OZURDEX- dexamethasone implant
OZURDEX by
Drug Labeling and Warnings
OZURDEX by is a Prescription medication manufactured, distributed, or labeled by Allergan, Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use OZURDEX® safely and effectively. See full prescribing information for OZURDEX®.
OZURDEX® (dexamethasone intravitreal implant)
For Intravitreal Injection
Initial U.S. Approval: 1958
INDICATIONS AND USAGE
OZURDEX® is a corticosteroid indicated for:
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
Intravitreal implant containing dexamethasone 0.7 mg in the NOVADUR® solid polymer drug delivery system. (3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- Intravitreal injections have been associated with endophthalmitis, eye inflammation, increased intraocular pressure, and retinal detachments. Patients should be monitored following the injection. (5.1)
- Use of corticosteroids may produce posterior subcapsular cataracts, increased intraocular pressure, glaucoma, and may enhance the establishment of secondary ocular infections due to bacteria, fungi, or viruses. (5.2)
ADVERSE REACTIONS
In controlled studies, the most common adverse reactions reported by 20–70% of patients were cataract, increased intraocular pressure and conjunctival hemorrhage. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Allergan at 1-800-678-1605 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.See 17 for PATIENT COUNSELING INFORMATION.
Revised: 5/2018
- Intravitreal injections have been associated with endophthalmitis, eye inflammation, increased intraocular pressure, and retinal detachments. Patients should be monitored following the injection. (5.1)
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Retinal Vein Occlusion
1.2 Posterior Segment Uveitis
1.3 Diabetic Macular Edema
2 DOSAGE AND ADMINISTRATION
2.1 General Dosing Information
2.2 Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
4.1 Ocular or Periocular Infections
4.2 Glaucoma
4.3 Torn or Ruptured Posterior Lens Capsule
4.4 Hypersensitivity
5 WARNINGS AND PRECAUTIONS
5.1 Intravitreal Injection-related Effects
5.2 Steroid-related Effects
6 ADVERSE REACTIONS
6.1 Clinical Studies Experience
6.2 Postmarketing Experience
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1
INDICATIONS AND USAGE
1.1 Retinal Vein Occlusion
OZURDEX® (dexamethasone intravitreal implant) is indicated for the treatment of macular edema following branch retinal vein occlusion (BRVO) or central retinal vein occlusion (CRVO).
-
2
DOSAGE AND ADMINISTRATION
2.2 Administration
The intravitreal injection procedure should be carried out under controlled aseptic conditions which include the use of sterile gloves, a sterile drape, and a sterile eyelid speculum (or equivalent). Adequate anesthesia and a broad-spectrum microbicide applied to the periocular skin, eyelid and ocular surface are recommended to be given prior to the injection.
Remove the foil pouch from the carton and examine for damage. Then, open the foil pouch over a sterile field and gently drop the applicator on a sterile tray. Carefully remove the cap from the applicator. Hold the applicator in one hand and pull the safety tab straight off the applicator. Do not twist or flex the tab. The long axis of the applicator should be held parallel to the limbus, and the sclera should be engaged at an oblique angle with the bevel of the needle up (away from the sclera) to create a shelved scleral path. The tip of the needle is advanced within the sclera for about 1 mm (parallel to the limbus), then re-directed toward the center of the eye and advanced until penetration of the sclera is completed and the vitreous cavity is entered. The needle should not be advanced past the point where the sleeve touches the conjunctiva.
Slowly depress the actuator button until an audible click is noted. Before withdrawing the applicator from the eye, make sure that the actuator button is fully depressed and has locked flush with the applicator surface. Remove the needle in the same direction as used to enter the vitreous.
Following the intravitreal injection, patients should be monitored for elevation in intraocular pressure and for endophthalmitis. Monitoring may consist of a check for perfusion of the optic nerve head immediately after the injection, tonometry within 30 minutes following the injection, and biomicroscopy between two and seven days following the injection. Patients should be instructed to report any symptoms suggestive of endophthalmitis without delay.
