VIZIMPRO- dacomitinib tablet, film coated
Vizimpro by
Drug Labeling and Warnings
Vizimpro by is a Prescription medication manufactured, distributed, or labeled by Pfizer Laboratories Div Pfizer Inc, Pharmacia & Upjohn Company LLC, Pfizer Ireland Pharmaceuticals Unlimited Company, Pfizer Manufacturing Deutschland GmbH. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use VIZIMPRO safely and effectively. See full prescribing information for VIZIMPRO.
VIZIMPRO® (dacomitinib) tablets, for oral use
Initial U.S. Approval: 2018INDICATIONS AND USAGE
VIZIMPRO is a kinase inhibitor indicated for the first-line treatment of patients with metastatic non-small cell lung cancer (NSCLC) with epidermal growth factor receptor (EGFR) exon 19 deletion or exon 21 L858R substitution mutations as detected by an FDA-approved test. (1)
DOSAGE AND ADMINISTRATION
Recommended Dosage: 45 mg orally once daily with or without food. (2.2)
DOSAGE FORMS AND STRENGTHS
Tablets: 15 mg, 30 mg, and 45 mg. (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
- Interstitial Lung Disease (ILD): Permanently discontinue VIZIMPRO if ILD is confirmed. (5.1)
- Diarrhea: Withhold and reduce the dose of VIZIMPRO based on the severity. (2.3, 5.2)
- Dermatologic Adverse Reactions: Withhold and reduce the dose of VIZIMPRO based on the severity. (2.3, 5.3)
- Embryo-Fetal Toxicity: VIZIMPRO can cause fetal harm. Advise females of reproductive potential to use effective contraception. (5.4, 8.1, 8.3)
ADVERSE REACTIONS
Most common adverse reactions are (incidence >20%) diarrhea, rash, paronychia, stomatitis, decreased appetite, dry skin, decreased weight, alopecia, cough, and pruritus. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Pfizer, Inc. at 1-800-438-1985 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- Proton Pump Inhibitors (PPIs): Avoid use with VIZIMPRO; use locally-acting antacids or H2-receptor antagonist; administer VIZIMPRO at least 6 hours before or 10 hours after H2-receptor antagonist. (2.4, 7.1)
- CYP2D6 Substrates: Avoid concomitant use with VIZIMPRO where minimal increases in concentration of the CYP2D6 substrate may lead to serious or life-threatening toxicities (7.2)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 9/2018
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection
2.2 Recommended Dosage
2.3 Dosage Modifications for Adverse Reactions
2.4 Dosage Modifications for Acid-Reducing Agents
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Interstitial Lung Disease (ILD)
5.2 Diarrhea
5.3 Dermatologic Adverse Reactions
5.4 Embryo-Fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on VIZIMPRO
7.2 Effect of VIZIMPRO on CYP2D6 Substrates
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14. CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
VIZIMPRO is indicated for the first-line treatment of patients with metastatic non-small cell lung cancer (NSCLC) with epidermal growth factor receptor (EGFR) exon 19 deletion or exon 21 L858R substitution mutations as detected by an FDA-approved test [see Dosage and Administration (2.1)].
-
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection
Select patients for the first-line treatment of metastatic NSCLC with VIZIMPRO based on the presence of an EGFR exon 19 deletion or exon 21 L858R substitution mutation in tumor specimens. Information on FDA-approved tests for the detection of EGFR mutations in NSCLC is available at: http://www.fda.gov/CompanionDiagnostics.
2.2 Recommended Dosage
The recommended dosage of VIZIMPRO is 45 mg taken orally once daily, until disease progression or unacceptable toxicity occurs. VIZIMPRO can be taken with or without food [see Dosage and Administration (2.4) and Clinical Pharmacology (12.3)].
Take VIZIMPRO the same time each day. If the patient vomits or misses a dose, do not take an additional dose or make up a missed dose but continue with the next scheduled dose.
2.3 Dosage Modifications for Adverse Reactions
Reduce the dose of VIZIMPRO for adverse reactions as described in Table 1. Dosage modifications for specific adverse reactions are provided in Table 2.
Table 1. VIZIMPRO Recommended Dose Reductions for Adverse Reactions Dose Level Dose (Once Daily) First dose reduction 30 mg Second dose reduction 15 mg Table 2. VIZIMPRO Dosage Modifications for Adverse Reactions Adverse Reaction Severity* Dosage Modification - * National Cancer Institute Common Terminology Criteria for Adverse Events Version 4.03.
Interstitial lung disease (ILD) [see Warnings and Precautions (5.1)] Any Grade - Permanently discontinue VIZIMPRO.
Diarrhea [see Warnings and Precautions (5.2)] Grade 2 - Withhold VIZIMPRO until recovery to less than or equal to Grade 1; then resume VIZIMPRO at the same dose level.
- For recurrent Grade 2 diarrhea, withhold until recovery to less than or equal to Grade 1; then resume VIZIMPRO at a reduced dose.
Grade 3 or 4 - Withhold VIZIMPRO until recovery to less than or equal to Grade 1; then resume VIZIMPRO at a reduced dose.
Dermatologic Adverse Reactions [see Warnings and Precautions (5.3)] Grade 2 - Withhold VIZIMPRO for persistent dermatologic adverse reactions; upon recovery to less than or equal to Grade 1, resume VIZIMPRO at the same dose level.
- For recurrent persistent Grade 2 dermatologic adverse reactions, withhold until recovery to less than or equal to Grade 1; then resume VIZIMPRO at a reduced dose.
Grade 3 or 4 - Withhold VIZIMPRO until recovery to less than or equal to Grade 1; then resume VIZIMPRO at a reduced dose.
Other Grade 3 or 4 - Withhold VIZIMPRO until recovery to less than or equal to Grade 2; then resume VIZIMPRO at a reduced dose.
2.4 Dosage Modifications for Acid-Reducing Agents
Avoid the concomitant use of proton pump inhibitors (PPIs) while taking VIZIMPRO. As an alternative to PPIs, use locally-acting antacids or if using an histamine 2 (H2)-receptor antagonist, administer VIZIMPRO at least 6 hours before or 10 hours after taking an H2-receptor antagonist [see Drug Interactions (7.1) and Clinical Pharmacology (12.3)].
