SIMVASTATIN tablet, film coated SIMVASTATIN tablet, film coated
SIMVASTATIN by
Drug Labeling and Warnings
SIMVASTATIN by is a Prescription medication manufactured, distributed, or labeled by Legacy Pharmaceutical Packaging, LLC. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use SIMVASTATIN TABLETS safely and effectively. See full prescribing information for SIMVASTATIN TABLETS.
SIMVASTATIN tablets, for oral use
Initial U.S. Approval: 1991RECENT MAJOR CHANGES
Dosage and Administration
Chinese Patients Taking Lipid-Modifying Doses (greater than or equal to 1 g/day Niacin) of Niacin-Containing Products - removal (2.7) 3/2019
Warnings and Precautions
Myopathy/Rhabdomyolisis (5.1) 3/2019
INDICATIONS AND USAGE
Simvastatin tablets USP are an HMG-CoA reductase inhibitor (statin) indicated as an adjunctive therapy to diet to:
- Reduce the risk of total mortality by reducing CHD deaths and reduce the risk of non-fatal myocardial infarction, stroke, and the need for revascularization procedures in patients at high risk of coronary events. (1.1)
- Reduce elevated total-C, LDL-C, Apo B, TG and increase HDL-C in patients with primary hyperlipidemia (heterozygous familial and nonfamilial) and mixed dyslipidemia. (1.2)
- Reduce elevated TG in patients with hypertriglyceridemia and reduce TG and VLDL-C in patients with primary dysbetalipoproteinemia. (1.2)
- Reduce total-C and LDL-C in adult patients with homozygous familial hypercholesterolemia. (1.2)
- Reduce elevated total-C, LDL-C, and Apo B in boys and postmenarchal girls, 10 to 17 years of age with heterozygous familial hypercholesterolemia after failing an adequate trial of diet therapy. (1.2, 1.3)
Limitations of Use
Simvastatin tablets USP has not been studied in Fredrickson Types I and V dyslipidemias. (1.4)
DOSAGE AND ADMINISTRATION
- Dose range is 5 to 40 mg/day. (2.1)
- Recommended usual starting dose is 10 or 20 mg once a day in the evening. (2.1)
- Recommended starting dose for patients at high risk of CHD is 40 mg/day. (2.1)
- Due to the increased risk of myopathy, including rhabdomyolysis, use of the 80-mg dose of simvastatin tablets USP should be restricted to patients who have been taking simvastatin 80 mg chronically (e.g., for 12 months or more) without evidence of muscle toxicity. (2.2)
- Patients who are currently tolerating the 80-mg dose of simvastatin tablets USP who need to be initiated on an interacting drug that is contraindicated or is associated with a dose cap for simvastatin should be switched to an alternative statin with less potential for the drug-drug interaction. (2.2)
- Due to the increased risk of myopathy, including rhabdomyolysis, associated with the 80-mg dose of simvastatin tablets USP, patients unable to achieve their LDL-C goal utilizing the 40-mg dose of simvastatin tablets USP should not be titrated to the 80-mg dose, but should be placed on alternative LDL-C-lowering treatment(s) that provides greater LDL-C lowering. (2.2)
- Adolescents (10 to 17 years of age) with HeFH: starting dose is 10 mg/day; maximum recommended dose is 40 mg/day. (2.5)
DOSAGE FORMS AND STRENGTHS
Tablets: 5 mg; 10 mg; 20 mg; 40 mg; 80 mg (3)
CONTRAINDICATIONS
- Concomitant administration of strong CYP3A4 inhibitors. (4,5.1)
- Concomitant administration of gemfibrozil, cyclosporine, or danazol. (4, 5.1)
- Hypersensitivity to any component of this medication. (4,6.2)
- Active liver disease, which may include unexplained persistent elevations in hepatic transaminase levels. (4,5.2)
- Women who are pregnant or may become pregnant. (4,8.1)
- Nursing mothers. (4, 8.3)
WARNINGS AND PRECAUTIONS
- Patients should be advised of the increased risk of myopathy including rhabdomyolysis with the 80-mg dose. (5.1)
- Skeletal muscle effects (e.g., myopathy and rhabdomyolysis): Risks increase with higher doses and concomitant use of certain medicines. Predisposing factors include advanced age (≥65), female gender, uncontrolled hypothyroidism, and renal impairment. Rare cases of rhabdomyolysis with acute renal failure secondary to myoglobinuria have been reported. (4, 5.1, 8.5, 8.6)
- Patients should be advised to report promptly any unexplained and/or persistent muscle pain, tenderness, or weakness. Simvastatin therapy should be discontinued immediately if myopathy is diagnosed or suspected. See Drug Interaction table. (5.1)
- Liver enzyme abnormalities: Persistent elevations in hepatic transaminases can occur. Check liver enzyme tests before initiating therapy and as clinically indicated thereafter. (5.2)
ADVERSE REACTIONS
Most common adverse reactions (incidence ≥5.0%) are: upper respiratory infection, headache, abdominal pain, constipation, and nausea. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Lupin Pharmaceuticals, Inc. at 1-800-399-2561 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
Drug Interactions Associated with Increased Risk of Myopathy/Rhabdomyolysis (2.3, 2.4, 4, 5.1, 7.1, 7.2, 7.3, 12.3)
Interacting Agents
Prescribing Recommendations
Strong CYP3A4 inhibitors (e.g., itraconazole, ketoconazole, posaconazole, voriconazole, erythromycin, clarithromycin, telithromycin, HIV protease inhibitors, boceprevir, telaprevir, nefazodone, cobicistat containing products), gemfibrozil, cyclosporine, danazol
Contraindicated with simvastatin
Niacin (≥1 g/day)
For Chinese patients, not recommended with simvastatin
Verapamil, diltiazem, drone darone
Do not exceed 10 mg simvastatin daily
Amiodarone, amlodipine, ranolazine
Do not exceed 20 mg simvastatin daily
Lomitapide
For patients with HoFH, do not exceed 20 mg simvastatin daily*
Grapefruit juice
Avoid grapefruit juice
* For patients with HoFH who have been taking 80 mg simvastatin chronically (e.g., for 12 months or more) without evidence of muscle toxicity, do not exceed 40 mg simvastatin when taking lomitapide.
- Other Lipid-lowering Medications: Use with other fibrate products increases the risk of adverse skeletal muscle effects. Caution should be used when prescribing with simvastatin. (5.1, 7.2)
- Coumarin anticoagulants: Concomitant use with simvastatin prolongs INR. Achieve stable INR prior to starting simvastatin. Monitor INR frequently until stable upon initiation or alteration of simvastatin therapy. (7.6)
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 12/2019
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
RECENT MAJOR CHANGES
1 INDICATIONS AND USAGE
1.1 Reductions in Risk of CHD Mortality and Cardiovascular Events
1.2 Hyperlipidemia
1.3 Adolescent Patients with Heterozygous Familial Hypercholesterolemia (HeFH)
1.4 Limitations of Use
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosing
2.2 Restricted Dosing for 80 mg
2.3 Coadministration with Other Drugs
2.4 Patients with Homozygous Familial Hypercholesterolemia
2.5 Adolescents (10 to 17 years of age) with Heterozygous Familial Hypercholesterolemia
2.6 Patients with Renal Impairment
2.7 Chinese Patients Taking Lipid-Modifying Doses (greater than or equal to 1 g/day Niacin) of Niacin-Containing Products
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Myopathy/Rhabdomyolysis
5.2 Liver Dysfunction
5.3 Endocrine Function
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Strong CYP3A4 Inhibitors, Cyclosporine, or Danazol
7.2 Lipid-Lowering Drugs That Can Cause Myopathy When Given Alone
7.3 Amiodarone, Dronedarone, Ranolazine, or Calcium Channel Blockers
7.4 Niacin
7.5 Digoxin
7.6 Coumarin Anticoagulants
7.7 Colchicine
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.3 Nursing Mothers
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
8.8 Chinese Patients
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 ANIMAL PHARMACOLOGY AND OR TOXICOLOGY
14 CLINICAL STUDIES
14.1 Clinical Studies in Adults
14.2 Clinical Studies in Adolescents
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
17.1 Muscle Pain
17.2 Liver Enzymes
17.3 Pregnancy
17.4 Breastfeeding
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
Therapy with lipid-altering agents should be only one component of multiple risk factor intervention in individuals at significantly increased risk for atherosclerotic vascular disease due to hypercholesterolemia. Drug therapy is indicated as an adjunct to diet when the response to a diet restricted in saturated fat and cholesterol and other nonpharmacologic measures alone has been inadequate. In patients with coronary heart disease (CHD) or at high risk of CHD, simvastatin tablets USP can be started simultaneously with diet.
1.1 Reductions in Risk of CHD Mortality and Cardiovascular Events
In patients at high risk of coronary events because of existing coronary heart disease, diabetes, peripheral vessel disease, history of stroke or other cerebrovascular disease, simvastatin tablets USP are indicated to:
- Reduce the risk of total mortality by reducing CHD deaths.
- Reduce the risk of non-fatal myocardial infarction and stroke.
- Reduce the need for coronary and non-coronary revascularization procedures.
1.2 Hyperlipidemia
Simvastatin tablets USP are indicated to:
- Reduce elevated total cholesterol (total-C), low-density lipoprotein cholesterol (LDL-C), apolipoprotein B (Apo B), and triglycerides (TG), and to increase high-density lipoprotein cholesterol (HDL-C) in patients with primary hyperlipidemia (Fredrickson type IIa, heterozygous familial and nonfamilial) or mixed dyslipidemia (Fredrickson type IIb).
- Reduce elevated TG in patients with hypertriglyceridemia (Fredrickson type lV hyperlipidemia).
- Reduce elevated TG and VLDL-C in patients with primary dysbetalipoproteinemia (Fredrickson type III hyperlipidemia).
- Reduce total-C and LDL-C in patients with homozygous familial hypercholesterolemia (HoFH) as an adjunct to other lipid-lowering treatments (e.g., LDL apheresis) or if such treatments are unavailable.
1.3 Adolescent Patients with Heterozygous Familial Hypercholesterolemia (HeFH)
Simvastatin tablets USP are indicated as an adjunct to diet to reduce total-C, LDL-C, and Apo B levels in adolescent boys and girls who are at least one year post-menarche, 10 to 17 years of age, with HeFH, if after an adequate trial of diet therapy the following findings are present:
1. LDL cholesterol remains ≥190 mg/dL; or
2. LDL cholesterol remains ≥160 mg/dL and- There is a positive family history of premature cardiovascular disease (CVD) or
- Two or more other CVD risk factors are present in the adolescent patient.
The minimum goal of treatment in pediatric and adolescent patients is to achieve a mean LDL-C <130 mg/dL. The optimal age at which to initiate lipid-lowering therapy to decrease the risk of symptomatic adulthood CAD has not been determined.
