Deferasirox by Xiromed, LLC / Piramal Healthcare UK Limited / Piramal Pharma Limited DEFERASIROX tablet
Deferasirox by
Drug Labeling and Warnings
Deferasirox by is a Prescription medication manufactured, distributed, or labeled by Xiromed, LLC, Piramal Healthcare UK Limited, Piramal Pharma Limited. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use DEFERASIROX TABLETS safely and effectively. See full prescribing information for DEFERASIROX TABLETS.
DEFERASIROX tablets, for oral use.
Initial U.S. Approval: 2005
WARNING: RENAL FAILURE, HEPATIC FAILURE, AND GASTROINTESTINAL HEMORRHAGE
See full prescribing information for complete boxed warning.
Deferasirox may cause serious and fatal:
- acute kidney injury, including acute renal failure requiring dialysis and renal tubular toxicity including Fanconi syndrome (5.1)
- hepatic toxicity, including failure (5.2)
- gastrointestinal hemorrhage (5.3)
Deferasirox therapy requires close patient monitoring, including laboratory tests of renal and hepatic function. (5)
RECENT MAJOR CHANGES
Indications and Usage, Limitations of Use (1.3) 7/2019
INDICATIONS AND USAGE
Deferasirox is an iron chelator indicated for the treatment of chronic iron overload due to blood transfusions in patients 2 years of age and older. (1.1)
Deferasirox tablets are indicated for the treatment of chronic iron overload in patients 10 years of age and older with non-transfusion-dependent thalassemia (NTDT) syndromes, and with a liver iron (Fe) concentration (LIC) of at least 5 mg Fe per gram of dry weight (Fe/g dw) and a serum ferritin greater than 300 mcg/L. This indication is approved under accelerated approval based on a reduction of liver iron concentrations (to less than 5 mg Fe/g dw) and serum ferritin levels. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials. (1.2)
Limitations of Use:
The safety and efficacy of deferasirox when administered with other iron chelation therapy have not been established. (1.3)
DOSAGE AND ADMINISTRATION
- Transfusional Iron Overload: Initial dose for patients with estimated glomerular filtration rate (eGFR) greater than 60 mL/min/1.73 m2 is 14 mg per kg (calculated to nearest whole tablet) once daily. (2.1)
- NTDT Syndromes: Initial dose for patients with eGFR greater than 60 mL/min/1.73 m2 is 7 mg per kg (calculated to nearest whole tablet) once daily. (2.2)
- See Full Prescribing information for information regarding monitoring, administration, and dose-reductions for organ impairment. (2.1, 2.2, 2.3, 2.4)
DOSAGE FORMS AND STRENGTHS
Tablets: 90 mg, 360 mg. (3)
CONTRAINDICATIONS
- Estimated GFR less than 40 mL/min/1.73 m2(4)
- Patients with poor performance status. (4)
- Patients with high-risk MDS. (4)
- Patients with advanced malignancies. (4)
- Patients with platelet counts less than 50 x 109/L. (4)
- Known hypersensitivity to deferasirox or any component of Deferasirox tablets. (4)
WARNINGS AND PRECAUTIONS
- Acute Kidney Injury: Measure serum creatinine in duplicate before starting therapy. Monitor renal function during deferasirox therapy and reduce dose or interrupt therapy for toxicity. (2.1, 2.4, 5.1)
- Hepatic Toxicity: Monitor hepatic function. Reduce dose or interrupt therapy for toxicity. (5.2)
- Fatal and Nonfatal Gastrointestinal Bleeding, Ulceration, and Irritation: Risk may be greater in patients who are taking deferasirox in combination with drugs that have known ulcerogenic or hemorrhagic potential. (5.3)
- Bone Marrow Suppression: Neutropenia, agranulocytosis, worsening anemia, and thrombocytopenia, including fatal events; monitor blood counts during deferasirox therapy. Interrupt therapy for toxicity. (5.4)
- Age-related Risk of Toxicity: Monitor elderly and pediatric patients closely for toxicity. (5.5)
- Hypersensitivity Reactions: Discontinue deferasirox for severe reactions and institute medical intervention. (5.7)
- Severe Skin Reactions including Stevens-Johnson Syndrome (SJS), toxic epidermal necrolysis (TEN) and drug reaction with eosinophilia and systemic symptoms (DRESS): Discontinue deferasirox. (5.8)
ADVERSE REACTIONS
In patients with transfusional iron overload, the most frequently occurring (greater than 5%) adverse reactions are diarrhea, vomiting, nausea, abdominal pain, skin rashes, and increases in serum creatinine. In deferasirox-treated patients with NTDT syndromes, the most frequently occurring (greater than 5%) adverse reactions are diarrhea, rash and nausea. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Xiromed, LLC. at 1-844-XIROMED (1-844-947-6633) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch .
DRUG INTERACTIONS
- Do not take deferasirox with aluminum-containing antacid preparations. (7.1)
- Deferasirox increases the exposure of repaglinide. Consider repaglinide dose reduction and monitor blood glucose levels. (7.3)
- Avoid the use of deferasirox with theophylline as theophylline levels could be increased. (7.4)
- Deferasirox increases exposure of busulfan. Monitor plasma concentrations of busulfan when coadministered with deferasirox to allow dose adjustment of busulfan, as needed. (7.7)
USE IN SPECIFIC POPULATIONS
- Lactation: Advise women not to breastfeed. (8.2)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 4/2020
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: RENAL FAILURE, HEPATIC FAILURE, AND GASTROINTESTINAL HEMORRHAGE
1 INDICATIONS & USAGE
1.1 Treatment of Chronic Iron Overload Due to Blood Transfusions (Transfusional Iron Overload)
1.2 Treatment of Chronic Iron Overload in Non-Transfusion-Dependent Thalassemia Syndromes
1.3 Limitations of Use
2 DOSAGE & ADMINISTRATION
2.1 Transfusional Iron Overload
2.2 Iron Overload in Non-Transfusion-Dependent Thalassemia Syndromes
2.3 Administration
2.4 Use in Patients with Baseline Hepatic or Renal Impairment
2.5 Dose Modifications for Decreases in Renal Function while on Deferasirox
2.6 Dose Modifications Based on Concomitant Medications
3 DOSAGE FORMS & STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Acute Kidney Injury, Including Acute Renal Failure Requiring Dialysis and Renal Tubular Toxicity Including Fanconi Syndrome
5.2 Hepatic Toxicity and Failure
5.3 Gastrointestinal (GI) Ulceration, Hemorrhage, and Perforation
5.4 Bone Marrow Suppression
5.5 Age-Related Risk of Toxicity
5.6 Overchelation
5.7 Hypersensitivity
5.8 Severe Skin Reactions
5.9 Skin Rash
5.10 Auditory and Ocular Abnormalities
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Aluminum-Containing Antacid Preparations
7.2 Agents Metabolized by CYP3A4
7.3 Agents Metabolized by CYP2C8
7.4 Agents Metabolized by CYP1A2
7.5 Agents Inducing UDP-glucuronosyltransferase (UGT) Metabolism
7.6 Bile Acid Sequestrants
7.7 Busulfan
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: RENAL FAILURE, HEPATIC FAILURE, AND GASTROINTESTINAL HEMORRHAGE
Renal Failure
- Deferasirox can cause acute renal failure and death, particularly in patients with comorbidities and those who are in the advanced stages of their hematologic disorders.
- Evaluate baseline renal function prior to starting or increasing deferasirox dosing in all patients. Deferasirox is contraindicated in adult and pediatric patients with eGFR less than 40 mL/min/1.73 m2. Measure serum creatinine in duplicate prior to initiation of therapy. Monitor renal function at least monthly. For patients with baseline renal impairment or increased risk of acute renal failure, monitor renal function weekly for the first month, then at least monthly. Reduce the starting dose in patients with preexisting renal disease. During therapy, increase the frequency of monitoring and modify the dose for patients with an increased risk of renal impairment, including use of concomitant nephrotoxic drugs, and pediatric patients with volume depletion or overchelation [see Dosage and Administration (2.1, 2.4, 2.5), Warnings and Precautions (5.1) Adverse Reactions (6.1, 6.2)].
Hepatic Failure
- Deferasirox can cause hepatic injury including hepatic failure and death.
- Measure serum transaminases and bilirubin in all patients prior to initiating treatment, every 2 weeks during the first month, and at least monthly thereafter.
- Avoid use of deferasirox in patients with severe (Child-Pugh C) hepatic impairment and reduce the dose in patients with moderate (Child-Pugh B) hepatic impairment [see Dosage and Administration (2.4), Warnings and Precautions (5.2)].