-
3 DOSAGE FORMS AND STRENGTHS
Tablets:
- 45 mg: blue film-coated, immediate release, round biconvex tablet, debossed with "Pfizer" on one side and "DCB45" on the other side.
- 30 mg: blue film-coated, immediate release, round biconvex tablet, debossed with "Pfizer" on one side and "DCB30" on the other side.
- 15 mg: blue film-coated, immediate release, round biconvex tablet, debossed with "Pfizer" on one side and "DCB15" on the other side.
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Interstitial Lung Disease (ILD)
Severe and fatal ILD/pneumonitis occurred in patients treated with VIZIMPRO and occurred in 0.5% of the 394 VIZIMPRO-treated patients; 0.3% of cases were fatal.
Monitor patients for pulmonary symptoms indicative of ILD/pneumonitis. Withhold VIZIMPRO and promptly investigate for ILD in patients who present with worsening of respiratory symptoms which may be indicative of ILD (e.g., dyspnea, cough, and fever). Permanently discontinue VIZIMPRO if ILD is confirmed [see Adverse Reactions (6.1)].
5.2 Diarrhea
Severe and fatal diarrhea occurred in patients treated with VIZIMPRO. Diarrhea occurred in 86% of the 394 VIZIMPRO-treated patients; Grade 3 or 4 diarrhea was reported in 11% of patients and 0.3% of cases were fatal.
Withhold VIZIMPRO for Grade 2 or greater diarrhea until recovery to less than or equal to Grade 1 severity, then resume VIZIMPRO at the same or a reduced dose depending on the severity of diarrhea [see Dosage and Administration (2.3) and Adverse Reactions (6.1)]. Promptly initiate anti-diarrheal treatment (loperamide or diphenoxylate hydrochloride with atropine sulfate) for diarrhea.
5.3 Dermatologic Adverse Reactions
Rash and exfoliative skin reactions occurred in patients treated with VIZIMPRO. Rash occurred in 78% of the 394 VIZIMPRO-treated patients; Grade 3 or 4 rash was reported in 21% of patients. Exfoliative skin reactions of any severity were reported in 7% of patients. Grade 3 or 4 exfoliative skin reactions were reported in 1.8% of patients.
Withhold VIZIMPRO for persistent Grade 2 or any Grade 3 or 4 dermatologic adverse reaction until recovery to less than or equal to Grade 1 severity, then resume VIZIMPRO at the same or a reduced dose depending on the severity of the dermatologic adverse reaction [see Dosage and Administration (2.3) and Adverse Reactions (6.1)]. The incidence and severity of rash and exfoliative skin reactions may increase with sun exposure. At the time of initiation of VIZIMPRO, initiate use of moisturizers and appropriate measures to limit sun exposure. Upon development of Grade 1 rash, initiate treatment with topical antibiotics and topical steroids. Initiate oral antibiotics for Grade 2 or more severe dermatologic adverse reactions.
5.4 Embryo-Fetal Toxicity
Based on findings from animal studies and its mechanism of action, VIZIMPRO can cause fetal harm when administered to a pregnant woman. In animal reproduction studies, oral administration of dacomitinib to pregnant rats during the period of organogenesis resulted in an increased incidence of post-implantation loss and reduced fetal body weight at doses resulting in exposures near the exposure at the 45 mg human dose. The absence of EGFR signaling has been shown to result in embryolethality as well as post-natal death in animals. Advise pregnant women of the potential risk to the fetus. Advise females of reproductive potential to use effective contraception during treatment with VIZIMPRO and for at least 17 days after the final dose [see Use in Specific Populations (8.1 and 8.3)].
-
6 ADVERSE REACTIONS
The following adverse drug reactions are described elsewhere in the labeling:
- Interstitial Lung Disease [see Warnings and Precautions (5.1)]
- Diarrhea [see Warnings and Precautions (5.2)]
- Dermatologic Adverse Reactions [see Warnings and Precautions (5.3)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data in the Warnings and Precautions section reflect exposure to VIZIMPRO in 394 patients with first-line or previously treated NSCLC with EGFR exon 19 deletion or exon 21 L858R substitution mutations who received VIZIMPRO at the recommended dose of 45 mg once daily in 4 randomized, active-controlled trials [ARCHER 1050 (N=227), Study A7471009 (N=38), Study A7471011 (N=83), and Study A7471028 (N=16)] and one single-arm trial [Study A7471017 (N=30)]. The median duration of exposure to VIZIMPRO was 10.8 months (range 0.07–68) [see Warnings and Precautions (5)].
The data described below reflect exposure to VIZIMPRO in 227 patients with EGFR mutation-positive, metastatic NSCLC enrolled in a randomized, active-controlled trial (ARCHER 1050); 224 patients received gefitinib 250 mg orally once daily in the active control arm [see Clinical Studies (14)]. Patients were excluded if they had a history of ILD, interstitial pneumonitis, or brain metastases. The median duration of exposure to VIZIMPRO was 15 months (range 0.07–37).
The most common (>20%) adverse reactions in patients treated with VIZIMPRO were diarrhea (87%), rash (69%), paronychia (64%), stomatitis (45%), decreased appetite (31%), dry skin (30%), decreased weight (26%), alopecia (23%), cough (21%), and pruritus (21%).
Serious adverse reactions occurred in 27% of patients treated with VIZIMPRO. The most common (≥1%) serious adverse reactions were diarrhea (2.2%) and interstitial lung disease (1.3%). Dose interruptions occurred in 57% of patients treated with VIZIMPRO. The most frequent (>5%) adverse reactions leading to dose interruptions were rash (23%), paronychia (13%), and diarrhea (10%). Dose reductions occurred in 66% of patients treated with VIZIMPRO. The most frequent (>5%) adverse reactions leading to dose reductions were rash (29%), paronychia (17%), and diarrhea (8%).
Adverse reactions leading to permanent discontinuation of VIZIMPRO occurred in 18% of patients. The most common (>0.5%) adverse reactions leading to permanent discontinuation of VIZIMPRO were: rash (2.6%), interstitial lung disease (1.8%), stomatitis (0.9%), and diarrhea (0.9%).
