COMIRNATY- covid-19 vaccine, mrna injection, suspension
Comirnaty by
Drug Labeling and Warnings
Comirnaty by is a Other medication manufactured, distributed, or labeled by Pfizer Laboratories Div Pfizer Inc, Pfizer Inc, Pfizer Manufacturing Belgium NV, Wyeth BioPharma Division of Wyeth Pharmaceuticals LLC, Pfizer Ireland Pharmaceuticals, BioNTech Innovative Manufacturing Services GmbH, BioNTech Manufacturing GmbH, BioNTech Manufacturing Marburg GmbH, Pharmacia & Upjohn Company LLC, Exela Pharma Sciences, LLC.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use COMIRNATY safely and effectively. See full prescribing information for COMIRNATY.
COMIRNATY® (COVID-19 Vaccine, mRNA) injectable suspension, for intramuscular use
2025-2026 Formula
Initial U.S. Approval: 2021RECENT MAJOR CHANGES
INDICATIONS AND USAGE
COMIRNATY is a vaccine indicated for active immunization to prevent coronavirus disease 2019 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). (1)
COMIRNATY is approved for use in individuals who are:
- 65 years of age and older, or
- 5 years through 64 years of age with at least one underlying condition that puts them at high risk for severe outcomes from COVID-19. (1)
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONS
Known history of a severe allergic reaction (e.g., anaphylaxis) to any component of COMIRNATY. (4)
WARNINGS AND PRECAUTIONS
- Analyses of postmarketing data from use of authorized or approved mRNA COVID-19 vaccines, including COMIRNATY, have demonstrated increased risks of myocarditis and pericarditis, with onset of symptoms typically in the first week following vaccination. The observed risk has been highest in males 12 years through 24 years of age. (5.2)
- Syncope (fainting) may occur in association with administration of injectable vaccines, including COMIRNATY. Procedures should be in place to avoid injury from fainting. (5.3)
ADVERSE REACTIONS
- Participants 12 years of age and older: The most commonly reported adverse reactions (≥10%) after a dose of COMIRNATY were pain at the injection site (up to 90.5%), fatigue (up to 77.5%), headache (up to 75.5%), chills (up to 49.2%), muscle pain (up to 45.5%), joint pain (up to 27.5%), fever (up to 24.3%), injection site swelling (up to 11.8%), and injection site redness (up to 10.4%). (6.1)
-
Participants 5 years through 11 years of age: The most commonly reported adverse reactions (≥5%) following any dose were pain at the injection site (up to 83.8%), fatigue (up to 51.9%), headache (up to 38.4%), injection site redness (up to 25.9%), injection site swelling (up to 20.0%), muscle pain (up to 18.1%), chills (up to 13.3%), fever (up to 7.8%), and joint pain (up to 7.6%). (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Pfizer Inc. at 1-800-438-1985 or VAERS at 1-800-822-7967 or http://vaers.hhs.gov.
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 2/2026
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Preparation for Administration
2.2 Administration Information
2.3 Vaccination Schedule
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Management of Acute Allergic Reactions
5.2 Myocarditis and Pericarditis
5.3 Syncope
5.4 Altered Immunocompetence
5.5 Limitation of Effectiveness
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Immunocompromised Individuals
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Adults and Adolescents 12 Years of Age and Older
14.2 Children 5 Years Through 11 Years of Age
15 REFERENCES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
COMIRNATY is a vaccine indicated for active immunization to prevent coronavirus disease 2019 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2).
COMIRNATY is approved for use in individuals who are:
- 65 years of age and older, or
- 5 years through 64 years of age with at least one underlying condition that puts them at high risk for severe outcomes from COVID-19.
-
2 DOSAGE AND ADMINISTRATION
For intramuscular use.
2.1 Preparation for Administration
COMIRNATY Single-Dose Prefilled Syringes for Individuals 65 Years of Age and Older and Individuals 12 Years Through 64 Years of Age with at Least One Underlying Condition that Puts Them at High Risk for Severe Outcomes from COVID-19:
- Verify that the label on the prefilled syringe states 2025-2026 Formula.
- If prefilled syringe has been frozen, discard.
- Do not shake.
- Remove tip cap by slowly turning the cap counterclockwise while holding the Luer lock and attach a sterile needle. Use immediately. If COMIRNATY cannot be used immediately, it must be used within 4 hours.
COMIRNATY Single-Dose Vials for Individuals 5 Years Through 11 Years of Age with at Least One Underlying Condition that Puts Them at High Risk for Severe Outcomes from COVID-19:
- Verify that the label on the vial states 2025-2026 Formula.
- If vial is frozen, thaw vial in the refrigerator [2ºC to 8ºC (35ºF to 46ºF) for up to 2 hours] or at room temperature [up to 25ºC (77ºF) for 30 minutes] [see How Supplied/Storage and Handling (16)].
- Prior to use, mix by inverting vial gently 10 times. Do not shake.
- Withdraw a single 0.3 mL dose using a sterile needle and syringe.
- Discard vial and any excess volume.
2.2 Administration Information
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. For the prefilled syringe, the vaccine will be a white to off-white suspension and for the vial, the vaccine will be clear to slightly opalescent suspension. Do not administer if vaccine is discolored or contains particulate matter.
Administer the 0.3 mL dose intramuscularly immediately after preparation. For the prefilled syringe, administer the entire volume to deliver a single 0.3 mL dose.
-
3 DOSAGE FORMS AND STRENGTHS
COMIRNATY is an injectable suspension.
- For individuals 5 years through 11 years of age: a single dose is 0.3 mL supplied in vials with blue caps and labeled with blue borders.
- For individuals 12 years of age and older: a single dose is 0.3 mL supplied in prefilled syringes labeled with gray borders.
-
4 CONTRAINDICATIONS
Do not administer COMIRNATY to individuals with known history of a severe allergic reaction (e.g., anaphylaxis) to any component of COMIRNATY [see Description (11)] or to individuals who had a severe allergic reaction (e.g., anaphylaxis) following a previous dose of a Pfizer-BioNTech COVID-19 vaccine.
-
5 WARNINGS AND PRECAUTIONS
5.1 Management of Acute Allergic Reactions
Appropriate medical treatment must be immediately available to manage potential anaphylactic reactions following administration of COMIRNATY.
5.2 Myocarditis and Pericarditis
Analyses of postmarketing data from use of authorized or approved mRNA COVID-19 vaccines, including COMIRNATY, have demonstrated increased risks of myocarditis and pericarditis, with onset of symptoms typically in the first week following vaccination. The observed risk has been highest in males 12 years through 24 years of age.
Based on analyses of commercial health insurance claims data from inpatient and outpatient settings, the estimated unadjusted incidence of myocarditis and/or pericarditis during the period 1 through 7 days following administration of the 2023-2024 Formula of mRNA COVID-19 vaccines was approximately 8 cases per million doses in individuals 6 months through 64 years of age and approximately 27 cases per million doses in males 12 through 24 years of age.
Although some individuals with myocarditis and/or pericarditis following administration of mRNA COVID-19 vaccines have required intensive care support, available data suggest that individuals typically have resolution of symptoms within a few days with conservative management.
Follow-up information on cardiovascular outcomes in hospitalized patients who had been diagnosed with COVID-19 vaccine-associated myocarditis is available from a longitudinal retrospective observational study. Most of these patients had received a two-dose primary series of an mRNA COVID-19 vaccine prior to their diagnosis. In this study, at a median follow-up of approximately 5 months postvaccination, persistence of abnormal cardiac magnetic resonance imaging (CMR) findings that are a marker for myocardial injury was common. The clinical and prognostic significance of these CMR findings is not known1 [see Adverse Reactions (6.2)].
Information is not yet available about potential long-term sequelae of myocarditis or pericarditis following administration of mRNA COVID-19 vaccines.
The Centers for Disease Control and Prevention (CDC) has published considerations related to myocarditis and pericarditis after vaccination, including for vaccination of individuals with a history of myocarditis or pericarditis (https://www.cdc.gov/vaccines/covid-19/clinical-considerations/myocarditis.html).
5.3 Syncope
Syncope (fainting) may occur in association with administration of injectable vaccines, including COMIRNATY. Procedures should be in place to avoid injury from fainting.
5.4 Altered Immunocompetence
Immunocompromised persons, including individuals receiving immunosuppressant therapy, may have a diminished immune response to COMIRNATY [see Use in Specific Populations (8.6)].
-
6 ADVERSE REACTIONS
An overview of clinical studies contributing to the safety assessment of COMIRNATY is provided in Table 1 and Table 2. Participants in these clinical studies received a single dose or a 2-dose series administered 3 weeks apart (referred to as a primary series) and subsequent doses referred to as booster doses.
Table 1: Clinical Studies – Adults and Adolescents 12 Years of Age and Older Abbreviation: SARS-CoV-2 = severe acute respiratory syndrome coronavirus 2. - * COMIRNATY encoding the viral spike (S) glycoprotein of SARS-CoV-2 Wuhan-Hu-1 strain [Original, 30 mcg nucleoside−modified messenger RNA (modRNA)].
- † Received COMIRNATY during placebo-control period.
- ‡ Vaccine encoding the viral spike (S) glycoprotein of SARS-CoV-2 Wuhan-Hu-1 strain (Original) and Omicron variant lineages BA.4 and BA.5 (Omicron BA.4/BA.5), previously authorized as Pfizer-BioNTech COVID-19 Vaccine, Bivalent (30 mcg modRNA).
- § Influenza Vaccine (Afluria® Quadrivalent).
Study
Age Group
Vaccine Strain Composition
Dosing
Number of Participants
Primary Series
Study 1
(NCT04380701)
18 through 55 years of age
Original*
Primary series
60
Study 2
(NCT04368728)
16 years of age and older
Original*
Primary series
22,026†
12 through 15 years of age
Original*
Primary series
1,131†
Booster Dose
Study 4
(NCT04955626)
16 years of age and older
Original*
1st booster
5,081†
Study 2
(NCT04368728)
18 through 55 years of age
Original*
1st booster
306
Study 4
(NCT04955626)
12 through 17 years of age
Original*
1st booster
65
Study 2
(NCT04368728)
12 through 15 years of age
Original*
1st booster
825
Study 5
(NCT05472038)
12 years of age and older
Original and Omicron BA.4/BA.5‡
2nd booster
726
Concomitant Administration
Study 8
(NCT05310084)
18 through 64 years of age
Original*
2nd booster administered alone or concomitantly with Influenza Vaccine§
1,128
Table 2: Clinical Studies – Children 5 Years Through 11 Years of Age Abbreviation: SARS-CoV-2 = severe acute respiratory syndrome coronavirus 2. - * COMIRNATY encoding the viral spike (S) glycoprotein of SARS-CoV-2 Wuhan-Hu-1 strain (Original, 10 mcg modRNA).
- † Received COMIRNATY during placebo-control period.
- ‡ Vaccine encoding the viral spike (S) glycoprotein of SARS-CoV-2 Wuhan-Hu-1 strain (Original) and Omicron variant lineages BA.4 and BA.5 (Omicron BA.4/BA.5), previously authorized as Pfizer-BioNTech COVID-19 Vaccine, Bivalent (10 mcg modRNA).
- § COMIRNATY encoding the viral spike (S) glycoprotein of SARS-CoV-2 Omicron XBB.1.5 (10 mcg modRNA).
Study
Age Group
Vaccine Strain Composition
Dosing
Number of Participants
Primary Series
Study 3
(NCT04816643)
5 through 11 years of age
Original*
Primary series
3,109†
Booster Dose
Study 3
(NCT04816643)
5 through 11 years of age
Original*
1st booster
2,408
Study 6
(NCT05543616)
5 through 11 years of age
Original and Omicron BA.4/BA.5‡
2nd booster
113
Single Dose
Study 6
(NCT05543616)
5 through 11 years of age
Omicron XBB.1.5§
Single dose
310
Participants 12 years of age and older: The most commonly reported adverse reactions (≥10%) after a dose of COMIRNATY were pain at the injection site (up to 90.5%), fatigue (up to 77.5%), headache (up to 75.5%), chills (up to 49.2%), muscle pain (up to 45.5%), joint pain (up to 27.5%), fever (up to 24.3%), injection site swelling (up to 11.8%), and injection site redness (up to 10.4%).
Participants 5 years through 11 years of age: The most commonly reported adverse reactions (≥5%) following any dose were pain at the injection site (up to 83.8%), fatigue (up to 51.9%), headache (up to 38.4%), injection site redness (up to 25.9%), injection site swelling (up to 20.0%), muscle pain (up to 18.1%), chills (up to 13.3%), fever (up to 7.8%), and joint pain (up to 7.6%).
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a vaccine cannot be directly compared to rates in the clinical trials of another vaccine and may not reflect the rates observed in practice.
Adults and Adolescents 12 Years of Age and Older
Two-Dose Series (Original Monovalent) in Vaccine-Naïve Individuals 16 Years of Age and Older
The safety of a 2-dose primary series of COMIRNATY was evaluated in participants 12 years of age and older in 2 clinical studies conducted in Germany (Study 1), United States, Argentina, Brazil, Turkey, South Africa, and Germany (Study 2). Study BNT162-01 (Study 1) was a Phase 1/2, 2-part, dose-escalation trial that enrolled 60 participants, 18 through 55 years of age and 36 participants, 56 through 85 years of age. Study 2 was a Phase 1/2/3 multicenter, randomized, saline placebo-controlled, double-blinded (Phase 2/3), dose-finding, vaccine candidate-selection and efficacy study that enrolled approximately 44,000 participants 16 years of age or older (22,026 COMIRNATY; 22,021 placebo).
Study 2 included 200 participants with confirmed stable human immunodeficiency virus (HIV) infection. Confirmed stable HIV infection was defined as documented viral load <50 copies/mL and CD4 count >200 cells/mm3 within 6 months before enrollment, and on stable antiretroviral therapy for at least 6 months. HIV-positive participants are included in the safety population but are summarized separately in the safety analyses.
In Study 2, participants 16 years and older in the reactogenicity subset were monitored using an electronic diary for solicited local and systemic reactions and use of antipyretic medication after each vaccination. Participants were also monitored for unsolicited adverse events throughout the study (from Dose 1 through 1 month [all unsolicited adverse events] or through 6 months [serious adverse events] after the last vaccination). Tables 3 through 6 present the frequency and severity of solicited local and systemic reactions, respectively, within 7 days following Dose 1 or Dose 2 of COMIRNATY.
At the time of the analysis of Study 2 with a data cutoff of March 13, 2021, there were 25,651 (58.2%) participants (13,031 COMIRNATY; 12,620 placebo) 16 years of age and older followed for ≥4 months after the second dose.
Demographic characteristics in Study 2 were generally similar with regard to age, sex, race, and ethnicity among participants who received COMIRNATY and those who received placebo. Overall, among the total participants who received either COMIRNATY or placebo, 50.9% were male, 49.1% were female, 79.3% were 16 through 64 years of age, 20.7% were 65 years of age and older, 82.0% were White, 9.6% were Black or African American, 25.9% were Hispanic/Latino, 4.3% were Asian, and 1.0% were American Indian or Alaska Native.
Local and Systemic Solicited Adverse Reactions
In participants 16 through 55 years of age after receiving Dose 2, the mean duration of pain at the injection site was 2.5 days (range 1 to 70 days), for redness 2.2 days (range 1 to 9 days), and for swelling 2.1 days (range 1 to 8 days) for participants in the COMIRNATY group.
In participants 56 years of age and older after receiving Dose 2, the mean duration of pain at the injection site was 2.4 days (range 1 to 36 days), for redness 3.0 days (range 1 to 34 days), and for swelling 2.6 days (range 1 to 34 days) for participants in the COMIRNATY group.
Table 3: Study 2 – Frequency and Percentages of Participants With Solicited Local Reactions, by Maximum Severity, Within 7 Days After Each Dose – Participants 16 Through 55 Years of Age – Reactogenicity Subset of the Safety Population* Notes: Reactions were collected in the electronic diary (e-diary) from Day 1 to Day 7 after vaccination.
No Grade 4 solicited local reactions were reported in participants 16 through 55 years of age.- * Randomized participants in the safety analysis population who received at least 1 dose of the study intervention. Participants with chronic, stable HIV infection were excluded.
- † Vaccine encoding the viral spike (S) glycoprotein of SARS-CoV-2 Wuhan-Hu-1 strain (Original).
- ‡ N = Number of participants reporting at least 1 yes or no response for the specified reaction after the specified dose. The N for each reaction was the same, therefore, this information was included in the column header.
- § n = Number of participants with the specified reaction.
- ¶ Mild: >2.0 to ≤5.0 cm; Moderate: >5.0 to ≤10.0 cm; Severe: >10.0 cm.
- # Mild: does not interfere with activity; Moderate: interferes with activity; Severe: prevents daily activity.
COMIRNATY†
Dose 1
N‡=2899
n§ (%)
Placebo
Dose 1
N‡=2908
n§ (%)
COMIRNATY†
Dose 2
N‡=2682
n§ (%)
Placebo
Dose 2
N‡=2684
n§ (%)
Redness¶
Any (>2.0 cm)
156 (5.4)
28 (1.0)
151 (5.6)
18 (0.7)
Mild
113 (3.9)
19 (0.7)
90 (3.4)
12 (0.4)
Moderate
36 (1.2)
6 (0.2)
50 (1.9)
6 (0.2)
Severe
7 (0.2)
3 (0.1)
11 (0.4)
0
Swelling¶
Any (>2.0 cm)
184 (6.3)
16 (0.6)
183 (6.8)
5 (0.2)
Mild
124 (4.3)
6 (0.2)
110 (4.1)
3 (0.1)
Moderate
54 (1.9)
8 (0.3)
66 (2.5)
2 (0.1)
Severe
6 (0.2)
2 (0.1)
7 (0.3)
0
Pain at the injection site#
Any
2426 (83.7)
414 (14.2)
2101 (78.3)
312 (11.6)
Mild
1464 (50.5)
391 (13.4)
1274 (47.5)
284 (10.6)
Moderate
923 (31.8)
20 (0.7)
788 (29.4)
28 (1.0)
Severe
39 (1.3)
3 (0.1)
39 (1.5)
0
Table 4: Study 2 – Frequency and Percentages of Participants With Solicited Systemic Reactions, by Maximum Severity, Within 7 Days After Each Dose – Participants 16 Through 55 Years of Age – Reactogenicity Subset of the Safety Population* Notes: Reactions and use of antipyretic or pain medication were collected in the electronic diary (e-diary) from Day 1 to Day 7 after each dose.
No Grade 4 solicited systemic reactions were reported in participants 16 through 55 years of age.- * Randomized participants in the safety analysis population who received at least 1 dose of the study intervention. Participants with chronic, stable HIV infection were excluded.
- † Vaccine encoding the viral spike (S) glycoprotein of SARS-CoV-2 Wuhan-Hu-1 strain (Original).
- ‡ N = Number of participants reporting at least 1 yes or no response for the specified reaction after the specified dose. The N for each reaction or use of antipyretic or pain medication was the same, therefore, this information was included in the column header.
- § n = Number of participants with the specified reaction.
- ¶ Mild: does not interfere with activity; Moderate: some interference with activity; Severe: prevents daily activity.
- # Mild: 1 to 2 times in 24 hours; Moderate: >2 times in 24 hours; Severe: requires intravenous hydration.
- Þ Mild: 2 to 3 loose stools in 24 hours; Moderate: 4 to 5 loose stools in 24 hours; Severe: 6 or more loose stools in 24 hours.
- ß Severity was not collected for use of antipyretic or pain medication.