Gastrointestinal Hemorrhage
- Deferasirox can cause gastrointestinal (GI) hemorrhages, which may be fatal, especially in elderly patients who have advanced hematologic malignancies and/or low platelet counts.
- Monitor patients and discontinue deferasirox for suspected GI ulceration or hemorrhage [see Warnings and Precautions (5.3)].
-
1 INDICATIONS & USAGE
1.1 Treatment of Chronic Iron Overload Due to Blood Transfusions (Transfusional Iron Overload)
Deferasirox tablets are indicated for the treatment of chronic iron overload due to blood transfusions (transfusional hemosiderosis) in patients 2 years of age and older.
1.2 Treatment of Chronic Iron Overload in Non-Transfusion-Dependent Thalassemia Syndromes
Deferasirox tablets are indicated for the treatment of chronic iron overload in patients 10 years of age and older with non-transfusion-dependent thalassemia (NTDT) syndromes and with a liver iron concentration (LIC) of at least 5 milligrams of iron per gram of liver dry weight (mg Fe/g dw) and a serum ferritin greater than 300 mcg/L. This indication is approved under accelerated approval based on a reduction of liver iron concentrations (to less than 5 mg Fe/g dw) and serum ferritin levels [see Clinical Studies (14)]. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
-
2 DOSAGE & ADMINISTRATION
2.1 Transfusional Iron Overload
Deferasirox therapy should only be considered when a patient has evidence of chronic transfusional iron overload. The evidence should include the transfusion of at least 100 mL/kg of packed red blood cells (e.g., at least 20 units of packed red blood cells for a 40 kg person or more in individuals weighing more than 40 kg), and a serum ferritin consistently greater than 1000 mcg/L.
Prior to starting therapy, or increasing dose, evaluate:
- Serum ferritin level
- Baseline renal function:
- Obtain serum creatinine in duplicate (due to variations in measurements)
- Calculate the estimated glomerular filtration rate (eGFR). Use a prediction equation appropriate for adult patients (e.g., CKD-EPI, MDRD method) and in pediatric patients (e.g., Schwartz equations).
- Obtain urinalyses and serum electrolytes to evaluate renal tubular function [see Dosage and Administration (2.4), Warnings and Precautions (5.1)].
- Serum transaminases and bilirubin [see Dosage and Administration (2.4), Warnings and Precautions (5.2)]
- Baseline auditory and ophthalmic examinations [see Warnings and Precautions (5.10)]
Initiating Therapy:
The recommended initial dose of deferasirox for patients 2 years of age and older with eGFR greater than 60 mL/min/1.73 m2 is 14 mg per kg body weight orally, once daily. Calculate doses (mg per kg per day) to the nearest whole tablet. Changes in weight of pediatric patients over time must be taken into account when calculating the dose.
During Therapy:
- Monitor serum ferritin monthly and adjust the dose of deferasirox, if necessary, every 3 to 6 months based on serum ferritin trends.
- Use the minimum effective dose to achieve a trend of decreasing ferritin
- Make dose adjustments in steps of 3.5 or 7 mg per kg and tailor adjustments to the individual patient’s response and therapeutic goals.
- In patients not adequately controlled with doses of 21 mg per kg (e.g., serum ferritin levels persistently above 2500 mcg/L and not showing a decreasing trend over time), doses of up to 28 mg per kg may be considered. Doses above 28 mg per kg are not recommended [see Warnings and Precautions (5.6)].
- Adjust dose based on serum ferritin levels
- If the serum ferritin falls below 1000 mcg/L at 2 consecutive visits, consider dose reduction, especially if the deferasirox dose is greater than 17.5 mg/kg/day [see Adverse Reactions (6.1)].
- If the serum ferritin falls below 500 mcg/L, interrupt deferasirox therapy and continue monthly monitoring.
- Evaluate the need for ongoing chelation therapy for patients whose conditions no longer require regular blood transfusions.
- Use the minimum effective dose to maintain iron burden in the target range [see Warnings and Precautions (5.6)].
- Monitor blood counts, liver function, renal function and ferritin monthly [see Warnings and Precautions (5.1, 5.2, 5.4)].
- Interrupt deferasirox for pediatric patients who have acute illnesses, which can cause volume depletion, such as vomiting, diarrhea, or prolonged decreased oral intake, and monitor more frequently. Resume therapy as appropriate, based on assessments of renal function, when oral intake and volume status are normal [see Dosage and Administration (2.4, 2.5), Warnings and Precautions (5.1), Use in Specific Populations (8.4), Clinical Pharmacology (12.3)].
2.2 Iron Overload in Non-Transfusion-Dependent Thalassemia Syndromes
Deferasirox therapy should only be considered when a patient with NTDT syndrome has an LIC of at least 5 mg Fe/g dw and a serum ferritin greater than 300 mcg/L.
Prior to starting therapy, obtain:
-
LIC by liver biopsy or by an FDA-cleared or approved method for identifying patients for treatment with deferasirox therapy
-
Serum ferritin level on at least 2 measurements 1-month apart [see Clinical Studies (14)]
-
Baseline renal function:
- Obtain serum creatinine in duplicate (due to variations in measurements)
- Calculate the estimated glomerular filtration rate (eGFR). Use a prediction equation appropriate for adult patients (e.g., CKD-EPI, MDRD method) and in pediatric patients (e.g., Schwartz equations).
- Obtain urinalyses and serum electrolytes to evaluate renal tubular function [see Dosage and Administration (2.4), Warnings and Precautions (5.1)].
-
Serum transaminases and bilirubin [see Dosage and Administration (2.4), Warnings and Precautions (5.2)]
-
Baseline auditory and ophthalmic examinations [see Warnings and Precautions (5.10)]
Initiating Therapy:
-
The recommended initial dose of deferasirox for patients with eGFR greater than 60 mL/min/1.73 m2 is 7 mg per kg body weight orally once daily. Calculate doses (mg per kg per day) to the nearest whole tablet.
-
If the baseline LIC is greater than 15 mg Fe/g dw, consider increasing the dose to 14 mg/kg/day after 4 weeks.
During Therapy:
-
Monitor serum ferritin monthly. Interrupt treatment when serum ferritin is less than 300 mcg/L and obtain an LIC to determine whether the LIC has fallen to less than 3 mg Fe/g dw.
-
Use the minimum effective dose to achieve a trend of decreasing ferritin.
-
Monitor LIC every 6 months.
-
After 6 months of therapy, if the LIC remains greater than 7 mg Fe/g dw, increase the dose of deferasirox to a maximum of 14 mg/kg/day. Do not exceed a maximum of 14 mg/kg/day.
-
If after 6 months of therapy, the LIC is 3 to 7 mg Fe/g dw, continue treatment with deferasirox at no more than 7 mg/kg/day.
-
When the LIC is less than 3 mg Fe/g dw, interrupt treatment with deferasirox and continue to monitor the LIC.
-
Monitor blood counts, liver function, renal function and ferritin monthly [see Warnings and Precautions (5.1, 5.2, 5.4)].
-
Increase monitoring frequency for pediatric patients who have acute illness, which can cause volume depletion, such as vomiting, diarrhea, or prolonged decreased oral intake. Consider dose interruption until oral intake and volume status are normal [see Dosage and Administration (2.4, 2.5), Warnings and Precautions (5.1), Use in Specific Populations (8.4), Clinical Pharmacology (12.3)].
Restart treatment when the LIC rises again to more than 5 mg Fe/g dw.
2.3 Administration
Swallow deferasirox tablets once daily with water or other liquids, preferably at the same time each day. Take deferasirox tablets on an empty stomach or with a light meal (contains less than 7% fat content and approximately 250 calories). Examples of light meals include 1 whole wheat English muffin, 1 packet jelly (0.5 ounces), and skim milk (8 fluid ounces) or a turkey sandwich (2 oz. turkey on whole wheat bread w/ lettuce, tomato, and 1 packet mustard). Do not take deferasirox tablets with aluminum-containing antacid products [see Drug Interactions (7.1)]. For patients who have difficulty swallowing whole tablets, deferasirox tablets may be crushed and mixed with soft foods (e.g., yogurt or applesauce) immediately prior to use and administered orally. Commercial crushers with serrated surfaces should be avoided for crushing a single 90 mg tablet. The dose should be immediately and completely consumed and not stored for future use.
For patients who are currently on chelation therapy with Exjade® tablets for oral suspension and converting to deferasirox, the dose should be about 30% lower, rounded to the nearest whole tablet. The table below provides additional information on dosing conversion to deferasirox tablets.
