EVENITY- romosozumab-aqqg injection, solution
Evenity by
Drug Labeling and Warnings
Evenity by is a Prescription medication manufactured, distributed, or labeled by Amgen Inc. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use EVENITY safely and effectively. See full prescribing information for EVENITY.
EVENITY™ (romosozumab-aqqg) injection, for subcutaneous use
Initial U.S. Approval: 2019
WARNING: POTENTIAL RISK OF MYOCARDIAL INFARCTION, STROKE AND CARDIOVASCULAR DEATH
See full prescribing information for complete boxed warning.
EVENITY may increase the risk of myocardial infarction, stroke and cardiovascular death. (5.1)
EVENITY should not be initiated in patients who have had a myocardial infarction or stroke within the preceding year. Consider whether the benefits outweigh the risks in patients with other cardiovascular risk factors. (5.1)
If a patient experiences a myocardial infarction or stroke during therapy, EVENITY should be discontinued. (5.1)
INDICATIONS AND USAGE
EVENITY is a sclerostin inhibitor indicated for the treatment of osteoporosis in postmenopausal women at high risk for fracture, defined as a history of osteoporotic fracture, or multiple risk factors for fracture; or patients who have failed or are intolerant to other available osteoporosis therapy. (1)
Limitations of Use: Limit duration of use to 12 monthly doses. If osteoporosis therapy remains warranted, continued therapy with an anti-resorptive agent should be considered. (1.2)
DOSAGE AND ADMINISTRATION
- Two separate subcutaneous injections are needed to administer the total dose of 210 mg. Inject two syringes, one after the other. (2.1)
- Should be administered by a healthcare provider. (2.1)
- Administer 210 mg subcutaneously once every month for 12 doses in the abdomen, thigh, or upper arm. (2.2)
- Adequately supplement calcium and vitamin D during treatment. (2.2)
DOSAGE FORMS AND STRENGTHS
Injection: 105 mg/1.17 mL solution in a single-use prefilled syringe. A full dose of EVENITY requires two single-use prefilled syringes. (3)
WARNINGS AND PRECAUTIONS
- Major Adverse Cardiac Events (MACE): Monitor for symptoms of MI and stroke and seek prompt medical attention if symptoms occur. (5.1)
- Hypersensitivity: Hypersensitivity reactions, including angioedema, erythema multiforme, dermatitis, rash, and urticaria. Discontinue EVENITY if a clinically significant allergic reaction occurs. (5.2)
- Hypocalcemia: Adequately supplement calcium and vitamin D during treatment with EVENITY. (5.3)
- Osteonecrosis of the Jaw: Monitor for symptoms. Consider discontinuation of therapy based on benefit-risk assessment. (5.4)
- Atypical Femoral Fracture: Evaluate new or unusual thigh, hip, or groin pain to rule out an incomplete femur fracture. (5.5)
ADVERSE REACTIONS
The most common adverse reactions (≥ 5%) reported with EVENITY in clinical trials were arthralgia and headache. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Amgen Inc. at 1-800-77-AMGEN (1-800-772-6436) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
To report SUSPECTED ADVERSE REACTIONS, contact Amgen Inc. at 1-800-77-AMGEN (1-800-772-6436) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 12/2019
- Two separate subcutaneous injections are needed to administer the total dose of 210 mg. Inject two syringes, one after the other. (2.1)
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: POTENTIAL RISK OF MYOCARDIAL INFARCTION, STROKE AND CARDIOVASCULAR DEATH
1 INDICATIONS AND USAGE
1.1 Treatment of Postmenopausal Women with Osteoporosis at High Risk for Fracture
1.2 Limitations of Use
2 DOSAGE AND ADMINISTRATION
2.1 Important Dosage and Administration Instructions
2.2 Recommended Dosage
2.3 Preparation and Administration Instructions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Major Adverse Cardiac Events (MACE)
5.2 Hypersensitivity Reactions
5.3 Hypocalcemia
5.4 Osteonecrosis of the Jaw
5.5 Atypical Subtrochanteric and Diaphyseal Femoral Fractures
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Immunogenicity
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.7 Renal Impairment
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and Pharmacology
14 CLINICAL STUDIES
14.1 Treatment of Osteoporosis in Postmenopausal Women
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage and Handling
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: POTENTIAL RISK OF MYOCARDIAL INFARCTION, STROKE AND CARDIOVASCULAR DEATH
- EVENITY may increase the risk of myocardial infarction, stroke, and cardiovascular death [see Warnings and Precautions (5.1)]. EVENITY should not be initiated in patients who have had a myocardial infarction or stroke within the preceding year. Consider whether the benefits outweigh the risks in patients with other cardiovascular risk factors. If a patient experiences a myocardial infarction or stroke during therapy, EVENITY should be discontinued.
-
1
INDICATIONS AND USAGE
1.1 Treatment of Postmenopausal Women with Osteoporosis at High Risk for Fracture
EVENITY is indicated for the treatment of osteoporosis in postmenopausal women at high risk for fracture, defined as a history of osteoporotic fracture, or multiple risk factors for fracture; or patients who have failed or are intolerant to other available osteoporosis therapy.
1.2 Limitations of Use
The anabolic effect of EVENITY wanes after 12 monthly doses of therapy. Therefore, the duration of EVENITY use should be limited to 12 monthly doses. If osteoporosis therapy remains warranted, continued therapy with an anti-resorptive agent should be considered [see Dosage and Administration (2.2) and Clinical Studies (14.1)].
-
2
DOSAGE AND ADMINISTRATION
2.1 Important Dosage and Administration Instructions
- Two separate syringes (and two separate subcutaneous injections) are needed to administer the total dose of 210 mg of EVENITY. Inject two 105 mg/1.17 mL prefilled syringes, one after the other.
- EVENITY should be administered by a healthcare provider.
2.2 Recommended Dosage
- The recommended dose of EVENITY is 210 mg administered subcutaneously in the abdomen, thigh or upper arm. Administer EVENITY once every month.
- The treatment duration for EVENITY is 12 monthly doses.
- Patients should be adequately supplemented with calcium and vitamin D during treatment with EVENITY [see Warnings and Precautions (5.3) and Clinical Studies (14.1)].
- If the EVENITY dose is missed, administer as soon as it can be rescheduled. Thereafter, EVENITY can be scheduled every month from the date of the last dose.
2.3 Preparation and Administration Instructions
Step 1: Prior to Administration:
- Remove two syringes from the carton.
- Visually inspect EVENITY for particles and discoloration prior to administration. EVENITY is a clear to opalescent, colorless to light yellow solution. Do not use if the solution is cloudy or discolored or contains particles.
