OJJAARA- momelotinib tablet
Ojjaara by
Drug Labeling and Warnings
Ojjaara by is a Prescription medication manufactured, distributed, or labeled by GlaxoSmithKline LLC. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use OJJAARA safely and effectively. See full prescribing information for OJJAARA.
OJJAARA (momelotinib) tablets, for oral use
Initial U.S. Approval: 2023RECENT MAJOR CHANGES
INDICATIONS AND USAGE
OJJAARA is a kinase inhibitor indicated for the treatment of intermediate or high‑risk myelofibrosis (MF), including primary MF or secondary MF [post‑polycythemia vera (PV) and post‑essential thrombocythemia (ET)], in adults with anemia. (1)
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
Tablets: 100 mg, 150 mg, 200 mg. (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
- Risk of Infections: Do not initiate OJJAARA in patients with an active infection. Monitor for signs and symptoms of infection, including reactivation of hepatitis B, and initiate appropriate treatment promptly. (5.1)
- Thrombocytopenia and Neutropenia: Manage by dose reduction or interruption. (5.2)
- Hepatotoxicity: Obtain liver tests before initiation of and periodically throughout treatment with OJJAARA. (5.3)
- Severe Cutaneous Adverse Reactions (SCARs): Monitor for signs and symptoms, and interrupt OJJAARA until etiology of reaction has been determined. (5.4)
- Major Adverse Cardiovascular Events (MACE): Monitor for symptoms, evaluate and treat promptly. (5.5)
- Thrombosis: Evaluate and treat symptoms of thrombosis promptly. (5.6)
- Malignancies: Monitor for development of secondary malignancies, particularly in current or past smokers. (5.7)
- Symptom Exacerbation Following Interruption or Discontinuation of Treatment: Manage with supportive care and consider restarting OJJAARA. (5.8)
ADVERSE REACTIONS
The most common adverse reactions (≥20% in either study) are thrombocytopenia, hemorrhage, bacterial infection, fatigue, dizziness, diarrhea, and nausea. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact GlaxoSmithKline at 1-888-825-5249 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 3/2026
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
2.2 Laboratory Monitoring for Safety
2.3 Dosage Modification for Hepatic Impairment
2.4 Dosage Modification for Adverse Reactions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Risk of Infections
5.2 Thrombocytopenia and Neutropenia
5.3 Hepatotoxicity
5.4 Severe Cutaneous Adverse Reactions (SCARs)
5.5 Major Adverse Cardiovascular Events (MACE)
5.6 Thrombosis
5.7 Malignancies
5.8 Symptom Exacerbation Following Interruption or Discontinuation of Treatment
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on OJJAARA
7.2 Effect of OJJAARA on Other Drugs
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
The recommended dosage of OJJAARA is 200 mg orally once daily. OJJAARA may be taken with or without food.
Swallow OJJAARA tablets whole. Do not cut, crush, or chew tablets.
If a dose of OJJAARA is missed, the next scheduled dose should be taken the following day.
2.2 Laboratory Monitoring for Safety
Obtain the following blood tests prior to starting treatment with OJJAARA, periodically during treatment, and as clinically indicated:
2.3 Dosage Modification for Hepatic Impairment
The recommended starting dosage in patients with severe hepatic impairment (Child‑Pugh Class C) is 150 mg orally once daily [see Use in Specific Populations (8.6)]. No dose adjustment is recommended for patients with mild or moderate hepatic impairment.
2.4 Dosage Modification for Adverse Reactions
Manage hematologic and non‑hematologic adverse reactions as described in Table 1.
Table 1: Dose Modifications for OJJAARA-Related Adverse Reactions ALT = alanine transaminase; AST = aspartate transaminase; ULN = upper limit of normal. a Reinitiate or escalate treatment up to starting dosage as clinically appropriate. b May reinitiate treatment at 100 mg if previously dosed at 100 mg. c If baseline >2 × ULN. d If baseline >1.5 × ULN. e Graded using the National Cancer Institute Common Terminology Criteria for Adverse Events per (CTCAE). Thrombocytopenia
Dose Modificationa
Baseline Platelet Count
Platelet Count
≥100 × 109/L
20 × 109/L to <50 × 109/L
Reduce daily dose by 50 mg from the last given dose.
<20 × 109/L
Interrupt treatment until platelets recover to 50 × 109/L.
Restart OJJAARA at a daily dose of 50 mg below the last given dose.b
≥50 × 109/L to <100 × 109/L
<20 × 109/L
Interrupt treatment until platelets recover to 50 × 109/L.
Restart OJJAARA at a daily dose of 50 mg below the last given dose.b
<50 × 109/L
<20 × 109/L
Interrupt treatment until platelets recover to baseline.
Restart OJJAARA at a daily dose of 50 mg below the last given dose.b
Neutropenia
Dose Modificationa
Absolute neutrophil count (ANC) <0.5 × 109/L
Interrupt treatment until ANC ≥0.75 × 109/L.
Restart OJJAARA at a daily dose of 50 mg below the last given dose.b
Hepatotoxicity
(unless other apparent causes)
Dose Modificationa
ALT and/or AST >5 × ULN (or >5 × baseline, if baseline is abnormal) and/or total bilirubin >2 × ULN (or >2 × baseline, if baseline is abnormal)
Interrupt treatment until AST and ALT ≤2 × ULN or baselinec and total bilirubin ≤1.5 × ULN or baseline.d
Restart OJJAARA at a daily dose of 50 mg below the last given dose.b
If reoccurrence of ALT or AST elevations >5 × ULN, permanently discontinue OJJAARA.
Other Non‑Hematologic
Dose Modificationa
Grade 3 or highere
Interrupt treatment until the toxicity resolves to Grade 1 or lower (or baseline).
Restart OJJAARA at a daily dose of 50 mg below the last given dose.b
Discontinue OJJAARA in patients unable to tolerate 100 mg once daily.
-
3 DOSAGE FORMS AND STRENGTHS
100 mg round tablet – brown with an underlined “M” debossed on one side and “100” on the other side.
150 mg triangular tablet – brown with an underlined “M” debossed on one side and “150” on the other side.
200 mg capsule‑shaped tablet – brown with an underlined “M” debossed on one side and “200” on the other side.
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Risk of Infections
Serious (including fatal) infections (e.g., bacterial and viral, including COVID‑19) occurred in 13% of patients treated with OJJAARA. Infections regardless of grade occurred in 38% of patients treated with OJJAARA [see Adverse Reactions (6.1)]. Delay starting therapy with OJJAARA until active infections have resolved. Monitor patients receiving OJJAARA for signs and symptoms of infection and initiate appropriate treatment promptly.
Hepatitis B Reactivation
Hepatitis B viral load (HBV‑DNA titer) increases, with or without associated elevations in alanine transaminase (ALT) or aspartate transaminase (AST), have been reported in patients with chronic hepatitis B virus (HBV) infection taking Janus Kinase (JAK) inhibitors, including OJJAARA. The effect of OJJAARA on viral replication in patients with chronic HBV infection is unknown. In patients with HBV infections, check hepatitis B serologies prior to starting OJJAARA. If HBsAg and/or anti‑HBc antibody is positive, consider consultation with a hepatologist regarding monitoring for reactivation versus prophylactic hepatitis B therapy. Patients with chronic HBV infection who receive OJJAARA should have their chronic HBV infection treated and monitored according to clinical HBV guidelines.
5.2 Thrombocytopenia and Neutropenia
OJJAARA can cause thrombocytopenia and neutropenia [see Adverse Reactions (6.1)].
New or worsening thrombocytopenia, with platelet count less than 50 × 109/L, was observed in 20% of patients treated with OJJAARA. Eight percent of patients treated with OJJAARA had baseline platelet counts less than 50 × 109/L.
Severe neutropenia, absolute neutrophil count (ANC) less than 0.5 × 109/L, was observed in 2% of patients treated with OJJAARA.
Assess CBCs, including platelet and neutrophil counts, before initiating treatment and periodically during treatment as clinically indicated. Interrupt dosing or reduce the dose for thrombocytopenia or neutropenia [see Dosage and Administration (2.4)].
