FONDAPARINUX SODIUM injection
Fondaparinux Sodium by
Drug Labeling and Warnings
Fondaparinux Sodium by is a Prescription medication manufactured, distributed, or labeled by Dr. Reddy's Laboratories Limited, Gland Pharma Limited. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use FONDAPARINUX SODIUM INJECTION safely and effectively. See full prescribing information for FONDAPARINUX SODIUM INJECTION.
FONDAPARINUX SODIUM injection for subcutaneous use
Initial U.S. Approval: 2001WARNING: SPINAL/EPIDURAL HEMATOMAS
See full prescribing information for complete boxed warning.
Epidural or spinal hematomas may occur in patients who are anticoagulated with low molecular weight heparins (LMWH), heparinoids, or fondaparinux sodium and are receiving neuraxial anesthesia or undergoing spinal puncture.
These hematomas may result in long-term or permanent paralysis. Consider these risks when scheduling patients for spinal procedures. Factors that can increase the risk of developing epidural or spinal hematomas in these patients include:
· use of indwelling epidural catheters
· concomitant use of other drugs that affect hemostasis, such as non-steroidal anti-inflammatory drugs (NSAIDs), platelet inhibitors, or other anticoagulants
· a history of traumatic or repeated epidural or spinal puncture
· a history of spinal deformity or spinal surgery
Monitor patients frequently for signs and symptoms of neurologic impairment. If neurologic compromise is noted, urgent treatment is necessary.
Consider the benefit and risks before neuraxial intervention in patients anticoagulated or to be anticoagulated for thromboprophylaxis. [see Warnings and Precautions (5.1) and Drug Interactions (7)]
INDICATIONS AND USAGE
Fondaparinux sodium injection is a Factor Xa inhibitor (anticoagulant) indicated for:
- Prophylaxis of deep vein thrombosis (DVT) in patients undergoing hip fracture surgery (including extended prophylaxis), hip replacement surgery, knee replacement surgery, or abdominal surgery. (1.1)
- Treatment of DVT or acute pulmonary embolism (PE) when administered in conjunction with warfarin. (1.2, 1.3)
DOSAGE AND ADMINISTRATION
- Prophylaxis of deep vein thrombosis: Fondaparinux sodium 2.5 mg subcutaneously once daily after hemostasis has been established. The initial dose should be given no earlier than 6 to 8 hours after surgery and continued for 5 to 9 days. For patients undergoing hip fracture surgery, extended prophylaxis up to 24 additional days is recommended. (2.1, 2.2)
- Treatment of deep vein thrombosis and pulmonary embolism: Fondaparinux sodium 5 mg (body weight <50 kg), 7.5 mg (50 to 100 kg), or 10 mg (>100 kg) subcutaneously once daily. Treatment should continue for at least 5 days until INR 2 to 3 achieved with warfarin sodium. (2.3)
Do not use as intramuscular injection. For subcutaneous use, do not mix with other injections or infusions.
DOSAGE FORMS AND STRENGTHS
Injection: Single-dose, prefilled syringes containing 2.5 mg, 5 mg, 7.5 mg, or 10 mg of fondaparinux sodium. (3)
CONTRAINDICATIONS
Fondaparinux sodium injection is contraindicated in the following conditions: (4)
- Severe renal impairment (creatinine clearance <30 mL/min) in prophylaxis or treatment of venous thromboembolism.
- Active major bleeding. Bacterial endocarditis.
- Thrombocytopenia associated with a positive in vitro test for anti-platelet antibody in the presence of fondaparinux sodium.
- Body weight <50 kg (venous thromboembolism prophylaxis only).
- History of serious hypersensitivity reaction (e.g., angioedema, anaphylactoid/anaphylactic reactions) to fondaparinux sodium.
WARNINGS AND PRECAUTIONS
- Spinal or epidural hematomas, which may result in long-term or permanent paralysis, can occur (5.1)
- Patients taking fondaparinux sodium injection with risk factors for bleeding are at increased risk of hemorrhage. (5.2)
- Bleeding risk is increased in renal impairment and in patients with low body weight <50 kg (5.3, 5.4)
- Thrombocytopenia can occur with administration of fondaparinux sodium. (5.5)
- Periodic routine complete blood counts (including platelet counts), serum creatinine level, and stool occult blood tests are recommended. (5.6)
ADVERSE REACTIONS
The most serious adverse reactions associated with the use of fondaparinux sodium are bleeding complications. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Dr. Reddy’s Laboratories, Inc. at 1-888-375-3784 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch
DRUG INTERACTIONS
Discontinue agents that may enhance the risk of hemorrhage prior to initiation of therapy with fondaparinux sodium unless essential. If co-administration is necessary,monitor patients closely for hemorrhage. (7)
USE IN SPECIFIC POPULATIONS
- Safety and effectiveness of fondaparinux sodium in pediatric patients have not been established. Because the risk for bleeding during treatment with fondaparinux sodium is increased in adults who weigh <50 kg, bleeding may be a particular safety concern for use of fondaparinux sodium in the pediatric population. (4, 5.4)
- Because elderly patients are more likely to have reduced renal function, fondaparinux sodiumshould be used with caution in these patients. (8.5)
- The risk of bleeding is increased with reduced renal or hepatic function. (8.6, 8.7)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 1/2020
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: SPINAL/EPIDURAL HEMATOMAS
1 INDICATIONS AND USAGE
1.1 Prophylaxis of Deep Vein Thrombosis
1.2 Treatment of Acute Deep Vein Thrombosis
1.3 Treatment of Acute Pulmonary Embolism
2 DOSAGE AND ADMINISTRATION
2.1 Deep Vein Thrombosis Prophylaxis Following Hip Fracture, Hip Replacement, and Knee Replacement Surgery
2.2 Deep Vein Thrombosis Prophylaxis Following Abdominal Surgery
2.3 Deep Vein Thrombosis and Pulmonary Embolism Treatment
2.4 Hepatic Impairment
2.5 Instructions for Use
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Neuraxial Anesthesia and Post-operative Indwelling Epidural Catheter Use
5.2 Hemorrhage
5.3 Renal Impairment and Bleeding Risk
5.4 Body Weight <50 kg and Bleeding Risk
5.5 Thrombocytopenia
5.6 Monitoring: Laboratory Tests
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Local Reactions
6.3 Elevations of Serum Aminotransferases
6.4 Other Adverse Reactions
6.5 Postmarketing Experience
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.4 Special Populations
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment Of Fertility
14 CLINICAL STUDIES
14.1 Prophylaxis of Thromboembolic Events Following Hip Fracture Surgery
14.2 Extended Prophylaxis of Thromboembolic Events Following Hip Fracture Surgery
14.3 Prophylaxis of Thromboembolic Events Following Hip Replacement Surgery
14.4 Prophylaxis of Thromboembolic Events Following Knee Replacement Surgery
14.5 Prophylaxis of Thromboembolic Events Following Abdominal Surgery in Patients at Risk for Thromboembolic Complications
14.6 Treatment of Deep Vein Thrombosis
14.7 Treatment of Pulmonary Embolism
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
17.1 Patient Advice
17.2 FDA-Approved Patient Labeling
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: SPINAL/EPIDURAL HEMATOMAS
Epidural or spinal hematomas may occur in patients who are anticoagulated with low molecular weight heparins (LMWH), heparinoids, or fondaparinux sodium and are receiving neuraxial anesthesia or undergoing spinal puncture. These hematomas may result in long-term or permanent paralysis. Consider these risks when scheduling patients for spinal procedures. Factors that can increase the risk of developing epidural or spinal hematomas in these patients include:
- use of indwelling epidural catheters
- concomitant use of other drugs that affect hemostasis, such as non-steroidal anti-inflammatory drugs (NSAIDs), platelet inhibitors, or other anticoagulants
- a history of traumatic or repeated epidural or spinal puncture
- a history of spinal deformity or spinal surgery
- Optimal timing between the administration of fondaparinux sodium and neuraxial procedures is not known.
Monitor patients frequently for signs and symptoms of neurologic impairment. If neurologic compromise is noted, urgent treatment is necessary.
Consider the benefit and risks before neuraxial intervention in patients anticoagulated or to be anticoagulated for thromboprophylaxis. [see Warnings and Precautions (5.1) and Drug Interactions (7)]
-
1 INDICATIONS AND USAGE
1.1 Prophylaxis of Deep Vein Thrombosis
Fondaparinux sodium injection is indicated for the prophylaxis of deep vein thrombosis (DVT), which may lead to pulmonary embolism (PE):
- in patients undergoing hip fracture surgery, including extended prophylaxis;
- in patients undergoing hip replacement surgery;
- in patients undergoing knee replacement surgery;
- in patients undergoing abdominal surgery who are at risk for thromboembolic complications.
-
2 DOSAGE AND ADMINISTRATION
Do not mix other medications or solutions with fondaparinux sodium injection. Administer fondaparinux sodium injection only subcutaneously. Discard unused portion.
2.1 Deep Vein Thrombosis Prophylaxis Following Hip Fracture, Hip Replacement, and Knee Replacement Surgery
In patients undergoing hip fracture, hip replacement, or knee replacement surgery, the recommended dose of fondaparinux sodium injection is 2.5 mg administered by subcutaneous injection once daily after hemostasis has been established. Administer the initial dose no earlier than 6 to 8 hours after surgery. Administration of fondaparinux sodium injection earlier than 6 hours after surgery increases the risk of major bleeding. The usual duration of therapy is 5 to 9 days; up to 11 days of therapy was administered in clinical trials.
In patients undergoing hip fracture surgery, an extended prophylaxis course of up to 24 additional days is recommended. In patients undergoing hip fracture surgery, a total of 32 days (peri-operative and extended prophylaxis) was administered in clinical trials. [see Warnings and Precautions (5.6), Adverse Reactions (6), and Clinical Studies (14)]
2.2 Deep Vein Thrombosis Prophylaxis Following Abdominal Surgery
In patients undergoing abdominal surgery, the recommended dose of fondaparinux sodium injection is 2.5 mg administered by subcutaneous injection once daily after hemostasis has been established. Administer the initial dose no earlier than 6 to 8 hours after surgery. Administration of fondaparinux sodium injection earlier than 6 hours after surgery increases the risk of major bleeding. The usual duration of administration is 5 to 9 days, and up to 10 days of fondaparinux sodium injection was administered in clinical trials.
