COSELA- trilaciclib injection, powder, lyophilized, for solution
COSELA by
Drug Labeling and Warnings
COSELA by is a Prescription medication manufactured, distributed, or labeled by G1 Therapeutics, Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use COSELA safely and effectively. See full prescribing information for COSELA.
COSELA® (trilaciclib) for injection, for intravenous use
Initial U.S. Approval: 2021RECENT MAJOR CHANGES
Dosage and Administration (2.2) 8/2023 INDICATIONS AND USAGE
COSELA is a kinase inhibitor indicated to decrease the incidence of chemotherapy-induced myelosuppression in adult patients when administered prior to a platinum/etoposide-containing regimen or topotecan-containing regimen for extensive-stage small cell lung cancer. (1)
DOSAGE AND ADMINISTRATION
COSELA is for intravenous use only.
The recommended dose of COSELA is 240 mg/m2 as a 30-minute intravenous infusion completed no more than 4 hours prior to the start of chemotherapy on each day chemotherapy is administered. (2.1)
Reduce dose in patients with moderate or severe hepatic impairment. (2.2)
See Full Prescribing Information for instructions on preparation and administration. (2.3)
DOSAGE FORMS AND STRENGTHS
For injection: 300 mg of trilaciclib as a lyophilized cake in a single-dose vial. (3)
CONTRAINDICATIONS
Patients with a history of serious hypersensitivity reactions to COSELA. (4)
WARNINGS AND PRECAUTIONS
- Injection-Site Reactions, Including Phlebitis and Thrombophlebitis: Monitor for signs and symptoms of injection-site reactions, including phlebitis and thrombophlebitis during infusion. Stop infusion and permanently discontinue COSELA for severe or life-threatening reactions. (5.1)
- Acute Drug Hypersensitivity Reactions: Monitor for signs and symptoms of acute drug hypersensitivity reactions, including edema (facial, eye, and tongue), urticaria, pruritus, and anaphylactic reactions. Withhold COSELA for moderate reactions, and permanently discontinue for severe or life-threatening reactions. (5.2)
- Interstitial Lung Disease (ILD)/Pneumonitis: Patients treated with CDK4/6 inhibitors should be monitored for pulmonary symptoms indicative of ILD/pneumonitis. Interrupt and evaluate patients with new or worsening symptoms suspected to be due to ILD/pneumonitis. Permanently discontinue COSELA in patients with recurrent symptomatic or severe/life-threatening ILD/pneumonitis. (5.3)
- Embryo-Fetal Toxicity: Can cause fetal harm. Advise patients of the potential risk to a fetus and to use effective contraception. (5.4)
ADVERSE REACTIONS
The most common adverse reactions (≥10% of patients with ≥2% difference in incidence compared to placebo) were fatigue, hypocalcemia, hypokalemia, hypophosphatemia, aspartate aminotransferase increased, headache, and pneumonia. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact G1 Therapeutics, Inc., at 1-800-790-4189 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
Certain OCT2, MATE1, and MATE-2K substrates: Avoid concomitant use with certain OCT2, MATE1, and MATE-2K substrates where minimal concentration changes may lead to serious or life-threatening toxicities. (7.1)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 8/2023
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
2.2 Dose Modification
2.3 Preparation and Administration Instructions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Injection-Site Reactions, Including Phlebitis and Thrombophlebitis
5.2 Acute Drug Hypersensitivity Reactions
5.3 Interstitial Lung Disease/Pneumonitis
5.4 Embryo-Fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 Effect of COSELA on Other Drugs, Certain OCT2, MATE1, and MATE-2K Substrates
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage and Handling
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
The recommended dose of COSELA is 240 mg/m2 per dose. Administer as a 30-minute intravenous infusion completed no more than 4 hours prior to the start of chemotherapy on each day chemotherapy is administered.
The interval between doses of COSELA on sequential days should not be greater than 28 hours.
2.2 Dose Modification
Dose Modification for Adverse Reactions
Withhold, discontinue, or alter the administration of COSELA to manage adverse reactions as described in Table 1 [see Warnings and Precautions (5)].
Table 1: Recommended Actions for Adverse Reactions Adverse Reaction Severity Grade* Recommended Action * National Cancer Institute – Common Terminology Criteria for Adverse Events (NCI-CTCAE) Version 4.03
Injection-site reactions including phlebitis and thrombophlebitis Grade 1: Tenderness with or without symptoms (e.g., warmth, erythema, itching) Interrupt or slow infusion of COSELA. If 0.9% Sodium Chloride Injection, USP is being used as a diluent/flush, consider changing to 5% Dextrose Injection, USP as appropriate for subsequent infusions. Grade 2: Pain; lipodystrophy; edema; phlebitis Interrupt infusion of COSELA. If pain not severe, follow instructions for Grade 1. Otherwise, stop infusion in extremity and rotate site of infusion to site in alternative extremity. If 0.9% Sodium Chloride Injection, USP is being used as a diluent/flush, consider changing to 5% Dextrose Injection, USP as appropriate for subsequent infusions. Central access may also be considered. Grade 3: Ulceration or necrosis; severe tissue damage; operative intervention indicated.
OR
Grade 4: Life-threatening consequences; urgent interventions indicated.Stop infusion and permanently discontinue COSELA. Acute drug hypersensitivity reactions Grade 2: Moderate; minimal, local, or noninvasive intervention indicated; limiting Activities of Daily Living (ADL). Stop infusion and hold COSELA until recovery to Grade ≤1 or baseline, then consider resuming COSELA. If Grade 2 recurs, permanently discontinue COSELA. Grade 3: Severe or medically significant but not immediately life-threatening; hospitalization or prolongation of hospitalization indicated; disabling; limiting self-care ADL.