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosing
The usual dosage range is 5 to 40 mg/day. In patients with CHD or at high risk of CHD, simvastatin tablets USP can be started simultaneously with diet. The recommended usual starting dose is 10 or 20 mg once a day in the evening. For patients at high risk for a CHD event due to existing CHD, diabetes, peripheral vessel disease, history of stroke or other cerebrovascular disease, the recommended starting dose is 40 mg/day. Lipid determinations should be performed after 4 weeks of therapy and periodically thereafter.
2.2 Restricted Dosing for 80 mg
Due to the increased risk of myopathy, including rhabdomyolysis, particularly during the first year of treatment, use of the 80-mg dose of simvastatin tablets USP should be restricted to patients who have been taking simvastatin 80 mg chronically (e.g., for 12 months or more) without evidence of muscle toxicity [see WARNINGS AND PRECAUTIONS (5.1)].
Patients who are currently tolerating the 80-mg dose of simvastatin tablets USP who need to be initiated on an interacting drug that is contraindicated or is associated with a dose cap for simvastatin should be switched to an alternative statin with less potential for the drug-drug interaction.
Due to the increased risk of myopathy, including rhabdomyolysis, associated with the 80-mg dose of simvastatin tablets USP, patients unable to achieve their LDL-C goal utilizing the 40-mg dose of simvastatin tablets USP should not be titrated to the 80-mg dose, but should be placed on alternative LDL-C-lowering treatment(s) that provides greater LDL-C lowering.
2.3 Coadministration with Other Drugs
Patients taking Verapamil, Diltiazem, or Dronedarone
- The dose of simvastatin tablets USP should not exceed 10 mg/day[see WARNINGS AND PRECAUTIONS (5.1), DRUG INTERACTIONS (7.3),and CLINICAL PHARMACOLOGY (12.3)].
Patients taking Amiodarone, Amlodipine or Ranolazine
2.4 Patients with Homozygous Familial Hypercholesterolemia
The recommended dosage is 40 mg/day in the evening [see DOSAGE AND ADMINISTRATION, Restricted Dosing for 80 mg (2.2)]. Simvastatin tablets USP should be used as an adjunct to other lipid-lowering treatments (e.g., LDL apheresis) in these patients or if such treatments are unavailable.
Simvastatin exposure is approximately doubled with concomitant use of lomitapide; therefore, the dose of simvastatin tablets USP should be reduced by 50% if initiating lomitapide. Simvastatin tablets USP dosage should not exceed 20 mg/day (or 40 mg/day for patients who have previously taken simvastatin tablets USP 80 mg/day chronically, e.g., for 12 months or more, without evidence of muscle toxicity) while taking lomitapide.
2.5 Adolescents (10 to 17 years of age) with Heterozygous Familial Hypercholesterolemia
The recommended usual starting dose is 10 mg once a day in the evening. The recommended dosing range is 10 to 40 mg/day; the maximum recommended dose is 40 mg/day. Doses should be individualized according to the recommended goal of therapy [see NCEP Pediatric Panel Guidelines1 and CLINICAL STUDIES (14.2)]. Adjustments should be made at intervals of 4 weeks or more.
2.6 Patients with Renal Impairment
Because simvastatin tablets USP do not undergo significant renal excretion, modification of dosage should not be necessary in patients with mild to moderate renal impairment. However, caution should be exercised when simvastatin tablets USP are administered to patients with severe renal impairment; such patients should be started at 5 mg/day and be closely monitored [see WARNINGS AND PRECAUTIONS (5.1) and CLINICAL PHARMACOLOGY (12.3)].
2.7 Chinese Patients Taking Lipid-Modifying Doses (greater than or equal to 1 g/day Niacin) of Niacin-Containing Products
Because of an increased risk for myopathy in Chinese patients taking simvastatin 40 mg coadministered with lipid-modifying doses (greater than or equal to 1 g/day niacin) of niacin-containing products, caution should be used when treating Chinese patients with simvastatin doses exceeding 20 mg/day coadministered with lipid-modifying doses of niacin-containing products. Because the risk for myopathy is dose-related, Chinese patients should not receive simvastatin 80 mg coadministered with lipid-modifying doses of niacin-containing products. The cause of the increased risk of myopathy is not known. It is also unknown if the risk for myopathy with coadministration of simvastatin with lipid-modifying doses of niacin-containing products observed in Chinese patients applies to other Asian patients [see WARNINGS AND PRECAUTIONS (5.1)].
-
3 DOSAGE FORMS AND STRENGTHS
- Simvastatin tablets 5 mg are tan colored, round, biconvex, film-coated tablets debossed with 'LL' on one side and 'C01' on the other side.
- Simvastatin tablets 10 mg are peach colored, oval shaped, biconvex, film-coated tablets debossed with 'LL' on one side and 'C02' on the other side.
- Simvastatin tablets 20 mg are tan colored, oval shaped, biconvex, film-coated tablets debossed with 'LL' on one side and 'C03' on the other side.
- Simvastatin tablets 40 mg are brick red colored, round shaped, biconvex, film-coated tablets debossed with 'LL'on one side and 'C04' on the other side.
- Simvastatin tablets 80 mg are brick red colored, capsule shaped, biconvex, film-coated tablets debossed with 'LL'on one side and 'C05' on the other side.
-
4 CONTRAINDICATIONS
Simvastatin is contraindicated in the following conditions:
- Concomitant administration of strong CYP3A4 inhibitors (e.g., itraconazole, ketoconazole, posaconazole, voriconazole, HIV protease inhibitors, boceprevir, telaprevir, erythromycin, clarithromycin, telithromycin, nefazodone, and cobicistat-containing products) [see WARNINGS AND PRECAUTIONS (5.1)] .
- Concomitant administration of gemfibrozil, cyclosporine, or danazol [see WARNINGS AND PRECAUTIONS (5.1)] .
- Hypersensitivity to any component of this medication [see ADVERSE REACTIONS (6.2)] .
- Active liver disease, which may include unexplained persistent elevations in hepatic transaminase levels [see WARNINGS AND PRECAUTIONS (5.2)] .
- Women who are pregnant or may become pregnant. Serum cholesterol and triglycerides increase during normal pregnancy, and cholesterol or cholesterol derivatives are essential for fetal development. Because HMG-CoA reductase inhibitors (statins) decrease cholesterol synthesis and possibly the synthesis of other biologically active substances derived from cholesterol, simvastatin may cause fetal harm when administered to a pregnant woman. Atherosclerosis is a chronic process and the discontinuation of lipid-lowering drugs during pregnancy should have little impact on the outcome of long-term therapy of primary hypercholesterolemia. There are no adequate and well-controlled studies of use with simvastatin during pregnancy; however, in rare reports congenital anomalies were observed following intrauterine exposure to statins. In rat and rabbit animal reproduction studies, simvastatin revealed no evidence of teratogenicity. Simvastatin should be administered to women of childbearing age only when such patients are highly unlikely to conceive. If the patient becomes pregnant while taking this drug, simvastatin should be discontinued immediately, and the patient should be apprised of the potential hazard to the fetus [see USE IN SPECIFIC POPULATIONS (8.1)] .
- Nursing mothers. It is not known whether simvastatin is excreted into human milk; however, a small amount of another drug in this class does pass into breast milk. Because statins have the potential for serious adverse reactions in nursing infants, women who require treatment with simvastatin should not breastfeed their infants [see USE IN SPECIFIC POPULATIONS (8.3)] .
-
5 WARNINGS AND PRECAUTIONS
5.1 Myopathy/Rhabdomyolysis
Simvastatin occasionally causes myopathy manifested as muscle pain, tenderness or weakness with creatine kinase (CK) above ten times the upper limit of normal (ULN). Myopathy sometimes takes the form of rhabdomyolysis with or without acute renal failure secondary to myoglobinuria, and rare fatalities have occurred. The risk of myopathy is increased by elevated plasma levels of simvastatin and simvastatin acid. Predisposing factors for myopathy include advanced age (≥65 years), female gender, uncontrolled hypothyroidism, and renal impairment.
The risk of myopathy, including rhabdomyolysis, is dose related. In a clinical trial database in which 41,413 patients were treated with simvastatin, 24,747 (approximately 60%) of whom were enrolled in studies with a median follow-up of at least 4 years, the incidence of myopathy was approximately 0.03% and 0.08% at 20 and 40 mg/day, respectively. The incidence of myopathy with 80 mg (0.61%) was disproportionately higher than that observed at the lower doses. In these trials, patients were carefully monitored and some interacting medicinal products were excluded.
In a clinical trial in which 12,064 patients with a history of myocardial infarction were treated with simvastatin (mean follow-up 6.7 years), the incidence of myopathy (defined as unexplained muscle weakness or pain with a serum creatine kinase [CK] >10 times upper limit of normal [ULN]) in patients on 80 mg/day was approximately 0.9% compared with 0.02% for patients on 20 mg/day. The incidence of rhabdomyolysis (defined as myopathy with a CK >40 times ULN) in patients on 80 mg/day was approximately 0.4% compared with 0% for patients on 20 mg/day. The incidence of myopathy, including rhabdomyolysis, was highest during the first year and then notably decreased during the subsequent years of treatment. In this trial, patients were carefully monitored and some interacting medicinal products were excluded.
The risk of myopathy, including rhabdomyolysis, is greater in patients on simvastatin 80 mg compared with other statin therapies with similar or greater LDL-C-lowering efficacy and compared with lower doses of simvastatin. Therefore, the 80-mg dose of simvastatin should be used only in patients who have been taking simvastatin 80 mg chronically (e.g., for 12 months or more) without evidence of muscle toxicity [see DOSAGE AND ADMINISTRATION, Restricted Dosing for 80 mg (2.2)]. If, however, a patient who is currently tolerating the 80-mg dose of simvastatin needs to be initiated on an interacting drug that is contraindicated or is associated with a dose cap for simvastatin, that patient should be switched to an alternative statin with less potential for the drug-drug interaction. Patients should be advised of the increased risk of myopathy, including rhabdomyolysis, and to report promptly any unexplained muscle pain, tenderness or weakness. If symptoms occur, treatment should be discontinued immediately. [see WARNINGS AND PRECAUTIONS (5.2)].
There have been rare reports of immune-mediated necrotizing myopathy (IMNM), an autoimmune myopathy, associated with statin use. IMNM is characterized by: proximal muscle weakness and elevated serum creatine kinase, which persist despite discontinuation of statin treatment; muscle biopsy showing necrotizing myopathy without significant inflammation; improvement with immunosuppressive agents.
All patients starting therapy with simvastatin, or whose dose of simvastatin is being increased, should be advised of the risk of myopathy, including rhabdomyolysis, and told to report promptly any unexplained muscle pain, tenderness or weakness particularly if accompanied by malaise or fever or if muscle signs and symptoms persist after discontinuing simvastatin. Simvastatin therapy should be discontinued immediately if myopathy is diagnosed or suspected. In most cases, muscle symptoms and CK increases resolved when treatment was promptly discontinued. Periodic CK determinations may be considered in patients starting therapy with simvastatin or whose dose is being increased, but there is no assurance that such monitoring will prevent myopathy.