COMIRNATY†
Dose 1
N‡=2899
n§ (%)
Placebo
Dose 1
N‡=2908
n§ (%)
COMIRNATY†
Dose 2
N‡=2682
n§ (%)
Placebo
Dose 2
N‡=2684
n§ (%)
Fever
≥38.0℃
119 (4.1)
25 (0.9)
440 (16.4)
11 (0.4)
≥38.0℃ to 38.4℃
86 (3.0)
16 (0.6)
254 (9.5)
5 (0.2)
>38.4℃ to 38.9℃
25 (0.9)
5 (0.2)
146 (5.4)
4 (0.1)
>38.9℃ to 40.0℃
8 (0.3)
4 (0.1)
39 (1.5)
2 (0.1)
>40.0℃
0
0
1 (0.0)
0
Fatigue¶
Any
1431 (49.4)
960 (33.0)
1649 (61.5)
614 (22.9)
Mild
760 (26.2)
570 (19.6)
558 (20.8)
317 (11.8)
Moderate
630 (21.7)
372 (12.8)
949 (35.4)
283 (10.5)
Severe
41 (1.4)
18 (0.6)
142 (5.3)
14 (0.5)
Headache¶
Any
1262 (43.5)
975 (33.5)
1448 (54.0)
652 (24.3)
Mild
785 (27.1)
633 (21.8)
699 (26.1)
404 (15.1)
Moderate
444 (15.3)
318 (10.9)
658 (24.5)
230 (8.6)
Severe
33 (1.1)
24 (0.8)
91 (3.4)
18 (0.7)
Chills¶
Any
479 (16.5)
199 (6.8)
1015 (37.8)
114 (4.2)
Mild
338 (11.7)
148 (5.1)
477 (17.8)
89 (3.3)
Moderate
126 (4.3)
49 (1.7)
469 (17.5)
23 (0.9)
Severe
15 (0.5)
2 (0.1)
69 (2.6)
2 (0.1)
Vomiting#
Any
34 (1.2)
36 (1.2)
58 (2.2)
30 (1.1)
Mild
29 (1.0)
30 (1.0)
42 (1.6)
20 (0.7)
Moderate
5 (0.2)
5 (0.2)
12 (0.4)
10 (0.4)
Severe
0
1 (0.0)
4 (0.1)
0
DiarrheaÞ
Any
309 (10.7)
323 (11.1)
269 (10.0)
205 (7.6)
Mild
251 (8.7)
264 (9.1)
219 (8.2)
169 (6.3)
Moderate
55 (1.9)
58 (2.0)
44 (1.6)
35 (1.3)
Severe
3 (0.1)
1 (0.0)
6 (0.2)
1 (0.0)
New or worsened muscle pain¶
Any
664 (22.9)
329 (11.3)
1055 (39.3)
237 (8.8)
Mild
353 (12.2)
231 (7.9)
441 (16.4)
150 (5.6)
Moderate
296 (10.2)
96 (3.3)
552 (20.6)
84 (3.1)
Severe
15 (0.5)
2 (0.1)
62 (2.3)
3 (0.1)
New or worsened joint pain¶
Any
342 (11.8)
168 (5.8)
638 (23.8)
147 (5.5)
Mild
200 (6.9)
112 (3.9)
291 (10.9)
82 (3.1)
Moderate
137 (4.7)
55 (1.9)
320 (11.9)
61 (2.3)
Severe
5 (0.2)
1 (0.0)
27 (1.0)
4 (0.1)
Use of antipyretic or pain medicationß
805 (27.8)
398 (13.7)
1213 (45.2)
320 (11.9)
Table 5: Study 2 – Frequency and Percentages of Participants With Solicited Local Reactions, by Maximum Severity, Within 7 Days After Each Dose – Participants 56 Years of Age and Older – Reactogenicity Subset of the Safety Population* Notes: Reactions were collected in the electronic diary (e-diary) from Day 1 to Day 7 after vaccination.
No Grade 4 solicited local reactions were reported in participants 56 years of age and older.- * Randomized participants in the safety analysis population who received at least 1 dose of the study intervention. Participants with chronic, stable HIV infection were excluded.
- † Vaccine encoding the viral spike (S) glycoprotein of SARS-CoV-2 Wuhan-Hu-1 strain (Original).
- ‡ N = Number of participants reporting at least 1 yes or no response for the specified reaction after the specified dose. The N for each reaction was the same, therefore, the information was included in the column header.
- § n = Number of participants with the specified reaction.
- ¶ Mild: >2.0 to ≤5.0 cm; Moderate: >5.0 to ≤10.0 cm; Severe: >10.0 cm.
- # Mild: does not interfere with activity; Moderate: interferes with activity; Severe: prevents daily activity.
COMIRNATY†
Dose 1
N‡=2008
n§ (%)
Placebo
Dose 1
N‡=1989
n§ (%)
COMIRNATY†
Dose 2
N‡=1860
n§ (%)
Placebo
Dose 2
N‡=1833
n§ (%)
Redness¶
Any (>2.0 cm)
106 (5.3)
20 (1.0)
133 (7.2)
14 (0.8)
Mild
71 (3.5)
13 (0.7)
65 (3.5)
10 (0.5)
Moderate
30 (1.5)
5 (0.3)
58 (3.1)
3 (0.2)
Severe
5 (0.2)
2 (0.1)
10 (0.5)
1 (0.1)
Swelling¶
Any (>2.0 cm)
141 (7.0)
23 (1.2)
145 (7.8)
13 (0.7)
Mild
87 (4.3)
11 (0.6)
80 (4.3)
5 (0.3)
Moderate
52 (2.6)
12 (0.6)
61 (3.3)
7 (0.4)
Severe
2 (0.1)
0
4 (0.2)
1 (0.1)
Pain at the injection site#
Any (>2.0 cm)
1408 (70.1)
185 (9.3)
1230 (66.1)
143 (7.8)
Mild
1108 (55.2)
177 (8.9)
873 (46.9)
138 (7.5)
Moderate
296 (14.7)
8 (0.4)
347 (18.7)
5 (0.3)
Severe
4 (0.2)
0
10 (0.5)
0
Table 6: Study 2 – Frequency and Percentages of Participants With Solicited Systemic Reactions, by Maximum Severity, Within 7 Days After Each Dose – Participants 56 Years of Age and Older – Reactogenicity Subset of the Safety Population* Notes: Reactions and use of antipyretic or pain medication were collected in the electronic diary (e-diary) from Day 1 to Day 7 after each dose.
The only Grade 4 solicited systemic reaction reported in participants 56 years of age and older was fatigue.- * Randomized participants in the safety analysis population who received at least 1 dose of the study intervention. Participants with chronic, stable HIV infection were excluded.
- † Vaccine encoding the viral spike (S) glycoprotein of SARS-CoV-2 Wuhan-Hu-1 strain (Original).
- ‡ N = Number of participants reporting at least 1 yes or no response for the specified reaction after the specified dose. N for each reaction or use of antipyretic or pain medication was the same, therefore was included in the column header.
- § n = Number of participants with the specified reaction.
- ¶ Mild: does not interfere with activity; Moderate: some interference with activity; Severe: prevents daily activity; Grade 4 reactions were defined in the clinical study protocol as emergency room visit or hospitalization for severe fatigue, severe headache, severe chills, severe muscle pain, or severe joint pain.
- # Mild: 1 to 2 times in 24 hours; Moderate: >2 times in 24 hours; Severe: requires intravenous hydration; Grade 4 emergency visit or hospitalization for severe vomiting.
- Þ Mild: 2 to 3 loose stools in 24 hours; Moderate: 4 to 5 loose stools in 24 hours; Severe: 6 or more loose stools in 24 hours; Grade 4: emergency room or hospitalization for severe diarrhea.
- ß Severity was not collected for use of antipyretic or pain medication.
COMIRNATY†
Dose 1
N‡=2008
n§ (%)
Placebo
Dose 1
N‡=1989
n§ (%)
COMIRNATY†
Dose 2
N‡=1860
n§ (%)
Placebo
Dose 2
N‡=1833
n§ (%)
Fever
≥38.0℃
26 (1.3)
8 (0.4)
219 (11.8)
4 (0.2)
≥38.0℃ to 38.4℃
23 (1.1)
3 (0.2)
158 (8.5)
2 (0.1)
>38.4℃ to 38.9℃
2 (0.1)
3 (0.2)
54 (2.9)
1 (0.1)
>38.9℃ to 40.0℃
1 (0.0)
2 (0.1)
7 (0.4)
1 (0.1)
>40.0℃
0
0
0
0
Fatigue¶
Any
677 (33.7)
447 (22.5)
949 (51.0)
306 (16.7)
Mild
415 (20.7)
281 (14.1)
391 (21.0)
183 (10.0)
Moderate
259 (12.9)
163 (8.2)
497 (26.7)
121 (6.6)
Severe
3 (0.1)
3 (0.2)
60 (3.2)
2 (0.1)
Grade 4
0
0
1 (0.1)
0
Headache¶
Any
503 (25.0)
363 (18.3)
733 (39.4)
259 (14.1)
Mild
381 (19.0)
267 (13.4)
464 (24.9)
189 (10.3)
Moderate
120 (6.0)
93 (4.7)
256 (13.8)
65 (3.5)
Severe
2 (0.1)
3 (0.2)
13 (0.7)
5 (0.3)
Chills¶
Any
130 (6.5)
69 (3.5)
435 (23.4)
57 (3.1)
Mild
102 (5.1)
49 (2.5)
229 (12.3)
45 (2.5)
Moderate
28 (1.4)
19 (1.0)
185 (9.9)
12 (0.7)
Severe
0
1 (0.1)
21 (1.1)
0
Vomiting#
Any
10 (0.5)
9 (0.5)
13 (0.7)
5 (0.3)
Mild
9 (0.4)
9 (0.5)
10 (0.5)
5 (0.3)
Moderate
1 (0.0)
0
1 (0.1)
0
Severe
0
0
2 (0.1)
0
DiarrheaÞ
Any
168 (8.4)
130 (6.5)
152 (8.2)
102 (5.6)
Mild
137 (6.8)
109 (5.5)
125 (6.7)
76 (4.1)
Moderate
27 (1.3)
20 (1.0)
25 (1.3)
22 (1.2)
Severe
4 (0.2)
1 (0.1)
2 (0.1)
4 (0.2)
New or worsened muscle pain¶
Any
274 (13.6)
165 (8.3)
537 (28.9)
99 (5.4)
Mild
183 (9.1)
111 (5.6)
229 (12.3)
65 (3.5)
Moderate
90 (4.5)
51 (2.6)
288 (15.5)
33 (1.8)
Severe
1 (0.0)
3 (0.2)
20 (1.1)
1 (0.1)
New or worsened joint pain¶
Any
175 (8.7)
124 (6.2)
353 (19.0)
72 (3.9)
Mild
119 (5.9)
78 (3.9)
183 (9.8)
44 (2.4)
Moderate
53 (2.6)
45 (2.3)
161 (8.7)
27 (1.5)
Severe
3 (0.1)
1 (0.1)
9 (0.5)
1 (0.1)
Use of antipyretic or pain medicationß
382 (19.0)
224 (11.3)
688 (37.0)
170 (9.3)
In participants with chronic, stable HIV infection the frequencies of solicited local and systemic adverse reactions were similar to or lower than those observed for all participants 16 years of age and older.
Unsolicited Adverse Events
Overall, 11,253 (51.1%) participants 16 years of age and older in the COMIRNATY group and 11,316 (51.4%) participants in the placebo group had follow-up time between ≥4 months to <6 months after Dose 2 in the blinded placebo-controlled follow-up period with an additional 1,778 (8.1%) and 1,304 (5.9%) with ≥6 months of blinded follow-up time in the COMIRNATY and placebo groups, respectively.
A total of 12,006 (54.5%) participants originally randomized to COMIRNATY had ≥6 months total (blinded and unblinded) follow-up after Dose 2.
In an analysis of all unsolicited adverse events reported following any dose, through 1 month after Dose 2, in participants 16 years of age and older (N = 43,847; 21,926 COMIRNATY group vs. 21,921 placebo group), those assessed as adverse reactions not already captured by solicited local and systemic reactions were nausea (274 vs. 87), malaise (130 vs. 22), lymphadenopathy (83 vs. 7), asthenia (76 vs. 25), decreased appetite (39 vs. 9), hyperhidrosis (31 vs. 9), lethargy (25 vs. 6), and night sweats (17 vs. 3).
In analyses of all unsolicited adverse events in Study 2 from Dose 1 up to the participant unblinding date, 58.2% of study participants had at least 4 months of follow-up after Dose 2. Among participants 16 through 55 years of age who received at least 1 dose of study vaccine, 12,995 of whom received COMIRNATY and 13,026 of whom received placebo, unsolicited adverse events were reported by 4,396 (33.8%) participants in the COMIRNATY group and 2,136 (16.4%) participants in the placebo group. In a similar analysis in participants 56 years of age and older that included 8,931 COMIRNATY recipients and 8,895 placebo recipients, unsolicited adverse events were reported by 2,551 (28.6%) participants in the COMIRNATY group and 1,432 (16.1%) participants in the placebo group. Among participants with confirmed stable HIV infection that included 100 COMIRNATY recipients and 100 placebo recipients, unsolicited adverse events were reported by 29 (29%) participants in the COMIRNATY group and 15 (15%) participants in the placebo group. The higher frequency of reported unsolicited adverse events among COMIRNATY recipients compared with placebo recipients was primarily attributed to events that are consistent with adverse reactions solicited among participants in the reactogenicity subset (Table 5 and Table 6).
Throughout the placebo-controlled safety follow-up period, Bell’s palsy (facial paralysis) was reported by 4 participants in the COMIRNATY group and 2 participants in the placebo group. Onset of facial paralysis was Day 37 after Dose 1 (participant did not receive Dose 2) and Days 3, 9, and 48 after Dose 2. In the placebo group the onset of facial paralysis was Day 32 and Day 102. Currently available information is insufficient to determine a causal relationship with the vaccine. In the analysis of blinded, placebo-controlled follow-up, there were no other notable patterns or numerical imbalances between treatment groups for specific categories of non-serious adverse events (including other neurologic or neuro-inflammatory, and thrombotic events) that would suggest a causal relationship to COMIRNATY. In the analysis of unblinded follow-up, there were no notable patterns of specific categories of non-serious adverse events that would suggest a causal relationship to COMIRNATY.
Serious Adverse Events
Participants 16 through 55 years of age in Study 2 who had received at least 1 dose of vaccine or placebo (COMIRNATY = 12,995; placebo = 13,026), reported serious adverse events from Dose 1 up to the participant unblinding date in ongoing follow-up as follows: 103 (0.8%) COMIRNATY recipients and 117 (0.9%) placebo recipients. In a similar analysis, in participants 56 years of age and older (8,931 COMIRNATY group and 8,895 placebo group), serious adverse events were reported by 165 (1.8%) COMIRNATY recipients and 151 (1.7%) placebo recipients who received at least 1 dose of COMIRNATY or placebo, respectively. In these analyses, 58.2% of study participants had at least 4 months of follow-up after Dose 2. Among participants with confirmed stable HIV infection serious adverse events from Dose 1 up to the participant unblinding date in ongoing follow-up were reported by 2 (2%) COMIRNATY recipients and 2 (2%) placebo recipients.
In the analysis of blinded, placebo-controlled follow-up, there were no notable patterns between treatment groups for specific categories of serious adverse events (including neurologic, neuro-inflammatory, and thrombotic events) that would suggest a causal relationship to COMIRNATY. In the analysis of unblinded follow-up, there were no notable patterns of specific categories of serious adverse events that would suggest a causal relationship to COMIRNATY.
Two-Dose Series (Original Monovalent) in Vaccine-Naïve Adolescents 12 Through 15 Years of Age
Study 2 was a Phase 1/2/3 multicenter, randomized, saline placebo-controlled, double-blinded (Phase 2/3), dose-finding, vaccine candidate-selection and efficacy study. In Study 2, 2,260 adolescents (1,131 COMIRNATY; 1,129 placebo) were 12 through 15 years of age. At the time of the analysis of the ongoing Study 2 with a data cutoff of September 2, 2021, there were 1,559 (69.0%) adolescents (786 COMIRNATY and 773 placebo) 12 through 15 years of age followed for ≥4 months after the second dose.
Demographic characteristics in Study 2 were generally similar with regard to age, sex, race, and ethnicity among adolescents who received COMIRNATY and those who received placebo. Overall, among the adolescents who received COMIRNATY, 50.1% were male and 49.9% were female, 85.8% were White, 4.6% were Black or African American, 11.7% were Hispanic/Latino, 6.4% were Asian, and 0.4% were American Indian/Alaska Native.
In Study 2, participants 12 through 15 years of age in the reactogenicity subset were monitored using an electronic diary for solicited local and systemic reactions and use of antipyretic medication after each vaccination. Participants were also monitored for unsolicited adverse events throughout the study (from Dose 1 through 1 month [all unsolicited adverse events] or through 6 months [serious adverse events] after the last vaccination). Table 7 and Table 8 present the frequency and severity of solicited local and systemic reactions, respectively, within 7 days following Dose 1 or Dose 2 of COMIRNATY.
Local and Systemic Solicited Adverse Reactions
In adolescents 12 through 15 years of age after receiving Dose 2, the mean duration of pain at the injection site was 2.5 days (range 1 to 11 days), for redness 1.8 days (range 1 to 5 days), and for swelling 1.6 days (range 1 to 5 days) in the COMIRNATY group.
Table 7: Study 2 – Frequency and Percentages of Adolescents With Solicited Local Reactions, by Maximum Severity, Within 7 Days After Each Dose – Adolescents 12 Through 15 Years of Age – Safety Population* Note: Reactions were collected in the electronic diary (e-diary) from Day 1 to Day 7 after vaccination. - * Randomized participants in the safety analysis population who received at least 1 dose of the study intervention.
- † Vaccine encoding the viral spike (S) glycoprotein of SARS-CoV-2 Wuhan-Hu-1 strain (Original).
- ‡ N = Number of participants reporting at least 1 yes or no response for the specified reaction after the specified dose.
- § n = Number of participants with the specified reaction.
- ¶ Mild: >2.0 to ≤5.0 cm; Moderate: >5.0 to ≤10.0 cm; Severe: >10.0 cm.
- # Mild: does not interfere with activity; Moderate: interferes with activity; Severe: prevents daily activity.
COMIRNATY†
Dose 1
N‡=1127
n§ (%)
Placebo
Dose 1
N‡=1127
n§ (%)
COMIRNATY†
Dose 2
N‡=1097
n§ (%)
Placebo
Dose 2
N‡=1078
n§ (%)
Redness¶
Any (>2 cm)
65 (5.8)
12 (1.1)
55 (5.0)
10 (0.9)
Mild
44 (3.9)
11 (1.0)
29 (2.6)
8 (0.7)
Moderate
20 (1.8)
1 (0.1)
26 (2.4)
2 (0.2)
Severe
1 (0.1)
0 (0.0)
0 (0.0)
0 (0.0)
Swelling¶
Any (>2 cm)
78 (6.9)
11 (1.0)
54 (4.9)
6 (0.6)
Mild
55 (4.9)
9 (0.8)
36 (3.3)
4 (0.4)
Moderate
23 (2.0)
2 (0.2)
18 (1.6)
2 (0.2)
Severe
0 (0.0)
0 (0.0)
0 (0.0)
0 (0.0)
Pain at the injection site#
Any
971 (86.2)
263 (23.3)
866 (78.9)
193 (17.9)
Mild
467 (41.4)
227 (20.1)
466 (42.5)
164 (15.2)
Moderate
493 (43.7)
36 (3.2)
393 (35.8)
29 (2.7)
Severe
11 (1.0)
0 (0.0)
7 (0.6)
0 (0.0)
Table 8: Study 2 – Frequency and Percentages of Adolescents With Solicited Systemic Reactions, by Maximum Severity, Within 7 Days After Each Dose – Adolescents 12 Through 15 Years of Age – Safety Population* Note: Events and use of antipyretic or pain medication were collected in the electronic diary (e-diary) from Day 1 to Day 7 after each dose. - * Randomized participants in the safety analysis population who received at least 1 dose of the study intervention.
- † Vaccine encoding the viral spike (S) glycoprotein of SARS-CoV-2 Wuhan-Hu-1 strain (Original).
- ‡ N = Number of participants reporting at least 1 yes or no response for the specified event after the specified dose.
- § n = Number of participants with the specified reaction.
- ¶ Mild: does not interfere with activity; Moderate: some interference with activity; Severe: prevents daily activity.
- # Mild: 1 to 2 times in 24 hours; Moderate: >2 times in 24 hours; Severe: requires intravenous hydration.
- Þ Mild: 2 to 3 loose stools in 24 hours; Moderate: 4 to 5 loose stools in 24 hours; Severe: 6 or more loose stools in 24 hours.
- ß Severity was not collected for use of antipyretic or pain medication.