Tables 3 and 4 summarize the most common adverse reactions and laboratory abnormalities, respectively, in ARCHER 1050. ARCHER 1050 was not designed to demonstrate a statistically significant difference in adverse reaction rates for VIZIMPRO or for gefitinib for any adverse reaction or laboratory value listed in Table 3 or 4.
Table 3. Adverse Reactions Occurring in ≥10% of Patients Receiving VIZIMPRO in ARCHER 1050* Adverse Reaction VIZIMPRO
(N=227)Gefitinib
(N=224)All Grades†
%Grades 3 and 4
%All Grades
%Grades 3 and 4
%- * National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) v4.03.
- † Grades 1 through 5 are included in All Grades.
- ‡ One Grade 5 (fatal) event in the VIZIMPRO arm.
- § Stomatitis includes mucosal inflammation and stomatitis.
- ¶ Rash includes dermatitis acneiform, rash, and rash maculo-papular.
- # Paronychia includes nail infection, nail toxicity, onychoclasis, onycholysis, onychomadesis, paronychia.
- Þ Dry skin includes dry skin, xerosis.
- ß Pruritus includes pruritus, pruritus generalized, rash pruritic.
- à Nasal mucosal disorder includes epistaxis, nasal inflammation, nasal mucosal disorder, nasal mucosal ulcer, rhinitis.
Gastrointestinal Diarrhea‡ 87 8 56 0.9 Stomatitis§ 45 4.4 19 0.4 Nausea 19 1.3 22 0.4 Constipation 13 0 14 0 Mouth ulceration 12 0 6 0 Skin and Subcutaneous Tissue Rash¶ 69 23 47 0.4 Paronychia# 64 8 21 1.3 Dry skinÞ 30 1.8 19 0.4 Alopecia 23 0.4 13 0 Pruritusß 21 0.9 15 1.3 Palmar-plantar erythrodysesthesia syndrome 15 0.9 3.1 0 Dermatitis 11 1.8 4 0.4 Metabolism and Nutrition Decreased appetite 31 3.1 25 0.4 Decreased weight 26 2.2 17 0.4 Respiratory Cough 21 0 19 0.4 Nasal mucosal disorderà 19 0 4.9 0 Dyspnea 13 2.2 13 1.8 Upper respiratory tract infection 12 1.3 13 0 Chest pain 10 0 14 0 Eye Conjunctivitis 19 0 4 0 Musculoskeletal Pain in extremity 14 0 12 0 Musculoskeletal pain 12 0.9 13 0 General Asthenia 13 2.2 13 1.3 Psychiatric Insomnia 11 0.4 15 0 Additional adverse reactions (All Grades) that were reported in <10% of patients who received VIZIMPRO in ARCHER 1050 include:
General: fatigue 9%
Skin and subcutaneous tissue: skin fissures 9%, hypertrichosis 1.3%, skin exfoliation/exfoliative skin reactions 3.5%
Gastrointestinal: vomiting 9%
Nervous system: dysgeusia 7%
Respiratory: interstitial lung disease 2.6%
Ocular: keratitis 1.8%
Metabolism and nutrition: dehydration 1.3%
Table 4. Laboratory Abnormalities Worsening from Baseline in >20% of Patients in ARCHER 1050* Laboratory Test Abnormality† VIZIMPRO Gefitinib Change from Baseline All Grades
(%)Change from Baseline to Grade 3 or Grade 4
(%)Change from Baseline All Grades
(%)Change from Baseline to Grade 3 or Grade 4
(%)ALT=alanine aminotransferase; AST=aspartate aminotransferase. - * NCI CTCAE v4.03, except for increased creatinine which only includes patients with creatinine increase based on upper limit of normal definition.
- † Based on the number of patients with available baseline and at least one on-treatment laboratory test.
Hematology Anemia 44 0.9 26 2.7 Lymphopenia 42 6 35 2.7 Chemistry Hypoalbuminemia 44 0 34 0 Increased ALT 40 1.4 63 13 Hyperglycemia 36 1.0 38 2.5 Increased AST 35 0.5 57 8 Hypocalcemia 33 1.4 28 2.0 Hypokalemia 29 7 18 2.0 Hyponatremia 26 2.9 20 1.5 Increased creatinine 24 0 16 0.5 Increased alkaline phosphatase 22 0.5 21 2.0 Hypomagnesemia 22 0.5 9 0 Hyperbilirubinemia 16 0.5 22 0.5 -
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on VIZIMPRO
Concomitant use with a PPI decreases dacomitinib concentrations, which may reduce VIZIMPRO efficacy. Avoid the concomitant use of PPIs with VIZIMPRO. As an alternative to PPIs, use locally-acting antacids or an H2-receptor antagonist. Administer VIZIMPRO at least 6 hours before or 10 hours after taking an H2-receptor antagonist [see Dosage and Administration (2.4) and Clinical Pharmacology (12.3)].
7.2 Effect of VIZIMPRO on CYP2D6 Substrates
Concomitant use of VIZIMPRO increases the concentration of drugs that are CYP2D6 substrates [see Clinical Pharmacology (12.3)] which may increase the risk of toxicities of these drugs. Avoid concomitant use of VIZIMPRO with CYP2D6 substrates where minimal increases in concentration of the CYP2D6 substrate may lead to serious or life-threatening toxicities.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings from animal studies and its mechanism of action, VIZIMPRO can cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)]. There are no available data on VIZIMPRO use in pregnant women. In animal reproduction studies, oral administration of dacomitinib to pregnant rats during the period of organogenesis resulted in an increased incidence of post-implantation loss and reduced fetal body weight at doses resulting in exposures near the exposure at the 45 mg human dose (see Data). The absence of EGFR signaling has been shown to result in embryolethality as well as post-natal death in animals (see Data). Advise pregnant women of the potential risk to a fetus [see Use in Special Populations (8.3)].
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
Daily oral administration of dacomitinib to pregnant rats during the period of organogenesis resulted in an increased incidence of post-implantation loss, maternal toxicity, and reduced fetal body weight at 5 mg/kg/day (approximately 1.2 times the exposure based on area under the curve [AUC] at the 45 mg human dose).
