CARVEDILOL tablet, film coated
Carvedilol by
Drug Labeling and Warnings
Carvedilol by is a Prescription medication manufactured, distributed, or labeled by Aurolife Pharma LLC. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use carvedilol safely and effectively. See full prescribing information for carvedilol tablets, USP.
Carvedilol Tablets, USP
Initial U.S. Approval: 1995
RECENT MAJOR CHANGES
INDICATIONS AND USAGE
DOSAGE AND ADMINISTRATION
Take with food. Individualize dosage and monitor during up-titration. (2)
- Left ventricular dysfunction following myocardial infarction: Start at 6.25 mg twice daily and increase to 12.5 mg then 25 mg twice daily after intervals of 3 to 10 days. A lower starting dose or slower titration may be used. (2.2)
- Hypertension: Start at 6.25 mg twice daily and increase if needed for blood pressure control to 12.5 mg then 25 mg twice daily over intervals of 1 to 2 weeks. (2.3)
DOSAGE FORMS AND STRENGTHS
Tablets: 3.125 mg, 6.25 mg, 12.5 mg, or 25 mg (3)
CONTRAINDICATIONS
- Bronchial asthma or related bronchospastic conditions (4)
- Second- or third-degree AV block (4)
- Sick sinus syndrome (4)
- Severe bradycardia (unless permanent pacemaker in place) (4)
- Patients in cardiogenic shock or decompensated heart failure requiring the use of IV inotropic therapy. (4)
- Severe hepatic impairment (2.4, 4)
- History of serious hypersensitivity reaction (e.g., Stevens-Johnson syndrome, anaphylactic reaction, angioedema) to any component of this medication or other medications containing carvedilol. (4)
WARNINGS AND PRECAUTIONS
- Acute exacerbation of coronary artery disease upon cessation of therapy: Do not abruptly discontinue. (5.1)
- Bradycardia, hypotension, worsening heart failure/fluid retention may occur. Reduce the dose as needed. (5.2, 5.3, 5.4)
- Non-allergic bronchospasm (e.g., chronic bronchitis and emphysema): Avoid β-blockers. (4) However, if deemed necessary, use with caution and at lowest effective dose. (5.5)
- Diabetes: Monitor glucose as β-blockers may mask symptoms of hypoglycemia or worsen hyperglycemia. (5.6)
ADVERSE REACTIONS
Most common adverse events (6.1):
- Left ventricular dysfunction following myocardial infarction (≥10%): Dizziness, fatigue, hypotension, diarrhea, hyperglycemia, asthenia, bradycardia, weight increase
- Hypertension (≥5%): Dizziness
To report SUSPECTED ADVERSE REACTIONS, contact Aurobindo Pharma USA, Inc. at 1-866-850-2876 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.DRUG INTERACTIONS
- CYP P450 2D6 enzyme inhibitors may increase and rifampin may decrease carvedilol levels. (7.1, 7.5)
- Hypotensive agents (e.g., reserpine, MAO inhibitors, clonidine) may increase the risk of hypotension and/or severe bradycardia. (7.2)
- Cyclosporine or digoxin levels may increase. (7.3, 7.4)
- Both digitalis glycosides and β-blockers slow atrioventricular conduction and decrease heart rate. Concomitant use can increase the risk of bradycardia. (7.4)
- Amiodarone may increase carvedilol levels resulting in further slowing of the heart rate or cardiac conduction. (7.6)
- Verapamil- or diltiazem-type calcium channel blockers may affect ECG and/or blood pressure. (7.7)
- Insulin and oral hypoglycemics action may be enhanced. (7.8)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 6/2011
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.2 Left Ventricular Dysfunction Following Myocardial Infarction
1.3 Hypertension
2 DOSAGE AND ADMINISTRATION
2.2 Left Ventricular Dysfunction Following Myocardial Infarction
2.3 Hypertension
2.4 Hepatic Impairment
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Cessation of Therapy
5.2 Bradycardia
5.3 Hypotension
5.4 Heart Failure/Fluid Retention
5.5 Non-allergic Bronchospasm
5.6 Glycemic Control in Type 2 Diabetes
5.7 Peripheral Vascular Disease
5.8 Deterioration of Renal Function
5.9 Major Surgery
5.10 Thyrotoxicosis
5.11 Pheochromocytoma
5.12 Prinzmetal’s Variant Angina
5.13 Risk of Anaphylactic Reaction
5.14 Intraoperative Floppy Iris Syndrome
6 ADVERSE REACTIONS
6.1 Clinical Studies Experience
6.2 Laboratory Abnormalities
6.3 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 CYP2D6 Inhibitors and Poor Metabolizers
7.2 Hypotensive Agents
7.3 Cyclosporine
7.4 Digitalis Glycosides
7.5 Inducers/Inhibitors of Hepatic Metabolism
7.6 Amiodarone
7.7 Calcium Channel Blockers
7.8 Insulin or Oral Hypoglycemics
7.9 Anesthesia
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.3 Nursing Mothers
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.4 Specific Populations
12.5 Drug-Drug Interactions
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.2 Left Ventricular Dysfunction Following Myocardial Infarction
14.3 Hypertension
14.4 Hypertension With Type 2 Diabetes Mellitus
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
17.1 Patient Advice
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
1.2 Left Ventricular Dysfunction Following Myocardial Infarction
Carvedilol tablets, USP are indicated to reduce cardiovascular mortality in clinically stable patients who have survived the acute phase of a myocardial infarction and have a left ventricular ejection fraction of ≤40% (with or without symptomatic heart failure) [see Clinical Studies (14.2)].1.3 Hypertension
Carvedilol tablets, USP are indicated for the management of essential hypertension [see Clinical Studies (14.3, 14.4)]. It can be used alone or in combination with other antihypertensive agents, especially thiazide-type diuretics [see Drug Interactions (7.2)]. -
2 DOSAGE AND ADMINISTRATION
Carvedilol tablets should be taken with food to slow the rate of absorption and reduce the incidence of orthostatic effects.2.2 Left Ventricular Dysfunction Following Myocardial Infarction
DOSAGE MUST BE INDIVIDUALIZED AND MONITORED DURING UP-TITRATION. Treatment with carvedilol tablets may be started as an inpatient or outpatient and should be started after the patient is hemodynamically stable and fluid retention has been minimized. It is recommended that carvedilol tablets be started at 6.25 mg twice daily and increased after 3 to 10 days, based on tolerability, to 12.5 mg twice daily, then again to the target dose of 25 mg twice daily. A lower starting dose may be used (3.125 mg twice daily) and/or the rate of up-titration may be slowed if clinically indicated (e.g., due to low blood pressure or heart rate, or fluid retention). Patients should be maintained on lower doses if higher doses are not tolerated. The recommended dosing regimen need not be altered in patients who received treatment with an IV or oral β-blocker during the acute phase of the myocardial infarction.2.3 Hypertension
DOSAGE MUST BE INDIVIDUALIZED. The recommended starting dose of carvedilol tablets is 6.25 mg twice daily. If this dose is tolerated, using standing systolic pressure measured about 1 hour after dosing as a guide, the dose should be maintained for 7 to 14 days, and then increased to 12.5 mg twice daily if needed, based on trough blood pressure, again using standing systolic pressure one hour after dosing as a guide for tolerance. This dose should also be maintained for 7 to 14 days and can then be adjusted upward to 25 mg twice daily if tolerated and needed. The full antihypertensive effect of carvedilol tablets is seen within 7 to 14 days. Total daily dose should not exceed 50 mg.