EXJADE®
Tablets for oral suspension
(white round tablet)
Deferasirox Tablets
(film coated blue oval tablet)
Transfusion-Dependent Iron Overload
Starting Dose
20 mg/kg/day
14 mg/kg/day
Titration Increments
5-10 mg/kg
3.5-7 mg/kg
Maximum Dose
40 mg/kg/day
28 mg/kg/day
Non-Transfusion-Dependent Thalassemia Syndromes
Starting Dose
10 mg/kg/day
7 mg/kg/day
Titration Increments
5-10 mg/kg
3.5-7 mg/kg
Maximum Dose
20 mg/kg/day
14 mg/kg/day
2.4 Use in Patients with Baseline Hepatic or Renal Impairment
Patients with Baseline Hepatic Impairment
Mild (Child-Pugh A) Hepatic Impairment: No dose adjustment is necessary.
Moderate (Child-Pugh B) Hepatic Impairment: Reduce the starting dose by 50%.
Severe (Child-Pugh C) Hepatic Impairment: Avoid deferasirox tablets [see Warnings and Precautions (5.2), Use in Specific Populations (8.7)].
Patients with Baseline Renal Impairment
Do not use deferasirox in adult or pediatric patients with eGFR less than 40 mL/min/1.73 m2[see Dosage and Administration (2.5), Contraindications (4)].
For patients with renal impairment (eGFR 40-60 mL/min/1.73 m2), reduce the starting dose by 50% [see Use in Specific Populations (8.6)].
Exercise caution in pediatric patients with eGFR between 40 and 60 mL/minute/1.73 m2. If treatment is needed, use the minimum effective dose and monitor renal function frequently. Individualize dose titration based on improvement in renal injury [see Use in Specific Populations (8.6)].
2.5 Dose Modifications for Decreases in Renal Function while on Deferasirox
Deferasirox is contraindicated in patients with eGFR less than 40 mL/minute/1.73 m2[see Contraindications (4)]
For decreases in renal function while receiving deferasirox [see Warnings and Precautions (5.1)], modify the dose as follows:
Transfusional Iron Overload
Adults:
- If the serum creatinine increases by 33% or more above the average baseline measurement, repeat the serum creatinine within 1 week, and if still elevated by 33% or more, reduce the dose by 7 mg per kg.
Pediatric Patients (ages 2 years-17 years):
- Reduce the dose by 7 mg per kg if eGFR decreases by greater than 33% below the average baseline measurement and repeat eGFR within 1 week
- Interrupt deferasirox for acute illnesses, which can cause volume depletion, such as vomiting, diarrhea, or prolonged decreased oral intake, and monitor more frequently. Resume therapy as appropriate, based on assessments of renal function, when oral intake and volume status are normal. Avoid use of other nephrotoxic drugs [see Warnings and Precautions (5.1)].
- In the setting of decreased renal function, evaluate the risk benefit profile of continued deferasirox use. Use the minimum effective deferasirox dose and monitor renal function more frequently, by evaluating tubular and glomerular function. Titrate dosing based on renal injury. Consider dose reduction or interruption and less nephrotoxic therapies until improvement of renal function. If signs of renal tubular or glomerular injury occur in the presence of other risk factors such as volume depletion, reduce or interrupt deferasirox to prevent severe and irreversible renal injury [see Warnings and Precautions (5.1)].
All Patients (regardless of age):
- Discontinue therapy for eGFR less than 40 mL/min/1.73 m2[see Contraindications (4)].
Non-Transfusion-Dependent Thalassemia Syndromes
Adults:
-
If the serum creatinine increases by 33% or more above the average baseline measurement, repeat the serum creatinine within 1 week, and if still elevated by 33% or more, interrupt therapy if the dose is 3.5 mg per kg, or reduce by 50% if the dose is 7 or 14 mg per kg.
Pediatric Patients (ages 10 years -17 years):
-
Reduce the dose by 3.5 mg per kg if eGFR decreases by greater than 33% below the average baseline measurement and repeat the eGFR within 1 week.
-
Increase monitoring frequency for pediatric patients who have acute illnesses, which can cause volume depletion, such as vomiting, diarrhea, or prolonged decreased oral intake. Consider dose interruption until oral intake and volume status are normal. Avoid use of other nephrotoxic drugs [see Warnings and Precautions (5.1)].
-
In the setting of decreased renal function, evaluate the risk benefit profile of continued deferasirox use. Use the minimum effective deferasirox dose and monitor renal function more frequently, by evaluating tubular and glomerular function. Titrate dosing based on renal injury. Consider dose reduction or interruption and less nephrotoxic therapies until improvement of renal function. If signs of renal tubular or glomerular injury occur in the presence of other risk factors such as volume depletion, reduce or interrupt deferasirox to prevent severe and irreversible renal injury [see Warnings and Precautions (5.1)].
All Patients (regardless of age):
- Discontinue therapy for eGFR less than 40 mL/min/1.73 m2 [see Contraindications (4)].
2.6 Dose Modifications Based on Concomitant Medications
UDP-glucuronosyltransferases (UGT) Inducers
Concomitant use of UGT inducers decreases systemic exposure. Avoid the concomitant use of strong UGT inducers (e.g., rifampicin, phenytoin, phenobarbital, ritonavir). If you must administer deferasirox tablets with a strong UGT inducer, consider increasing the initial dose by 50%, and monitor serum ferritin levels and clinical responses for further dose modification [see Dosage and Administration (2.1, 2.2), Drug Interactions (7.5)].
Bile Acid Sequestrants
Concomitant use of bile acid sequestrants decreases systemic exposure. Avoid the concomitant use of bile acid sequestrants (e.g., cholestyramine, colesevelam, colestipol). If you must administer deferasirox tablets with a bile acid sequestrant, consider increasing the initial dose by 50%, and monitor serum ferritin levels and clinical responses for further dose modification [see Dosage and Administration (2.1, 2.2), Drug Interactions (7.6)].
-
3 DOSAGE FORMS & STRENGTHS
- 90 mg deferasiroxtablets
Light blue, oval shaped, biconvex, beveled edges, film coated tablet, debossed with “90” on one side and plain on other side.
- 360 mg deferasiroxtablets
Dark blue, oval shaped, biconvex, beveled edges, film coated tablet, debossed with “360” on one side and plain on other side.
-
4 CONTRAINDICATIONS
Deferasirox is contraindicated in patients with:
- Estimated GFR less than 40 mL/min/1.73 m2[see Dosage and Administration (2.5), Warnings and Precautions (5.1)];
- Poor performance status [see Warnings and Precautions (5.1,5.3)];
- High-risk myelodysplastic syndromes (this patient population was not studied and is not expected to benefit from chelation therapy);
- Advanced malignancies [see Warnings and Precautions (5.1, 5.3)];
- Platelet counts less than 50 x 109/L [see Warnings and Precautions (5.3, 5.4)];
- Known hypersensitivity to deferasirox or any component of deferasirox[see Warnings and Precautions (5.7), Adverse Reactions (6.2)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Acute Kidney Injury, Including Acute Renal Failure Requiring Dialysis and Renal Tubular Toxicity Including Fanconi Syndrome
Deferasirox is contraindicated in patients with eGFR less than 40 mL/min/1.73 m2. Exercise caution in pediatric patients with eGFR between 40 and 60 mL/minute/1.73 m2. If treatment is needed, use the minimum effective dose and monitor renal function frequently. Individualize dose titration based on improvement in renal injury [see Use in Specific Populations (8.6)]. For patients with renal impairment (eGFR 40-60 mL/min/1.73 m2) reduce the starting dose by 50% [see Dosage and Administration (2.4, 2.5), Use in Specific Populations (8.6)].
Deferasirox can cause acute kidney injury including renal failure requiring dialysis that has resulted in fatal outcomes. Based on postmarketing experience most fatalities have occurred in patients with multiple comorbidities and who were in advanced stages of their hematological disorders. In the clinical trials, adults and pediatric deferasirox-treated patients with no preexisting renal disease experienced dose-dependent mild, non- progressive increases in serum creatinine and proteinuria. Preexisting renal disease and concomitant use of other nephrotoxic drugs may increase the risk of acute kidney injury in adult and pediatric patients. Acute illnesses associated with volume depletion and overchelation may increase the risk of acute kidney injury in pediatric patients. In pediatric patients, small decreases in eGFR can result in increases in deferasirox exposure, particularly in younger patients with body surface area typical of patients less than age 7 years. This can lead to a cycle of worsening renal function and further increases in Exjade® exposure, unless the dose is reduced or interrupted. Renal tubular toxicity, including acquired Fanconi syndrome, has been reported in patients treated with deferasirox, most commonly in pediatric patients with beta-thalassemia and serum ferritin levels less than 1,500 mcg/L [see Warnings and Precautions (5.6), Adverse Reactions (6.1, 6.2), Use in Special Populations (8.4), Clinical Pharmacology (12.3)].