Disruption or depletion of EGFR in mouse models has shown EGFR is critically important in reproductive and developmental processes including blastocyst implantation, placental development, and embryo-fetal/post-natal survival and development. Reduction or elimination of embryo-fetal or maternal EGFR signaling in mice can prevent implantation, and can cause embryo-fetal loss during various stages of gestation (through effects on placental development), developmental anomalies, early death in surviving fetuses, and adverse developmental outcomes in multiple organs in embryos/neonates.
8.2 Lactation
Risk Summary
There is no information regarding the presence of dacomitinib or its metabolites in human milk or their effects on the breastfed infant or on milk production. Because of the potential for serious adverse reactions in breastfed infants from VIZIMPRO, advise women not to breastfeed during treatment with VIZIMPRO and for at least 17 days after the last dose.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Verify the pregnancy status of females of reproductive potential prior to initiating VIZIMPRO [see Use in Specific Populations (8.1)].
Contraception
VIZIMPRO can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
8.5 Geriatric Use
Of the total number of patients (N=394) in five clinical studies with EGFR mutation-positive NSCLC who received VIZIMPRO at a dose of 45 mg orally once daily [ARCHER 1050 (N=227), Study A7471009 (N=38), Study A7471011 (N=83), Study A7471028 (N=16), and Study A7471017 (N=30)] 40% were 65 years of age and older.
Exploratory analyses across this population suggest a higher incidence of Grade 3 and 4 adverse reactions (67% versus 56%, respectively), more frequent dose interruptions (53% versus 45%, respectively), and more frequent discontinuations (24% versus 10%, respectively) for adverse reactions in patients 65 years or older as compared to those younger than 65 years.
8.6 Renal Impairment
No dose adjustment is recommended for patients with mild or moderate renal impairment (creatinine clearance [CLcr] 30 to 89 mL/min estimated by Cockcroft-Gault). The recommended dose of VIZIMPRO has not been established for patients with severe renal impairment (CLcr <30 mL/min) [see Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
No dose adjustment is recommended in patients with mild (total bilirubin ≤ upper limit of normal [ULN] with AST > ULN or total bilirubin > 1 to 1.5 × ULN with any AST) or moderate (total bilirubin > 1.5 to 3 × ULN and any AST) hepatic impairment. The recommended dose of VIZIMPRO has not been established for patients with severe hepatic impairment (total bilirubin > 3 to 10 × ULN and any AST) [see Clinical Pharmacology (12.3)].
-
11 DESCRIPTION
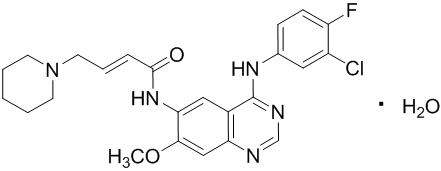
Dacomitinib is an oral kinase inhibitor with a molecular formula of C24H25ClFN5O2 ∙ H2O and a molecular weight of 487.95 Daltons. The chemical name is: (2E)-N-{4-[(3-Chloro-4-fluorophenyl)amino]-7-methoxyquinazolin-6-yl}-4-(piperidin-1-yl)but-2-enamide monohydrate and its structural formula is:
Dacomitinib is a white to pale yellow powder.
VIZIMPRO tablets contain 45, 30, or 15 mg of dacomitinib with the following inactive ingredients in the tablet core; lactose monohydrate, microcrystalline cellulose, sodium starch glycolate, and magnesium stearate. The film coating consists of Opadry II® Blue 85F30716 containing: Polyvinyl alcohol – partially hydrolyzed, Talc, Titanium dioxide, Macrogol/PEG 3350, and FD&C Blue #2/Indigo Carmine Aluminum Lake.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Dacomitinib is an irreversible inhibitor of the kinase activity of the human EGFR family (EGFR/HER1, HER2, and HER4) and certain EGFR activating mutations (exon 19 deletion or the exon 21 L858R substitution mutation). In vitro dacomitinib also inhibited the activity of DDR1, EPHA6, LCK, DDR2, and MNK1 at clinically relevant concentrations.
Dacomitinib demonstrated dose-dependent inhibition of EGFR and HER2 autophosphorylation and tumor growth in mice bearing subcutaneously implanted human tumor xenografts driven by HER family targets including mutated EGFR. Dacomitinib also exhibited antitumor activity in orally-dosed mice bearing intracranial human tumor xenografts driven by EGFR amplifications.
12.2 Pharmacodynamics
Cardiac Electrophysiology
The effect of dacomitinib on the QT interval corrected for heart rate (QTc) was evaluated using time-matched electrocardiograms (ECGs) evaluating the change from baseline and corresponding pharmacokinetic data in 32 patients with advanced NSCLC. Dacomitinib had no large effect on QTc (i.e., >20 ms) at maximum dacomitinib concentrations achieved with VIZIMPRO 45 mg orally once daily.
12.3 Pharmacokinetics
The maximum dacomitinib plasma concentration (Cmax) and AUC at steady state increased proportionally over the dose range of VIZIMPRO 2 mg to 60 mg orally once daily (0.04 to 1.3 times the recommended dose) across dacomitinib studies in patients with cancer. At a dose of 45 mg orally once daily, the geometric mean [coefficient of variation (CV%)] Cmax was 108 ng/mL (35%) and the AUC0–24h was 2213 ng∙h/mL (35%) at steady state in a dose-finding clinical study conducted in patients with solid tumors. Steady state was achieved within 14 days following repeated dosing and the estimated geometric mean (CV%) accumulation ratio was 5.7 (28%) based on AUC.
Absorption
The mean absolute bioavailability of dacomitinib is 80% after oral administration. The median dacomitinib time to reach maximum concentration (Tmax) occurred at approximately 6.0 hours (range 2.0 to 24 hours) after a single oral dose of VIZIMPRO 45 mg in patients with cancer.
Distribution
The geometric mean (CV%) volume of distribution of dacomitinib (Vss) was 1889 L (18%). In vitro binding of dacomitinib to human plasma proteins is approximately 98% and is independent of drug concentrations from 250 ng/mL to 1000 ng/mL.
Elimination
Following a single 45 mg oral dose of VIZIMPRO in patients with cancer, the mean (CV%) plasma half-life of dacomitinib was 70 hours (21%), and the geometric mean (CV%) apparent plasma clearance of dacomitinib was 24.9 L/h (36%).