Many of the patients who have developed rhabdomyolysis on therapy with simvastatin have had complicated medical histories, including renal insufficiency usually as a consequence of long-standing diabetes mellitus. Such patients merit closer monitoring. Simvastatin therapy should be discontinued if markedly elevated CPK levels occur or myopathy is diagnosed or suspected. Simvastatin therapy should also be temporarily withheld in any patient experiencing an acute or serious condition predisposing to the development of renal failure secondary to rhabdomyolysis, e.g., sepsis; hypotension; major surgery; trauma; severe metabolic, endocrine, or electrolyte disorders; or uncontrolled epilepsy.
Drug Interactions
The risk of myopathy and rhabdomyolysis is increased by elevated plasma levels of simvastatin and simvastatin acid. Simvastatin is metabolized by the cytochrome P450 isoform 3A4. Certain drugs which inhibit this metabolic pathway can raise the plasma levels of simvastatin and may increase the risk of myopathy. These include itraconazole, ketoconazole, posaconazole, voriconazole, the macrolide antibiotics erythromycin and clarithromycin, and the ketolide antibiotic telithromycin, HIV protease inhibitors, boceprevir, telaprevir, the antidepressant nefazodone, cobicistat-containing products, or grapefruit juice [see CLINICAL PHARMACOLOGY (12.3)]. Combination of these drugs with simvastatin is contraindicated. If short-term treatment with strong CYP3A4 inhibitor is unavoidable, therapy with simvastatin must be suspended during the course of treatment [see CONTRAINDICATIONS (4) and DRUG INTERACTIONS (7.1)].
The combined use of simvastatin with gemfibrozil, cyclosporine, or danazol is contraindicated [see CONTRAINDICATIONS (4) and DRUG INTERACTIONS (7.1 and 7.2)].
Caution should be used when prescribing other fibrates with simvastatin, as these agents can cause myopathy when given alone and the risk is increased when they are coadministered [see DRUG INTERACTIONS (7.2)].
Cases of myopathy, including rhabdomysis, have been reported with simvastatin coadministered with colchicine, and caution should be exercised when prescribing simvastatin with colchicine [see DRUG INTERACTIONS (7.7)].
The benefits of the combined use of simvastatin with the following drugs should be carefully weighed against the potential risks of combinations: other lipid-lowering drugs (fibrates or, for patients with HoFH, lomitapide), amiodarone, dronedarone, verapamil, diltiazem, amlodipine, or ranolazine [see DOSAGE AND ADMINISTRATION (2.4), DRUG INTERACTIONS (7.3)].
Cases of myopathy, including rhabdomyolysis, have been observed with simvastatin coadministered with lipid-modifying doses (≥1 g/day niacin) of niacin-containing products. [see DRUG INTERACTION (7.4)].
Prescribing recommendations for interacting agents are summarized in TABLE 1 [see also DOSAGE AND ADMINISTRATION (2.3, 2.4), DRUG INTERACTIONS (7), CLINICAL PHARMACOLOGY (12.3)].
TABLE 1 Drug Interactions Associated with Increased Risk of Myopathy/Rhabdomyolysis - * For patients with HoFH who have been taking 80 mg simvastatin chronically (e.g., for 12 months or more) without evidence of muscle toxicity, do not exceed 40 mg simvastatin when taking lomitapide.
Interacting Agents
Prescribing Recommendations
Strong CYP3A4 Inhibitors, e.g.:
Contraindicated with simvastatin
Itraconazole
Ketoconazole
Posaconazole
Voriconazole
Erythromycin
Clarithromycin
Telithromycin
HIV protease inhibitors
Boceprevir
Telaprevir
Nefazodone
Cobicistat-containing products
Gemfibrozil
Cyclosporine
Danazol
Verapamil
Diltiazem
Dronedarone
Do not exceed 10 mg simvastatin daily
Amiodarone
Amlodipine
Ranolazine
Do not exceed 20 mg simvastatin daily
Lomitapide
For patients with HoFH, do not exceed 20 mg simvastatin daily*
Grapefruit juice
Avoid grapefruit juice
5.2 Liver Dysfunction
Persistent increases (to more than 3X the ULN) in serum transaminases have occurred in approximately 1% of patients who received simvastatin in clinical studies. When drug treatment was interrupted or discontinued in these patients, the transaminase levels usually fell slowly to pretreatment levels. The increases were not associated with jaundice or other clinical signs or symptoms. There was no evidence of hypersensitivity.
In the Scandinavian Simvastatin Survival Study (4S) [see CLINICAL STUDIES (14.1)], the number of patients with more than one transaminase elevation to >3X ULN, over the course of the study, was not significantly different between the simvastatin and placebo groups (14 [0.7%] vs. 12 [0.6%]). Elevated transaminases resulted in the discontinuation of 8 patients from therapy in the simvastatin group (n=2,221) and 5 in the placebo group (n=2,223). Of the 1,986 simvastatin treated patients in 4S with normal liver function tests (LFTs) at baseline, 8 (0.4%) developed consecutive LFT elevations to >3X ULN and/or were discontinued due to transaminase elevations during the 5.4 years (median follow-up) of the study. Among these 8 patients, 5 initially developed these abnormalities within the first year. All of the patients in this study received a starting dose of 20 mg of simvastatin; 37% were titrated to 40 mg.
In 2 controlled clinical studies in 1,105 patients, the 12-month incidence of persistent hepatic transaminase elevation without regard to drug relationship was 0.9% and 2.1% at the 40- and 80-mg dose, respectively. No patients developed persistent liver function abnormalities following the initial 6 months of treatment at a given dose.
It is recommended that liver function tests be performed before the initiation of treatment, and thereafter when clinically indicated. There have been rare postmarketing reports of fatal and non-fatal hepatic failure in patients taking statins, including simvastatin. If serious liver injury with clinical symptoms and/or hyperbilirubinemia or jaundice occurs during treatment with simvastatin, promptly interrupt therapy. If an alternate etiology is not found do not restart simvastatin. Note that ALT may emanate from muscle, therefore ALT rising with CK may indicate myopathy [see WARNINGS AND PRECAUTIONS (5.1)].
The drug should be used with caution in patients who consume substantial quantities of alcohol and/or have a past history of liver disease. Active liver diseases or unexplained transaminase elevations are contraindications to the use of simvastatin.
Moderate (less than 3X ULN) elevations of serum transaminases have been reported following therapy with simvastatin. These changes appeared soon after initiation of therapy with simvastatin, were often transient, were not accompanied by any symptoms and did not require interruption of treatment.
-
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
Because clinical studies are conducted under widely varying conditions, adverse reaction rates observed in the clinical studies of a drug cannot be directly compared to rates in the clinical studies of another drug and may not reflect the rates observed in practice.
In the pre-marketing controlled clinical studies and their open extensions (2,423 patients with median duration of follow-up of approximately 18 months), 1.4% of patients were discontinued due to adverse reactions. The most common adverse reactions that led to treatment discontinuation were: gastrointestinal disorders (0.5%), myalgia (0.1%), and arthralgia (0.1%). The most commonly reported adverse reactions (incidence ≥5%) in simvastatin controlled clinical trials were: upper respiratory infections (9.0%), headache (7.4%), abdominal pain (7.3%), constipation (6.6%), and nausea (5.4%).
Scandinavian Simvastatin Survival Study
In 4S involving 4,444 (age range 35 to 71 years, 19% women, 100% Caucasians) treated with 20 to 40 mg/day of simvastatin (n=2,221) or placebo (n=2,223) over a median of 5.4 years, adverse reactions reported in ≥2% of patients and at a rate greater than placebo are shown in TABLE 2.
TABLE 2 Adverse Reactions Reported Regardless of Causality by ≥ 2% of Patients Treated with Simvastatin and Greater than Placebo in 4S
Simvastatin
Placebo
(N = 2,221)
(N = 2,223)
%
%
Body as a Whole
Edema/swelling
2.7
2.3
Abdominal pain
5.9
5.8
Cardiovascular System Disorders
Atrial fibrillation
5.7
5.1
Digestive System Disorders
Constipation
2.2
1.6
Gastritis
4.9
3.9
Endocrine Disorders
Diabetes mellitus
4.2
3.6
Musculoskeletal Disorders
Myalgia
3.7
3.2
Nervous System/ Psychiatric Disorders
Headache
2.5
2.1
Insomnia
4.0
3.8
Vertigo
4.5
4.2
Respiratory System Disorders
Bronchitis
6.6
6.3
Sinusitis
2.3
1.8
Skin / Skin Appendage Disorders
Eczema
4.5
3.0
Urogenital System Disorders
Infection, urinary tract
3.2
3.1
Heart Protection Study
In the Heart Protection Study (HPS), involving 20,536 patients (age range 40 to 80 years, 25% women, 97% Caucasians, 3% other races) treated with simvastatin 40 mg/day (n=10,269) or placebo (n=10,267) over a mean of 5 years, only serious adverse reactions and discontinuations due to any adverse reactions were recorded. Discontinuation rates due to adverse reactions were 4.8% in patients treated with simvastatin compared with 5.1% in patients treated with placebo. The incidence of myopathy/rhabdomyolysis was <0.1% in patients treated with simvastatin.
Other Clinical Studies
In a clinical trial in which 12,064 patients with a history of myocardial infarction were treated with simvastatin (mean follow-up 6.7 years), the incidence of myopathy (defined as unexplained muscle weakness or pain with a serum creatine kinase [CK] >10 times upper limit of normal [ULN] in patients on 80 mg/day was approximately 0.9% compared with 0.02% for patients on 20 mg/day. The incidence of rhabdomyolysis (defined as myopathy with a CK >40 times ULN) in patients on 80 mg/day was approximately 0.4% compared with 0% for patients on 20 mg/day. The incidence of myopathy, including rhabdomyolysis, was highest during the first year and then notably decreased during the subsequent years of treatment. In this trial, patients were carefully monitored and some interacting medicinal products were excluded.
Other adverse reactions reported in clinical trials were: diarrhea, rash, dyspepsia, flatulence, and asthenia.
Laboratory Tests
Marked persistent increases of hepatic transaminases have been noted [see WARNINGS AND PRECAUTIONS (5.2)]. Elevated alkaline phosphatase and γ-glutamyl transpeptidase have also been reported. About 5% of patients had elevations of CK levels of 3 or more times the normal value on one or more occasions. This was attributable to the noncardiac fraction of CK [see WARNINGS AND PRECAUTIONS (5.1)].