Each applicator can only be used for the treatment of a single eye. If the contralateral eye requires treatment, a new applicator must be used, and the sterile field, syringe, gloves, drapes, and eyelid speculum should be changed before OZURDEX® is administered to the other eye.
- 3 DOSAGE FORMS AND STRENGTHS
-
4
CONTRAINDICATIONS
4.1 Ocular or Periocular Infections
OZURDEX® (dexamethasone intravitreal implant) is contraindicated in patients with active or suspected ocular or periocular infections including most viral diseases of the cornea and conjunctiva, including active epithelial herpes simplex keratitis (dendritic keratitis), vaccinia, varicella, mycobacterial infections, and fungal diseases.
4.2 Glaucoma
OZURDEX® is contraindicated in patients with glaucoma, who have cup to disc ratios of greater than 0.8.
4.3 Torn or Ruptured Posterior Lens Capsule
OZURDEX® is contraindicated in patients whose posterior lens capsule is torn or ruptured because of the risk of migration into the anterior chamber. Laser posterior capsulotomy in pseudophakic patients is not a contraindication for OZURDEX® use.
4.4 Hypersensitivity
OZURDEX® is contraindicated in patients with known hypersensitivity to any components of this product [see Adverse Reactions (6)].
-
5
WARNINGS AND PRECAUTIONS
5.1 Intravitreal Injection-related Effects
Intravitreal injections, including those with OZURDEX®, have been associated with endophthalmitis, eye inflammation, increased intraocular pressure, and retinal detachments. Patients should be monitored regularly following the injection [see Patient Counseling Information (17)].
5.2 Steroid-related Effects
Use of corticosteroids including OZURDEX® may produce posterior subcapsular cataracts, increased intraocular pressure, and glaucoma. Use of corticosteroids may enhance the establishment of secondary ocular infections due to bacteria, fungi, or viruses [see Adverse Reactions (6.1)].
Corticosteroids are not recommended to be used in patients with a history of ocular herpes simplex because of the potential for reactivation of the viral infection.
-
6
ADVERSE REACTIONS
6.1 Clinical Studies Experience
Because clinical studies are conducted under widely varying conditions, adverse reaction rates observed in the clinical studies of a drug cannot be directly compared to rates in the clinical studies of another drug and may not reflect the rates observed in practice.
Adverse reactions associated with ophthalmic steroids including OZURDEX® include elevated intraocular pressure, which may be associated with optic nerve damage, visual acuity and field defects, posterior subcapsular cataract formation, secondary ocular infection from pathogens including herpes simplex, and perforation of the globe where there is thinning of the cornea or sclera.
Retinal Vein Occlusion and Posterior Segment Uveitis
The following information is based on the combined clinical trial results from 3 initial, randomized, 6-month, sham-controlled studies (2 for retinal vein occlusion and 1 for posterior segment uveitis):
Table 1: Adverse Reactions Reported by Greater than 2% of Patients MedDRA Term
OZURDEX®
N=497 (%)Sham
N=498 (%)Intraocular pressure increased 125 (25%) 10 (2%) Conjunctival hemorrhage 108 (22%) 79 (16%) Eye pain 40 (8%) 26 (5%) Conjunctival hyperemia 33 (7%) 27 (5%) Ocular hypertension 23 (5%) 3 (1%) Cataract 24 (5%) 10 (2%) Vitreous detachment 12 (2%) 8 (2%) Headache 19 (4%) 12 (2%) Increased IOP with OZURDEX® peaked at approximately week 8. During the initial treatment period, 1% (3/421) of the patients who received OZURDEX® required surgical procedures for management of elevated IOP.
Following a second injection of OZURDEX® in cases where a second injection was indicated, the overall incidence of cataracts was higher after 1 year.
In a 2 year observational study, among patients who received >2 injections, the most frequent adverse reaction was cataract 54% (n= 96 out of 178 phakic eyes at baseline). Other frequent adverse reactions from the 283 treated eyes, regardless of lens status at baseline, were increased IOP 24% (n = 68) and vitreous hemorrhage 6.0% (n = 17).