Metabolism
Hepatic metabolism is the main route of clearance of dacomitinib, with oxidation and glutathione conjugation as the major pathways. Following oral administration of a single 45 mg dose of [14C] dacomitinib, the most abundant circulating metabolite was O-desmethyl dacomitinib, which had similar in vitro pharmacologic activity as dacomitinib. The steady-state plasma trough concentration of O-desmethyl dacomitinib ranges from 7.4% to 19% of the parent. In vitro studies indicated that cytochrome P450 (CYP) 2D6 was the major isozyme involved in the formation of O-desmethyl dacomitinib, while CYP3A4 contributed to the formation of other minor oxidative metabolites.
Specific Populations
Patients with Renal Impairment
Based on population pharmacokinetic analyses, mild (60 mL/min ≤ CLcr <90 mL/min; N=590) and moderate (30 mL/min ≤ CLcr <60 mL/min; N=218) renal impairment did not alter dacomitinib pharmacokinetics, relative to the pharmacokinetics in patients with normal renal function (CLcr ≥90 mL/min; N=567). The pharmacokinetics of dacomitinib has not been adequately characterized in patients with severe renal impairment (CLcr <30 mL/min) (N=4) or studied in patients requiring hemodialysis.
Patients with Hepatic Impairment
In a dedicated hepatic impairment trial, following a single oral dose of 30 mg VIZIMPRO, dacomitinib exposure (AUCinf and Cmax) was unchanged in subjects with mild hepatic impairment (Child-Pugh A; N=8) and decreased by 15% and 20%, respectively in subjects with moderate hepatic impairment (Child-Pugh B; N=9) when compared to subjects with normal hepatic function (N=8). Based on this trial, mild and moderate hepatic impairment had no clinically important effects on pharmacokinetics of dacomitinib. In addition, based on a population pharmacokinetic analysis of 1381 patients, in which 158 patients had mild hepatic impairment (total bilirubin ≤ ULN and AST > ULN, or total bilirubin > 1 to 1.5 × ULN with any AST) and 5 patients had moderate hepatic impairment (total bilirubin > 1.5 to 3 × ULN and any AST), no effects on pharmacokinetics of dacomitinib were observed. The effect of severe hepatic impairment (total bilirubin > 3 to 10 × ULN and any AST) on dacomitinib pharmacokinetics is unknown [see Use in Specific Populations (8.7)].
Drug Interaction Studies
Clinical Studies
Effect of Acid-Reducing Agents on Dacomitinib
Coadministration of a single 45 mg dose of VIZIMPRO with multiple doses of rabeprazole (a proton pump inhibitor) decreased dacomitinib Cmax by 51% and AUC0–96h by 39% [see Dosage and Administration (2.4) and Drug Interactions (7.1)].
Coadministration of VIZIMPRO with a local antacid (Maalox® Maximum Strength, 400 mg/5 mL) did not cause clinically relevant changes dacomitinib concentrations [see Dosage and Administration (2.4) and Drug Interactions (7.1)].
The effect of H2 receptor antagonists on dacomitinib pharmacokinetics has not been studied [see Dosage and Administration (2.4) and Drug Interactions (7.1)].
Effect of Strong CYP2D6 Inhibitors on Dacomitinib
Coadministration of a single 45 mg dose of VIZIMPRO with multiple doses of paroxetine (a strong CYP2D6 inhibitor) in healthy subjects increased the total AUClast of dacomitinib plus its active metabolite (O-desmethyl dacomitinib) in plasma by approximately 6%, which is not considered clinically relevant.
Effect of Dacomitinib on CYP2D6 Substrates
Coadministration of a single 45 mg oral dose of VIZIMPRO increased dextromethorphan (a CYP2D6 substrate) Cmax by 9.7-fold and AUClast by 9.6-fold [see Drug Interactions (7.2)].
In Vitro Studies
Effect of Dacomitinib and O-desmethyl Dacomitinib on CYP Enzymes: Dacomitinib and its metabolite O-desmethyl dacomitinib do not inhibit CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, or CYP3A4/5. Dacomitinib does not induce CYP1A2, CYP2B6, or CYP3A4.
Effect of Dacomitinib on Uridine 5' diphospho-glucuronosyltransferase (UGT) Enzymes: Dacomitinib inhibits UGT1A1. Dacomitinib does not inhibit UGT1A4, UGT1A6, UGT1A9, UGT2B7, or UGT2B15.
Effect of Dacomitinib on Transporter Systems: Dacomitinib is a substrate for the membrane transport protein P-glycoprotein (P-gp) and Breast Cancer Resistance Protein (BCRP). Dacomitinib inhibits P-gp, BCRP, and organic cation transporter (OCT)1. Dacomitinib does not inhibit organic anion transporters (OAT)1 and OAT3, OCT2, organic anion transporting polypeptide (OATP)1B1, and OATP1B3.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been performed with VIZIMPRO.
Dacomitinib was not mutagenic in a bacterial reverse mutation (Ames) assay or clastogenic in an in vitro human lymphocyte chromosome aberration assay or clastogenic or aneugenic in an in vivo rat bone marrow micronucleus assay.
Daily oral administration of dacomitinib at doses ≥ 0.5 mg/kg/day to female rats (approximately 0.14 times the exposure based on AUC at the 45 mg human dose) resulted in reversible epithelial atrophy in the cervix and vagina. Oral administration of dacomitinib at 2 mg/kg/day to male rats (approximately 0.6 times the human exposure based on AUC at the 45 mg clinical dose) resulted in reversible decreased secretion in the prostate gland.
-
14. CLINICAL STUDIES
The efficacy of VIZIMPRO was demonstrated in a randomized, multicenter, multinational, open-label study (ARCHER 1050; [NCT01774721]). Patients were required to have unresectable, metastatic NSCLC with no prior therapy for metastatic disease or recurrent disease with a minimum of 12 months disease-free after completion of systemic therapy; an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1; EGFR exon 19 deletion or exon 21 L858R substitution mutations. EGFR mutation status was prospectively determined by local laboratory or commercially available tests (e.g., therascreen® EGFR RGQ PCR and cobas® EGFR Mutation Test).