2.3 Deep Vein Thrombosis and Pulmonary Embolism Treatment
In patients with acute symptomatic DVT and in patients with acute symptomatic PE, the recommended dose of fondaparinux sodium injection is 5 mg (body weight <50 kg), 7.5 mg (body weight 50 to 100 kg), or 10 mg (body weight >100 kg) by subcutaneous injection once daily (fondaparinux sodium treatment regimen). Initiate concomitant treatment with warfarin sodium as soon as possible, usually within 72 hours. Continue treatment with fondaparinux sodium injection for at least 5 days and until a therapeutic oral anticoagulant effect is established (INR 2 to 3). The usual duration of administration of fondaparinux sodium injection is 5 to 9 days; up to 26 days of fondaparinux sodium injection was administered in clinical trials. [See Warnings and Precautions (5.6), Adverse Reactions (6), and Clinical Studies (14)]
2.4 Hepatic Impairment
No dose adjustment is recommended in patients with mild to moderate hepatic impairment, based upon single-dose pharmacokinetic data. Pharmacokinetic data are not available for patients with severe hepatic impairment. Patients with hepatic impairment may be particularly vulnerable to bleeding during fondaparinux sodium therapy. Observe these patients closely for signs and symptoms of bleeding. [see Clinical Pharmacology (12.4)]
2.5 Instructions for Use
Fondaparinux sodium injection is provided in a single-dose, prefilled syringe affixed with an active needle protection system. Fondaparinux sodium is administered by subcutaneous injection. It must not be administered by intramuscular injection. Fondaparinux sodium injection is intended for use under a physician’s guidance. Patients may self-inject only if their physician determines that it is appropriate and the patients are trained in subcutaneous injection techniques. Prior to administration, visually inspect fondaparinux sodium injection to ensure the solution is clear and free of particulate matter. The following instructions are specific to the Preventis TM injection system and may differ from the directions for other injection systems.
To avoid the loss of drug when using the prefilled syringe, do not expel the air bubble from the syringe before the injection. Administration should be made in the fatty tissue, alternating injection sites (e.g., between the left and right anterolateral or the left and right posterolateral abdominal wall).
To administer fondaparinux sodium:
STEP 1:
- Wipe the surface of the injection site with an alcohol swab.
- Remove the needle shield by pulling it straight off the syringe (Figure 1).
- Discard the needle shield.

STEP 2:
- Do not try to remove the air bubbles from the syringe before giving the injection.
- Pinch a fold of skin at the injection site between your thumb and forefinger and hold it throughout the injection time.
- Hold the syringe with your thumb on the top pad of the plunger and keep your index and middle fingers on the finger-grips of the syringe barrel. Pay attention to avoid pricking yourself with the exposed needle.
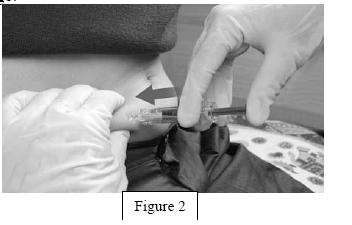
- Insert the full length of the syringe needle perpendicularly into the skin fold held between the thumb and forefinger (Figure 2).
- Push the plunger to the bottom of the syringe. This will ensure you have injected all the contents of the syringe.
STEP 3:
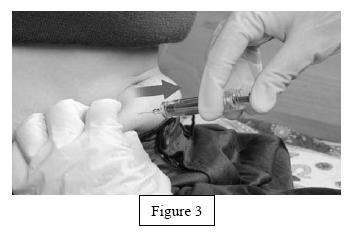
- Remove the syringe from the injection site keeping your finger on the plunger (Figure 3).
STEP 4:
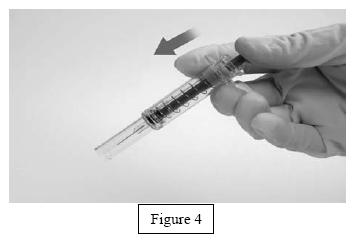
- Orient the needle away from you and others, and activate the safety shield by firmly pushing the plunger. The protective sleeve will automatically cover the needle and an audible "click" will be heard to confirm shield activation (Figure 4).
STEP 5:
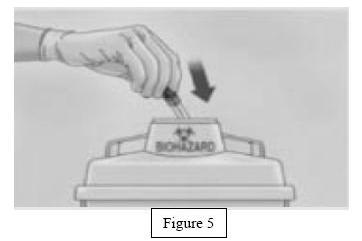
- Immediately discard the syringe into the sharps container (Figure 5).
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
Fondaparinux sodium injection is contraindicated in the following conditions:
- Severe renal impairment (creatinine clearance [CrCl] <30 mL/min). [see Warnings and Precautions (5.3) and Use in Specific Populations (8.6)]
- Active major bleeding.
- Bacterial endocarditis.
- Thrombocytopenia associated with a positive in vitro test for anti-platelet antibody in the presence of fondaparinux sodium.
- Body weight <50 kg (venous thromboembolism [VTE] prophylaxis only) [see Warnings and Precautions(5.4)].
- History of serious hypersensitivity reaction (e.g., angioedema, anaphylactoid/anaphylactic reactions) to fondaparinux sodium.
-
5 WARNINGS AND PRECAUTIONS
5.1 Neuraxial Anesthesia and Post-operative Indwelling Epidural Catheter Use
Spinal or epidural hematomas, which may result in long-term or permanent paralysis, can occur with the use of anticoagulants and neuraxial (spinal/epidural) anesthesia or spinal puncture. The risk of these events may be higher with post-operative use of indwelling epidural catheters or concomitant use of other drugs affecting hemostasis such as NSAIDs [see Boxed Warning]. In the postmarketing experience, epidural or spinal hematoma has been reported in association with the use of fondaparinux sodium by subcutaneous (SC) injection. Optimal timing between the administration of fondaparinux sodium and neuraxial procedures is not known. Monitor patients undergoing these procedures for signs and symptoms of neurologic impairment such as midline back pain, sensory and motor deficits (numbness, tingling, or weakness in lower limbs), and bowel or bladder dysfunction. Consider the potential risks and benefits before neuraxial intervention in patients anticoagulated or who may be anticoagulated for thromboprophylaxis.
5.2 Hemorrhage
Fondaparinux sodium increases the risk of hemorrhage in patients at risk for bleeding, including conditions such as congenital or acquired bleeding disorders, active ulcerative and angiodysplastic gastrointestinal disease, hemorrhagic stroke, uncontrolled arterial hypertension, diabetic retinopathy, or shortly after brain, spinal, or ophthalmological surgery. Cases of elevated aPTT temporally associated with bleeding events have been reported following administration of fondaparinux sodium (with or without concomitant administration of other anticoagulants) [See Adverse Reactions (6.5)].
Do not administer agents that enhance the risk of hemorrhage with fondaparinux sodium unless essential for the management of the underlying condition, such as vitamin K antagonists for the treatment of VTE. If co-administration is essential, closely monitor patients for signs and symptoms of bleeding.
Do not administer the initial dose of fondaparinux sodium earlier than 6 to 8 hours after surgery. Administration earlier than 6 hours after surgery increases risk of major bleeding [see Dosage and Administration (2) and Adverse Reactions (6.1)].
5.3 Renal Impairment and Bleeding Risk
Fondaparinux sodium increases the risk of bleeding in patients with impaired renal function due to reduced clearance [see Clinical Pharmacology (12.4)].
The incidence of major bleeding by renal function status reported in clinical trials of patients receiving fondaparinux sodium for VTE surgical prophylaxis is provided in Table 1. In these patient populations, the following is recommended:
- Do not use fondaparinux sodium for VTE prophylaxis and treatment in patients with CrCl <30 mL/min [seeContraindications (4)].
- Fondaparinux sodium may cause prolonged anticoagulation in patients with CrCl 30 to 50 mL/min.
Table 1. Incidence of Major Bleeding in Patients Treated With Fondaparinux Sodium by Renal Function Status for Surgical Prophylaxis and Treatment of Deep Vein Thrombosis (DVT) and Pulmonary Embolism (PE)
Population Timing of Dose Degree of Renal Impairment Normal % (n/N) Mild % (n/N) Moderate % (n/N) Severe % (n/N) CrCl (mL/min) ≥80 ≥50 - <80 ≥30 - <50 <30 Orthopedic surgerya Overall 1.6%
(25/1,565)2.4%
(31/1,288)3.8%
(19/504)4.8%
(4/83)6-8 hours
after surgery1.8%
(16/905)2.2%
(15/675)2.3%
(6/265)0%
(0/40)Abdominal surgery Overall 2.1%
(13/606)3.6%
(22/613)6.7%
(12/179)7.1%
(1/14)6-8 hours
after surgery2.1%
(10/467)3.3%
(16/481)5.8%
(8/137)7.7%
(1/13)DVT and PE Treatment 0.4%
(4/1,132)1.6%
(12/733)2.2%
(7/318)7.3%
(4/55)Assess renal function periodically in patients receiving fondaparinux sodium. Discontinue the drug immediately in patients who develop severe renal impairment while on therapy. After discontinuation of fondaparinux sodium, its anticoagulant effects may persist for 2 to 4 days in patients with normal renal function (i.e., at least 3 to 5 half-lives). The anticoagulant effects of fondaparinux sodium may persist even longer in patients with renal impairment [see Clinical Pharmacology (12.4)].
5.4 Body Weight <50 kg and Bleeding Risk
Fondaparinux sodium increases the risk for bleeding in patients who weigh less than 50 kg, compared to patients with higher weights.
In patients who weigh less than 50 kg:
- Do not administer fondaparinux sodium as prophylactic therapy for patients undergoing hip fracture, hip replacement, or knee replacement surgery and abdominal surgery [see Contraindications (4)].
During the randomized clinical trials of VTE prophylaxis in the peri-operative period following hip fracture, hip replacement, or knee replacement surgery and abdominal surgery, major bleeding occurred at a higher rate among patients with a body weight <50 kg compared to those with a body weight >50 kg (5.4% versus 2.1% in patients undergoing hip fracture, hip replacement, or knee replacement surgery; 5.3% versus 3.3% in patients undergoing abdominal surgery).
5.5 Thrombocytopenia
Thrombocytopenia can occur with the administration of fondaparinux sodium. Thrombocytopenia of any degree should be monitored closely. Discontinue fondaparinux sodium if the platelet count falls below 100,000/mm3. Moderate thrombocytopenia (platelet counts between 100,000/mm3 and 50,000/mm3) occurred at a rate of 3.0% in patients given fondaparinux sodium 2.5 mg in the peri-operative hip fracture, hip replacement, or knee replacement surgery and abdominal surgery clinical trials. Severe thrombocytopenia (platelet counts less than 50,000/mm3) occurred at a rate of 0.2% in patients given fondaparinux sodium 2.5 mg in these clinical trials. During extended prophylaxis, no cases of moderate or severe thrombocytopenia were reported.
Moderate thrombocytopenia occurred at a rate of 0.5% in patients given the fondaparinux sodium treatment regimen in the DVT and PE treatment clinical trials. Severe thrombocytopenia occurred at a rate of 0.04% in patients given the fondaparinux sodium treatment regimen in the DVT and PE treatment clinical trials.
Occurrences of thrombocytopenia with thrombosis that manifested similar to heparin-induced thrombocytopenia have been reported with the use of fondaparinux sodium in postmarketing experience. [see Adverse Reactions (6.5)]
5.6 Monitoring: Laboratory Tests
Routine coagulation tests such as Prothrombin Time (PT) and Activated Partial Thromboplastin Time (aPTT) are relatively insensitive measures of the activity of fondaparinux sodium and international standards of heparin or LMWH are not calibrators to measure anti-Factor Xa activity of fondaparinux sodium. If unexpected changes in coagulation parameters or major bleeding occur during therapy with fondaparinux sodium, discontinue fondaparinux sodium. In postmarketing experience, isolated occurrences of aPTT elevations have been reported following administration of fondaparinux sodium [see Adverse Reactions (6.5)].
Periodic routine complete blood counts (including platelet count), serum creatinine level, and stool occult blood tests are recommended during the course of treatment with fondaparinux sodium.