COMIRNATY†
Dose 1
N‡=1127
n§ (%)
Placebo
Dose 1
N‡=1127
n§ (%)
COMIRNATY†
Dose 2
N‡=1097
n§ (%)
Placebo
Dose 2
N‡=1078
n§ (%)
Fever
≥38.0℃
114 (10.1)
12 (1.1)
215 (19.6)
7 (0.6)
≥38.0℃ to 38.4℃
74 (6.6)
8 (0.7)
107 (9.8)
5 (0.5)
>38.4℃ to 38.9℃
29 (2.6)
2 (0.2)
83 (7.6)
1 (0.1)
>38.9℃ to 40.0℃
10 (0.9)
2 (0.2)
25 (2.3)
1 (0.1)
>40.0℃
1 (0.1)
0 (0.0)
0 (0.0)
0 (0.0)
Fatigue¶
Any
677 (60.1)
457 (40.6)
726 (66.2)
264 (24.5)
Mild
278 (24.7)
250 (22.2)
232 (21.1)
133 (12.3)
Moderate
384 (34.1)
199 (17.7)
468 (42.7)
127 (11.8)
Severe
15 (1.3)
8 (0.7)
26 (2.4)
4 (0.4)
Headache¶
Any
623 (55.3)
396 (35.1)
708 (64.5)
264 (24.5)
Mild
361 (32.0)
256 (22.7)
302 (27.5)
170 (15.8)
Moderate
251 (22.3)
131 (11.6)
384 (35.0)
93 (8.6)
Severe
11 (1.0)
9 (0.8)
22 (2.0)
1 (0.1)
Chills¶
Any
311 (27.6)
109 (9.7)
455 (41.5)
74 (6.9)
Mild
195 (17.3)
82 (7.3)
221 (20.1)
53 (4.9)
Moderate
111 (9.8)
25 (2.2)
214 (19.5)
21 (1.9)
Severe
5 (0.4)
2 (0.2)
20 (1.8)
0 (0.0)
Vomiting#
Any
31 (2.8)
10 (0.9)
29 (2.6)
12 (1.1)
Mild
30 (2.7)
8 (0.7)
25 (2.3)
11 (1.0)
Moderate
0 (0.0)
2 (0.2)
4 (0.4)
1 (0.1)
Severe
1 (0.1)
0 (0.0)
0 (0.0)
0 (0.0)
DiarrheaÞ
Any
90 (8.0)
82 (7.3)
65 (5.9)
44 (4.1)
Mild
77 (6.8)
72 (6.4)
59 (5.4)
39 (3.6)
Moderate
13 (1.2)
10 (0.9)
6 (0.5)
5 (0.5)
Severe
0 (0.0)
0 (0.0)
0 (0.0)
0 (0.0)
New or worsened muscle pain¶
Any
272 (24.1)
148 (13.1)
355 (32.4)
90 (8.3)
Mild
125 (11.1)
88 (7.8)
152 (13.9)
51 (4.7)
Moderate
145 (12.9)
60 (5.3)
197 (18.0)
37 (3.4)
Severe
2 (0.2)
0 (0.0)
6 (0.5)
2 (0.2)
New or worsened joint pain¶
Any
109 (9.7)
77 (6.8)
173 (15.8)
51 (4.7)
Mild
66 (5.9)
50 (4.4)
91 (8.3)
30 (2.8)
Moderate
42 (3.7)
27 (2.4)
78 (7.1)
21 (1.9)
Severe
1 (0.1)
0 (0.0)
4 (0.4)
0 (0.0)
Use of antipyretic or pain medicationß
413 (36.6)
111 (9.8)
557 (50.8)
95 (8.8)
Unsolicited Adverse Events
In Study 2, 2,260 adolescents (1,131 COMIRNATY; 1,129 placebo) were 12 through 15 years of age. Of these, 634 (56.1%) participants in the COMIRNATY group and 629 (55.7%) participants in the placebo group had follow-up time between ≥4 months to <6 months after Dose 2 in the blinded placebo-controlled follow-up period with an additional 152 (13.4%) and 144 (12.8%) with ≥6 months of blinded follow-up time in the COMIRNATY and placebo groups, respectively.
A total of 1,113 (98.4%) participants 12 through 15 years of age originally randomized to COMIRNATY had ≥6 months total (blinded and unblinded) follow-up after Dose 2. An analysis of all unsolicited adverse events in Study 2 from Dose 1 up to the participant unblinding date was conducted. Among participants 12 through 15 years of age who received at least 1 dose of study vaccine, unsolicited adverse events were reported by 95 (8.4%) participants in the COMIRNATY group and 113 (10.0%) participants in the placebo group.
In an analysis of all unsolicited adverse events reported during blinded follow-up from Dose 1 through 1 month after Dose 2, in adolescents 12 to 15 years of age, those assessed as adverse reactions not already captured by solicited local and systemic reactions were lymphadenopathy (9 vs. 2), and nausea (5 vs. 2).
In the analysis of blinded, placebo-controlled follow-up, there were no other notable patterns or numerical imbalances between treatment groups for specific categories of unsolicited adverse events (including other neurologic or neuro-inflammatory, and thrombotic events) that would suggest a causal relationship to COMIRNATY. In the analysis of unblinded follow-up, there were no notable patterns of specific categories of non-serious adverse events that would suggest a causal relationship to COMIRNATY.
Serious Adverse Events
In Study 2, among participants 12 through 15 years of age who had received at least 1 dose of vaccine or placebo (COMIRNATY = 1,131; placebo = 1,129), serious adverse events from Dose 1 up to the participant unblinding date in ongoing follow-up were reported by 10 (0.9%) COMIRNATY recipients and 2 (0.2%) placebo recipients. In these analyses, 69.0% of study participants had at least 4 months of follow-up after Dose 2. In the analysis of blinded, placebo-controlled follow-up, there were no notable patterns between treatment groups for specific categories of serious adverse events (including neurologic, neuro-inflammatory, and thrombotic events) that would suggest a causal relationship to COMIRNATY. In the analysis of unblinded follow-up, there were no notable patterns of specific categories of serious adverse events that would suggest a causal relationship to COMIRNATY.
Single Dose (Original Monovalent) in Vaccine-Experienced Individuals 12 Years of Age and Older
16 Years of Age and Older
In Study 4, a double-blind placebo-controlled booster study, 5,081 participants 16 years of age and older recruited from Study 2 received a booster dose of COMIRNATY 10.8 months (median time, range of 5.0 to 12.6 months) after completing the primary series of COMIRNATY series and had a median follow-up time of 2.9 months based on data up to the cutoff date of February 8, 2022. The median age of participants who received COMIRNATY or placebo was 53.0 years (range 16 through 87 years of age), 49.1% were male and 50.9% were female, 79.0% were White, 14.9% were Hispanic/Latino, 9.2% were Black or African American, 5.5% were Asian, and 1.7% were American Indian/Alaska Native.
Adverse reactions reported in participants receiving a booster dose of COMIRNATY were similar to those previously observed in participants receiving COMIRNATY as part of the primary series. Lymphadenopathy occurred in 141 (2.8%) participants who received a booster dose of COMIRNATY and in 83 (0.4%) participants who received COMIRNATY as a primary series.
18 Years Through 55 Years of Age
A subset of 306 Study 2 Phase 2/3 participants 18 through 55 years of age received a booster dose of COMIRNATY 6.8 months (median time, range 4.8 to 8.0 months) after completing the primary series. These participants had a median follow-up time of 8.3 months up to a data cutoff date of November 22, 2021. Among the 306 participants, the median age was 42 years (range 19 through 55 years of age), 45.8% were male and 54.2% were female, 81.4% were White, 27.8% were Hispanic/Latino, 9.2% were Black or African American, 5.2% were Asian, and 0.7% were American Indian/Alaska Native.
Adverse reactions reported in participants receiving a booster dose of COMIRNATY were similar to those previously observed in participants receiving COMIRNATY as part of the primary series. Lymphadenopathy occurred in 16 (5.2%) of participants who received a booster dose of COMIRNATY and 83 (0.4%) in participants who received COMIRNATY as a primary series.
12 Years Through 17 Years of Age
A subset of 65 Study 4 participants 12 through 17 years of age received a booster dose of COMIRNATY 13.3 months (median time, range 6.5 to 16.9 months) after completing the primary series and had a median follow-up time of 5.6 months up to a data cutoff date of July 14, 2022. The median age of participants was 14 years (range 12 through 17 years of age), 49.2% were male and 50.8% were female, 76.9% were White, 16.9% were Hispanic/Latino, 13.8% were Black or African American, 7.7% were Asian, and 1.5% were American Indian/Alaska Native.
Adverse reactions reported in participants receiving a booster dose of COMIRNATY were similar to those previously observed in participants receiving COMIRNATY as part of the primary series. There were no cases of lymphadenopathy reported in participants who received a booster dose of COMIRNATY.
12 Years Through 15 Years of Age
A subset of 825 Study 2 Phase 2/3 participants 12 through 15 years of age received a booster dose of COMIRNATY 11.2 months (median time, range 6.3 to 20.1 months) after completing the primary series and had a median follow-up time of 9.5 months up to a data cutoff date of November 3, 2022. The median age of participants was 14.0 years (range 13 through 15 years of age), 49.3% were male and 50.7% were female, 83.5% were White, 10.8% were Hispanic/Latino, 4.6% were Black or African American, 7.5% were Asian, and 0.4% were American Indian/Alaska Native.
Adverse reactions reported in participants receiving a booster dose of COMIRNATY were similar to those previously observed in participants receiving COMIRNATY as part of the primary series. Lymphadenopathy occurred in 8 (1.0%) participants who received a booster dose of COMIRNATY and in 9 (0.8%) participants who received COMIRNATY as a primary series.
Single Dose (Bivalent Original and BA.4/BA.5) in Vaccine-Experienced Individuals 12 Years of Age and Older
A subset of 107 Study 5 Phase 2/3 participants 12 through 17 years of age, 313 participants 18 through 55 years of age and 306 participants 56 years of age and older previously vaccinated with a 2-dose primary series and 1 booster dose of COMIRNATY, went on to receive a second booster dose with Pfizer-BioNTech COVID-19 Vaccine, Bivalent.
Participants received a second booster dose 11.1 months (median time; range 5.4 to 16.9 months) after receiving the first booster dose and had a median follow-up time of 1.5 months up to a data cutoff date of October 31, 2022. The median age was 48.0 years, 42.7% were male, 57.3% were female, 80.6% were White, 11.4% were Hispanic/Latino, 5.9% were Asian, and 11.4% were Black or African American.
Local and Systemic Solicited Adverse Reactions
Table 9 and Table 10 present the frequency and severity of reported solicited local reactions and systemic reactions, respectively, within 7 days of a second booster dose of Pfizer-BioNTech COVID-19 Vaccine, Bivalent.
In participants 12 years of age and older who received a second booster dose, the mean duration of injection site pain was 2.1 to 2.4 days (range 1 to 11 days), injection site redness was 1.5 to 2.5 days (range 1 to 4 days), and injection site swelling was 1.3 to 1.9 days (range 1 to 4 days), respectively.
Table 9: Study 5 – Frequency and Percentages of Participants With Solicited Local Reactions, by Maximum Severity, Within 7 Days After a Second Booster Dose – Participants 12 Years of Age and Older – Safety Population Note: Adverse Reactions were collected in the electronic diary (e-diary) from day of vaccination (Day 1) through Day 7 after the study vaccination. - * Vaccine encoding the viral spike (S) glycoprotein of SARS-CoV-2 Wuhan-Hu-1 strain (Original) and Omicron variant lineages BA.4 and BA.5 (Omicron BA.4/BA.5).
- † N = Number of participants reporting at least 1 yes or no response for the specified reaction after the study vaccination.
- ‡ n = Number of participants with the specified adverse reaction.
- § N = 310 for redness and pain at injection site in participants 18 through 55 years of age; N = 301 for pain at injection site in participants 56 years of age and older.
- ¶ Mild: >2.0 to 5.0 cm; Moderate: >5.0 to 10.0 cm; Severe: >10.0 cm.
- # Mild: does not interfere with activity; Moderate: interferes with activity; Severe: prevents daily activity.
Pfizer-BioNTech COVID-19 Vaccine, Bivalent*
12 Through 17 Years of Age
N†=107
n‡ (%)
18 Through 55 Years of Age
n‡ (%)
56 Years of Age and Older
n‡ (%)
Redness¶
Any (>2 cm)
6 (5.6)
21 (6.8%)
11 (3.7%)
Mild
4 (3.7)
16 (5.2%)
7 (2.3)
Moderate
2 (1.9)
5 (1.6)
4 (1.3%)
Severe
0
0
0
Swelling¶
Any (>2 cm)
8 (7.5)
23 (7.4%)
8 (2.7)
Mild
6 (5.6)
19 (6.1%)
5 (1.7)
Moderate
2 (1.9)
4 (1.3)
3 (1.0)
Severe
0
0
0
Pain at the injection site#
Any
75 (70.1)
236 (76.1)
172 (57.1)
Mild
45 (42.1)
178 (57.4)
147 (48.8)
Moderate
29 (27.1)
58 (18.7)
24 (8.0)
Severe
1 (0.9)
0
1 (0.3)
Table 10: Study 5 – Frequency and Percentages of Participants With Solicited Systemic Adverse Reactions, by Maximum Severity, Within 7 Days After a Second Booster Dose – Participants 12 Years of Age and Older – Safety Population Note: Adverse reactions and use of antipyretic or pain medication were collected in the electronic diary (e-diary) from day of vaccination (Day 1) through Day 7 after the study vaccination. - * Vaccine encoding the viral spike (S) glycoprotein of SARS-CoV-2 Wuhan-Hu-1 strain (Original) and Omicron variant lineages BA.4 and BA.5 (Omicron BA.4/BA.5).
- † N = Number of participants reporting at least 1 yes or no response for the specified adverse reaction after the study vaccination.
- ‡ n = Number of participants with the specified adverse reaction.
- § N = 301 for fever, fatigue and diarrhea in participants 56 years of age and older.
- ¶ Mild: does not interfere with activity; Moderate: some interference with activity; Severe: prevents daily activity.
- # Mild: 1 to 2 times in 24 hours; Moderate: >2 times in 24 hours; Severe: requires intravenous hydration.
- Þ Mild: 2 to 3 loose stools in 24 hours; Moderate: 4 to 5 loose stools in 24 hours; Severe: 6 or more loose stools in 24 hours.
- ß Severity was not collected for use of antipyretic or pain medication.
Pfizer-BioNTech COVID-19 Vaccine, Bivalent*
12 Through 17 Years of Age
N†=107
n‡ (%)
18 Through 55 Years of Age
N†=309
n‡ (%)
56 Years of Age and Older
n‡ (%)
Fever
≥38.0℃
10 (9.3)
15 (4.9)
14 (4.7)
≥38.0℃ to 38.4℃
7 (6.5)
9 (2.9)
10 (3.3)
>38.4℃ to 38.9℃
2 (1.9)
6 (1.9)
3 (1.0)
>38.9℃ to 40.0℃
1 (0.9)
0
0
>40.0℃
0
0
0
Fatigue¶
Any
72 (67.3)
189 (61.2)
116 (38.5)
Mild
27 (25.2)
83 (26.9)
56 (18.6)
Moderate
45 (42.1)
100 (32.4)
56 (18.6)
Severe
0
6 (1.9)
4 (1.3)
Headache¶
Any
54 (50.5)
144 (46.6)
92 (30.7)
Mild
28 (26.2)
87 (28.2)
62 (20.7)
Moderate
26 (24.3)
55 (17.8)
30 (10.0)
Severe
0
2 (0.6)
0
Chills¶
Any
25 (23.4)
68 (22.0)
36 (12.0)
Mild
19 (17.8)
38 (12.3)
21 (7.0)
Moderate
6 (5.6)
28 (9.1)
14 (4.7)
Severe
0
2 (0.6)
1 (0.3)
Vomiting#
Any
3 (2.8)
6 (1.9)
2 (0.7)
Mild
3 (2.8)
5 (1.6)
2 (0.7)
Moderate
0
1 (0.3)
0
Severe
0
0
0
DiarrheaÞ
Any
7 (6.5)
33 (10.7)
29 (9.6)
Mild
7 (6.5)
27 (8.7)
23 (7.6)
Moderate
0
5 (1.6)
6 (2.0)
Severe
0
1 (0.3)
0
New or worsened muscle pain¶
Any
28 (26.2)
94 (30.4)
54 (18.0)
Mild
12 (11.2)
47 (15.2)
30 (10.0)
Moderate
16 (15.0)
47 (15.2)
24 (8.0)
Severe
0
0
0
New or worsened joint pain¶
Any
13 (12.1)
46 (14.9)
36 (12.0)
Mild
9 (8.4)
21 (6.8)
20 (6.7)
Moderate
4 (3.7)
25 (8.1)
16 (5.3)
Severe
0
0
0
Use of antipyretic or pain medicationß
36 (33.6)
105 (34.0)
74 (24.7)
Unsolicited Adverse Events
Among participants 12 years of age and older, unsolicited adverse events were reported by 48 (6.6%) participants who received a second booster dose through 1 month after the booster dose. Lymphadenopathy occurred in 7 (1.0%) participants.
Concomitant Administration of COMIRNATY (Original Monovalent) With Influenza Vaccine in Adults 18 Years Through 64 Years of Age
In Study 8, a Phase 3 study, participants 18 through 64 years of age who received COMIRNATY concomitantly administered with Influenza Vaccine (Afluria Quadrivalent) followed 1 month later by saline placebo (n = 564) were compared with participants who received influenza vaccine with saline placebo followed 1 month later by COMIRNATY (n = 564).
Demographic characteristics in Study 8 among the participants in the concomitant administration and separate administration groups were similar with regard to age, sex, race, and ethnicity. Among the 564 participants in the concomitant administration group, the median age was 39.0 years (range 18 through 64 years of age), 36.9% were male and 63.1% were female, 79.1% were White, 12.9% were Asian, and 0.9% were Hispanic/Latino.
Solicited local and systemic adverse reactions were reported more frequently by participants who received COMIRNATY concomitantly with influenza vaccine, compared with participants who received COMIRNATY alone. The most common adverse reactions reported in the concomitant administration group and after COMIRNATY alone were injection site pain (COMIRNATY injection site) (86.2% and 84.4%, respectively), fatigue (64.0% and 50.8%, respectively), and headache (47.2% and 37.8%, respectively).
Children 5 Years Through 11 Years of Age
Two-Dose Series (Original Monovalent) in Vaccine-Naïve Children 5 Years Through 11 Years of Age
Study 3 is a Phase 1/2/3 multicenter, randomized, dose-finding, open label (Phase 1) and multinational, placebo controlled (saline placebo), observer-blind, immunogenicity and efficacy (Phase 2/3) study that has evaluated 4,695 participants 5 through 11 years of age, of whom 3,109 participants received COMIRNATY and 1,538 participants received placebo in Phase 2/3.
Demographic characteristics were generally similar with regard to age, sex, race, and ethnicity among participants who received COMIRNATY and those who received placebo. Overall, among the 4,647 participants who received at least 1 dose of COMIRNATY or placebo, 51.4% were male and 48.6% were female, 77.5% were White, 6.0% were Black or African American, 17.0% were Hispanic/Latino, 8.1% were Asian, and 0.4% were American Indian/Alaska Native.
In an analysis of Study 3 (Phase 2/3), 4,632 participants 5 through 11 years of age who received a 2-dose primary series [3,100 COMIRNATY; 1,532 placebo] have been followed a median of 1.9 months (range 0.1 to 7.5 months) after the second dose in the blinded placebo-controlled follow-up period up to the cutoff date of May 20, 2022.
In Study 3, participants 5 years through 11 years of age in the reactogenicity subset were monitored using an electronic diary for solicited local and systemic adverse reactions and use of antipyretic medication after each vaccination. Participants were also monitored for unsolicited adverse events throughout the study (from Dose 1 through 1 month [all unsolicited adverse events] or through 6 months [serious adverse events] after the last vaccination). Table 11 and Table 12 present the frequency and severity of solicited local and systemic reactions, respectively, within 7 days following Dose 1 or Dose 2 of COMIRNATY.
Solicited Local and Systemic Adverse Reactions
The mean duration of pain at the injection site after Dose 2 was 2.3 days (range 1 to 37 days), for redness 2.0 days (range 1 to 10 days), and for swelling 2.2 days (range 1 to 16 days) for children in the COMIRNATY group in the blinded placebo-controlled follow-up period up to the cutoff date of May 20, 2022.
Table 11: Study 3 – Frequency and Percentages of Participants With Solicited Local Reactions, by Maximum Severity, Within 7 Days After Each Dose – Children 5 Through 11 Years of Age – Safety Population* Note: Reactions were collected in an electronic diary (e-diary) from Day 1 to Day 7 after vaccination. - * Randomized participants who received at least 1 dose of the study intervention.