5.3 Hepatotoxicity
Two of the 993 patients with MF who received at least one dose of OJJAARA in clinical trials experienced reversible drug‑induced liver injury. Overall, new or worsening elevations of ALT and AST (all grades) occurred in 23% and 24%, respectively, of patients treated with OJJAARA; Grade 3 and 4 transaminase elevations occurred in 1% and 0.5% of patients, respectively. New or worsening elevations of total bilirubin occurred in 16% of patients treated with OJJAARA. All total bilirubin elevations were Grades 1‑2. The median time to onset of any grade transaminase elevation was 2 months, with 75% of cases occurring within 4 months.
Delay starting therapy in patients presenting with uncontrolled acute and chronic liver disease until apparent causes have been investigated and treated as clinically indicated. When initiating OJJAARA, refer to dosing in patients with hepatic impairment [see Dosage and Administration (2.3)].
Monitor liver tests at baseline, every month for 6 months during treatment, then periodically as clinically indicated. If increases in ALT, AST or bilirubin related to treatment are suspected, modify OJJAARA dosage based upon Table 1 [see Dosage and Administration (2.4)].
5.4 Severe Cutaneous Adverse Reactions (SCARs)
Severe cutaneous adverse reactions, including toxic epidermal necrolysis (TEN), have been observed in some patients treated with OJJAARA.
If signs or symptoms of severe cutaneous reactions occur, interrupt OJJAARA until the etiology of the reaction has been determined. Consider early consultation with a dermatologist for evaluation and management.
If etiology is considered to be associated with OJJAARA, permanently discontinue and do not reintroduce OJJAARA in patients who have experienced SCARs or other life‑threatening cutaneous reactions during OJJAARA treatment.
5.5 Major Adverse Cardiovascular Events (MACE)
Another JAK inhibitor increased the risk of MACE, including cardiovascular death, myocardial infarction, and stroke [compared with those treated with tumor necrosis factor (TNF) blockers] in patients with rheumatoid arthritis, a condition for which OJJAARA is not indicated.
Consider the benefits and risks for the individual patient prior to initiating or continuing therapy with OJJAARA, particularly in patients who are current or past smokers and patients with other cardiovascular risk factors. Inform patients receiving OJJAARA of the symptoms of serious cardiovascular events and the steps to take if they occur.
5.6 Thrombosis
Another JAK inhibitor increased the risk of thrombosis, including deep venous thrombosis, pulmonary embolism, and arterial thrombosis (compared with those treated with TNF blockers) in patients with rheumatoid arthritis, a condition for which OJJAARA is not indicated.
Evaluate patients with symptoms of thrombosis and treat appropriately.
5.7 Malignancies
Another JAK inhibitor increased the risk of lymphoma and other malignancies excluding nonmelanoma skin cancer (NMSC) (compared with those treated with TNF blockers) in patients with rheumatoid arthritis, a condition for which OJJAARA is not indicated. Current or past smokers were at increased risk.
Consider the benefits and risks for the individual patient prior to initiating or continuing therapy with OJJAARA, particularly in patients with a known malignancy (other than a successfully treated NMSC), patients who develop a malignancy, and patients who are current or past smokers.
5.8 Symptom Exacerbation Following Interruption or Discontinuation of Treatment
Following discontinuation of JAK inhibitors, including OJJAARA, signs and symptoms from myeloproliferative neoplasms may flare. Some patients with MF have experienced one or more of the following after discontinuing JAK inhibitors: fever, respiratory distress, hypotension, disseminated intravascular coagulation, or multi‑organ failure.
If one or more of these signs and symptoms occur after discontinuation of OJJAARA, evaluate for and treat any intercurrent illness and consider restarting OJJAARA. Instruct patients not to interrupt or discontinue therapy without consulting their healthcare provider. When discontinuing or interrupting therapy for reasons other than potentially life-threatening toxicities, consider tapering the dose of OJJAARA gradually rather than discontinuing abruptly.
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Risk of Infections and Hepatitis B Reactivation [see Warnings and Precautions (5.1)]
- Thrombocytopenia and Neutropenia [see Warnings and Precautions (5.2)]
- Hepatotoxicity [see Warnings and Precautions (5.3)]
- Severe Cutaneous Adverse Reactions [see Warnings and Precautions (5.4)]
- Major Adverse Cardiovascular Events [see Warnings and Precautions (5.5)]
- Thrombosis [see Warnings and Precautions (5.6)]
- Malignancies [see Warnings and Precautions (5.7)]
- Symptom Exacerbation Following Interruption or Discontinuation of Treatment [see Warnings and Precautions (5.8)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of OJJAARA was evaluated in 215 patients in 2 clinical trials (MOMENTUM and SIMPLIFY‑1 anemic subgroup [hemoglobin (Hb) <10 g/dL]) [see Clinical Studies (14)].
MOMENTUM
Patients in the MOMENTUM trial had been previously treated with a JAK inhibitor and were randomly assigned 2:1 to receive double‑blind OJJAARA 200 mg orally once daily (n = 130) or danazol 300 mg orally twice daily (n = 65) for 24 weeks, after which they were eligible to receive open‑label OJJAARA in an extended treatment phase. Among patients who received OJJAARA, 72% were exposed for 24 weeks or longer and 52% were exposed for 48 weeks or longer [see Clinical Studies (14)].
Serious adverse reactions occurred in 35% of patients who received OJJAARA during the randomized treatment period of the MOMENTUM trial; the most common serious adverse reactions (≥2%) included bacterial infection (8%), viral infection (5%), hemorrhage (4%), acute kidney injury (3%), pneumonia (3%), pyrexia (3%), thrombosis (3%), syncope (2%), thrombocytopenia (2%), and renal and urinary tract infection (2%). Fatal adverse reactions occurred in 12% of patients who received OJJAARA; the most common (≥2%) fatal adverse reaction was viral infection (5%).
Permanent discontinuation of OJJAARA due to an adverse reaction occurred in 18% of patients during the randomized treatment period of the MOMENTUM trial. Adverse reactions that resulted in permanent discontinuation (≥2%) included viral infection (2%) and thrombocytopenia (2%). Dosage reduction or treatment interruption due to an adverse reaction occurred in 34% of patients. Adverse reactions requiring dosage reduction and/or treatment interruption (≥2%) included thrombocytopenia (13%), bacterial infection (2%), diarrhea (2%), and neutropenia (2%).
Among the 130 patients treated with OJJAARA during the randomized treatment period of MOMENTUM, the most common adverse reactions (≥20%) were thrombocytopenia, diarrhea, hemorrhage, and fatigue (Table 2).
Table 2: Adverse Reactions Occurring in ≥5% of Patients Receiving OJJAARA during Randomized Treatment in MOMENTUM a Study was not designed to evaluate meaningful comparisons of the incidence of adverse reactions across treatment groups. b Adverse reactions graded using CTCAE v.5. c Grouped term includes other related terms. d Excludes opportunistic infections. Adverse Reaction
OJJAARA
n = 130
Danazola
n = 65
All Gradesb
%
Grade ≥3
%
All Grades
%
Grade ≥3
%
Thrombocytopeniac
28
22
17
12
Diarrheac
22
0
9
2
Hemorrhagec
22
2
18
8
Fatiguec
21
2
20
5
Nauseac
16
2
9
3
Bacterial infectionc,d
15
8
18
8
Abdominal painc
13
1
18
3
Viral infectionc,d
12
5
3
0
Pruritusc
11
2
11
0
Elevated liver enzymesc
10
2
9
3
Pyrexiac
10
2
8
0
Coughc
8
0
5
0
Paresthesiac
8
1
2
0
Dizzinessc
8
2
2
0
Vomitingc
8
1
0
0
Rashc
6
0
11
0
Renal and urinary tract infectionc,d
6
2
11
5
Arrhythmiac
5
1
6
2
Neutropenia
5
5
3
3
SIMPLIFY‑1
Patients in the SIMPLIFY‑1 trial were JAK inhibitor naïve and randomly assigned 1:1 to receive double‑blind OJJAARA 200 mg orally once daily (n = 215) or ruxolitinib 5 to 20 mg orally twice daily (n = 217). Upon completion of the double‑blind treatment phase, all patients were eligible to receive OJJAARA during the open‑label phase. The safety of OJJAARA was evaluated in the population of patients with MF who were anemic at study entry. SIMPLIFY‑1 enrolled 180 anemic patients who received OJJAARA (n = 85) or ruxolitinib (n = 95). Among these anemic patients who received OJJAARA, 78% were exposed for 24 weeks or longer and 61% were exposed for 48 weeks or longer [see Clinical Studies (14)].