Diabetic Macular Edema
The following information is based on the combined clinical trial results from 2 randomized, 3-year, sham-controlled studies in patients with diabetic macular edema. Discontinuation rates due to the adverse reactions listed in Table 2 were 3% in the OZURDEX® group and 1% in the Sham group. The most common ocular (study eye) and non-ocular adverse reactions are shown in Tables 2 and 3:
Table 2: Ocular Adverse Reactions Reported by ≥ 1% of Patients and Non-ocular Adverse Reactions Reported by ≥ 5% of Patients MedDRA Term
OZURDEX®
N=324 (%)Sham
N=328 (%)Ocular Cataract1 166/2432 (68%) 49/230 (21%) Conjunctival hemorrhage 73 (23%) 44 (13%) Visual acuity reduced 28 (9%) 13 (4%) Conjunctivitis 19 (6%) 8 (2%) Vitreous floaters 16 (5%) 6 (2%) Conjunctival edema 15 (5%) 4 (1%) Dry eye 15 (5%) 7 (2%) Vitreous detachment 14 (4%) 8 (2%) Vitreous opacities 11 (3%) 3 (1%) Retinal aneurysm 10 (3%) 5 (2%) Foreign body sensation 7 (2%) 4 (1%) Corneal erosion 7 (2%) 3 (1%) Keratitis 6 (2%) 3 (1%) Anterior Chamber Inflammation 6 (2%) 0 (0%) Retinal tear 5 (2%) 2 (1%) Eyelid ptosis 5 (2%) 2 (1%) Non-ocular Hypertension 41 (13%) 21 (6%) Bronchitis 15 (5%) 8 (2%) 1 Includes cataract, cataract nuclear, cataract subcapsular, lenticular opacities in patients who were phakic at baseline. Among these patients, 61% of OZURDEX® subjects vs. 8% of sham-controlled subjects underwent cataract surgery.
2 243 of the 324 OZURDEX® subjects were phakic at baseline; 230 of 328 sham-controlled subjects were phakic at baseline.
Increased Intraocular Pressure
Table 3: Summary of Elevated Intraocular Pressure (IOP) Related Adverse Reactions IOP Treatment: N (%) OZURDEX®
N=324Sham
N=328IOP elevation ≥10 mm Hg from Baseline at any visit 91 (28%) 13 (4%) ≥30 mm Hg IOP at any visit 50 (15%) 5 (2%) Any IOP lowering medication 136 (42%) 32 (10%) Any surgical intervention for elevated IOP * 4 (1.2%) 1 (0.3%) * OZURDEX®: 1 surgical trabeculectomy for steroid-induced IOP increase, 1 surgical trabeculectomy for iris neovascularization,
1 laser iridotomy, 1 surgical iridectomy
Sham: 1 laser iridotomy
The increase in mean IOP was seen with each treatment cycle, and the mean IOP generally returned to baseline between treatment cycles (at the end of the 6 month period) shown below:
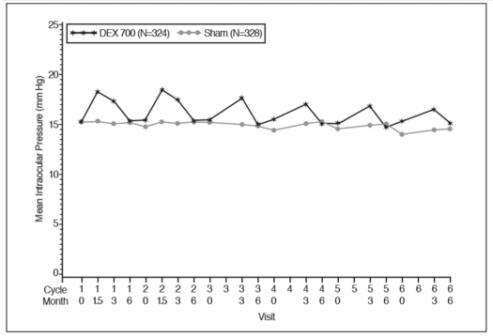
Figure 1: Mean IOP during the study
Cataracts and Cataract Surgery
At baseline, 243 of the 324 OZURDEX® subjects were phakic; 230 of 328 sham-controlled subjects were phakic. The incidence of cataract development in patients who had a phakic study eye was higher in the OZURDEX® group (68%) compared with Sham (21%). The median time of cataract being reported as an adverse event was approximately 15 months in the OZURDEX® group and 12 months in the Sham group. Among these patients, 61% of OZURDEX® subjects vs. 8% of sham-controlled subjects underwent cataract surgery, generally between Month 18 and Month 39 (Median Month 21 for OZURDEX® group and 20 for Sham) of the studies.