OR
Grade 4: Life-threatening consequences; urgent intervention indicated.Permanently discontinue COSELA. Interstitial lung disease/pneumonitis Grade 2 (symptomatic) Hold COSELA until recovery to Grade ≤1 or baseline, then consider resuming COSELA.
If Grade 2 recurs, permanently discontinue COSELA.Grade 3: Severe symptoms; limiting self-care ADL; oxygen indicated.
OR
Grade 4: Life-threatening respiratory compromise; urgent intervention indicated (e.g., tracheotomy or intubation)Permanently discontinue COSELA. Other toxicities Grade 3: Severe or medically significant but not immediately life-threatening; hospitalization or prolongation of hospitalization indicated; disabling; limiting self-care ADL. Hold COSELA until recovery to Grade ≤1 or baseline, then consider resuming COSELA.
If Grade 3 recurs, permanently discontinue COSELA.Grade 4: Life-threatening consequences; urgent intervention indicated. Permanently discontinue COSELA. Dose Modifications for Hepatic Impairment
Reduce the dose of COSELA to 170 mg/m2 in patients with moderate or severe hepatic impairment (Child-Pugh classes B and C). No dose adjustment is required for patients with mild hepatic impairment (Child-Pugh class A) [see Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)].
2.3 Preparation and Administration Instructions
Reconstitute and further dilute COSELA prior to intravenous infusion as outlined below. Use aseptic technique for reconstitution and dilution.
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit.
Reconstitution of COSELA
- Calculate the COSELA dose based on the patient's body surface area (BSA), the total volume of reconstituted COSELA solution required, and the number of COSELA vials needed.
- Reconstitute each 300 mg vial with 19.5 mL of 0.9% Sodium Chloride Injection, USP or 5% Dextrose Injection, USP using a sterile syringe to obtain a concentration of 15 mg/mL of trilaciclib.
- Gently swirl the vial for up to 3 minutes until the sterile lyophilized cake is completely dissolved. Do not shake.
- Inspect the reconstituted solution for discoloration and particulate matter. Reconstituted COSELA solution should be a clear, yellow solution. Do not use if the reconstituted solution is discolored, cloudy, or contains visible particulates.
- Reconstituted solution in the vial can be stored at 20°C to 25°C (68°F to 77°F) for up to 4 hours prior to transfer to the infusion bag. Do not refrigerate or freeze.
- Discard any unused portion after use.
Dilution of Reconstituted COSELA Solution
- Withdraw the required volume from the vial(s) of reconstituted COSELA solution and dilute into an intravenous infusion bag containing 0.9% Sodium Chloride Injection, USP or 5% Dextrose Injection, USP. The final concentration of the diluted COSELA solution should be between 0.5 mg/mL and 3 mg/mL.
- Mix diluted solution by gentle inversion. Do not shake.
- The diluted COSELA solution for infusion is a clear, yellow solution.
- If not used immediately, store the diluted COSELA solution in the intravenous infusion bag as specified in Table 2. Discard if storage time exceeds these limits. Do not refrigerate or freeze.
Table 2: Diluted COSELA Solution Storage Conditions a To ensure product stability, do not exceed specified storage durations.
Intravenous Infusion Bag Material Diluent Diluted COSELA Storage Durationa Polyvinyl chloride (PVC),
Ethylene vinyl acetate (EVA),
Polyolefin (PO), or Polyolefin/polyamide (PO/PA)5% Dextrose for Injection, USP Up to 12 hours at 20°C to 25°C (68°F to 77°F) PVC, EVA, or PO 0.9% Sodium Chloride Injection, USP Up to 8 hours at 20°C to 25°C (68°F to 77°F) PO/PA 0.9% Sodium Chloride Injection, USP Up to 4 hours at 20°C to 25°C (68°F to 77°F) Administration
- Administer diluted COSELA solution as a 30-minute intravenous infusion completed no more than 4 hours prior to the start of chemotherapy.
- Diluted COSELA solution must be administered with an infusion set, including an in-line filter (0.2 or 0.22 micron). Compatible in-line filters include polyethersulfone (PES), polyvinylidene fluoride (PVDF), and cellulose acetate (CA).
- Do not administer diluted COSELA solution with a polytetrafluorethylene (PTFE) in-line filter as it is not compatible. PTFE is acceptable for use in air vent filters.
- Do not co-administer other drugs through the same infusion line.
- Do not co-administer other drugs through a central access device unless the device supports co-administration of incompatible drugs.
- Upon completion of infusion of diluted COSELA solution, the infusion line/cannula must be flushed with at least 20 mL sterile 0.9% Sodium Chloride Injection, USP or 5% Dextrose Injection, USP.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
COSELA is contraindicated in patients with a history of serious hypersensitivity reactions to trilaciclib. Reactions have included anaphylaxis [see Warnings and Precautions (5.2)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Injection-Site Reactions, Including Phlebitis and Thrombophlebitis
COSELA administration can cause injection-site reactions including phlebitis and thrombophlebitis. Injection-site reactions including phlebitis and thrombophlebitis occurred in 56 (21%) of 272 patients receiving COSELA in clinical trials, including Grade 2 (10%) and Grade 3 (0.4%) adverse reactions (ARs). The median time to onset from start of COSELA was 15 days (range 1 to 542) and from the preceding dose of COSELA was 1 day (1 to 15).The median duration was 1 day (range 1 to 151 for the resolved cases). Injection-site reactions including phlebitis and thrombophlebitis resolved in 49 (88%) of the 56 patients and led to discontinuation of treatment in 3 (1%) of the 272 patients.