Serious adverse reactions occurred in 28% of the anemic patients who received OJJAARA during the randomized treatment period of the SIMPLIFY‑1 trial; the most common serious adverse reactions (≥2%) included bacterial infection (7%), pneumonia (6%), heart failure (4%) arrhythmia (2%), and respiratory failure (2%). A fatal adverse reaction (bacterial infection) occurred in 1 patient who received OJJAARA.
Permanent discontinuation of OJJAARA due to an adverse reaction occurred in 19% of the anemic patients during the randomized treatment period of the SIMPLIFY‑1 trial. Adverse reactions that resulted in permanent discontinuation of OJJAARA (≥2%) included bacterial infection (2%), dizziness (2%), fatigue (2%), hypotension (2%), and thrombocytopenia (2%). Dosage reductions or treatment interruptions of OJJAARA due to an adverse reaction occurred in 21% of patients. Adverse reactions requiring dosage reduction and/or treatment interruption (≥2%) were thrombocytopenia (8%), pneumonia (4%), bacterial infection (2%), abdominal pain (2%), elevated liver enzymes (2%), and hypotension (2%).
Among the 85 anemic patients treated with OJJAARA during the randomized treatment period of SIMPLIFY‑1, the most common adverse reactions (≥20%) were dizziness, fatigue, bacterial infection, hemorrhage, thrombocytopenia, diarrhea, and nausea (Table 3).
Table 3: Adverse Reactions Occurring in ≥5% of Anemic Patients Receiving OJJAARA during Randomized Treatment in SIMPLIFY-1 a Study was not designed to evaluate meaningful comparisons of the incidence of adverse reactions across treatment groups. b Adverse reactions graded using CTCAE v.4.03. c Grouped term includes other related terms. d Excludes opportunistic infections. Adverse Reactions
OJJAARA
n = 85
Baseline Hb <10 g/dL
Ruxolitiniba
n = 95
Baseline Hb <10 g/dL
All Gradesb
%
Grade ≥3
%
All Grades
%
Grade ≥3
%
Dizzinessc
24
1
15
2
Fatiguec
22
0
25
1
Bacterial infectionc,d
21
8
12
2
Hemorrhagec
21
1
18
2
Thrombocytopeniac
21
11
34
6
Diarrheac
20
1
20
1
Nauseac
20
0
3
1
Abdominal painc
18
1
14
1
Coughc
14
0
11
0
Hypotensionc
14
2
0
0
Pain in extremity
12
0
5
0
Pyrexiac
12
1
11
0
Rashc
12
0
3
0
Renal and urinary tract infectionc,d
12
1
4
0
Elevated liver enzymesc
11
4
9
0
Headachec
11
0
16
0
Peripheral edema
11
0
8
0
Arrhythmiac
8
2
2
1
Paresthesiac
8
0
3
0
Pneumoniac
8
8
5
3
Vomitingc
8
0
5
0
Back pain
7
1
2
0
Viral infectionc,d
6
0
13
2
Vitamin B1 deficiency
6
0
7
0
Other Adverse Reactions
Clinically relevant adverse reactions occurring in <5% of anemic patients in the MOMENTUM and SIMPLIFY‑1 studies include:
Eye Disorders: Blurred vision.
Infections and Infestations: Fungal infection (excludes opportunistic infections).
Musculoskeletal and Connective Tissue Disorders: Arthralgia.
Nervous System Disorders: Neuralgia, peripheral neuropathy, peripheral motor neuropathy, polyneuropathy, syncope.
Vascular Disorders: Flushing.
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of OJJAARA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Immune System Disorders: Hypersensitivity.
Skin and Subcutaneous Tissue Disorders: Erythema multiforme, toxic epidermal necrolysis (TEN).
-
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on OJJAARA
Organic Anion Transporting Polypeptide (OATP)1B1/B3 Inhibitors
Momelotinib is an OATP1B1/B3 substrate. Concomitant use with an OATP1B1/B3 inhibitor increases momelotinib maximal concentrations (Cmax) and area under the concentration‑time curve (AUC), which may increase the risk of adverse reactions with OJJAARA [see Clinical Pharmacology (12.3)]. Monitor patients concomitantly receiving an OATP1B1/B3 inhibitor for adverse reactions and consider OJJAARA dose modifications [see Dosage and Administration (2.4)].
7.2 Effect of OJJAARA on Other Drugs
Breast Cancer Resistance Protein (BCRP) Substrates
Momelotinib is a BCRP inhibitor. OJJAARA may increase exposure of BCRP substrates, which may increase the risk of BCRP substrate adverse reactions [see Clinical Pharmacology (12.3)]. When administered concomitantly with OJJAARA, initiate rosuvastatin (BCRP substrate) at 5 mg and do not increase to more than 10 mg once daily. Dose adjustment of other BCRP substrates may also be needed. Follow approved product information recommendations for other BCRP substrates.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Available data on the use of OJJAARA in pregnant women are insufficient to determine whether there is a drug‑associated risk for major birth defects or miscarriage. Based on animal reproduction studies conducted in rats and rabbits, momelotinib may cause embryo‑fetal toxicity at exposures lower than the expected exposure in patients receiving 200 mg once daily (see Data). OJJAARA should only be used during pregnancy if the expected benefits to the mother outweigh the potential risks to the fetus.
The background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data: In an embryofetal development study, pregnant rats received momelotinib 2, 6 or 12 mg/kg/day orally, during the period of organogenesis (Gestation Day 6 to 17). Embryo‑fetal toxicity (embryonic death, soft tissue anomalies, skeletal variations, and lower mean fetal body weights) was observed at 12 mg/kg (in the presence of maternal toxicity). Skeletal variations were observed (in the absence of maternal toxicity) at 6 mg/kg/day at exposures 3.5 times the exposure at the recommended human dose of 200 mg daily based on combined momelotinib and M21 (a major human metabolite) AUC. No developmental toxicity was observed at 2 mg/kg/day at exposures equivalent to the recommended dose (based on combined momelotinib and M21 AUC).
In an embryofetal developmental study, pregnant rabbits received momelotinib at 7.5, 30 or 60 mg/kg/day orally during the period of organogenesis (Gestation Day 7 to 20). Momelotinib was associated with maternal toxicity at 60 mg/kg/day, which resulted in reduced mean fetal weight, delayed bone ossification, and an abortion at less than the exposure at the recommended dose (based on combined momelotinib and M21 AUC). No developmental toxicity was observed at lower doses tested in rabbits.
In a pre- and post‑natal development study, pregnant rats received momelotinib 2, 6 or 12 mg/kg/day orally from organogenesis through lactation (Gestation Day 6 to lactation Day 20). Decreased pup body weights and embryo‑lethality were observed in the dams administered 6 and 12 mg/kg/day. Pup survival was significantly reduced in the 12 mg/kg/day group from birth to Day 4 of lactation. Momelotinib exposure in dams at 12 mg/kg and 6 mg/kg were approximately 2 times the exposure at the recommended dose (based on combined momelotinib and M21 AUC). The exposure in dams at the No Observed Adverse Effect Level (NOAEL) dose of 2 mg/kg was less than the exposure at the recommended dose (based on combined momelotinib and M21 AUC).
8.2 Lactation
Risk Summary
There are no data on the presence of momelotinib or its metabolites in human milk, the effects on the breastfed child, or the effects on milk production. It is not known whether OJJAARA is excreted in human milk. Momelotinib was present in rat pups following nursing from treated dams with adverse effects observed in the offspring. When a drug is present in animal milk, it is likely that the drug will be present in human milk. Because of the potential for serious adverse reactions in a breastfed child, patients should not breastfeed during treatment with OJJAARA, and for at least 1 week after the last dose of OJJAARA.
Data
Animal Data: In a pre- and post‑natal development study, momelotinib was administered orally to rats during the lactation period; the drug was detected in plasma of nursing pups, which adversely affected pup survival.
8.3 Females and Males of Reproductive Potential
Contraception
Advise females of reproductive potential who are not pregnant to use highly effective contraception during therapy and for at least 1 week after the last dose of OJJAARA.