Patients were randomized (1:1) to receive VIZIMPRO 45 mg orally once daily or gefitinib 250 mg orally once daily until disease progression or unacceptable toxicity. Randomization was stratified by region (Japanese versus mainland Chinese versus other East Asian versus non-East Asian), and EGFR mutation status (exon 19 deletions versus exon 21 L858R substitution mutation). The major efficacy outcome measure was progression-free survival (PFS) as determined by blinded Independent Radiologic Central (IRC) review per RECIST v1.1. Additional efficacy outcome measures were overall response rate (ORR), duration of response (DoR), and overall survival (OS).
A total of 452 patients were randomized to receive VIZIMPRO (N=227) or gefitinib (N=225). The demographic characteristics were 60% female; median age 62 years (range: 28 to 87), with 40% aged 65 years and older; and 23% White, 77% Asian, and less than 1% Black. Prognostic and tumor characteristics were ECOG performance status 0 (30%) or 1 (70%); 59% with exon 19 deletion and 41% with exon 21 L858R substitution; Stage IIIB (8%) and Stage IV (92%); 64% were never smokers; and 1% received prior adjuvant or neoadjuvant therapy.
ARCHER 1050 demonstrated a statistically significant improvement in PFS as determined by the IRC. Results are summarized in Table 5 and Figures 1 and 2.
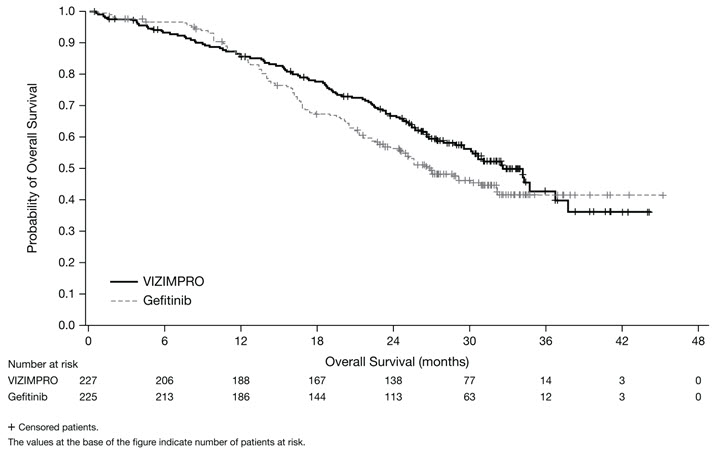
The hierarchical statistical testing order was PFS followed by ORR and then OS. No formal testing of OS was conducted since the formal comparison of ORR was not statistically significant.
Table 5. Efficacy Results in ARCHER 1050 VIZIMPRO
N=227Gefitinib
N=225CI=confidence interval; DoR=duration of response; HR=hazard ratio; IRC=Independent Radiologic Central; N/n=total number; PFS=progression-free survival. - * From stratified Cox Regression.
- † Based on the stratified log-rank test.
- ‡ Based on the stratified Cochran-Mantel-Haenszel test.
Progression-Free Survival (per IRC) Number of patients with event, n (%) 136 (59.9%) 179 (79.6%) Median PFS in months (95% CI) 14.7 (11.1, 16.6) 9.2 (9.1, 11.0) HR (95% CI)* 0.59 (0.47, 0.74) p-value† <0.0001 Overall Response Rate (per IRC) Overall Response Rate % (95% CI) 75% (69, 80) 72% (65, 77) p-value‡ 0.39 Duration of Response in Responders (per IRC) Median DoR in months (95% CI) 14.8 (12.0, 17.4) 8.3 (7.4, 9.2) Figure 1. Kaplan-Meier Curve for PFS per IRC Review in ARCHER 1050
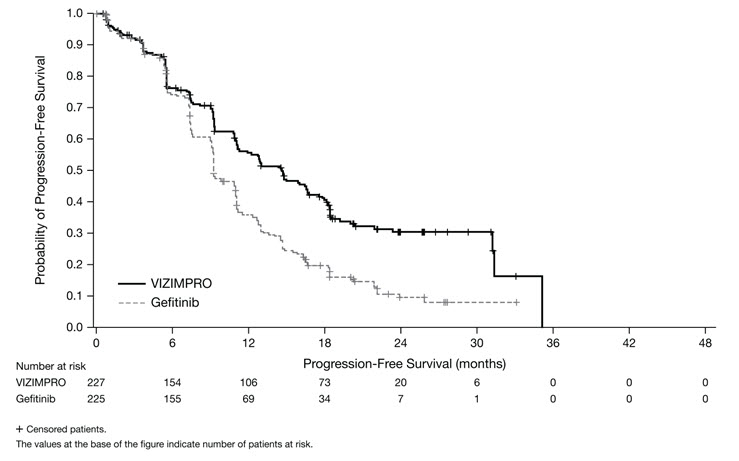
Figure 2. Kaplan-Meier Curve for OS in ARCHER 1050
-
16 HOW SUPPLIED/STORAGE AND HANDLING
VIZIMPRO is supplied in strengths and package configurations as described in Table 6 below:
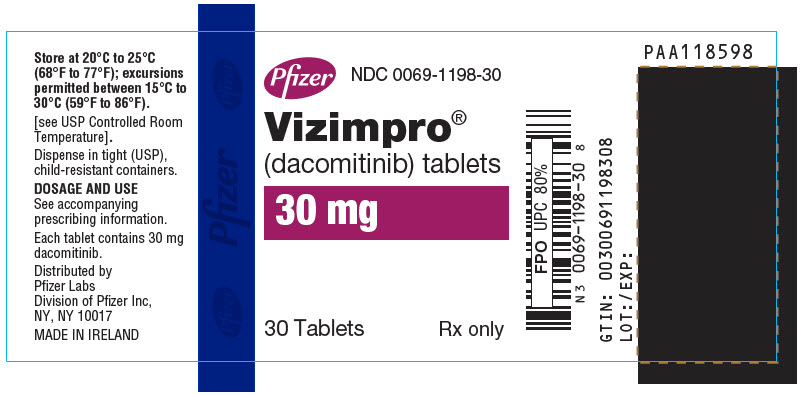
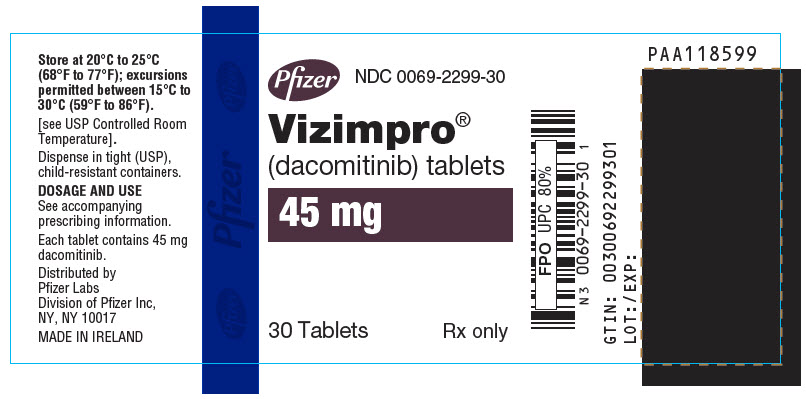
Table 6. VIZIMPRO Strengths and Package Configurations VIZIMPRO Tablets Package Configuration Tablet Strength (mg) NDC Tablet Description 30-Count Bottle with a child-resistant closure 15 0069-0197-30 Blue film-coated, immediate release, round biconvex tablet, debossed with "Pfizer" on one side and "DCB15" on the other side. 30-Count Bottle with a child-resistant closure 30 0069-1198-30 Blue film-coated immediate release, round biconvex tablet, debossed with "Pfizer" on one side and "DCB30" on the other side. 30-Count Bottle with a child-resistant closure 45 0069-2299-30 Blue film-coated immediate release, round biconvex tablet, debossed with "Pfizer" on one side and "DCB45" on the other side. -
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Interstitial Lung Disease (ILD)
- Advise patients of the risks of severe or fatal ILD, including pneumonitis. Advise patients to contact their healthcare provider immediately to report new or worsening respiratory symptoms [see Warnings and Precautions (5.1)].