The anti-Factor Xa activity of fondaparinux sodium can be measured by anti-Xa assay using the appropriate calibrator (fondaparinux). The activity of fondaparinux sodium is expressed in milligrams (mg) of the fondaparinux and cannot be compared with activities of heparin or low molecular weight heparins. [see Clinical Pharmacology (12.2, 12.3)]
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
Spinal or epidural hematomas [see Warnings and Precautions (5.1)]
Hemorrhage [see Warnings and Precautions (5.2)]
Renal impairment and bleeding risk [see Warnings and Precautions (5.3)]
Body weight <50 kg and bleeding risk [see Warnings and Precautions (5.4)]
Thrombocytopenia [see Warnings and Precautions (5.5)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The adverse reaction information below is based on data from 8,877 patients exposed to fondaparinux sodium in controlled trials of hip fracture, hip replacement, major knee, or abdominal surgeries, and DVT and PE treatment.
Hemorrhage
During administration of fondaparinux sodium, the most common adverse reactions were bleeding complications [see Warnings and Precautions (5.2) ].
Hip Fracture, Hip Replacement, and Knee Replacement Surgery
The rates of major bleeding events reported during 3 active-controlled peri-operative VTE prophylaxis trials with enoxaparin sodium in hip fracture, hip replacement, or knee replacement surgery (N = 3,616) and in an extended VTE prophylaxis trial (n = 327) with fondaparinux sodium 2.5 mg are provided in Table 2.
Table 2. Bleeding Across Randomized, Controlled Hip Fracture, Hip Replacement, and Knee Replacement Surgery Studies
Peri-Operative Prophylaxis (Day 1 to Day 7 ± 1 post-surgery) Extended Prophylaxis (Day 8 to Day 28 ± 2 post-surgery) Fondaparinux Sodium 2.5 mg SC
once daily
N = 3,616Enoxaparin Sodium
a, b
N = 3,956Fondaparinux Sodium 2.5 mg SC
once daily
N = 327Placebo SC
once daily
N = 329Major bleedingc 96 (2.7%) 75 (1.9%) 8 (2.4%) 2 (0.6%) Hip fracture 18/831 (2.2%) 19/842 (2.3%) 8/327 (2.4%) 2/329 (0.6%) Hip replacement 67/2,268 (3.0%) 55/2,597 (2.1%) — — Knee replacement 11/517 (2.1%) 1/517 (0.2%) — — Fatal bleeding 0 (0.0%) 1 (<0.1%) 0 (0.0%) 0 (0.0%) Non-fatal bleeding at critical site 0 (0.0%) 1 (<0.1%) 0 (0.0%) 0 (0.0%) Re-operation due to bleeding 12 (0.3%) 10 (0.3%) 2 (0.6%) 2 (0.6%) BI ≥2d 84 (2.3%) 63 (1.6%) 6 (1.8%) 0 (0.0%) Minor bleedinge 109 (3.0%) 116 (2.9%) 5 (1.5%) 2 (0.6%) a Enoxaparin sodium dosing regimen: 30 mg every 12 hours or 40 mg once daily.
b Not approved for use in patients undergoing hip fracture surgery.
c Major bleeding was defined as clinically overt bleeding that was (1) fatal, (2) bleeding at critical site (e.g. intracranial, retroperitoneal, intraocular, pericardial, spinal, or into adrenal gland), (3) associated with re-operation at operative site, or (4) with a bleeding index (BI) ≥2.
d BI ≥2: Overt bleeding associated only with a bleeding index (BI) ≥2 calculated as [number of whole blood or packed red blood cell units transfused + [(pre-bleeding) – (post-bleeding)] hemoglobin (g/dL) values].
e Minor bleeding was defined as clinically overt bleeding that was not major.
A separate analysis of major bleeding across all randomized, controlled, peri-operative, prophylaxis clinical studies of hip fracture, hip replacement, or knee replacement surgery according to the time of the first injection of fondaparinux sodium after surgical closure was performed in patients who received fondaparinux sodium only post-operatively. In this analysis, the incidences of major bleeding were as follows: <4 hours was 4.8% (5/104), 4 to 6 hours was 2.3% (28/1,196), 6 to 8 hours was 1.9% (38/1,965). In all studies, the majority (≥75%) of the major bleeding events occurred during the first 4 days after surgery.
Abdominal Surgery: In a randomized study of patients undergoing abdominal surgery, fondaparinux sodium 2.5 mg once daily (n = 1,433) was compared with dalteparin 5,000 IU once daily (n = 1,425). Bleeding rates are shown in Table 3.
Table 3. Bleeding in the Abdominal Surgery Study
Fondaparinux Sodium2.5 mg SC
once dailyDalteparin Sodium 5,000 IU SC
once dailyN = 1,433 N = 1,425 Major bleedinga 49 (3.4%) 34 (2.4%) Fatal bleeding 2 (0.1%) 2 (0.1%) Non-fatal bleeding at critical site 0 (0.0%) 0 (0.0%) Other non-fatal major bleeding Surgical site Non-surgical site 38 (2.7%) 9 (0.6%) 26 (1.8%) 6 (0.4%) Minor bleedingb 31 (2.2%) 23 (1.6%) a Major bleeding was defined as bleeding that was (1) fatal, (2) bleeding at the surgical site leading to intervention, (3) non-surgical bleeding at a critical site (e.g. intracranial, retroperitoneal, intraocular, pericardial, spinal, or into adrenal gland), or leading to an intervention, and/or with a bleeding index (BI) ≥2.
b Minor bleeding was defined as clinically overt bleeding that was not major.
The rates of major bleeding according to the time interval following the first fondaparinux sodium injection were as follows: <6 hours was 3.4% (9/263) and 6 to 8 hours was 2.9% (32/1112).
Treatment of Deep Vein Thrombosis and Pulmonary Embolism:
The rates of bleeding events reported during a dose-response trial (n = 111) and an active controlled trial with enoxaparin sodium in DVT treatment (n = 1,091) and an active-controlled trial with heparin in PE treatment (n = 1,092) with fondaparinux sodium injection are provided in Table 4.
Table 4. Bleedinga in Deep Vein Thrombosis and Pulmonary Embolism Treatment Studies
Fondaparinux Sodium
N = 2,294Enoxaparin Sodium
N = 1,101HeparinaPTT adjusted IV
N = 1,092Major bleeding b 28 (1.2%) 13 (1.2%) 12 (1.1%) Fatal bleeding 3 (0.1%) 0 (0.0%) 1 (0.1%) Non-fatal bleeding at a critical site 3 (0.1%) 0 (0.0%) 2 (0.2%) Intracranial bleeding 3 (0.1%) 0 (0.0%) 1 (0.1%) Retro-peritoneal bleeding 0 (0.0%) 0 (0.0%) 1 (0.1%) Other clinically overt bleeding c 22 (1.0%) 13 (1.2%) 10 (0.9%) Minor bleeding d 70 (3.1%) 33 (3.0%) 57 (5.2%) a Bleeding rates are during the study drug treatment period (approximately 7 days). Patients were also treated with vitamin K antagonists initiated within 72 hours after the first study drug administration.
b Major bleeding was defined as clinically overt: –and/or contributing to death – and/or in a critical organ including intracranial, retroperitoneal, intraocular, spinal, pericardial, or adrenal gland – and/or associated with a fall in hemoglobin level ≥2 g/dL – and/or leading to a transfusion ≥2 units of packed red blood cells or whole blood.
c Clinically overt bleeding with a 2 g/dL fall in hemoglobin and/or leading to transfusion of PRBC or whole blood ≥2 units.
d Minor bleeding was defined as clinically overt bleeding that was not major.
6.2 Local Reactions
Local irritation (injection site bleeding, rash, and pruritus) may occur following subcutaneous injection of fondaparinux sodium.
6.3 Elevations of Serum Aminotransferases
In the peri-operative prophylaxis randomized clinical trials of 7 ± 2 days, asymptomatic increases in aspartate (AST) and alanine (ALT) aminotransferase levels greater than 3 times the upper limit of normal were reported in 1.7% and 2.6% of patients, respectively, during treatment with fondaparinux sodium 2.5 mg once daily versus 3.2% and 3.9% of patients, respectively, during treatment with enoxaparin sodium 30 mg every 12 hours or 40 mg once daily enoxaparin sodium. These elevations are reversible and may be associated with increases in bilirubin. In the extended prophylaxis clinical trial, no significant differences in AST and ALT levels between fondaparinux sodium 2.5 mg and placebo-treated patients were observed.
In the DVT and PE treatment clinical trials, asymptomatic increases in AST and ALT levels greater than 3 times the upper limit of normal of the laboratory reference range were reported in 0.7% and 1.3% of patients, respectively, during treatment with fondaparinux sodium. In comparison, these increases were reported in 4.8% and 12.3% of patients, respectively, in the DVT treatment trial during treatment with enoxaparin sodium 1 mg/kg every 12 hours and in 2.9% and 8.7% of patients, respectively, in the PE treatment trial during treatment with aPTT adjusted heparin.
Since aminotransferase determinations are important in the differential diagnosis of myocardial infarction, liver disease, and pulmonary emboli, elevations that might be caused by drugs like fondaparinux sodium should be interpreted with caution.
6.4 Other Adverse Reactions
Other adverse reactions that occurred during treatment with fondaparinux sodium in clinical trials with patients undergoing hip fracture, hip replacement, or knee replacement surgery are provided in Table 5.
Table 5. Adverse Reactions Across Randomized, Controlled, Hip Fracture Surgery, Hip Replacement Surgery, and Knee Replacement Surgery Studies
Adverse Reactions Peri-Operative Prophylaxis (Day 1 to Day 7 ± 1 post-surgery) Extended Prophylaxis (Day 8 to Day 28 ± 2 post-surgery) Fondaparinux Sodium 2.5 mg SC once daily Enoxaparin Sodiuma,b Fondaparinux Sodium 2.5 mg SC once daily Placebo SC once daily N = 3,616 N = 3,956 N = 327 N = 329 Anemia 707 (19.6%) 670 (16.9%) 5 (1.5%) 4 (1.2%) Insomnia 179 (5.0%) 214 (5.4%) 3 (0.9%) 1 (0.3%) Wound drainage increased 161 (4.5%) 184 (4.7%) 2 (0.6%) 0 (0.0%) Hypokalemia 152 (4.2%) 164 (4.1%) 0 (0.0%) 0 (0.0%) Dizziness 131 (3.6%) 165 (4.2%) 2 (0.6%) 0 (0.0%) Purpura 128 (3.5%) 137 (3.5%) 0 (0.0%) 0 (0.0%) Hypotension 126 (3.5%) 125 (3.2%) 1 (0.3%) 0 (0.0%) Confusion 113 (3.1%) 132 (3.3%) 4 (1.2%) 1 (0.3%) Bullous eruptionc 112 (3.1%) 102 (2.6%) 0 (0.0%) 1 (0.3%) Hematoma 103 (2.8%) 109 (2.8%) 7 (2.1%) 1 (0.3%) Post-operative hemorrhage 85 (2.4%) 69 (1.7%) 2 (0.6%) 2 (0.6%) a Enoxaparin sodium dosing regimen: 30 mg every 12 hours or 40 mg once daily.
b Not approved for use in patients undergoing hip fracture surgery.
c Localized blister coded as bullous eruption.