- † Vaccine encoding the viral spike (S) glycoprotein of SARS-CoV-2 Wuhan-Hu-1 strain (Original).
- ‡ N = Number of participants reporting at least 1 yes or no response for the specified reaction after the specified dose.
- § n = Number of participants with the specified reaction.
- ¶ Mild: ≥0.5 to <2.0 cm; Moderate: >2.0 to <7.0 cm; Severe: >7.0 cm.
- # Mild: does not interfere with activity; Moderate: interferes with activity; Severe: prevents daily activity.
COMIRNATY†
Dose 1
N‡=3096
n§ (%)
Placebo
Dose 1
N‡=1531 to 1532
n§ (%)
COMIRNATY†
Dose 2
N‡=3064
n§ (%)
Placebo
Dose 2
N‡=1521 to 1522
n§ (%)
Redness¶
Any (≥0.5 cm)
434 (14.0)
91 (5.9)
575 (18.8)
79 (5.2)
Mild
287 (9.3)
78 (5.1)
315 (10.3)
57 (3.7)
Moderate
146 (4.7)
11 (0.7)
257 (8.4)
20 (1.3)
Severe
1 (0.0)
2 (0.1)
3 (0.1)
2 (0.1)
Swelling¶
Any (≥0.5 cm)
320 (10.3)
46 (3.0)
450 (14.7)
41 (2.7)
Mild
177 (5.7)
28 (1.8)
247 (8.1)
30 (2.0)
Moderate
142 (4.6)
18 (1.2)
203 (6.6)
11 (0.7)
Severe
1 (0.0)
0
0
0
Pain at the injection site#
Any
2258 (72.9)
482 (31.5)
2181 (71.2)
434 (28.5)
Mild
1810 (58.5)
434 (28.3)
1642 (53.6)
389 (25.6)
Moderate
442 (14.3)
48 (3.1)
533 (17.4)
44 (2.9)
Severe
6 (0.2)
0
6 (0.2)
1 (0.1)
Table 12: Study 3 – Frequency and Percentages of Participants With Solicited Systemic Reactions, by Maximum Severity, Within 7 Days After Each Dose – Children 5 Through 11 Years of Age – Safety Population* Note: Events and use of antipyretic or pain medication were collected in an electronic diary (e-diary) from Day 1 to Day 7 after each dose. - * Randomized participants who received at least 1 dose of the study intervention.
- † Vaccine encoding the viral spike (S) glycoprotein of SARS-CoV-2 Wuhan-Hu-1 strain (Original).
- ‡ N = Number of participants reporting at least 1 yes or no response for the specified event after the specified dose.
- § n = Number of participants with the specified reaction.
- ¶ Mild: does not interfere with activity; Moderate: some interference with activity; Severe: prevents daily activity.
- # Mild: 1 to 2 times in 24 hours; Moderate: >2 times in 24 hours; Severe: requires intravenous hydration.
- Þ Mild: 2 to 3 loose stools in 24 hours; Moderate: 4 to 5 loose stools in 24 hours; Severe: 6 or more loose stools in 24 hours.
- ß Severity was not collected for use of antipyretic or pain medication.
COMIRNATY†
Dose 1
N‡=3096
n§ (%)
Placebo
Dose 1
N‡=1531 to 1532
n§ (%)
COMIRNATY†
Dose 2
N‡=3064
n§ (%)
Placebo
Dose 2
N‡=1521 to 1522
n§ (%)
Fever
≥38.0℃
64 (2.1)
21 (1.4)
193 (6.3)
21 (1.4)
≥38.0℃ to 38.4℃
37 (1.2)
10 (0.7)
101 (3.3)
13 (0.9)
>38.4℃ to 38.9℃
22 (0.7)
9 (0.6)
70 (2.3)
5 (0.3)
>38.9℃ to 40.0℃
4 (0.1)
2 (0.1)
21 (0.7)
3 (0.2)
>40.0℃
1 (0.0)
0
1 (0.0)
0
Fatigue¶
Any
1067 (34.5)
496 (32.4)
1200 (39.2)
383 (25.2)
Mild
702 (22.7)
323 (21.1)
665 (21.7)
230 (15.1)
Moderate
360 (11.6)
171 (11.2)
508 (16.6)
149 (9.8)
Severe
5 (0.2)
2 (0.1)
27 (0.9)
4 (0.3)
Headache¶
Any
703 (22.7)
372 (24.3)
870 (28.4)
284 (18.7)
Mild
530 (17.1)
275 (18.0)
576 (18.8)
201 (13.2)
Moderate
170 (5.5)
91 (5.9)
286 (9.3)
82 (5.4)
Severe
3 (0.1)
6 (0.4)
8 (0.3)
1 (0.1)
Chills¶
Any
174 (5.6)
84 (5.5)
301 (9.8)
66 (4.3)
Mild
138 (4.5)
69 (4.5)
205 (6.7)
52 (3.4)
Moderate
36 (1.2)
15 (1.0)
94 (3.1)
13 (0.9)
Severe
0
0
2 (0.1)
1 (0.1)
Vomiting#
Any
63 (2.0)
30 (2.0)
62 (2.0)
27 (1.8)
Mild
52 (1.7)
28 (1.8)
56 (1.8)
22 (1.4)
Moderate
11 (0.4)
2 (0.1)
5 (0.2)
5 (0.3)
Severe
0
0
1 (0.0)
0
DiarrheaÞ
Any
198 (6.4)
75 (4.9)
166 (5.4)
76 (5.0)
Mild
184 (5.9)
72 (4.7)
149 (4.9)
70 (4.6)
Moderate
14 (0.5)
3 (0.2)
15 (0.5)
6 (0.4)
Severe
0
0
2 (0.1)
0
New or worsened muscle pain¶
Any
289 (9.3)
126 (8.2)
368 (12.0)
104 (6.8)
Mild
206 (6.7)
96 (6.3)
245 (8.0)
68 (4.5)
Moderate
82 (2.6)
30 (2.0)
122 (4.0)
36 (2.4)
Severe
1 (0.0)
0
1 (0.0)
0
New or worsened joint pain¶
Any
106 (3.4)
70 (4.6)
159 (5.2)
57 (3.7)
Mild
71 (2.3)
56 (3.7)
103 (3.4)
42 (2.8)
Moderate
35 (1.1)
14 (0.9)
56 (1.8)
15 (1.0)
Severe
0
0
0
0
Use of antipyretic or pain medicationß
436 (14.1)
135 (8.8)
601 (19.6)
111 (7.3)
Unsolicited Adverse Events
In the following analyses of Study 3 in participants 5 through 11 years of age, 3,109 participants received COMIRNATY and 1,538 participants received placebo. Among those who received 2 doses of COMIRNATY or placebo, 1,185 participants in the COMIRNATY group and 575 participants in the placebo group had follow-up time ≥4 to <6 months and 296 participants in the COMIRNATY group and 150 participants in the placebo group had follow-up time of >6 months in the blinded placebo-controlled follow-up period.
Among participants who received at least 1 dose of study vaccine, unsolicited adverse events were reported by 333 (10.7%) participants in the COMIRNATY group and 150 (9.8%) participants in the placebo group.
In an analysis of all unsolicited adverse events reported following administration of Dose 1 to one month after administration of Dose 2, the adverse reactions (excluding reactions reported as solicited adverse reactions) in participants who received COMIRNATY compared with participants who received placebo were lymphadenopathy (n=23; 0.7% vs. n=4; 0.3%), nausea (n=7; 0.2% vs. n=3; 0.2%), decreased appetite (n=3; 0.1% vs. n=2; 0.1%), malaise (n=2; 0.1% vs. n=0), and night sweats (n=1; 0.0% vs. n=0).
Serious Adverse Events
Serious adverse events, from administration of Dose 1 to the participant unblinding date, were reported in 8 (0.3%) COMIRNATY recipients and in 2 (0.1%) placebo recipients. No serious adverse events were considered related to vaccination.
Single Dose (Original Monovalent) in Vaccine-Experienced Children 5 Years Through 11 Years of Age
In Phase 2/3 of Study 3, 2,408 participants 5 years through 11 years of age received a first booster dose of COMIRNATY at a median of 7.9 months (range 5.3 to 19.4 months) after completing the primary series. These participants had a median safety follow-up of 6.4 months from vaccination through the data cutoff date of February 28, 2023. The median age was 8.0 years (range 5 through 11 years of age), 50.5% were male and 49.5% were female, 76.3% were White, 5.9% were Black or African American, 16.9% were Hispanic/Latino, 8.2% were Asian, and 0.5% were American Indian/Alaska Native.
Solicited Local and Systemic Adverse Reactions
The frequency of solicited adverse reactions reported in participants receiving a booster dose of COMIRNATY were generally consistent with those reported in pediatric participants receiving COMIRNATY as part of the two-dose series.
Unsolicited Adverse Events
Lymphadenopathy occurred in 46 (1.9%) participants who received a booster dose of COMIRNATY and in 23 (0.7%) participants who received COMIRNATY as a primary series.
Serious Adverse Events
Serious adverse events from study vaccination through 6 months after study vaccination were reported by 10 (0.4%) COMIRNATY recipients. No serious adverse events were considered related to vaccination.
Single Dose (Bivalent Original and BA.4/BA.5) in Vaccine-Experienced Children 5 Years Through 11 Years of Age
In Study 6, 113 participants 5 years through 11 years of age previously vaccinated with a 2-dose primary series and 1 booster dose of COMIRNATY received a second booster (fourth dose) with Pfizer-BioNTech COVID-19 Vaccine, Bivalent (Original and Omicron BA.4/BA.5). Participants received a second booster with Pfizer-BioNTech COVID-19, Bivalent 2.6 to 8.5 months after receiving their third dose with COMIRNATY and had a median follow-up time of 6.3 months (range 1.1 to 6.8 months) up to a data cutoff date of April 20, 2023. The median age of participants was 9 years (range 5 through 11 years of age), 50.4% were male and 49.6% were female, 58.4% were White, 20.4% were Hispanic/Latino, 19.5% were multiracial, 11.5% were Asian, and 8.0% were Black or African American.
Solicited Local and Systemic Adverse Reactions
The frequency of solicited adverse reactions reported in participants receiving a second booster dose of Pfizer-BioNTech COVID-19 Vaccine, Bivalent (Original and BA.4/BA.5) were generally consistent with those reported in pediatric participants receiving COMIRNATY.
Unsolicited Adverse Events
Lymphadenopathy was reported in 1 (0.9%) participant who received Pfizer-BioNTech COVID-19 Vaccine, Bivalent (Original and BA.4/BA.5).
Serious Adverse Events
No serious adverse events were reported.
Single Dose (Monovalent XBB.1.5) in Vaccine-Naïve Children 5 Years Through 11 Years of Age
In a subset of Study 6, the safety of a single dose of COMIRNATY (encoding the viral spike (S) glycoprotein of SARS-CoV-2 Omicron XBB.1.5) was evaluated in 310 COVID-19 vaccine-naïve participants 5 through 11 years of age. Participants had a median follow-up time of 6.4 months (range 1.7 to 6.9 months). The median age of participants was 7.0 years (range 5 through 11 years of age), 47.1% were male and 52.9% were female, 41.3% were White, 52.9% were Black or African American, 52.3% were Hispanic or Latino, 1.9% were Asian, and 0.3% were American Indian/Alaska Native.
Solicited Local and Systemic Adverse Reactions
The frequency of solicited adverse reactions reported in participants who received a single dose of COMIRNATY, monovalent (XBB.1.5) were generally consistent with those previously reported by participants receiving COMIRNATY.
Unsolicited Adverse Events
In an analysis of all unsolicited adverse events through 1 month after study vaccination, unsolicited adverse events were reported by 11 (3.5%) COMIRNATY recipients. The adverse reaction not already captured by solicited local and systemic reactions was decreased appetite (n=1; 0.3%).
Serious Adverse Events
Serious adverse events from study vaccination through 6 months after study vaccination were reported by 3 (1.0%) COMIRNATY recipients. No serious adverse events were considered related to vaccination.
6.2 Postmarketing Experience
The following adverse reactions have been identified during postmarketing use of COMIRNATY, Pfizer-BioNTech COVID-19 Vaccine and Pfizer-BioNTech COVID-19 Vaccine, Bivalent. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to vaccine exposure.
Cardiac disorders: myocarditis, pericarditis
Gastrointestinal disorders: diarrhea, vomiting
Immune system disorders: severe allergic reactions, including anaphylaxis, and other hypersensitivity reactions (e.g., rash, pruritus, urticaria, angioedema)
Musculoskeletal and connective tissue disorders: pain in extremity (arm)
Nervous system disorders: syncope, dizziness, febrile seizures (in children 5 through 11 years of age)Cardiovascular Outcomes in Patients Diagnosed With mRNA COVID-19 Vaccine-associated Myocarditis
In a longitudinal retrospective observational cohort study across 38 hospitals in the U.S., information on cardiovascular outcomes was collected on 333 patients 5 through 29 years of age who had been diagnosed with COVID-19 vaccine-associated myocarditis. Among these patients, 322 were confirmed to have received an mRNA COVID-19 vaccine encoding the S glycoprotein of the Original SARS-CoV-2. Of 331 patients, 278 had onset of symptoms following the second dose of a primary series, 33 following the first dose of a primary series, and 20 following a first booster dose1.
Among 307 patients who had been diagnosed with COVID-19 vaccine-associated myocarditis for whom follow-up information was available, 89 reported cardiac symptoms at a median follow-up of 91 days (interquartile range 25-186 days) postvaccination1.
Initial gadolinium-enhanced cardiac magnetic resonance imaging (CMR) was performed on 216 patients, of whom 177 had late gadolinium enhancement (LGE), a marker of myocardial injury. Among 161 patients who had LGE on initial CMR and who had a follow-up gadolinium-enhanced CMR at a median follow-up of 159 days (interquartile range 78-253 days), 98 had persistence of LGE. Overall, the severity of LGE decreased during follow-up. The clinical and prognostic significance of these CMR findings is not known1.
Limitations of this study include potential selection bias towards patients with more severe myocarditis who are more likely to be hospitalized and have CMR, variability in diagnostic testing, and variability in follow-up1.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
All pregnancies have a risk of birth defect, loss, or other adverse outcomes. In the US general population, the estimated background risk of major birth defects, miscarriage and pre-eclampsia in clinically recognized pregnancies is 2% to 4%, 15% to 20%, and 5% to 7%, respectively. There are no available data with COMIRNATY use in pregnant women before 24 weeks gestation to inform about risks for major birth defects and miscarriage.
A developmental toxicity study has been performed in female rats administered the equivalent of a single human dose of COMIRNATY [encoding the viral spike (S) glycoprotein of SARS-CoV-2 Wuhan-Hu-1 strain (Original)] on 4 occasions, twice prior to mating and twice during gestation. These studies revealed no evidence of harm to the fetus due to the vaccine (see Data).
Clinical Considerations
Disease-Associated Maternal and/or Embryo/Fetal Risk
Pregnant individuals infected with SARS-CoV-2 are at increased risk of severe COVID-19 compared with non-pregnant individuals.
Human Data
In Study 9, a randomized, placebo-controlled trial (NCT04754594), COVID-19 vaccine naïve, pregnant women 18 years of age and older received 2 doses of COMIRNATY (Original Monovalent) (n=173 received at least 1 dose) or placebo (n=173 received at least 1 dose), approximately 21 days apart, with the first dose administered at 24 to 34 weeks gestation (i.e., late second trimester and early third trimester of pregnancy). The trial was conducted in Brazil, South Africa, Spain, United Kingdom, and the US. The trial failed to reach target accrual (originally designed to be 4,000 participants), as it was terminated prematurely, resulting in uncertainty regarding differences in outcomes.
In an analysis which excluded participants with HIV infection, a numerical imbalance in pre-eclampsia in COMIRNATY recipients (6 of 161, 3.7%) compared with placebo recipients (2 of 163, 1.2%) was observed in this study. These data are insufficient to establish or exclude a causal relationship between COMIRNATY and pre-eclampsia.
Major or minor congenital anomalies or chromosomal abnormalities were reported in 10 of 156 (6.4%) infants born to women in the COMIRNATY group and 7 of 159 (4.4%) infants born to women in the placebo group (see Table 13). Some infants had more than one event listed in the table. The available trial data are insufficient to establish or exclude vaccine-associated risk of congenital anomalies because COMIRNATY was not administered in the first trimester (the period when the risk of major congenital anomalies is highest). In addition, predisposing factors including genetic risk factors and potential maternal risk factors were reported in some cases in the COMIRNATY group.
Descriptive immunogenicity analyses of geometric mean [neutralizing] titers (GMT) in pregnant participants compared with GMTs from non-pregnant participants enrolled in another study (Study 2) were inconclusive due to study limitations including a small sample size.
Table 13: Study 9 – Number and Percentage of Infants With Major and Minor Congenital Anomalies and Chromosomal Abnormalities Following Maternal Exposure to COMIRNATY or Placebo at or After 24 Weeks of Gestation - * N = number of infants born to women who received at least 1 dose of the study intervention; infants born to maternal participants living with HIV were excluded from this analysis.
- † n = number of infants with event, some infants had more than 1 event.
- ‡ These anomalies occur in the development of a fetus prior to Week 24 of gestation.
- § Among infants born to women in the COMIRNATY group, atrial septal defect (ASD) occurred in 1 infant with Trisomy 21, 1 infant with central nervous system anomalies (see footnote f), 1 infant with a maternal history of congenital heart disease, and 1 infant with significant risks for ASD (maternal alcohol and tobacco use during pregnancy).
- ¶ Among infants born to women in the COMIRNATY group, microcephaly occurred in 1 infant with Trisomy 21 and 1 infant with significant risks for microcephaly (gestational hypertension, thyroid disease, and alcohol use during pregnancy).
- # In 1 infant born to a woman in the COMIRNATY group, the following central nervous system anomalies were observed: lissencephaly and bilateral chorioretinitis.
- Þ Among infants born to women in the COMIRNATY group, patent ductus arteriosus (PDA) occurred in 1 infant who also had Trisomy 21.
- ß Among infants born to women in the COMIRNATY group, vesicoureteral reflux occurred in the infant who also had DiGeorge’s syndrome (with family history of DiGeorge’s syndrome).
- à These abnormalities occur at the time of conception.
- è In 1 infant born to a woman in the COMIRNATY group, mucopolysaccharidosis and congenital rubella syndrome were reported.
- ð In 1 infant born to a woman in the COMIRNATY group with Trisomy 21 who presented with ASD, PDA and microcephaly, only Trisomy 21 was reported as an adverse event and the remaining manifestations were not reported as separate adverse events by the investigator.
Congenital Anomalies and Chromosomal Abnormalities
COMIRNATY
N*=156
n† (%)
Placebo
N*=159
n† (%)
Major Congenital Anomalies‡
6 (3.8)
2 (1.3)
Atrial septal defects
4 (2.6)§
1 (0.6)
Microcephaly
2 (1.3)¶
0
Central nervous system anomalies
1 (0.6)#
0
Patent ductus arteriosus
1 (0.6)Þ
1 (0.6)
Ventricular septal defect
0
1 (0.6)
Vesicoureteral reflux
1 (0.6)ß
0
Chromosomal Abnormalitiesà
3 (1.9)
0
DiGeorge’s syndrome
1 (0.6)ß
0
Mucopolysaccharidosis
1 (0.6)è
0
Trisomy 21
0
Minor Congenital Anomalies‡
3 (1.9)
5 (3.1)
Ankyloglossia congenital
1 (0.6)
4 (2.5)
Congenital skin dimples
0
2 (1.3)
Phimosis
0
1 (0.6)
Polydactyly
1 (0.6)
0
Syndactyly
1 (0.6)
0
Other
1 (0.6)
0
Congenital rubella syndrome
1 (0.6)è
0
Animal Data
In a developmental toxicity study, 0.06 mL of a vaccine formulation containing the same quantity of nucleoside-modified messenger ribonucleic acid (mRNA) (30 mcg of modRNA) and other ingredients included in a single human dose of COMIRNATY [encoding the viral spike (S) glycoprotein of SARS-CoV-2 Wuhan-Hu-1 strain (Original)] was administered to female rats by the intramuscular route on 4 occasions: 21 and 14 days prior to mating, and on gestation days 9 and 20. No vaccine-related adverse effects on female fertility, fetal development, or postnatal development were reported in the study.