Adolescent Patients (ages 10 to 17 years)
In a 48-week, controlled study in adolescent boys and girls who were at least 1 year post-menarche, 10 to 17 years of age (43.4% female, 97.7% Caucasians, 1.7% Hispanics, 0.6% Multiracial) with heterozygous familial hypercholesterolemia (n=175), treated with placebo or simvastatin (10 to 40 mg daily), the most common adverse reactions observed in both groups were upper respiratory infection, headache, abdominal pain, and nausea [see USE IN SPECIFIC POPULATIONS (8.4) and CLINICAL STUDIES (14.2)].
6.2 Postmarketing Experience
Because the below reactions are reported voluntarily from a population of uncertain size, it is generally not possible to reliably estimate their frequency or establish a causal relationship to drug exposure. The following additional adverse reactions have been identified during postapproval use of simvastatin: pruritus, alopecia, a variety of skin changes (e.g., nodules, discoloration, dryness of skin/mucous membranes, changes to hair/nails), dizziness, muscle cramps, myalgia, pancreatitis, paresthesia, peripheral neuropathy, vomiting, anemia, erectile dysfunction, interstitial lung disease, rhabdomyolysis, hepatitis/jaundice, fatal and non-fatal hepatic failure and depression.
There have been rare reports of immune-mediated necrotizing myopathy associated with statin use [see WARNINGS AND PRECAUTIONS (5.1)].
An apparent hypersensitivity syndrome has been reported rarely which has included some of the following features: anaphylaxis, angioedema, lupus erythematous-like syndrome, polymyalgia rheumatica, dermatomyositis, vasculitis, purpura, thrombocytopenia, leukopenia, hemolytic anemia, positive ANA, ESR increase, eosinophilia, arthritis, arthralgia, urticaria, asthenia, photosensitivity, fever, chills, flushing, malaise, dyspnea, toxic epidermal necrolysis, erythema multiforme, including Stevens-Johnson syndrome.
There have been rare postmarketing reports of cognitive impairment (e.g., memory loss, forgetfulness, amnesia, memory impairment, confusion) associated with statin use. These cognitive issues have been reported for all statins. The reports are generally nonserious, and reversible upon statin discontinuation, with variable times to symptoms onset (1day to years) and symptoms resolution (median of 3 weeks).
-
7 DRUG INTERACTIONS
7.1 Strong CYP3A4 Inhibitors, Cyclosporine, or Danazol
Strong CYP3A4 inhibitors
Simvastatin, like several other inhibitors of HMG-CoA reductase, is a substrate of CYP3A4. Simvastatin is metabolized by CYP3A4 but has no CYP3A4 inhibitory activity; therefore it is not expected to affect the plasma concentrations of other drugs metabolized by CYP3A4.
Elevated plasma levels of HMG-CoA reductase inhibitory activity increases the risk of myopathy and rhabdomyolysis, particularly with higher doses of simvastatin [see WARNINGS AND PRECAUTIONS (5.1) and CLINICAL PHARMACOLOGY (12.3)]. Concomitant use of drugs labeled as having a strong inhibitory effect on CYP3A4 is contraindicated [see CONTRAINDICATIONS (4)]. If treatment with itraconazole, ketoconazole, posaconazole, voriconazole, erythromycin, clarithromycin or telithromycin is unavoidable, therapy with simvastatin must be suspended during the course of treatment.
Cyclosporine or Danazol
The risk of myopathy, including rhabdomyolysis is increased by concomitant administration of cyclosporine or danazol. Therefore, concomitant use of these drugs is contraindicated [see CONTRAINDICATIONS (4), WARNINGS AND PRECAUTIONS (5.1) and CLINICAL PHARMACOLOGY (12.3)].
7.2 Lipid-Lowering Drugs That Can Cause Myopathy When Given Alone
Gemfibrozil
Contraindicated with simvastatin [see CONTRAINDICATIONS (4) and WARNINGS AND PRECAUTIONS (5.1)].
Other fibrates
Caution should be used when prescribing with simvastatin [see WARNINGS AND PRECAUTIONS (5.1)].
7.3 Amiodarone, Dronedarone, Ranolazine, or Calcium Channel Blockers
The risk of myopathy, including rhabdomyolysis, is increased by concomitant administration of amiodarone, dronedarone, ranolazine, or calcium channel blockers such as verapamil, diltiazem, or amlodipine [see DOSAGE AND ADMINISTRATION (2.3) and WARNINGS AND PRECAUTIONS (5.1), and TABLE 3 in CLINICAL PHARMACOLOGY (12.3)].
7.4 Niacin
Cases of myopathy/rhabdomyolysis have been observed with simvastatin coadministered with lipid-modifying doses (≥1 g/day niacin) of niacin-containing products. The risk of myopathy is greater in Chinese patients. In a clinical trial (median follow-up 3.9 years) involving patients at high risk of cardiovascular disease and with well-controlled LDL-C levels on simvastatin 40 mg/day with or without ezetimibe 10 mg/day, there was no incremental benefit on cardiovascular outcomes with the addition of lipid-modifying doses (≥1 g/day) of niacin. Coadministration of simvastatin with lipid- modifying doses (≥1 g/day) of niacin is not recommended in Chinese patients. It is unknown if this risk applies to other Asian patients [see WARNINGS AND PRECAUTIONS (5.1) and USE IN SPECIFIC POPULATIONS (8.8)].
7.5 Digoxin
In one study, concomitant administration of digoxin with simvastatin resulted in a slight elevation in digoxin concentrations in plasma. Patients taking digoxin should be monitored appropriately when simvastatin is initiated [see CLINICAL PHARMACOLOGY (12.3)].
7.6 Coumarin Anticoagulants
In two clinical studies, one in normal volunteers and the other in hypercholesterolemic patients, simvastatin 20 to 40 mg/day modestly potentiated the effect of coumarin anticoagulants: the prothrombin time, reported as International Normalized Ratio (INR), increased from a baseline of 1.7 to 1.8 and from 2.6 to 3.4 in the volunteer and patient studies, respectively. With other statins, clinically evident bleeding and/or increased prothrombin time has been reported in a few patients taking coumarin anticoagulants concomitantly. In such patients, prothrombin time should be determined before starting simvastatin and frequently enough during early therapy to ensure that no significant alteration of prothrombin time occurs. Once a stable prothrombin time has been documented, prothrombin times can be monitored at the intervals usually recommended for patients on coumarin anticoagulants. If the dose of simvastatin is changed or discontinued, the same procedure should be repeated. Simvastatin therapy has not been associated with bleeding or with changes in prothrombin time in patients not taking anticoagulants.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Category X [See CONTRAINDICATIONS (4)].
Simvastatin is contraindicated in women who are or may become pregnant. Lipid lowering drugs offer no benefit during pregnancy, because cholesterol and cholesterol derivatives are needed for normal fetal development. Atherosclerosis is a chronic process, and discontinuation of lipid-lowering drugs during pregnancy should have little impact on long-term outcomes of primary hypercholesterolemia therapy. There are no adequate and well-controlled studies of use with simvastatin during pregnancy; however, there are rare reports of congenital anomalies in infants exposed to statins in utero. Animal reproduction studies of simvastatin in rats and rabbits showed no evidence of teratogenicity. Serum cholesterol and triglycerides increase during normal pregnancy, and cholesterol or cholesterol derivatives are essential for fetal development. Because statins decrease cholesterol synthesis and possibly the synthesis of other biologically active substances derived from cholesterol, simvastatin may cause fetal harm when administered to a pregnant woman. If simvastatin is used during pregnancy or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to the fetus.
There are rare reports of congenital anomalies following intrauterine exposure to statins. In a review2 of approximately 100 prospectively followed pregnancies in women exposed to simvastatin or another structurally related statin, the incidences of congenital anomalies, spontaneous abortions, and fetal deaths/stillbirths did not exceed those expected in the general population. However, the study was only able to exclude a 3- to 4-fold increased risk of congenital anomalies over the background rate. In 89% of these cases, drug treatment was initiated prior to pregnancy and was discontinued during the first trimester when pregnancy was identified.
Simvastatin was not teratogenic in rats or rabbits at doses (25, 10 mg/kg/day, respectively) that resulted in 3 times the human exposure based on mg/m2 surface area. However, in studies with another structurally-related statin, skeletal malformations were observed in rats and mice.
Women of childbearing potential, who require treatment with simvastatin for a lipid disorder, should be advised to use effective contraception. For women trying to conceive, discontinuation of simvastatin should be considered. If pregnancy occurs, simvastatin should be immediately discontinued.
8.3 Nursing Mothers
It is not known whether simvastatin is excreted in human milk. Because a small amount of another drug in this class is excreted in human milk and because of the potential for serious adverse reactions in nursing infants, women taking simvastatin should not nurse their infants. A decision should be made whether to discontinue nursing or discontinue drug, taking into account the importance of the drug to the mother [see CONTRAINDICATIONS (4)].
8.4 Pediatric Use
Safety and effectiveness of simvastatin in patients 10 to 17 years of age with heterozygous familial hypercholesterolemia have been evaluated in a controlled clinical trial in adolescent boys and in girls who were at least 1 year post-menarche. Patients treated with simvastatin had an adverse reaction profile similar to that of patients treated with placebo. Doses greater than 40 mg have not been studied in this population. In this limited controlled study, there was no significant effect on growth or sexual maturation in the adolescent boys or girls, or on menstrual cycle length in girls [see DOSAGE AND ADMINISTRATION (2.5), ADVERSE REACTIONS (6.1), CLINICAL STUDIES (14.2)]. Adolescent females should be counseled on appropriate contraceptive methods while on simvastatin therapy [see CONTRAINDICATIONS (4) and USE IN SPECIFIC POPULATIONS (8.1)]. Simvastatin has not been studied in patients younger than 10 years of age, nor in premenarchal girls.
8.5 Geriatric Use
Of the 2,423 patients who received simvastatin in Phase III clinical studies and the 10,269 patients in the Heart Protection Study who received simvastatin, 363 (15%) and 5,366 (52%), respectively were ≥65 years old. In HPS, 615 (6%) were ≥75 years old. No overall differences in safety or effectiveness were observed between these subjects and younger subjects, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out. Since advanced age (≥65 years) is a predisposing factor for myopathy, simvastatin should be prescribed with caution in the elderly [see CLINICAL PHARMACOLOGY, (12.3)].
A pharmacokinetic study with simvastatin showed the mean plasma level of statin activity to be approximately 45% higher in elderly patients between 70 to 78 years of age compared with patients between 18 to 30 years of age. In 4S, 1,021 (23%) of 4,444 patients were 65 or older. Lipid-lowering efficacy was at least as great in elderly patients compared with younger patients, and simvastatin significantly reduced total mortality and CHD mortality in elderly patients with a history of CHD. In HPS, 52% of patients were elderly (4,891 patients 65 to 69 years and 5,806 patients 70 years or older). The relative risk reductions of CHD death, non-fatal MI, coronary and non-coronary revascularization procedures, and stroke were similar in older and younger patients [see CLINICAL STUDIES (14.1)]. In HPS, among 32,145 patients entering the active run-in period, there were 2 cases of myopathy/rhabdomyolysis; these patients were aged 67 and 73. Of the 7 cases of myopathy/rhabdomyolysis among 10,269 patients allocated to simvastatin, 4 were aged 65 or more (at baseline), of whom one was over 75. There were no overall differences in safety between older and younger patients in either 4S or HPS.