The most common adverse reaction in the abdominal surgery trial was post-operative wound infection (4.9%), and the most common adverse reaction in the VTE treatment trials was epistaxis (1.3%).
6.5 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of fondaparinux sodium. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
In the postmarketing experience, epidural or spinal hematoma has been reported in association with the use of fondaparinux sodium by subcutaneous (SC) injection [see Warnings and Precautions (5.1)]. Occurrences of thrombocytopenia with thrombosis that manifested similar to heparin-induced thrombocytopenia have been reported in the postmarketing experience and cases of elevated aPTT temporally associated with bleeding events have been reported following administration of fondaparinux sodium (with or without concomitant administration of other anticoagulants) [see Warnings and Precautions (5.5)].
Serious allergic reactions, including angioedema, anaphylactoid/anaphylactic reactions have been reported with the use of fondaparinux sodium [see Contraindications (4)].
-
7 DRUG INTERACTIONS
In clinical studies performed with fondaparinux sodium, the concomitant use of oral anticoagulants (warfarin), platelet inhibitors (acetylsalicylic acid), NSAIDs (piroxicam), and digoxin did not significantly affect the pharmacokinetics/pharmacodynamics of fondaparinux sodium. In addition, fondaparinux sodium neither influenced the pharmacodynamics of warfarin, acetylsalicylic acid, piroxicam, and digoxin, nor the pharmacokinetics of digoxin at steady state.
Agents that may enhance the risk of hemorrhage should be discontinued prior to initiation of therapy with fondaparinux sodium unless these agents are essential. If co-administration is necessary, monitor patients closely for hemorrhage. [See Warnings and Precautions (5.2)]
In an in vitro study in human liver microsomes, inhibition of CYP2A6 hydroxylation of coumarin by fondaparinux (200 micromolar i.e., 350 mg/L) was 17 to 28%. Inhibition of the other isozymes evaluated (CYPs 1A2, 2C9, 2C19, 2D6, 3A4, and 3E1) was 0 to 16%. Since fondaparinux does not markedly inhibit CYP450s (CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2D6, CYP2E1, or CYP3A4) in vitro, fondaparinux sodium is not expected to significantly interact with other drugs in vivo by inhibition of metabolism mediated by these isozymes.
Since fondaparinux sodium does not bind significantly to plasma proteins other than ATIII, no drug interactions by protein-binding displacement are expected.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Available data from published literature and postmarketing reports have not reported a clear association with fondaparinux sodium and adverse developmental outcomes. Fondaparinux sodium plasma concentrations obtained from four women treated with fondaparinux sodium injection during pregnancy and their newborn infants demonstrated low placental transfer of fondaparinux sodium (see Data). There are risks to the mother associated with untreated venous thromboembolism in pregnancy and a risk of hemorrhage in the mother and fetus associated with use of anticoagulants (see Clinical Considerations). In animal reproduction studies, there was no evidence of adverse developmental outcomes when fondaparinux sodium was administered to pregnant rats and rabbits during organogenesis at doses 32 and 65 times, respectively, the recommended human dose based on body surface area.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Clinical Considerations
Disease-associated maternal and/or embryo/fetal risk
Pregnancy confers an increased risk for thromboembolism that is higher for women with underlying thromboembolic disease and certain high-risk pregnancy conditions. Published data describe that women with a previous history of venous thrombosis are at high risk for recurrence during pregnancy.
Fetal/Neonatal adverse reactions
Fondaparinux sodium has been demonstrated to cross the placenta in humans (see Data). Use of anticoagulants, including fondaparinux sodium, may increase the risk of bleeding in the fetus and neonate. Monitor neonates for bleeding [see Warnings and Precautions (5.2, 5.4, 5.6)].
Labor or delivery
All patients receiving anticoagulants, including pregnant women, are at risk for bleeding. Fondaparinux sodium use during labor or delivery in women who are receiving neuraxial anesthesia may result in epidural or spinal hematomas. Pregnant women receiving fondaparinux sodium should be carefully monitored for evidence of bleeding or unexpected changes in coagulation parameters. Consideration for use of a shorter acting anticoagulant should be specifically addressed as delivery approaches [see Warnings and Precautions (5.1, 5.6)].
Data
Human Data
In a study of five pregnant women treated with fondaparinux sodium during the third trimester of pregnancy at a dose of 2.5 mg/day, four of the women had elevated anti-factor Xa activity noted in the cord blood. Anti-factor Xa clotting times in these four cases were between 37.5 and 50.9 seconds. The patient who did not have elevated anti-factor Xa activity had received only one dose of fondaparinux sodium 22 hours prior to delivery. The concentration of fondaparinux sodium in umbilical cord plasma was approximately 1/10th the level of fondaparinux sodium in maternal plasma. None of the infants experienced adverse effects.
Animal Data
Embryo-fetal development studies have been conducted with fondaparinux sodium in pregnant rats at subcutaneous doses up to 10 mg/kg/day (about 32 times the recommended human dose based on body surface area) administered from days 6 to 17 of gestation and pregnant rabbits at subcutaneous doses up to 10 mg/kg/day (about 65 times the recommended human dose based on body surface area) administered from days 6 to 18 of gestation. These studies have revealed no evidence of adverse developmental outcomes when fondaparinux sodium was administered to pregnant rats and rabbits during organogenesis. Additionally, there were no effects on pre and postnatal development in a study conducted in rats at subcutaneous doses up to 10 mg/kg/day (about 32 times the recommended human dose based on body surface area).
8.2 Lactation
Risk Summary
There are no data on the presence of fondaparinux sodium in human milk, or the effects on milk production. Limited clinical data during lactation preclude a clear determination of the risk of fondaparinux sodium to an infant during lactation; therefore, the developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for fondaparinux sodium and any potential adverse effects on the breastfed infant from fondaparinux sodium or from the underlying maternal condition.
8.4 Pediatric Use
Safety and effectiveness of fondaparinux sodium in pediatric patients have not been established. Because risk for bleeding during treatment with fondaparinux sodium is increased in adults who weigh <50 kg, bleeding may be a particular safety concern for use of fondaparinux sodium in the pediatric population [see Warnings and Precautions (5.4) ].
8.5 Geriatric Use
In clinical trials the efficacy of fondaparinux sodium in the elderly (65 years or older) was similar to that seen in patients younger than 65 years; however, serious adverse events increased with age. When using fondaparinux sodium in elderly patients, paying particular attention to dosing directions and concomitant medications (especially anti-platelet medication). [see Warnings and Precautions (5.2)]
Fondaparinux sodium is substantially excreted by the kidney, and the risk of adverse reactions to fondaparinux sodium may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, assess renal function prior to fondaparinux sodium administration. [see Contraindications (4), Warnings and Precautions (5.3), and Clinical Pharmacology (12.4)]
In the peri-operative hip fracture, hip replacement, or knee replacement surgery clinical trials with patients receiving fondaparinux sodium 2.5 mg, serious adverse events increased with age for patients receiving fondaparinux sodium. The incidence of major bleeding in clinical trials of fondaparinux sodium by age is provided in Table 6.
Table 6. Incidence of Major Bleeding in Patients Treated With Fondaparinux Sodium by Age
Age <65 years% (n/N) 65 to 74 years% (n/N) ≥75 years% (n/N) Orthopedic surgerya Extended prophylaxis 1.8% (23/1,253)1.9% (1/52) 2.2% (24/1,111)1.4% (1/71) 2.7% (33/1,277)2.9% (6/204) Abdominal surgery 3.0% (19/644) 3.2% (16/507) 5.0% (14/282) DVT and PE treatment 0.6% (7/1,151) 1.6% (9/560) 2.1% (12/583) a Includes hip fracture, hip replacement, and knee replacement surgery prophylaxis.
8.6 Renal Impairment
Patients with impaired renal function are at increased risk of bleeding due to reduced clearance of fondaparinux sodium [see Contraindications (4) and Warnings and Precautions (5.3) ]. Assess renal function periodically in patients receiving fondaparinux sodium. Discontinue fondaparinux sodium immediately in patients who develop severe renal impairment while on therapy. After discontinuation of fondaparinux sodium, its anticoagulant effects may persist for 2 to 4 days in patients with normal renal function (i.e., at least 3 to 5 half-lives). The anticoagulant effects of fondaparinux sodium may persist even longer in patients with renal impairment [see Clinical Pharmacology (12.4)].
8.7 Hepatic Impairment
Following a single, subcutaneous dose of 7.5 mg of fondaparinux sodium in patients with moderate hepatic impairment (Child-Pugh Category B) compared to subjects with normal liver function, changes from baseline in aPTT, PT/INR, and antithrombin III were similar in the two groups. However, a higher incidence of hemorrhage was observed in subjects with moderate hepatic impairment than in normal subjects, especially mild hematomas at the blood sampling or injection site. The pharmacokinetics of fondaparinux have not been studied in patients with severe hepatic impairment. [see Dosage and Administration (2.4) and Clinical Pharmacology (12.4)]
-
10 OVERDOSAGE
There is no known antidote for fondaparinux sodium. Overdose of fondaparinux sodium may lead to hemorrhagic complications. Discontinue treatment and initiate appropriate therapy if bleeding complications associated with overdosage occur.
Data obtained in patients undergoing chronic intermittent hemodialysis suggest that clearance of fondaparinux sodium can increase by 20% during hemodialysis.
-
11 DESCRIPTION
Fondaparinux sodium injection is a sterile solution containing fondaparinux sodium. It is a synthetic and specific inhibitor of activated Factor X (Xa). Fondaparinux sodium is methyl O-2-deoxy-6-O-sulfo-2-(sulfoamino)-α-D-glucopyranosyl-(1→4)-O-β-D-glucopyranuronosyl-(1→4)-O-2-deoxy-3,6-di-O-sulfo-2-(sulfoamino)-α-D-glucopyranosyl-(1→4)-O-2-Osulfo-α-L-idopyranuronosyl-(1→4)-2-deoxy-6-O-sulfo-2-(sulfoamino)-α-D-glucopyranoside, decasodium salt.
The molecular formula of fondaparinux sodium is C31H43N3Na10O49S8 and its molecular weight is 1728. The structural formula is provided below:
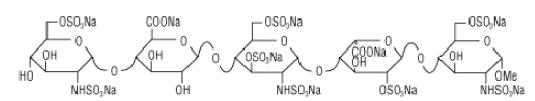
Fondaparinux sodium is supplied as a sterile, preservative-free injectable solution for subcutaneous use.
Each single-dose, prefilled syringe of fondaparinux sodium, affixed with an active needle protection system, contains 2.5 mg of fondaparinux sodium, USP in 0.5 mL, 5.0 mg of fondaparinux sodium, USP in 0.4 mL, 7.5 mg of fondaparinux sodium, USP in 0.6 mL, or 10.0 mg of fondaparinux sodium, USP in 0.8 mL of an isotonic solution of sodium chloride, water for injection. Also contain hydrochloric acid and sodium hydroxide as pH adjusters. The final drug product is a clear and colorless to slightly yellow liquid with a pH between 5.5 and 8.0.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
The antithrombotic activity of fondaparinux sodium is the result of antithrombin III (ATIII)-mediated selective inhibition of Factor Xa. By selectively binding to ATIII, fondaparinux sodium potentiates (about 300 times) the innate neutralization of Factor Xa by ATIII. Neutralization of Factor Xa interrupts the blood coagulation cascade and thus inhibits thrombin formation and thrombus development.