- Do not use the syringe if
○ any part appears cracked or broken
○ the gray needle cap is missing or not securely attached
○ the expiration date printed on the label has passed
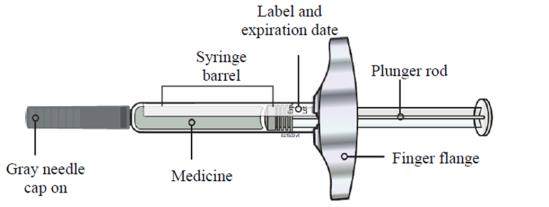
- Always hold the prefilled syringe by the syringe barrel to remove the syringe from the tray. See Figure A.
○ Do not grasp the plunger rod.
○ Do not grasp the gray needle cap.
○ Do not remove the gray needle cap until you are ready to inject.
- Allow EVENITY to sit at room temperature for at least 30 minutes before injecting. Do not warm in any other way [see How Supplied/Storage and Handling (16)].
Step 2: Select the Injection Site and Prepare the Syringe
Prepare and clean two injection sites, one for each of the two injections. See Figure B.
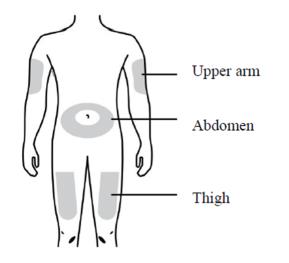
The recommended subcutaneous injection sites include:
- The thigh
- Abdomen, except for a two-inch area right around the navel
- Outer area of upper arm
Clean the injection sites with alcohol wipes. Let the skin dry.
- Choose a different site each time you give an injection. If you want to use the same injection site, make sure it is not the same spot on the injection site you used for a previous injection.
- Do not inject into areas where the skin is tender, bruised, red, or hard. Avoid injecting into areas with scars or stretch marks.
Choose the first syringe. Pull the gray needle cap straight off and away from your body when you are ready to inject. See Figure C.
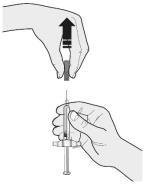
- Do not put the gray needle cap back onto the syringe.
Step 3: Inject EVENITY
Insert needle and inject all the liquid subcutaneously. Do not administer into muscle or blood vessel. See Figure D.
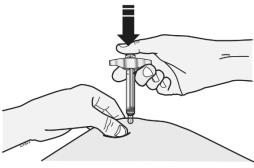
When done, gently lift the syringe off of the skin.
Step 4: Syringe and Needle Cap Disposal
Immediately dispose of the syringe and needle cap in the nearest sharps container.
Important: Repeat all steps with the second syringe to inject the full dose.
- Two separate syringes (and two separate subcutaneous injections) are needed to administer the total dose of 210 mg of EVENITY. Inject two 105 mg/1.17 mL prefilled syringes, one after the other.
- 3 DOSAGE FORMS AND STRENGTHS
-
4
CONTRAINDICATIONS
EVENITY is contraindicated in patients with:
- Hypocalcemia. Pre-existing hypocalcemia must be corrected prior to initiating therapy with EVENITY [see Warnings and Precautions (5.3), Adverse Reactions (6.1) and Use in Specific Populations (8.7)].
- A history of systemic hypersensitivity to romosozumab or to any component of the product formulation. Reactions have included angioedema, erythema multiforme, and urticaria [see Warnings and Precautions (5.2) and Adverse Reactions (6.1)].
- Hypocalcemia. Pre-existing hypocalcemia must be corrected prior to initiating therapy with EVENITY [see Warnings and Precautions (5.3), Adverse Reactions (6.1) and Use in Specific Populations (8.7)].
-
5
WARNINGS AND PRECAUTIONS
5.1 Major Adverse Cardiac Events (MACE)
In a randomized controlled trial in postmenopausal women, there was a higher rate of major adverse cardiac events (MACE), a composite endpoint of cardiovascular death, nonfatal myocardial infarction and nonfatal stroke, in patients treated with EVENITY compared to those treated with alendronate [see Boxed Warning and Adverse Reactions (6.1)].
EVENITY should not be initiated in patients who have had a myocardial infarction or stroke within the preceding year. Consider whether the benefits outweigh the risks in patients with other cardiovascular risk factors. Monitor for signs and symptoms of myocardial infarction and stroke and instruct patients to seek prompt medical attention if symptoms occur. If a patient experiences a myocardial infarction or stroke during therapy, EVENITY should be discontinued.
5.2 Hypersensitivity Reactions
Hypersensitivity reactions, including angioedema, erythema multiforme, dermatitis, rash, and urticaria have occurred in EVENITY-treated patients. If an anaphylactic or other clinically significant allergic reaction occurs, initiate appropriate therapy and discontinue further use of EVENITY [see Contraindications (4) and Adverse Reactions (6.1)].
5.3 Hypocalcemia
Hypocalcemia has occurred in patients receiving EVENITY. Correct hypocalcemia prior to initiating EVENITY [see Contraindications (4), Adverse Reactions (6.1) and Use in Specific Populations (8.7)].
Monitor patients for signs and symptoms of hypocalcemia. Patients should be adequately supplemented with calcium and vitamin D while on EVENITY [see Dosage and Administration (2.2) and Clinical Studies (14.1)].
Patients with severe renal impairment (estimated glomerular filtration rate [eGFR] 15 to 29 mL/min/1.73 m2) or receiving dialysis are at greater risk of developing hypocalcemia. Monitor serum calcium and adequately supplement patients who have severe renal impairment or are receiving dialysis with calcium and vitamin D. Instruct patients with severe renal impairment, including those receiving dialysis, about the symptoms of hypocalcemia and the importance of maintaining calcium levels with adequate calcium and vitamin D supplementation.
5.4 Osteonecrosis of the Jaw
Osteonecrosis of the jaw (ONJ), which can occur spontaneously, is generally associated with tooth extraction and/or local infection with delayed healing, and has been reported in patients receiving EVENITY. A routine oral examination should be performed by the prescriber prior to initiation of EVENITY treatment. Concomitant administration of drugs associated with ONJ (chemotherapy, bisphosphonates, denosumab, angiogenesis inhibitors, and corticosteroids) may increase the risk of developing ONJ. Other risk factors for ONJ include cancer, radiotherapy, poor oral hygiene, pre-existing dental disease or infection, anemia, and coagulopathy [see Adverse Reactions (6.1)].
For patients requiring invasive dental procedures, clinical judgment of the treating physician and/or oral surgeon should guide the management plan of each patient based on benefit-risk assessment. Patients who are suspected of having or who develop ONJ while on EVENITY should receive care by a dentist or an oral surgeon. In these patients, dental surgery to treat ONJ may exacerbate the condition. Discontinuation of EVENITY should be considered based on benefit-risk assessment.
5.5 Atypical Subtrochanteric and Diaphyseal Femoral Fractures
Atypical low-energy or low trauma fractures of the femoral shaft have been reported in patients receiving EVENITY [see Adverse Reactions (6.1)]. These fractures can occur anywhere in the femoral shaft from just below the lesser trochanter to above the supracondylar flare and are transverse or short oblique in orientation without evidence of comminution. Causality has not been established as these fractures also occur in osteoporotic patients who have not been treated.