8.2 Lactation
Risk Summary
It is not known whether COMIRNATY is excreted in human milk. Data are not available to assess the effects of COMIRNATY on the breastfed infant or on milk production/excretion. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for COMIRNATY and any potential adverse effects on the breastfed child from COMIRNATY or from the underlying maternal condition. For preventive vaccines, the underlying maternal condition is susceptibility to disease prevented by the vaccine.
8.4 Pediatric Use
Safety and effectiveness of COMIRNATY in individuals 5 years through 17 years of age with at least one underlying condition that puts them at high risk for severe outcomes from COVID-19 is based on safety and effectiveness data in this age group and in adults [see Adverse Reactions (6) and Clinical Studies (14)].
The safety and effectiveness of COMIRNATY in individuals younger than 5 years of age have not been established. Evidence from clinical studies in individuals 6 months through 4 years of age strongly suggests that a single dose of COMIRNATY would be ineffective in individuals younger than 6 months of age.
8.5 Geriatric Use
Of the total number of COMIRNATY recipients in Study 2 as of March 13, 2021 (N = 22,026), 20.7% (n = 4,552) were 65 years of age and older and 4.2% (n = 925) were 75 years of age and older [see Clinical Studies (14.1)]. In Study 4, of 5,081 recipients who received COMIRNATY as the first booster dose, 23.1% (n = 1,175) were 65 years of age and older and 5.2% (n = 265) were 75 years of age and older. In Study 5, of 726 recipients who received Pfizer-BioNTech COVID-19 Vaccine, Bivalent as the second booster dose, 21.9% (n = 159) were 65 years of age and older and 4.8% (n = 35) were 75 years of age and older [see Clinical Studies (14.1)]. No overall differences in safety or effectiveness were observed between these recipients and younger recipients.
8.6 Immunocompromised Individuals
In an open-label descriptive study (NCT04895982, Study 10), 87 immunocompromised participants 5 years of age and older received up to 4 age-appropriate doses of COMIRNATY (Original Monovalent) in a 4-dose regimen (an unapproved dosing regimen), with the first 2 doses separated by approximately 21 days, a third dose approximately 28 days after Dose 2, and a fourth dose administered approximately 3 to 6 months after Dose 3. The study evaluated safety and immunogenicity in participants 5 years through 11 years of age (n=65), 12 years through 17 years (n=15), and 18 years of age and older (n=7) with the following immunocompromising conditions: autoimmune inflammatory disorders on immunomodulatory therapy (n=31), solid organ transplant (n=25), stem cell transplant (n=29), non-small cell lung cancer (n=1), or hemodialysis (n=1). Study completion rates were 83.1% (54/65), 53.3% (8/15), and 57.1% (4/7) for the respective age groups. The trial was stopped early due to challenges with study enrollment.
Overall, 58.6% were male, 82.8% were White, 20.7% were Hispanic/Latino, 6.9% were Black or African American, 2.3% were Asian, 1.1% were American Indian or Alaska Native, 2.3% were Multiracial, and race and ethnicity was not reported by 4.6% of participants. The median age of participants in age groups 5 years through 11 years, 12 through 17 years, and 18 years of age and older was 9.0, 12.0, and 40.0 years, respectively.
The safety profile in immunocompromised participants 5 years of age and older who received COMIRNATY (Original Monovalent) was similar to that in immunocompetent participants in other clinical studies [see Adverse Reactions (6.1)]. There were no deaths in the study and no serious adverse events were assessed as related to vaccination.
Seroresponse rates (defined as achieving a ≥4-fold rise in neutralizing titers from baseline) were evaluated in all participants with at least one valid immunogenicity result after vaccination, regardless of prior SARS-CoV-2 infection status.
Among participants 5 years through 11 years of age: At 1 month after Dose 3, seroresponse rate was 81.7% [95% confidence interval (CI): 69.6%, 90.5%] (n=60). At 1 month after Dose 4, seroresponse rate was 96.0% (95% CI: 86.3%, 99.5%) (n=50). At 6 months after Dose 4, seroresponse rate was 87.8% (95% CI: 75.2%, 95.4%) (n=49).
Among participants 12 years through 17 years of age: At 1 month after Dose 3, seroresponse rate was 92.9% (95% CI: 66.1%, 99.8%) (n=14). At 1 month after Dose 4, seroresponse rate was 100.0% (95% CI: 66.4%, 100.0%) (n=9). At 6 months after Dose 4, seroresponse rate was 100.0% (95% CI: 63.1%, 100.0%) (n=8).
Among participants 18 years of age and older: At 1 month after Dose 3, seroresponse rate was 50.0% (95% CI: 11.8%, 88.2%) (n=6). At 1 month after Dose 4, seroresponse rate was 75.0% (95% CI: 19.4%, 99.4%) (n=4). At 6 months after Dose 4, seroresponse rate was 33.3% (95% CI: 0.8%, 90.6%) (n=3).
The limitations of the study data include small sample sizes, particularly in the 12 through 17 years and ≥18 years age groups, and early study termination due to enrollment challenges. The data from this study do not establish the effectiveness of COMIRNATY in immunocompromised individuals.
The Centers for Disease Control and Prevention has published considerations related to COVID-19 vaccination guidance for people who are immunocompromised.
-
11 DESCRIPTION
COMIRNATY (COVID-19 Vaccine, mRNA) is a sterile injectable suspension for intramuscular use.
Single-Dose Prefilled Syringe for Individuals 65 Years of Age and Older and Individuals 12 Years Through 64 Years of Age with at Least One Underlying Condition that Puts Them at High Risk for Severe Outcomes from COVID-19:
Each 0.3 mL dose of COMIRNATY (2025-2026 Formula) is formulated to contain 30 mcg of a nucleoside-modified messenger RNA (modRNA) encoding the viral spike (S) glycoprotein of SARS-CoV-2 Omicron variant sublineage LP.8.1.
Each 0.3 mL dose of COMIRNATY also includes the following ingredients: lipids (0.43 mg ((4-hydroxybutyl)azanediyl)bis(hexane-6,1-diyl)bis(2-hexyldecanoate), 0.05 mg 2-(polyethylene glycol 2000)-N,N-ditetradecylacetamide, 0.09 mg 1,2-distearoyl-sn-glycero-3-phosphocholine, and 0.19 mg cholesterol), 0.06 mg tromethamine, 0.4 mg tromethamine hydrochloride, and 31 mg sucrose.
Single-Dose Vial for Individuals 5 Years Through 11 Years of Age with at Least One Underlying Condition that Puts Them at High Risk for Severe Outcomes from COVID-19:
Each 0.3 mL dose of COMIRNATY (2025-2026 Formula) is formulated to contain 10 mcg of a modRNA encoding the viral spike (S) glycoprotein of SARS-CoV-2 Omicron variant sublineage LP.8.1.
Each 0.3 mL dose of COMIRNATY also includes the following ingredients: lipids (0.14 mg ((4-hydroxybutyl)azanediyl)bis(hexane-6,1-diyl)bis(2-hexyldecanoate), 0.02 mg 2-(polyethylene glycol 2000)-N,N-ditetradecylacetamide, 0.03 mg 1,2-distearoyl-sn-glycero-3-phosphocholine, and 0.06 mg cholesterol), 0.06 mg tromethamine, 0.4 mg tromethamine hydrochloride, and 31 mg sucrose.
COMIRNATY does not contain preservatives.
The vial stoppers are not made with natural rubber latex.
The prefilled syringe tip cap and plunger stopper are not made with natural rubber latex.
- 12 CLINICAL PHARMACOLOGY
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
COMIRNATY has not been evaluated for the potential to cause carcinogenicity, genotoxicity, or impairment of male fertility. In a developmental toxicity study in rats with COMIRNATY [encoding the viral spike (S) glycoprotein of SARS-CoV-2 Wuhan-Hu-1 strain (Original)] there were no vaccine-related effects on female fertility [see Use in Specific Populations (8.1)].
-
14 CLINICAL STUDIES
14.1 Adults and Adolescents 12 Years of Age and Older
Efficacy of Two-Dose Series (Original Monovalent) in Vaccine-Naïve Individuals 16 Years of Age and Older
Study 2 is an ongoing, multicenter, multinational, randomized, placebo-controlled, observer-blind, dose-finding, vaccine candidate–selection, and efficacy study in participants 12 years of age and older. Randomization was stratified by age: 12 through 15 years of age, 16 through 55 years of age, or 56 years of age and older, with a minimum of 40% of participants in the ≥56-year stratum. The study excluded participants who were immunocompromised and those who had previous clinical or microbiological diagnosis of COVID-19. Participants with preexisting stable disease, defined as disease not requiring significant change in therapy or hospitalization for worsening disease during the 6 weeks before enrollment, were included as were participants with known stable infection with HIV, hepatitis C virus (HCV), or hepatitis B virus (HBV).
In Study 2, based on data accrued through March 13, 2021, approximately 44,000 participants 12 years of age and older were randomized equally and received 2 doses of COMIRNATY or placebo. Participants are planned to be followed for up to 24 months, for assessments of safety and efficacy against COVID-19.
Overall, among the total participants who received COMIRNATY or placebo, 51.4% and 50.3% were male and 48.6% and 49.7% were female, 79.1% and 79.2% were 16 through 64 years of age, 20.9% and 20.8% were 65 years of age and older, 81.9% and 82.1% were White, 9.5% and 9.6% were Black or African American, 1.0% and 0.9% were American Indian or Alaska Native, 4.4% and 4.3% were Asian, 0.3% and 0.2% Native Hawaiian or other Pacific Islander, 25.6% and 25.4% were Hispanic/Latino, 73.9% and 74.1% were non-Hispanic/Latino, 0.5% and 0.5% did not report ethnicity, 46.0% and 45.7% had comorbidities [participants who have 1 or more comorbidities that increase the risk of severe COVID-19 disease: defined as subjects who had at least 1 of the Charlson comorbidity index category or body mass index (BMI) ≥30 kg/m2], respectively. The mean age at vaccination was 49.8 and 49.7 years and median age was 51.0 and 51.0 in participants who received COMIRNATY or placebo, respectively.
Efficacy Against COVID-19
The population for the analysis of the protocol pre-specified primary efficacy endpoint included 36,621 participants 12 years of age and older (18,242 in the COMIRNATY group and 18,379 in the placebo group) who did not have evidence of prior infection with SARS-CoV-2 through 7 days after the second dose. The population in the protocol pre-specified primary efficacy analysis included all participants 12 years of age and older who had been enrolled from July 27, 2020, and followed for the development of COVID-19 through November 14, 2020. Participants 18 through 55 years of age and 56 years of age and older began enrollment from July 27, 2020, 16 through 17 years of age began enrollment from September 16, 2020, and 12 through 15 years of age began enrollment from October 15, 2020.
For participants without evidence of SARS-CoV-2 infection prior to 7 days after Dose 2, vaccine efficacy against confirmed COVID-19 occurring at least 7 days after Dose 2 was 95.0% (95% credible interval: 90.3, 97.6), which met the pre-specified success criterion. The case split was 8 COVID-19 cases in the COMIRNATY group compared to 162 COVID-19 cases in the placebo group.
The population for the updated vaccine efficacy analysis included participants 16 years of age and older who had been enrolled from July 27, 2020, and followed for the development of COVID-19 during blinded placebo-controlled follow-up through March 13, 2021, representing up to 6 months of follow-up after Dose 2. There were 12,796 (60.8%) participants in the COMIRNATY group and 12,449 (58.7%) in the placebo group followed for ≥4 months after Dose 2 in the blinded placebo-controlled follow-up period.
SARS-CoV-2 variants of concern identified from COVID-19 cases for this age group from this data cutoff include B.1.1.7 (Alpha) and B.1.351 (Beta). Representation of identified variants among cases in vaccine versus placebo recipients did not suggest decreased vaccine effectiveness against these variants.
The updated vaccine efficacy information is presented in Table 14.
Table 14: Vaccine Efficacy – First COVID-19 Occurrence From 7 Days After Dose 2, by Age Subgroup – Participants 16 Years of Age and Older Without Evidence of Infection and Participants With or Without Evidence of Infection Prior to 7 Days After Dose 2 – Evaluable Efficacy (7 Days) Population During the Placebo-Controlled Follow-up Period Abbreviations: CI = confidence interval; NAAT = nucleic acid amplification test; SARS-CoV-2 = severe acute respiratory syndrome coronavirus 2.
Note: Confirmed cases were determined by Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and at least 1 symptom consistent with COVID-19 (symptoms included: fever; new or increased cough; new or increased shortness of breath; chills; new or increased muscle pain; new loss of taste or smell; sore throat; diarrhea; vomiting).- * Participants who had no evidence of past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative at Visit 1 and SARS-CoV-2 not detected by NAAT [nasal swab] at Visits 1 and 2) and had negative NAAT (nasal swab) at any unscheduled visit prior to 7 days after Dose 2 were included in the analysis.
- † Vaccine encoding the viral spike (S) glycoprotein of SARS-CoV-2 Wuhan-Hu-1 strain (Original).
- ‡ N = Number of participants in the specified group.
- § n1 = Number of participants meeting the endpoint definition.
- ¶ Total surveillance time in 1,000 person-years for the given endpoint across all participants within each group at risk for the endpoint. Time period for COVID-19 case accrual is from 7 days after Dose 2 to the end of the surveillance period.
- # n2 = Number of participants at risk for the endpoint.
- Þ Two-sided confidence interval (CI) for vaccine efficacy is derived based on the Clopper and Pearson method adjusted to the surveillance time.
First COVID-19 occurrence from 7 days after Dose 2 in participants without evidence of prior SARS-CoV-2 infection*
Subgroup
Vaccine Efficacy %
(95% CIÞ)All participants
77
6.092 (19,711)833
5.857 (19,741)91.1
(88.8, 93.1)16 through 64 years
70
4.859 (15,519)709
4.654 (15,515)90.5
(87.9, 92.7)65 years and older
7
1.233 (4192)124
1.202 (4226)94.5
(88.3, 97.8)First COVID-19 occurrence from 7 days after Dose 2 in participants with or without* evidence of prior SARS-CoV-2 infection
Subgroup
Vaccine Efficacy %
(95% CIÞ)All participants
81
6.340 (20,533)854
6.110 (20,595)90.9
(88.5, 92.8)16 through 64 years
74
5.073 (16,218)726
4.879 (16,269)90.2
(87.5, 92.4)65 years and older
7
1.267 (4315)128
1.232 (4326)94.7
(88.7, 97.9)Subgroup analyses of vaccine efficacy (although limited by small numbers of cases in some subgroups) did not suggest meaningful differences in efficacy across sex, ethnic groups, geographies, or for participants with obesity or medical comorbidities associated with high risk of severe COVID-19.
Efficacy Against Severe COVID-19
Efficacy analyses of secondary efficacy endpoints supported the benefit of COMIRNATY in preventing severe COVID-19. Vaccine efficacy against severe COVID-19 is presented only for participants with or without prior SARS-CoV-2 infection (Table 15) as the COVID-19 case counts in participants without prior SARS-CoV-2 infection were the same as those in participants with or without prior SARS-CoV-2 infection in both the COMIRNATY and placebo groups.
Table 15: Vaccine Efficacy – First Severe COVID-19 Occurrence in Participants 16 Years of Age and Older With or Without* Prior SARS-CoV-2 Infection Based on Protocol† or Centers for Disease Control and Prevention (CDC)‡ Definition From 7 Days After Dose 2 – Evaluable Efficacy (7 Days) Population During the Placebo-Controlled Follow-up Abbreviations: CI = confidence interval; NAAT = nucleic acid amplification test; SARS-CoV-2 = severe acute respiratory syndrome coronavirus 2.
Note: Confirmed cases were determined by Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and at least 1 symptom consistent with COVID-19 (symptoms included: fever; new or increased cough; new or increased shortness of breath; chills; new or increased muscle pain; new loss of taste or smell; sore throat; diarrhea; vomiting).- * Participants who had no evidence of past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative at Visit 1 and SARS-CoV-2 not detected by NAAT [nasal swab] at Visits 1 and 2) and had negative NAAT (nasal swab) at any unscheduled visit prior to 7 days after Dose 2 were included in the analysis.
- † Severe illness from COVID-19 is defined in the protocol as confirmed COVID-19 and presence of at least 1 of the following:
Clinical signs at rest indicative of severe systemic illness (respiratory rate ≥30 breaths per minute, heart rate ≥125 beats per minute, saturation of oxygen ≤93% on room air at sea level, or ratio of arterial oxygen partial pressure to fractional inspired oxygen <300 mm Hg);
Respiratory failure [defined as needing high flow oxygen, noninvasive ventilation, mechanical ventilation, or extracorporeal membrane oxygenation (ECMO)];
Evidence of shock (systolic blood pressure <90 mm Hg, diastolic blood pressure <60 mm Hg, or requiring vasopressors);
Significant acute renal, hepatic, or neurologic dysfunction;
Admission to an Intensive Care Unit;
Death.- ‡ Severe illness from COVID-19 as defined by CDC is confirmed COVID-19 and presence of at least 1 of the following:
Hospitalization;
Admission to the Intensive Care Unit;
Intubation or mechanical ventilation;
Death.- § Vaccine encoding the viral spike (S) glycoprotein of SARS-CoV-2 Wuhan-Hu-1 strain (Original).
- ¶ n1 = Number of participants meeting the endpoint definition.
- # Total surveillance time in 1,000 person-years for the given endpoint across all participants within each group at risk for the endpoint. Time period for COVID-19 case accrual is from 7 days after Dose 2 to the end of the surveillance period.
- Þ n2 = Number of participants at risk for the endpoint.
- ß Two-side confidence interval (CI) for vaccine efficacy is derived based on the Clopper and Pearson method adjusted to the surveillance time.
Vaccine Efficacy – First Severe COVID-19 Occurrence
Vaccine Efficacy %
(95% CIß)7 days after Dose 2ß
1
6.353 (20,540)21
6.237 (20,629)95.3
(70.9, 99.9)Vaccine Efficacy – First Severe COVID-19 Occurrence Based on CDC Definition
Vaccine Efficacy %
(95% CIß)7 days after Dose 2ß
0
6.345 (20,513)31
6.225 (20,593)100
(87.6, 100.0)Efficacy and Immunogenicity of Two-Dose Series (Original Monovalent) in Vaccine-Naïve Adolescents 12 Through 15 Years of Age
A descriptive efficacy analysis of Study 2 has been performed in 2,260 adolescents 12 through 15 years of age evaluating confirmed COVID-19 cases accrued up to a data cutoff date of September 2, 2021.
The vaccine efficacy information in adolescents 12 through 15 years of age is presented in Table 16.
Table 16: Vaccine Efficacy – First COVID-19 Occurrence From 7 Days After Dose 2: Without Evidence of Infection and With or Without Evidence of Infection Prior to 7 Days After Dose 2 – Blinded Placebo-Controlled Follow-up Period, Adolescents 12 Through 15 Years of Age Evaluable Efficacy (7 Days) Population Abbreviations: CI = confidence interval; NAAT = nucleic acid amplification test; SARS-CoV-2 = severe acute respiratory syndrome coronavirus 2.
Note: Confirmed cases were determined by Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and at least 1 symptom consistent with COVID-19 (symptoms included: fever; new or increased cough; new or increased shortness of breath; chills; new or increased muscle pain; new loss of taste or smell; sore throat; diarrhea; vomiting).- * Participants who had no evidence of past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative at Visit 1 and SARS-CoV-2 not detected by NAAT [nasal swab] at Visits 1 and 2) and had negative NAAT (nasal swab) at any unscheduled visit prior to 7 days after Dose 2 were included in the analysis.
- † Vaccine encoding the viral spike (S) glycoprotein of SARS-CoV-2 Wuhan-Hu-1 strain (Original).
- ‡ N = Number of participants in the specified group.
- § n1 = Number of participants meeting the endpoint definition.
- ¶ Total surveillance time in 1,000 person-years for the given endpoint across all participants within each group at risk for the endpoint. Time period for COVID-19 case accrual is from 7 days after Dose 2 to the end of the surveillance period.
- # n2 = Number of participants at risk for the endpoint.
- Þ Two-side confidence interval (CI) for vaccine efficacy is derived based on the Clopper and Pearson method adjusted for surveillance time.
- ß The only SARS-CoV-2 variant of concern identified from COVID-19 cases in this age group from this data cutoff was B.1.1.7 (Alpha).