Monitor patients for signs and symptoms of injection-site reactions, phlebitis, and thrombophlebitis, including infusion-site pain and erythema during infusion. For mild (Grade 1) to moderate (Grade 2) injection-site reactions, flush line/cannula with at least 20 mL of sterile 0.9% Sodium Chloride Injection, USP or 5% Dextrose Injection, USP after end of infusion. For severe (Grade 3) or life-threatening (Grade 4) injection-site reactions, stop infusion and permanently discontinue COSELA [see Dosage and Administration (2.2)].
5.2 Acute Drug Hypersensitivity Reactions
COSELA administration can cause acute drug hypersensitivity reactions, including facial edema and urticaria. Acute drug hypersensitivity reactions occurred in 16 (6%) of 272 patients receiving COSELA in clinical trials, including Grade 2 reactions (2%). One patient experienced a Grade 2 anaphylactic reaction 4 days after receiving COSELA, which resolved with epinephrine, and treatment with COSELA was continued. The median time to onset from start of COSELA was 77 days (range 2 to 256) and from the preceding dose of COSELA was 1 day (range 1 to 28). The median duration was 6 days (range 1 to 69 for the resolved cases). Acute drug hypersensitivity reactions resolved in 12 (75%) of the 16 patients.
Monitor patients for signs and symptoms of acute drug hypersensitivity reactions including facial, eye, and tongue edema, urticaria, pruritus, and anaphylactic reactions. For moderate (Grade 2) acute drug hypersensitivity reactions, stop infusion and hold COSELA until the adverse reaction recovers to Grade ≤1. For severe (Grade 3) or life-threatening (Grade 4) acute drug hypersensitivity reactions, stop infusion and permanently discontinue COSELA [see Dosage and Administration (2.2)].
5.3 Interstitial Lung Disease/Pneumonitis
Severe, life-threatening, or fatal interstitial lung disease (ILD) and/or pneumonitis can occur in patients treated with cyclin-dependent kinases (CDK)4/6 inhibitors, the same drug class as COSELA. ILD/pneumonitis occurred in 1 (0.4%) of 272 patients receiving COSELA in clinical trials. The adverse reaction was Grade 3 and reported 2 months after discontinuing COSELA, in a patient receiving a confounding medication. The adverse reaction did not resolve.
Monitor patients for pulmonary symptoms indicative of ILD/pneumonitis such as cough, dyspnea, and hypoxia. For recurrent moderate (Grade 2) ILD/pneumonitis, permanently discontinue COSELA. For severe (Grade 3) or life-threatening (Grade 4) ILD/pneumonitis, permanently discontinue COSELA [see Dosage and Administration (2.2)].
5.4 Embryo-Fetal Toxicity
Based on its mechanism of action, COSELA can cause fetal harm when administered to a pregnant woman. Females of reproductive potential should use an effective method of contraception during treatment with COSELA and for at least 3 weeks after the final dose [see Use in Specific Populations (8.1, 8.3)].
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the label:
- Injection-Site Reactions, including phlebitis and thrombophlebitis [see Warnings and Precautions (5.1)]
- Acute Drug Hypersensitivity Reactions [see Warnings and Precautions (5.2)]
- ILD/Pneumonitis [see Warnings and Precautions (5.3)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
The safety of COSELA was evaluated in Studies 1, 2, and 3 [see Clinical Studies (14)]. Patients received COSELA 240 mg/m2 by 30-minute intravenous infusion prior to chemotherapy on each chemotherapy day. The data described in this section reflect exposure to COSELA among 240 patients (122 patients in the trilaciclib group and 118 patients in the placebo group) being treated for extensive stage-small cell lung cancer (ES-SCLC) in 3 randomized, double-blind, placebo-controlled trials: 32 patients with treatment naïve ES-SCLC received carboplatin (AUC 5 Day 1) + etoposide (100 mg/m2 Days 1-3) every 21 days; 58 received carboplatin (AUC 5 Day 1) + etoposide (100 mg/m2 Days 1-3) every 21 days + atezolizumab (1200 mg on Day 1) every 21 days; 32 patients with previously treated ES-SCLC received topotecan (1.5 mg/m2 Days 1-5) every 21 days.
Study 1: COSELA Prior to Etoposide, Carboplatin, and Atezolizumab (E/P/A)
Patients with newly diagnosed ES-SCLC not previously treated with chemotherapy
Study 1 (G1T28-05; NCT03041311) was an international, randomized (1:1), double-blind, placebo-controlled study of COSELA or placebo administered prior to treatment with etoposide, carboplatin, and atezolizumab (E/P/A) for patients with newly diagnosed ES-SCLC not previously treated with chemotherapy. The data presented below are for the 105 patients who received study treatment.
Eighty-five percent of patients receiving COSELA and 91% receiving placebo completed 4 cycles of induction therapy.
Study 2: COSELA Prior to Etoposide and Carboplatin (E/P)
Patients with newly diagnosed ES-SCLC not previously treated with chemotherapy
Study 2 (G1T28-02; NCT02499770) was an international, randomized (1:1), double-blind, placebo-controlled study of COSELA or placebo administered prior to treatment with etoposide and carboplatin (E/P) for patients with newly diagnosed ES-SCLC not previously treated with chemotherapy. The data presented below are for the 75 patients who received study treatment.
Seventy-six percent of patients in the COSELA group and 87% of patients in the placebo group completed at least 4 cycles of therapy. The median duration of treatment was 6 cycles in each treatment group.