Concomitant administration with a diuretic can be expected to produce additive effects and exaggerate the orthostatic component of carvedilol action.2.4 Hepatic Impairment
Carvedilol tablets should not be given to patients with severe hepatic impairment [see Contraindications (4)]. -
3 DOSAGE FORMS AND STRENGTHS
Carvedilol tablets 3.125 mg are white to off-white, oval shaped, film-coated tablets debossed with ‘E’ on one side and ‘01’ on the other side. The 6.25 mg are white to off-white, oval shaped, film-coated tablets debossed with ‘E’ on one side and ‘02’ on the other side. The 12.5 mg are white to off-white, oval shaped, film-coated tablets debossed with ‘E’ on one side and ‘03’ on the other side. The 25 mg are white to off-white, oval shaped, film-coated tablets debossed with ‘E’ on one side and ‘04’ on the other side. -
4 CONTRAINDICATIONS
Carvedilol tablets are contraindicated in the following conditions:
- Bronchial asthma or related bronchospastic conditions. Deaths from status asthmaticus have been reported following single doses of carvedilol tablets.
- Second- or third-degree AV block
- Sick sinus syndrome
- Severe bradycardia (unless a permanent pacemaker is in place)
- Patients with cardiogenic shock or who have decompensated heart failure requiring the use of intravenous inotropic therapy. Such patients should first be weaned from intravenous therapy before initiating carvedilol tablets.
- Patients with severe hepatic impairment
- Patients with a history of a serious hypersensitivity reaction (e.g., Stevens-Johnson syndrome, anaphylactic reaction, angioedema) to any component of this medication or other medications containing carvedilol.
-
5 WARNINGS AND PRECAUTIONS
5.1 Cessation of Therapy
Patients with coronary artery disease, who are being treated with carvedilol, should be advised against abrupt discontinuation of therapy. Severe exacerbation of angina and the occurrence of myocardial infarction and ventricular arrhythmias have been reported in angina patients following the abrupt discontinuation of therapy with β-blockers. The last 2 complications may occur with or without preceding exacerbation of the angina pectoris. As with other β-blockers, when discontinuation of carvedilol is planned, the patients should be carefully observed and advised to limit physical activity to a minimum. Carvedilol should be discontinued over 1 to 2 weeks whenever possible. If the angina worsens or acute coronary insufficiency develops, it is recommended that carvedilol be promptly reinstituted, at least temporarily. Because coronary artery disease is common and may be unrecognized, it may be prudent not to discontinue therapy with carvedilol abruptly even in patients treated only for hypertension or heart failure.5.2 Bradycardia
In clinical trials, carvedilol caused bradycardia in about 2% of hypertensive patients, and 6.5% of myocardial infarction patients with left ventricular dysfunction. If pulse rate drops below 55 beats/minute, the dosage should be reduced.5.3 Hypotension
Postural hypotension occurred in 1.8% and syncope in 0.1% of hypertensive patients, primarily following the initial dose or at the time of dose increase and was a cause for discontinuation of therapy in 1% of patients.
In the CAPRICORN study of survivors of an acute myocardial infarction, hypotension or postural hypotension occurred in 20.2% of patients receiving carvedilol compared to 12.6% of placebo patients. Syncope was reported in 3.9% and 1.9% of patients, respectively. These events were a cause for discontinuation of therapy in 2.5% of patients receiving carvedilol, compared to 0.2% of placebo patients.
Starting with a low dose, administration with food, and gradual up-titration should decrease the likelihood of syncope or excessive hypotension [see Dosage and Administration (2.2, 2.3)]. During initiation of therapy, the patient should be cautioned to avoid situations such as driving or hazardous tasks, where injury could result should syncope occur.5.4 Heart Failure/Fluid Retention
Worsening heart failure or fluid retention may occur during up-titration of carvedilol. If such symptoms occur, diuretics should be increased and the carvedilol dose should not be advanced until clinical stability resumes [see Dosage and Administration (2)]. Occasionally it is necessary to lower the carvedilol dose or temporarily discontinue it. Such episodes do not preclude subsequent successful titration of, or a favorable response to, carvedilol.5.5 Non-allergic Bronchospasm
Patients with bronchospastic disease (e.g., chronic bronchitis and emphysema) should, in general, not receive β-blockers. Carvedilol may be used with caution, however, in patients who do not respond to, or cannot tolerate, other antihypertensive agents. It is prudent, if carvedilol is used, to use the smallest effective dose, so that inhibition of endogenous or exogenous β-agonists is minimized.
In clinical trials of patients with bronchospastic disease were enrolled if they did not require oral or inhaled medication to treat their bronchospastic disease. In such patients, it is recommended that carvedilol be used with caution. The dosing recommendations should be followed closely and the dose should be lowered if any evidence of bronchospasm is observed during up-titration.5.6 Glycemic Control in Type 2 Diabetes
In general, β-blockers may mask some of the manifestations of hypoglycemia, particularly tachycardia. Nonselective β-blockers may potentiate insulin-induced hypoglycemia and delay recovery of serum glucose levels. Patients subject to spontaneous hypoglycemia, or diabetic patients receiving insulin or oral hypoglycemic agents, should be cautioned about these possibilities.
Studies designed to examine the effects of carvedilol on glycemic control in patients with diabetes and heart failure have not been conducted.
In a study designed to examine the effects of carvedilol on glycemic control in a population with mild-to-moderate hypertension and well-controlled type 2 diabetes mellitus, carvedilol had no adverse effect on glycemic control, based on HbA1c measurements [see Clinical Studies (14.4)].5.7 Peripheral Vascular Disease
β-blockers can precipitate or aggravate symptoms of arterial insufficiency in patients with peripheral vascular disease. Caution should be exercised in such individuals.5.8 Deterioration of Renal Function
Rarely, use of carvedilol in patients with heart failure has resulted in deterioration of renal function. Patients at risk appear to be those with low blood pressure (systolic blood pressure <100 mm Hg), ischemic heart disease and diffuse vascular disease, and/or underlying renal insufficiency. Renal function has returned to baseline when carvedilol was stopped. In patients with these risk factors it is recommended that renal function be monitored during up-titration of carvedilol and the drug discontinued or dosage reduced if worsening of renal function occurs.5.9 Major Surgery
Chronically administered beta-blocking therapy should not be routinely withdrawn prior to major surgery; however, the impaired ability of the heart to respond to reflex adrenergic stimuli may augment the risks of general anesthesia and surgical procedures.5.10 Thyrotoxicosis
β-adrenergic blockade may mask clinical signs of hyperthyroidism, such as tachycardia. Abrupt withdrawal of β-blockade may be followed by an exacerbation of the symptoms of hyperthyroidism or may precipitate thyroid storm.5.11 Pheochromocytoma
In patients with pheochromocytoma, an α-blocking agent should be initiated prior to the use of any β-blocking agent. Although carvedilol has both α- and β-blocking pharmacologic activities, there has been no experience with its use in this condition. Therefore, caution should be taken in the administration of carvedilol to patients suspected of having pheochromocytoma.5.12 Prinzmetal’s Variant Angina
Agents with non-selective β-blocking activity may provoke chest pain in patients with Prinzmetal’s variant angina. There has been no clinical experience with carvedilol in these patients although the α-blocking activity may prevent such symptoms. However, caution should be taken in the administration of carvedilol to patients suspected of having Prinzmetal’s variant angina.5.13 Risk of Anaphylactic Reaction
While taking β-blockers, patients with a history of severe anaphylactic reaction to a variety of allergens may be more reactive to repeated challenge, either accidental, diagnostic, or therapeutic. Such patients may be unresponsive to the usual doses of epinephrine used to treat allergic reaction.5.14 Intraoperative Floppy Iris Syndrome
Intraoperative Floppy Iris Syndrome (IFIS) has been observed during cataract surgery in some patients treated with alpha-1 blockers (carvedilol is an alpha/beta blocker). This variant of small pupil syndrome is characterized by the combination of a flaccid iris that billows in response to intraoperative irrigation currents, progressive intraoperative miosis despite preoperative dilation with standard mydriatic drugs, and potential prolapse of the iris toward the phacoemulsification incisions. The patient’s ophthalmologist should be prepared for possible modifications to the surgical technique, such as utilization of iris hooks, iris dilator rings, or viscoelastic substances. There does not appear to be a benefit of stopping alpha-1 blocker therapy prior to cataract surgery. -
6 ADVERSE REACTIONS
6.1 Clinical Studies Experience
Carvedilol has been evaluated for safety in patients with left ventricular dysfunction following myocardial infarction and in hypertensive patients. The observed adverse event profile was consistent with the pharmacology of the drug and the health status of the patients in the clinical trials. Adverse events reported for each of these patient populations are provided below. Excluded are adverse events considered too general to be informative, and those not reasonably associated with the use of the drug because they were associated with the condition being treated or are very common in the treated population. Rates of adverse events were generally similar across demographic subsets (men and women, elderly and non-elderly, blacks and non-blacks).