Atypical femoral fractures most commonly occur with minimal or no trauma to the affected area. They may be bilateral and many patients report prodromal pain in the affected area, usually presenting as dull, aching thigh pain, weeks to months before a complete fracture occurs.
During EVENITY treatment, patients should be advised to report new or unusual thigh, hip, or groin pain. Any patient who presents with thigh or groin pain should be suspected of having an atypical fracture and should be evaluated to rule out an incomplete femur fracture. Patient presenting with an atypical femur fracture should also be assessed for symptoms and signs of fracture in the contralateral limb. Interruption of EVENITY therapy should be considered based on benefit-risk assessment [see Clinical Studies (14)].
-
6
ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections of the label:
- Major adverse cardiac events [see Boxed Warning and Warnings and Precautions (5.1)]
- Hypersensitivity [see Contraindications (4) and Warnings and Precautions (5.2)]
- Hypocalcemia [see Contraindications (4) and Warnings and Precautions (5.3)]
- Osteonecrosis of the Jaw [see Warnings and Precautions (5.4)]
- Atypical Subtrochanteric and Diaphyseal Femoral Fractures [see Warnings and Precautions (5.5)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of EVENITY for the treatment of postmenopausal osteoporosis was evaluated in a multicenter, randomized, double-blind, placebo-controlled study (Study 1, NCT01575834) of 7180 postmenopausal women aged 55 to 90 years (mean age of 71 years). A total of 3581 and 3576 women received at least one dose of EVENITY and placebo, respectively, administered once every month during the 12-month double-blind study period. Women received at least 500 mg calcium and 600 international units of vitamin D supplementation daily and 77% received a loading dose of 50,000 to 60,000 international units of vitamin D within one week of randomization (if serum 25-hydroxyvitamin D concentrations were 40 ng/mL or less).
The safety of EVENITY for the treatment of postmenopausal osteoporosis in patients at high risk of fracture was evaluated in a multicenter, randomized, double-blind, alendronate-controlled study (Study 2, NCT01631214) of 4093 postmenopausal women aged 55 to 90 years (mean age of 74 years). A total of 2040 and 2014 women received at least one dose of EVENITY and alendronate, respectively, during the 12-month double-blind study period. Women received at least 500 mg calcium and 600 international units vitamin D supplementation daily and 74% received a loading dose of 50,000 to 60,000 international units of vitamin D within one week of randomization (if serum 25-hydroxyvitamin D concentrations were 40 ng/mL or less).
In Study 1, during the 12-month double-blind treatment period, the incidence of all-cause mortality was 0.7% (24/3576) in the placebo group and 0.8% (29/3581) in the EVENITY group. The incidence of nonfatal serious adverse events was 8.3% in the placebo group and 9.1% in the EVENITY group. The percentage of patients who withdrew from the study due to adverse events was 1.1% in the placebo group and 1.1% in the EVENITY group. The most common adverse reactions reported with EVENITY (greater than or equal to 5% and at a higher incidence than placebo) were arthralgia and headache. The most common adverse reaction leading to discontinuation of EVENITY was arthralgia (6 subjects [0.2%] in the placebo group and 5 subjects [0.1%] in the EVENITY group).
In Study 2, during the 12-month double-blind treatment period, the incidence of all-cause mortality was 1.1% (22/2014) in the alendronate group and 1.5% (30/2040) in the EVENITY group. The incidence of nonfatal serious adverse events was 13.3% in the alendronate group and 11.9% in the EVENITY group. The percentage of patients who withdrew from the study due to adverse events was 1.2% in the alendronate group and 1.2% in the EVENITY group. The most common adverse reactions reported with EVENITY (greater than or equal to 5%) were arthralgia and headache.
Table 1 outlines the most common adverse reactions occurring in greater than or equal to 2% of EVENITY treated women in at least one study.
Table 1 Adverse Reactions Occurring in ≥ 2% of EVENITY-Treated Women in at Least One Study (Studies 1 and 2) Study 1 Study 2 Preferred Term Placebo
(N = 3576)
n (%)EVENITY
(N = 3581)
n (%)Alendronate
(N = 2014)
n (%)EVENITY
(N = 2040)
n (%)Arthralgia 434 (12.1) 468 (13.1) 194 (9.6) 166 (8.1) Headache 208 (5.8) 235 (6.6) 110 (5.5) 106 (5.2) Muscle spasms 140 (3.9) 163 (4.6) 81 (4.0) 70 (3.4) Edema peripheral 67 (1.9) 86 (2.4) 38 (1.9) 34 (1.7) Asthenia 79 (2.2) 84 (2.3) 53 (2.6) 50 (2.5) Neck pain 54 (1.5) 80 (2.2) 42 (2.1) 34 (1.7) Insomnia 68 (1.9) 72 (2.0) 36 (1.8) 34 (1.7) Paresthesia 62 (1.7) 72 (2.0) 34 (1.7) 29 (1.4) The adverse reactions described below are from the 12-month treatment periods of Study 1 (placebo-controlled) and Study 2 (alendronate-controlled).
Major Adverse Cardiac Events (MACE)
During the 12-month double-blind treatment period of the placebo-controlled trial (Study 1), myocardial infarction occurred in 9 (0.3%) women in the EVENITY group and 8 (0.2%) women in the placebo group; stroke occurred in 8 (0.2%) women in the EVENITY group and 10 (0.3%) women in the placebo group. These events occurred in patients with and without a history of myocardial infarction or stroke. Cardiovascular death occurred in 17 (0.5%) women in the EVENITY group and 15 (0.4%) women in the placebo group. The number of women with positively adjudicated MACE was 30 (0.8%) in the EVENITY group and 29 (0.8%) in the placebo group, yielding a hazard ratio of 1.03 (95% confidence interval [0.62, 1.72]) for EVENITY compared to placebo.
During the 12-month double-blind treatment period of the active-controlled trial (Study 2), myocardial infarction occurred in 16 (0.8%) women in the EVENITY group and 5 (0.2%) women in the alendronate group; stroke occurred in 13 (0.6%) women in the EVENITY group and 7 (0.3%) women in the alendronate group. These events occurred in patients with and without a history of myocardial infarction or stroke. Cardiovascular death occurred in 17 (0.8%) women in the EVENITY group and 12 (0.6%) women in the alendronate group. The number of women with positively adjudicated MACE was 41 (2.0%) in the EVENITY group and 22 (1.1%) in the alendronate group, yielding a hazard ratio of 1.87 (95% confidence interval [1.11, 3.14]) for EVENITY compared to alendronate [see Boxed Warning and Warnings and Precautions (5.1)].