Evaluate renal glomerular and tubular function before initiating therapy or increasing the dose. Use prediction equations validated for use in adult and pediatric patients to estimate GFR. Obtain serum electrolytes and urinalysis in all patients to evaluate renal tubular function [see Dosage and Administration (2.1, 2.2)].
Monitor all patients for changes in eGFR and for renal tubular toxicity weekly during the first month after initiation or modification of therapy and at least monthly thereafter. Monitor serum ferritin monthly to evaluate for overchelation. Use the minimum dose to establish and maintain a low iron burden. Monitor renal function more frequently in patients with preexisting renal disease or decreased renal function. In pediatric patients, interrupt deferasirox during acute illnesses, which can cause volume depletion such as vomiting, diarrhea, or prolonged decreased oral intake, and monitor renal function more frequently. Promptly correct fluid deficits to prevent renal injury. Resume therapy as appropriate, based on assessments of renal function, when oral intake and volume status are normal [see Dosage and Administration (2.5), Warnings and Precautions (5.6), Adverse Reactions (6.1, 6.2), Use in Specific Populations (8.4)].
5.2 Hepatic Toxicity and Failure
Deferasirox can cause hepatic injury, fatal in some patients. In Study 1, 4 patients (1.3%) discontinued deferasirox because of hepatic toxicity (drug-induced hepatitis in 2 patients and increased serum transaminases in 2 additional patients). Hepatic toxicity appears to be more common in patients greater than 55 years of age. Hepatic failure was more common in patients with significant comorbidities, including liver cirrhosis and multiorgan failure [see Adverse Reactions (6.1)].
Acute liver injury and failure, including fatal outcomes, have occurred in pediatric deferasirox-treated patients. Liver failure occurred in association with acute kidney injury in pediatric patients at risk for overchelation during a volume-depleting event. Interrupt deferasirox therapy when acute liver injury or acute kidney injury is suspected and during volume depletion. Monitor liver and renal function more frequently in pediatric patients who are receiving deferasirox in the 14-28 mg/kg/day range and when iron burden is approaching normal. Use the minimum effective dose to achieve and maintain a low iron burden [see Dosage and Administration (2.5), Warnings and Precautions (5.6), Adverse Reactions (6.1)].
Measure transaminases (AST and ALT) and bilirubin in all patients before the initiation of treatment and every 2 weeks during the first month and at least monthly thereafter. Consider dose modifications or interruption of treatment for severe or persistent elevations.
Avoid the use of deferasirox in patients with severe (Child-Pugh C) hepatic impairment. Reduce the starting dose in patients with moderate (Child-Pugh B) hepatic impairment [see Dosage and Administration (2.4), Use in Specific Populations (8.7)]. Patients with mild (Child-Pugh A) or moderate (Child-Pugh B) hepatic impairment may be at higher risk for hepatic toxicity.
5.3 Gastrointestinal (GI) Ulceration, Hemorrhage, and Perforation
GI hemorrhage, including deaths, has been reported, especially in elderly patients who had advanced hematologic malignancies and/or low platelet counts. Nonfatal upper GI irritation, ulceration and hemorrhage have been reported in patients, including children and adolescents, receiving deferasirox [see Adverse Reactions (6.1)]. Monitor for signs and symptoms of GI ulceration and hemorrhage during deferasirox therapy, and promptly initiate additional evaluation and treatment if a serious GI adverse event is suspected. The risk of gastrointestinal hemorrhage may be increased when administering deferasirox in combination with drugs that have ulcerogenic or hemorrhagic potential, such as nonsteroidal anti-inflammatory drugs (NSAIDs), corticosteroids, oral bisphosphonates, or anticoagulants. There have been reports of ulcers complicated with gastrointestinal perforation (including fatal outcome) [see Adverse Reactions (6.2)].
5.4 Bone Marrow Suppression
Neutropenia, agranulocytosis, worsening anemia, and thrombocytopenia, including fatal events, have been reported in patients treated with deferasirox. Preexisting hematologic disorders may increase this risk. Monitor blood counts in all patients. Interrupt treatment with deferasirox in patients who develop cytopenias until the cause of the cytopenia has been determined. deferasirox is contraindicated in patients with platelet counts below 50 x 109/L.
5.5 Age-Related Risk of Toxicity
Elderly Patients
Deferasirox has been associated with serious and fatal adverse reactions in the postmarketing setting among adults, predominantly in elderly patients. Monitor elderly patients treated with deferasirox more frequently for toxicity [see Use in Specific Populations (8.5)].
Pediatric Patients
Deferasirox has been associated with serious and fatal adverse reactions in pediatric patients in the postmarketing setting. These events were frequently associated with volume depletion or with continued Exjade® doses in the 20-40 mg/kg/day range equivalent to 14-28 mg/kg/day deferasirox when body iron burden was approaching or in the normal range. Interrupt deferasirox in patients with volume depletion, and resume deferasirox when renal function and fluid volume have normalized. Monitor liver and renal function more frequently during volume depletion and in patients receiving deferasirox in the 14-28 mg/kg/day range when iron burden is approaching the normal range. Use the minimum effective dose to achieve and maintain a low iron burden [see Dosage and Administration (2.4), Warnings and Precautions (5.6), Use in Specific Populations (8.4)].
5.6 Overchelation
For patients with transfusional iron overload, measure serum ferritin monthly to assess for possible overchelation of iron. An analysis of pediatric patients treated with Exjade® in pooled clinical trials (n = 158), found a higher rate of renal adverse events among patients receiving doses greater than 25 mg/kg/day equivalent to 17.5 mg/kg/day deferasirox, while their serum ferritin values were less than 1000 mcg/L. Consider dose reduction or closer monitoring of renal and hepatic function, and serum ferritin levels during these periods. Use the minimum effective dose to maintain a low-iron burden [see Adverse Reactions (6.1), Use in Specific Populations (8.4)].
If the serum ferritin falls below 1000 mcg/L at 2 consecutive visits, consider dose reduction, especially if the deferasirox dose is greater than 17.5 mg/kg/day [see Adverse Reactions (6.1)]. If the serum ferritin falls below 500 mcg/L, interrupt therapy with deferasirox and continue monthly monitoring. Evaluate the need for ongoing chelation for patients whose conditions do not require regular blood transfusions. Use the minimum effective dose to maintain iron burden in the target range. Continued administration of deferasirox in the 14 to 28 mg/kg/day range, when the body iron burden is approaching or within the normal range can result in life threatening adverse events [see Dosage and Administration (2.1)].
For patients with NTDT, measure LIC by liver biopsy or by using an FDA-cleared or approved method for monitoring patients receiving deferasirox therapy every 6 months on treatment. Interrupt deferasirox administration when the LIC is less than 3 mg Fe/g dw. Measure serum ferritin monthly, and if the serum ferritin falls below 300 mcg/L, interrupt deferasirox and obtain a confirmatory LIC [see Clinical Studies (14)].
5.7 Hypersensitivity
Deferasirox may cause serious hypersensitivity reactions (such as anaphylaxis and angioedema), with the onset of the reaction usually occurring within the first month of treatment [see Adverse Reactions (6.2)]. If reactions are severe, discontinue deferasirox and institute appropriate medical intervention. Deferasirox is contraindicated in patients with known hypersensitivity to deferasirox products and should not be reintroduced in patients who have experienced previous hypersensitivity reactions on deferasirox products due to the risk of anaphylactic shock.
5.8 Severe Skin Reactions
Severe cutaneous adverse reactions (SCARs), including Stevens-Johnson syndrome (SJS), toxic epidermal necrolysis (TEN) and drug reaction with eosinophilia and systemic symptoms (DRESS) which could be life- threatening or fatal have been reported during deferasirox therapy [see Adverse Reactions (6.1, 6.2)]. Cases of erythema multiforme have been observed. Advise patients of the signs and symptoms of severe skin reactions, and closely monitor. If any severe skin reactions are suspected, discontinue deferasirox immediately and do not reintroduce deferasirox therapy.