Diarrhea
- Advise patients to contact their healthcare provider at the first signs of diarrhea. Advise patients that intravenous hydration and/or anti-diarrheal medication (e.g., loperamide) may be required to manage diarrhea [see Warnings and Precautions (5.2)].
Dermatologic Adverse Reactions
- Advise patients to initiate use of moisturizers and to minimize sun exposure with protective clothing and use of sunscreen at the time of initiation of VIZIMPRO. Advise patients to contact their healthcare provider immediately to report new or worsening rash, erythematous and exfoliative reactions [see Warning and Precautions (5.3)].
Drug Interactions
- Advise patients to avoid use of PPIs while taking VIZIMPRO. Short-acting antacids or H2 receptor antagonists may be used if needed. Advise patients to take VIZIMPRO at least 6 hours before or 10 hours after taking an H2-receptor antagonist [see Drug Interactions (7.1)].
Embryo-Fetal Toxicity
- Advise females of reproductive potential that VIZIMPRO can result in fetal harm and to use effective contraception during treatment with VIZIMPRO and for 17 days after the last dose of VIZIMPRO. Advise females of reproductive potential to contact their healthcare provider with a known or suspected pregnancy [see Use in Specific Populations (8.1 and 8.3)].
Lactation
- Advise women not to breastfeed during treatment with VIZIMPRO and for 17 days after the last dose of VIZIMPRO [see Use in Specific Populations (8.2)].
- SPL UNCLASSIFIED SECTION
-
PATIENT PACKAGE INSERT
This Patient Information has been approved by the U.S. Food and Drug Administration. Issued: September 2018 PATIENT INFORMATION
VIZIMPRO® (vih-ZIM-pro)
(dacomitinib)
tabletsWhat is VIZIMPRO?
VIZIMPRO is a prescription medicine used to treat non-small cell lung cancer (NSCLC) that has spread to other parts of the body (metastatic):- As your first treatment if your tumor has certain types of abnormal epidermal growth factor receptor (EGFR) gene(s).
It is not known if VIZIMPRO is safe and effective in children.Before taking VIZIMPRO, tell your healthcare provider about all your medical conditions, including if you: - have frequent diarrhea.
- have a history of lung or breathing problems other than lung cancer.
- are pregnant, or plan to become pregnant. VIZIMPRO can harm your unborn baby.
-
- Your healthcare provider should do a pregnancy test before you start treatment with VIZIMPRO.
- You should use effective birth control (contraception) during treatment and for at least 17 days after your last dose of VIZIMPRO. Talk to your healthcare provider about birth control methods that may be right for you during this time.
- Tell your healthcare provider right away if you become pregnant during your treatment with VIZIMPRO.
- are breastfeeding or plan to breastfeed. It is not known if VIZIMPRO passes into your breast milk. Do not breastfeed during treatment and for at least 17 days after your last dose of VIZIMPRO. Talk to your healthcare provider about the best way to feed your baby during this time.
How should I take VIZIMPRO? - Take VIZIMPRO exactly as your healthcare provider tells you.
- Take your dose at approximately the same time each day.
- Your healthcare provider may change your dose, temporarily stop, or permanently stop treatment with VIZIMPRO if you have side effects.
- Take VIZIMPRO 1 time each day with or without food.
- If you take an antacid or H2 blocker medicine during treatment with VIZIMPRO, take your dose of VIZIMPRO at least 6 hours before or 10 hours after taking the antacid or H2 blocker medicine. Do not change your dose or stop taking VIZIMPRO unless your healthcare provider tells you.
- If you vomit or miss a dose of VIZIMPRO, do not take another dose or make up for the missed dose. Take your next dose at your regular time.
What should I avoid during treatment with VIZIMPRO? - Minimize exposure to sunlight. VIZIMPRO can cause skin reactions. See "What are the possible side effects of VIZIMPRO?"
What are the possible side effects of VIZIMPRO?
VIZIMPRO may cause serious side effects, including:- Lung or breathing problems. VIZIMPRO may cause severe inflammation of the lung that may lead to death. Symptoms may be similar to those symptoms from lung cancer. Tell your healthcare provider right away if you have any new or worsening lung symptoms, including trouble breathing or shortness of breath, cough, or fever.
- Diarrhea. Diarrhea is common during treatment with VIZIMPRO, and can be severe and lead to death. Diarrhea can cause you to lose too much body fluid (dehydration). Your healthcare provider may tell you to start drinking more fluids or start taking your anti-diarrheal medicines. Tell your healthcare provider right away, if you have any loose stools or have stools more often than is normal for you.