First COVID-19 occurrence from 7 days after Dose 2 in adolescents 12 through 15 years of age without evidence of prior SARS-CoV-2 infection*
Vaccine Efficacy %
(95% CIÞ)Adolescents
12 through 15 years of age0
0.343 (1043)28
0.322 (1019)100.0
(86.8, 100.0)First COVID-19 occurrence from 7 days after Dose 2 in adolescents 12 through 15 years of age with or without evidence of prior SARS-CoV-2 infection
Vaccine Efficacy %
(95% CIÞ)Adolescents
12 through 15 years of age0
0.362 (1098)30ß
0.345 (1088)100.0
(87.5, 100.0)In Study 2, an analysis of SARS-CoV-2 50% neutralizing titers (NT50) 1 month after Dose 2 in a randomly selected subset of participants demonstrated noninferior immune responses (within 1.5-fold) comparing adolescents 12 through 15 years of age to participants 16 through 25 years of age who had no serological or virological evidence of past SARS-CoV-2 infection up to 1 month after Dose 2 (Table 17).
Table 17: Summary of Geometric Mean Ratio for 50% Neutralizing Titer – Comparison of Adolescents 12 Through 15 Years of Age to Participants 16 Through 25 Years of Age (Immunogenicity Subset) – Participants Without Evidence of Infection up to 1 Month After Dose 2 – Dose 2 Evaluable Immunogenicity Population Abbreviations: CI = confidence interval; GMR = geometric mean ratio; GMT = geometric mean titer; LLOQ = lower limit of quantitation; NAAT = nucleic acid amplification test; NT50 = 50% neutralizing titer; SARS-CoV-2 = severe acute respiratory syndrome coronavirus 2.
Note: Participants who had no serological or virological evidence (up to 1 month after receipt of the last dose) of past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative at Visit 1 and SARS-CoV-2 not detected by NAAT [nasal swab] at Visits 1 and 2), and had negative NAAT (nasal swab) at any unscheduled visit up to 1 month after Dose 2 were included in the analysis.- * Vaccine encoding the viral spike (S) glycoprotein of SARS-CoV-2 Wuhan-Hu-1 strain (Original).
- † n = Number of participants with valid and determinate assay results for the specified assay at the given dose/sampling time point.
- ‡ Protocol-specified timing for blood sample collection.
- § GMTs and 2-sided 95% CIs were calculated by exponentiating the mean logarithm of the titers and the corresponding CIs (based on the Student t distribution). Assay results below the LLOQ were set to 0.5 × LLOQ.
- ¶ GMRs and 2-sided 95% CIs were calculated by exponentiating the mean difference of the logarithms of the titers (Group 1 [12 through 15 years of age] – Group 2 [16 through 25 years of age]) and the corresponding CI (based on the Student t distribution).
- # Noninferiority is declared if the lower bound of the 2-sided 95% CI for the GMR is greater than 0.67.
- Þ SARS-CoV-2 NT50 were determined using the SARS-CoV-2 mNeonGreen Virus Microneutralization Assay. The assay uses a fluorescent reporter virus derived from the USA_WA1/2020 strain and virus neutralization is read on Vero cell monolayers. The sample NT50 is defined as the reciprocal serum dilution at which 50% of the virus is neutralized.
COMIRNATY*
12 Through 15 Years
n†=19016 Through 25 Years
n†=17012 Through 15 Years/
16 Through 25 YearsAssay
Time Point‡
Met Noninferiority Objective#
(Y/N)SARS-CoV-2 neutralization assay - NT50 (titer)Þ
1 month after Dose 2
1253.6
(1117.7, 1406.1)708.1
(625.9, 801.1)1.77
(1.50, 2.09)Y
Immunogenicity of Single Dose (Original Monovalent) in Vaccine-Experienced Adults 18 Years Through 55 Years of Age
Effectiveness of a booster dose of COMIRNATY was based on an assessment of 50% neutralizing antibody titers (NT50) against SARS-CoV-2 reference strain (USA_WA1/2020) in Study 2 participants 18 through 55 years of age (n = 200 – 212) who had no serological or virological evidence of past SARS‑CoV‑2 infection up to 1 month after the booster vaccination. Analyses of NT50 1 month after the booster dose compared with 1 month after the primary series demonstrated noninferiority for both geometric mean ratio (GMR) [3.26 (97.5% CI: 2.76, 3.86)] and difference in seroresponse rates (percentage) [4.5% (97.5% CI: 1.0, 7.9)]. Seroresponse for a participant was defined as achieving a ≥4-fold rise in NT50 from baseline (before primary series).
Immunogenicity of a Single Dose (Bivalent Original and BA.4/BA.5) in Vaccine-Experienced Individuals 12 Years of Age and Older
In an analysis of a subset from Study 5, 105 participants 12 through 17 years of age, 297 participants 18 through 55 years of age, and 286 participants 56 years of age and older who had previously received a 2-dose primary series and 1 booster dose with COMIRNATY received a second booster dose of Pfizer-BioNTech COVID-19 Vaccine, Bivalent. In participants 12 through 17 years of age, 18 through 55 years of age, and 56 years of age and older, 75.2%, 71.7% and 61.5% were positive for SARS-CoV-2 at baseline, respectively.
Analyses of NT50 against Omicron BA.4/BA.5 and against reference strain among participants 56 years of age and older who received a second booster dose of Pfizer-BioNTech COVID-19 Vaccine, Bivalent in Study 5 compared with a subset of participants from Study 4 who received a second booster dose of COMIRNATY demonstrated superiority of Pfizer-BioNTech COVID-19 Vaccine, Bivalent to COMIRNATY based on GMR and noninferiority based on difference in seroresponse rates with respect to anti-Omicron BA.4/BA.5 response, and noninferiority of anti-reference strain immune response based on GMR (Table 18 and Table 19).
Analyses of NT50 against Omicron BA.4/BA.5 among participants 18 through 55 years of age compared with participants 56 years of age and older who received a second booster dose of Pfizer-BioNTech COVID-19 Vaccine, Bivalent in Study 5 demonstrated noninferiority of anti-Omicron BA.4/BA.5 response among participants 18 through 55 years of age to participants 56 years of age and older for both GMR and difference in seroresponse rates (Table 18 and Table 19).
The study also assessed the level of NT50 against the anti-Omicron BA.4/BA.5 and original SARS-CoV-2 strains pre-vaccination and 1 month after vaccination in participants who received a second booster dose (Table 20).
Table 18: Geometric Mean Titer Ratios – Study 5 COMIRNATY – Participants With or Without Evidence of Infection – Evaluable Immunogenicity Population Abbreviations: GMT = geometric mean titer; LLOQ = lower limit of quantitation; N-binding = SARS-CoV-2 nucleoprotein–binding; NAAT = nucleic acid amplification test; NT50 = 50% neutralizing titer; SARS-CoV-2 = severe acute respiratory syndrome coronavirus 2. - * Protocol-specified timing for blood sample collection.
- † Vaccine encoding the viral spike (S) glycoprotein of SARS-CoV-2 Wuhan-Hu-1 strain (Original) and Omicron variant lineages BA.4 and BA.5 (Omicron BA.4/BA.5).
- ‡ Vaccine encoding the viral spike (S) glycoprotein of SARS-CoV-2 Wuhan-Hu-1 strain (Original).
- § n = Number of participants with valid and determinate assay results for the specified assay at the given sampling time point.
- ¶ GMTs and 2-sided 95% CIs were calculated by exponentiating the mean logarithm of the titers and the corresponding CIs (based on the Student t distribution). Assay results below the LLOQ were set to 0.5 × LLOQ.
- # GMRs and 2-sided 95% CIs were calculated by exponentiating the difference of LS means and corresponding CIs based on analysis of logarithmically transformed neutralizing titers using a linear regression model with terms of baseline neutralizing titer (log scale) and vaccine group or age group.
- Þ SARS-CoV-2 NT50 were determined using a validated 384-well assay platform (original strain [USA_WA1/2020, isolated in January 2020] and Omicron B.1.1.529 subvariant BA.4/BA.5).
- ß Noninferiority is declared if the lower bound of the 2-sided 95% CI for the GMR is greater than 0.67.
- à Superiority is declared if the lower bound of the 2-sided 95% CI for the GMR is greater than 1.
- è Noninferiority is declared if the lower bound of the 2-sided 95% CI for the GMR is greater than 0.67 and the point estimate of the GMR is ≥0.8.
SARS-CoV-2 Neutralization Assay
Sampling Time Point*
Pfizer-BioNTech COVID-19 Vaccine, Bivalent†
Study 5
COMIRNATY‡
Subset of Study 4
Age Group Comparison
Vaccine Group Comparison
18 Through 55 Years of Age
56 Years of Age and Older
56 Years of Age and Older
Pfizer-BioNTech COVID-19 Vaccine, Bivalent
18 Through 55 Years of Age/≥56 Years of Age
≥56 Years of Age
n§
GMT¶
(95% CI¶)
n§
GMT¶
(95% CI¶)
n§
GMT¶
(95% CI¶)
Omicron BA.4/BA.5 - NT50 (titer)Þ
1 Month
297
4455.9
(3851.7, 5154.8)
284
4158.1
(3554.8, 4863.8)
282
938.9
(802.3, 1098.8)
0.98
(0.83, 1.16)ß
2.91
(2.45, 3.44)à
Reference Strain –
NT50 (titer)Þ
1 Month
-
-
286
16250.1
(14499.2, 18212.4)
289
10415.5
(9366.7, 11581.8)
-
1.38
(1.22, 1.56)è
Table 19: Difference in Percentages of Participants With Seroresponse – Pfizer-BioNTech COVID-19 Vaccine, Bivalent from Study 5 and COMIRNATY from Subset of Study 4 – Participants With or Without Evidence of Infection – Evaluable Immunogenicity Population Abbreviations: LLOQ = lower limit of quantitation; NT50 = 50% neutralizing titer; SARS-CoV-2 = severe acute respiratory syndrome coronavirus 2.
Note: Seroresponse is defined as achieving a ≥4-fold rise from baseline. If the baseline measurement is below the LLOQ, a postvaccination assay result ≥4 × LLOQ is considered a seroresponse.- * Vaccine encoding the viral spike (S) glycoprotein of SARS-CoV-2 Wuhan-Hu-1 strain (Original) and Omicron variant lineages BA.4 and BA.5 (Omicron BA.4/BA.5).
- † Vaccine encoding the viral spike (S) glycoprotein of SARS-CoV-2 Wuhan-Hu-1 strain (Original).
- ‡ Protocol-specified timing for blood sample collection.
- § N = Number of participants with valid and determinate assay results for the specified assay at both the pre-vaccination time point and the given sampling time point. This value is the denominator for the percentage calculation.
- ¶ n = Number of participants with seroresponse for the given assay at the given sampling time point.
- # Exact 2-sided CI, based on the Clopper and Pearson method.
- Þ Difference in proportions, expressed as a percentage.
- ß 2-sided CI based on the Miettinen and Nurminen method stratified by baseline neutralizing titer category (<median, ≥ median) for the difference in proportions. The median of baseline neutralizing titers was calculated based on the pooled data in 2 comparator groups.
- à SARS-CoV-2 NT50 were determined using a validated 384-well assay platform (Omicron B.1.1.529 subvariant BA.4/BA.5).
- è Noninferiority is declared if the lower bound of the 2-sided 95% CI for the difference in percentages of participants with seroresponse is >-10%.
- ð Noninferiority is declared if the lower bound of the 2-sided 95% CI for the difference in percentages of participants with seroresponse is >-5%.
Pfizer-BioNTech COVID-19 Vaccine, Bivalent*
Study 5
COMIRNATY†
Subset of Study 4
Age Group Comparison
Vaccine Group Comparison
18 Through 55 Years of Age
56 Years of Age and Older
56 Years of Age and Older
Pfizer-BioNTech COVID-19 Vaccine, Bivalent*
18 Through 55 Years of Age/≥56 Years of Age
≥56 Years of Age
SARS-CoV-2 Neutralization Assay
Sampling Time Point‡
n§
N¶ (%)
(95% CI#)
n§
N¶ (%)
(95% CI#)
n§
N¶ (%)
(95% CI#)
Omicron BA.4/BA.5 - NT50 (titer)à
1 Month
294
180 (61.2)
(55.4, 66.8)
282
188 (66.7)
(60.8, 72.1)
273
127 (46.5)
(40.5, 52.6)
-3.03
(-9.68, 3.63)è
26.77
(19.59, 33.95)ð
Table 20: Geometric Mean Titers – Pfizer-BioNTech COVID-19 Vaccine, Bivalent Groups Subset of Study 5 – Prior to and 1 Month After Second Booster – Participants 12 Years of Age and Older – Evaluable Immunogenicity Population Abbreviations: GMT = geometric mean titer; LLOQ = lower limit of quantitation; N-binding = SARS-CoV-2 nucleoprotein–binding; NAAT = nucleic acid amplification test; NT50 = 50% neutralizing titer; SARS-CoV-2 = severe acute respiratory syndrome coronavirus 2. - * Protocol-specified timing for blood sample collection.
- † Vaccine encoding the viral spike (S) glycoprotein of SARS-CoV-2 Wuhan-Hu-1 strain (Original) and Omicron variant lineages BA.4 and BA.5 (Omicron BA.4/BA.5).
- ‡ n = Number of participants with valid and determinate assay results for the specified assay at the given sampling time point.
- § GMTs and 2-sided 95% CIs were calculated by exponentiating the mean logarithm of the titers and the corresponding CIs (based on the Student t distribution). Assay results below the LLOQ were set to 0.5 × LLOQ.
- ¶ SARS-CoV-2 NT50 were determined using a validated 384-well assay platform (original strain [USA_WA1/2020, isolated in January 2020] and Omicron B.1.1.529 subvariant BA.4/BA.5).
SARS-CoV-2 Neutralization Assay
Sampling Time Point*
Pfizer-BioNTech COVID-19 Vaccine, Bivalent†
12 Through 17 Years of Age
18 Through 55 Years of Age
56 Years of Age and Older
n‡
GMT§
(95% CI§)
n‡
GMT§
(95% CI§)
n‡
GMT§
(95% CI§)
Omicron BA.4/BA.5 - NT50 (titer)¶
Pre-
vaccination
104
1105.8
(835.1, 1464.3)
294
569.6
(471.4, 688.2)
284
458.2
(365.2, 574.8)
1 Month
105
8212.8
(6807.3, 9908.7)
297
4455.9
(3851.7, 5154.8)
284
4158.1
(3554.8, 4863.8)
Reference strain - NT50 (titer)¶
Pre-vaccination
105
6863.3
(5587.8, 8430.1)
296
4017.3
(3430.7, 4704.1)
284
3690.6
(3082.2, 4419.0)
1 Month
105
23641.3
(20473.1, 27299.8)
296
16323.3
(14686.5, 18142.6)
286
16250.1
(14499.2, 18212.4)
Immunogenicity of a Single Dose (Bivalent Alpha and Delta) in Seropositive, Vaccine-Naïve Adults 18 Years of Age and Older
In a post-hoc analysis in a subset of participants 18 through 85 years of age enrolled in Study 7 (NCT05004181), immunogenicity of a single dose of a Pfizer-BioNTech bivalent COVID-19 vaccine containing equal quantities of modRNA (30 mcg total) encoding the viral spike (S) glycoprotein for the Alpha and Delta SARS-CoV-2 variants [not authorized or approved in the U.S., hereafter referred to as bivalent vaccine (Alpha and Delta)] was assessed in COVID-19 vaccine-naïve participants with evidence of prior SARS-CoV-2 infection (n = 262) compared with participants without prior SARS-CoV-2 infection who received 2 doses of COMIRNATY in Study 2 (n = 275). Among Study 7 participants, 253 were from study sites in South Africa and 9 were from study sites in the U.S. The immunogenicity of the bivalent Alpha and Delta vaccine is relevant to COMIRNATY because these vaccines are manufactured using the same process with differences only in the encoded spike proteins.
Table 21 presents demographic characteristics for participants in the immunogenicity analysis set.
Table 21: Demographic Characteristics – Subset of Participants from Study 7 and Study 2 – Reference Strain Neutralization – Immunogenicity Analysis Set - * N = Number of participants in the specified group. This value is the denominator for the percentage calculations.
- † n = Number of participants with the specified characteristic.
- ‡ Vaccine encoding the viral spike (S) glycoprotein of SARS-CoV-2 Wuhan-Hu-1 strain (Original).
- § Includes multiracial and not reported.
Study 7
Single Dose of Bivalent Vaccine (Alpha and Delta)
With Evidence of Prior Infection
(N*=262)
N† (%)
Study 2
Two Doses of COMIRNATY‡
Without Evidence of Infection
(N*=275)
N† (%)
Sex
Male
109 (41.6)
113 (41.1)
Female
153 (58.4)
162 (58.9)
Age at Vaccination (Years)
Mean (SD)
42.9 (16.21)
42.7 (16.08)
Median
41.0
40.0
Min, max
(18,84)
(18, 84)
Race
White
4 (1.5)
230 (83.6)
Black or African American
169 (64.5)
25 (9.1)
American Indian or Alaska Native
0
2 (0.7)
Asian
0
7 (2.5)
Other§
89 (34.0)
11 (4.0)
Ethnicity
Hispanic or Latino
5 (1.9)
83 (30.2)
Not Hispanic or Latino
255 (97.3)
192 (69.8)
Not reported
2 (0.8)
0
The objective of this analysis was to assess noninferiority with respect to level of 50% neutralizing titer (NT50) and to the seroresponse rate to the reference strain induced by a single dose of the bivalent Alpha and Delta vaccine in COVID-19 vaccine-naïve participants with evidence of prior infection relative to participants without evidence of SARS-CoV-2 infection who received 2 doses of COMIRNATY.
Noninferiority of the reference strain immune response with respect to level of NT50 was met, as the lower bound of the 2-sided 95% CI for the geometric mean ratio (GMR) was >0.67 (Table 22). Noninferiority of the seroresponse rate to the reference strain was not met, as the lower bound of the 2-sided 95% CIs for the difference in seroresponse rate of reference strain was -10.04%, below the noninferiority margin of -10% (Table 23).
Table 22: Geometric Mean Ratios – Single Dose of Bivalent Vaccine (Alpha and Delta) in Vaccine-Naïve Participants from Study 7 With Evidence of Prior SARS-CoV-2 Infection Compared With 2 Doses of COMIRNATY in a Subset of Participants from Study 2 Without Evidence of SARS-CoV-2 Infection – Reference Strain Neutralization – Immunogenicity Analysis Set Abbreviations: CI = confidence interval; GMR = geometric mean ratio; GMT = geometric mean titer; LLOQ = lower limit of quantitation; N-binding = SARS-CoV-2 nucleoprotein–binding; NAAT = nucleic acid amplification test; NT50 = 50% neutralizing titer; SARS-CoV-2 = severe acute respiratory syndrome coronavirus 2. - * Participants with positive N-binding antibody result at baseline, positive NAAT result prior to vaccination, or medical history or adverse event of COVID-19 prior to vaccination.
- † Protocol-specified timing for blood sample collection.
- ‡ Vaccine encoding the viral spike (S) glycoprotein of SARS-CoV-2 Wuhan-Hu-1 strain (Original).
- § Participants who had no serological or virological evidence (up to the 1-month post–Dose 2 blood sample collection) of past SARS-CoV-2 infection (i.e., negative N-binding antibody [serum] result at the Dose 1 and 1-month post–Dose 2 visits, negative NAAT [nasal swab] at the Dose 1 and Dose 2 visits, and any unscheduled visit [up to the 1-month post–Dose 2 blood sample collection]) and had no medical history of COVID-19 were included in the analysis.
- ¶ n = Number of participants with valid and determinate assay results for the specified assay at the given sampling time point.
- # GMTs and 2-sided 95% CIs were calculated by exponentiating the mean logarithm of the titers and the corresponding CIs (based on the Student t distribution). Assay results below the LLOQ were set to 0.5 × LLOQ.
- Þ GMRs and 2-sided 95% CIs were calculated by exponentiating the difference of LS means and corresponding CIs based on the analysis of logarithmically transformed neutralizing titers using a linear regression model with terms of age, sex, and group. Assay results below the LLOQ were set to 0.5 × LLOQ.
- ß SARS-CoV-2 NT50 were determined using a validated 384-well assay platform (original strain [USA_WA1/2020, isolated in January 2020]).
- à Noninferiority is declared if the lower bound of the 2-sided 95% CI for the GMR is greater than 0.67.