8.4 Pediatric Use
The safety and effectiveness of OJJAARA in pediatric patients have not been established.
8.5 Geriatric Use
There were 275 patients aged 65 years and older in the clinical studies for MF [see Clinical Studies (14)]. Of the total number of OJJAARA‑treated patients in these studies, 163/216 (75%) were aged 65 years and older, and 63/216 (29%) were aged 75 years and older. No overall differences in safety or effectiveness of OJJAARA have been observed between patients aged 65 years and older and younger adult patients.
8.6 Hepatic Impairment
The recommended starting dose of OJJAARA in patients with severe hepatic impairment (Child‑Pugh C) is 150 mg orally once daily [see Dosage and Administration (2.3)]. No dose modification is recommended for patients with mild hepatic impairment (Child‑Pugh A) or moderate hepatic impairment (Child‑Pugh B).
Momelotinib is extensively metabolized [see Clinical Pharmacology (12.3)]. Momelotinib exposure increased with severe hepatic impairment (Child‑Pugh C). No clinically significant changes in momelotinib exposure were observed in subjects with mild hepatic impairment (Child‑Pugh A) or moderate hepatic impairment (Child‑Pugh B) [see Clinical Pharmacology (12.3)].
-
10 OVERDOSAGE
There is no known antidote for overdose with OJJAARA. If overdose is suspected, the patient should be monitored for signs or symptoms of adverse reactions or effects, and appropriate supportive treatment should be instituted immediately. Further management should be as clinically indicated. Hemodialysis is not expected to enhance the elimination of momelotinib.
Consider contacting the Poison Help line (1‑800‑222‑1222) or a medical toxicologist for additional overdose management recommendations.
-
11 DESCRIPTION
OJJAARA contains momelotinib dihydrochloride monohydrate, which is a kinase inhibitor with the chemical name N‑(Cyanomethyl)-4-(2-{[4-(morpholin-4-yl)phenyl]amino}pyrimidin-4-yl)benzamide dihydrochloride monohydrate. It has a molecular formula of C23H22N6O2●2HCl●H2O, molecular weight of 505.40 and the following structural formula:
Momelotinib dihydrochloride monohydrate is a light yellow to brown to reddish-brown solid and is slightly soluble in water and insoluble in aqueous buffers across a pH range of 2.1 to 9. Momelotinib free base has a molecular formula of C23H22N6O2 and a molecular weight of 414.47.
OJJAARA (momelotinib) tablets are for oral administration. Each tablet contains 100 mg, 150 mg, or 200 mg of momelotinib, which is equivalent to 121.94 mg, 182.91 mg, or 243.88 mg, respectively, of momelotinib dihydrochloride monohydrate as the active ingredient. The core of each tablet contains the following inactive ingredients: lactose monohydrate, magnesium stearate, microcrystalline cellulose, propyl gallate, silicon dioxide, and sodium starch glycolate. The film coating of each tablet contains the following inactive ingredients: polyethylene glycol, polyvinyl alcohol, red iron oxide, talc, titanium dioxide, and yellow iron oxide.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Momelotinib is an inhibitor of wild type Janus Kinase 1 and 2 (JAK1/JAK2) and mutant JAK2V617F, which contribute to signaling of a number of cytokines and growth factors that are important for hematopoiesis and immune function. Momelotinib and its major human circulating metabolite, M21, have higher inhibitory activity for JAK2 compared to JAK3 and tyrosine kinase 2 (TYK2). Momelotinib and M21 additionally inhibit activin A receptor type 1 (ACVR1), also known as activin receptor like kinase 2 (ALK2), which produces subsequent inhibition of liver hepcidin expression and increased iron availability resulting in increased red blood cell production. MF is a myeloproliferative neoplasm associated with constitutive activation and dysregulated JAK signaling that contributes to inflammation and hyperactivation of ACVR1. JAK signaling recruits and activates STAT (signal transducers and activation of transcription) proteins resulting in nuclear localization and subsequent regulation of gene transcription.
12.2 Pharmacodynamics
Momelotinib inhibited STAT3 phosphorylation in whole blood from patients with MF. Maximal inhibition of STAT3 phosphorylation occurred 2 hours after momelotinib dosing and inhibition persisted for at least 6 hours [see Clinical Pharmacology (12.1)]. Iron availability and erythropoiesis was assessed by analysis of circulating hepcidin concentrations. An acute and sustained reduction of circulating hepcidin was observed for the duration of the 24‑week administration of momelotinib to patients with MF [see Clinical Pharmacology (12.1)].
Cardiac Electrophysiology
Momelotinib did not prolong the QT interval to any clinically relevant extent at 4 times the highest recommended dosage of 200 mg.
12.3 Pharmacokinetics
Momelotinib pharmacokinetic parameters are presented as mean (%CV) and were derived in patients with MF unless otherwise specified.
The momelotinib steady‑state Cmax is 479 ng/mL (61%) and AUC is 3,288 ngh/mL (60%) at the maximum recommended dosage. Momelotinib exposure (i.e., Cmax and AUC) increases dose proportionally from 100 mg to 300 mg (0.5 to 1.5 times the maximum recommended dosage), but less than dose‑proportional at doses from 400 mg to 800 mg (2 to 4 times the maximum recommended dosage). There is no clinically significant accumulation.
Absorption
Median time to momelotinib Cmax (Tmax) at steady state is 2 hours (Q1: 1 hour; Q3: 3 hours) post dose.
Effect of Food
No clinically significant differences in momelotinib pharmacokinetics were observed following administration of either a high‑fat meal (800 kcal; 50% fat) or low‑fat meal (400 kcal; 20% fat) in healthy subjects.
Distribution
Momelotinib steady state apparent volume of distribution is 984 L (118%). Momelotinib plasma protein binding is approximately 91% in healthy volunteers.
Elimination
The elimination half‑life of momelotinib and the M21 metabolite is 4 to 8 hours. Momelotinib clearance is 103 L/h (87%).
Metabolism: Momelotinib is metabolized by multiple cytochrome P450 (CYP) enzymes including CYP3A4 (36%), CYP2C8 (19%), CYP2C9 (17%), CYP2C19 (19%), and CYP1A2 (9%).
M21 is an active human metabolite that has approximately 40% of the pharmacological activity of the parent. M21 is formed by CYP followed by aldehyde oxidase metabolism of momelotinib. The mean M21 to momelotinib ratio for AUC ranged from 1.4 to 2.1.
Excretion: Following a single oral dose of radiolabeled momelotinib, 69% (13% unchanged) of radioactivity was excreted in feces and 28% (<1% unchanged) in urine. Approximately 12% of the administered dose was excreted in urine as M21.
Specific Populations
No clinically significant differences in momelotinib and M21 pharmacokinetics were observed based on age (range: 28 to 92 years), race (83% White, 6% Asian, 2% Black), sex (60% male), weight (range: 34 kg to 138 kg), renal impairment (eGFR: 16.4 mL/min/1.73 m2 to above 120 mL/min/1.73 m2), or mild or moderate hepatic impairment (Child‑Pugh A or B). The effect of end stage renal disease receiving dialysis on momelotinib pharmacokinetics is unknown.
Patients with Hepatic Impairment: Momelotinib Cmax increased by 13% and AUC increased by 97% in subjects with severe hepatic impairment (Child‑Pugh C). The M21 metabolite Cmax decreased by 76% and AUC decreased by 48% in subjects with severe hepatic impairment (Child‑Pugh C).
Drug Interaction Studies
Clinical Studies
OATP1B1/1B3 Inhibitors: Momelotinib Cmax increased by 40% and AUC increased by 57% following concomitant use with a single dose of a OATP1B1/1B3 inhibitor (rifampin). The M21 metabolite Cmax increased by 6% and AUC increased by 12%.
BCRP Substrates: A BCRP substrate (rosuvastatin) Cmax increased by 220% and AUC increased by 170% following concomitant use of a single dose of rosuvastatin at 10 mg with multiple doses of momelotinib (200 mg once daily).
Other Drugs: No clinically significant differences in momelotinib and M21metabolite pharmacokinetics were observed when used concomitantly with a strong CYP3A4 inducer (tested with multiple‑dose rifampin), a strong CYP3A4 inhibitor (ritonavir), or an acid reducing agent (omeprazole, a proton pump inhibitor).