Left Ventricular Dysfunction Following Myocardial Infarction: Carvedilol has been evaluated for safety in survivors of an acute myocardial infarction with left ventricular dysfunction in the CAPRICORN trial which involved 969 patients who received carvedilol and 980 who received placebo. Approximately 75% of the patients received carvedilol for at least 6 months and 53% received carvedilol for at least 12 months. Patients were treated for an average of 12.9 months and 12.8 months with carvedilol and placebo, respectively.
The following adverse events were reported with a frequency of >1% but ≤3% and more frequently with carvedilol: Flu syndrome, cerebrovascular accident, peripheral vascular disorder, hypotonia, depression, gastrointestinal pain, arthritis, and gout. The overall rates of discontinuations due to adverse events were similar in both groups of patients. In this database, the only cause of discontinuation >1%, and occurring more often on carvedilol was hypotension (1.5% on carvedilol, 0.2% on placebo).
Hypertension: Carvedilol has been evaluated for safety in hypertension in more than 2,193 patients in U.S. clinical trials and in 2,976 patients in international clinical trials. Approximately 36% of the total treated population received carvedilol for at least 6 months. Most adverse events reported during therapy with carvedilol were of mild to moderate severity. In U.S. controlled clinical trials directly comparing carvedilol in doses up to 50 mg (n = 1,142) to placebo (n = 462), 4.9% of patients receiving carvedilol discontinued for adverse events versus 5.2% of placebo patients. Although there was no overall difference in discontinuation rates, discontinuations were more common in the carvedilol group for postural hypotension (1% versus 0). The overall incidence of adverse events in U.S. placebo-controlled trials increased with increasing dose of carvedilol. For individual adverse events this could only be distinguished for dizziness, which increased in frequency from 2% to 5% as total daily dose increased from 6.25 mg to 50 mg.
Table 1 shows adverse events in U.S. placebo-controlled clinical trials for hypertension that occurred with an incidence of ≥1% regardless of causality, and that were more frequent in drug-treated patients than placebo-treated patients.
Table 1. Adverse Events (%) Occurring in U.S. Placebo-Controlled Hypertension Trials (Incidence ≥1%, Regardless of Causality)* Carvedilol Placebo (n = 1,142) (n = 462) *Shown are events with rate >1% rounded to nearest integer.
Cardiovascular
Bradycardia
2
—
Postural hypotension
2
—
Peripheral edema
1
—
Central Nervous System
Dizziness
6
5
Insomnia
2
1
Gastrointestinal
Diarrhea
2
1
Hematologic
Thrombocytopenia
1
—
Metabolic
Hypertriglyceridemia
1
—
Dyspnea and fatigue were also reported in these studies, but the rates were equal or greater in patients who received placebo.
The following adverse events not described above were reported as possibly or probably related to carvedilol in worldwide open or controlled trials with carvedilol in patients with hypertension.
Incidence >0.1% to ≤1%
Cardiovascular: Peripheral ischemia, tachycardia.
Central and Peripheral Nervous System: Hypokinesia.
Gastrointestinal: Bilirubinemia, increased hepatic enzymes (0.2% of hypertension patients were discontinued from therapy because of increases in hepatic enzymes) [see Adverse Reactions (6.2)].
Psychiatric: Nervousness, sleep disorder, aggravated depression, impaired concentration, abnormal thinking, paroniria, emotional lability.
Respiratory System: Asthma [see Contraindications (4)].
Reproductive, male: Decreased libido.
Skin and Appendages: Pruritus, rash erythematous, rash maculopapular, rash psoriaform, photosensitivity reaction.
Special Senses: Tinnitus.
Urinary System: Micturition frequency increased.
Autonomic Nervous System: Dry mouth, sweating increased.
Metabolic and Nutritional: Hypokalemia, hypertriglyceridemia.
Hematologic: Anemia, leukopenia.
The following events were reported in ≤0.1% of patients and are potentially important: Complete AV block, bundle branch block, myocardial ischemia, cerebrovascular disorder, convulsions, migraine, neuralgia, paresis, anaphylactoid reaction, alopecia, exfoliative dermatitis, amnesia, GI hemorrhage, bronchospasm, pulmonary edema, decreased hearing, respiratory alkalosis, increased BUN, decreased HDL, pancytopenia, and atypical lymphocytes.6.2 Laboratory Abnormalities
Reversible elevations in serum transaminases (ALT or AST) have been observed during treatment with carvedilol. Rates of transaminase elevations (2- to 3-times the upper limit of normal) observed during controlled clinical trials have generally been similar between patients treated with carvedilol and those treated with placebo. However, transaminase elevations, confirmed by rechallenge, have been observed with carvedilol. In a long-term, placebo-controlled trial in severe heart failure, patients treated with carvedilol had lower values for hepatic transaminases than patients treated with placebo, possibly because improvements in cardiac function induced by carvedilol led to less hepatic congestion and/or improved hepatic blood flow.
Carvedilol has not been associated with clinically significant changes in serum potassium, total triglycerides, total cholesterol, HDL cholesterol, uric acid, blood urea nitrogen, or creatinine. No clinically relevant changes were noted in fasting serum glucose in hypertensive patients.6.3 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of carvedilol. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Reports of aplastic anemia and severe skin reactions (Stevens-Johnson syndrome, toxic epidermal necrolysis, and erythema multiforme) have been rare and received only when carvedilol was administered concomitantly with other medications associated with such reactions. Rare reports of hypersensitivity reactions (e.g., anaphylactic reaction, angioedema, and urticaria) have been received for carvedilol tablets and carvedilol phosphate extended-release capsules, including cases occurring after the initiation of carvedilol phosphate extended-release capsules in patients previously treated with carvedilol. Urinary incontinence in women (which resolved upon discontinuation of the medication) and interstitial pneumonitis have been reported rarely. -
7 DRUG INTERACTIONS
7.1 CYP2D6 Inhibitors and Poor Metabolizers
Interactions of carvedilol with potent inhibitors of CYP2D6 isoenzyme (such as quinidine, fluoxetine, paroxetine, and propafenone) have not been studied, but these drugs would be expected to increase blood levels of the R(+) enantiomer of carvedilol [see Clinical Pharmacology (12.3)]. Retrospective analysis of side effects in clinical trials showed that poor 2D6 metabolizers had a higher rate of dizziness during up-titration, presumably resulting from vasodilating effects of the higher concentrations of the α-blocking R(+) enantiomer.7.2 Hypotensive Agents
Patients taking both agents with β-blocking properties and a drug that can deplete catecholamines (e.g., reserpine and monoamine oxidase inhibitors) should be observed closely for signs of hypotension and/or severe bradycardia.
