. Discuss with women of childbearing age whether they are pregnant, might be pregnant, or are trying to become pregnant. Advise women of childbearing potential of the need for effective contraception during treatment with MAYZENT and for 10 days after stopping MAYZENT. Advise a female patient to immediately inform that prescriber if she is pregnant or planning to become pregnant [see Warnings and Precautions (5.7) and Use in Specific Populations (8.1, 8.3)].
Posterior Reversible Encephalopathy Syndrome
Advise patients to immediately report to their healthcare provider any symptoms involving sudden onset of severe headache, altered mental status, visual disturbances, or seizure. Inform patients that delayed treatment could lead to permanent neurological sequelae [see Warnings and Precautions (5.8)].
Severe Increase in Disability After Stopping MAYZENT
Inform patients that severe increase in disability has been reported after discontinuation of another sphingosine 1-phosphate (S1P) receptor modulator like MAYZENT. Advise patients to contact their physician if they develop worsening symptoms of MS following discontinuation of MAYZENT [see Warnings and Precautions (5.10)].
Immune System Effects After Stopping MAYZENT
Advise patients that MAYZENT continues to have effects, such as lowering effects on peripheral lymphocyte count, for up to 3-4 weeks after the last dose [see Warnings and Precautions (5.11)].
Storage and Handling
Instruct patients to store any unopened containers of MAYZENT in a refrigerator. Inform patients that opened starter packs may be stored at room temperature for 1 week and opened bottles may be stored at room temperature for 1 month [see How Supplied/Storage and Handling (16.2)].
Distributed by:
Novartis Pharmaceuticals Corporation
East Hanover, New Jersey 07936MAYZENT is a registered trademark of Novartis AG
© Novartis
T2019-45
-
MEDICATION GUIDE
This Medication Guide has been approved by the U.S. Food and Drug Administration Issued: March 2019 MEDICATION GUIDE
MAYZENT (Māʹzĕnt)
(siponimod)
tablets, for oral useWhat is the most important information I should know about MAYZENT?
1. MAYZENT may cause serious side effects, including: Slow heart rate (bradycardia or bradyarrhythmia) when you start taking MAYZENT. MAYZENT can cause your heart rate to slow down, especially after you take your first dose. You should have a test to check the electrical activity of your heart called an electrocardiogram (ECG) before you take your first dose of MAYZENT.
During the initial updosing period (4 days for the 1 mg daily dose or 5 days for the 2 mg daily dose), if you miss 1 or more doses of MAYZENT, you need to restart the updosing. Call your healthcare provider if you miss a dose of MAYZENT. See “How should I take MAYZENT?”
2. Infections. MAYZENT can increase your risk of serious infections that can be life-threatening and cause death. MAYZENT lowers the number of white blood cells (lymphocytes) in your blood. This will usually go back to normal within 3 to 4 weeks of stopping treatment. Your healthcare provider should review a recent blood test of your white blood cells before you start taking MAYZENT.
Call your healthcare provider right away if you have any of these symptoms of an infection during treatment with MAYZENT and for 3 to 4 weeks after your last dose of MAYZENT:- fever
- tiredness
- body aches
- chills
- nausea
- vomiting
- headache with fever, neck stiffness, sensitivity to light, nausea, confusion (these may be symptoms of meningitis, an infection of the lining around your brain and spine)
3. A problem with your vision called macular edema. Macular edema can cause some of the same vision symptoms as a multiple sclerosis (MS) attack (optic neuritis). You may not notice any symptoms with macular edema. If macular edema happens, it usually starts in the first 1 to 4 months after your start taking MAYZENT. Your healthcare provider should test your vision before you start taking MAYZENT and any time you notice vision changes during treatment with MAYZENT. Your risk of macular edema is higher if you have diabetes or have had an inflammation of your eye called uveitis.
Call your healthcare provider right away if you have any of the following:- blurriness or shadows in the center of your vision
- a blind spot in the center of your vision
- sensitivity to light
- unusually colored (tinted) vision
See "What are possible side effects of MAYZENT?" for more information about side effects. What is MAYZENT?
MAYZENT is a prescription medicine that is used to treat relapsing forms of multiple sclerosis, to include clinically isolated syndrome, relapsing-remitting disease, and active secondary progressive disease, in adults.
It is not known if MAYZENT is safe and effective in children.Who should not take MAYZENT?
Do not take MAYZENT if you:- have a CYP2C9*3/*3 genotype. Before starting treatment with MAYZENT, your CYP2C9 genotype should be determined by your healthcare provider. Ask your healthcare provider if you are not sure.
- have had a heart attack, chest pain called unstable angina, stroke or mini-stroke (transient ischemic attack or TIA), or certain types of heart failure in the last 6 months
- have certain types of heart block or irregular or abnormal heartbeat (arrhythmia), unless you have a pacemaker
What should I tell my healthcare provider before taking MAYZENT?