No clinically significant differences in the pharmacokinetics of a CYP3A4 substrate (midazolam) were observed when used concomitantly with momelotinib.
In Vitro Studies
Cytochrome P450 (CYP) Enzymes: Momelotinib is a weak, reversible, time‑independent inhibitor of CYP2B6 but does not inhibit CYP1A2, 2C8, 2C9, 2C19, 2D6, or 3A4/5. The M21 metabolite does not inhibit any of these CYP enzymes.
Momelotinib and M21 do not induce CYP3A4, CYP2C8, CYP2C9, or P‑glycoprotein (P‑gp).
Uridine diphosphate‑glucuronosyltransferase (UGT): Momelotinib is an inhibitor of UGT1A1 and UGT1A9. The M21 metabolite is an inducer of UGT1A1.
Transporter Systems: Momelotinib and M21 are substrates in vitro for efflux transporters, P‑gp and BCRP, and hepatic uptake transporters, OATP1B1/1B3. Momelotinib is an inhibitor of BCRP.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
The carcinogenicity potential of momelotinib was assessed in rasH2 transgenic mice and Sprague‑Dawley rats. There was no evidence of tumorigenicity in male or female mice that received momelotinib doses up to 100 mg/kg/day for 26 weeks. In a 2‑year oral carcinogenicity study in Sprague‑Dawley rats, momelotinib caused benign Leydig cell tumors at a dose of 15 mg/kg/day (17 times the maximum recommended dose based on combined momelotinib and M21 AUC). The increase in Leydig cell adenomas was considered related to a rat‑specific phenomenon (i.e., prolactin‑dependent Leydig cell tumorigenesis).
Momelotinib was not mutagenic in a bacterial reverse mutation assay, or clastogenic in an in vitro chromosomal aberration assay with human peripheral blood lymphocytes or in vivo in a rat bone marrow micronucleus assay.
In fertility studies in rats, momelotinib was administered for at least 70 days (males) and 14 days (females) prior to cohabitation and up to the implantation day (gestation Day 7) at doses of 5, 25, and 68 mg/kg/day. Momelotinib reduced sperm concentration and motility and reduced testes and seminal vesicle weights at 25 mg/kg/day or greater (approximately 13 times the recommended dose based on combined momelotinib and M21 AUC) leading to reduced fertility at 68 mg/kg/day. In females, momelotinib reduced ovarian function (reproductive cycles and ovulation) at 68 mg/kg/day and decreased the number of pregnant females and increased pre- and post‑implantation loss with most pregnant rats having total litter loss at 25 mg/kg/day or greater. Exposures at the NOAEL in male and female rats at 5 mg/kg/day are approximately 3‑times the recommended dose (based on combined momelotinib and M21 AUC).
-
14 CLINICAL STUDIES
The efficacy of OJJAARA in the treatment of adults with intermediate 1, intermediate 2, or high‑risk MF, including primary MF, post‑PV MF or post‑ET MF, as defined by the Dynamic International Prognostic Scoring System (DIPSS) or International Prognostic Scoring System (IPSS) for MF, was established in the MOMENTUM trial and in a subpopulation of adults with anemia in the SIMPLIFY‑1 trial. All patients received a starting dosage of OJJAARA 200 mg once daily. Eligible patients had baseline platelet count of ≥25 × 109/L in MOMENTUM and ≥50 × 109/L in SIMPLIFY‑1.
MOMENTUM
MOMENTUM (NCT04173494) was a double‑blind, 2:1 randomized, active‑controlled trial in 195 symptomatic and anemic adults with MF who had previously received an approved JAK inhibitor therapy. Patients were treated with OJJAARA 200 mg once daily or danazol 300 mg twice daily for 24 weeks, then switched to open‑label treatment with OJJAARA.
The median age was 71 years (range 38 to 86 years) with 79% of patients aged 65 years and older, and 63% of patients were male. Overall, 81% of patients were White, 9% of patients were Asian, 2% of patients were Black, and 6% of patients were Hispanic or Latino. Sixty‑four percent of patients had primary MF, 19% had post‑PV MF, and 17% had post‑ET MF. Five percent of patients had intermediate‑1 risk, 57% had intermediate‑2 risk, and 35% had high‑risk disease. Within the 8 weeks prior to treatment, 79% of patients had received red blood cell (RBC) transfusions (median of 4 RBC units; interquartile range: 1‑6). At baseline, 13% and 15% of patients were transfusion independent (defined as no red blood cell transfusions in the 12 weeks before the first dose and Hb ≥8 g/dL) in the OJJAARA and danazol groups, respectively. The baseline median Hb count was 8 g/dL and the median platelet count was 96 × 109/L (range 24 × 109/L to 733 × 109/L). The baseline median palpable spleen length was 11 cm below the left costal margin; the median central spleen volume measured by magnetic resonance imaging (MRI) or computed tomography (CT) was 2,105 cm3 (range 609 cm3 to 9,717 cm3).
Symptoms were measured using the Myelofibrosis Symptom Assessment Form (MFSAF v4.0) diary. The MFSAF v4.0 patient diary, completed throughout the randomized treatment period, captured the core symptoms of MF: fatigue (weariness and tiredness), night sweats (or feeling hot or flushed), itching, abdominal discomfort (feeling pressure or bloating), pain under ribs on left side, feeling of fullness after beginning to eat, and bone pain. For each item, symptom scores, ranging from 0 (absent) to 10 (worst imaginable), were added to create a daily Total Symptom Score (maximum score of 70). At baseline, the mean MFSAF v4.0 Total Symptom Score was 28 in the OJJAARA group and 26 in the danazol group.
The efficacy of OJJAARA in the treatment of patients with primary or secondary MF and anemia was established based on a significantly higher percentage of patients treated with OJJAARA compared to danazol achieving a MFSAF v4.0 Total Symptom Score reduction of 50% or more at Week 24 compared with their own baseline score (Table 4). Other endpoints included transfusion independence, spleen volume response, MFSAF v4.0 Total Symptom Score change from baseline, and percentage of patients with no transfusions.
Table 4: Percent of Patients Achieving Symptom Reduction, Transfusion Independence, and Spleen Volume Reduction at Week 24 in MOMENTUM OJJAARA
n = 130Danazol
n = 65P‑value a Analyses stratified by baseline MFSAF v4.0 Total Symptom Score (<22 vs. ≥22), baseline palpable spleen length below the left costal margin (<12 vs. ≥12 cm), and baseline red blood cell or whole blood units transfused in the 8‑week period before randomization (0, 1‑4, ≥5 units). b Non‑inferiority difference between OJJAARA response rate and 80% of danazol response rate. c Least squares means and difference are reported. d Eight subjects treated with OJJAARA and 3 subjects treated with danazol had no transfusion, but discontinued treatment prior to Week 24. Patients with MFSAF v4.0 Total Symptom Score Reduction of 50% or more, n (%)
32 (25%)
6 (9%)
<0.01
Treatment differencea
(95% CI)
16%
(6, 26)
Patients with Transfusion Independence (no transfusion or Hb <8 g/dL between Weeks 12 and 24), n (%)
39 (30%)
13 (20%)
0.023
Non‑inferiority treatment differencea, b
(95% CI)
14%
(2, 25)
Patients with Spleen Volume Reduction by 25% or More, n (%)
51 (39%)
4 (6%)
<0.0001
Treatment differencea
(95% CI)
33%
(23, 44)
MFSAF v4.0 Total Symptom Score Change from Baselineb (SE)
-9.4 (1.1)
-3.1 (1.6)
0.001
Treatment differencec
(95% CI)
-6.2
(-10, -2.4)
Patients with Spleen Volume Reduction by 35% or More, n (%)
29 (22%)
2 (3%)
0.001
Treatment differencea
(95% CI)
18%
(10, 27)
Patients with No Transfusionsc, d (during the 24‑week treatment period), n (%)
46 (35%)
11 (17%)
0.001
Treatment differencea
(95% CI)
17%
(8, 26)
Figure 1 shows the percentage of patients treated with OJJAARA and danazol who achieved a 50% or greater reduction from baseline for each individual symptom in the MFSAF v4.0.