5.9 Skin Rash
Rashes may occur during deferasirox treatment [see Adverse Reactions (6.1)]. For rashes of mild to moderate severity, deferasirox may be continued without dose adjustment, since the rash often resolves spontaneously. In severe cases, interrupt treatment with deferasirox. Reintroduction at a lower dose with escalation may be considered after resolution of the rash.
5.10 Auditory and Ocular Abnormalities
Auditory disturbances (high frequency hearing loss, decreased hearing), and ocular disturbances (lens opacities, cataracts, elevations in intraocular pressure, and retinal disorders) were reported at a frequency of less than 1% with deferasirox therapy in the clinical studies. The frequency of auditory adverse events irrespective of causality was increased among pediatric patients, who received Exjade® doses greater than 25 mg/kg/day equivalent to 17.5 mg/kg/day deferasirox when serum ferritin was less than 1000 mcg/L [see Warnings and Precautions (5.6)].
Perform auditory and ophthalmic testing (including slit lamp examinations and dilated fundoscopy) before starting deferasirox treatment and thereafter at regular intervals (every 12 months). If disturbances are noted, monitor more frequently. Consider dose reduction or interruption.
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are also discussed in other sections of the labeling:
- Acute Kidney Injury, Including Acute Renal Failure Requiring Dialysis, and Renal Tubular Toxicity Including Fanconi Syndrome [see Warnings and Precautions (5.1)]
- Hepatic Toxicity and Failure [see Warnings and Precautions (5.2)]
- Gastrointestinal (GI) Hemorrhage [see Warnings and Precautions (5.3)]
- Bone Marrow Suppression [see Warnings and Precautions (5.4)]
- Hypersensitivity [see Warnings and Precautions (5.7)]
- Severe Skin Reactions [see Warnings and Precautions (5.8)]
- Skin Rash [see Warnings and Precautions (5.9)]
- Auditory and Ocular Abnormalities [see Warnings and Precautions (5.10)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice. Deferasirox was evaluated in healthy volunteer trials. Currently, there are no clinical data in patients with deferasirox tablets. Deferasirox contains the same active ingredient as Exjade® (deferasirox) tablets for oral suspension. The following adverse reactions have been reported with Exjade® tablets for oral suspension.
Transfusional Iron Overload
A total of 700 adult and pediatric patients were treated with deferasirox for 48 weeks in premarketing studies. These included 469 patients with beta-thalassemia, 99 with rare anemias, and 132 with sickle cell disease. Of these patients, 45% were male, 70% were Caucasian and 292 patients were less than 16 years of age. In the sickle cell disease population, 89% of patients were black. Median treatment duration among the sickle cell patients was 51 weeks. Of the 700 patients treated, 469 (403 beta-thalassemia and 66 rare anemias) were entered into extensions of the original clinical protocols. In ongoing extension studies, median durations of treatment were 88 to 205 weeks.
Six hundred twenty-seven patients with MDS were enrolled across 5 uncontrolled trials. These studies varied in duration from 1 to 5 years. The discontinuation rate across studies in the first year was 46% (AEs 20%, withdrawal of consent 10%, death 8%, other 4%, lab abnormalities 3%, and lack of efficacy 1%). Among 47 patients enrolled in the study of 5-year duration, 10 remained on deferasirox at the completion of the study.
Table 1 displays adverse reactions occurring in greater than 5% of deferasirox-treated beta-thalassemia patients (Study 1), sickle cell disease patients (Study 3), and patients with MDS (MDS pool). Abdominal pain, nausea, vomiting, diarrhea, skin rashes, and increases in serum creatinine were the most frequent adverse reactions reported with a suspected relationship to deferasirox. Gastrointestinal symptoms, increases in serum creatinine, and skin rash were dose related.
Table 1. Adverse Reactions *Occurring in > 5% of Deferasirox-treated Patients in Study 1, Study 3, and MDS Pool
Study 1
(Beta-thalassemia)
Study 3
(Sickle Cell Disease)
MDS Pool
Adverse Reactions
Deferasirox
N = 296
n (%)
Deferoxamine
N = 290
n (%)
Deferasirox
N = 132
n (%)
Deferoxamine
N = 63
n (%)
Deferasirox
N = 627
n (%)
Abdominal Pain**
63 (21)
41 (14)
37 (28)
9 (14)
145 (23)
Diarrhea
35 (12)
21 (7)
26 (20)
3 (5)
297 (47)
Creatinine
Increased***
33 (11)
0 (0)
9 (7)
0
89 (14)
Nausea
31 (11)
14 (5)
30 (23)
7 (11)
161 (26)
Vomiting
30 (10)
28 (10)
28 (21)
10 (16)
83 (13)
Rash
25 (8)
9 (3)
14 (11)
3 (5)
83 (13)
*Adverse reaction frequencies are based on adverse events reported regardless of relationship to study drug.
**Includes ‘abdominal pain’, ‘abdominal pain lower’, and ‘abdominal pain upper’ which were reported as adverse events.
***Includes ‘blood creatinine increased’ and ‘blood creatinine abnormal’, which were reported as adverse events. See also Table 2.
In Study 1, a total of 113 (38%) patients treated with deferasirox had increases in serum creatinine greater than 33% above baseline on 2 separate occasions (Table 2) and 25 (8%) patients required dose reductions. Increases in serum creatinine appeared to be dose related [see Warnings and Precautions (5.1)]. In this study, 17 (6%) patients treated with deferasirox developed elevations in SGPT/ALT levels greater than 5 times the ULN at 2 consecutive visits. Of these, 2 patients had liver biopsy proven drug-induced hepatitis and both discontinued deferasirox therapy [see Warnings and Precautions (5.2)]. An additional 2 patients, who did not have elevations in SGPT/ALT greater than 5 times the ULN, discontinued deferasirox because of increased SGPT/ALT. Increases in transaminases did not appear to be dose related. Adverse reactions that led to discontinuations included abnormal liver function tests (2 patients) and drug-induced hepatitis (2 patients), skin rash, glycosuria/proteinuria, Henoch Schönlein purpura, hyperactivity/insomnia, drug fever, and cataract (1 patient each).
In Study 3, a total of 48 (36%) patients treated with deferasirox had increases in serum creatinine greater than 33% above baseline on 2 separate occasions (Table 2) [see Warnings and Precautions (5.1)]. Of the patients who experienced creatinine increases in Study 3, 8 deferasirox-treated patients required dose reductions. In this study, 5 patients in the deferasirox group developed elevations in SGPT/ALT levels greater than 5 times the ULN at 2 consecutive visits and 1 patient subsequently had deferasirox permanently discontinued. Four additional patients discontinued due to adverse reactions with a suspected relationship to study drug, including diarrhea, pancreatitis associated with gallstones, atypical tuberculosis, and skin rash.
In the MDS pool, in the first year, a total of 229 (37%) patients treated with deferasirox had increases in serum creatinine greater than 33% above baseline on 2 consecutive occasions (Table 2) and 8 (3.5%) patients permanently discontinued [see Warnings and Precautions (5.1)]. A total of 5 (0.8%) patients developed SGPT/ALT levels greater than 5 times the ULN at 2 consecutive visits. The most frequent adverse reactions that led to discontinuation included increases in serum creatinine, diarrhea, nausea, rash, and vomiting. Death was reported in the first year in 52 (8%) of patients [see Clinical Studies (14)].
Table 2. Number (%) of Patients with Increases in Serum Creatinine or SGPT/ALT in Study 1, Study 3, and MDS Pool
Study 1 (Beta-thalassemia)
Study 3
(Sickle Cell Disease)
MDS Pool
Laboratory Parameter
Deferasirox
N = 296
n (%)
Deferoxamine
N = 290
n (%)
Deferasirox
N = 132
n (%)
Deferoxamine
N = 63
n (%)
Deferasirox
N = 627
n (%)
Serum Creatinine
Creatinine increase > 33% at 2 consecutive post-baseline visits113 (38)
41 (14)
48 (36)
14 (22)
229 (37)
Creatinine increase > 33% and > ULN at 2 consecutive post- baseline visits
7 (2)
1 (0)
3 (2)
2 (3)
126 (20)
SGPT/ALT
SGPT/ALT > 5 x
ULN at 2 post- baseline visits
25 (8)
7 (2)
2 (2)
0
9 (1)
SGPT/ALT > 5 x
ULN at 2 consecutive post-baseline visits
17 (6)
5 (2)
5 (4)
0
5 (1)
Non-Transfusion-Dependent Thalassemia Syndromes
In Study 5, 110 patients with NTDT received 1 year of treatment with deferasirox 5 or 10 mg/kg/day and 56 patients received placebo in a double-blind, randomized trial. In Study 6, 130 of the patients who completed Study 5 were treated with open-label deferasirox at 5, 10, or 20 mg/kg/day (depending on the baseline LIC) for 1 year [see Clinical Studies (14)]. Table 3 displays adverse reactions occurring in greater than 5% in any group. The most frequent adverse reactions with a suspected relationship to study drug were nausea, rash, and diarrhea.