6.2 Postmarketing Experience
The following reactions have been identified during post-marketing use of OZURDEX® in clinical practice. Because they are reported voluntarily from a population of unknown size, estimates of frequency cannot be made. The reactions, which have been chosen for inclusion due to either their seriousness, frequency of reporting, possible causal connection to OZURDEX®, or a combination of these factors, include: complication of device insertion (implant misplacement), device dislocation with or without corneal edema, endophthalmitis, hypotony of the eye (associated with vitreous leakage due to injection), and retinal detachment.
-
8
USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no adequate and well-controlled studies with OZURDEX® in pregnant women. Topical ocular administration of dexamethasone in mice and rabbits during the period of organogenesis produced cleft palate and embryofetal death in mice, and malformations of the abdominal wall/intestines and kidneys in rabbits at doses 5 and 4 times higher than the recommended human ophthalmic dose (RHOD) of OZURDEX® (0.7 milligrams dexamethasone), respectively.
In the US general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Data
Animal Data
Topical ocular administration of 0.15% dexamethasone (0.75 mg/kg/day) on gestational days 10 to 13 produced embryofetal lethality and a high incidence of cleft palate in mice. A dose of 0.75 mg/kg/day in the mouse is approximately 5 times an OZURDEX® injection in humans (0.7 mg dexamethasone) on a mg/m2 basis. In rabbits, topical ocular administration of 0.1% dexamethasone throughout organogenesis (0.20 mg/kg/day, on gestational day 6 followed by 0.13 mg/kg/day on gestational days 7-18) produced intestinal anomalies, intestinal aplasia, gastroschisis and hypoplastic kidneys. A dose of 0.13 mg/kg/day in the rabbit is approximately 4 times an OZURDEX® injection in humans (0.7 mg dexamethasone) on a mg/m2 basis. A no-observed-adverse-effect-level (NOAEL) was not identified in the mouse or rabbits studies.
8.2 Lactation
Risk Summary
Systemically administered corticosteroids are present in human milk and can suppress growth and interfere with endogenous corticosteroid production or cause other unwanted effects. There is no information regarding the presence of dexamethasone in human milk, the effects on the breastfed infants, or the effects on milk producton to inform risk of OZURDEX® to an infant during lactation. The developmental and health benefits of breastfeeding should be considered, along with the mother’s clinical need for OZURDEX® and any potential adverse effects on the breastfed child from OZURDEX®.
-
11
DESCRIPTION
OZURDEX® is an intravitreal implant containing 0.7 mg (700 mcg) dexamethasone in the NOVADUR® solid polymer sustained-release drug delivery system. OZURDEX® is preloaded into a single-use, DDS® applicator to facilitate injection of the rod-shaped implant directly into the vitreous. The NOVADUR® system contains poly (D,L-lactide-co-glycolide) PLGA intravitreal polymer matrix without a preservative. The chemical name for dexamethasone is Pregna-1,4-diene-3,20-dione, 9-fluoro-11,17,21-trihydroxy-16-methyl-, (11β,16α)-. Its structural formula is:
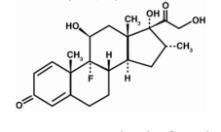
MW 392.47; molecular formula: C22H29FO5
Dexamethasone occurs as a white to cream-colored crystalline powder having not more than a slight odor, and is practically insoluble in water and very soluble in alcohol.
The PLGA matrix slowly degrades to lactic acid and glycolic acid.
-
12
CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Dexamethasone, a corticosteroid, has been shown to suppress inflammation by inhibiting multiple inflammatory cytokines resulting in decreased edema, fibrin deposition, capillary leakage and migration of inflammatory cells.
12.3 Pharmacokinetics
Plasma concentrations were obtained from 21 patients with macular edema due to branch retinal vein occlusion (BRVO) and central retinal vein occlusion (CRVO), and 21 patients with diabetic macular edema (DME) prior to dosing and at 4 to 5 additional post-dose timepoints on Days 1, 7, 21, 30, 45, 60, and 90 following the administration of the first intravitreal implant containing 0.7 mg dexamethasone. In RVO and DME patients, the majority of plasma dexamethasone concentrations were below the lower limit of quantitation (LLOQ = 50 pg/mL). Plasma dexamethasone concentrations from 12% of samples were above the LLOQ, ranging from 52 pg/mL to 102 pg/mL. Plasma dexamethasone concentration did not appear to be related to age, body weight, or sex of patients.