Figure 1: Percent of Patients Achieving a 50% or Greater Reduction in Individual MFSAF v4.0 Symptom Scores at Week 24a in MOMENTUM
BID = twice a day; QD = once daily.
a Thirty‑six (27.7%) subjects treated with OJJAARA and 27 (41.5%) subjects treated with danazol discontinued treatment prior to Week 24.
SIMPLIFY‑1
SIMPLIFY‑1 (NCT01969838) was a double‑blind, randomized, active‑controlled trial in 432 adults with MF who had not previously received a JAK inhibitor. Patients were treated with OJJAARA 200 mg once daily or ruxolitinib adjusted dose twice daily for 24 weeks. Patients were eligible to switch to open‑label OJJAARA after 24 weeks (without tapering of the JAK inhibitor received during the randomization period). The baseline characteristics and efficacy results provided are for the subset of patients who had anemia (Hb <10 g/dL) at baseline (n = 181).
The median age was 68 years (range 25 to 86 years) with 67% of patients aged 65 years and older, and 59% of patients were male. Eighty‑one percent of patients were White, 8% of patients were Asian, 1% of patients were Black, and 2% of patients were Hispanic or Latino. Sixty‑three percent of patients had primary MF, 13% had post‑PV MF, and 24% had post‑ET MF. Four percent of patients had intermediate‑1 risk, 25% had intermediate‑2 risk, and 71% had high‑risk disease. At baseline, 29% and 44% of patients were transfusion independent in the groups treated with OJJAARA or ruxolitinib, respectively. The baseline median Hb measurement was 8.8 g/dL and the median platelet count was 193 × 109/L (range 54 × 109/L to 2,865 × 109/L). Median palpable spleen length at baseline was 12 cm below the left costal margin; the median spleen volume at baseline (measured by MRI or CT) was 1,843 cm3 (range 352 cm3 to 9,022 cm3).
The efficacy of OJJAARA in the treatment of patients with MF in SIMPLIFY‑1 was based on spleen volume response (reduction by 35% or greater). A numerically lower percent of patients treated with OJJAARA (25%) achieved a Total Symptom Score reduction of 50% or more at Week 24 compared with ruxolitinib (36%).
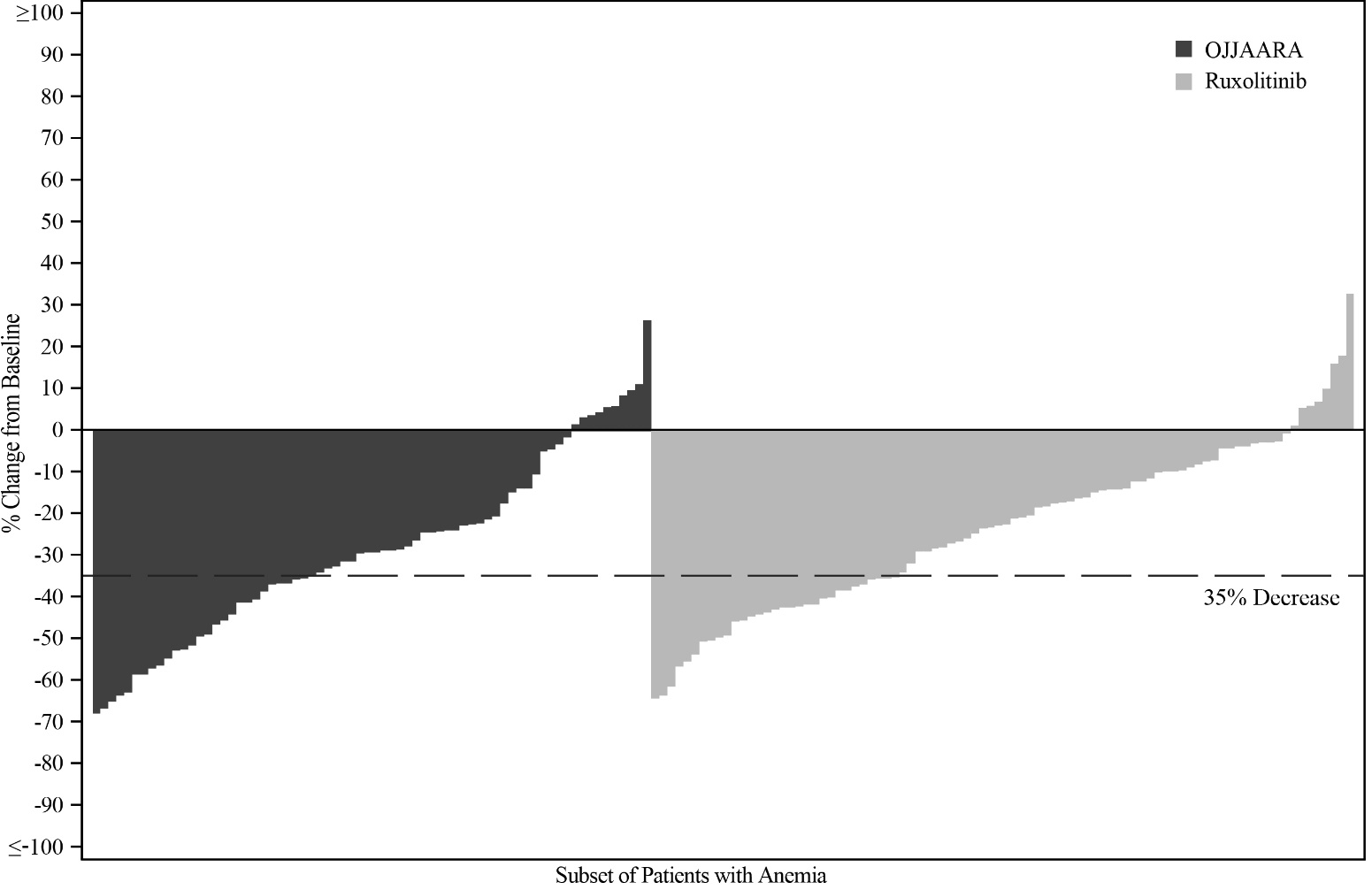
The spleen volume reduction results are presented in Table 5.
Table 5: Percent of Patientsa Achieving 35% or Greater Reduction from Baseline in Spleen Volume at Week 24 in SIMPLIFY-1 a Subset of patients with anemia (Hb <10 g/dL) at baseline. OJJAARA
n = 86
Ruxolitinib
n = 95
Patients with Spleen Volume Reduction by 35% or More, n (%)
(95% CI)
27 (31.4%)
(21.8, 42.3)
31 (32.6%)
(23.4, 43.0)
Figure 2 shows the percent change from baseline in spleen volume for each patient at Week 24 in SIMPLIFY‑1.
Figure 2: Percent Change from Baseline in Spleen Volume for Each Patient at Week 24 in SIMPLIFY‑1a, b
-
a Subset of patients with anemia (Hb <10 g/dL) at baseline.
b Missing data rates for OJJAARA and ruxolitinib were 19% and 8%.
-
a Subset of patients with anemia (Hb <10 g/dL) at baseline.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
OJJAARA (momelotinib) tablets are available as follows:
Table 6: OJJAARA Presentations NDC Number
Strength
Description
Tablets per Bottle
NDC: 81864-103-30
100 mg
Round‑shaped brown film‑coated tablet with “M” on one side and “100” on the other side.
30
NDC: 81864-102-30
150 mg
Triangular‑shaped brown film‑coated tablet with “M” on one side and “150” on the other side.
30
NDC: 81864-101-30
200 mg
Capsule‑shaped brown film‑coated tablet with “M” on one side and “200” on the other side.
30
Store at 20°C to 25°C (68°F to 77°F); excursions permitted to 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature].
Dispense to patient in original bottle only. Store in original bottle to protect from moisture. Replace cap securely each time after opening. Do not discard desiccant.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA approved patient labeling (Patient Information).
Infections
Inform patients that OJJAARA can increase the risk of infections (including COVID‑19) and instruct them to promptly report to their healthcare provider any signs and symptoms of infection [see Warnings and Precautions (5.1)].
Thrombocytopenia and Neutropenia
Inform patients that OJJAARA can cause thrombocytopenia and neutropenia, and of the need to monitor CBC, including platelet and neutrophil counts, before and during treatment. Advise patients to observe for and report any bleeding to their healthcare provider [see Warnings and Precautions (5.2)].