Fondaparinux sodium does not inactivate thrombin (activated Factor II) and has no known effect on platelet function. At the recommended dose, fondaparinux sodium does not affect fibrinolytic activity or bleeding time.
12.2 Pharmacodynamics
Anti-Xa Activity: The pharmacodynamics/pharmacokinetics of fondaparinux sodium are derived from fondaparinux plasma concentrations quantified via anti-Factor Xa activity. Only fondaparinux can be used to calibrate the anti-Xa assay. (The international standards of heparin or LMWH are not appropriate for this use.) As a result, the activity of fondaparinux sodium is expressed as milligrams (mg) of the fondaparinux calibrator. The anti-Xa activity of the drug increases with increasing drug concentration, reaching maximum values in approximately three hours.
12.3 Pharmacokinetics
Absorption: Fondaparinux sodium administered by subcutaneous injection is rapidly and completely absorbed (absolute bioavailability is 100%). Following a single subcutaneous dose of fondaparinux sodium 2.5 mg in young male subjects, Cmax of 0.34 mg/L is reached in approximately 2 hours. In patients undergoing treatment with fondaparinux sodium injection 2.5 mg, once daily, the peak steady-state plasma concentration is, on average, 0.39 to 0.50 mg/L and is reached approximately 3 hours post-dose. In these patients, the minimum steady-state plasma concentration is 0.14 to 0.19 mg/L. In patients with symptomatic deep vein thrombosis and pulmonary embolism undergoing treatment with fondaparinux sodium injection 5 mg (body weight <50 kg), 7.5 mg (body weight 50 to 100 kg), and 10 mg (body weight >100 kg) once daily, the body-weight-adjusted doses provide similar mean steady-state peaks and minimum plasma concentrations across all body weight categories. The mean peak steady-state plasma concentration is in the range of 1.20 to 1.26 mg/L. In these patients, the mean minimum steady-state plasma concentration is in the range of 0.46 to 0.62 mg/L.
Distribution: In healthy adults, intravenously or subcutaneously administered fondaparinux sodium distributes mainly in blood and only to a minor extent in extravascular fluid as evidenced by steady state and non-steady state apparent volume of distribution of 7 to 11 L. Similar fondaparinux distribution occurs in patients undergoing elective hip surgery or hip fracture surgery. In vitro, fondaparinux sodium is highly (at least 94%) and specifically bound to antithrombin III (ATIII) and does not bind significantly to other plasma proteins (including platelet Factor 4 [PF4]) or red blood cells.
Metabolism: In vivo metabolism of fondaparinux has not been investigated since the majority of the administered dose is eliminated unchanged in urine in individuals with normal kidney function.
Elimination: In individuals with normal kidney function, fondaparinux is eliminated in urine mainly as unchanged drug. In healthy individuals up to 75 years of age, up to 77% of a single subcutaneous or intravenous fondaparinux dose is eliminated in urine as unchanged drug in 72 hours. The elimination half-life is 17 to 21 hours.
12.4 Special Populations
Renal Impairment: Fondaparinux elimination is prolonged in patients with renal impairment since the major route of elimination is urinary excretion of unchanged drug. In patients undergoing prophylaxis following elective hip surgery or hip fracture surgery, the total clearance of fondaparinux is approximately 25% lower in patients with mild renal impairment (CrCl 50 to 80 mL/min), approximately 40% lower in patients with moderate renal impairment (CrCl 30 to 50 mL/min), and approximately 55% lower in patients with severe renal impairment (<30 mL/min) compared to patients with normal renal function. A similar relationship between fondaparinux clearance and extent of renal impairment was observed in DVT treatment patients. [see Contraindications (4) and Warnings and Precautions (5.3)]
Hepatic Impairment: Following a single, subcutaneous dose of 7.5 mg of fondaparinux sodium in patients with moderate hepatic impairment (Child-Pugh Category B), Cmax and AUC were decreased by 22% and 39%, respectively, compared to subjects with normal liver function. The changes from baseline in pharmacodynamic parameters, such as aPTT, PT/INR, and antithrombin III, were similar in normal subjects and in patients with moderate hepatic impairment. Based on these data, no dosage adjustment is recommended in these patients. However, a higher incidence of hemorrhage was observed in subjects with moderate hepatic impairment than in normal subjects [see Use in Specific Populations (8.7)]. The pharmacokinetics of fondaparinux have not been studied in patients with severe hepatic impairment. [see Dosage and Administration (2.4)]
Pediatric: The pharmacokinetics of fondaparinux have not been investigated in pediatric patients. [seeContraindications(4), Warnings and Precautions (5.4), and Pediatric Use (8.4)]
Geriatric: Fondaparinux elimination is prolonged in patients older than 75 years. In studies evaluating fondaparinux sodium 2.5 mg prophylaxis in hip fracture surgery or elective hip surgery, the total clearance of fondaparinux was approximately 25% lower in patients older than 75 years as compared to patients younger than 65 years. A similar relationship between fondaparinux clearance and age was observed in DVT treatment patients. [see Use in Specific Populations (8.5)]
Patients Weighing Less Than 50 kg: Total clearance of fondaparinux sodium is decreased by approximately 30% in patients weighing less than 50 kg [see Dosage and Administration (2.3) and Contraindications (4)].
Gender: The pharmacokinetic properties of fondaparinux sodium are not significantly affected by gender.
Race: Pharmacokinetic differences due to race have not been studied prospectively. However, studies performed in Asian (Japanese) healthy subjects did not reveal a different pharmacokinetic profile compared to Caucasian healthy subjects. Similarly, no plasma clearance differences were observed between black and Caucasian patients undergoing orthopedic surgery.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment Of Fertility
No long-term studies in animals have been performed to evaluate the carcinogenic potential of fondaparinux sodium.
Fondaparinux sodium was not genotoxic in the Ames test, the mouse lymphoma cell (L5178Y/TK+/-) forward mutation test, the human lymphocyte chromosome aberration test, the rat hepatocyte unscheduled DNA synthesis (UDS) test, or the rat micronucleus test.
At subcutaneous doses up to 10 mg/kg/day (about 32 times the recommended human dose based on body surface area), fondaparinux sodium was found to have no effect on fertility and reproductive performance of male and female rats.
-
14 CLINICAL STUDIES
14.1 Prophylaxis of Thromboembolic Events Following Hip Fracture Surgery
In a randomized, double-blind, clinical trial in patients undergoing hip fracture surgery, fondaparinux sodium 2.5 mg SC once daily was compared to enoxaparin sodium 40 mg SC once daily, which is not approved for use in patients undergoing hip fracture surgery. A total of 1,711 patients were randomized and 1,673 were treated. Patients ranged in age from 17 to 101 years (mean age 77 years) with 25% men and 75% women. Patients were 99% Caucasian, 1% other races. Patients with multiple traumas affecting more than one organ system, serum creatinine level more than 2 mg/dL (180 micromol/L), or platelet count less than 100,000/mm3 were excluded from the trial. Fondaparinux sodium was initiated after surgery in 88% of patients (mean 6 hours) and enoxaparin sodium was initiated after surgery in 74% of patients (mean 18 hours). For both drugs, treatment was continued for 7 ± 2 days. The primary efficacy endpoint, venous thromboembolism (VTE), was a composite of documented deep vein thrombosis (DVT) and/or documented symptomatic pulmonary embolism (PE) reported up to Day 11. The efficacy data are provided in Table 7 and demonstrate that under the conditions of the trial fondaparinux sodium was associated with a VTE rate of 8.3% compared with a VTE rate of 19.1% for enoxaparin sodium for a relative risk reduction of 56% (95% CI: 39%, 70%; P <0.001). Major bleeding episodes occurred in 2.2% of patients receiving fondaparinux sodium and 2.3% of enoxaparin sodium patients [see Adverse Reactions (6.1)].
Table 7. Efficacy of Fondaparinux Sodium in the Peri-operative Prophylaxis of Thromboembolic Events Following Hip Fracture Surgery
Endpoint Peri-operative Prophylaxis (Day 1 to Day 7 ± 2 post-surgery) Fondaparinux Sodium 2.5 mg SC once daily Enoxaparin Sodium 40 mg SC once daily n/Na % (95% CI) n/Na % (95% CI) VTE 52/626 8.3%b (6.3, 10.8) 119/624 19.1% (16.1, 22.4) All DVT 49/624 7.9%b (5.9, 10.2) 117/623 18.8% (15.8, 22.1) Proximal DVT 6/650 0.9%b (0.3, 2.0) 28/646 4.3% (2.9, 6.2) Symptomatic PE 3/831 0.4%c (0.1, 1.1) 3/840 0.4% (0.1, 1.0) a N = all evaluable hip fracture surgery patients. Evaluable patients were those who were treated and underwent the appropriate surgery (i.e., hip fracture surgery of the upper third of the femur), with an adequate efficacy assessment up to Day 11.
b P value versus enoxaparin sodium <0.001.
c P value versus enoxaparin sodium: NS.
14.2 Extended Prophylaxis of Thromboembolic Events Following Hip Fracture Surgery
In a noncomparative, unblinded manner, 737 patients undergoing hip fracture surgery were initially treated during the peri-operative period with fondaparinux sodium 2.5 mg once daily for 7 ± 1 days. Eighty-one (81) of the 737 patients were not eligible for randomization into the 3-week double-blind period. Three hundred twenty-six (326) patients and 330 patients were randomized to receive fondaparinux sodium 2.5 mg once daily or placebo, respectively, in or out of the hospital for 21 ± 2 days. Patients ranged in age from 23 to 96 years (mean age 75 years) and were 29% men and 71% women. Patients were 99% Caucasian and 1% other races. Patients with multiple traumas affecting more than one organ system or serum creatinine level more than 2 mg/dL (180 micromol/L) were excluded from the trial. The primary efficacy endpoint, venous thromboembolism (VTE), was a composite of documented deep vein thrombosis (DVT) and/or documented symptomatic pulmonary embolism (PE) reported for up to 24 days following randomization. The efficacy data are provided in Table 8 and demonstrate that extended prophylaxis with fondaparinux sodium was associated with a VTE rate of 1.4% compared with a VTE rate of 35.0% for placebo for a relative risk reduction of 95.9% (95% CI = [98.7; 87.1], P <0.0001). Major bleeding rates during the 3-week extended prophylaxis period for fondaparinux sodium occurred in 2.4% of patients receiving fondaparinux sodium and 0.6% of placebo-treated patients [see Adverse Reactions (6.1)].