Study 3: COSELA Prior to Topotecan
Patients with ES-SCLC previously treated with chemotherapy
Study 3 (G1T28-03; NCT02514447) was an international, randomized (2:1), double-blind, placebo-controlled study of COSELA or placebo administered prior to treatment with topotecan for patients with ES-SCLC previously treated with chemotherapy. The data presented below are for the 60 patients who received study treatment with the 1.5 mg/m2 dose of topotecan.
Thirty-eight percent of patients receiving COSELA and 29% of patients receiving placebo completed 5 or more cycles of therapy. The median duration of treatment was 3 cycles in each treatment group.
Integrated Safety Analysis
The adverse reaction summary presented in Table 3 are pooled safety results from Studies 1, 2, and 3. The patients included in the pooling are those randomized patients that received at least 1 dose of COSELA (122 patients) or placebo (118 patients).
Seventy-one percent of patients receiving COSELA and 78% of patients receiving placebo completed at least 4 cycles of therapy. The median duration of treatment was the same (4 cycles) for patients receiving COSELA and placebo.
Serious adverse reactions occurred in 30% of patients receiving COSELA. Serious adverse reactions reported in >3% of patients who received COSELA included respiratory failure, hemorrhage, and thrombosis.
Permanent discontinuation due to an adverse reaction occurred in 9% of patients who received COSELA. Adverse reactions leading to permanent discontinuation of any study treatment for patients receiving COSELA included pneumonia (2%), asthenia (2%), injection-site reaction, thrombocytopenia, cerebrovascular accident, ischemic stroke, infusion-related reaction, respiratory failure, and myositis (<1% each).
Fatal adverse reactions were observed in 5% of patients receiving COSELA. Fatal adverse reactions for patients receiving COSELA included pneumonia (2%), respiratory failure (2%), acute respiratory failure (<1%), hemoptysis (<1%), and cerebrovascular accident (<1%).
Infusion interruptions due to an adverse reaction occurred in 4.1% of patients who received COSELA.
The most common adverse reactions (≥10%) were fatigue, hypocalcemia, hypokalemia, hypophosphatemia, aspartate aminotransferase increased, headache, and pneumonia. The most frequently reported Grade ≥3 adverse reaction (≥5%) in patients receiving COSELA occurring at the same or higher incidence than in patients receiving placebo was hypophosphatemia.
The most common adverse reactions reported in at least 5% of patients receiving COSELA with a ≥2% higher incidence compared to patients receiving placebo are shown in Table 3.
Table 3: Adverse Reactions in ≥5% Patients with SCLC Receiving COSELA (with ≥2% Higher Incidence in COSELA Compared to Placebo) Adverse Reaction COSELA
(N=122)Placebo
(N=118)All
Gradesa (%)Grade ≥3 (%) All
Gradesa (%)Grade ≥3 (%) a Graded per NCI CTCAE v4.03
b Hypocalcemia=calcium decreased (lab) or treatment-emergent adverse event (TEAE) preferred term 'Hypocalcemia'
c Hypokalemia=potassium decreased (lab) or TEAE preferred terms 'Hypokalemia,' 'Blood potassium decreased'
d Hypophosphatemia=phosphate decreased (lab) or TEAE preferred terms 'Hypophosphatemia,' 'Blood phosphorus decreased'
e Aspartate aminotransferase increased=aspartate aminotransferase increased (lab) or TEAE preferred term 'Blood aspartate aminotransferase increased'
Fatigue 34 3 27 2 Hypocalcemiab 24 <1 21 <1 Hypokalemiac 22 6 18 3 Hypophosphatemiad 21 7 16 2 Aspartate aminotransferase increasede 17 <1 14 <1 Headache 13 0 9 0 Pneumonia 10 7 8 7 Rash 9 <1 6 0 Infusion-related reaction 8 0 2 0 Edema peripheral 7 0 4 <1 Abdominal pain upper 7 0 3 0 Thrombosis 7 3 2 2 Hyperglycemia 6 2 3 0 Grade 3/4 hematological adverse reactions occurring in patients treated with COSELA and placebo included neutropenia (32% and 69%), febrile neutropenia (3% and 9%), anemia (16% and 34%), thrombocytopenia (18% and 33%), leukopenia (4% and 17%), and lymphopenia (<1% and <1%), respectively.
-
7 DRUG INTERACTIONS
7.1 Effect of COSELA on Other Drugs, Certain OCT2, MATE1, and MATE-2K Substrates
COSELA is an inhibitor of OCT2, MATE1, and MATE-2K. Co-administration of COSELA may increase the concentration or net accumulation of OCT2, MATE1, and MATE-2K substrates in the kidney (e.g., dofetilide, dalfampridine, and cisplatin) [see Clinical Pharmacology (12.3)].
Refer to the prescribing information for these concomitant medications for assessing the benefit and risk of concomitant use of COSELA.
Table 4: Potentially Significant Drug Interactions with COSELA Drugs Recommendations Comments Dofetilide The potential benefits of taking COSELA concurrently with dofetilide should be considered against the risk of QT interval prolongation. Increased dofetilide blood levels may occur in patients who are also receiving COSELA. Increased plasma concentrations of dofetilide may cause serious ventricular arrhythmias associated with QT interval prolongation, including torsade de pointes. Dalfampridine The potential benefits of taking COSELA concurrently with dalfampridine should be considered against the risk of seizures in these patients. Increased dalfampridine blood levels may occur in patients who are also receiving COSELA. Elevated levels of dalfampridine increase the risk of seizure. Cisplatin Closely monitor for nephrotoxicity. Concurrent treatment with COSELA may increase the exposure and alter the net accumulation of cisplatin in the kidney, which may associate with dose-related nephrotoxicity. -
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on the mechanism of action, COSELA can cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12)]. There are no available human or animal data on COSELA use to evaluate for a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes. Advise pregnant women of the potential risk to a fetus.