In an in vitro metabolism study, following the incubation of [14C]-dexamethasone with human cornea, iris-ciliary body, choroid, retina, vitreous humor, and sclera tissues for 18 hours, no metabolites were observed.
- 13 NONCLINICAL TOXICOLOGY
-
14
CLINICAL STUDIES
Retinal Vein Occlusion
The efficacy of OZURDEX® for the treatment of macular edema following branch retinal vein occlusion (BRVO) or central retinal vein occlusion (CRVO) was assessed in two, multicenter, double-masked, randomized, parallel studies.
Following a single injection, OZURDEX® demonstrated the following clinical results for the percent of patients with ≥ 15 letters of improvement from baseline in best-corrected visual acuity (BCVA):
Table 4: Number (Percent) of Patients with ≥ 15 Letters Improvement from Baseline in BCVA Study Day Study 1 Study 2 OZURDEX® N=201 Sham
N=202p-value* OZURDEX® N=226 Sham
N=224p-value* Day 30 40 (20%) 15 (7%) < 0.01 51 (23%) 17 (8%) < 0.01 Day 60 58 (29%) 21 (10%) < 0.01 67 (30%) 27 (12%) < 0.01 Day 90 45 (22%) 25 (12%) < 0.01 48 (21%) 31 (14%) 0.039 Day 180 39 (19%) 37 (18%) 0.780 53 (24%) 38 (17%) 0.087 *P-values were based on the Pearson’s chi-square test.
In each individual study and in a pooled analysis, time to achieve ≥ 15 letters (3-line) improvement in BCVA cumulative response rate curves were significantly faster with OZURDEX® compared to sham (p < 0.01), with OZURDEX® treated patients achieving a 3-line improvement in BCVA earlier than sham-treated patients.
The onset of a ≥ 15 letter (3-line) improvement in BCVA with OZURDEX® occurs within the first two months after implantation in approximately 20-30% of subjects. The duration of effect persists approximately one to three months after onset of this effect.
Posterior Segment Uveitis
The efficacy of OZURDEX® was assessed in a single, multicenter, masked, randomized study of 153 patients with non-infectious uveitis affecting the posterior segment of the eye.
After a single injection, the percent of patients reaching a vitreous haze score of 0 (where a score of 0 represents no inflammation) was statistically significantly greater for patients receiving OZURDEX® versus sham at week 8 (primary time point) (47% versus 12%). The percent of patients achieving a 3-line improvement from baseline BCVA was 43% for patients receiving OZURDEX® versus 7% for sham at week 8.
Diabetic Macular Edema
The efficacy of OZURDEX® for the treatment of diabetic macular edema was assessed in two, multicenter, masked, randomized, sham-controlled studies. Subjects were to be evaluated for retreatment eligibility every three months starting from Month 6 but could only receive successive treatments at least 6 months apart. Retreatment was based on physician’s discretion after examination including Optical Coherence Tomography. Patients in the OZURDEX® arm received an average of 4 treatments during the 36 months.
The primary endpoint was the proportion of patients with 15 or more letters improvement in BCVA from baseline at Month 39 or final visit for subjects who exited the study at or prior to Month 36. The Month 39 extension was included to accommodate the evaluation of safety and efficacy outcomes for subjects who received re-treatment at Month 36. Only fourteen percent of the study patients completed the Month 39 visit (16.8% from OZURDEX® and 12.2% from Sham).