Concomitant administration of clonidine with agents with β-blocking properties may potentiate blood-pressure- and heart-rate-lowering effects. When concomitant treatment with agents with β-blocking properties and clonidine is to be terminated, the β-blocking agent should be discontinued first. Clonidine therapy can then be discontinued several days later by gradually decreasing the dosage.7.3 Cyclosporine
Modest increases in mean trough cyclosporine concentrations were observed following initiation of carvedilol treatment in 21 renal transplant patients suffering from chronic vascular rejection. In about 30% of patients, the dose of cyclosporine had to be reduced in order to maintain cyclosporine concentrations within the therapeutic range, while in the remainder no adjustment was needed. On the average for the group, the dose of cyclosporine was reduced about 20% in these patients. Due to wide interindividual variability in the dose adjustment required, it is recommended that cyclosporine concentrations be monitored closely after initiation of carvedilol therapy and that the dose of cyclosporine be adjusted as appropriate.7.4 Digitalis Glycosides
Both digitalis glycosides and β-blockers slow atrioventricular conduction and decrease heart rate. Concomitant use can increase the risk of bradycardia. Digoxin concentrations are increased by about 15% when digoxin and carvedilol are administered concomitantly. Therefore, increased monitoring of digoxin is recommended when initiating, adjusting, or discontinuing carvedilol [see Clinical Pharmacology (12.5)].7.5 Inducers/Inhibitors of Hepatic Metabolism
Rifampin reduced plasma concentrations of carvedilol by about 70% [see Clinical Pharmacology (12.5)]. Cimetidine increased AUC by about 30% but caused no change in Cmax [see Clinical Pharmacology (12.5)].7.6 Amiodarone
Amiodarone, and its metabolite desethyl amiodarone, inhibitors of CYP2C9 and P-glycoprotein, increased concentrations of the S(-)-enantiomer of carvedilol by at least 2-fold [see Clinical Pharmacology (12.5)]. The concomitant administration of amiodarone or other CYP2C9 inhibitors such as fluconazole with carvedilol may enhance the β-blocking properties of carvedilol resulting in further slowing of the heart rate or cardiac conduction. Patients should be observed for signs of bradycardia or heart block, particularly when one agent is added to pre-existing treatment with the other.7.7 Calcium Channel Blockers
Conduction disturbance (rarely with hemodynamic compromise) has been observed when carvedilol is coadministered with diltiazem. As with other agents with β-blocking properties, if carvedilol is to be administered with calcium channel blockers of the verapamil or diltiazem type, it is recommended that ECG and blood pressure be monitored.7.8 Insulin or Oral Hypoglycemics
Agents with β-blocking properties may enhance the blood-sugar-reducing effect of insulin and oral hypoglycemics. Therefore, in patients taking insulin or oral hypoglycemics, regular monitoring of blood glucose is recommended [see Warnings and Precautions (5.6)].7.9 Anesthesia
If treatment with carvedilol is to be continued perioperatively, particular care should be taken when anesthetic agents which depress myocardial function, such as ether, cyclopropane, and trichloroethylene, are used [see Overdosage (10)]. -
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Category C. Studies performed in pregnant rats and rabbits given carvedilol revealed increased post-implantation loss in rats at doses of 300 mg/kg/day (50 times the maximum recommended human dose [MRHD] as mg/m2) and in rabbits at doses of 75 mg/kg/day (25 times the MRHD as mg/m2). In the rats, there was also a decrease in fetal body weight at the maternally toxic dose of 300 mg/kg/day (50 times the MRHD as mg/m2), which was accompanied by an elevation in the frequency of fetuses with delayed skeletal development (missing or stunted 13th rib). In rats the no-observed-effect level for developmental toxicity was 60 mg/kg/day (10 times the MRHD as mg/m2); in rabbits it was 15 mg/kg/day (5 times the MRHD as mg/m2). There are no adequate and well-controlled studies in pregnant women. Carvedilol should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.8.3 Nursing Mothers
It is not known whether this drug is excreted in human milk. Studies in rats have shown that carvedilol and/or its metabolites (as well as other β-blockers) cross the placental barrier and are excreted in breast milk. There was increased mortality at one week post-partum in neonates from rats treated with 60 mg/kg/day (10 times the MRHD as mg/m2) and above during the last trimester through day 22 of lactation. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from β-blockers, especially bradycardia, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother. The effects of other α- and β-blocking agents have included perinatal and neonatal distress.8.4 Pediatric Use
Effectiveness of carvedilol in patients younger than 18 years of age has not been established.
In a double-blind trial, 161 children (mean age 6 years, range 2 months to 17 years; 45% less than 2 years old) with chronic heart failure [NYHA class II to IV, left ventricular ejection fraction <40% for children with a systemic left ventricle (LV), and moderate-severe ventricular dysfunction qualitatively by echo for those with a systemic ventricle that was not an LV] who were receiving standard background treatment were randomized to placebo or to 2 dose levels of carvedilol. These dose levels produced placebo-corrected heart rate reduction of 4 to 6 heart beats per minute, indicative of β-blockade activity. Exposure appeared to be lower in pediatric subjects than adults. After 8 months of follow-up, there was no significant effect of treatment on clinical outcomes. Adverse reactions in this trial that occurred in greater than 10% of patients treated with carvedilol and at twice the rate of placebo-treated patients included chest pain (17% versus 6%), dizziness (13% versus 2%), and dyspnea (11% versus 0%).8.5 Geriatric Use
Of the 975 myocardial infarction patients randomized to carvedilol in the CAPRICORN trial, 48% (468) were 65 years of age or older, and 11% (111) were 75 years of age or older.
Of the 2,065 hypertensive patients in U.S. clinical trials of efficacy or safety who were treated with carvedilol, 21% (436) were 65 years of age or older. Of 3,722 patients receiving carvedilol in hypertension clinical trials conducted worldwide, 24% were 65 years of age or older.
With the exception of dizziness in hypertensive patients (incidence 8.8% in the elderly versus 6% in younger patients), no overall differences in the safety or effectiveness (see Figure 2) were observed between the older subjects and younger subjects in each of these populations. Similarly, other reported clinical experience has not identified differences in responses between the elderly and younger subjects, but greater sensitivity of some older individuals cannot be ruled out. -
10 OVERDOSAGE
Overdosage may cause severe hypotension, bradycardia, cardiac insufficiency, cardiogenic shock, and cardiac arrest. Respiratory problems, bronchospasms, vomiting, lapses of consciousness, and generalized seizures may also occur.
The patient should be placed in a supine position and, where necessary, kept under observation and treated under intensive-care conditions. Gastric lavage or pharmacologically induced emesis may be used shortly after ingestion. The following agents may be administered:
for excessive bradycardia: Atropine, 2 mg IV.
to support cardiovascular function: Glucagon, 5 to 10 mg IV rapidly over 30 seconds, followed by a continuous infusion of 5 mg/hour; sympathomimetics (dobutamine, isoprenaline, adrenaline) at doses according to body weight and effect.
If peripheral vasodilation dominates, it may be necessary to administer adrenaline or noradrenaline with continuous monitoring of circulatory conditions. For therapy-resistant bradycardia, pacemaker therapy should be performed. For bronchospasm, β-sympathomimetics (as aerosol or IV) or aminophylline IV should be given. In the event of seizures, slow IV injection of diazepam or clonazepam is recommended.
NOTE: In the event of severe intoxication where there are symptoms of shock, treatment with antidotes must be continued for a sufficiently long period of time consistent with the 7- to 10-hour half-life of carvedilol.