The background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. However, the background risk of major birth defects is 2% to 4% and of miscarriage is 15% to 20% of clinically recognized pregnancies in the United States general population.
8.2 Lactation
Risk Summary
There are no data on the presence of trilaciclib in either human or animal milk, the effects on the breastfed child or the effects on milk production. Because of the potential for serious adverse reactions in breastfed children, advise lactating women to not breastfeed while taking COSELA and for at least 3 weeks after the last dose.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Based on its mechanism of action, COSELA can cause fetal harm if administered to a pregnant woman [see Use in Specific Populations (8.1)]. Pregnancy testing is recommended for females of reproductive potential prior to initiating COSELA.
Contraception
COSELA can cause fetal harm when administered to pregnant women [see Use in Specific Populations (8.1)]. Advise female patients of reproductive potential to use effective contraception during treatment with COSELA and for at least 3 weeks after the final dose.
Infertility
No studies have been performed in humans to evaluate the effects of COSELA on fertility in either sex.
Based on animal toxicology studies, COSELA may impair fertility in females of reproductive potential [see Nonclinical Toxicology (13)].
8.5 Geriatric Use
In the pooled efficacy dataset from Studies 1, 2, and 3, 46% of 123 patients randomized to COSELA were ≥65 years of age, and 49% of 119 patients randomized to placebo were ≥65 years of age. No overall differences in safety or effectiveness of COSELA were observed between these patients and younger patients.
8.6 Hepatic Impairment
Reduce the dose of COSELA to 170 mg/m2 in patients with moderate or severe hepatic impairment (Child-Pugh classes B and C). No dose adjustment is required in patients with mild hepatic impairment (Child-Pugh class A) [see Dosage and Administration (2.2)].
Trilaciclib is mainly metabolized in the liver. Trilaciclib exposure increased with moderate and severe hepatic impairment (Child-Pugh classes B and C) [see Clinical Pharmacology (12.3)].
-
11 DESCRIPTION
COSELA for injection contains trilaciclib dihydrochloride, a kinase inhibitor.
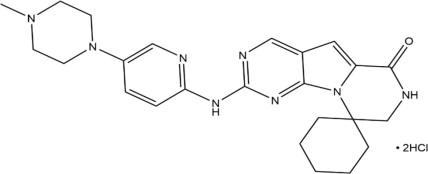
The chemical name for trilaciclib is 2'-{[5-(4-methylpiperazin-1-yl)pyridin-2-yl]amino}-7',8'-dihydro-6'H-spiro[cyclohexane-1,9'-pyrazino[1',2':1,5]pyrrolo[2,3-d]pyrimidin]-6'-one.
Trilaciclib dihydrochloride is a water-soluble yellow solid, with molecular formula of C24H30N8O2HCl, a molecular weight of 519.48 g/mol (Free base: 446.56 g/mol), and the following chemical structure:
COSELA (trilaciclib) for injection is a sterile, preservative-free, yellow lyophilized cake in a single-dose vial for intravenous infusion after reconstitution and dilution.
Each single-dose vial contains the equivalent of 300 mg of trilaciclib (provided as 349 mg of trilaciclib dihydrochloride) and the following inactive ingredients: citric acid monohydrate (75.6 mg) and mannitol (300 mg); hydrochloric acid and sodium hydroxide to adjust pH.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Trilaciclib is a transient inhibitor of CDK 4 and 6. Hematopoietic stem and progenitor cells (HSPCs) in the bone marrow give rise to circulating neutrophils, RBCs, and platelets. HSPC proliferation is dependent on CDK4/6 activity.
12.2 Pharmacodynamics
Bone Marrow
Trilaciclib exhibited dose-dependent inhibition of CD45+/CD3+ lymphocyte proliferation following administration of single-dose COSELA 96 or 192 mg/m2 (0.4 or 0.8 times the approved recommended dose) in healthy subjects.
Trilaciclib increased the percentage of cells arrested in G1 up to 32 hours post-infusion for all bone marrow progenitor subsets evaluated (hematopoietic stem cell/multipotent progenitor, oligopotent progenitor, monocyte lineage, granulocyte lineage, erythroid lineage, and megakaryocyte lineage) following a single dose of COSELA 192 mg/m2 (0.8 times the approved recommended dose) in healthy subjects. Partial recovery of the total bone marrow with resumption of proliferation of the bone marrow progenitor subsets was observed by 32 hours post-dose. This transient G1 arrest of hematopoietic stem cells contributed to the myeloprotective effect of trilaciclib.
Cardiac Electrophysiology
COSELA is associated with dose-dependent and delayed increase in the QTc interval. The underlying mechanism of the delayed QT effect is unknown. At the clinical dose of 240 mg/m2, COSELA did not have a clinically relevant effect on QTc (i.e., >10 msec). QTc prolongation was observed at higher doses.
12.3 Pharmacokinetics
The maximum concentration (Cmax) increased proportionally whereas the total plasma exposure (AUC0-last) increased slightly greater than proportional over a dosage range of trilaciclib 200 mg/m2 to 700 mg/m2 (0.83 to 2.9 times the approved recommended dose). There was no accumulation of trilaciclib following repeated dosing.