Table 3. Adverse Reactions Occurring in Greater Than 5% in NTDT Patients
Any adverse reaction
Study 5
Study 6
Deferasirox
Placebo
Deferasirox
N = 110
N = 56
N = 130
n (%)
n (%)
n (%)
31 (28)
9 (16)
27 (21)
Nausea
7 (6)
4 (7)
2 (2)
Rash
7 (6)
1 (2)
2 (2)
Diarrhea
5 (5)
1 (2)
7 (5)
In Study 5, 1 patient in the placebo 10 mg/kg/day group experienced an ALT increase to greater than 5 times ULN and greater than 2 times baseline (Table 4). Three deferasirox-treated patients (all in the 10 mg/kg/day group) had 2 consecutive serum creatinine level increases greater than 33% from baseline and greater than ULN. Serum creatinine returned to normal in all 3 patients (in 1 spontaneously and in the other 2 after drug interruption). Two additional cases of ALT increase and 2 additional cases of serum creatinine increase were observed in the 1-year extension of Study 5.
Table 4. Number (%) of NTDT Patients with Increases in Serum Creatinine or SGPT/ALT
Laboratory Parameter
Study 5
Study 6
Deferasirox
Placebo
Deferasirox
N = 110
N = 56
N = 130
n (%)
n (%)
n (%)
Serum creatinine (> 33% increase from baseline and > ULN at ≥ 2 consecutive post-baseline values)
3 (3)
0
2 (2)
SGPT/ALT (> 5 x ULN and > 2 x baseline)
1 (1)
1 (2)
2 (2)
Proteinuria
In clinical studies, urine protein was measured monthly. Intermittent proteinuria (urine protein/creatinine ratio greater than 0.6 mg/mg) occurred in 18.6% of deferasirox-treated patients compared to 7.2% of deferoxamine treated patients in Study 1 [see Warnings and Precautions (5.1)].
Other Adverse Reactions
In the population of more than 5,000 patients with transfusional iron overload, who have been treated with deferasirox during clinical trials, adverse reactions occurring in 0.1% to 1% of patients included gastritis, edema, sleep disorder, pigmentation disorder, dizziness, anxiety, maculopathy, cholelithiasis, pyrexia, fatigue, laryngeal pain, cataract, hearing loss, gastrointestinal hemorrhage, gastric ulcer (including multiple ulcers), duodenal ulcer, renal tubular disorder (Fanconi syndrome), and acute pancreatitis (with and without underlying biliary conditions). Adverse reactions occurring in 0.01% to 0.1% of patients included optic neuritis, esophagitis, erythema multiforme, and drug reaction with eosinophilia and systemic symptoms (DRESS).
Adverse reactions, which most frequently led to dose interruption or dose adjustment during clinical trials were rash, gastrointestinal disorders, infections, increased serum creatinine, and increased serum transaminases.
Pooled Analysis of Pediatric Clinical Trial Data
A nested case control analysis was conducted within a deferasirox pediatric-pooled clinical trial dataset to evaluate the effects of dose and serum ferritin level, separately and combined, on kidney function. Among 1213 children (aged 2 to 15 years) with transfusion-dependent thalassemia, 162 cases of acute kidney injury (eGFR < 90 mL/min/1.73 m2) and 621 matched-controls with normal kidney function (eGFR > 120 mL/min/1.73 m2) were identified. The primary findings were:
- A 26% increased risk of acute kidney injury was observed with each 5 mg/kg increase in daily Exjade® dosage equivalent to 3.5 mg/kg deferasirox, starting at 20 mg/kg/day equivalent to 14 mg/kg/day deferasirox (95% CI: 1.08-1.48).
- A 25% increased risk for acute kidney injury was observed with each 250 mcg/L decrease in serum ferritin starting at 1250 mcg/L (95% CI: 1.01-1.56).
- Among pediatric patients with a serum ferritin < 1000 mcg/L, those who received Exjade® dosage > 30 mg/kg/day, equivalent to 21 mg/kg/day deferasirox compared to those who received lower dosages, had a higher risk for acute kidney injury (OR = 4.47, 95% CI: 1.25-15.95), consistent with overchelation.
In addition, a cohort-based analysis of adverse events was conducted in the deferasirox pediatric pooled clinical trial data. Pediatric patients who received Exjade® dose > 25 mg/kg/day equivalent to 17.5 mg/kg/day deferasirox when their serum ferritin was < 1000 mcg/L (n = 158), had a 6-fold greater rate of renal adverse events (IRR = 6.00, 95% CI: 1.75-21.36) and a 2-fold greater rate of dose interruptions (IRR = 2.06, 95% CI: 1.33-3.17) compared to the time-period prior to meeting these simultaneous criteria. Adverse events of special interest (cytopenia, renal, hearing, and gastrointestinal disorders) occurred 1.9-fold more frequently when these simultaneous criteria were met, compared to preceding time-periods (IRR = 1.91, 95% CI: 1.05-3.48) [see Warnings and Precautions (5.6)].
6.2 Postmarketing Experience
The following adverse reactions have been spontaneously reported during post-approval use of deferasirox in the transfusional iron overload setting. Because these reactions are reported voluntarily from a population of uncertain size, in which patients may have received concomitant medication, it is not always possible to reliably estimate frequency or establish a causal relationship to drug exposure.
Skin and Subcutaneous Tissue Disorders: Stevens-Johnson syndrome (SJS), leukocytoclastic vasculitis, urticaria, alopecia, toxic epidermal necrolysis (TEN)
Immune System Disorders: hypersensitivity reactions (including anaphylactic reaction and angioedema)
Renal and Urinary Disorders: acute renal failure, tubulointerstitial nephritis
Hepatobiliary Disorders: hepatic failure
Gastrointestinal Disorders: gastrointestinal perforation
Blood and Lymphatic System Disorders: worsening anemia
5-Year Pediatric RegistryIn a 5-year observational study, 267 pediatric patients 2 to < 6 years of age (at enrollment) with transfusional hemosiderosis received deferasirox. Of the 242 patients who had pre- and post-baseline eGFR measurements, 116 (48%) patients had a decrease in eGFR of ≥ 33% observed at least once. Twenty-one (18%) of these 116 patients with decreased eGFR had a dose interruption, and 15 (13%) of these 116 patients had a dose decrease within 30 days. Adverse events leading to permanent discontinuation from the study included liver injury (n = 11), vomiting (n = 2), renal tubular disorder (n = 1), proteinuria (n = 1), hematuria (n = 1), upper gastrointestinal hemorrhage (n = 1), abdominal pain (n = 1), and hypokalemia (n = 1).
-
7 DRUG INTERACTIONS
7.1 Aluminum-Containing Antacid Preparations
The concomitant administration of deferasirox and aluminum-containing antacid preparations has not been formally studied. Although deferasirox has a lower affinity for aluminum than for iron, do not take deferasirox with aluminum-containing antacid preparations.
7.2 Agents Metabolized by CYP3A4
Deferasirox may induce CYP3A4 resulting in a decrease in CYP3A4 substrate concentration when these drugs are coadministered. Closely monitor patients for signs of reduced effectiveness when deferasirox is administered with drugs metabolized by CYP3A4 (e.g., alfentanil, aprepitant, budesonide, buspirone, conivaptan, cyclosporine, darifenacin, darunavir, dasatinib, dihydroergotamine, dronedarone, eletriptan, eplerenone, ergotamine, everolimus, felodipine, fentanyl, hormonal contraceptive agents, indinavir, fluticasone, lopinavir, lovastatin, lurasidone, maraviroc, midazolam, nisoldipine, pimozide, quetiapine, quinidine, saquinavir, sildenafil, simvastatin, sirolimus, tacrolimus, tolvaptan, tipranavir, triazolam, ticagrelor, and vardenafil) [see Clinical Pharmacology (12.3)].
7.3 Agents Metabolized by CYP2C8
Deferasirox inhibits CYP2C8 resulting in an increase in CYP2C8 substrate (e.g., repaglinide and paclitaxel) concentration when these drugs are coadministered. If deferasirox and repaglinide are used concomitantly, consider decreasing the dose of repaglinide and perform careful monitoring of blood glucose levels. Closely monitor patients for signs of exposure related toxicity when deferasirox is coadministered with other CYP2C8 substrates [see Clinical Pharmacology (12.3)].