Hypersensitivity Reactions
Across both trials, hypersensitivity reactions were reported in 364 (6.5%) women in the EVENITY group and 365 (6.5%) women in the control group. Reported reactions included angioedema (3 [< 0.1%] women in the EVENITY group vs. 3 [< 0.1%] women in the control group), erythema multiforme (1 [< 0.1%] woman in the EVENITY group vs. no woman in the control group), dermatitis (32 [0.6%] women in the EVENITY group vs. 42 [0.8%] women in the control group), rash (60 [1.1%] women in the EVENITY group vs. 53 [0.9%] women in the control group), and urticaria (23 [0.4%] women in the EVENITY group vs. 27 [0.5%] women in the control group). Although angioedema, dermatitis and urticaria were not reported at a higher incidence with EVENITY than control, there were cases of angioedema, dermatitis and urticaria that were determined to be related to EVENITY use [see Contraindications (4) and Warnings and Precautions (5.2)].
Hypocalcemia
Across both trials, adverse events of hypocalcemia occurred in 2 EVENITY-treated women and in 1 woman in the control group. Decreases in albumin-adjusted serum calcium to below the lower limit of the reference range (8.3 mg/dL) were reported in 14 (0.2%) women in the EVENITY group and 10 (0.2%) women in the control group. No patient receiving EVENITY developed serum calcium less than 7.5 mg/dL. The nadir in albumin-adjusted serum calcium occurred by month 1 after EVENITY dosing in patients with normal renal function [see Contraindications (4) and Warnings and Precautions (5.3)].
Injection Site Reactions
Across both trials, injection site reactions occurred in 278 (4.9%) women in the EVENITY group and 157 (2.8%) women in the control group. The most common injection site reactions were pain (94 [1.7%] women in the EVENITY group; 70 [1.3%] women in the control group) and erythema (80 [1.4%] women in the EVENITY group and 14 [0.3%] women in the control group). Injection site reactions resulted in discontinuation of treatment in 7 (0.1%) EVENITY-treated patients and 3 (< 0.1%) patients in the control group.
Osteonecrosis of the Jaw
Across both trials, osteonecrosis of the jaw occurred in one patient during treatment with EVENITY [see Warnings and Precautions (5.4)].
Atypical Subtrochanteric and Diaphyseal Fractures
Across both trials, atypical femoral fracture occurred in one patient during treatment with EVENITY [see Warnings and Precautions (5.5)].
6.2 Immunogenicity
As with all therapeutic proteins, there is potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors, including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies in the studies described below with the incidence of antibodies in other studies or to other romosozumab products may be misleading.
The immunogenicity of EVENITY was evaluated using an immunoassay for the detection of anti-romosozumab-aqqg antibodies. An in vitro biological assay was performed to detect neutralizing antibodies for those subjects whose sera tested positive for anti-romosozumab-aqqg antibodies.
Among 5914 postmenopausal women treated with EVENITY 210 mg monthly, 18.1% of subjects developed antibodies to romosozumab-aqqg. Of the subjects who developed antibodies to romosozumab-aqqg, 4.7% had antibodies that were classified as neutralizing. Development of antibodies to romosozumab-aqqg was associated with lower serum romosozumab-aqqg concentrations [see Clinical Pharmacology (12.3)]. Antibodies to romosozumab-aqqg were generally not associated with changes in the efficacy or safety of EVENITY.
- Major adverse cardiac events [see Boxed Warning and Warnings and Precautions (5.1)]
-
8
USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
EVENITY is not indicated for use in women of reproductive potential. In animal reproduction studies, weekly administration of romosozumab-aqqg to pregnant rats during the period of organogenesis at exposures greater than 31 times the clinical exposure produced skeletal abnormalities in the offspring. Administration of romosozumab-aqqg to rats prior to mating and through to the end of lactation produced minimal to slight decreases in femoral bone mineral density and/or cortical circumferences in the offspring at 1.4 to 54 times the expected exposure in humans [see Data].
Data
Animal Data
Reproductive and developmental effects of romosozumab-aqqg were assessed in the rat in a preliminary and definitive embryo-fetal development study, a combined fertility and embryo-development study, and a pre- and postnatal development study.
Skeletal malformations including syndactyly and polydactyly occurred in 1 out of 75 litters across all rat reproductive toxicity studies, in the litter of a dam given weekly subcutaneous romosozumab-aqqg doses of 300 mg/kg (equivalent to at least 31 times the clinical exposure observed in humans following a monthly subcutaneous dose of 210 mg, based on area under the concentration-time curve [AUC] comparison).
In the offspring of female rats given weekly romosozumab-aqqg doses from 6 weeks before cohabitation through mating and lactation, femoral periosteal and endocortical circumferences were slightly decreased at 10, 60, and 300 mg/kg (equivalent to 1.4, 18, and 54 times the clinical exposure following a monthly subcutaneous dose of 210 mg, based on AUC comparison). Cortical thickness was increased at 300 mg/kg (equivalent to 54 times expected clinical exposure). Femoral metaphyseal bone mineral density was slightly decreased at 60 and 300 mg/kg (equivalent to 18 and 54 times expected clinical exposure).
8.2 Lactation
Risk Summary
EVENITY is not indicated for use in women of reproductive potential. In animal studies where pregnant rats were given weekly doses of romosozumab-aqqg from 6 weeks before cohabitation through mating and lactation at 10, 60, or 300 mg/kg (equivalent to 1.4, 18 or 54 times the clinical exposure following a monthly subcutaneous dose of 210 mg, based on AUC comparison), romosozumab-aqqg was dose-dependently present in the serum of offspring on postnatal day 21 at 0.01 to 2.4 times maternal exposure due to gestational and/or lactational exposure.
8.4 Pediatric Use
Safety and effectiveness of EVENITY have not been established in pediatric patients.
8.5 Geriatric Use
Of the 6544 postmenopausal women with osteoporosis in the clinical studies of EVENITY, 5234 (80%) were age 65 years and over and 2390 (37%) were age 75 years and over. No overall differences in safety or efficacy were observed between these subjects and younger subjects, and other reported clinical experience has not identified differences in response between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
8.7 Renal Impairment
No dose adjustment is required in patients with renal impairment.
Patients with severe renal impairment (estimated glomerular filtration rate [eGFR] 15 to 29 mL/min/1.73 m2 by MDRD equation) or receiving dialysis are at greater risk of developing hypocalcemia [see Contraindications (4), Warnings and Precautions (5.3) and Adverse Reactions (6.1)]. Monitor calcium concentrations and adequately supplement calcium and vitamin D in patients who have severe renal impairment or are receiving dialysis.
-
11
DESCRIPTION
Romosozumab-aqqg is a humanized monoclonal antibody (IgG2) produced in a mammalian cell line (Chinese Hamster Ovary) by recombinant DNA technology that binds to and inhibits sclerostin. Romosozumab-aqqg has an approximate molecular weight of 149 kDa.
EVENITY (romosozumab-aqqg) injection is supplied as a sterile, preservative-free, clear to opalescent, colorless to light yellow solution for subcutaneous injection in a single-use prefilled syringe.