Table 8. Efficacy of Fondaparinux Sodium Injection in the Extended Prophylaxis of Thromboembolic Events Following Hip Fracture Surgery
Endpoint Extended Prophylaxis (Day 8 to Day 28 ± 2 post-surgery) Fondaparinux Sodium2.5 mg SC once daily Placebo SC once daily n/Na % (95% CI) n/Na % (95% CI) VTE 3/208 1.4%b (0.3, 4.2) 77/220 35.0% (28.7, 41.7) All DVT 3/208 1.4%b (0.3, 4.2) 74/218 33.9% (27.7, 40.6) Proximal DVT 2/221 0.9%b (0.1, 3.2) 35/222 15.8% (11.2, 21.2) Symptomatic VTE (all) 1/326 0.3%c (0.0, 1.7) 9/330 2.7% (1.3, 5.1) Symptomatic PE 0/326 0.0%d (0.0, 1.1) 3/330 0.9% (0.2, 2.6) a N = all randomized evaluable hip fracture surgery patients. Evaluable patients were those who were treated in the post-randomization period, with an adequate efficacy assessment for up to 24 days following randomization.
b P value versus placebo <0.001
c P value versus placebo = 0.021.
d P value versus placebo = NS.
14.3 Prophylaxis of Thromboembolic Events Following Hip Replacement Surgery
In 2 randomized, double-blind, clinical trials in patients undergoing hip replacement surgery, fondaparinux sodium 2.5 mg SC once daily was compared to either enoxaparin sodium 30 mg SC every 12 hours (Study 1) or to enoxaparin sodium 40 mg SC once a day (Study 2). In Study 1, a total of 2,275 patients were randomized and 2,257 were treated. Patients ranged in age from 18 to 92 years (mean age 65 years) with 48% men and 52% women. Patients were 94% Caucasian, 4% black, <1% Asian, and 2% others. In Study 2, a total of 2,309 patients were randomized and 2,273 were treated. Patients ranged in age from 24 to 97 years (mean age 65 years) with 42% men and 58% women. Patients were 99% Caucasian, and 1% other races. Patients with serum creatinine level more than 2 mg/dL (180 micromol/L), or platelet count less than 100,000/mm3 were excluded from both trials. In Study 1, fondaparinux sodium was initiated 6 ± 2 hours (mean 6.5 hours) after surgery in 92% of patients and enoxaparin sodium was initiated 12 to 24 hours (mean 20.25 hours) after surgery in 97% of patients. In Study 2, fondaparinux sodium was initiated 6 ± 2 hours (mean 6.25 hours) after surgery in 86% of patients and enoxaparin sodium was initiated 12 hours before surgery in 78% of patients. The first post-operative enoxaparin sodium dose was given within 12 hours after surgery in 60% of patients and 12 to 24 hours after surgery in 35% of patients with a mean of 13 hours. For both studies, both study treatments were continued for 7 ± 2 days. The efficacy data are provided in Table 9. Under the conditions of Study 1, fondaparinux sodium was associated with a VTE rate of 6.1% compared with a VTE rate of 8.3% for enoxaparin sodium for a relative risk reduction of 26% (95% CI: -11%, 53%; P = NS). Under the conditions of Study 2, fondaparinux sodium was associated with a VTE rate of 4.1% compared with a VTE rate of 9.2% for enoxaparin sodium for a relative risk reduction of 56% (95% CI: 33%, 73%; P <0.001). For the 2 studies combined, the major bleeding episodes occurred in 3.0% of patients receiving fondaparinux sodium and 2.1% of enoxaparin sodium patients [see Adverse Reactions (6.1)].
Table 9. Efficacy of Fondaparinux Sodium in the Prophylaxis of Thromboembolic Events Following Hip Replacement Surgery
Endpoint Study 1 n/Na% (95% CI) Study 2 n/Na% (95% CI) Fondaparinux Sodium 2.5 mg SC
once dailyEnoxaparin Sodium 30 mg SC every
12 hrFondaparinux Sodium 2.5 mg SC
once dailyEnoxaparin Sodium 40 mg SC
once dailyVTEb 48/787
6.1%c (4.5, 8.0)66/797
8.3% (6.5, 10.4)37/908
4.1%e (2.9, 5.6)85/919
9.2% (7.5, 11.3)All DVT 44/784
5.6%d (4.1, 7.5)65/796
8.2% (6.4, 10.3)36/908
4.0%e (2.8, 5.4)83/918
9.0% (7.3, 11.1)Proximal DVT 14/816
1.7%c (0.9, 2.9)10/830
1.2% (0.6, 2.2)6/922
0.7%f (0.2, 1.4)23/927
2.5% (1.6, 3.7)Symptomatic PE 5/1,126
0.4%c (0.1, 1.0)1/1,128
0.1% (0.0, 0.5)2/1,129
0.2%c (0.0, 0.6)2/1,123
0.2% (0.0, 0.6)a N = all evaluable hip replacement surgery patients. Evaluable patients were those who were treated and underwent the appropriate surgery (i.e., hip replacement surgery), with an adequate efficacy assessment up to Day 11.
b VTE was a composite of documented DVT and/or documented symptomatic PE reported up to Day 11.
c P value versus enoxaparin sodium: NS.
d P value versus enoxaparin sodium in study 1: <0.05.
e P value versus enoxaparin sodium in study 2: <0.001.
f P value versus enoxaparin sodium in study 2: <0.01.
14.4 Prophylaxis of Thromboembolic Events Following Knee Replacement Surgery
In a randomized, double-blind, clinical trial in patients undergoing knee replacement surgery (i.e., surgery requiring resection of the distal end of the femur or proximal end of the tibia), fondaparinux sodium 2.5 mg SC once daily was compared to enoxaparin sodium 30 mg SC every 12 hours. A total of 1,049 patients were randomized and 1,034 were treated. Patients ranged in age from 19 to 94 years (mean age 68 years) with 41% men and 59% women. Patients were 88% Caucasian, 8% black, <1% Asian, and 3% others. Patients with serum creatinine level more than 2 mg/dL (180 micromol/L), or platelet count less than 100,000/mm3 were excluded from the trial. Fondaparinux sodium was initiated 6 ± 2 hours (mean 6.25 hours) after surgery in 94% of patients, and enoxaparin sodium was initiated 12 to 24 hours (mean 21 hours) after surgery in 96% of patients. For both drugs, treatment was continued for 7 ± 2 days. The efficacy data are provided in Table 10 and demonstrate that under the conditions of the trial, fondaparinux sodium was associated with a VTE rate of 12.5% compared with a VTE rate of 27.8% for enoxaparin sodium for a relative risk reduction of 55% (95% CI: 36%, 70%; P <0.001). Major bleeding episodes occurred in 2.1% of patients receiving fondaparinux sodium and 0.2% of enoxaparin sodium patients [see Adverse Reactions (6.1)].
Table 10. Efficacy of Fondaparinux Sodium in the Prophylaxis of Thromboembolic Events Following Knee Replacement Surgery
Endpoint Fondaparinux Sodium2.5 mg SC once daily Enoxaparin Sodium 30 mg SC every 12 hours n/Na % (95% CI) n/Na % (95% CI) VTEb 45/361 12.5%c (9.2, 16.3) 101/363 27.8% (23.3, 32.7) All DVT 45/361 12.5%c (9.2, 16.3) 98/361 27.1% (22.6, 32.0) Proximal DVT 9/368 2.4%d (1.1, 4.6) 20/372 5.4% (3.3, 8.2) Symptomatic PE 1/517 0.2%d (0.0, 1.1) 4/517 0.8% (0.2, 2.0) aN = all evaluable knee replacement surgery patients. Evaluable patients were those who were treated and underwent the appropriate surgery (i.e., knee replacement surgery), with an adequate efficacy assessment up to Day 11.
b VTE was a composite of documented DVT and/or documented symptomatic PE reported up to Day 11.
c P value versus enoxaparin sodium <0.001. d P value versus enoxaparin sodium: NS
14.5 Prophylaxis of Thromboembolic Events Following Abdominal Surgery in Patients at Risk for Thromboembolic Complications
Abdominal surgery patients at risk included the following: Those undergoing surgery under general anesthesia lasting longer than 45 minutes who are older than 60 years with or without additional risk factors; and those undergoing surgery under general anesthesia lasting longer than 45 minutes who are older than 40 years with additional risk factors. Risk factors included neoplastic disease, obesity, chronic obstructive pulmonary disease, inflammatory bowel disease, history of deep vein thrombosis (DVT) or pulmonary embolism (PE), or congestive heart failure.
In a randomized, double-blind, clinical trial in patients undergoing abdominal surgery, fondaparinux sodium 2.5 mg SC once daily started postoperatively was compared to dalteparin sodium 5,000 IU SC once daily, with one 2,500 IU SC preoperative injection and a 2,500 IU SC first postoperative injection. A total of 2,927 patients were randomized and 2,858 were treated. Patients ranged in age from 17 to 93 years (mean age 65 years) with 55% men and 45% women. Patients were 97% Caucasian, 1% black, 1% Asian, and 1% others. Patients with serum creatinine level more than 2 mg/dL (180 micromol/L), or platelet count less than 100,000/mm3 were excluded from the trial. Sixty-nine percent (69%) of study patients underwent cancer-related abdominal surgery. Study treatment was continued for 7 ± 2 days. The efficacy data are provided in Table 11 and demonstrate that prophylaxis with fondaparinux sodium was associated with a VTE rate of 4.6% compared with a VTE rate of 6.1% for dalteparin sodium (P = NS).
Table 11. Efficacy of Fondaparinux Sodium In Prophylaxis of Thromboembolic Events Following Abdominal Surgery
Endpoint Fondaparinux Sodium 2.5 mg SC once daily Dalteparin Sodium 5,000 IU SC once daily n/Na % (95% CI) n/Na % (95% CI) VTEb 47/1,027 4.6%c (3.4, 6.0) 62/1,021 6.1% (4.7, 7.7) All DVT 43/1,024 4.2% (3.1, 5.6) 59/1,018 5.8% (4.4, 7.4) Proximal DVT 5/1,076 0.5% (0.2, 1.1) 5/1,077 0.5% (0.2, 1.1) Symptomatic VTE 6/1,465 0.4% (0.2, 0.9) 5/1,462 0.3% (0.1, 0.8) a N = all evaluable abdominal surgery patients. Evaluable patients were those who were randomized and had an adequate efficacy assessment up to Day 10; non-treated patients and patients who did not undergo surgery did not get a VTE assessment.
b VTE was a composite of venogram positive DVT, symptomatic DVT, non-fatal PE and/or fatal PE reported up to Day 10.
c P value versus dalteparin sodium: NS.
14.6 Treatment of Deep Vein Thrombosis
In a randomized, double-blind, clinical trial in patients with a confirmed diagnosis of acute symptomatic DVT without PE, fondaparinux sodium 5 mg (body weight <50 kg), 7.5 mg (body weight 50 to 100 kg), or 10 mg (body weight >100 kg) SC once daily (fondaparinux sodium treatment regimen) was compared to enoxaparin sodium 1 mg/kg SC every 12 hours. Almost all patients started study treatment in hospital. Approximately 30% of patients in both groups were discharged home from the hospital while receiving study treatment. A total of 2,205 patients were randomized and 2,192 were treated. Patients ranged in age from 18 to 95 years (mean age 61 years) with 53% men and 47% women. Patients were 97% Caucasian, 2% black, and 1% other races. Patients with serum creatinine level more than 2 mg/dL (180 micromol/L), or platelet count less than 100,000/mm3 were excluded from the trial. For both groups, treatment continued for at least 5 days with a treatment duration range of 7 ± 2 days, and both treatment groups received vitamin K antagonist therapy initiated within 72 hours after the first study drug administration and continued for 90 ± 7 days, with regular dose adjustments to achieve an INR of 2 to 3. The primary efficacy endpoint was confirmed, symptomatic, recurrent VTE reported up to Day 97. The efficacy data are provided in Table 12.