Hepatotoxicity
Inform patients that OJJAARA can cause hepatotoxicity, and of the need to monitor liver blood tests before and during treatment [see Warnings and Precautions (5.3)].
Severe Cutaneous Adverse Reactions (SCARs)
Inform patients that SCARs have been observed in some patients treated with OJJAARA and instruct them to promptly report any signs and symptoms of SCARs to their healthcare provider [see Warnings and Precautions (5.4)].
Major Adverse Cardiovascular Events (MACE)
Advise patients that events of MACE including myocardial infarction, stroke, and cardiovascular death have been reported in clinical studies with another JAK inhibitor used to treat rheumatoid arthritis, a condition for which OJJAARA is not indicated. Advise patients, especially current or past smokers and patients with other cardiovascular risk factors, to be alert for the development of signs and symptoms of cardiovascular events and to report them to their healthcare provider [see Warnings and Precautions (5.5)].
Thrombosis
Advise patients that events of deep vein thrombosis (DVT) and pulmonary embolism (PE) have been reported in clinical studies with another JAK‑inhibitor used to treat rheumatoid arthritis, a condition for which OJJAARA is not indicated. Advise patients to tell their healthcare provider if they develop any signs or symptoms of a DVT or PE [see Warnings and Precautions (5.6)].
Malignancies
Advise patients, especially current or past smokers, that lymphoma and other malignancies (excluding non‑melanoma skin cancers (NMSC) have been reported in clinical studies with another JAK inhibitor used to treat rheumatoid arthritis, a condition for which OJJAARA is not indicated [see Warnings and Precautions (5.7)].
Symptom Exacerbation Following Interruption or Discontinuation of Treatment
Inform patients that after discontinuation of OJJAARA, signs and symptoms from myeloproliferative neoplasms may return. Instruct patients not to interrupt or discontinue OJJAARA without consulting their healthcare provider [see Warnings and Precautions (5.8)].
Pregnancy
- Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females to inform their prescriber of a known or suspected pregnancy [see Use in Specific Populations (8.1)].
- Advise females of reproductive potential who are not pregnant to use highly effective contraception during therapy and for 1 week after the last dose of OJJAARA [see Use in Specific Populations (8.3)].
Lactation
Advise patients not to breastfeed during treatment with OJJAARA and for at least 1 week after the last dose of OJJAARA [see Use in Specific Populations (8.2)].
Trademarks are owned by or licensed to the GSK group of companies.
GlaxoSmithKline
Durham, NC 27701©2026 GSK group of companies or its licensor.
OJJ:3PI
-
PATIENT PACKAGE INSERT
PATIENT INFORMATION
OJJAARA (oh-JAR-uh)
(momelotinib)
tablets, for oral useWhat is OJJAARA?
OJJAARA is a prescription medicine used to treat adults with certain types of myelofibrosis (MF) who have anemia.
It is not known if OJJAARA is safe and effective in children.
Before taking OJJAARA, tell your healthcare provider about all of your medical conditions, including if you:
- have an infection. See “What are the possible side effects of OJJAARA?”
- have or have had hepatitis B
- have or have had liver problems
- have had a heart attack, or have or have had other heart problems, or stroke
- have or have had a blood clot
- smoke or were a smoker in the past
- have or have had any other cancers
- are pregnant or plan to become pregnant. OJJAARA may harm your unborn baby.
-
Females who are able to become pregnant:
- o Use highly effective birth control (contraception) during treatment and for at least 1 week after the last dose of OJJAARA.
- o Tell your healthcare provider right away if you think you are pregnant or become pregnant during treatment with OJJAARA.
- are breastfeeding or plan to breastfeed. It is not known if OJJAARA passes into your breast milk. You should not breastfeed during treatment and for at least 1 week after the last dose of OJJAARA. Talk to your healthcare provider about the best way to feed your baby during this time.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. Taking OJJAARA with certain other medicines may affect the amount of OJJAARA or the other medicines in your blood and may increase your risk of side effects.
Know the medicines you take. Keep a list of the medicines you take to show your healthcare provider and pharmacist when you get a new medicine.
How should I take OJJAARA?
- Take OJJAARA exactly as your healthcare provider tells you to take it.
- Do not change your dose or stop taking OJJAARA without first talking to your healthcare provider. If you stop treatment with OJJAARA, symptoms of your condition may return. See “What are the possible side effects of OJJAARA?”
- Take OJJAARA by mouth 1 time each day.
- Take OJJAARA with or without food.
- Swallow OJJAARA tablets whole. Do not cut, crush, or chew tablets.
- If you miss a dose of OJJAARA, skip the missed dose and take your next dose the following day at your regularly scheduled time. Do not take 2 doses at the same time to make up for the missed dose.
- If you take too much OJJAARA, call your healthcare provider or Poison Help line at 1-800-222-1222, or go to the nearest emergency room right away and take your bottle of OJJAARA with you.
What are the possible side effects of OJJAARA?
OJJAARA may cause serious side effects, including:
- Risk of Infections. People who take OJJAARA may develop serious infections that can lead to death, such as bacterial and viral infections, including COVID‑19. If you have an active infection, your healthcare provider should not start treatment with OJJAARA until your infection is gone. If you have had hepatitis B for a long time (chronic), OJJAARA may cause your hepatitis B to become active again.
- Your healthcare provider will monitor you and treat you for any infections that you get during treatment with OJJAARA.
- Tell your healthcare provider right away if you develop any of the following symptoms of infection:
- o fever
- o chills
- o cough
- o breathing problems
- o diarrhea
- o vomiting
- o pain or burning feeling when passing urine
-
Low platelet and white blood cell counts. OJJAARA may cause new or worsening low platelet and white blood cell counts. Low platelet counts may increase your risk for bleeding and low white blood cell counts may increase your risk for infection.
Tell your healthcare provider right away if you have any signs of bleeding during treatment with OJJAARA, including:
- o unusual bleeding
- o black or tarry stools
- o bruising
- Liver problems. OJJAARA may cause new or worsening increased liver enzymes and bilirubin in your blood. Tell your healthcare provider if you develop any of the following signs or symptoms of liver problems:
- o tiredness
- o loss of appetite
- o pain in your right upper stomach area (abdomen)
- o dark urine
- o yellowing of your skin or the white part of your eyes
-
Severe skin reactions. Severe skin reactions that can be life‑threatening have happened with OJJAARA.
Tell your healthcare provider or get medical help right away if you get any of the following signs or symptoms of severe skin reactions, with or without fever:
- o rash that keeps getting worse
- o severe rash
- o reddened skin
- o flu‑like symptoms
- o skin pain or burning
- o blistering of the lips, eyes, or mouth
- o blisters on the skin
- o skin peeling
-
Major cardiovascular events such as heart attack, stroke, and death. Major cardiac events have happened, especially in people with cardiac risk factors and who are current or past smokers, while taking another Janus kinase (JAK) inhibitor to treat rheumatoid arthritis. OJJAARA is in the JAK family of medicines.
Get emergency help right away if you have any symptoms of a heart attack or stroke during treatment with OJJAARA, including:- o discomfort in your chest that lasts for more than a few minutes, or that goes away and comes back
- o severe tightness, pain, pressure, or heaviness in your chest, throat, neck, or jaw
- o pain or discomfort in your arms, back, neck, jaw, or stomach
- o shortness of breath with or without chest discomfort
- o breaking out in a cold sweat
- o nausea or vomiting
- o feeling lightheaded
- o weakness in one part or on one side of your body
- o slurred speech
-
Blood clots. Blood clots in the veins of the legs (deep vein thrombosis [DVT]) and lungs (pulmonary embolism [PE]) have happened in some people taking another JAK inhibitor to treat rheumatoid arthritis, and may be life‑threatening. Tell your healthcare provider if you have had blood clots in the veins of your legs or lungs in the past.
Tell your healthcare provider right away if you have any signs and symptoms of blood clots during treatment with OJJAARA, including:- o swelling, pain, or tenderness in one or both legs
- o sudden, unexplained chest pain
- o shortness of breath or difficulty breathing
- New cancers. New cancers, including lymphoma and other cancers, except non‑melanoma skin cancer, have happened in some people taking another JAK inhibitor to treat rheumatoid arthritis. The risk of new cancers is further increased in people who smoke or who smoked in the past.