Distribution
The in vitro human plasma protein binding of trilaciclib is 69% and appeared independent of trilaciclib concentration from 0.75 to 3.0 μg/mL. The blood/plasma ratio ranged from 1.21 to 1.53 for trilaciclib across concentrations of 0.5 μg/mL to 50 μg/mL in vitro. The volume of distribution at steady state was 1130 L.
Elimination
The mean terminal half-life of trilaciclib is approximately 14 hours. Clearance was estimated to be 158 L/hr.
Metabolism
Trilaciclib undergoes extensive metabolism. Trilaciclib is the predominant circulating compound in plasma following intravenous administration, representing ~50% of plasma total radioactivity. In vitro studies indicated that trilaciclib is primarily metabolized by aldehyde oxidase (AO), cytochrome P450 (CYP) 3A4 and CYP2C8.
Excretion
After a single dose of radiolabeled trilaciclib 192 mg/m2 (0.8 times the approved recommended dosage), approximately 79.1% of the dose was recovered in feces (7% unchanged) and 14% was recovered in urine (2% unchanged).
Trilaciclib is eliminated mainly via the fecal route, with a small contribution of the renal route.
Specific Populations
No clinically significant differences in the pharmacokinetics of trilaciclib were observed based on age (range: 19 to 80 years), sex, race, mild to moderate renal impairment (30 to 89 mL/min/1.73 m2 measured by estimated glomerular filtration rate [eGFR]), or mild hepatic impairment (total bilirubin ≤ULN and AST >ULN, or total bilirubin >1.0 to 1.5 × ULN, irrespective of AST). The effect of severe renal impairment (<30 mL/min/1.73 m2), end stage renal disease, or dialysis on trilaciclib pharmacokinetics has not been studied.
Subjects with Hepatic Impairment
Trilaciclib unbound AUCinf increased by 40% and 63%, respectively, in subjects with moderate and severe hepatic impairment (Child-Pugh classes B and C). No clinically significant differences were observed in trilaciclib systemic exposures in subjects with mild hepatic impairment [see Dosage and Administration (2.2) and Use in Specific Populations (8.6)].
Drug Interaction Studies
Clinical Studies
Cytochrome P450 (CYP) Enzymes: There were no clinically significant differences in trilaciclib pharmacokinetics when used concomitantly with itraconazole (strong CYP3A inhibitor) or rifampin (strong CYP3A inducer). There were no clinically significant differences in midazolam (CYP3A substrate) pharmacokinetics when used concomitantly with trilaciclib.
Transporter Systems: Concomitant use of trilaciclib increased metformin (OCT2, MATE1, and MATE-2K substrate) AUCinf and Cmax by approximately 65% and 81%, respectively. Renal clearance of metformin was decreased by 37%. There were no clinically significant differences in topotecan (MATE1 and MATE-2K substrate) pharmacokinetics when used concomitantly with trilaciclib.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis studies have not been conducted with trilaciclib.
Trilaciclib was negative for mutagenic potential in a bacterial reverse mutation (Ames) assay and negative for clastogenic potential in an in vitro histone H2AX phosphorylation assay in primary human fibroblasts. Trilaciclib increased the frequency of micronucleus formation in human lymphocytes in vitro. Clastogenic potential of trilaciclib was not assessed in vivo.
Fertility studies have not been performed to evaluate the effects of trilaciclib. Treatment with trilaciclib in female rats and dogs resulted in reductions in mean ovary and uterus weights at clinically relevant exposures, which were reversible after a two-week drug-free recovery period.
-
14 CLINICAL STUDIES
The efficacy of COSELA was established in three randomized, double-blind, placebo-controlled trials in patients with extensive stage-small cell lung cancer (ES-SCLC). Study 1 (G1T28-05; NCT03041311) enrolled adult patients receiving carboplatin, etoposide, and atezolizumab for newly diagnosed ES-SCLC. Study 2 (G1T28-02; NCT02499770) enrolled adult patients receiving etoposide/carboplatin for newly diagnosed ES-SCLC. Study 3 (G1T28-03; NCT02514447) enrolled adult patients receiving topotecan for previously treated ES-SCLC.
During Cycle 1, all three studies prohibited primary prophylactic granulocyte-colony stimulating factor (G-CSF) and erythropoiesis-stimulating agent (ESA) use. Both ESAs and prophylactic G-CSF were allowed from Cycle 2 onwards as clinically indicated. Therapeutic G-CSF, RBC, and platelet transfusions were allowed at any time during the studies as clinically indicated.
Study 1: COSELA Prior to Etoposide, Carboplatin, and Atezolizumab (E/P/A)
Patients with newly diagnosed ES-SCLC not previously treated with chemotherapy
Study 1 (G1T28-05) was a randomized (1:1), double-blind, placebo-controlled study of COSELA or placebo administered prior to treatment with etoposide, carboplatin, and atezolizumab (E/P/A) for patients with newly diagnosed ES-SCLC not previously treated with chemotherapy.
A total of 107 patients were randomized to receive COSELA (n=54) or placebo (n=53) prior to administration of E/P/A; patients were stratified by Eastern Cooperative Oncology Group (ECOG) performance status (0 to 1 vs 2) and the presence of brain metastases. Carboplatin (AUC 5) and atezolizumab (1200 mg) were administered on Day 1 and etoposide (100 mg/m2) and COSELA (240 mg/m2) or placebo were administered on Days 1, 2, and 3 of a 21-day cycle for a maximum of 4 cycles (induction). After induction, maintenance atezolizumab (1200 mg) monotherapy on Day 1 of a 21-day cycle continued until disease progression or unacceptable toxicity. COSELA was not administered during maintenance.