Because advanced age (≥65 years) is a predisposing factor for myopathy, including rhabdomyolysis, simvastatin should be prescribed with caution in the elderly. In a clinical trial of patients treated with simvastatin 80 mg/day, patients ≥65 years of age had an increased risk of myopathy, including rhabdomyolysis, compared to patients <65 years of age [see WARNING AND PRECAUTIONS (5.1) and CLINICAL PHARMACOLOGY (12.3)].
8.6 Renal Impairment
Caution should be exercised when simvastatin is administered to patients with severe renal impairment [see DOSAGE AND ADMINISTRATION (2.6)].
8.7 Hepatic Impairment
Simvastatin is contraindicated in patients with active liver disease which may include unexplained persistent elevations in hepatic transaminase levels [see CONTRAINDICATIONS (4) and WARNINGS AND PRECAUTIONS (5.2)].
8.8 Chinese Patients
In a clinical trial in which patients at high risk of cardiovascular disease were treated with simvastatin 40 mg/day (median follow-up 3.9 years), the incidence of myopathy was approximately 0.05% for non- Chinese patients (n=7367) compared with 0.24% for Chinese patients (n=5468). The incidence of myopathy for Chinese patients on simvastatin 40 mg/day or ezetimibe/simvastatin 10/40 mg/day coadministered with extended-release niacin 2 g/day was 1.24%.
Chinese patients may be at higher risk for myopathy, monitor patients appropriately. Coadministration of simvastatin with lipid-modifying doses (≥1 g/day niacin) of niacin-containing products is not recommended in Chinese patients [see WARNINGS AND PRECAUTIONS (5.1), DRUG INTERACTIONS (7.4)].
-
10 OVERDOSAGE
Significant lethality was observed in mice after a single oral dose of 9 g/m2. No evidence of lethality was observed in rats or dogs treated with doses of 30 and 100 g/m2, respectively. No specific diagnostic signs were observed in rodents. At these doses the only signs seen in dogs were emesis and mucoid stools.
A few cases of overdosage with simvastatin have been reported; the maximum dose taken was 3.6 g. All patients recovered without sequelae. Supportive measures should be taken in the event of an overdose. The dialyzability of simvastatin and its metabolites in man is not known at present.
-
11 DESCRIPTION
Simvastatin is a lipid-lowering agent that is derived synthetically from a fermentation product of Aspergillus terreus. After oral ingestion, simvastatin, which is an inactive lactone, is hydrolyzed to the corresponding β-hydroxyacid form. This is an inhibitor of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase. This enzyme catalyzes the conversion of HMG-CoA to mevalonate, which is an early and rate-limiting step in the biosynthesis of cholesterol.
Simvastatin is butanoic acid, 2,2-dimethyl-,1,2,3,7,8,8a-hexahydro-3,7-dimethyl-8-[2-(tetrahydro-4-hydroxy-6-oxo-2H-pyran-2-yl)-ethyl]-1-naphthalenyl ester, [1S-[1α,3α,7β,8β(2S*,4S*),-8aβ]]. The empirical formula of simvastatin is C25H38O5 and its molecular weight is 418.57. Its structural formula is:
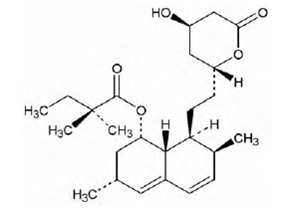
Simvastatin is a white to off-white, nonhygroscopic, crystalline powder that is practically insoluble in water, and freely soluble in chloroform, methanol and ethanol.
Simvastatin tablets USP for oral administration contain either 5 mg, 10 mg, 20 mg, 40 mg or 80 mg of simvastatin and the following inactive ingredients: ascorbic acid, citric acid, hydroxy propyl cellulose, hypromellose, iron oxides, lactose monohydrate, magnesium stearate, microcrystalline cellulose, pregelatinised starch, talc and titanium dioxide. Butylated hydroxyanisole is added as a preservative.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Simvastatin is a prodrug and is hydrolyzed to its active β-hydroxyacid form, simvastatin acid, after administration. Simvastatin is a specific inhibitor of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase, the enzyme that catalyzes the conversion of HMG-CoA to mevalonate, an early and rate limiting step in the biosynthetic pathway for cholesterol. In addition, simvastatin reduces VLDL and TG and increases HDL-C.
12.2 Pharmacodynamics
Epidemiological studies have demonstrated that elevated levels of total-C, LDL-C, as well as decreased levels of HDL-C are associated with the development of atherosclerosis and increased cardiovascular risk. Lowering LDL-C decreases this risk. However, the independent effect of raising HDL-C or lowering TG on the risk of coronary and cardiovascular morbidity and mortality has not been determined.
12.3 Pharmacokinetics
Simvastatin is a lactone that is readily hydrolyzed in vivo to the corresponding β-hydroxyacid, a potent inhibitor of HMG-CoA reductase. Inhibition of HMG-CoA reductase is the basis for an assay in pharmacokinetic studies of the β-hydroxyacid metabolites (active inhibitors) and, following base hydrolysis, active plus latent inhibitors (total inhibitors) in plasma following administration of simvastatin.
Following an oral dose of 14C-labeled simvastatin in man, 13% of the dose was excreted in urine and 60% in feces. Plasma concentrations of total radioactivity (simvastatin plus 14C-metabolites) peaked at 4 hours and declined rapidly to about 10% of peak by 12 hours postdose. Since simvastatin undergoes extensive first-pass extraction in the liver, the availability of the drug to the general circulation is low (<5%).
Both simvastatin and its β-hydroxyacid metabolite are highly bound (approximately 95%) to human plasma proteins. Rat studies indicate that when radiolabeled simvastatin was administered, simvastatin derived radioactivity crossed the blood-brain barrier.
The major active metabolites of simvastatin present in human plasma are the β-hydroxyacid of simvastatin and its 6′-hydroxy, 6′-hydroxymethyl, and 6′-exomethylene derivatives. Peak plasma concentrations of both active and total inhibitors were attained within 1.3 to 2.4 hours postdose. While the recommended therapeutic dose range is 5 to 40 mg/day, there was no substantial deviation from linearity of AUC of inhibitors in the general circulation with an increase in dose to as high as 120 mg. Relative to the fasting state, the plasma profile of inhibitors was not affected when simvastatin was administered immediately before an American Heart Association recommended low-fat meal.
In a study including 16 elderly patients between 70 and 78 years of age who received simvastatin 40 mg/day, the mean plasma level of HMG-CoA reductase inhibitory activity was increased approximately 45% compared with 18 patients between 18 to 30 years of age. Clinical study experience in the elderly (n=1522), suggests that there were no overall differences in safety between elderly and younger patients [see USE IN SPECIFIC POPULATIONS (8.5)].
Kinetic studies with another statin, having a similar principal route of elimination, have suggested that for a given dose level higher systemic exposure may be achieved in patients with severe renal insufficiency (as measured by creatinine clearance).
Simvastatin acid is a substrate of the transport protein OATP1B1. Concomitant administration of medicinal products that are inhibitors of the transport protein OATP1B1 may lead to increased plasma concentrations of simvastatin acid and an increased risk of myopathy. For example, cyclosporine has been shown to increase the AUC of statins; although the mechanism is not fully understood, the increase in AUC for simvastatin acid is presumably due, in part, to inhibition of CYP3A4 and/or OATP1B1.
The risk of myopathy is increased by high levels of HMG-CoA reductase inhibitory activity in plasma. Inhibitors of CYP3A4 can raise the plasma levels of HMG-CoA reductase inhibitory activity and increase the risk of myopathy [see WARNINGS AND PRECAUTIONS (5.1) and DRUG INTERACTIONS (7.1)].
TABLE 3 Effect of Coadministered Drugs or Grapefruit Juice on Simvastatin Systemic Exposure - * Results based on a chemical assay except results with propranolol as indicated.
- † Results could be representative of the following CYP3A4 inhibitors: ketoconazole, erythromycin, clarithromycin, HIV protease inhibitors, and nefazodone.
- ‡ Simvastatin acid refers to the β-hydroxyacid of simvastatin.
- § The effect of amounts of grapefruit juice between those used in these two studies on simvastatin pharmacokinetics has not been studied.
- ¶ Double-strength: one can of frozen concentrate diluted with one can of water. Grapefruit juice was administered TID for 2 days, and 200 mL together with single dose simvastatin and 30 and 90 minutes following single dose simvastatin on Day 3.
- # Single-strength: one can of frozen concentrate diluted with 3 cans of water. Grapefruit juice was administered with breakfast for 3 days, and simvastatin was administered in the evening on Day 3.
- Þ Because Chinese patients have an increased risk for myopathy with simvastatin coadministered with lipid-modifying doses (≥ 1 g/day niacin) of niacin-containing products, and the risk is dose-related, Chinese patients should not receive simvastatin 80 mg coadministered with lipid-modifying doses of niacin-containing products [see WARNINGS AND PRECAUTIONS (5.1) and DRUG INTERACTIONS (7.4)].