Two 105 mg/1.17 mL single-use prefilled syringes are required to administer the recommended 210 mg dose of EVENITY [see Dosage and Administration (2.1)]. Each single-use prefilled syringe delivers 1.17 mL of solution containing 105 mg of romosozumab-aqqg, acetate (3.8 mg), calcium (0.61 mg), polysorbate 20 (0.07 mg) and sucrose (70 mg) in Water for Injection, USP, and sodium hydroxide to a pH of 5.2.
-
12
CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
EVENITY inhibits the action of sclerostin, a regulatory factor in bone metabolism. EVENITY increases bone formation and, to a lesser extent, decreases bone resorption. Animal studies showed that romosozumab-aqqg stimulates new bone formation on trabecular and cortical bone surfaces by stimulating osteoblastic activity resulting in increases in trabecular and cortical bone mass and improvements in bone structure and strength [see Nonclinical Toxicology (13.2) and Clinical Studies (14.1)].
12.2 Pharmacodynamics
In postmenopausal women with osteoporosis, EVENITY increased the bone formation marker procollagen type 1 N-telopeptide (P1NP) with a peak increase from baseline of approximately 145% compared to placebo 2 weeks after initiating treatment, followed by a return to concentrations seen with placebo at month 9 and a decline from baseline to approximately 15% below the concentration change seen with placebo at month 12.
EVENITY decreased the bone resorption marker type 1 collagen C-telopeptide (CTX) with a maximal reduction from baseline of approximately 55% compared to placebo 2 weeks after initiating treatment. CTX remained below concentrations seen with placebo and was approximately 25% below the concentration change seen with placebo at month 12.
After discontinuation of EVENITY, P1NP levels returned to baseline within 12 months; CTX increased above baseline levels within 3 months and returned toward baseline levels by month 12.
12.3 Pharmacokinetics
Administration of a single dose of 210 mg EVENITY in healthy volunteers resulted in a mean (standard deviation [SD]) maximum romosozumab-aqqg serum concentration (Cmax) of 22.2 (5.8) mcg/mL and a mean (SD) AUC of 389 (127) mcg*day/mL. Steady-state concentrations were achieved by month 3 following the monthly administration of 210 mg to postmenopausal women. The mean trough serum romosozumab-aqqg concentrations at months 3, 6, 9, and 12 ranged from 8 to 13 mcg/mL.
Romosozumab-aqqg exhibited nonlinear pharmacokinetics with exposure increasing greater than dose proportionally (e.g., 550-fold increase in mean AUCinf for the 100-fold increase in subcutaneous doses ranging from 0.1 to 10 mg/kg [0.03 to 3.3 times the approved recommended dosage for a 70 kg woman).
Absorption
The median time to maximum romosozumab-aqqg concentration (Tmax) is 5 days (range: 2 to 7 days).
Distribution
The estimated volume of distribution at steady-state is approximately 3.92 L.
Elimination
Romosozumab-aqqg exhibited nonlinear pharmacokinetics with the clearance of romosozumab-aqqg decreasing as the dose increased. The estimated mean systemic clearance (CL/F) of romosozumab-aqqg was 0.38 mL/hr/kg, following a single subcutaneous administration of 3 mg/kg (the approved recommended dosage for a 70 kg woman). The mean effective t1/2 was 12.8 days after 3 doses of 3 mg/kg (the approved recommended dosage for a 70 kg woman) every 4 weeks.
Metabolism
The metabolic pathway of romosozumab-aqqg has not been characterized. As a humanized IgG2 monoclonal antibody, romosozumab-aqqg is expected to be degraded into small peptides and amino acids via catabolic pathways in a manner similar to endogenous IgG.
Anti-Product Antibody Formation Affecting Pharmacokinetics
Development of anti-romosozumab-aqqg antibodies was associated with reduced serum romosozumab-aqqg concentrations. The presence of anti-romosozumab-aqqg antibodies led to decreased mean romosozumab-aqqg concentrations up to 22%. The presence of neutralizing antibodies led to decreased mean romosozumab-aqqg concentrations up to 63% [see Adverse Reactions (6.2)].
Specific Populations
No clinically significant differences in the pharmacokinetics of romosozumab-aqqg were observed based on age (20-89 years), sex, race, disease state (low bone mass or osteoporosis), prior exposure to alendronate, or renal impairment including end-stage renal disease (ESRD) requiring dialysis. The effect of ESRD not requiring dialysis on the pharmacokinetics of romosozumab-aqqg is unknown.
Body Weight
The exposure of romosozumab-aqqg decreases with increasing body weight.
-
13
NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity
In a rat carcinogenicity study, once-weekly romosozumab-aqqg doses of 3, 10 or 50 mg/kg were administered by subcutaneous injection to Sprague-Dawley rats from 8 weeks up to 98 weeks of age, resulting in systemic exposures that were up to 19 times the systemic exposure observed in humans following a monthly subcutaneous dose of 210 mg EVENITY (based on AUC comparison). Romosozumab-aqqg caused a dose-dependent increase in bone mass with trabecular and cortical bone thickening at all doses. There were no effects of romosozumab-aqqg on mortality and romosozumab-aqqg did not cause significant increases in tumor incidence in male or female rats.
Mutagenicity
Mutagenesis has not been evaluated, as monoclonal antibodies are not expected to alter DNA or chromosomes.
Impairment of Fertility
No effects on fertility were observed in male and female rats given subcutaneous romosozumab-aqqg doses up to 300 mg/kg (up to 54 times the systemic exposure observed in humans following a monthly subcutaneous dose of 210 mg EVENITY, based on AUC comparison). No effects were noted in reproductive organs in rats and cynomolgus monkeys dosed subcutaneously for 6 months with weekly doses up to 100 mg/kg (exposures up to 37 and 90 times, respectively, the systemic exposure observed in humans administered monthly subcutaneous doses of 210 mg based on AUC comparison).
13.2 Animal Toxicology and Pharmacology
No adverse effects were noted in rats and monkeys after 26 once-weekly subcutaneous romosozumab-aqqg doses up to 100 mg/kg, equivalent to systemic exposures of 37 and 90 times, respectively, the systemic exposure observed in humans following a monthly subcutaneous dose of 210 mg EVENITY (based on AUC comparison).
Bone safety studies of up to 12-month duration were conducted in ovariectomized rats and monkeys with once-weekly romosozumab-aqqg doses yielding exposures ranging from 1 to 21 times the systemic exposure in humans given monthly doses of 210 mg, based on AUC comparison. Romosozumab-aqqg increased bone mass and improved cancellous bone microarchitecture and cortical bone geometry by increasing bone formation on periosteal, endocortical, and trabecular surfaces, and decreasing bone resorption on trabecular and endocortical surfaces. The increases in bone mass were significantly correlated with increases in bone strength. In rats and monkeys, bone quality was maintained at all skeletal sites at doses ranging from 1 to 21 times human exposure, and slightly improved in vertebrae at 19 to 21 times human exposure. There was no evidence of mineralization defects, osteoid accumulation, or woven bone formation.