Table 5: Visual Acuity outcomes at Month 39 (All randomized subjects with LOCFc) Study Outcomes OZURDEX® Sham
Estimated Difference (95% CI) 1a
Mean (SD) Baseline BCVA (Letters) 56 (10)
57 (9)
Median (range) Baseline BCVA (Letters) 59 (34-95) 58 (34-74) Gain of ≥15 letters in BCVA (n(%)) 34 (21%) 19 (12%) 9.3% (1.4%, 17.3%) Loss of ≥15 letters in BCVA (n(%)) 15 (9%) 17 (10%) -1.1% (-7.5%, 5.3%) Mean change in BCVA (SD) 4.1 (13.9) 0.9 (11.9) 3.2 (0.4, 5.9) 2b
Mean (SD) Baseline BCVA (Letters) 55 (10)
56 (9)
Median (range) Baseline BCVA (Letters) 58 (34-72) 58 (36-82) Gain of ≥15 letters in BCVA (n(%)) 30 (18%) 16 (10%) 8.4% (0.9%, 15.8%) Loss of ≥15 letters in BCVA (n(%)) 30 (18%) 18 (11%) 7.1% (-0.5%, 14.7%) Mean change in BCVA (SD) 0.4 (17.5) 0.8 (13.6) -0.7 (-4.1, 2.6) aStudy 1: OZURDEX®, N=163; Sham, N=165
bStudy 2: OZURDEX®, N=165; Sham, N=163
c14% (16.8% from OZURDEX® and 12.2% from Sham) of patients had BCVA outcome at Month 39, for the remaining patients, the data at Month 36 or earlier was carried forward.
Visual acuity outcomes by lens status (Phakic or Pseudophakic) at different visits are presented in Figure 2 and Figure 3. The occurrence of cataracts impacted visual acuity during the study. The visual acuity improvement from baseline increases during a treatment cycle, peaks at approximately 3 Months posttreatment and diminishes thereafter. Patients who were pseudophakic at baseline achieved greater mean BCVA change from baseline at the final study visit.
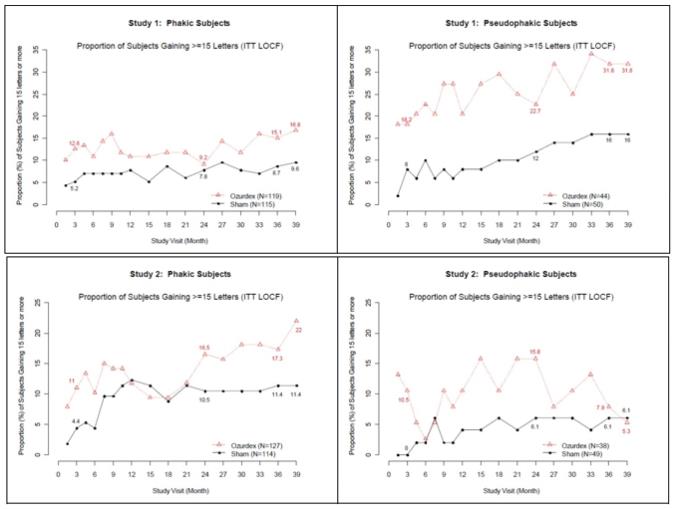
Figure 2: Proportion of Subjects with ≥ 15 Letters Improvement from Baseline BCVA in the Study Eye
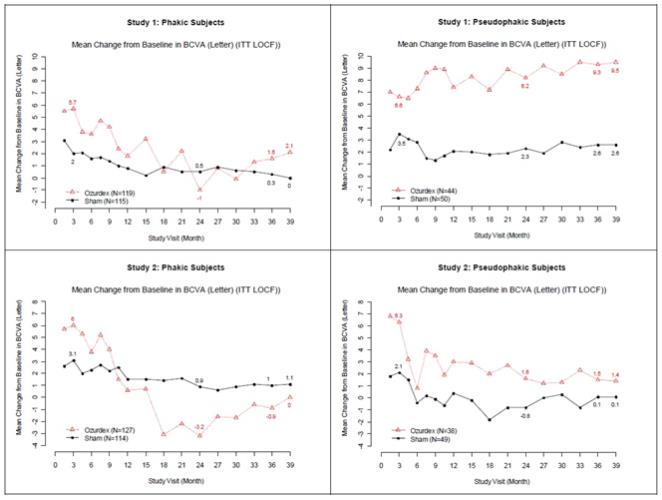
Figure 3: Mean BCVA Change from Baseline
The best corrected visual acuity outcomes for the Pseudophakic and Phakic subgroups from Studies 1 and 2 at Month 39 are presented in Table 6.