Table 12. Efficacy of Fondaparinux Sodium in the Treatment of Deep Vein Thrombosis (All Randomized)
Endpoint Fondaparinux Sodium5, 7.5, or 10 mg SC once daily
N = 1,098Enoxaparin Sodium1 mg/kg SC every 12 hours
N = 1,107n % (95% CI) n % (95% CI) Total VTEa 43 3.9% (2.8, 5.2) 45 4.1% (3.0, 5.4) DVT only 18 1.6% (1.0, 2.6) 28 2.5% (1.7, 3.6) Non-fatal PE 20 1.8% (1.1, 2.8) 12 1.1% (0.6, 1.9) Fatal PE 5 0.5% (0.1, 1.1) 5 0.5% (0.1, 1.1) a VTE was a composite of symptomatic recurrent non-fatal VTE or fatal PE reported up to Day 97. The 95% confidence interval for the treatment difference for total VTE was: (-1.8% to 1.5%).
During the initial treatment period, 18 (1.6%) of patients treated with fondaparinux sodium and 10 (0.9%) of patients treated with enoxaparin sodium had a VTE endpoint (95% CI for the treatment difference [fondaparinux sodium-enoxaparin sodium] for VTE rates: -0.2%; 1.7%).
14.7 Treatment of Pulmonary Embolism
In a randomized, open-label, clinical trial in patients with a confirmed diagnosis of acute symptomatic PE, with or without DVT, fondaparinux sodium 5 mg (body weight <50 kg), 7.5 mg (body weight 50 to 100 kg), or 10 mg (body weight >100 kg) SC once daily (fondaparinux sodium treatment regimen) was compared to heparin IV bolus (5,000 USP units) followed by a continuous IV infusion adjusted to maintain 1.5 to 2.5 times aPTT control value. Patients with a PE requiring thrombolysis or surgical thrombectomy were excluded from the trial. All patients started study treatment in hospital. Approximately 15% of patients were discharged home from the hospital while receiving fondaparinux sodium therapy. A total of 2,213 patients were randomized and 2,184 were treated. Patients ranged in age from 18 to 97 years (mean age 62 years) with 44% men and 56% women. Patients were 94% Caucasian, 5% black, and 1% other races. Patients with serum creatinine level more than 2 mg/dL (180 micromol/L), or platelet count less than 100,000/mm3 were excluded from the trial. For both groups, treatment continued for at least 5 days with a treatment duration range 7 ± 2 days, and both treatment groups received vitamin K antagonist therapy initiated within 72 hours after the first study drug administration and continued for 90 ± 7 days, with regular dose adjustments to achieve an INR of 2 to 3. The primary efficacy endpoint was confirmed, symptomatic, recurrent VTE reported up to Day 97. The efficacy data are provided in Table 13.
Table 13. Efficacy of Fondaparinux Sodium in the Treatment of Pulmonary Embolism (All Randomized)
Endpoint Fondaparinux Sodium5, 7.5, or 10 mg SC once dailyN = 1,103 HeparinaPTT adjusted IVN = 1,110 n % (95% CI) n % (95% CI) Total VTEa 42 3.8% (2.8, 5.1) 56 5.0% (3.8, 6.5) DVT only 12 1.1% (0.6, 1.9) 17 1.5% (0.9, 2.4) Non-fatal PE 14 1.3% (0.7, 2.1) 24 2.2% (1.4, 3.2) Fatal PE 16 1.5% (0.8, 2.3) 15 1.4% (0.8, 2.2) a VTE was a composite of symptomatic recurrent non-fatal VTE or fatal PE reported up to Day 97. The 95% confidence interval for the treatment difference for total VTE was: (-3.0% to 0.5%).
During the initial treatment period, 12 (1.1%) of patients treated with fondaparinux sodium and 19 (1.7%) of patients treated with heparin had a VTE endpoint (95% CI for the treatment difference [fondaparinux sodium-heparin] for VTE rates: -1.6%; 0.4%).
-
16 HOW SUPPLIED/STORAGE AND HANDLING
Fondaparinux sodium injection is available in the following strengths and package sizes:
2.5 mg fondaparinux sodium in 0.5 mL single dose prefilled syringe, affixed with a 27-gauge x ½-inch needle with a blue plunger rod.
2 Single Unit Syringes NDC: 55111-678-02
Single Unit Syringes NDC: 55111-678-10
5 mg fondaparinux sodium in 0.4 mL single dose prefilled syringe, affixed with a 27-gauge x ½-inch needle with an orange plunger rod.
2 Single Unit Syringes NDC: 55111-679-02
Single Unit Syringes NDC: 55111-679-10
7.5 mg fondaparinux sodium in 0.6 mL single dose prefilled syringe, affixed with a 27-gauge x ½-inch needle with a magenta plunger rod.
2 Single Unit Syringes NDC: 55111-680-02
Single Unit Syringes NDC: 55111-680-10
10 mg fondaparinux sodium in 0.8 mL single dose prefilled syringe, affixed with a 27-gauge x ½-inch needle with a violet plunger rod.
2 Single Unit Syringes NDC: 55111-681-02
Single Unit Syringes NDC: 55111-681-10
Store at 20° to 25°C (68° to 77°F) [see USP Controlled Room Temperature].
Discard unused portion.
Pharmacist: Dispense a Patient Information Leaflet with each prescription.
-
17 PATIENT COUNSELING INFORMATION
See FDA-Approved Patient Labeling (17.2)
17.1 Patient Advice
If the patients have had neuraxial anesthesia or spinal puncture, and particularly, if they are taking concomitant NSAIDS, platelet inhibitors, or other anticoagulants, they should be informed to watch for signs and symptoms of spinal or epidural hematomas, such as back pain, tingling, numbness (especially in the lower limbs), muscular weakness, and stool or urine incontinence. If any of these symptoms occur, the patients should contact his or her physician immediately.
The use of aspirin and other NSAIDS may enhance the risk of hemorrhage. Their use should be discontinued prior to fondaparinux sodium therapy whenever possible; if co-administration is essential, the patient’s clinical and laboratory status should be closely monitored. [see Drug Interactions (7)]
If patients must self-administer fondaparinux sodium (e.g., if fondaparinux sodium is used at home), they should be advised of the following:
- Fondaparinux sodium should be given by subcutaneous injection. Patients must be instructed in the proper technique for administration
- The most important risk with fondaparinux sodium administration is bleeding. Patients should be counseled on signs and symptoms of possible bleeding.
- It may take them longer than usual to stop bleeding.
- They may bruise and/or bleed more easily when they are treated with fondaparinux sodium. They should report any unusual bleeding, bruising, or signs of thrombocytopenia (such as a rash of dark red spots under the skin) to their physician [see Warnings and Precautions (5.2, 5.5) ].
- To tell their physicians and dentists they are taking fondaparinux sodium and/or any other product known to affect bleeding before any surgery is scheduled and before any new drug is taken [see Warnings and Precautions (5.2) ].
- To tell their physicians and dentists of all medications they are taking, including those obtained without a prescription, such as aspirin or other NSAIDs. [see Drug Interactions (7)]
Keep out of the reach of children.
-
PATIENT PACKAGE INSERT
PATIENT INFORMATION
Fondaparinux (fon-da-PEH-rih-nux) Sodium Injection
for subcutaneous use
What is the most important information I should know about fondaparinux sodium injection?
Fondaparinux sodium injection may cause serious side effects, including:
Spinal or epidural blood clots (hematoma). People who take a blood thinner medicine (anticoagulant) like fondaparinux sodium injection, and have medicine injected into their spinal and epidural area, or have a spinal puncture have a risk of forming a blood clot that can cause long-term or permanent loss of the ability to move (paralysis). Your risk of developing a spinal or epidural blood clot is higher if:
- a thin tube called an epidural catheter is placed in your back to give you certain medicine.
- you take NSAIDs or a medicine to prevent blood from clottingyou have a history of difficult or repeated epidural or spinal punctures
- you have a history of problems with your spine or have had surgery on your spine.
If you take fondaparinux sodium injection and receive spinal anesthesia or have a spinal puncture, your doctor should watch you closely for symptoms of spinal or epidural blood clots. Tell your doctor right away if you have back pain, tingling, numbness, muscle weakness (especially in your legs and feet), loss of control of the bowels or bladder (incontinence).
Because the risk of bleeding may be higher, tell your doctor before taking fondaparinux sodium if you:
- are also taking certain other medicines that affect blood clotting such as aspirin, an NSAID (for example, ibuprofen or naproxen), clopidogrel, or warfarin sodium.
- have bleeding problems.
- had problems in the past with pain medication given through the spine.
- have had surgery to your spine.
- have a spinal deformity.
What is fondaparinux sodiuminjection?
Fondaparinux sodium injection is a prescription medicine that is used to:
help prevent blood clots from forming in people who have had certain surgeries of the hip, knee, or the stomach area (abdominal surgery)
treat people who have blood clots in their legs or blood clots that travel to their lungs along with the blood thinner medicine warfarin.
It is not known if fondaparinux sodium injection is safe and effective for use in children younger than 18 years of age.
Who should not take fondaparinux sodiuminjection?
Do not take fondaparinux sodium injection if you:
- have certain kidney problems
- have active bleeding problems
- have an infection in your heart
- have low platelet counts and if you test positive for a certain antibody while you are taking fondaparinux sodium injection.
- weigh less than 110 pounds (50 kg) to prevent blood clots from surgery. See, “What are the possible side effects of fondaparinux sodium injection?”
- had a serious allergic reaction to fondaparinux sodium injection
What should I tell my doctor before taking fondaparinux sodiuminjection?
Before taking fondaparinux sodium injection tell your doctor about all of your medical conditions, including if you:
- have had any bleeding problems (such as stomach ulcers)
- have had a stroke
- have had recent surgeries, including eye surgery
- have diabetic eye disease
- have kidney or liver problems
- have uncontrolled high blood pressure
- are pregnant or plan to become pregnant. Fondaparinux sodium injection may harm your unborn baby. If you are pregnant, talk to your doctor about the best way for you to prevent or treat blood clots.
- are breastfeeding or plan to breastfeed. It is not known if fondaparinux sodium passes into breast milk. You and your doctor should decide if you will breastfeed during treatment with fondaparinux sodium injection.
Tell your doctor about all the medicines you take including prescriptions and over-the-counter medicines, vitamins, and herbal supplements. Some medicines can increase your risk of bleeding.
See “What is the most important information I should know about how fondaparinux sodiuminjection?” Do not start taking any new medicines without first talking to your doctor.
Tell all your doctors and dentist that you take fondaparinux sodium injection, especially if you need to have any kind of surgery or a dental procedure. Keep a list of your medicines and show it to all your doctors and pharmacist before you start a new medicine.
How should I take fondaparinux sodiuminjection?
- See the detailed Instructions for Use that comes with fondaparinux sodium injection for information about to give an fondaparinux sodium injection.
- If your doctor tells you that you may give yourself injections of fondaparinux sodium injection at home, you will be shown how to give the injections first before you do them on your own.
- Take fondaparinux sodium injection exactly as your doctor tells you to. Fondaparinux sodium is given by injection under the skin (subcutaneous injection).