Study 7
Single Dose of Bivalent Vaccine (Alpha and Delta)
With Evidence of Prior Infection*
3 Weeks After Dose 1†
Study 2
Two Doses of COMIRNATY‡
Without Evidence of Infection§
1 Month After Dose 2†
Bivalent Vaccine (Alpha and Delta) With Evidence of Prior Infection†/
COMIRNATY Without Evidence of Infection§
SARS-CoV-2 Neutralization Assay
n¶
GMT#
(95% CI#)
n¶
GMT#
(95% CI#)
GMRÞ
(95% CIÞ)
Reference strain - NT50 (titer)ß
262
17404.2
(15485.1, 19561.1)
275
1328.1
(1183.1, 1491.0)
13.12
(11.14, 15.45)à
Table 23: Difference in Percentages of Participants With Seroresponse – Bivalent Vaccine (Alpha and Delta) in Vaccine-Naïve Participants from Study 7 With Evidence of Prior SARS-CoV-2 Infection Compared With 2 Doses of COMIRNATY in a Subset of Participants from Study 2 Without Evidence of Prior SARS-CoV-2 Infection – Reference Strain Neutralization – Immunogenicity Analysis Set Abbreviations: CI = confidence interval; N-binding = SARS-CoV-2 nucleoprotein–binding; NAAT = nucleic acid amplification test; NT50 = 50% neutralizing titer; SARS-CoV-2 = severe acute respiratory syndrome coronavirus 2. - * Participants with positive N-binding antibody result at baseline, positive NAAT result prior to vaccination, or medical history or adverse event of COVID-19 prior to vaccination.
- † Protocol-specified timing for blood sample collection.
- ‡ Vaccine encoding the viral spike (S) glycoprotein of SARS-CoV-2 Wuhan-Hu-1 strain (Original).
- § Participants who had no serological or virological evidence (up to the 1-month post–Dose 2 blood sample collection) of past SARS-CoV-2 infection (i.e., negative N-binding antibody [serum] result at the Dose 1 and 1-month post–Dose 2 visits, negative NAAT [nasal swab] at the Dose 1 and Dose 2 visits, and any unscheduled visit [up to the 1-month post–Dose 2 blood sample collection]) and had no medical history of COVID-19 were included in the analysis.
- ¶ N = Number of participants with valid and determinate assay results for the specified assay at both the pre-vaccination time point and the given sampling time point. This value is the denominator for the percentage calculation.
- # n = Number of participants with seroresponse for the given assay at the given sampling time point.
- Þ Exact 2-sided CI, based on the Clopper and Pearson method.
- ß Adjusted difference in proportions estimated using minimum risk weights and stratified by sex and age group (18 to 55 years, 56 to 85 years), expressed as a percentage.
- à 2-sided CI based on the Newcombe method stratified by sex and age group (18 to 55 years, 56 to 85 years) with minimum risk weights for the difference in proportions.
- è SARS-CoV-2 NT50 were determined using a validated 384-well assay platform (original strain [USA_WA1/2020, isolated in January 2020]).
- ð Noninferiority is declared if the lower bound of the 2-sided 95% CI for the difference in percentages of participants with seroresponse is >-10%.
Study 7
Bivalent Vaccine (Alpha and Delta) With Evidence of Prior Infection*
3 Weeks After Dose 1†
Study 2
COMIRNATY‡
Without Evidence of Prior Infection§
1 Month After Dose 2†
Bivalent Vaccine (Alpha and Delta)
With Evidence of Prior Infection†
Minus
COMIRNATY
Without Evidence of Prior Infection§
SARS-CoV-2 Neutralization Assay
N¶
n# (%)
(95% CIÞ)
N¶
n# (%)
(95% CIÞ)
Difference %ß
95% CIà
Reference strain – NT50 (titer)è
260
223 (85.8)
(80.9, 89.8)
275
249 (90.5)
(86.5, 93.7)
-4.55
(-10.04, 0.83)ð
Concomitant Administration of COMIRNATY (Original Monovalent) With Influenza Vaccine in Adults 18 Years Through 64 Years of Age
In Study 8, a Phase 3 multicenter, randomized, observer-blind study, 1,134 participants 18 through 64 years of age who had received 3 doses of COMIRNATY at least 3 months prior were randomized in a 1:1 ratio to receive either COMIRNATY concomitantly administered with Influenza Vaccine (Afluria Quadrivalent) followed 1 month later by placebo (Group 1, n = 568) or influenza vaccine with placebo followed 1 month later with COMIRNATY (Group 2, n = 566).
Full-length spike (S)-binding IgG responses to COMIRNATY and influenza strain-specific hemagglutination inhibition (HAI) titers were assessed 1-month postvaccination in each group.
The non-inferiority criteria (lower bound of the 2-sided 95% CI >0.67) for the comparison of concomitant administration versus separate administration were met. The GMC ratio of full-length S-binding IgG levels of SARS-CoV-2 Wuhan-Hu-1 strain (Original) (Group 1/Group 2) was 0.83 [95% CI: 0.77, 0.89]. The GMT ratio (Group 1/Group 2) for the 4 strain-specific influenza HAI titers were H1N1 A/Victoria: 0.95 [95% CI: 0.83, 1.09]; H3N2 A/Darwin: 0.96 [95% CI: 0.85, 1.09]; B/Austria: 0.89 [95% CI: 0.77, 1.04]; B/Phuket: 1.00 [95% CI: 0.89, 1.13].
SARS-CoV-2 Wuhan-Hu-1 strain (Original) neutralizing GMTs were descriptively assessed in a subset of participants, 100 participants from Group 1 and 100 participants from Group 2.
The SARS-CoV-2 neutralization assay (NT50 titer) GMTs increased from baseline to 1 month after vaccination with COMIRNATY from 2,755.9 to 6,773.9 in Group 1 and from 2,421.2 to 7,886.6 in Group 2.
14.2 Children 5 Years Through 11 Years of Age
Efficacy and Immunogenicity of Two-Dose Series (Original Monovalent) in Vaccine-Naïve Children 5 Years Through 11 Years of Age
An efficacy analysis of Study 3 has been performed in 4,051 participants 5 years through 11 years of age without evidence of infection prior to 7 days after Dose 2. This analysis evaluated confirmed symptomatic COVID-19 cases in the placebo-controlled blinded follow-up period accrued up to a data cutoff date of 20 May 2022.
In Study 3, participants 5 years through 11 years of age were enrolled in the United States, Spain, Finland, and Poland. Among participants 5 years through 11 years of age without evidence of prior infection with SARS-CoV-2 through 7 days after Dose 2, who received COMIRNATY (n=2,703) or placebo (n=1,348) in the evaluable efficacy population, 51.2% and 51.0% were male, 48.8% and 49.0% were female, 76.3% and 77.2% were White, 5.5% and 6.0% were Black or African American, 0.5% and 0.3% were American Indian or Alaska Native, 8.9% and 8.5% were Asian, 0.3% and 0% were Native Hawaiian or other Pacific Islander, 8.5% and 7.9% were multiracial or not reported, 15.1% and 15.7% were Hispanic/Latino, 84.8% and 84.3% were non-Hispanic/Latino, and 0.1% and 0% did not report ethnicity, respectively. In the evaluable efficacy population, 25.7% and 25.9% of participants had 1 or more comorbidities that increase the risk of severe COVID-19 disease: defined as participants who had at least 1 of the pre-specified comorbidities based on MMWR 69(32);1081-1088 and/or obesity (BMI ≥95th percentile), respectively. The median age of participants at vaccination was 8.0 years with a range of 5 through 11 years of age in both the COMIRNATY and placebo groups.
The overall vaccine efficacy results and subgroup analysis in participants 5 years through 11 years of age without evidence of prior SARS-CoV-2 infection are presented in Table 24. None of the cases met criteria for severe COVID-19 or multisystem inflammatory syndrome in children (MIS-C).
Table 24: Overall Vaccine Efficacy and Subgroup Analysis* – First COVID-19 Occurrence From 7 Days After Dose 2 – Study 3 Participants Without Evidence of Infection Prior to 7 Days After Dose 2 – Evaluable Efficacy Population Abbreviations: BMI = body mass index; MMWR = Morbidity and Mortality Weekly Report; NAAT = nucleic acid amplification test; N-binding = SARS-CoV-2 nucleoprotein-binding; SARS-CoV-2 = severe acute respiratory syndrome coronavirus 2.
Note: Participants who had no serological or virological evidence (prior to 7 days after receipt of Dose 2) of past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative at Dose 1 visit, SARS-CoV-2 not detected by NAAT [nasal swab] at Dose 1 and Dose 2 study visits, and negative NAAT [nasal swab] result at any unscheduled visit prior to 7 days after receipt of Dose 2) and had no medical history of COVID- 19 were included in the analysis.- * Subgroup analysis was not a prespecified hypothesis, and the results may not necessarily be valid inferences about vaccine efficacy.
- † N = number of participants in the specified group.
- ‡ n1 = Number of participants meeting the endpoint definition.
- § Total surveillance time in 1000 person-years for the given endpoint across all participants within each group at risk for the endpoint. Time period for COVID-19 case accrual is from 7 days after Dose 2 to the end of the surveillance period.
- ¶ n2 = Number of participants at risk for the endpoint.
- # Two-sided 95% confidence interval (CI) for VE is derived based on the Clopper and Pearson method adjusted for surveillance time.
- Þ Number of participants who have 1 or more comorbidities that increase the risk of severe COVID-19 disease: defined as participants who had at least one of the prespecified comorbidities based on MMWR Morb Mortal Wkly Rep. 2020;69(32):1081-8 and/or obesity (BMI ≥95th percentile).
First COVID-19 Occurrence From 7 Days after Dose 2
COMIRNATY
(N†=2703)
Cases
n1‡
Surveillance Time§
(n2¶)
Placebo
(N†=1348)
Cases
n1‡
Vaccine Efficacy (%)
(95% CI#)
Overall
10
0.591 (2640)
42
0.292 (1309)
88.2
(76.2, 94.7)
Participants with at least one comorbidity of interestÞ
Yes
2
0.150 (676)
13
0.075 (336)
92.3*
(66.0, 99.2)
No
8
0.441 (1964)
29
0.217 (973)
86.4*
(69.5, 94.6)
SARS-CoV-2 50% neutralizing antibody titers (NT50) 1 month after the two-dose series were compared between randomly selected subsets of participants 5 years through 11 years of age from Study 3 (Phase 2/3) and participants 16 years through 25 years of age from efficacy Study 2 (Phase 2/3), using a microneutralization assay against the reference strain (USA_WA1/2020). The primary immunobridging analyses compared the geometric mean titers (using a geometric mean ratio [GMR]) and the seroresponse (defined as achieving at least 4-fold rise in SARS-CoV-2 NT50 from before Dose 1) rate percentages in the evaluable immunogenicity population of participants without evidence of prior SARS-CoV-2 infection up to 1 month after Dose 2 in each group. The pre-specified noninferiority criteria were met for both the GMR and the seroresponse difference (Table 25 and Table 26).
Table 25: SARS-CoV-2 GMTs (NT50) at 1 Month After Primary Series – Immunobridging Subset – Participants 5 Through 11 Years of Age (Study 3) and Participants 16 Through 25 Years of Age (Study 2) – Without Evidence of SARS-CoV-2 Infection up to 1 Month After Dose 2 – Evaluable Immunogenicity Population Abbreviations: CI = confidence interval; GMR = geometric mean ratio; GMT = geometric mean titer; LLOQ = lower limit of quantitation; NAAT = nucleic acid amplification test; NT50 = 50% neutralizing titer; SARS-CoV-2 = severe acute respiratory syndrome coronavirus 2.
Note: Participants who had no serological or virological evidence (up to 1 month post-Dose 2 blood sample collection) of past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative at pre-Dose 1 and 1 month after Dose 2, SARS-CoV-2 not detected by NAAT [nasal swab] at pre-Dose 1 and pre-Dose 2, and negative NAAT (nasal swab) at any unscheduled visit up to 1 month after Dose 2 blood collection) and had no medical history of COVID-19 were included in the analysis.- * Vaccine encoding the viral spike (S) glycoprotein of SARS-CoV-2 Wuhan-Hu-1 strain (Original).
- † GMT ratio and 2-sided 95% CIs were calculated by exponentiating the mean difference of the logarithms of the titers (5 through 11 years of age minus 16 through 25 years of age) and the corresponding CI (based on the Student t distribution).
- ‡ Noninferiority is declared if the lower bound of the 2-sided 95% CI for the GMT ratio is greater than 0.67 and the point estimate of the GMR is ≥0.8.
- § n = Number of participants with valid and determinate assay results for the specified assay at the given dose/sampling time point.
- ¶ Protocol-specified timing for blood sample collection.
- # GMTs and 2-sided 95% CIs were calculated by exponentiating the mean logarithm of the titers and the corresponding CIs (based on the Student t distribution). Assay results below the LLOQ were set to 0.5 × LLOQ.
- Þ SARS-CoV-2 NT50 were determined using the SARS-CoV-2 mNeonGreen Virus Microneutralization Assay. The assay uses a fluorescent reporter virus derived from the USA_WA1/2020 strain and virus neutralization is read on Vero cell monolayers. The sample NT50 is defined as the reciprocal serum dilution at which 50% of the virus is neutralized.
COMIRNATY*
GMT Ratio (95% CI)
5 Through 11 Years of Age
n§=264
16 Through 25 Years of Age
n§=253
Assay
Time Point¶
GMT#
(95% CI#)
GMT#
(95% CI#)
SARS-CoV-2 neutralization assay − NT50 (titer)Þ
1 month after Dose 2
1197.6
(1106.1, 1296.6)
1146.5
(1045.5, 1257.2)
1.04
(0.93, 1.18)
Table 26: Difference in Percentages of Participants With Seroresponse at 1 Month After Primary Series – Immunobridging Subset – Participants 5 Through 11 Years of Age (Study 3) and Participants 16 Through 25 Years of Age (Study 2) Without Evidence of Infection up to 1 Month After Dose 2 – Evaluable Immunogenicity Population Abbreviations: CI = confidence interval; GMR = geometric mean ratio; LLOQ = lower limit of quantitation; NAAT = nucleic acid amplification test; N-binding = SARS-CoV-2 nucleoprotein–binding; NT50 = 50% neutralizing titer 50; SARS-CoV-2 = severe acute respiratory syndrome coronavirus 2.
Note: Seroresponse is defined as achieving a ≥4-fold rise from baseline (before Dose 1). If the baseline measurement is below the LLOQ, a postvaccination assay result ≥4 × LLOQ is considered a seroresponse.
Note: Participants who had no serological or virological evidence (up to 1 month post-Dose 2 blood sample collection) of past SARS-CoV-2 infection (i.e., N-binding antibody [serum] negative at pre-Dose 1 and 1 month after Dose 2, SARS-CoV-2 not detected by NAAT [nasal swab] at pre-Dose 1 and pre-Dose 2, and negative NAAT (nasal swab) at any unscheduled visit up to 1 month after Dose 2 blood collection) and had no medical history of COVID-19 were included in the analysis.- * Vaccine encoding the viral spike (S) glycoprotein of SARS-CoV-2 Wuhan-Hu-1 strain (Original).
- † Difference in proportions, expressed as a percentage (5 through 11 years of age minus 16 through 25 years of age).
- ‡ 2-sided CI, based on the Miettinen and Nurminen method for the difference in proportions, expressed as a percentage.
- § Noninferiority is declared if the lower bound of the 2-sided 95% CI for the difference in proportions is greater than -10.0% provided that the immunobridging criteria based on GMR were met.
- ¶ N = Number of participants with valid and determinate assay results both before vaccination and at 1 month after Dose 2. These values are the denominators for the percentage calculations.
- # Protocol-specified timing for blood sample collection.
- Þ n = Number of participants with seroresponse for the given assay at the given dose/sampling time point.
- ß Exact 2-sided CI based on the Clopper and Pearson method.
- à SARS-CoV-2 NT50 were determined using the SARS-CoV-2 mNeonGreen Virus Microneutralization Assay. The assay uses a fluorescent reporter virus derived from the USA_WA1/2020 strain and virus neutralization is read on Vero cell monolayers. The sample NT50 is defined as the reciprocal serum dilution at which 50% of the virus is neutralized.
COMIRNATY*
Difference in Seroresponse Rates %† (95% CI‡)
(5 Through 11 Years of Age minus 16 Through 25 Years of Age)§
5 Through 11 Years of Age
N¶=264
16 Through 25 Years of Age
N¶=253
Assay
Time Point#
nÞ (%)
(95% CIß)
nÞ (%)
(95% CIß)
SARS-CoV-2 neutralization assay − NT50 (titer)à
1 month after Dose 2
262 (99.2)
(97.3, 99.9)
251 (99.2)
(97.2, 99.9)
0.0
(-2.0, 2.2)
Immunogenicity of a Single Dose (Original Monovalent) in Vaccine-Experienced Children 5 Years Through 11 Years of Age
In Study 3, immunogenicity of a first booster dose of COMIRNATY administered after the second dose of the two-dose series with COMIRNATY was evaluated in 67 study participants 5 through 11 years of age who had no serological or virological evidence of past SARS-CoV-2 infection up to 1 month after the booster dose. Using a microneutralization assay against the reference strain of SARS-CoV-2 (USA_WA1/2020), the NT50 GMT at 1 month after the booster dose (2,720.9 [95% CI: 2,280.1, 3,247.0]) was increased compared with before the booster dose (271.0 [95% CI: 229.1, 320.6]).
Immunogenicity of a Single Dose (Monovalent XBB.1.5) in Vaccine-Naïve Children 5 Years Through 11 Years of Age
In an analysis of a subset of Study 6, immunogenicity of a single dose of COMIRNATY (encoding the viral spike (S) glycoprotein of SARS-CoV-2 Omicron XBB.1.5) in COVID-19 vaccine-naïve participants 5 through 11 years of age (n=285) was compared with participants 12 years of age and older who had received at least 3 prior U.S.-authorized mRNA COVID-19 vaccine doses and then received a single dose of COMIRNATY (encoding the viral spike (S) glycoprotein of SARS-CoV-2 Omicron XBB.1.5, 30 mcg of modRNA) in a subset of study C4591054 (Study 13; NCT 05997290) (n=302). Study 13 evaluated safety and immunogenicity of a single dose of COMIRNATY administered to COVID-19 vaccine-experienced participants 12 years of age and older. In Study 6 and Study 13, 98.9% and 99.3% of participants had evidence of prior SARS-CoV-2 infection at baseline, respectively.
Study 6 was conducted in Brazil, Puerto Rico, South Africa and United States. Among the COVID-19 vaccine−naïve participants 5 through 11 years of age in Study 6 (n=285) and COVID-19 vaccine-experienced participants 12 years of age and older in Study 13 (n=302) who received a single dose of COMIRNATY in the evaluable immunogenicity population, 46.3% and 41.7% were male, 53.7% and 58.3% were female, 41.1% and 79.1% were White, 53.0% and 12.9% were Black or African American, 0.4% and 0% were American Indian or Alaska Native, 2.1% and 5.0% were Asian, 0% and 0.3% were Native Hawaiian or other Pacific Islander, 3.5% and 2.6% were multiracial, not reported, or unknown, 53.3% and 19.2% were Hispanic/Latino, and 46.7% and 80.1% were non-Hispanic/Latino, respectively. At study vaccination, the median age of participants in Study 6 was 7.0 years (range 5 through 11 years of age) and the median age of participants in Study 13 was 53.5 years (range 12 through 82 years).
The primary immunobridging analyses compared the geometric mean titers (using a geometric mean ratio [GMR]) and the seroresponse (defined as achieving at least 4-fold rise from baseline) rates in the COVID-19 vaccine-naïve participants 5 through 11 years of age to COVID-19 vaccine-experienced participants 12 years of age and older. The noninferiority criteria were met for both the GMR and the seroresponse rate percentages (Table 27 and Table 28).
Table 27: Geometric Mean Ratio – Study 6 to Study 13 – Participants at 1 Month After Study Vaccination – Evaluable Immunogenicity Population Abbreviations: CI = confidence interval; GMR = geometric mean ratio; GMT = geometric mean titer; LLOQ = lower limit of quantitation; LS = least square; NT50 = 50% neutralizing titer; SARS-CoV-2 = severe acute respiratory syndrome coronavirus 2. - * Vaccine encoding the viral spike (S) glycoprotein of SARS-CoV-2 Omicron XBB.1.5.
- † n = Number of participants with valid and determinate assay results for the specified assay at the given sampling time point.