Before taking MAYZENT, tell your healthcare provider about all of your medical conditions, including if you:- have an irregular or abnormal heartbeat
- a history of stroke or other diseases related to blood vessels in the brain
- breathing problems, including during your sleep
- a fever or infection, or you are unable to fight infections due to a disease or taking medicines that lower your immune system. Tell your healthcare provider if you have had chicken pox or have received the vaccine for chicken pox. Your healthcare provider may do a blood test for chicken pox virus. You may need to get the full course of vaccine for chicken pox and then wait 1 month before you start taking MAYZENT.
- have slow heart rate
- have liver problems
- have diabetes
- have eye problems, especially an inflammation of the eye called uveitis
- have high blood pressure
- are pregnant or plan to become pregnant. MAYZENT may harm your unborn baby. Talk to your healthcare provider right away if you become pregnant while taking MAYZENT or if you become pregnant within 10 days after you stop taking MAYZENT.
- If you are a woman who can become pregnant, you should use effective birth control during your treatment with MAYZENT and for at least 10 days after you stop taking MAYZENT.
- are breastfeeding or plan to breastfeed. It is not known if MAYZENT passes into your breast milk. Talk to your healthcare provider about the best way to feed your baby if you take MAYZENT.
Tell your healthcare provider about all the medicines you take, including prescription medicines, over-the-counter medicines, vitamins, and herbal supplements. Especially tell your healthcare provider if you:
- take medicines to control your heart rhythm (antiarrhythmics), or blood pressure (antihypertensives), or heart beat (such as calcium channel blockers or beta-blockers)
- take medicines that affect your immune system, such as beta-interferon or glatiramer acetate, or any of these medicines that you took in the past
- have recently received a live vaccine. You should avoid receiving live vaccines during treatment with MAYZENT. MAYZENT should be stopped 1 week before and for 4 weeks after receiving a live vaccine. If you receive a live vaccine, you may get the infection the vaccine was meant to prevent. Vaccines may not work as well when given during treatment with MAYZENT.
Know the medicines you take. Keep a list of your medicines with you to show your healthcare provider and pharmacist when you get a new medicine.
Using MAYZENT and other medicines together may affect each other causing serious side effects.
How should I take MAYZENT?
The daily maintenance dose of MAYZENT is either 1 mg or 2 mg, depending on your CYP2C9 genotype. Ask your healthcare provider if you are not sure about your daily maintenance dose.
Start your treatment with MAYZENT using the following titration schedule:
For the 1 mg daily maintenance dose:
Day 1
Day 2
Day 3
Day 4
Day 5 and every day after
Tablets a day
1 x 0.25 mg tablet
1 x 0.25 mg tablet
2 x 0.25 mg tablet
3 x 0.25 mg tablet
4 x 0.25 mg tablet
For the 2 mg daily maintenance dose, use the starter pack:
Day 1
Day 2
Day 3
Day 4
Day 5
Day 6 and every day after
Tablets a day
1 x 0.25 mg tablet
1 x 0.25 mg tablet
2 x 0.25 mg tablet
3 x 0.25 mg tablet
5 x 0.25 mg tablet
1 x 2 mg tablet
- Take MAYZENT exactly as your healthcare provider tells you. Do not change your dose or stop taking MAYZENT unless your healthcare provider tells you to.
- Take MAYZENT 1 time each day.
- Take MAYZENT with or without food.
- If you miss 1 or more doses of MAYZENT during the initial dose titration, you need to restart the medication.
- If you miss a dose of MAYZENT after the initial dose-titration, take it as soon as you remember.
- If MAYZENT treatment is stopped for 4 days in a row, treatment has to be restarted with the titration.
- Do not stop taking MAYZENT without talking with your healthcare provider first.
What are the possible side effects of MAYZENT?
MAYZENT may cause serious side effects, including:- See "What is the most important information I should know about MAYZENT?"
- increased blood pressure. Your healthcare provider should check your blood pressure during treatment with MAYZENT.
- liver problems. MAYZENT may cause liver problems. Your healthcare provider should do blood tests to check your liver before you start taking MAYZENT. Call your healthcare provider right away if you have any of the following symptoms of liver problems:
◦ nausea
◦ vomiting
◦ stomach pain
◦ tiredness◦ loss of appetite
◦ your skin or the whites of your eyes turn yellow
◦ dark urine
- breathing problems. Some people who take MAYZENT have shortness of breath. Call your healthcare provider right away if you have new or worsening breathing problems.
- swelling and narrowing of the blood vessels in your brain. A condition called PRES (Posterior Reversible Encephalopathy Syndrome) has happened with drugs in the same class. Symptoms of PRES usually get better when you stop taking MAYZENT. However, if left untreated, it may lead to a stroke. Call your healthcare provider right away if you have any of the following symptoms:
◦ sudden severe headache
◦ sudden confusion◦ sudden loss of vision or other changes in your vision
◦ seizure
- severe worsening of multiple sclerosis after stopping MAYZENT. When MAYZENT is stopped, symptoms of MS may return and become worse compared to before or during treatment. Always talk to your doctor before you stop taking MAYZENT for any reason. Tell your healthcare provider if you have worsening symptoms of MS after stopping MAYZENT.