-
Worsening of symptoms after interruption or stopping treatment. Signs and symptoms of myelofibrosis may worsen after you stop treatment with OJJAARA. Do not interrupt or stop treatment with OJJAARA without talking to your healthcare provider. If you need to stop treatment with OJJAARA, your healthcare provider may give you instructions for slowly stopping treatment.
Tell your healthcare provider right away if you get any of the following signs and symptoms after stopping treatment with OJJAARA:
- o fever
- o trouble breathing
- o feeling dizzy or lightheaded
- o weakness
- o night sweats
- o pain in your left upper stomach (abdomen) area or left shoulder
Your healthcare provider will do blood tests before you start treatment with OJJAARA and during treatment to check for side effects.
Your healthcare provider may change your dose, temporarily stop, or permanently stop treatment with OJJAARA if you get certain side effects.
The most common side effects of OJJAARA include:
- low platelet count
- bleeding
- bacterial infection
- tiredness
- dizziness
- diarrhea
- nausea
These are not all of the possible side effects of OJJAARA.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1‑800‑FDA‑1088.
How should I store OJJAARA?
- Store OJJAARA at room temperature between 68°F to 77°F (20°C to 25°C).
- Keep OJJAARA in its original bottle. The OJJAARA bottle contains a desiccant packet to help keep your tablets dry (protect from moisture). Keep the desiccant in the bottle.
- Tightly close the OJJAARA bottle after you take your dose.
Keep OJJAARA and all medicines out of the reach of children.
General information about the safe and effective use of OJJAARA.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use OJJAARA for a condition for which it was not prescribed. Do not give OJJAARA to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about OJJAARA that is written for health professionals.
What are the ingredients in OJJAARA?
Active ingredient: momelotinib dihydrochloride monohydrate
Inactive ingredients: lactose monohydrate, magnesium stearate, microcrystalline cellulose, propyl gallate, silicon dioxide, and sodium starch glycolate
Film coating: polyethylene glycol, polyvinyl alcohol, red iron oxide, talc, titanium dioxide, and yellow iron oxide
Trademarks are owned by or licensed to the GSK group of companies.
GlaxoSmithKline, Durham, NC 27701
For more information about OJJAARA, call 1-888-825-5249 or visit our website at www.OJJAARA.com.
©2026 GSK group of companies or its licensor.
OJJ:3PIL
- This Patient Information has been approved by the U.S. Food and Drug Administration Revised: March 2026
-
PRINCIPAL DISPLAY PANEL
PRINICPAL DISPLAY PANEL
NDC: 81864-103-30
Ojjaara
(momelotinib)tablets
100 mg
Rx Only
GSK
30 Tablets
Each tablet contains 100 mg tablet of momelotinib equivalent to 121.94 mg of momelotinib dihydrochloride monohydrate. Swallow tablets whole.
Do not cut, crush, or chew tablets.
Store at 20°C to 25°C (68°F to 77°F); Excursions permitted between 15°C to 30°C (59°F to 86°F). (see USP Controlled Room Temperature). Dispense and store in original bottle with desiccant to protect from moisture.
Do not accept if safety seal under cap is missing or broken.
Recommended dosage: See Prescribing Information
Keep out of reach of children.
For current patient information visit epi-pla.org
Trademarks owned or licensed by GSK.
Mfd for: GSK Durham, NC 27701
Product of Sweden and Switzerland
©2026 GSK or licensor.
Rev. 3/26
MCK-163127-01
-
PRINCIPAL DISPLAY PANEL
PRINICPAL DISPLAY PANEL
NDC: 81864-102-30
Ojjaara
(momelotinib)tablets
150 mg
Rx Only
GSK
30 Tablets
Each tablet contains 150 mg of momelotinib equivalent to 182.91 mg of momelotinib dihydrochloride monohydrate. Swallow tablets whole.
Do not cut, crush, or chew tablets.
Store at 20°C to 25°C (68°F to 77°F); Excursions permitted between 15°C to 30°C (59°F to 86°F). (see USP Controlled Room Temperature). Dispense and store in original bottle with desiccant to protect from moisture.
Do not accept if safety seal under cap is missing or broken.
Recommended dosage: See Prescribing Information
Keep out of reach of children.
For current patient information visit epi-pla.org
Trademarks owned or licensed by GSK.
Mfd for: GSK Durham, NC 27701
Product of Sweden and Switzerland
©2026 GSK or licensor.
Rev. 3/26
MCK-163257-01
-
PRINCIPAL DISPLAY PANEL
PRINICPAL DISPLAY PANEL
NDC: 81864-101-30
Ojjaara
(momelotinib) tablets
200 mg
Rx Only
GSK
30 Tablets
Each tablet contains 200 mg of momelotinib is equivalent to 243.88 mg of momelotinib dihydrochloride monohydrate. Swallow tablets whole.
Do not cut, crush, or chew tablets.
Store at 20°C to 25°C (68°F to 77°F); Excursions permitted between 15°C to 30°C (59°F to 86°F). (see USP Controlled Room Temperature). Dispense and store in original bottle with desiccant to protect from moisture.
Do not accept if safety seal under cap is missing or broken.
Recommended dosage: See Prescribing Information
Keep out of reach of children.
For current patient information visit epi-pla.org
Trademarks owned or licensed by GSK.
Mfd for: GSK Durham, NC 27701
Product of Sweden and Switzerland
©20265 GSK or licensor.
Rev. 3/26
MCK-163258-01
-
INGREDIENTS AND APPEARANCE
OJJAARA
momelotinib tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 81864-103 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength MOMELOTINIB DIHYDROCHLORIDE MONOHYDRATE (UNII: LDX8893L5D) (MOMELOTINIB - UNII:6O01GMS00P) MOMELOTINIB 100 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) SODIUM STARCH GLYCOLATE TYPE A CORN (UNII: AG9B65PV6B) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) MAGNESIUM STEARATE (UNII: 70097M6I30) PROPYL GALLATE (UNII: 8D4SNN7V92) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) TALC (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) FERRIC OXIDE RED (UNII: 1K09F3G675) Product Characteristics Color BROWN Score no score Shape ROUND Size 9mm Flavor Imprint Code M;;100 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 81864-103-30 30 in 1 BOTTLE; Type 0: Not a Combination Product 09/15/2023 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA216873 09/15/2023 OJJAARA
momelotinib tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 81864-102 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength MOMELOTINIB DIHYDROCHLORIDE MONOHYDRATE (UNII: LDX8893L5D) (MOMELOTINIB - UNII:6O01GMS00P) MOMELOTINIB 150 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) SODIUM STARCH GLYCOLATE TYPE A CORN (UNII: AG9B65PV6B) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) MAGNESIUM STEARATE (UNII: 70097M6I30) PROPYL GALLATE (UNII: 8D4SNN7V92) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) TALC (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) FERRIC OXIDE RED (UNII: 1K09F3G675) Product Characteristics Color BROWN Score no score Shape TRIANGLE Size 11mm Flavor Imprint Code M;;150 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 81864-102-30 30 in 1 BOTTLE; Type 0: Not a Combination Product 09/15/2023 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA216873 09/15/2023 OJJAARA
momelotinib tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 81864-101 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength MOMELOTINIB DIHYDROCHLORIDE MONOHYDRATE (UNII: LDX8893L5D) (MOMELOTINIB - UNII:6O01GMS00P) MOMELOTINIB 200 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) SODIUM STARCH GLYCOLATE TYPE A CORN (UNII: AG9B65PV6B) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) MAGNESIUM STEARATE (UNII: 70097M6I30) PROPYL GALLATE (UNII: 8D4SNN7V92) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) TALC (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) FERRIC OXIDE RED (UNII: 1K09F3G675) Product Characteristics Color BROWN Score no score Shape CAPSULE Size 15mm Flavor Imprint Code M;;200 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 81864-101-30 30 in 1 BOTTLE; Type 0: Not a Combination Product 09/15/2023 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA216873 09/15/2023 Labeler - GlaxoSmithKline LLC (167380711)





Trademark Results [Ojjaara]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 OJJAARA 90058874 not registered Live/Pending |
Sierra Oncology, Inc. 2020-07-17 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.