7.4 Agents Metabolized by CYP1A2
Deferasirox inhibits CYP1A2 resulting in an increase in CYP1A2 substrate (e.g., alosetron, caffeine, duloxetine, melatonin, ramelteon, tacrine, theophylline, tizanidine) concentration when these drugs are coadministered. An increase in theophylline plasma concentrations could lead to clinically significant theophylline induced CNS or other adverse reactions. Avoid the concomitant use of theophylline or other CYP1A2 substrates with a narrow therapeutic index (e.g., tizanidine) with deferasirox. Monitor theophylline concentrations and consider theophylline dose modification if you must coadminister theophylline with deferasirox. Closely monitor patients for signs of exposure related toxicity when deferasirox is coadministered with other drugs metabolized by CYP1A2 [see Clinical Pharmacology (12.3)].
7.5 Agents Inducing UDP-glucuronosyltransferase (UGT) Metabolism
Deferasirox is a substrate of UGT1A1 and to a lesser extent UGT1A3. The concomitant use of deferasirox with strong UGT inducers (e.g., rifampicin, phenytoin, phenobarbital, ritonavir) may result in a decrease in deferasirox efficacy due to a possible decrease in deferasirox concentration. Avoid the concomitant use of strong UGT inducers with deferasirox. Consider increasing the initial dose of deferasirox if you must coadminister these agents together [see Dosage and Administration (2.5), Clinical Pharmacology (12.3)].
7.6 Bile Acid Sequestrants
Avoid the concomitant use of bile acid sequestrants (e.g., cholestyramine, colesevelam, colestipol) with deferasirox due to a possible decrease in deferasirox concentration. If you must coadminister these agents together, consider increasing the initial dose of deferasirox [see Dosage and Administration (2.5), Clinical Pharmacology (12.3)].
7.7 Busulfan
Increased exposure of busulfan was observed with concomitant use with deferasirox. Monitor plasma concentrations of busulfan when coadministered with deferasirox to allow dose adjustment of busulfan as needed [see Clinical Pharmacology (12.3)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no studies with the use of deferasirox in pregnant women to inform drug-associated risks.
Administration of deferasirox to rats during pregnancy resulted in decreased offspring viability and an increase in renal anomalies in male offspring at doses that were about or less than the recommended human dose on a mg/m2basis. No fetal effects were noted in pregnant rabbits at doses equivalent to the human recommended dose on an mg/m2 basis. Deferasirox should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
The background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies had a background risk of birth defect, loss, or other adverse outcomes. However, the background risk in the U.S. general population of major birth defects is 2% to 4% and of miscarriage is 15% to 20% of clinically recognized pregnancies.
Data
Animal Data
In embryo-fetal developmental studies, pregnant rats and rabbits received oral deferasirox during the period of organogenesis at doses up to 100 mg/kg/day in rats and 50 mg/kg/day in rabbits (1.2 times the maximum recommended human dose (MRHD) on an mg/m2 basis). These doses resulted in maternal toxicity but no fetal harm was observed.
In a prenatal and postnatal developmental study, pregnant rats received oral deferasirox daily from organogenesis through lactation day 20 at doses of 10, 30, and 90 mg/kg/day (0.1, 0.3, and 1.0 times the MRHD on a mg/m2basis). Maternal toxicity, loss of litters, and decreased offspring viability occurred at 90 mg/kg/day (1.0 times the MRHD on a mg/m2basis), and increases in renal anomalies in male offspring occurred at 30 mg/kg/day (0.3 times the MRHD on a mg/m2basis).
8.2 Lactation
Risk Summary
No data are available regarding the presence of deferasirox or its metabolites in human milk, the effects of the drug on the breastfed child, or the effects of the drug on milk production. Deferasirox and its metabolites were excreted in rat milk. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in breastfeeding child from deferasirox and its metabolites, a decision should be made whether to discontinue breastfeeding or to discontinue the drug, taking into account the importance of the drug to the mother.
8.3 Females and Males of Reproductive Potential
Contraception
Counsel patients to use non-hormonal method(s) of contraception since deferasirox can render hormonal contraceptives ineffective [see Drug Interactions (7.2)].8.4 Pediatric Use
Transfusional Iron Overload
The safety and effectiveness of deferasirox have been established in pediatric patients 2 years of age and older for the treatment of transfusional iron overload [see Dosage and Administration (2.1)].
Safety and effectiveness have not been established in pediatric patients less than 2 years of age for the treatment of transfusional iron overload.
Pediatric approval for treatment of transfusional iron overload was based on clinical studies of 292 pediatric patients 2 years to less than 16 years of age with various congenital and acquired anemias. Seventy percent of these patients had beta-thalassemia [see Indications and Usage (1), Dosage and Administration (2.1), Clinical Studies (14)]. In those clinical studies, 173 children (ages 2 to < 12 years) and 119 adolescents (ages 12 to < 17 years) were exposed to deferasirox.
Iron Overload in Non-Transfusion-Dependent Thalassemia Syndromes
The safety and effectiveness of deferasirox has been established in patients 10 years of age and older for the treatment of chronic iron overload with non-transfusion-dependent thalassemia (NTDT) syndromes [see Dosage and Administration (2.2)].
Safety and effectiveness have not been established in patients less than 10 years of age with chronic iron overload in NTDT syndromes.
Pediatric approval for treatment of NTDT syndromes with liver iron (Fe) concentration (LIC) of at least 5 mg Fe per gram of dry weight and a serum ferritin greater than 300 mcg/L was based on 16 pediatric patients treated with deferasirox therapy (10 years to less than 16 years of age) with chronic iron overload and NTDT. Use of deferasirox in these age groups is supported by evidence from adequate and well-controlled studies of deferasirox in adult and pediatric patients [see Indications and Usage (1.2), Dosage and Administration (2.2), Clinical Studies (14)].
In general, risk factors for deferasirox-associated kidney injury include preexisting renal disease, volume depletion, overchelation, and concomitant use of other nephrotoxic drugs. Acute kidney injury, and acute liver injury and failure has occurred in pediatric patients. In a pooled safety analysis, pediatric patients with higher deferasirox exposures had a greater probability of renal toxicity and decreased renal function, resulting in increased deferasirox exposure and progressive renal toxicity/kidney injury. Higher rates of renal adverse events have been identified among pediatric patients receiving Exjade® doses greater than 25 mg/kg/day equivalent to 17.5 mg/kg/day deferasirox when their serum ferritin values were less than 1,000 mcg/L [see Dosage and Administration (2.5), Warnings and Precautions (5.1, 5.6), Adverse Reactions (6.1, 6.2)].
Monitor renal function by estimating GFR using an eGFR prediction equation appropriate for pediatric patients and evaluate renal tubular function. Monitor renal function more frequently in pediatric patients in the presence of renal toxicity risk factors, including episodes of dehydration, fever and acute illness that may result in volume depletion or decreased renal perfusion. Use the minimum effective dose [see Warnings and Precautions (5.1)].
Interrupt deferasirox in pediatric patients with transfusional iron overload, and consider dose interruption in pediatric patients with non-transfusion-dependent iron overload, for acute illnesses which can cause volume depletion, such as vomiting, diarrhea, or prolonged decreased oral intake, and monitor more frequently. Resume therapy as appropriate, based on assessments of renal function, when oral intake and volume status are normal. Evaluate the risk benefit profile of continued deferasirox use in the setting of decreased renal function. Avoid use of other nephrotoxic drugs [see Dosage and Administration (2.5), Warnings and Precautions (5.1)].
Juvenile Animal Toxicity Data
Renal toxicity was observed in adult mice, rats, and marmoset monkeys administered deferasirox at therapeutic doses. In a neonatal and juvenile toxicity study in rats, deferasirox was administered orally from postpartum Day 7 through 70, which equates to a human age range of term neonate through adolescence. Increased renal toxicity was identified in juvenile rats compared to adult rats at a dose based on mg/m2approximately 0.4 times the recommended dose of 20 mg/kg/day. A higher frequency of renal abnormalities was noted when deferasirox was administered to non-iron overloaded animals compared to iron overloaded animals.
Additional pediatric use information is approved for Novartis Pharmaceuticals Corporation’s JADENU® (deferasirox) tablets. However, due to Novartis Pharmaceuticals Corporation’s marketing exclusivity rights, this drug product is not labeled with that pediatric information.