- Skin reactions. Skin reactions are common with VIZIMPRO and can be severe. These skin reactions may include: dry skin, redness, rash, acne, itching, and peeling or blistering of your skin. Use moisturizers every day when taking VIZIMPRO. Use sunscreen and wear protective clothing that covers your skin, while exposed to sunlight, while you are taking VIZIMPRO. Your healthcare provider may prescribe other medicines to help skin reactions. Tell your healthcare provider right away about any worsening skin reactions.
- rash
- diarrhea
- mouth pain and sores
- nail inflammation
- common cold
- dry skin
- decreased appetite
- decreased weight
- dry, red, itchy eyes
- hair loss
- itching
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. How should I store VIZIMPRO? - Store VIZIMPRO at 20 °C to 25 °C (68 °F to 77 °F).
General information about the safe and effective use of VIZIMPRO.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use VIZIMPRO for a condition for which it was not prescribed. Do not give VIZIMPRO to other people, even if they have the same symptoms you have. It may harm them.
You can ask your pharmacist or healthcare provider for more information about VIZIMPRO that is written for health professionals.What are the ingredients in VIZIMPRO?
Active ingredient: dacomitinib
Inactive ingredients: lactose monohydrate, microcrystalline cellulose, sodium starch glycolate, and magnesium stearate.
Film coating contains: Opadry II® Blue 85F30716 containing: Polyvinyl alcohol – partially hydrolyzed, Talc, Titanium dioxide, Macrogol/PEG 3350, and FD&C Blue #2/Indigo Carmine Aluminum Lake.
LAB-1238-1.0
For more information, go to www.VIZIMPRO.com or call 1-800-438-1985. - PRINCIPAL DISPLAY PANEL - 15 mg Tablet Bottle Label
- PRINCIPAL DISPLAY PANEL - 30 mg Tablet Bottle Label
- PRINCIPAL DISPLAY PANEL - 45 mg Tablet Bottle Label
-
INGREDIENTS AND APPEARANCE
VIZIMPRO
dacomitinib tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0069-0197 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength DACOMITINIB (UNII: 5092U85G58) (DACOMITINIB ANHYDROUS - UNII:2XJX250C20) DACOMITINIB 15 mg Inactive Ingredients Ingredient Name Strength LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) SODIUM STARCH GLYCOLATE TYPE A POTATO (UNII: 5856J3G2A2) MAGNESIUM STEARATE (UNII: 70097M6I30) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) TALC (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) POLYETHYLENE GLYCOL 3350 (UNII: G2M7P15E5P) FD&C BLUE NO. 2 (UNII: L06K8R7DQK) Product Characteristics Color BLUE Score no score Shape ROUND Size 7mm Flavor Imprint Code Pfizer;DCB15 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0069-0197-30 30 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 10/04/2018 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA211288 10/04/2018 VIZIMPRO
dacomitinib tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0069-1198 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength DACOMITINIB (UNII: 5092U85G58) (DACOMITINIB ANHYDROUS - UNII:2XJX250C20) DACOMITINIB 30 mg Inactive Ingredients Ingredient Name Strength LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) SODIUM STARCH GLYCOLATE TYPE A POTATO (UNII: 5856J3G2A2) MAGNESIUM STEARATE (UNII: 70097M6I30) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) TALC (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) POLYETHYLENE GLYCOL 3350 (UNII: G2M7P15E5P) FD&C BLUE NO. 2 (UNII: L06K8R7DQK) Product Characteristics Color BLUE Score no score Shape ROUND Size 8mm Flavor Imprint Code Pfizer;DCB30 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0069-1198-30 30 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 10/04/2018 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA211288 10/04/2018 VIZIMPRO
dacomitinib tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0069-2299 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength DACOMITINIB (UNII: 5092U85G58) (DACOMITINIB ANHYDROUS - UNII:2XJX250C20) DACOMITINIB 45 mg Inactive Ingredients Ingredient Name Strength LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) SODIUM STARCH GLYCOLATE TYPE A POTATO (UNII: 5856J3G2A2) MAGNESIUM STEARATE (UNII: 70097M6I30) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) TALC (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) POLYETHYLENE GLYCOL 3350 (UNII: G2M7P15E5P) FD&C BLUE NO. 2 (UNII: L06K8R7DQK) Product Characteristics Color BLUE Score no score Shape ROUND Size 9mm Flavor Imprint Code Pfizer;DCB45 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0069-2299-30 30 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 10/04/2018 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA211288 10/04/2018 Labeler - Pfizer Laboratories Div Pfizer Inc (134489525) Establishment Name Address ID/FEI Business Operations Pfizer Pharmaceuticals LLC 829084552 ANALYSIS(0069-0197, 0069-1198, 0069-2299) , LABEL(0069-0197, 0069-1198, 0069-2299) , PACK(0069-0197, 0069-1198, 0069-2299) Establishment Name Address ID/FEI Business Operations Pharmacia and Upjohn Company LLC 618054084 ANALYSIS(0069-0197, 0069-1198, 0069-2299) , LABEL(0069-0197, 0069-1198, 0069-2299) , PACK(0069-0197, 0069-1198, 0069-2299) Establishment Name Address ID/FEI Business Operations Pfizer Ireland Pharmaceuticals 985104227 ANALYSIS(0069-0197, 0069-1198, 0069-2299) , API MANUFACTURE(0069-0197, 0069-1198, 0069-2299) Establishment Name Address ID/FEI Business Operations Pfizer Manufacturing Deutschland GmbH 341970073 ANALYSIS(0069-0197, 0069-1198, 0069-2299) , LABEL(0069-0197, 0069-1198, 0069-2299) , MANUFACTURE(0069-0197, 0069-1198, 0069-2299) , PACK(0069-0197, 0069-1198, 0069-2299)

Trademark Results [Vizimpro]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 VIZIMPRO 85979594 4401809 Live/Registered |
Pfizer Inc. 2010-12-08 |
 VIZIMPRO 85193224 not registered Dead/Abandoned |
PF PRISM C.V. 2010-12-08 |
 VIZIMPRO 77235981 not registered Dead/Abandoned |
Pfizer Inc. 2007-07-23 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.