Cases of overdosage with carvedilol alone or in combination with other drugs have been reported. Quantities ingested in some cases exceeded 1,000 milligrams. Symptoms experienced included low blood pressure and heart rate. Standard supportive treatment was provided and individuals recovered. -
11 DESCRIPTION
Carvedilol is a nonselective β-adrenergic blocking agent with α1-blocking activity. It is (±)-1-(Carbazol-4-yloxy)-3-[[2-(o-methoxyphenoxy)ethyl]amino]-2-propanol. Carvedilol is a racemic mixture with the following structure:
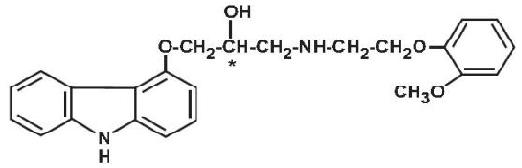
Carvedilol tablets, USP are white, oval, film-coated tablets containing 3.125 mg, 6.25 mg, 12.5 mg, or 25 mg of carvedilol. Inactive ingredients consist of lactose monohydrate, colloidal silicon dioxide, crospovidone, povidone, sucrose, magnesium stearate, polyethylene glycol 400, polysorbate 80, titanium dioxide, and hypromellose.
Carvedilol USP is a white to off-white powder with a molecular weight of 406.5 and a molecular formula of C24H26N2O4. It is freely soluble in dimethylsulfoxide; soluble in methylene chloride and methanol; sparingly soluble in 95% ethanol and isopropanol; slightly soluble in ethyl ether; and practically insoluble in water, gastric fluid (simulated, TS, pH 1.1), and intestinal fluid (simulated, TS without pancreatin, pH 7.5).
Meets USP Dissolution Test 2. -
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Carvedilol is a racemic mixture in which nonselective β-adrenoreceptor blocking activity is present in the S(-) enantiomer and α1-adrenergic blocking activity is present in both R(+) and S(-) enantiomers at equal potency. Carvedilol has no intrinsic sympathomimetic activity.12.2 Pharmacodynamics
Left Ventricular Dysfunction Following Myocardial Infarction: The basis for the beneficial effects of carvedilol in patients with left ventricular dysfunction following an acute myocardial infarction is not established.
Hypertension: The mechanism by which β-blockade produces an antihypertensive effect has not been established.
β-adrenoreceptor blocking activity has been demonstrated in animal and human studies showing that carvedilol (1) reduces cardiac output in normal subjects; (2) reduces exercise- and/or isoproterenol-induced tachycardia; and (3) reduces reflex orthostatic tachycardia. Significant β-adrenoreceptor blocking effect is usually seen within 1 hour of drug administration.
α1-adrenoreceptor blocking activity has been demonstrated in human and animal studies, showing that carvedilol (1) attenuates the pressor effects of phenylephrine; (2) causes vasodilation; and (3) reduces peripheral vascular resistance. These effects contribute to the reduction of blood pressure and usually are seen within 30 minutes of drug administration.
Due to the α1-receptor blocking activity of carvedilol, blood pressure is lowered more in the standing than in the supine position, and symptoms of postural hypotension (1.8%), including rare instances of syncope, can occur. Following oral administration, when postural hypotension has occurred, it has been transient and is uncommon when carvedilol is administered with food at the recommended starting dose and titration increments are closely followed [see Dosage and Administration (2)].
In hypertensive patients with normal renal function, therapeutic doses of carvedilol decreased renal vascular resistance with no change in glomerular filtration rate or renal plasma flow. Changes in excretion of sodium, potassium, uric acid, and phosphorus in hypertensive patients with normal renal function were similar after carvedilol and placebo.
Carvedilol has little effect on plasma catecholamines, plasma aldosterone, or electrolyte levels, but it does significantly reduce plasma renin activity when given for at least 4 weeks. It also increases levels of atrial natriuretic peptide.12.3 Pharmacokinetics
Carvedilol is rapidly and extensively absorbed following oral administration, with absolute bioavailability of approximately 25% to 35% due to a significant degree of first-pass metabolism. Following oral administration, the apparent mean terminal elimination half-life of carvedilol generally ranges from 7 to 10 hours. Plasma concentrations achieved are proportional to the oral dose administered. When administered with food, the rate of absorption is slowed, as evidenced by a delay in the time to reach peak plasma levels, with no significant difference in extent of bioavailability. Taking carvedilol with food should minimize the risk of orthostatic hypotension.
Carvedilol is extensively metabolized. Following oral administration of radiolabelled carvedilol to healthy volunteers, carvedilol accounted for only about 7% of the total radioactivity in plasma as measured by area under the curve (AUC). Less than 2% of the dose was excreted unchanged in the urine. Carvedilol is metabolized primarily by aromatic ring oxidation and glucuronidation. The oxidative metabolites are further metabolized by conjugation via glucuronidation and sulfation. The metabolites of carvedilol are excreted primarily via the bile into the feces. Demethylation and hydroxylation at the phenol ring produce 3 active metabolites with β-receptor blocking activity. Based on preclinical studies, the 4'-hydroxyphenyl metabolite is approximately 13 times more potent than carvedilol for β-blockade.
Compared to carvedilol, the 3 active metabolites exhibit weak vasodilating activity. Plasma concentrations of the active metabolites are about one-tenth of those observed for carvedilol and have pharmacokinetics similar to the parent.
Carvedilol undergoes stereoselective first-pass metabolism with plasma levels of R(+)-carvedilol approximately 2 to 3 times higher than S(-)-carvedilol following oral administration in healthy subjects. The mean apparent terminal elimination half-lives for R(+)-carvedilol range from 5 to 9 hours compared with 7 to 11 hours for the S(-)-enantiomer.
The primary P450 enzymes responsible for the metabolism of both R(+) and S(-)-carvedilol in human liver microsomes were CYP2D6 and CYP2C9 and to a lesser extent CYP3A4, 2C19, 1A2, and 2E1. CYP2D6 is thought to be the major enzyme in the 4’- and 5’-hydroxylation of carvedilol, with a potential contribution from 3A4. CYP2C9 is thought to be of primary importance in the O-methylation pathway of S(-)-carvedilol.
Carvedilol is subject to the effects of genetic polymorphism with poor metabolizers of debrisoquin (a marker for cytochrome P450 2D6) exhibiting 2- to 3-fold higher plasma concentrations of R(+)-carvedilol compared to extensive metabolizers. In contrast, plasma levels of S(-)-carvedilol are increased only about 20% to 25% in poor metabolizers, indicating this enantiomer is metabolized to a lesser extent by cytochrome P450 2D6 than R(+)-carvedilol. The pharmacokinetics of carvedilol do not appear to be different in poor metabolizers of S-mephenytoin (patients deficient in cytochrome P450 2C19).
Carvedilol is more than 98% bound to plasma proteins, primarily with albumin. The plasma-protein binding is independent of concentration over the therapeutic range. Carvedilol is a basic, lipophilic compound with a steady-state volume of distribution of approximately 115 L, indicating substantial distribution into extravascular tissues. Plasma clearance ranges from 500 to 700 mL/min.12.4 Specific Populations
Geriatric: Plasma levels of carvedilol average about 50% higher in the elderly compared to young subjects.
Hepatic Impairment: Compared to healthy subjects, patients with severe liver impairment (cirrhosis) exhibit a 4- to 7-fold increase in carvedilol levels. Carvedilol is contraindicated in patients with severe liver impairment.
Renal Impairment: Although carvedilol is metabolized primarily by the liver, plasma concentrations of carvedilol have been reported to be increased in patients with renal impairment. Based on mean AUC data, approximately 40% to 50% higher plasma concentrations of carvedilol were observed in hypertensive patients with moderate to severe renal impairment compared to a control group of hypertensive patients with normal renal function. However, the ranges of AUC values were similar for both groups. Changes in mean peak plasma levels were less pronounced, approximately 12% to 26% higher in patients with impaired renal function.