Coadministered Drug or
Grapefruit Juice
Dosing of Coadministered
Drug or Grapefruit Juice
Dosing of Simvastatin
Geometric Mean Ratio
(Ratio* with / without
coadministered drug)
No Effect = 1.00
AUC
Cmax
Contraindicated with simvastatin [see CONTRAINDICATIONS (4) and WARNINGS AND PRECAUTIONS (5.1)] Telithromycin†
200 mg QD for 4 days
80 mg
simvastatin acid‡
12
15
simvastatin
8.9
5.3
Nelfinavir†
1250 mg BID for 14 days
20 mg QD for 28 days
simvastatin acid‡
simvastatin
6
6.2
Itraconazole†
200 mg QD for 4 days
80 mg
simvastatin acid‡
13.1
simvastatin
13.1
Posaconazole
100 mg (oral suspension) QD for 13 days
40 mg
simvastatin acid
simvastatin
7.3
10.3
9.2
9.4
200 mg (oral suspension) QD for 13 days
40 mg
simvastatin acid
simvastatin
8.5
10.6
9.5
11.4
Gemfibrozil
600 mg BID for 3 days
40 mg
simvastatin acid
simvastatin
2.85
1.35
2.18
0.91
Avoid grapefruit juice with simvastatin [see WARNINGS AND PRECAUTIONS (5.1)] Grapefruit Juice§
(high dose)
200 mL of double-strength TID¶
60 mg single dose
simvastatin acid
simvastatin
7
16
Grapefruit Juice§ (low dose)
8 oz (about 237mL) of single-strength#
20 mg single dose
simvastatin acid
simvastatin
1.3
1.9
Avoid taking with >10 mg simvastatin, based on clinical and/or postmarketing experience [see WARNINGS AND PRECAUTIONS (5.1)] Verapamil SR
240 mg QD Days 1 to 7 then 240 mg BID on Days 8 to 10
80 mg on Day 10
simvastatin acid
simvastatin
2.3
2.5
2.4
2.1
Diltiazem
120 mg BID for 10 days
80 mg on Day 10
simvastatin acid
simvastatin
2.69
3.10
2.69
2.88
Diltiazem
120 mg BID for 14 days
20 mg on Day 14
simvastatin
4.6
3.6
Dronedarone
400 mg BID for 14 days
40 mg QD for 14 days
simvastatin acid
simvastatin
1.96
3.90
2.14
3.75
Avoid taking with >20 mg simvastatin, based on clinical and/or postmarketing experience [see WARNINGS AND PRECAUTIONS (5.1)] Amiodarone
400 mg QD for 3 days
40 mg on Day 3
simvastatin acid
simvastatin
1.75
1.76
1.72
1.79
Amlodipine
10 mg QD x 10 days
80 mg on Day 10
simvastatin acid
simvastatin
1.58
1.77
1.56
1.47
Ranolazine SR
1000 mg BID for 7 days
80 mg on Day 1 and Days 6 to 9
simvastatin acid
simvastatin
2.26
1.86
2.28
1.75
Avoid taking with >20 mg simvastatin (or 40 mg for patients who have previously taken 80 mg simvastatin chronically,
e.g., for 12 months or more, without evidence of muscle toxicity), based on clinical experience
Lomitapide
60 mg QD for 7 days
40 mg single dose
simvastatin acid
simvastatin
1.7
2
1.6
2
Lomitapide
10 mg QD for 7 days
20 mg single dose
simvastatin acid
simvastatin
1.4
1.6
1.4
1.7
No dosing adjustments required for the following:
Fenofibrate
160 mg QD x 14 days
80 mg QD on Days 8 to 14
simvastatin acid
simvastatin
0.64
0.89
0.89
0.83
Niacin
extended-releaseÞ2 g single dose
20 mg single dose
simvastatin acid
simvastatin
1.6
1.4
1.84
1.08
Propranolol
80 mg single dose
80 mg single dose
total inhibitor
0.79
↓ from 33.6 to 21.1 ng·eq/mL
active inhibitor
0.79
↓ from 7.0 to 4.7 ng·eq/mL
In a study of 12 healthy volunteers, simvastatin at the 80-mg dose had no effect on the metabolism of the probe cytochrome P450 isoform 3A4 (CYP3A4) substrates midazolam and erythromycin. This indicates that simvastatin is not an inhibitor of CYP3A4, and, therefore, is not expected to affect the plasma levels of other drugs metabolized by CYP3A4.
Coadministration of simvastatin (40 mg QD for 10 days) resulted in an increase in the maximum mean levels of cardioactive digoxin (given as a single 0.4 mg dose on day 10) by approximately 0.3 ng/mL.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
In a 72-week carcinogenicity study, mice were administered daily doses of simvastatin of 25, 100, and 400 mg/kg body weight, which resulted in mean plasma drug levels approximately 1, 4, and 8 times higher than the mean human plasma drug level, respectively (as total inhibitory activity based on AUC) after an 80-mg oral dose. Liver carcinomas were significantly increased in high-dose females and mid- and high-dose males with a maximum incidence of 90% in males. The incidence of adenomas of the liver was significantly increased in mid- and high-dose females. Drug treatment also significantly increased the incidence of lung adenomas in mid- and high-dose males and females. Adenomas of the Harderian gland (a gland of the eye of rodents) were significantly higher in high-dose mice than in controls. No evidence of a tumorigenic effect was observed at 25 mg/kg/day.
In a separate 92-week carcinogenicity study in mice at doses up to 25 mg/kg/day, no evidence of a tumorigenic effect was observed (mean plasma drug levels were 1 times higher than humans given 80 mg simvastatin as measured by AUC).
In a two-year study in rats at 25 mg/kg/day, there was a statistically significant increase in the incidence of thyroid follicular adenomas in female rats exposed to approximately 11 times higher levels of simvastatin than in humans given 80 mg simvastatin (as measured by AUC).
A second two-year rat carcinogenicity study with doses of 50 and 100 mg/kg/day produced hepatocellular adenomas and carcinomas (in female rats at both doses and in males at 100 mg/kg/day). Thyroid follicular cell adenomas were increased in males and females at both doses; thyroid follicular cell carcinomas were increased in females at 100 mg/kg/day. The increased incidence of thyroid neoplasms appears to be consistent with findings from other statins. These treatment levels represented plasma drug levels (AUC) of approximately 7 and 15 times (males) and 22 and 25 times (females) the mean human plasma drug exposure after an 80 milligram daily dose.
No evidence of mutagenicity was observed in a microbial mutagenicity (Ames) test with or without rat or mouse liver metabolic activation. In addition, no evidence of damage to genetic material was noted in an in vitro alkaline elution assay using rat hepatocytes, a V-79 mammalian cell forward mutation study, an in vitro chromosome aberration study in CHO cells, or an in vivo chromosomal aberration assay in mouse bone marrow.
There was decreased fertility in male rats treated with simvastatin for 34 weeks at 25 mg/kg body weight (4 times the maximum human exposure level, based on AUC, in patients receiving 80 mg/day); however, this effect was not observed during a subsequent fertility study in which simvastatin was administered at this same dose level to male rats for 11 weeks (the entire cycle of spermatogenesis including epididymal maturation). No microscopic changes were observed in the testes of rats from either study. At 180 mg/kg/day, (which produces exposure levels 22 times higher than those in humans taking 80 mg/day based on surface area, mg/m2), seminiferous tubule degeneration (necrosis and loss of spermatogenic epithelium) was observed. In dogs, there was drug-related testicular atrophy, decreased spermatogenesis, spermatocytic degeneration and giant cell formation at 10 mg/kg/day, (approximately 2 times the human exposure, based on AUC, at 80 mg/day). The clinical significance of these findings is unclear.
13.2 ANIMAL PHARMACOLOGY AND OR TOXICOLOGY
CNS Toxicity
Optic nerve degeneration was seen in clinically normal dogs treated with simvastatin for 14 weeks at 180 mg/kg/day, a dose that produced mean plasma drug levels about 12 times higher than the mean plasma drug level in humans taking 80 mg/day.
A chemically similar drug in this class also produced optic nerve degeneration (Wallerian degeneration of retinogeniculate fibers) in clinically normal dogs in a dose-dependent fashion starting at 60 mg/kg/day, a dose that produced mean plasma drug levels about 30 times higher than the mean plasma drug level in humans taking the highest recommended dose (as measured by total enzyme inhibitory activity). This same drug also produced vestibulocochlear Wallerian-like degeneration and retinal ganglion cell chromatolysis in dogs treated for 14 weeks at 180 mg/kg/day, a dose that resulted in a mean plasma drug level similar to that seen with the 60 mg/kg/day dose.
CNS vascular lesions, characterized by perivascular hemorrhage and edema, mononuclear cell infiltration of perivascular spaces, perivascular fibrin deposits and necrosis of small vessels were seen in dogs treated with simvastatin at a dose of 360 mg/kg/day, a dose that produced mean plasma drug levels that were about 14 times higher than the mean plasma drug levels in humans taking 80 mg/day. Similar CNS vascular lesions have been observed with several other drugs of this class.
There were cataracts in female rats after two years of treatment with 50 and 100 mg/kg/day (22 and 25 times the human AUC at 80 mg/day, respectively) and in dogs after three months at 90 mg/kg/day (19 times) and at two years at 50 mg/kg/day (5 times).
-
14 CLINICAL STUDIES
14.1 Clinical Studies in Adults
Reductions in Risk of CHD Mortality and Cardiovascular Events
In 4S, the effect of therapy with simvastatin on total mortality was assessed in 4,444 patients with CHD and baseline total cholesterol 212 to 309 mg/dL (5.5 to 8.0 mmol/L). In this multicenter, randomized, double-blind, placebo-controlled study, patients were treated with standard care, including diet, and either simvastatin 20 to 40 mg/day (n=2,221) or placebo (n=2,223) for a median duration of 5.4 years. Over the course of the study, treatment with simvastatin led to mean reductions in total-C, LDL-C and TG of 25%, 35%, and 10%, respectively, and a mean increase in HDL-C of 8%. Simvastatin significantly reduced the risk of mortality by 30% (p=0.0003, 182 deaths in the simvastatin group vs 256 deaths in the placebo group). The risk of CHD mortality was significantly reduced by 42% (p=0.00001, 111 vs 189 deaths). There was no statistically significant difference between groups in non-cardiovascular mortality. Simvastatin significantly decreased the risk of having major coronary events (CHD mortality plus hospital-verified and silent nonfatal myocardial infarction [MI]) by 34% (p<0.00001, 431 vs 622 patients with one or more events). The risk of having a hospital-verified non-fatal MI was reduced by 37%. Simvastatin significantly reduced the risk for undergoing myocardial revascularization procedures (coronary artery bypass grafting or percutaneous transluminal coronary angioplasty) by 37% (p<0.00001, 252 vs 383 patients). Simvastatin significantly reduced the risk of fatal plus non-fatal cerebrovascular events (combined stroke and transient ischemic attacks) by 28% (p=0.033, 75 vs 102 patients). Simvastatin reduced the risk of major coronary events to a similar extent across the range of baseline total and LDL cholesterol levels. Because there were only 53 female deaths, the effect of simvastatin on mortality in women could not be adequately assessed. However, simvastatin significantly lessened the risk of having major coronary events by 34% (60 vs 91 women with one or more event). The randomization was stratified by angina alone (21% of each treatment group) or a previous MI. Because there were only 57 deaths among the patients with angina alone at baseline, the effect of simvastatin on mortality in this subgroup could not be adequately assessed. However, trends in reduced coronary mortality, major coronary events and revascularization procedures were consistent between this group and the total study cohort. Additionally, simvastatin resulted in similar decreases in relative risk for total mortality, CHD mortality, and major coronary events in elderly patients (≥65 years), compared with younger patients.