-
14
CLINICAL STUDIES
14.1 Treatment of Osteoporosis in Postmenopausal Women
Study 1 (NCT01575834) was a randomized, double-blind, placebo-controlled study of postmenopausal women aged 55 to 90 years (mean age of 71 years) with bone mineral density (BMD) T-score less than or equal to −2.5 at the total hip or femoral neck. Women were randomized to receive subcutaneous injections of either EVENITY (N = 3589) or placebo (N = 3591) for 12 months. At baseline, 18% of women had a vertebral fracture. After the 12-month treatment period, women in both arms transitioned to open-label anti-resorptive therapy (denosumab) for 12 months while remaining blinded to their initial treatment. Women received 500 to 1000 mg calcium and 600 to 800 international units vitamin D supplementation daily. The coprimary efficacy endpoints were new vertebral fracture at month 12 and month 24.
Effect on Fractures
EVENITY significantly reduced the incidence of new vertebral fractures through month 12 compared to placebo. In addition, the significant reduction in fracture risk persisted through the second year in women who received EVENITY during the first year and transitioned to denosumab compared to those who transitioned from placebo to denosumab (see Table 2).
Table 2 Effect of EVENITY on the Incidence and Risk of Fractures in Study 1 Proportion of Women with Fractures Absolute Risk
Reduction
(%)
(95% CI)aRelative Risk
Reduction
(%)
(95% CI)ap-valueb At Month 12 Placebo
(N = 3591)EVENITY
(N = 3589)New vertebral fracture 1.8% 0.5% 1.3
(0.8, 1.8)73
(53, 84)< 0.001 At Month 24 Placebo Followed by
Denosumab
(N = 3591)EVENITY Followed by
Denosumab
(N = 3589)New vertebral fracture 2.5% 0.6% 1.9
(1.3, 2.5)75
(60, 84)< 0.001 N = Number of subjects randomized
a. Absolute and relative risk reduction are based on the Mantel-Haenszel method adjusting for age and prevalent vertebral fracture strata.
b. P-value is based on logistic regression model adjusting for age and prevalent vertebral fracture strata.
EVENITY significantly reduced the incidence of clinical fracture (a composite endpoint of symptomatic vertebral fracture and nonvertebral fracture) at 12 months. However, 88% of these clinical fractures were nonvertebral fractures and the incidence of nonvertebral fractures was not statistically significantly different when comparing EVENITY-treated women to placebo-treated women at month 12 or month 24.
Effect on Bone Mineral Density (BMD)
EVENITY significantly increased BMD at the lumbar spine, total hip, and femoral neck compared with placebo at month 12. The treatment differences in BMD were 12.7% at the lumbar spine, 5.8% at the total hip, and 5.2% at the femoral neck.
Following the transition from EVENITY to denosumab at month 12, BMD continued to increase through month 24. In patients who transitioned from placebo to denosumab, BMD also increased with denosumab use. The differences in BMD achieved at month 12 between EVENITY and placebo patients were overall maintained at month 24, when comparing patients who transitioned from EVENITY to denosumab to those who transitioned from placebo to denosumab. There was no evidence of differences in effects on BMD at the lumbar spine or total hip across subgroups defined by baseline age, baseline BMD, or geographic region.
After EVENITY discontinuation, BMD returns to approximately baseline levels within 12 months in the absence of follow-on antiresorptive therapy [see Indications and Usage (1.2)].
Bone Histology and Histomorphometry
A total of 154 transiliac crest bone biopsy specimens were obtained from 139 postmenopausal women with osteoporosis at month 2, month 12, and/or month 24. All of these biopsies were adequate for qualitative histology and 138 (90%) were adequate for full quantitative histomorphometry assessment. Qualitative histology assessments from women treated with EVENITY showed normal bone architecture and quality at all time points. There was no evidence of woven bone, mineralization defects, or marrow fibrosis.
Histomorphometry assessments on biopsies at months 2 and 12 compared the effect of EVENITY with placebo (15 specimens at month 2 and 39 specimens at month 12 in the EVENITY group, 14 specimens at month 2 and 31 specimens at month 12 in the placebo group). At month 2 in women treated with EVENITY, histomorphometric indices of bone formation at trabecular and endocortical surfaces were increased. These effects on bone formation were accompanied by a decrease in indices of bone resorption. At month 12, both bone formation and resorption indices were decreased with EVENITY, while bone volume, and trabecular and cortical thickness were increased.
Study 2 (NCT01631214) was a randomized, double-blind, alendronate-controlled study of postmenopausal women aged 55 to 90 years (mean age of 74 years) with BMD T-score less than or equal to −2.5 at the total hip or femoral neck and either one moderate or severe vertebral fracture or two mild vertebral fractures, or BMD T-score less than or equal to -2.0 at the total hip or femoral neck and either two moderate or severe vertebral fractures or a history of a proximal femur fracture. Women were randomized (1:1) to receive either monthly subcutaneous injections of EVENITY (N = 2046) or oral alendronate 70 mg weekly (N = 2047) for 12 months, with 500 to 1000 mg calcium and 600 to 800 international units vitamin D supplementation daily. After the 12-month treatment period, women in both arms transitioned to open-label alendronate 70 mg weekly while remaining blinded to their initial treatment.
This was an event driven trial. The two primary efficacy endpoints were the incidence of morphometric vertebral fracture at 24 months and time to the first clinical fracture through the primary analysis period, which ended when at least 330 subjects had a clinical fracture and all subjects had completed the 24-month visit. Clinical fracture was a composite endpoint of nonvertebral fracture and symptomatic vertebral fracture.
Effect on Fractures
EVENITY significantly reduced the incidence of new vertebral fracture at 24 months (see Table 3).
Table 3 Effect of EVENITY on the Incidence of New Vertebral Fractures in Study 2 Proportion of Women with
Fracture (%)Risk Reduction p-value b Alendronate
Alone
(N = 2047)EVENITY
Followed by
Alendronate
(N = 2046)Absolute Risk
Reduction (%)
(95% CI)aRelative Risk
Reduction (%)
(95% CI)aNew vertebral fracture
through Month 248.0% 4.1% 4.0 (2.5, 5.6) 50 (34, 62) <0.001 N = Number of subjects randomized
a. Absolute and relative risk reductions are based on the Mantel-Haenszel method adjusting for age strata, baseline total hip BMD T-score (≤ -2.5, > -2.5), and presence of severe vertebral fracture at baseline.
b. P-value is based on logistic regression model for new vertebral fracture) adjusting for age strata, baseline total hip BMD T-score, and presence of severe vertebral fracture at baseline.EVENITY significantly reduced the risk of clinical fracture through the end of the primary analysis period (see Table 4). This was an event-driven trial and the duration of follow-up varied across subjects. The median duration of subject follow-up for the primary analysis period was 33 months. Subjects with nonvertebral fracture comprised 83% of the subjects with clinical fracture during the primary analysis period.