Consistent with its high degree of plasma protein-binding, carvedilol does not appear to be cleared significantly by hemodialysis.12.5 Drug-Drug Interactions
Since carvedilol undergoes substantial oxidative metabolism, the metabolism and pharmacokinetics of carvedilol may be affected by induction or inhibition of cytochrome P450 enzymes.
Amiodarone: In a pharmacokinetic study conducted in 106 Japanese patients with heart failure, coadministration of small loading and maintenance doses of amiodarone with carvedilol resulted in at least a 2-fold increase in the steady-state trough concentrations of S(-)-carvedilol [see Drug Interactions (7.6)].
Cimetidine: In a pharmacokinetic study conducted in 10 healthy male subjects, cimetidine (1,000 mg/day) increased the steady-state AUC of carvedilol by 30% with no change in Cmax [see Drug Interactions (7.5)].
Digoxin: Following concomitant administration of carvedilol (25 mg once daily) and digoxin (0.25 mg once daily) for 14 days, steady-state AUC and trough concentrations of digoxin were increased by 14% and 16%, respectively, in 12 hypertensive patients [see Drug Interactions (7.4)].
Glyburide: In 12 healthy subjects, combined administration of carvedilol (25 mg once daily) and a single dose of glyburide did not result in a clinically relevant pharmacokinetic interaction for either compound.
Hydrochlorothiazide: A single oral dose of carvedilol 25 mg did not alter the pharmacokinetics of a single oral dose of hydrochlorothiazide 25 mg in 12 patients with hypertension. Likewise, hydrochlorothiazide had no effect on the pharmacokinetics of carvedilol.
Rifampin: In a pharmacokinetic study conducted in 8 healthy male subjects, rifampin (600 mg daily for 12 days) decreased the AUC and Cmax of carvedilol by about 70% [see Drug Interactions (7.5)].
Torsemide: In a study of 12 healthy subjects, combined oral administration of carvedilol 25 mg once daily and torsemide 5 mg once daily for 5 days did not result in any significant differences in their pharmacokinetics compared with administration of the drugs alone.
Warfarin: Carvedilol (12.5 mg twice daily) did not have an effect on the steady-state prothrombin time ratios and did not alter the pharmacokinetics of R(+)- and S(-)-warfarin following concomitant administration with warfarin in 9 healthy volunteers. -
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
In 2-year studies conducted in rats given carvedilol at doses up to 75 mg/kg/day (12 times the MRHD when compared on a mg/m2 basis) or in mice given up to 200 mg/kg/day (16 times the MRHD on a mg/m2 basis), carvedilol had no carcinogenic effect.
Carvedilol was negative when tested in a battery of genotoxicity assays, including the Ames and the CHO/HGPRT assays for mutagenicity and the in vitro hamster micronucleus and in vivo human lymphocyte cell tests for clastogenicity.
At doses ≥200 mg/kg/day (≥32 times the MRHD as mg/m2) carvedilol was toxic to adult rats (sedation, reduced weight gain) and was associated with a reduced number of successful matings, prolonged mating time, significantly fewer corpora lutea and implants per dam, and complete resorption of 18% of the litters. The no-observed-effect dose level for overt toxicity and impairment of fertility was 60 mg/kg/day (10 times the MRHD as mg/m2). -
14 CLINICAL STUDIES
14.2 Left Ventricular Dysfunction Following Myocardial Infarction
CAPRICORN was a double-blind study comparing carvedilol and placebo in 1,959 patients with a recent myocardial infarction (within 21 days) and left ventricular ejection fraction of ≤40%, with (47%) or without symptoms of heart failure. Patients given carvedilol received 6.25 mg twice daily, titrated as tolerated to 25 mg twice daily. Patients had to have a systolic blood pressure >90 mm Hg, a sitting heart rate >60 beats/minute, and no contraindication to β-blocker use. Treatment of the index infarction included aspirin (85%), IV or oral β-blockers (37%), nitrates (73%), heparin (64%), thrombolytics (40%), and acute angioplasty (12%). Background treatment included ACE inhibitors or angiotensin receptor blockers (97%), anticoagulants (20%), lipid-lowering agents (23%), and diuretics (34%). Baseline population characteristics included an average age of 63 years, 74% male, 95% Caucasian, mean blood pressure 121/74 mm Hg, 22% with diabetes, and 54% with a history of hypertension. Mean dosage achieved of carvedilol was 20 mg twice daily; mean duration of follow-up was 15 months.
All-cause mortality was 15% in the placebo group and 12% in the carvedilol group, indicating a 23% risk reduction in patients treated with carvedilol (95% CI 2 to 40%, p = 0.03), as shown in Figure 1. The effects on mortality in various subgroups are shown in Figure 2. Nearly all deaths were cardiovascular (which were reduced by 25% by carvedilol), and most of these deaths were sudden or related to pump failure (both types of death were reduced by carvedilol). Another study end point, total mortality and all-cause hospitalization, did not show a significant improvement.
There was also a significant 40% reduction in fatal or non-fatal myocardial infarction observed in the group treated with carvedilol (95% CI 11% to 60%, p = 0.01). A similar reduction in the risk of myocardial infarction was also observed in a meta-analysis of placebo-controlled trials of carvedilol in heart failure.
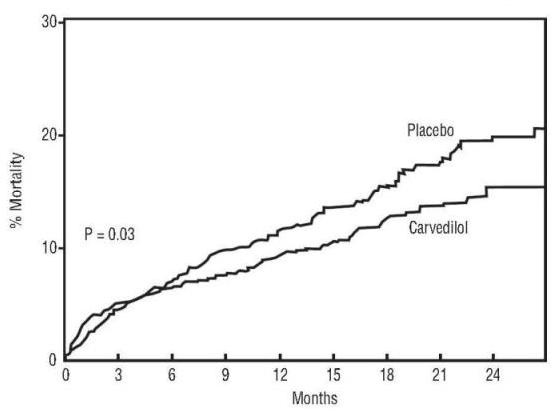
Figure 1. Survival Analysis for CAPRICORN (intent-to-treat)
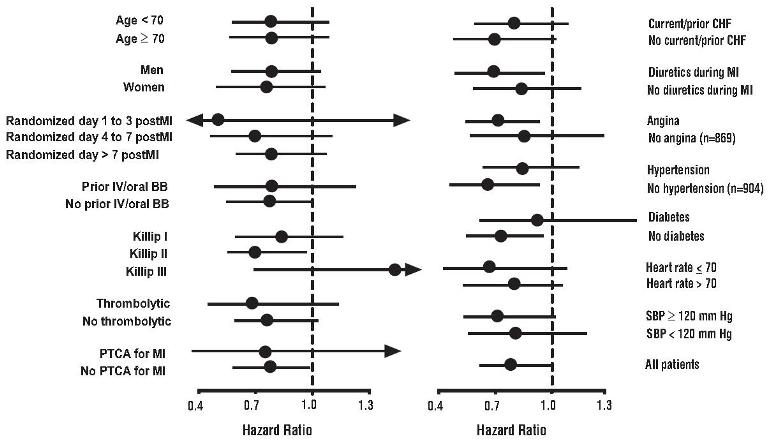
Figure 2. Effects on Mortality for Subgroups in CAPRICORN14.3 Hypertension
Carvedilol was studied in 2 placebo-controlled trials that utilized twice-daily dosing, at total daily doses of 12.5 to 50 mg. In these and other studies, the starting dose did not exceed 12.5 mg. At 50 mg/day, carvedilol reduced sitting trough (12-hour) blood pressure by about 9/5.5 mm Hg; at 25 mg/day the effect was about 7.5/3.5 mm Hg. Comparisons of trough to peak blood pressure showed a trough to peak ratio for blood pressure response of about 65%. Heart rate fell by about 7.5 beats/minute at 50 mg/day. In general, as is true for other β-blockers, responses were smaller in black than non-black patients. There were no age- or gender-related differences in response.