The Heart Protection Study (HPS) was a large, multi-center, placebo-controlled, double-blind study with a mean duration of 5 years conducted in 20,536 patients (10,269 on simvastatin 40 mg and 10,267 on placebo). Patients were allocated to treatment using a covariate adaptive method3 which took into account the distribution of 10 important baseline characteristics of patients already enrolled and minimized the imbalance of those characteristics across the groups. Patients had a mean age of 64 years (range 40 to 80 years), were 97% Caucasian and were at high risk of developing a major coronary event because of existing CHD (65%), diabetes (Type 2, 26%; Type 1, 3%), history of stroke or other cerebrovascular disease (16%), peripheral vessel disease (33%), or hypertension in males ≥65 years (6%). At baseline, 3,421 patients (17%) had LDL-C levels below 100 mg/dL, of whom 953 (5%) had LDL-C levels below 80 mg/dL; 7,068 patients (34%) had levels between 100 and 130 mg/dL; and 10,047 patients (49%) had levels greater than 130 mg/dL.
The HPS results showed that simvastatin 40 mg/day significantly reduced: total and CHD mortality; nonfatal MI, stroke, and revascularization procedures (coronary and non-coronary) (see TABLE 4).
TABLE 4 Summary of Heart Protection Study Results - * n = number of patients with indicated event
Endpoint
Simvastatin
Placebo
Risk
p-Value
(N=10,269)
(N=10,267)
Reduction (%)
n (%)*
n (%)*
(95% CI)
Primary
Mortality
1,328 (12.9)
1,507 (14.7)
13 (6 to 19)
p=0.0003
CHD mortality
587 (5.7)
707 (6.9)
18 (8 to 26)
p=0.0005
Secondary
Non-fatal MI
357 (3.5)
574 (5.6)
38 (30 to 46)
p<0.0001
Stroke
444 (4.3)
585 (5.7)
25 (15 to 34)
p<0.0001
Tertiary
Coronary revascularization
513 (5)
725 (7.1)
30 (22 to 38)
p<0.0001
Peripheral and other non-
coronary revascularization
450 (4.4)
532 (5.2)
16 (5 to 26)
p=0.006
Two composite endpoints were defined in order to have sufficient events to assess relative risk reductions across a range of baseline characteristics (see Figure 1). A composite of major coronary events (MCE) was comprised of CHD mortality and non-fatal MI (analyzed by time-to-first event; 898 patients treated with simvastatin had events and 1,212 patients on placebo had events). A composite of major vascular events (MVE) was comprised of MCE, stroke and revascularization procedures including coronary, peripheral and other non-coronary procedures (analyzed by time-to-first event; 2,033 patients treated with simvastatin had events and 2,585 patients on placebo had events). Significant relative risk reductions were observed for both composite endpoints (27% for MCE and 24% for MVE, p<0.0001). Treatment with simvastatin produced significant relative risk reductions for all components of the composite endpoints. The risk reductions produced by simvastatin in both MCE and MVE were evident and consistent regardless of cardiovascular disease related medical history at study entry (i.e., CHD alone; or peripheral vascular disease, cerebrovascular disease, diabetes or treated hypertension, with or without CHD), gender, age, creatinine levels up to the entry limit of 2.3 mg/dL, baseline levels of LDL-C, HDL-C, apolipoprotein B and A-1, baseline concomitant cardiovascular medications (i.e., aspirin, beta blockers, or calcium channel blockers), smoking status, alcohol intake, or obesity. Diabetics showed risk reductions for MCE and MVE due to simvastatin treatment regardless of baseline HbA1c levels or obesity with the greatest effects seen for diabetics without CHD.
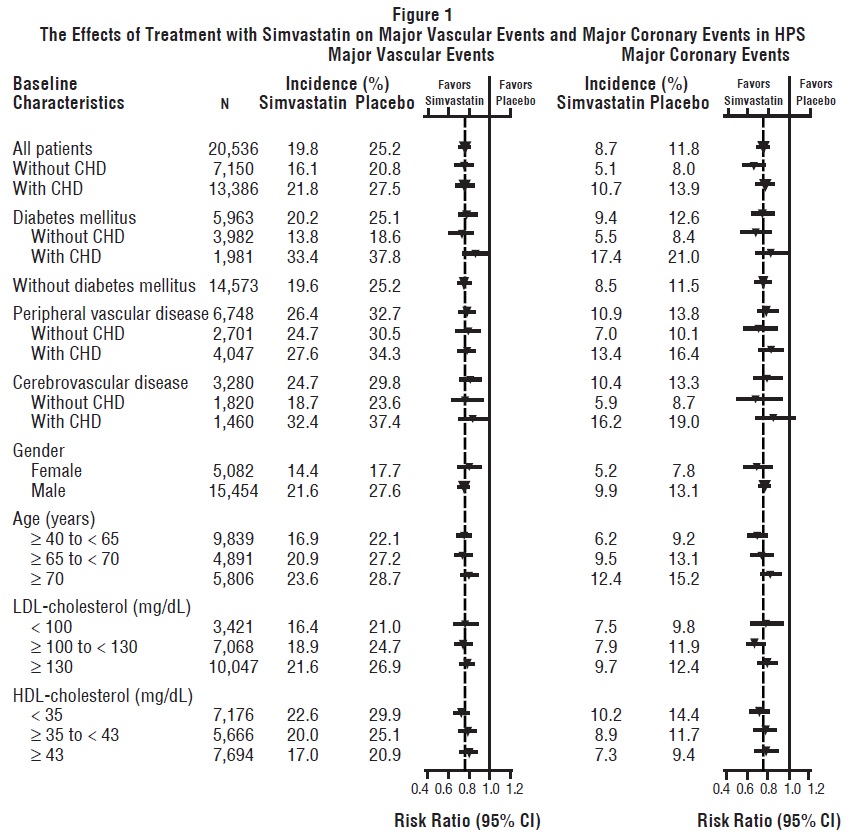
N = number of patients in each subgroup. The inverted triangles are point estimates of the relative risk, with their 95% confidence intervals represented as a line. The area of a triangle is proportional to the number of patients with MVE or MCE in the subgroup relative to the number with MVE or MCE, respectively, in the entire study population. The vertical solid line represents a relative risk of one. The vertical dashed line represents the point estimate of relative risk in the entire study population.
Angiographic Studies
In the Multicenter Anti-Atheroma Study, the effect of simvastatin on atherosclerosis was assessed by quantitative coronary angiography in hypercholesterolemic patients with CHD. In this randomized, double-blind, controlled study, patients were treated with simvastatin 20 mg/day or placebo. Angiograms were evaluated at baseline, two and four years. The co-primary study endpoints were mean change per-patient in minimum and mean lumen diameters, indicating focal and diffuse disease, respectively. Simvastatin significantly slowed the progression of lesions as measured in the Year 4 angiogram by both parameters, as well as by change in percent diameter stenosis. In addition, simvastatin significantly decreased the proportion of patients with new lesions and with new total occlusions.
Modifications of Lipid Profiles
Primary Hyperlipidemia (Fredrickson type lla and llb)
Simvastatin has been shown to be effective in reducing total-C and LDL-C in heterozygous familial and non-familial forms of hyperlipidemia and in mixed hyperlipidemia. Maximal to near maximal response is generally achieved within 4 to 6 weeks and maintained during chronic therapy. Simvastatin significantly decreased total-C, LDL-C, total-C/HDL-C ratio, and LDL-C/HDL-C ratio; simvastatin also decreased TG and increased HDL-C (see Table 5).
TABLE 5 Mean Response in Patients with Primary Hyperlipidemia and Combined (mixed) Hyperlipidemia (Mean Percent Change from Baseline After 6 to 24 Weeks) - * median percent change
- † mean baseline LDL-C 244 mg/dL and median baseline TG 168 mg/dL
- ‡ mean baseline LDL-C 188 mg/dL and median baseline TG 128 mg/dL
- § mean baseline LDL-C 226 mg/dL and median baseline TG 156 mg/dL
- ¶ 21% and 36% median reduction in TG in patients with TG ≤200 mg/dL and TG >200 mg/dL, respectively. Patients with TG >350 mg/dL were excluded
- # mean baseline LDL-C 156 mg/dL and median baseline TG 391 mg/dL.
TREATMENT
N
TOTAL-C
LDL-C
HDL-C
TG*
Lower Dose Comparative Study †
(Mean % Change at Week 6)
Simvastatin 5 mg q.p.m.
109
-19
-26
10
-12
Simvastatin 10 mg q.p.m.
110
-23
-30
12
-15
Scandinavian Simvastatin Survival Study ‡
(Mean % Change at Week 6)
Placebo
2223
-1
-1
0
-2
Simvastatin 20 mg q.p.m.
2221
-28
-38
8
-19
Upper Dose Comparative Study§
(Mean % Change Averaged at Weeks 18 and 24)
Simvastatin 40 mg q.p.m.
433
-31
-41
9
-18
Simvastatin 80 mg q.p.m.¶
664
-36
-47
8
-24
Multi-Center Combined Hyperlipidemia Study#
(Mean % Change at Week 6)
Placebo
125
1
2
3
-4
Simvastatin 40 mg q.p.m.
123
-25
-29
13
-28
Simvastatin 80 mg q.p.m.
124
-31
-36
16
-33
Hypertriglyceridemia (Fredrickson type IV)
The results of a subgroup analysis in 74 patients with type IV hyperlipidemia from a 130-patient, double-blind, placebo-controlled, 3-period crossover study are presented in TABLE 6.
TABLE 6 Six-week, Lipid-lowering Effects of Simvastatin in Type IV Hyperlipidemia Median Percent Change (25thand 75thpercentile) from Baseline* - * The median baseline values (mg/dL) for the patients in this study were: total-C = 254, LDL-C = 135, HDL-C = 36, TG = 404, VLDL-C = 83, and non-HDL-C = 215.
TREATMENT
N
Total-C
LDL-C
HDL-C
TG
VLDL-C
Non-HDL-C
Placebo
74
+2
(-7, +7)
+1
(-8, +14)
+3
(-3, +10)
-9
(-25, +13)
-7
(-25, +11)
+1
(-9, +8)
Simvastatin 40 mg/day
74
-25
(-34, -19)
-28
(-40, -17)
+11
(+5, +23)
-29
(-43, -16)
-37
(-54, -23)
-32
(-42, -23)
Simvastatin 80 mg/day
74
-32
(-38, -24)
-37
(-46, -26)
+15
(+5, +23)
-34
(-45, -18)
-41
(-57, -28)
-38
(-49, -32)
Dysbetalipoproteinemia (Fredrickson type III)
The results of a subgroup analysis in 7 patients with type III hyperlipidemia (dysbetalipoproteinemia) (apo E2/2) (VLDL-C/TG>0.25) from a 130-patient, double-blind, placebo-controlled, 3-period crossover study are presented in TABLE 7.
TABLE 7 Six-week, Lipid-lowering Effects of Simvastatin in Type III Hyperlipidemia Median Percent Change (min, max) from Baseline* - * The median baseline values (mg/dL) were: total-C = 324, LDL-C = 121, HDL-C = 31, TG = 411, VLDL-C = 170, and non-HDL-C = 291.