- If you miss a dose of fondaparinux sodium injection, take your dose as soon as you remember. Do not take 2 doses at the same time. ·
- If you take too much fondaparinux sodium injection, call your doctor right away.
What are possible side effects of fondaparinux sodium injection? Fondaparinux sodium can cause serious side effects. See “What is the most important information I should know about fondaparinux sodium injection?”
Severe bleeding Certain conditions can increase your risk for severe bleeding, including:
-some bleeding problems
-some gastrointestinal problems including ulcers
-some types of strokes
-uncontrolled high blood pressure
-diabetic eye disease
-soon after brain, spine, or eye surgery
- Certain kidney problems can also increase your risk of bleeding with fondaparinux sodium injection. Your doctor may check your kidney function during your treatment with fondaparinux sodium injection.
- Increased bleeding risk in people undergoing certain surgeries who weigh less than 110 pounds (50 kg).
- Low blood platelets (thrombocytopenia). Low blood platelets can happen when you take fondaparinux sodium injection. Platelets are blood cells that help your blood to clot normally. Your doctor may check your platelet counts during your treatment with fondaparinux sodium injection.
You may bruise or bleed more easily during your treatment with fondaparinux sodium injection, and it may take longer than usual for bleeding to stop. Tell your doctor if you have any signs or symptoms of bleeding, bruising, or a rash of dark spots under the skin during your treatment with fondaparinux sodium injection.
Tell your doctor if you have any bleeding, bruising, or a rash of dark spots under the skin (thrombocytopenia).
The most common side effects of fondaparinux sodium injection include:
bleeding problems
bleeding, rash, and itching at the injection site
(injection site reactions)
sleep problems (insomnia)
low red blood cell count (anemia)
increased wound drainage
low potassium in your blood (hypokalemia)
dizziness
purplish spots on skin (purpura)
low blood pressure (hypotension)
confusion
fluid-filled blisters (bullous eruption)
blood clots (hematoma)
severe bleeding after surgery (post-operative hemorrhage)
These are not all the possible side effects of fondaparinux sodium injection. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store fondaparinux sodium injection?
Store fondaparinux sodium injection at 20° to 25°C (68° to 77°F); [room temperature].
Safely, throw away fondaparinux sodium injection that is out of date or no longer needed.
Keep fondaparinux sodium injection and all medicines out of the reach of children.
General information about the safe and effective use of fondaparinux sodium injection
Medicines are sometimes prescribed for purposes other than those listed in Patient Information leaflets. Do not use fondaparinux sodium injection for a condition for which it was not prescribed. Do not give fondaparinux sodium injection to other people, even if they have the same symptoms that you have. It may harm them.
You can ask your doctor or pharmacist for information about fondaparinux sodium injection that is written for healthcare professionals.
What are the ingredients in fondaparinux sodiuminjection?
Active Ingredient: fondaparinux sodium
Inactive Ingredients: sodium chloride and water for injection. Also contain hydrochloric acid and sodium hydroxide as pH adjusters.
For more information about fondaparinux sodium injection, call 1-888-375-3784.
This Patient Information has been approved by the U.S Food and Drug Administration.
INSTRUCTIONS FOR USE
Fondaparinux (fon-da-PEH-rih-nux) Sodium Injection
for subcutaneous use
Be sure that you read, understand, and follow the step-by-step Instructions for Use, before you try to give yourself an injection of fondaparinux sodium for the first time and each time you get a new prescription. There may be new information. Talk to your doctor or pharmacist if you have any questions.
Do not use fondaparinux sodium injection if:
the solution appears discolored (the solution should normally appear clear)
you see any particles in the solution
the syringe is damaged
How should I give an injection of fondaparinux sodium?
Fondaparinux sodium is injected into a skin fold of the lower stomach area (abdomen). Do not inject fondaparinux sodium into muscle. Usually a doctor or nurse will give this injection to you. In some cases you may be taught how to do this yourself.

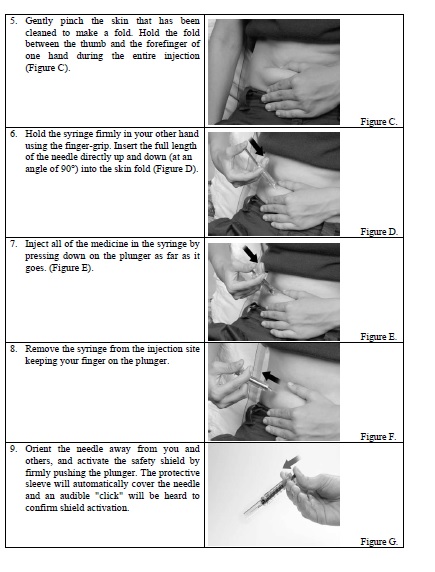

To reorder additional Patient Information Sheets contact Dr. Reddy’s Customer Service at 1-866-733-3952 or visit www.reddyfondaparinux.com
Rx Only
Manufactured by:
Gland Pharma Limited
D.P. Pally – 500 043 INDIA
Manufactured for:
Dr. Reddy’s Laboratories Limited
Bachupally - 500 090 INDIA
Revised: 0120
- PACKAGE LABEL.PRINCIPAL DISPLAY PANEL SECTION
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
FONDAPARINUX SODIUM
fondaparinux sodium injectionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 55111-678 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength fondaparinux sodium (UNII: X0Q6N9USOZ) (fondaparinux - UNII:J177FOW5JL) fondaparinux sodium 2.5 mg in 0.5 mL Inactive Ingredients Ingredient Name Strength sodium chloride (UNII: 451W47IQ8X) hydrochloric acid (UNII: QTT17582CB) sodium hydroxide (UNII: 55X04QC32I) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 55111-678-02 2 in 1 CARTON 07/14/2011 1 NDC: 55111-678-11 0.5 mL in 1 SYRINGE; Type 0: Not a Combination Product 2 NDC: 55111-678-10 10 in 1 CARTON 07/14/2011 2 NDC: 55111-678-11 0.5 mL in 1 SYRINGE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA091316 07/14/2011 FONDAPARINUX SODIUM
fondaparinux sodium injectionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 55111-679 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength fondaparinux sodium (UNII: X0Q6N9USOZ) (fondaparinux - UNII:J177FOW5JL) fondaparinux sodium 5 mg in 0.4 mL Inactive Ingredients Ingredient Name Strength sodium chloride (UNII: 451W47IQ8X) hydrochloric acid (UNII: QTT17582CB) sodium hydroxide (UNII: 55X04QC32I) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 55111-679-02 2 in 1 CARTON 07/14/2011 1 NDC: 55111-679-11 0.4 mL in 1 SYRINGE; Type 0: Not a Combination Product 2 NDC: 55111-679-10 10 in 1 CARTON 07/14/2011 2 NDC: 55111-679-11 0.4 mL in 1 SYRINGE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA091316 07/14/2011 FONDAPARINUX SODIUM
fondaparinux sodium injectionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 55111-680 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength fondaparinux sodium (UNII: X0Q6N9USOZ) (fondaparinux - UNII:J177FOW5JL) fondaparinux sodium 7.5 mg in 0.6 mL Inactive Ingredients Ingredient Name Strength sodium chloride (UNII: 451W47IQ8X) hydrochloric acid (UNII: QTT17582CB) sodium hydroxide (UNII: 55X04QC32I) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 55111-680-02 2 in 1 CARTON 07/14/2011 1 NDC: 55111-680-11 0.6 mL in 1 SYRINGE; Type 0: Not a Combination Product 2 NDC: 55111-680-10 10 in 1 CARTON 07/14/2011 2 NDC: 55111-680-11 0.6 mL in 1 SYRINGE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA091316 07/14/2011 FONDAPARINUX SODIUM
fondaparinux sodium injectionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 55111-681 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength fondaparinux sodium (UNII: X0Q6N9USOZ) (fondaparinux - UNII:J177FOW5JL) fondaparinux sodium 10 mg in 0.8 mL Inactive Ingredients Ingredient Name Strength sodium chloride (UNII: 451W47IQ8X) hydrochloric acid (UNII: QTT17582CB) sodium hydroxide (UNII: 55X04QC32I) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 55111-681-02 2 in 1 CARTON 07/14/2011 1 NDC: 55111-681-11 0.8 mL in 1 SYRINGE; Type 0: Not a Combination Product 2 NDC: 55111-681-10 10 in 1 CARTON 07/14/2011 2 NDC: 55111-681-11 0.8 mL in 1 SYRINGE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA091316 07/14/2011 Labeler - Dr. Reddy's Laboratories Limited (650562841) Establishment Name Address ID/FEI Business Operations Gland Pharma Limited 918601238 analysis(55111-678, 55111-679, 55111-680, 55111-681) , manufacture(55111-678, 55111-679, 55111-680, 55111-681)

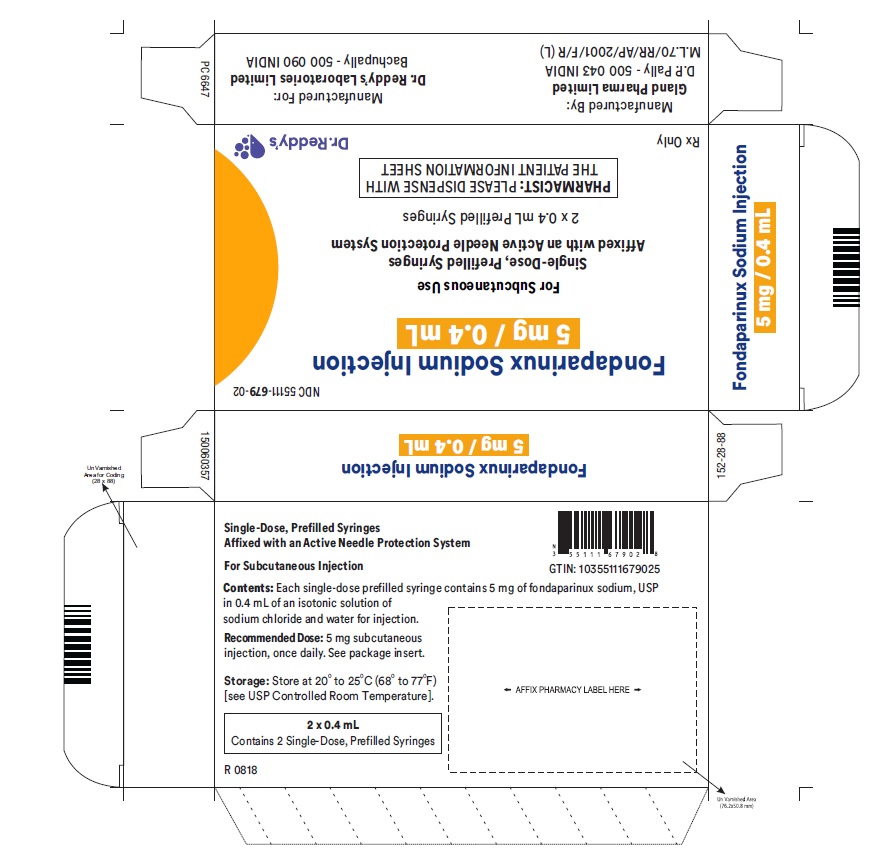


© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.