Table 4 Effect of EVENITY on the Risk of Clinical Fractures in Study 2 Proportion of Women with
Fracture (%)aHazard Ratio (95% CI)c p-valuec Alendronate Alone
(N = 2047)EVENITY Followed
by Alendronate
(N = 2046)Clinical fracture through
primary analysis periodb13.0% 9.7% 0.73 (0.61, 0.88) <0.001 N= Number of subjects randomized
a. % = number of subjects who had a clinical fracture through the primary analysis period/N*100%; the duration of follow-up varied across subjects.
b. Primary analysis period ended when clinical fracture events were confirmed for at least 330 subjects and all subjects completed the month 24 study visit. The median duration of follow-up for the primary analysis period was 33 months.
c. Hazard ratio and P-value are based on Cox proportional hazards model adjusting for age strata, baseline total hip BMD T-score, and presence of severe vertebral fracture at baseline
EVENITY followed by alendronate also significantly reduced the risk of nonvertebral fracture through the primary analysis period (with a median follow-up of 33 months), with a hazard ratio of 0.81 (95% CI: 0.66, 0.99; p = 0.04) compared to alendronate alone.
Effect on Bone Mineral Density (BMD)
EVENITY significantly increased BMD at the lumbar spine, total hip, and femoral neck compared with alendronate at month 12. The treatment differences in BMD were 8.7% at the lumbar spine, 3.3% at the total hip, and 3.2% at the femoral neck.
Twelve months of treatment with EVENITY followed by 12 months of treatment with alendronate significantly increased BMD compared with alendronate alone. The BMD increase with EVENITY over alendronate observed at month 12 was maintained at month 24. The treatment differences in BMD at month 24 were 8.1% at the lumbar spine, 3.8% at the total hip, and 3.8% at the femoral neck.
There was no evidence of differences in effects on BMD at the lumbar spine or total hip across subgroups defined by baseline age, baseline BMD, or geographic region.
-
16
HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
EVENITY (romosozumab-aqqg) injection is a clear to opalescent, colorless to light yellow solution for subcutaneous injection supplied in a single-use prefilled syringe.
Each single-use prefilled syringe contains 105 mg of EVENITY in a deliverable volume of 1.17 mL. To deliver a full dose, inject two 105 mg/1.17 mL EVENITY prefilled syringes, one after the other for a total dose of 210 mg.
- NDC: 55513-880-02: Carton of two 105 mg/1.17 mL single-use prefilled syringes.
The prefilled syringe is not made with natural rubber latex.
16.2 Storage and Handling
- Refrigerate EVENITY at 2°C to 8°C (36°F to 46°F) in the original carton to protect from light. Do not freeze. Do not shake.
- If removed from the refrigerator, EVENITY can be kept at room temperature up to 25°C (77°F) in the original carton and must be used within 30 days. If not used within 30 days, discard EVENITY.
- Do not expose EVENITY to temperatures above 25°C (77°F).
-
17
PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Major Adverse Cardiac Events
Advise patients to seek immediate medical attention if they experience signs or symptoms of a myocardial infarction or stroke [see Boxed Warning, Warnings and Precautions (5.1) and Adverse Reactions (6.1)].
Hypersensitivity Reactions
Advise patients to seek immediate medical attention if they experience signs or symptoms of a hypersensitivity reaction including angioedema, erythema multiforme, dermatitis, rash, and urticaria [see Warnings and Precautions (5.2) and Adverse Reactions (6.1)].
Calcium and Vitamin D Supplements to Prevent Hypocalcemia
Advise patients to take calcium and vitamin D supplements daily to reduce the risk of hypocalcemia. Advise patients to seek immediate medical attention for symptoms of hypocalcemia [see Dosage and Administration (2.1), Warnings and Precautions (5.3) and Adverse Reactions (6.1)].
Osteonecrosis of the Jaw
Advise patients to practice good oral hygiene during treatment with EVENITY and tell their dentist that they are receiving EVENITY before having dental work. [see Warnings and Precautions (5.4) and Adverse Reactions (6.1)].
Atypical Femoral Fracture
Advise patients to report signs and symptoms that could be consistent with impending atypical femoral fracture including new or unusual thigh, hip, or groin pain [see Warnings and Precautions (5.5) and Adverse Reactions (6.1)].
EVENITY™ (romosozumab-aqqg)
Manufactured by:
Amgen Inc.
One Amgen Center Drive
Thousand Oaks, California 91320-1799
US License No. 1080
Patent: http://pat.amgen.com/EVENITY/
© 2019 Amgen Inc. All rights reserved.
1xxxxxx- v2
-
MEDICATION GUIDE
MEDICATION GUIDE
EVENITY™ (E-ven-i-tee)
(romosozumab-aqqg)
injection, for subcutaneous useWhat is the most important information I should know about EVENITY?
EVENITY can cause serious side effects, including:
- increased risk of having a heart attack, stroke, or death from a cardiovascular (heart or blood vessel) problem. Call your healthcare provider or get emergency help right away if you have any of the symptoms listed below.
○ chest pain or pressure
○ shortness of breath
○ feeling light-headed or dizzy
○ Symptoms of stroke may include:
○ headache
○ numbness or weakness in face, arm, or legs
○ difficulty talking
○ changes in vision or loss of balance
Before you receive EVENITY, tell your healthcare provider if you have had a heart attack or stroke, especially if it has happened in the past year.
See “What are the possible side effects of EVENITY?” below for other side effects of EVENITY.What is EVENITY?
EVENITY is a prescription medicine used to:
- treat osteoporosis (thinning and weakening of bone) in women after menopause (“change of life”) who:
- are at high risk of fracture (broken bone), or
- cannot use another osteoporosis medicine or other osteoporosis medicines did not work well.
- are at high risk of fracture (broken bone), or
Do not receive EVENITY if you: - have been told by your healthcare provider that your blood calcium level is too low.
- are allergic to romosozumab or any of the ingredients in EVENITY. See the end of this Medication Guide for a complete list of ingredients in EVENITY.
Before receiving EVENITY, tell your healthcare provider about all your medical conditions, including if you:
- have a history of other heart or blood vessel problems
- have low blood calcium
- cannot take daily calcium and vitamin D
- have kidney problems or are on kidney dialysis
- plan to have dental surgery or teeth removed
How will I receive EVENITY? - EVENITY is an injection that will be given to you by your healthcare provider. EVENITY is injected under your skin (subcutaneous).
- You will receive an EVENITY dose (2 injections) 1 time every month for 12 doses.
- You should take calcium and vitamin D while you receive EVENITY.
- If you miss a dose of EVENITY, contact your healthcare provider as soon as possible to schedule your next dose. Your next dose of EVENITY should then be scheduled every month from the date of the last injection.
- You should take good care of your teeth and gums while you receive EVENITY. You should tell your dentist that you are receiving EVENITY before you have dental work.
What are the possible side effects of EVENITY?
EVENITY may cause serious side effects, including:
See “What is the most important information I should know about EVENITY?”