The most common side effects of MAYZENT include: - headache
- high blood pressure (hypertension)
- abnormal liver tests
Tell your healthcare provider if you have any side effects that bother you or that do not go away.
These are not all of the possible side effects of MAYZENT. For more information, ask your healthcare provider or pharmacist. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store MAYZENT?
Before opening:- MAYZENT 0.25 mg and 2 mg tablets should be stored in a refrigerator between 36°F to 46°F (2°C to 8°C).
- MAYZENT 0.25 mg tablets in the Starter Pack may be stored at room temperature, 68°F to 77°F (20°C to 25°C), for up to 1 week after opening.
- MAYZENT 0.25 mg and 2 mg tablets in bottles may be stored at room temperature, 68°F to 77°F (20°C to 25°C), for up to 1 month after opening.
General information about the safe and effective use of MAYZENT
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use MAYZENT for a condition for which it was not prescribed. Do not give MAYZENT to other people, even if they have the same symptoms you have. It may harm them. You can ask your pharmacist or healthcare provider for more information about MAYZENT that is written for health professionals.What are the ingredients in MAYZENT?
Active ingredient: siponimod
Inactive ingredients: colloidal silicon dioxide, crospovidone, glyceryl behenate, lactose monohydrate, microcrystalline cellulose, with a film coating containing iron oxides (black and red iron oxides for the 0.25 mg strength and red and yellow iron oxides for the 2 mg strength), lecithin (soy), polyvinyl alcohol, talc, titanium dioxide, and xanthan gum.
Distributed by: Novartis Pharmaceuticals Corporation, East Hanover, New Jersey 07936
For more information, go to www.pharma.us.novartis.com or call 1-888-669-6682.
T2019-46
-
PRINCIPAL DISPLAY PANEL
PRINCIPAL DISPLAY PANEL
NDC: 0078-0979-50
Rx only
MAYZENT®
(siponimod) tablets
0.25 mg
28 tablets
Dispense with accompanying
Medication Guide.NOVARTIS

-
PRINCIPAL DISPLAY PANEL
PRINCIPAL DISPLAY PANEL
NDC: 0078-0986-15
Rx only
MAYZENT®
(siponimod) tablets
2 mg
30 tablets
Dispense with accompanying
Medication Guide.NOVARTIS

-
INGREDIENTS AND APPEARANCE
MAYZENT
siponimod tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0078-0979 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength SIPONIMOD (UNII: RR6P8L282I) (SIPONIMOD - UNII:RR6P8L282I) SIPONIMOD 0.25 mg Inactive Ingredients Ingredient Name Strength LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) CROSPOVIDONE, UNSPECIFIED (UNII: 2S7830E561) GLYCERYL DIBEHENATE (UNII: R8WTH25YS2) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) TALC (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FERRIC OXIDE RED (UNII: 1K09F3G675) FERROSOFERRIC OXIDE (UNII: XM0M87F357) Product Characteristics Color RED (pale red) Score no score Shape ROUND Size 6mm Flavor Imprint Code IMPRINT;T Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0078-0979-50 28 in 1 BOTTLE; Type 0: Not a Combination Product 03/26/2019 2 NDC: 0078-0979-12 12 in 1 BLISTER PACK; Type 0: Not a Combination Product 03/26/2019 3 NDC: 0078-0979-92 28 in 1 BOTTLE; Type 0: Not a Combination Product 03/26/2019 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA209884 03/26/2019 MAYZENT
siponimod tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0078-0986 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength SIPONIMOD (UNII: RR6P8L282I) (SIPONIMOD - UNII:RR6P8L282I) SIPONIMOD 2 mg Inactive Ingredients Ingredient Name Strength LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) CROSPOVIDONE, UNSPECIFIED (UNII: 2S7830E561) GLYCERYL DIBEHENATE (UNII: R8WTH25YS2) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) TALC (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FERRIC OXIDE RED (UNII: 1K09F3G675) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) Product Characteristics Color YELLOW (pale yellow) Score no score Shape ROUND Size 6mm Flavor Imprint Code IMPRINT;II Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0078-0986-15 30 in 1 BOTTLE; Type 0: Not a Combination Product 03/26/2019 2 NDC: 0078-0986-93 30 in 1 BOTTLE; Type 0: Not a Combination Product 03/26/2019 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA209884 03/26/2019 Labeler - Novartis Pharmaceuticals Corporation (002147023)
Trademark Results [MAYZENT]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 MAYZENT 87816240 5846370 Live/Registered |
Novartis AG 2018-03-01 |
 MAYZENT 79261592 not registered Live/Pending |
NOVARTIS AG 2019-05-03 |
 MAYZENT 79156316 4714545 Live/Registered |
Novartis AG 2014-09-12 |
 MAYZENT 79043338 3441076 Dead/Cancelled |
Novartis AG 2007-08-31 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.