The study population characteristics were: median age 64 years (range: 45 to 83); 70% male; 97% white; 14% ECOG performance status 2; 28% with a history of brain metastases; 38% current smokers; 46% lactate dehydrogenase (LDH) >ULN.
The mean relative dose intensities (RDIs) for E/P/A in patients receiving COSELA were 93%, 95%, and 93%, respectively. The mean RDIs for E/P/A in patients receiving placebo were 88%, 89%, and 91%, respectively. Dose reductions of carboplatin occurred in 2% of patients receiving COSELA and in 25% of patients receiving placebo; dose reductions of etoposide occurred in 6% of patients receiving COSELA and in 26% of patients receiving placebo. No dose reduction was allowed for COSELA or atezolizumab.
The study demonstrated a statistically significantly shorter duration of severe neutropenia (DSN) in Cycle 1 (0 vs 4 days) and a lower proportion of patients with severe neutropenia (SN) (2% vs 49%) in patients receiving COSELA compared with placebo (Table 5). Nineteen percent of patients receiving COSELA had Grade 3 or 4 decreased hemoglobin compared with 28% of patients receiving placebo (adjusted relative risk 0.663 [95% CI: 0.336, 1.310]). The rate of RBC transfusions over time was 1.7/100 weeks for patients receiving COSELA and 2.6/100 weeks for patients receiving placebo (adjusted relative risk was not estimable). Six percent of patients receiving COSELA received erythropoiesis-stimulating agents (ESAs) compared with 11% of patients receiving placebo (adjusted relative risk 0.529 [95% CI: 0.145, 1.927]).
Table 5: Study 1: Myeloprotective Efficacy Results in Patients Treated with COSELA or Placebo Prior to Chemotherapy (Intent-to-Treat Analysis) Endpoint COSELA 240 mg/m2 (N=54) Placebo
(N=53)Treatment Effecta
(Mean Difference* or Adjusted Relative Risk)
(95% CI)Adjusted 1-sided p-valueb ANCOVA=analysis of covariance; CI=confidence interval; DSN=duration of severe neutropenia; G-CSF=granulocyte colony-stimulating factor; N=total number of patients in each treatment group; RBC=red blood cell; SD=standard deviation
a The following statistical models were used to assess treatment effects: non-parametric ANCOVA (DSN in Cycle 1); modified Poisson regression (occurrence of SN and RBC transfusion on/after 5 weeks); negative binomial regression (number of all-cause dose reductions). All models included the following as covariates: ECOG status, presence of brain metastases, and the corresponding baseline laboratory values.
b One-sided adjusted p-value obtained from a Hochberg-based gatekeeping procedure.
Primary Endpoints DSN in Cycle 1 (days): mean (SD) 0 (1.0) 4 (4.7) -3.6* (-4.9, -2.3) <0.0001 Number (%) of patients with severe neutropenia 1 (1.9%) 26 (49.1%) 0.038
(0.008, 0.195)<0.0001 Key Secondary Endpoints Number of all-cause dose reductions, event rate per cycle 0.021 0.085 0.242
(0.079, 0.742)0.0195 Number (%) of patients with RBC transfusion on/after 5 weeks 7 (13.0%) 11 (20.8%) 0.642
(0.294, 1.404)-- Number (%) of patients with G-CSF administration 16 (29.6%) 25 (47.2%) 0.646
(0.403, 1.034)-- Study 2: COSELA Prior to Etoposide and Carboplatin
Patients with newly diagnosed ES-SCLC not previously treated with chemotherapy
Study 2 (G1T28-02) was a randomized (1:1), double-blind, placebo-controlled evaluation of COSELA or placebo administered prior to treatment with etoposide and carboplatin (E/P) for patients with newly diagnosed ES-SCLC not previously treated with chemotherapy. A total of 77 patients were randomized to COSELA (n=39) or placebo (n=38) and stratified by ECOG performance status (0 to 1 vs 2). Carboplatin (AUC 5) was administered on Day 1 and etoposide (100 mg/m2) and COSELA (240 mg/m2) or placebo were administered on Days 1, 2, and 3 of a 21-day cycle until disease progression or unacceptable toxicity.
Ten percent of patients receiving COSELA had Grade 3 or 4 decreased hemoglobin compared with 18% of patients receiving placebo. The rate of RBC transfusions over time was 0.5/100 weeks for patients receiving COSELA and 1.9/100 weeks for patients receiving placebo. Three percent of patients receiving COSELA received ESAs compared with 5% of patients receiving placebo.
Table 6: Study 2: Myeloprotective Efficacy Results in Patients Treated with COSELA or Placebo Prior to Chemotherapy (Intent-to-Treat Analysis) Endpoint COSELA
240 mg/m2 (N=39)Placebo
(N=38)DSN=duration of severe neutropenia; G-CSF=granulocyte colony-stimulating factor; N=total number of patients in each treatment group; RBC=red blood cell; SD=standard deviation
DSN in Cycle 1 (days): mean (SD) 0 (0.5) 3 (3.9) Number (%) of patients with severe neutropenia 2 (5.1%) 16 (42.1%) Number of all-cause dose reductions, event rate per cycle 0.022 0.084 Number (%) of patients with RBC transfusion on/after 5 weeks 2 (5.1%) 9 (23.7%) Number (%) of patients with G-CSF administration 4 (10.3%) 24 (63.2%) Study 3: COSELA Prior to Topotecan
Patients with ES-SCLC previously treated with chemotherapy
Study 3 (G1T28-03) included a randomized, double-blind, placebo-controlled evaluation of COSELA or placebo administered prior to topotecan in patients with ES-SCLC previously treated with chemotherapy. A total of 61 patients were randomized to COSELA (n=32) or placebo (n=29). Patients were stratified by ECOG performance status (0 to 1 vs 2) and sensitivity to first-line treatment. Topotecan (1.5 mg/m2) and COSELA (240 mg/m2) or placebo were administered on Days 1-5 of a 21-day cycle. Treatment was administered until disease progression or unacceptable toxicity.