The peak antihypertensive effect occurred 1 to 2 hours after a dose. The dose-related blood pressure response was accompanied by a dose-related increase in adverse effects [see Adverse Reactions (6)].14.4 Hypertension With Type 2 Diabetes Mellitus
In a double-blind study (GEMINI), carvedilol, added to an ACE inhibitor or angiotensin receptor blocker, was evaluated in a population with mild-to-moderate hypertension and well-controlled type 2 diabetes mellitus. The mean HbA1c at baseline was 7.2%. Carvedilol was titrated to a mean dose of 17.5 mg twice daily and maintained for 5 months. Carvedilol had no adverse effect on glycemic control, based on HbA1c measurements (mean change from baseline of 0.02%, 95% CI -0.06 to 0.1, p = NS) [see Warnings and Precautions (5.6)].
-
16 HOW SUPPLIED/STORAGE AND HANDLING
Carvedilol Tablets USP, 3.125 mg are white to off-white, oval shaped, film-coated tablets debossed with ‘E’ on one side and ‘01’ on the other side.
Bottles of 100 NDC: 65862-142-01
Bottles of 500 NDC: 65862-142-05
Bottles of 1000 NDC: 65862-142-99
Carvedilol Tablets USP, 6.25 mg are white to off-white, oval shaped, film-coated tablets debossed with ‘E’ on one side and ‘02’ on the other side.
Bottles of 100 NDC: 65862-143-01
Bottles of 500 NDC: 65862-143-05
Bottles of 1000 NDC: 65862-143-99
Carvedilol Tablets USP, 12.5 mg are white to off-white, oval shaped, film-coated tablets debossed with ‘E’ on one side and ‘03’ on the other side.
Bottles of 100 NDC: 65862-144-01
Bottles of 500 NDC: 65862-144-05
Bottles of 1000 NDC: 65862-144-99
Carvedilol Tablets USP, 25 mg are white to off-white, oval shaped, film-coated tablets debossed with ‘E’ on one side and ‘04’ on the other side.
Bottles of 100 NDC: 65862-145-01
Bottles of 500 NDC: 65862-145-05
Bottles of 1000 NDC: 65862-145-99
Store at 20 to 25°C (68 to 77°F) [See USP Controlled Room Temperature]. Protect from moisture. Dispense in a tight, light-resistant container. -
17 PATIENT COUNSELING INFORMATION
See FDA-Approved Patient Labeling (17.2).17.1 Patient Advice
Patients taking carvedilol should be advised of the following:
- Patients should take carvedilol with food.
- Patients should not interrupt or discontinue using carvedilol without a physician’s advice.
- Patients should consult their physician if they experience signs or symptoms of worsening heart failure such as weight gain or increasing shortness of breath.
- Patients may experience a drop in blood pressure when standing, resulting in dizziness and, rarely, fainting. Patients should sit or lie down when these symptoms of lowered blood pressure occur.
- If experiencing dizziness or fatigue, patients should avoid driving or hazardous tasks.
- Patients should consult a physician if they experience dizziness or faintness, in case the dosage should be adjusted.
- Diabetic patients should report any changes in blood sugar levels to their physician.
- Contact lens wearers may experience decreased lacrimation.
-
17.2 FDA-Approved Patient Labeling
PATIENT INFORMATION
Carvedilol Tablets, USP
Read the Patient Information that comes with carvedilol tablets before you start taking them and each time you get a refill. There may be new information. This information does not take the place of talking with your doctor about your medical condition or your treatment. If you have any questions about carvedilol tablets, ask your doctor or pharmacist.
What are carvedilol tablets?
Carvedilol tablets are a prescription medicine that belongs to a group of medicines called “beta-blockers”.
Carvedilol tablets are used, often with other medicines, for the following conditions:
- To treat patients who had a heart attack that worsened how well the heart pumps
- To treat patients with high blood pressure (hypertension)
Carvedilol tablets are not approved for use in children under 18 years of age.
Who should not take carvedilol tablets?
Do not take carvedilol tablets if you:
- Have severe heart failure and are hospitalized in the intensive care unit or require certain intravenous medications that help support circulation (inotropic medications)
- Are prone to asthma or other breathing problems
- Have a slow heartbeat or a heart that skips a beat (irregular heartbeat)
- Have liver problems
- Are allergic to any of the ingredients in carvedilol tablets. The active ingredient is carvedilol. See the end of this leaflet for a list of all the ingredients in carvedilol tablets.
What should I tell my doctor before taking carvedilol tablets?
Tell your doctor about all of your medical conditions, including if you:
- Have asthma or other lung problems (such as bronchitis or emphysema)
- Have problems with blood flow in your feet and legs (peripheral vascular disease) carvedilol tablets can make some of your symptoms worse.
- Have diabetes
- Have thyroid problems
- Have a condition called pheochromocytoma
- Have had severe allergic reactions
- Are pregnant or trying to become pregnant. It is not known if carvedilol tablets are safe for your unborn baby. You and your doctor should talk about the best way to control your high blood pressure during pregnancy.
- Are breastfeeding. It is not known if carvedilol passes into your breast milk. You should not breastfeed while using carvedilol tablets.
- Are scheduled for surgery and will be given anesthetic agents
- Are scheduled for cataract surgery and have taken or are currently taking carvedilol tablets.
- Are taking prescription or non-prescription medicines, vitamins, and herbal supplements. Carvedilol tablets and certain other medicines can affect each other and cause serious side effects. Carvedilol tablets may affect the way other medicines work. Also, other medicines may affect how well carvedilol tablets work.
Keep a list of all the medicines you take. Show this list to your doctor and pharmacist before you start a new medicine.
How should I take carvedilol tablets?
It is important for you to take your medicine every day as directed by your doctor. If you stop taking carvedilol tablets suddenly, you could have chest pain and/or a heart attack. If your doctor decides that you should stop taking carvedilol tablets, your doctor may slowly lower your dose over a period of time before stopping them completely.
- Take carvedilol tablets exactly as prescribed. Your doctor will tell you how many tablets to take and how often. In order to minimize possible side effects, your doctor might begin with a low dose and then slowly increase the dose.
- Do not stop taking carvedilol tablets and do not change the amount of carvedilol tablets you take without talking to your doctor.
- Tell your doctor if you gain weight or have trouble breathing while taking carvedilol tablets.
- Take carvedilol tablets with food.
- If you miss a dose of carvedilol tablets, take your dose as soon as you remember, unless it is time to take your next dose. Take your next dose at the usual time. Do not take 2 doses at the same time.
- If you take too much carvedilol, call your doctor or poison control center right away.
What should I avoid while taking carvedilol tablets?
Carvedilol tablets can cause you to feel dizzy, tired, or faint. Do not drive a car, use machinery, or do anything that needs you to be alert if you have these symptoms.
What are possible side effects of carvedilol tablets?
- Low blood pressure (which may cause dizziness or fainting when you stand up). If these happen, sit or lie down right away and tell your doctor.
- Tiredness. If you feel tired or dizzy you should not drive, use machinery, or do anything that needs you to be alert.
- Slow heartbeat.
- Changes in your blood sugar. If you have diabetes, tell your doctor if you have any changes in your blood sugar levels.
- Carvedilol tablets may hide some of the symptoms of low blood sugar, especially a fast heartbeat.
- Carvedilol tablets may mask the symptoms of hyperthyroidism (overactive thyroid).
- Worsening of severe allergic reactions.
- Rare but serious allergic reactions (including hives or swelling of the face, lips, tongue, and/or throat that may cause difficulty in breathing or swallowing) have happened in patients who were on carvedilol tablets. These reactions can be life-threatening.
Other side effects of carvedilol tablets include shortness of breath, weight gain, diarrhea, and fewer tears or dry eyes that become bothersome if you wear contact lenses.