-
Serious allergic reactions. Serious allergic reactions have happened in people who receive EVENITY. Call your healthcare provider or go to the nearest emergency room right away if you have any symptoms of a serious allergic reaction. Symptoms of a serious allergic reaction may include:
○ rash
○ hives
○ swelling of the face, lips, mouth, tongue, or throat which may cause difficulty in swallowing or breathing
-
Low calcium levels in your blood (hypocalcemia). EVENITY may lower the calcium levels in your blood. Your low blood calcium should be treated before you receive EVENITY. Call your healthcare provider if you have symptoms of low blood calcium such as:
○ spasms, twitches, or cramps in your muscles
○ numbness or tingling in your fingers, toes or around your mouth
-
Severe jaw bone problems (osteonecrosis). Severe jaw bone problems may happen when you take EVENITY. Your healthcare provider should examine your mouth before you start EVENITY.
○ Your healthcare provider may tell you to see your dentist before you start EVENITY. Ask your healthcare provider or dentist about good mouth care.
-
Unusual thigh bone fractures. Symptoms of this type of fracture include:
○ new or unusual pain in your hip, groin, or thigh.
joint pain headache These are not all the possible side effects of EVENITY.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.How should I store EVENITY if I need to pick it up from a pharmacy?
- Keep EVENITY in a refrigerator at 36°F to 46°F (2°C to 8°C) in the original carton to protect it from light.
- Do not freeze EVENITY.
- Do not shake EVENITY.
- When you remove EVENITY from the refrigerator, EVENITY must be kept at room temperature up to 77°F (25°C) in the original carton and must be used within 30 days. EVENITY should be thrown away if taken out of the refrigerator and not used within 30 days.
- Do not keep EVENITY at temperatures above 77°F (25°C). Warm temperatures will affect how EVENITY works.
General information about EVENITY
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. You can ask your pharmacist or healthcare provider for information about EVENITY that is written for health professionals.What are the ingredients in EVENITY?
Active ingredient: romosozumab-aqqg
Inactive ingredients: acetate, calcium, polysorbate 20, sodium hydroxide, sucrose and water for injection.Manufactured by: Amgen Inc., One Amgen Center Drive, Thousand Oaks, CA 91320-1799
U.S. License No. 1080 © 2019 Amgen Inc. All rights reserved. 1xxxxxx – v1
For more information, go to www.evenity.com or call Amgen at 1-800-772-6436.This Medication Guide has been approved by the U.S. Food and Drug Administration. Revised: 04/2019
-
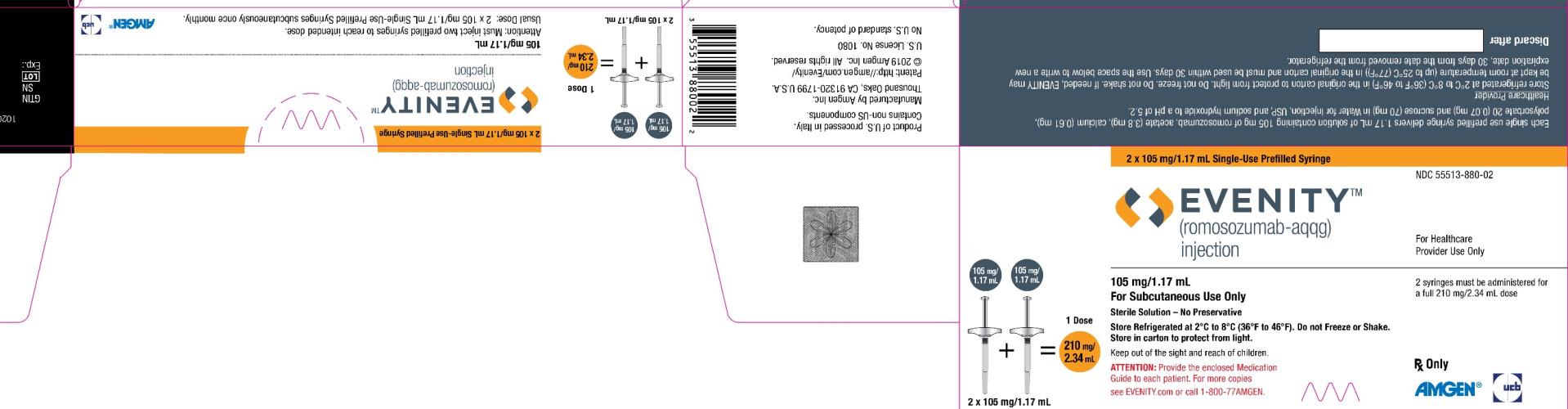
PRINCIPAL DISPLAY PANEL
PRINCIPAL DISPLAY PANEL
2 x 105 mg/1.17 mL Singe-Use Prefilled Syringe
NDC: 55513-880-02
EVENITY™
(romosozumab-aqqg)
injection
For Healthcare
Provider Use Only
105 mg/1.17 mL + 105 mg/1.17 mL = 1 Dose
210 mg/2.34 mL
2 x 105 mg/1.17 mL
105 mg/1.17 mL
For Subcutaneous Use Only
Sterile Solution – No Preservative
Store Refrigerated at 2°C to 8°C (36°F to 46°F). Do not Freeze or Shake.
Store in carton to protect from light.
Keep out of the sight and reach of children.
ATTENTION: Provide the enclose Medication
Guide to each patient. For more copies
see EVENITY.com or call 1-800-77AMGEN.
2 syringes must be administered for
a full 210 mg/2.34 mL dose
Rx Only
Amgen® UCB
-
INGREDIENTS AND APPEARANCE
EVENITY
romosozumab-aqqg injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 55513-880 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ROMOSOZUMAB (UNII: 3VHF2ZD92J) (ROMOSOZUMAB - UNII:3VHF2ZD92J) ROMOSOZUMAB 105 mg in 1.17 mL Inactive Ingredients Ingredient Name Strength ACETATE ION (UNII: 569DQM74SC) 3.8 mg in 1.17 mL CALCIUM (UNII: SY7Q814VUP) 0.61 mg in 1.17 mL POLYSORBATE 20 (UNII: 7T1F30V5YH) 0.07 mg in 1.17 mL SUCROSE (UNII: C151H8M554) 70 mg in 1.17 mL SODIUM HYDROXIDE (UNII: 55X04QC32I) WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 55513-880-02 2 in 1 CARTON 04/09/2019 1 NDC: 55513-880-01 1.17 mL in 1 SYRINGE; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761062 04/09/2019 Labeler - Amgen Inc (039976196)
Trademark Results [Evenity]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 EVENITY 88685775 not registered Live/Pending |
Amgen Inc. 2019-11-08 |
 EVENITY 87108839 not registered Dead/Abandoned |
Amgen Inc. 2016-07-19 |
 EVENITY 86670587 5781885 Live/Registered |
Amgen Inc. 2015-06-22 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.