- ‡ GMTs and 2-sided 95% CIs were calculated by exponentiating the mean logarithm of the titers and the corresponding CIs (based on the Student t distribution). Assay results below the LLOQ were set to 0.5 × LLOQ.
- § GMRs and 2-sided 95% CIs were calculated by exponentiating the difference of LS Means for the assay (Study 6, 5 through 11 years of age – Study 13, 12 years of age and older) and the corresponding CIs based on a linear regression model with baseline log-transformed neutralizing titers, postbaseline infection status, and vaccine group as covariates.
- ¶ SARS-CoV-2 NT50 were determined using a validated 384-well assay platform (Omicron subvariant XBB.1.5).
- # Noninferiority is declared if the lower bound of the 2-sided 95% CI for the GMR is greater than 0.67 and the point estimate of the GMR is ≥0.8.
Study 6
COVID-19 Vaccine-Naïve
5 Through 11 Years of Age
COMIRNATY*
Study 13
COVID-19 Vaccine-Experienced
≥12 Years of Age
COMIRNATY*
Study 6/Study 13
SARS-CoV-2 Neutralization Assay
n†
GMT‡
(95% CI‡)
n†
GMT‡
(95% CI‡)
GMR§
(95% CI§)
Omicron XBB.1.5 - NT50 (titer)¶
285
5930.5
(5283.8, 6656.4)
302
4006.4
(3438.3, 4668.4)
1.81
(1.51, 2.16)#
Table 28: Difference in Percentages of Participants With Seroresponse Between Study 6 and Study 13 Participants at 1 Month After the Study Vaccination – Evaluable Immunogenicity Population Abbreviations: CI = confidence interval; LLOQ = lower limit of quantitation; NT50 = 50% neutralizing titer; SARS-CoV-2 = severe acute respiratory syndrome coronavirus 2.
Note: Seroresponse is defined as achieving a ≥4-fold rise from baseline. If the baseline measurement is below the LLOQ, a postvaccination assay result ≥4 × LLOQ is considered a seroresponse.- * Vaccine encoding the viral spike (S) glycoprotein of SARS-CoV-2 Omicron XBB.1.5.
- † N = Number of participants with valid and determinate assay results for the specified assay both before vaccination and at the given sampling time point. This value is the denominator for the percentage calculations.
- ‡ n = Number of participants with seroresponse for the given assay at the given sampling time point.
- § Exact 2-sided 95% CI based on the Clopper and Pearson method.
- ¶ Adjusted difference in proportions based on the Miettinen and Nurminen method stratified by baseline neutralizing titer category (< median, ≥ median), expressed as a percentage (Study 6, 5 through 11 years of age – Study 13, 12 years of age and older). The median of baseline neutralizing titers was calculated based on the pooled data in 2 comparator groups.
- # 2-sided 95% CI, based on the Miettinen and Nurminen method for the difference in proportions stratified by baseline neutralizing titer category (< median, ≥ median), expressed as a percentage.
- Þ SARS-CoV-2 NT50 were determined using a validated 384-well assay platform (Omicron subvariant XBB.1.5).
- ß Noninferiority is declared if the lower bound of the 2-sided 95% CI for the adjusted difference in percentage of participants with seroresponse is greater than -10.0%.
Study 6
COVID-19 Vaccine-Naïve
5 Through 11 Years of Age COMIRNATY*
Study 13
COVID-19 Vaccine-Experienced
≥12 Years of Age COMIRNATY*
Difference
SARS-CoV-2 Neutralization Assay
N†
n‡ (%)
(95% CI§)
N†
n‡ (%)
(95% CI§)
%¶
95% CI#
Omicron XBB.1.5 - NT50 (titer)Þ
285
253 (88.8)
(84.5, 92.2)
300
231 (77.0)
(71.8, 81.6)
8.97
(3.91, 14.02)ß
-
15 REFERENCES
- 1. Jain SS, Anderson SA, Steele JM, et al. Cardiac manifestations and outcomes of COVID-19 vaccine-associated myocarditis in the young in the USA: longitudinal results from the Myocarditis After COVID Vaccination (MACiV) multicenter study. Lancet. 2024;76:1-13. https://doi.org/10.1016/j.eclinm.2024.102809
-
16 HOW SUPPLIED/STORAGE AND HANDLING
COMIRNATY is supplied as follows:
-
COMIRNATY Single-Dose Prefilled Syringes for Individuals 65 Years of Age and Older and Individuals 12 Years Through 64 Years of Age with at Least One Underlying Condition that Puts Them at High Risk for Severe Outcomes from COVID-19:
- o Carton of 10 single-dose prefilled syringes: NDC: 0069-2528-10
- o Single-dose prefilled syringe: NDC: 0069-2528-01
- o Supplied in prefilled syringes labeled with gray borders
-
COMIRNATY Single-Dose Vials for Individuals 5 Through 11 Years of Age with at Least One Underlying Condition that Puts Them at High Risk for Severe Outcomes from COVID-19:
- o Carton of 10 single-dose vials: NDC: 0069-2501-10
- o Single-dose vial: NDC: 0069-2501-01
- o Supplied in vials with blue caps and labeled with blue borders
During storage, minimize exposure to room light, and avoid exposure to direct sunlight and ultraviolet light.
Do not use the vaccine after the expiration date printed on the vials, prefilled syringes, and cartons.
Storage Prior to Use
Prefilled Syringes
Store COMIRNATY prefilled syringes refrigerated at 2°C to 8°C (35°F to 46°F). DO NOT FREEZE.
The total time out of refrigeration (at temperatures between 8°C and 25°C (46°F and 77°F)) must not exceed 12 hours.
Single-Dose Vials
COMIRNATY single-dose vials may arrive frozen at ultra-cold conditions in thermal containers with dry ice. Once received, frozen vials may be immediately transferred to the refrigerator at 2ºC to 8ºC (35ºF to 46ºF), thawed and stored for up to 10 weeks. The 10-week refrigerated expiry date should be recorded on the carton at the time of transfer. Cartons of 10 single-dose vials may take up to 2 hours to thaw at this temperature. Once thawed, they should not be refrozen.
Alternatively, single-dose vials may be stored in an ultra-low temperature freezer at -90ºC to -60ºC (-130ºF to -76ºF). Do not store vials at -25°C to -15°C (-13°F to 5°F).
Cartons of COMIRNATY single-dose vials may be received at 2°C to 8°C (35ºF to 46ºF), and they should be stored at 2°C to 8°C (35ºF to 46ºF). Check that the carton has been previously updated to reflect the 10-week refrigerated expiry date.
The total time out of refrigeration (at temperatures between 8°C and 25°C (46°F and 77°F)) must not exceed 12 hours.
-
COMIRNATY Single-Dose Prefilled Syringes for Individuals 65 Years of Age and Older and Individuals 12 Years Through 64 Years of Age with at Least One Underlying Condition that Puts Them at High Risk for Severe Outcomes from COVID-19:
-
17 PATIENT COUNSELING INFORMATION
Advise the vaccine recipient or caregiver to read the FDA-approved patient labeling.
Inform the vaccine recipient or caregiver of the potential benefits and risks of vaccination with COMIRNATY.
Advise the vaccine recipient or caregiver to report any adverse events to their healthcare provider or to the Vaccine Adverse Event Reporting System at 1-800-822-7967 and www.vaers.hhs.gov.
This product’s labeling may have been updated. For the most recent prescribing information, please visit https://fda.report/dailymed/.
- SPL UNCLASSIFIED SECTION
-
Patient Package Insert
INFORMATION FOR RECIPIENTS AND CAREGIVERS
COMIRNATY (Cuh-mir'-na-tee)
(COVID-19 VACCINE, mRNA)
(2025-2026 Formula)
This summary is not intended to take the place of talking with your or your child’s healthcare provider. If you have questions or would like more information, please talk with the healthcare provider.
What is COMIRNATY?
COMIRNATY is a vaccine to protect against COVID-19. COMIRNATY is for people who are:
- 65 years of age and older, or
- 5 years through 64 years of age at high risk for severe COVID-19.
Vaccination with COMIRNATY may not protect all people who receive the vaccine.
COMIRNATY does not contain SARS-CoV-2, the virus that causes COVID-19. COMIRNATY cannot give you or your child COVID-19.
Who should not get COMIRNATY?
You or your child should not get COMIRNATY if you or your child had:
- a severe allergic reaction after a previous dose of COMIRNATY or any Pfizer-BioNTech COVID-19 vaccine
- a severe allergic reaction to any ingredient in these vaccines (see What are the ingredients in COMIRNATY?).
Before getting COMIRNATY, tell the vaccination provider about all of your or your child’s medical conditions, including if you or your child:
- have any allergies
- had a severe allergic reaction after receiving a previous dose of any COVID-19 vaccine
- have had myocarditis (inflammation of the heart muscle) or pericarditis (inflammation of the lining outside the heart)
- have a fever
- have a bleeding disorder or are on a blood thinner
- are immunocompromised or are on a medicine that affects the immune system
- are pregnant or plan to become pregnant
- are breastfeeding
- have received another COVID-19 vaccine
- have ever fainted in association with an injection
How is COMIRNATY given?
COMIRNATY is given as an injection into the muscle.
What are the risks of COMIRNATY?
There is a remote chance that COMIRNATY could cause a severe allergic reaction. A severe allergic reaction would usually occur within a few minutes to 1 hour after getting a dose. For this reason, the vaccination provider may ask you or your child to stay at the place where you or your child received the vaccine for monitoring after vaccination. Signs of a severe allergic reaction can include:
- Difficulty breathing
- Swelling of the face and throat
- A fast heartbeat
- A bad rash all over the body
- Dizziness and weakness
Myocarditis (inflammation of the heart muscle) and pericarditis (inflammation of the lining outside the heart) have occurred in some people who have received mRNA COVID-19 vaccines, including COMIRNATY and Pfizer-BioNTech COVID-19 vaccines. Myocarditis and pericarditis following administration of mRNA COVID-19 vaccines have occurred most commonly in males 12 years through 24 years of age. In most of these people, symptoms began within a week following vaccination. Based on available data, estimated rates of myocarditis and/or pericarditis from 1 through 7 days after getting a dose of the 2023-2024 Formula of mRNA COVID-19 vaccines were approximately 8 cases per million doses in people 6 months through 64 years of age and approximately 27 cases per million doses in males 12 years through 24 years of age.
In most people who have had myocarditis or pericarditis after receiving an mRNA COVID-19 vaccine, symptoms have gone away a few days after receiving treatment with medicines used to reduce inflammation.
In a study, follow-up information was collected on people who developed myocarditis after receiving the original formula of a COVID-19 vaccine; most people had received an mRNA COVID-19 vaccine. Some people in the study reported having heart symptoms approximately 3 months after developing myocarditis. Some people in the study had heart MRIs (scans that show detailed images of the heart muscle) initially after developing myocarditis and again approximately 5 months later. The initial and follow-up heart MRIs commonly showed signs of injury to the heart muscle, with improvement over time in most people. It is not known if these heart MRI findings might predict long-term heart effects of myocarditis. Studies are underway to find out if there are long-term heart effects in people who have had myocarditis after receiving an mRNA COVID-19 vaccine.
You should seek medical attention right away if you or your child have any of the following symptoms after receiving COMIRNATY, particularly during the 2 weeks after receiving a dose of the vaccine:
- Chest pain
- Shortness of breath
- Feelings of having a fast-beating, fluttering, or pounding heart
These could be symptoms of myocarditis or pericarditis.
Additional symptoms, particularly in children, may include:
- Fainting
- Unusual and persistent fatigue or lack of energy
- Persistent vomiting
- Persistent pain in the abdomen
- Unusual and persistent cool, pale skin
Side effects that have been reported with COMIRNATY or Pfizer-BioNTech COVID-19 vaccines include:
- Severe allergic reactions
- Non-severe allergic reactions such as rash, itching, hives, or swelling of the face
- Myocarditis (inflammation of the heart muscle)
- Pericarditis (inflammation of the lining outside the heart)
- Injection site reactions: pain, swelling, redness, arm pain
- General side effects: tiredness, headache, muscle pain, chills, joint pain, fever, nausea, feeling unwell, swollen lymph nodes (lymphadenopathy), decreased appetite, diarrhea, vomiting, dizziness
- Fainting in association with injection of the vaccine
- Febrile seizures (convulsions during a fever) in children 5 through 11 years of age
These may not be all the possible side effects of COMIRNATY. Ask your or your child’s healthcare provider about any side effects that concern you.
Report vaccine side effects to FDA/CDC Vaccine Adverse Event Reporting System (VAERS). The VAERS toll-free number is 1-800-822-7967 or report online to https://vaers.hhs.gov/reportevent.html.
In addition, you can report side effects to Pfizer Inc. at 1-800-438-1985 or www.pfizersafetyreporting.com.
What if you or your child are pregnant or breastfeeding?
If you or your child are pregnant or breastfeeding, discuss your options with the healthcare provider.
What are the ingredients in COMIRNATY?
COMIRNATY contains the following ingredients:
- messenger ribonucleic acid (mRNA)
- lipids (((4-hydroxybutyl)azanediyl)bis(hexane-6,1-diyl)bis(2-hexyldecanoate), 2-(polyethylene glycol 2000)-N,N-ditetradecylacetamide, 1,2-distearoyl-sn-glycero-3-phosphocholine, and cholesterol)
- tromethamine
- tromethamine hydrochloride
- sucrose
COMIRNATY does not contain preservatives.
This Information for Recipients and Caregivers may have been updated. For the most recent Information for Recipients and Caregivers, please visit https://fda.report/dailymed/.
If you have questions, talk to your healthcare provider or visit www.COMIRNATY.com or call 1-877-VAX-CO19 (1-877-829-2619).

Manufactured for
BioNTech Manufacturing GmbH
An der Goldgrube 12
55131 Mainz, Germany
Manufactured by
Pfizer Inc., New York, NY 10001LAB-1587-7.0
US Govt. License No. 2229
Revised: 8/2025
-
PRINCIPAL DISPLAY PANEL - Glass Prefilled Syringe Label 2025-2026 Formula
NDC: 0069-2528-01
Rx onlyCOVID-19 Vaccine, mRNA
COMIRNATY®2025 – 2026 Formula
DO NOT FREEZE
1 dose of 0.3 mL
For intramuscular use.
US License No. 2229
BioNTech Manufacturing
GmbH & Pfizer Inc. -
PRINCIPAL DISPLAY PANEL - Glass Prefilled Syringe Carton 2025-2026 Formula
NDC: 0069-2528-10
COVID-19 Vaccine, mRNA
COMIRNATY®2025 – 2026 Formula
Injectable Suspension, for Intramuscular Use
10 Single-Dose Prefilled Syringes
Each prefilled syringe contains 1 dose of 0.3 mL
For 12 years through 64 years of age at
high-risk for severe COVID-19
For 65 years of age and olderDO NOT FREEZE
BIONTECH
Pfizer
Rx only -
PRINCIPAL DISPLAY PANEL - Glass Single-Dose Vial Label 2025-2026 Formula
COVID-19 Vaccine, mRNA
COMIRNATY®
Rx only2025 – 2026 Formula
DO NOT DILUTE
Vial contains 1 dose of 0.3 mL
For intramuscular use.
US License No. 2229
BioNTech Manufacturing GmbH & Pfizer Inc.NDC: 0069-2501-01
-
PRINCIPAL DISPLAY PANEL - Glass Single-Dose Vial Carton 2025-2026 Formula
NDC: 0069-2501-10
COVID-19 Vaccine, mRNA
COMIRNATY®2025 – 2026 Formula
Injectable Suspension, for Intramuscular Use
DO NOT DILUTE
10 Single-Dose Vials
Each vial contains 1 dose of 0.3 mL
For 5 years through 11 years of age at high-risk for severe COVID-19
BIONTECH
Pfizer
Rx only -
INGREDIENTS AND APPEARANCE
COMIRNATY
covid-19 vaccine, mrna injection, suspensionProduct Information Product Type VACCINE Item Code (Source) NDC: 0069-2528 Route of Administration INTRAMUSCULAR Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength BNT162b2 Omicron (LP.8.1) (UNII: 7V7MWD9BXL) (BNT162b2 Omicron (LP.8.1) - UNII:7V7MWD9BXL) BNT162b2 Omicron (LP.8.1) 0.042 mg in 0.418 mL Inactive Ingredients Ingredient Name Strength ((4-HYDROXYBUTYL)AZANEDIYL)BIS(HEXANE-6,1-DIYL)BIS(2-HEXYLDECANOATE) (UNII: AVX8DX713V) 0.598 mg in 0.418 mL 2-(MPEG 2000)-N,N-DITETRADECYLACETAMIDE (UNII: PJH39UMU6H) 0.075 mg in 0.418 mL 1,2-DISTEAROYL-SN-GLYCERO-3-PHOSPHOCHOLINE (UNII: 043IPI2M0K) 0.130 mg in 0.418 mL CHOLESTEROL (UNII: 97C5T2UQ7J) 0.259 mg in 0.418 mL SUCROSE (UNII: C151H8M554) 43.1 mg in 0.418 mL TROMETHAMINE (UNII: 023C2WHX2V) 0.084 mg in 0.418 mL TROMETHAMINE HYDROCHLORIDE (UNII: 383V75M34E) 0.552 mg in 0.418 mL WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0069-2528-10 10 in 1 CARTON 1 NDC: 0069-2528-01 0.418 mL in 1 SYRINGE, GLASS; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125742 08/27/2025 COMIRNATY
covid-19 vaccine, mrna injection, suspensionProduct Information Product Type VACCINE Item Code (Source) NDC: 0069-2501 Route of Administration INTRAMUSCULAR Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength BNT162b2 Omicron (LP.8.1) (UNII: 7V7MWD9BXL) (BNT162b2 Omicron (LP.8.1) - UNII:7V7MWD9BXL) BNT162b2 Omicron (LP.8.1) 0.0158 mg in 0.48 mL Inactive Ingredients Ingredient Name Strength ((4-HYDROXYBUTYL)AZANEDIYL)BIS(HEXANE-6,1-DIYL)BIS(2-HEXYLDECANOATE) (UNII: AVX8DX713V) 0.227 mg in 0.48 mL 2-(MPEG 2000)-N,N-DITETRADECYLACETAMIDE (UNII: PJH39UMU6H) 0.028 mg in 0.48 mL 1,2-DISTEAROYL-SN-GLYCERO-3-PHOSPHOCHOLINE (UNII: 043IPI2M0K) 0.048 mg in 0.48 mL CHOLESTEROL (UNII: 97C5T2UQ7J) 0.1 mg in 0.48 mL SUCROSE (UNII: C151H8M554) 49.44 mg in 0.48 mL TROMETHAMINE (UNII: 023C2WHX2V) 0.10 mg in 0.48 mL TROMETHAMINE HYDROCHLORIDE (UNII: 383V75M34E) 0.63 mg in 0.48 mL WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0069-2501-10 10 in 1 CARTON 1 NDC: 0069-2501-01 0.48 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125742 08/27/2025 Labeler - Pfizer Laboratories Div Pfizer Inc (134489525) Registrant - Pfizer Inc (113480771) Establishment Name Address ID/FEI Business Operations Pfizer Manufacturing Belgium NV 370156507 ANALYSIS(0069-2528, 0069-2501) , MANUFACTURE(0069-2528, 0069-2501) , PACK(0069-2528, 0069-2501) , LABEL(0069-2528, 0069-2501) Establishment Name Address ID/FEI Business Operations Wyeth BioPharma Division of Wyeth Pharmaceuticals LLC 174350868 ANALYSIS(0069-2528, 0069-2501) , API MANUFACTURE(0069-2528, 0069-2501) Establishment Name Address ID/FEI Business Operations Pfizer Ireland Pharmaceuticals Unlimited Company 985586408 ANALYSIS(0069-2528, 0069-2501) , API MANUFACTURE(0069-2528, 0069-2501) Establishment Name Address ID/FEI Business Operations Pharmacia & Upjohn Company LLC 618054084 ANALYSIS(0069-2501) , MANUFACTURE(0069-2501) , PACK(0069-2501) , LABEL(0069-2501)
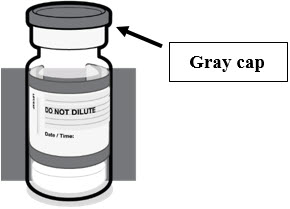



Trademark Results [Comirnaty]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 COMIRNATY 88942267 not registered Live/Pending |
BioNTech SE 2020-06-01 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.