Call your doctor if you have any side effects that bother you or don’t go away.
How should I store carvedilol tablets?
- Store carvedilol tablets at 20 to 25°C (68 to 77°F). Keep the tablets dry.
- Safely, throw away carvedilol tablets that are out of date or no longer needed.
- Keep carvedilol tablets and all medicines out of the reach of children.
General information about carvedilol tablets
Medicines are sometimes prescribed for conditions other than those described in patient information leaflets. Do not use carvedilol tablets for a condition for which it was not prescribed. Do not give carvedilol tablets to other people, even if they have the same symptoms you have. They may harm them.
This leaflet summarizes the most important information about carvedilol tablets. If you would like more information, talk with your doctor. You can ask your doctor or pharmacist for information about carvedilol tablets that is written for healthcare professionals. You can also find out more about carvedilol tablets by calling 1-866-850-2876. This call is free.
What are the ingredients in carvedilol tablets?
Active Ingredient: Carvedilol.
Inactive Ingredients: lactose monohydrate, colloidal silicon dioxide, crospovidone, povidone, sucrose, magnesium stearate, polyethylene glycol 400, polysorbate 80, titanium dioxide, and hypromellose.
Carvedilol tablets come in the following strengths: 3.125 mg, 6.25 mg, 12.5 mg, or 25 mg.
What is high blood pressure (hypertension)?
Blood pressure is the force of blood in your blood vessels when your heart beats and when your heart rests. You have high blood pressure when the force is too much. High blood pressure makes the heart work harder to pump blood through the body and causes damage to blood vessels. Carvedilol tablets can help your blood vessels relax so your blood pressure is lower. Medicines that lower blood pressure may lower your chance of having a stroke or heart attack.
Manufactured by:
Aurolife Pharma LLC
Dayton, NJ 08810
Manufactured for:
Aurobindo Pharma USA, Inc.
Dayton, NJ 08810
Revised: 04/2011 -
PACKAGE LABEL-PRINCIPAL DISPLAY PANEL - 3.125 mg (1000 Tablet Bottle)
NDC: 13107-142-99
Carvedilol Tablets, USP
3.125 mg
Pharmacist: Dispense the accompanying
Patient Information Leaflet to each patient.
Rx only 1000 Tablets
AUROBINDO

-
PACKAGE LABEL-PRINCIPAL DISPLAY PANEL - 6.25 mg (1000 Tablet Bottle)
NDC: 13107-143-99
Carvedilol Tablets, USP
6.25 mg
Pharmacist: Dispense the accompanying
Patient Information Leaflet to each patient.
Rx only 1000 Tablets
AUROBINDO

-
PACKAGE LABEL-PRINCIPAL DISPLAY PANEL - 12.5 mg (1000 Tablet Bottle)
NDC: 13107-144-99
Carvedilol Tablets, USP
12.5 mg
Pharmacist: Dispense the accompanying
Patient Information Leaflet to each patient.
Rx only 1000 Tablets
AUROBINDO

-
PACKAGE LABEL-PRINCIPAL DISPLAY PANEL - 25 mg (1000 Tablet Bottle)
NDC: 13107-145-99
Carvedilol Tablets, USP
25 mg
Pharmacist: Dispense the accompanying
Patient Information Leaflet to each patient.
Rx only 1000 Tablets
AUROBINDO

-
INGREDIENTS AND APPEARANCE
CARVEDILOL
carvedilol tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 13107-142 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength CARVEDILOL (UNII: 0K47UL67F2) (CARVEDILOL - UNII:0K47UL67F2) CARVEDILOL 3.125 mg Inactive Ingredients Ingredient Name Strength LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) CROSPOVIDONE (UNII: 68401960MK) POVIDONE K30 (UNII: U725QWY32X) SUCROSE (UNII: C151H8M554) MAGNESIUM STEARATE (UNII: 70097M6I30) POLYETHYLENE GLYCOL 400 (UNII: B697894SGQ) POLYSORBATE 80 (UNII: 6OZP39ZG8H) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) HYPROMELLOSE 2910 (5 MPA.S) (UNII: R75537T0T4) Product Characteristics Color WHITE (White to Off-white) Score no score Shape OVAL Size 6mm Flavor Imprint Code E;01 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 13107-142-01 100 in 1 BOTTLE 2 NDC: 13107-142-05 500 in 1 BOTTLE 3 NDC: 13107-142-99 1000 in 1 BOTTLE Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA078332 03/23/2009 CARVEDILOL
carvedilol tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 13107-143 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength CARVEDILOL (UNII: 0K47UL67F2) (CARVEDILOL - UNII:0K47UL67F2) CARVEDILOL 6.25 mg Inactive Ingredients Ingredient Name Strength LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) CROSPOVIDONE (UNII: 68401960MK) POVIDONE K30 (UNII: U725QWY32X) SUCROSE (UNII: C151H8M554) MAGNESIUM STEARATE (UNII: 70097M6I30) POLYETHYLENE GLYCOL 400 (UNII: B697894SGQ) POLYSORBATE 80 (UNII: 6OZP39ZG8H) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) HYPROMELLOSE 2910 (5 MPA.S) (UNII: R75537T0T4) Product Characteristics Color WHITE (White to Off-white) Score no score Shape OVAL Size 8mm Flavor Imprint Code E;02 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 13107-143-01 100 in 1 BOTTLE 2 NDC: 13107-143-05 500 in 1 BOTTLE 3 NDC: 13107-143-99 1000 in 1 BOTTLE Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA078332 03/23/2009 CARVEDILOL
carvedilol tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 13107-144 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength CARVEDILOL (UNII: 0K47UL67F2) (CARVEDILOL - UNII:0K47UL67F2) CARVEDILOL 12.5 mg Inactive Ingredients Ingredient Name Strength LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) CROSPOVIDONE (UNII: 68401960MK) POVIDONE K30 (UNII: U725QWY32X) SUCROSE (UNII: C151H8M554) MAGNESIUM STEARATE (UNII: 70097M6I30) POLYETHYLENE GLYCOL 400 (UNII: B697894SGQ) POLYSORBATE 80 (UNII: 6OZP39ZG8H) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) HYPROMELLOSE 2910 (5 MPA.S) (UNII: R75537T0T4) Product Characteristics Color WHITE (White to Off-white) Score no score Shape OVAL Size 11mm Flavor Imprint Code E;03 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 13107-144-01 100 in 1 BOTTLE 2 NDC: 13107-144-05 500 in 1 BOTTLE 3 NDC: 13107-144-99 1000 in 1 BOTTLE Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA078332 03/23/2009 CARVEDILOL
carvedilol tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 13107-145 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength CARVEDILOL (UNII: 0K47UL67F2) (CARVEDILOL - UNII:0K47UL67F2) CARVEDILOL 25 mg Inactive Ingredients Ingredient Name Strength LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) CROSPOVIDONE (UNII: 68401960MK) POVIDONE K30 (UNII: U725QWY32X) SUCROSE (UNII: C151H8M554) MAGNESIUM STEARATE (UNII: 70097M6I30) POLYETHYLENE GLYCOL 400 (UNII: B697894SGQ) POLYSORBATE 80 (UNII: 6OZP39ZG8H) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) HYPROMELLOSE 2910 (5 MPA.S) (UNII: R75537T0T4) Product Characteristics Color WHITE (White to Off-white) Score no score Shape OVAL Size 13mm Flavor Imprint Code E;04 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 13107-145-01 100 in 1 BOTTLE 2 NDC: 13107-145-05 500 in 1 BOTTLE 3 NDC: 13107-145-99 1000 in 1 BOTTLE Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA078332 03/23/2009 Labeler - Aurolife Pharma LLC (829084461)
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.