Thirty-eight percent of patients receiving COSELA had Grade 3 or 4 decreased hemoglobin compared with 59% of patients receiving placebo. The rate of RBC transfusions over time was 2.6/100 weeks for patients receiving COSELA and 6.3/100 weeks for patients receiving placebo. Three percent of patients receiving COSELA received ESAs compared with 21% of patients receiving placebo.
Table 7: Study 3: Myeloprotective Efficacy Results in Patients Treated with COSELA or Placebo Prior to Chemotherapy (Intent-to-Treat Analysis) Endpoint COSELA
240 mg/m2 (N=32)Placebo
(N=29)DSN=duration of severe neutropenia; G-CSF=granulocyte colony-stimulating factor; N=total number of patients in each treatment group; RBC=red blood cell; SD=standard deviation
Primary Endpoints DSN in Cycle 1 (days): mean (SD) 2 (3.9) 7 (6.2) Number (%) of patients with severe neutropenia 13 (40.6%) 22 (75.9%) Key Secondary Endpoints Number of all-cause dose reductions, event rate per cycle 0.051 0.116 Number (%) of patients with RBC transfusion on/after 5 weeks 10 (31.3%) 12 (41.4%) Number (%) of patients with G-CSF administration 16 (50.0%) 19 (65.5%) Number (%) of patients with platelet transfusion 8 (25.0%) 9 (31.0%) -
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
COSELA (trilaciclib) for injection is a yellow lyophilized cake supplied in a single-dose vial. Each carton (NDC: 73462-101-01) contains one 300 mg strength single-dose vial.
-
17 PATIENT COUNSELING INFORMATION
Injection-Site Reactions, Including Phlebitis and Thrombophlebitis
Inform patients of the signs and symptoms of injection-site reactions, including phlebitis and thrombophlebitis. Advise patients to contact their healthcare provider immediately for signs and symptoms of injection-site reactions, including phlebitis and thrombophlebitis [see Warnings and Precautions (5.1)].
Acute Drug Hypersensitivity Reactions
Advise patients to contact their healthcare provider immediately for signs and symptoms of acute drug hypersensitivity reactions including facial, eye, and tongue edema, urticaria, pruritis, and anaphylactic reactions [see Warnings and Precautions (5.2)].
Interstitial Lung Disease/Pneumonitis
Advise patients to immediately report new or worsening respiratory symptoms [see Warnings and Precautions (5.3) and Dosage and Administration (2.2)].
Embryo-Fetal Toxicity
Advise females of reproductive potential of the potential risk to a fetus and to inform their healthcare provider of a known or suspected pregnancy [see Use in Specific Populations (8.1)].
Advise females of reproductive potential to use effective contraception during treatment with COSELA and for at least 3 weeks after the final dose [see Use in Specific Populations (8.1, 8.3)].
Lactation
Advise women not to breastfeed during treatment with COSELA and for at least 3 weeks after the final dose of COSELA [see Use in Specific Populations (8.2)].
Drug Interactions
Advise patients to inform their healthcare providers of all concomitant medications, including prescription medicines, over-the-counter drugs, vitamins, and herbal products [see Drug Interactions (7)].
Distributed by:
G1 Therapeutics, Inc.
Durham, NC 27709COSELA® is a registered trademark of G1 Therapeutics, Inc.
©2023 G1 Therapeutics, Inc.
######
10003 Rev. 8/2023
-
PRINCIPAL DISPLAY PANEL
Principal Display Panel – 300 mg Carton Label
NDC: 73462-101-01
Rx only
Cosela®
(trilaciclib)
for injection300 mg/vial*
For Intravenous Infusion
After Reconstitution
and DilutionOne Single-Dose Vial
Discard Unused Portion
-
PRINCIPAL DISPLAY PANEL
Principal Display Panel – 300 mg Vial Label
NDC: 73462-101-01
Rx only
Cosela®
(trilaciclib)
for injection300 mg/vial*
For Intravenous Infusion After
Reconstitution and DilutionSingle-Dose Vial
-
INGREDIENTS AND APPEARANCE
COSELA
trilaciclib injection, powder, lyophilized, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 73462-101 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength trilaciclib dihydrochloride (UNII: 4BX07W725T) (trilaciclib - UNII:U6072DO9XG) trilaciclib 300 mg in 20 mL Inactive Ingredients Ingredient Name Strength citric acid monohydrate (UNII: 2968PHW8QP) mannitol (UNII: 3OWL53L36A) sodium hydroxide (UNII: 55X04QC32I) hydrochloric acid (UNII: QTT17582CB) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 73462-101-01 1 in 1 CARTON 02/12/2021 12/31/2028 1 20 mL in 1 VIAL, GLASS; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA214200 02/12/2021 12/31/2028 Labeler - G1 Therapeutics, Inc. (828732813)

Trademark Results [COSELA]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 COSELA 90455987 not registered Live/Pending |
G1 Therapeutics, Inc. 2021-01-08 |
 COSELA 90455982 not registered Live/Pending |
G1 Therapeutics, Inc. 2021-01-08 |
 COSELA 88942707 not registered Live/Pending |
Yiwu MiLa Trading Co., Ltd. 2020-06-01 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.