Table 6: Visual Acuity outcomes at Month 39 (Subgroup for pooled data with LOCFc) Subgroup
(Pooled)Outcomes OZURDEX® Sham
Estimated Difference (95% CI) aPseudophakic Gain of ≥15 letters in BCVA (n(%)) 16 (20%) 11 (11%) 8.4%
(-2.2%, 19.0%)Loss of ≥15 letters in BCVA (n(%)) 4 (5%) 7 (7%) -2.2% (-9.1%, 4.7%) Mean change in BCVA (SD) 5.8 (11.6) 1.4 (12.3) 4.2 (0.8, 7.6) bPhakic Gain of ≥15 letters in BCVA (n(%)) 48 (20%) 24 (11%) 9.0% (2.7%, 15.4%) Loss of ≥15 letters in BCVA (n(%)) 41 (17%) 28 (12%) 4.4% (-1.9%, 10.7%) Mean change in BCVA (SD) 1.0 (16.9) 0.6 (12.9) 0.3 (-2.4, 3.0) a Pseudophakic: OZURDEX®, N=82; Sham, N=99
b Phakic: OZURDEX®, N=246; Sham, N=229
c14% (16.8% from OZURDEX® and 12.2% from Sham) of patients had BCVA outcome at Month 39, for the remaining patients the data at Month 36 or earlier was used in the analysis.
-
16
HOW SUPPLIED/STORAGE AND HANDLING
OZURDEX® (dexamethasone intravitreal implant) 0.7 mg is supplied in a foil pouch with 1 single-use plastic applicator, NDC: 0023-3348-07.
Storage: Store at 15o-30oC (59o-86oF).
-
17
PATIENT COUNSELING INFORMATION
Steroid-related Effects
Advise patients that a cataract may occur after repeated treatment with OZURDEX®. If this occurs, advise patients that their vision will decrease, and they will need an operation to remove the cataract and restore their vision.
Advise patients that they may develop increased intraocular pressure with OZURDEX® treatment, and
the increased IOP will need to be managed with eye drops, and, rarely, with surgery.
Intravitreal Injection-related Effects
Advise patients that in the days following intravitreal injection of OZURDEX®, patients are at risk for potential complications including in particular, but not limited to, the development of endophthalmitis or elevated intraocular pressure.
When to Seek Physician Advice
Advise patients that if the eye becomes red, sensitive to light, painful, or develops a change in vision, they should seek immediate care from an ophthalmologist.
Driving and Using Machines
Inform patients that they may experience temporary visual blurring after receiving an intravitreal injection. Advise patients not to drive or use machines until this has been resolved.
Distributed by: Allergan USA, Inc.
Madison, NJ 07940© 2018 Allergan. All rights reserved.
All trademarks are the property of their respective owners.
Patented. See: www.allergan.com/patentsv1.0USPI3348
-
PRINCIPAL DISPLAY PANEL
NDC: 0023-3348-07
Ozurdex
(dexamethasone
intravitreal implant) 0.7 mg
For Intravitreal Injection Only
Rx Only
-
INGREDIENTS AND APPEARANCE
OZURDEX
dexamethasone implantProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0023-3348 Route of Administration INTRAVITREAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength DEXAMETHASONE (UNII: 7S5I7G3JQL) (DEXAMETHASONE - UNII:7S5I7G3JQL) DEXAMETHASONE 0.7 mg Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0023-3348-07 1 in 1 CARTON 09/01/2009 1 1 in 1 POUCH; Type 0: Not a Combination Product 2 NDC: 0023-3348-08 1 in 1 CARTON 09/01/2009 2 1 in 1 POUCH; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA022315 09/01/2009 Labeler - Allergan, Inc. (144796497)
Trademark Results [OZURDEX]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 OZURDEX 77717722 3773304 Live/Registered |
Allergan, Inc. 2009-04-20 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.