TREATMENT
N
Total-C
LDL-C + IDL
HDL-C
TG
VLDL-C + IDL
Non-HDL-C
Placebo
7
-8
(-24, +34)
-8
(-27, +23)
-2
(-21, +16)
+4
(-22, +90)
-4
(-28, +78)
-8
(-26, -39)
Simvastatin 40 mg/day
7
-50
(-66, -39)
-50
(-60, -31)
+7
(-8, +23)
-41
(-74, -16)
-58
(-90, -37)
-57
(-72, -44)
Simvastatin 80 mg/day
7
-52
(-55, -41)
-51
(-57, -28)
+7
(-5, +29)
-38
(-58, +2)
-60
(-72, -39)
-59
(-61, -46)
Homozygous Familial Hypercholesterolemia
In a controlled clinical study, 12 patients 15 to 39 years of age with homozygous familial hypercholesterolemia received simvastatin 40 mg/day in a single dose or in 3 divided doses, or 80 mg/day in 3 divided doses. In 11 patients with reductions in LDL-C, the mean LDL-C changes for the 40- and 80-mg doses were 14% (range 8% to 23%, median 12%) and 30% (range 14% to 46%, median 29%), respectively. One patient had an increase of 15% in LDL-C. Another patient with absent LDL-C receptor function had an LDL-C reduction of 41% with the 80-mg dose.
Endocrine Function
In clinical studies, simvastatin did not impair adrenal reserve or significantly reduce basal plasma cortisol concentration. Small reductions from baseline in basal plasma testosterone in men were observed in clinical studies with simvastatin, an effect also observed with other statins and the bile acid sequestrant cholestyramine. There was no effect on plasma gonadotropin levels. In a placebo-controlled, 12-week study there was no significant effect of simvastatin 80 mg on the plasma testosterone response to human chorionic gonadotropin. In another 24-week study, simvastatin 20 to 40 mg had no detectable effect on spermatogenesis. In 4S, in which 4,444 patients were randomized to simvastatin 20 to 40 mg/day or placebo for a median duration of 5.4 years, the incidence of male sexual adverse events in the two treatment groups was not significantly different. Because of these factors, the small changes in plasma testosterone are unlikely to be clinically significant. The effects, if any, on the pituitary-gonadal axis in pre-menopausal women are unknown.
14.2 Clinical Studies in Adolescents
In a double-blind, placebo-controlled study, 175 patients (99 adolescent boys and 76 post-menarchal girls) 10 to 17 years of age (mean age 14.1 years) with heterozygous familial hypercholesterolemia (HeFH) were randomized to simvastatin (n=106) or placebo (n=67) for 24 weeks (base study). Inclusion in the study required a baseline LDL-C level between 160 and 400 mg/dL and at least one parent with an LDL-C level >189 mg/dL. The dosage of simvastatin (once daily in the evening) was 10 mg for the first 8 weeks, 20 mg for the second 8 weeks, and 40 mg thereafter. In a 24-week extension, 144 patients elected to continue therapy with simvastatin 40 mg or placebo.
Simvastatin significantly decreased plasma levels of total-C, LDL-C, and Apo B (see TABLE 8). Results from the extension at 48 weeks were comparable to those observed in the base study.
TABLE 8 Lipid-Lowering Effects of Simvastatin in Adolescent Patients with Heterozygous Familial Hypercholesterolemia (Mean Percent Change from Baseline) - * median percent change
Dosage
Duration
N
Total-C
LDL-C
HDL-C
TG*
Apo B
Placebo
24 Weeks
67
% Change
from Baseline
(95% CI)
1.6
(-2.2, 5.3)
1.1
(-3.4, 5.5)
3.6
(-0.7, 8.0)
-3.2
(-11.8, 5.4)
-0.5
(-4.7, 3.6)
Mean baseline,
mg/dL (SD)
278.6
(51.8)
211.9
(49.0)
46.9
(11.9)
90.0
(50.7)
186.3
(38.1)
Simvastatin
24 Weeks
106
% Change
from Baseline
(95% CI)
-26.5
(-29.6, -23.3)
-36.8
(-40.5, -33.0)
8.3
(4.6, 11.9)
-7.9
(-15.8, 0.0)
-32.4
(-35.9, -29.0)
Mean baseline,
mg/dL (SD)
270.2
(44.0)
203.8
(41.5)
47.7
(9.0)
78.3
(46.0)
179.9
(33.8)
After 24 weeks of treatment, the mean achieved LDL-C value was 124.9 mg/dL (range: 64.0 to 289.0 mg/dL) in the simvastatin 40 mg group compared to 207.8 mg/dL (range: 128.0 to 334.0 mg/dL) in the placebo group.
The safety and efficacy of doses above 40 mg daily have not been studied in children with HeFH. The long-term efficacy of simvastatin therapy in childhood to reduce morbidity and mortality in adulthood has not been established.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
Simvastatin tablets USP, 20 mg are tan colored, oval shaped, biconvex, film-coated tablets, debossed with ‘LL’ on one side and ‘C03’ on the other side. They are supplied as follows:
Unit of Use Bottles of 30: NDC: 68645-470-54
Simvastatin tablets USP, 40 mg are brick red colored, round shaped, biconvex, film-coated tablets, debossed with 'LL' on one side and 'C04' on the other side. They are supplied as follows:
Unit of Use Bottles of 30: NDC: 68645-527-54
Storage
Store between 20° to 25°C (68° to 77°F) [See USP Controlled Room Temperature].
Preserve in tight container as defined in USP.
-
17 PATIENT COUNSELING INFORMATION
Patients should be advised to adhere to their National Cholesterol Education Program (NCEP)- recommended diet, a regular exercise program, and periodic testing of a fasting lipid panel.
Patients should be advised about substances they should not take concomitantly with simvastatin [see CONTRAINDICATONS (4) and WARNINGS AND PRECAUTIONS (5.1)]. Patients should also be advised to inform other healthcare professionals prescribing a new medication or increasing the dose of an existing medication that they are taking simvastatin.
17.1 Muscle Pain
All patients starting therapy with simvastatin should be advised of the risk of myopathy, including rhabdomyolysis, and told to report promptly any unexplained muscle pain, tenderness or weakness particularly if accompanied by malaise or fever or if these muscle signs or symptoms persist after discontinuing simvastatin. Patients using the 80-mg dose should be informed that the risk of myopathy, including rhabdomyolysis, is increased with use of the 80-mg dose. The risk of myopathy, including rhabdomyolysis, occurring with use of simvastatin is increased when taking certain types of medication or consuming grapefruit juice. Patients should discuss all medication, both prescription and over the counter, with their healthcare professional.
17.2 Liver Enzymes
It is recommended that liver function tests be performed before the initiation of simvastatin, and thereafter when clinically indicated. All patients treated with simvastatin should be advised to report promptly any symptoms that may indicate liver injury, including fatigue, anorexia, right upper abdominal discomfort, dark urine or jaundice.
17.3 Pregnancy
Women of childbearing age should be advised to use an effective method of birth control to prevent pregnancy while using simvastatin. Discuss future pregnancy plans with your patients, and discuss when to stop taking simvastatin if they are trying to conceive. Patients should be advised that if they become pregnant they should stop taking simvastatin and call their healthcare professional.
17.4 Breastfeeding
Women who are breastfeeding should not use simvastatin. Patients who have a lipid disorder and are breastfeeding should be advised to discuss the options with their healthcare professional.
1National Cholesterol Education Program (NCEP): Highlights of the Report of the Expert Panel on Blood Cholesterol Levels in Children and Adolescents. Pediatrics. 89(3):495-501. 1992.
2Manson, J.M., Freyssinges, C., Ducrocq, M.B., Stephenson, W.P., Postmarketing Surveillance of Lovastatin and Simvastatin Exposure During Pregnancy, Reproductive Toxicology, 10(6):439-446, 1996.
3D.R. Taves, Minimization: a new method of assigning patients to treatment and control groups. Clin. Pharmacol. Ther. 15 (1974), pp. 443-453.
Manufactured for:
Lupin Pharmaceuticals, Inc.
Baltimore, Maryland 21202
United StatesMADE IN INDIA.
Distributed by:
The Kroger Co.
Cincinnati, OH 45202Packaged by:
Legacy Pharmaceutical Packaging LLC
Earth City, MO 63045Revised: June 2019
PN: 20397-1
-
PRINCIPAL DISPLAY PANEL
NDC: 68645-470-54
Simvastatin Tablets USP 20 mg
Each film-coated tablet contains simvastatin USP 20 mg.
Rx only
30 Tablets

NDC: 68645-527-54
Simvastatin Tablets USP 40 mg
Each film-coated tablet contains simvastatin USP 40 mg.
Rx only
30 Tablets

-
INGREDIENTS AND APPEARANCE
SIMVASTATIN
simvastatin tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 68645-470(NDC:68180-479) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength SIMVASTATIN (UNII: AGG2FN16EV) (SIMVASTATIN - UNII:AGG2FN16EV) SIMVASTATIN 20 mg Inactive Ingredients Ingredient Name Strength ASCORBIC ACID (UNII: PQ6CK8PD0R) CITRIC ACID MONOHYDRATE (UNII: 2968PHW8QP) HYPROMELLOSES (UNII: 3NXW29V3WO) FERROSOFERRIC OXIDE (UNII: XM0M87F357) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) STARCH, CORN (UNII: O8232NY3SJ) TALC (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) BUTYLATED HYDROXYANISOLE (UNII: REK4960K2U) HYDROXYPROPYL CELLULOSE (1600000 WAMW) (UNII: RFW2ET671P) Product Characteristics Color BROWN Score no score Shape OVAL Size 11mm Flavor Imprint Code LL;C03 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 68645-470-54 30 in 1 BOTTLE; Type 0: Not a Combination Product 06/01/2007 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA078103 06/01/2007 SIMVASTATIN
simvastatin tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 68645-527(NDC:68180-464) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength SIMVASTATIN (UNII: AGG2FN16EV) (SIMVASTATIN - UNII:AGG2FN16EV) SIMVASTATIN 40 mg Inactive Ingredients Ingredient Name Strength ASCORBIC ACID (UNII: PQ6CK8PD0R) BUTYLATED HYDROXYANISOLE (UNII: REK4960K2U) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) CITRIC ACID MONOHYDRATE (UNII: 2968PHW8QP) FERROSOFERRIC OXIDE (UNII: XM0M87F357) HYDROXYPROPYL CELLULOSE (1600000 WAMW) (UNII: RFW2ET671P) HYPROMELLOSES (UNII: 3NXW29V3WO) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) STARCH, CORN (UNII: O8232NY3SJ) TALC (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) Product Characteristics Color RED (brick red) Score no score Shape ROUND (biconvex) Size 10mm Flavor Imprint Code LL;C04 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 68645-527-54 30 in 1 BOTTLE; Type 0: Not a Combination Product 12/02/2015 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA078103 12/02/2015 Labeler - Legacy Pharmaceutical Packaging, LLC (143213275)
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.