8.5 Geriatric Use
Four hundred thirty-one patients greater than or equal to 65 years of age were studied in clinical trials of deferasirox in the transfusional iron overload setting. Two hundred twenty-five of these patients were between 65 and 75 years of age while 206 were greater than or equal to 75 years of age. The majority of these patients had myelodysplastic syndrome (MDS) (n = 393). In these trials, elderly patients experienced a higher frequency of adverse reactions than younger patients. Monitor elderly patients for early signs or symptoms of adverse reactions that may require a dose adjustment. Elderly patients are at increased risk for toxicity due to the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy. Dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range. In elderly patients, including those with MDS, individualize the decision to remove accumulated iron based on clinical circumstances and the anticipated clinical benefit and risks of deferasirox tablets for oral suspension therapy.
8.6 Renal Impairment
Deferasirox is contraindicated in patients with eGFR less than 40 mL/min/1.73 m2[see Contraindications (4)]. For patients with renal impairment (eGFR 40 to 60 mL/min/1.73 m2), reduce the starting dose by 50% [see Dosage and Administration (2.4), Clinical Pharmacology (12.3)]. Exercise caution in pediatric patients with an eGFR between 40 and 60 mL/minute/1.73 m2[see Dosage and Administration (2.4)]. If treatment is needed, use the minimum effective dose with enhanced monitoring of glomerular and renal tubular function. Individualize dose titration based on improvement in renal injury [see Dosage and Administration (2.4, 2.5)].
Deferasirox can cause glomerular dysfunction, renal tubular toxicity, or both, and can result in acute renal failure. Monitor all patients closely for changes in eGFR and renal tubular dysfunction during deferasirox treatment. If either develops, consider dose reduction, interruption or discontinuation of deferasirox until glomerular or renal tubular function returns to baseline [see Dosage and Administration (2.4, 2.5), Warnings and Precautions (5.1)].
8.7 Hepatic Impairment
Avoid use in patients with severe (Child-Pugh C) hepatic impairment. For patients with moderate (Child-Pugh B) hepatic impairment, reduce the starting dose by 50%. Closely monitor patients with mild (Child-Pugh A) or moderate (Child-Pugh B) hepatic impairment for efficacy and adverse reactions that may require dose titration [see Dosage and Administration (2.4), Warnings and Precautions (5.2), Clinical Pharmacology (12.3)].
-
10 OVERDOSAGE
Cases of overdose (2 to 3 times the prescribed dose for several weeks) have been reported. In one case, this resulted in hepatitis which resolved without long-term consequences after a dose interruption. In one pediatric case, a dose of 2-3 times the prescribed dose for 6 days resulted in acute renal failure requiring hemofiltration and acute liver injury/failure, which were reversible with intensive care support. Single doses of deferasirox up to 80 mg per kg per day with the tablet for oral suspension formulation in iron-overloaded beta-thalassemic patients have been tolerated with nausea and diarrhea noted. In healthy subjects, single doses of up to 40 mg per kg per day with the tablet for oral suspension formulation were tolerated. There is no specific antidote for deferasirox. In case of overdose, induce vomiting and employ gastric lavage.
-
11 DESCRIPTION
Deferasirox is an iron-chelating agent provided as a tablet for oral use. Deferasirox is designated chemically as 4-[3,5-bis(2-hydroxyphenyl)-1H-1,2,4-triazol-1-yl]benzoic acid and has the following structural formula:
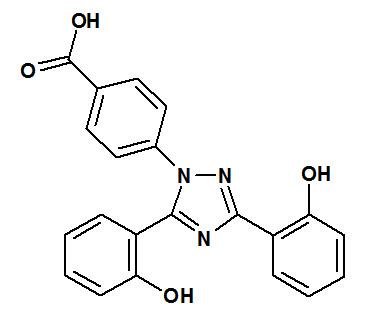
Deferasirox is a white to slightly yellow powder. It has a molecular formula C21H15N3O4and molecular weight of 373.4 g/mol. It is insoluble in water.
Deferasirox tablets contain 90 mg, or 360 mg deferasirox. Inactive ingredients include crospovidone, magnesium stearate, microcrystalline cellulose, poloxamer, povidone and talc. The film coating contains opadry blue which has ingredients FD&C blue, hypromellose, polyethylene glycol, talc and titanium dioxide.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Deferasirox is an orally active chelator that is selective for iron (as Fe3+). It is a tridentate ligand that binds iron with high affinity in a 2:1 ratio. Although deferasirox has very low affinity for zinc and copper, there are variable decreases in the serum concentration of these trace metals after the administration of deferasirox. The clinical significance of these decreases is uncertain.
12.2 Pharmacodynamics
Pharmacodynamic effects tested in an iron balance metabolic study with the tablet for oral suspension formulation showed that deferasirox (10, 20, and 40 mg per kg per day) was able to induce a mean net iron excretion (0.119, 0.329, and 0.445 mg Fe/kg body weight per day, respectively) within the clinically relevant range (0.1 to 0.5 mg per kg per day). Iron excretion was predominantly fecal.
An analysis of pooled pediatric clinical trial data found a statistically significant relationship between exposure and the probability of renal toxicity (increase in serum creatinine and urinary protein), resulting in a decrease in renal function. Decreases in renal function resulted in an increase in deferasirox exposure which may increase the probability of renal toxicity.
Cardiac Electrophysiology
At the maximum approved recommended dose, deferasirox does not prolong the QT interval to any clinically relevant extent.
12.3 Pharmacokinetics
Absorption
Based on studies in patients with the tablet for oral suspension, deferasirox is absorbed following oral administration with median times to maximum plasma concentration (Tmax) of about 1.5 to 4 hours. In healthy subjects, deferasirox showed comparable Tmax. The maximal concentrations (Cmax) and area under the curve (AUC0-24h, AUCτ) of deferasirox increase approximately linearly with dose after both single administration and under steady-state conditions. Exposure to deferasirox increased by an accumulation factor of 1.3 to 2.3 after multiple doses with the tablet for oral suspension formulation.
Tablets
The absolute bioavailability [as measured by area under the curve over time to infinity (AUCinf)] of deferasirox tablets for oral suspension is 70% compared to an intravenous dose. The bioavailability (as measured by AUCinf) of deferasirox tablets was 36% greater than with deferasirox tablets for oral suspension. After strength- adjustment, the mean AUCinfof deferasirox tablets (i.e., 360 mg strength) was similar to that of deferasirox tablets for oral suspension (i.e., 500 mg strength) under fasting conditions; however the mean Cmaxwas increased by 30%. The 30% increase in Cmaxobserved with deferasirox tablets is not clinically meaningful.
The administration of deferasirox tablets with a light meal (approximately 250 calories with fat content less than 7% of total calories) indicated that the AUCinfand Cmaxwere similar to that under fasting conditions. The administration of deferasirox tablets with a high-fat meal (approximately 1000 calories with fat content greater than 50% of total calories), increased AUCinfby 18% and Cmaxby 29% compared to that under fasting conditions [see Dosage and Administration (2.3)].
Distribution
Deferasirox is highly (~99%) protein bound almost exclusively to serum albumin. The percentage of deferasirox confined to the blood cells was 5% in humans. The volume of distribution at steady state (Vss) of deferasirox is 14.37 ± 2.69 L in adults
Metabolism
Glucuronidation is the main metabolic pathway for deferasirox, with subsequent biliary excretion. Deconjugation of glucuronidates in the intestine and subsequent reabsorption (enterohepatic recycling) is likely to occur. Deferasirox is mainly glucuronidated by UGT1A1 and to a lesser extent UGT1A3. CYP450-catalyzed (oxidative) metabolism of deferasirox appears to be minor in humans (about 8%). Deconjugation of glucuronide metabolites in the intestine and subsequent reabsorption (enterohepatic recycling) was confirmed in a healthy subjects study in which the administration of cholestyramine 12 g twice daily (strongly binds to deferasirox and its conjugates) 4 and 10 hours after a single dose of deferasirox resulted in a 45% decrease in deferasirox exposure (AUCinf) by interfering with the enterohepatic recycling of deferasirox.
ExcretionDeferasirox and metabolites are primarily (84% of the dose) excreted in the feces. Renal excretion of deferasirox and metabolites is minimal (8% of the dose). The mean elimination half-life (t1/2) ranged from 8 to 16 hours following oral administration.
Drug Interactions
Midazolam: The concomitant administration of deferasirox tablets for oral suspension and CYP3A4 probe substrate midazolam resulted in a decrease of midazolam Cmaxby 23% and AUCinfby 17%. In the clinical setting, this effect may be more pronounced, as the study was not adequately designed to conclusively assess the potential induction of CYP3A4 by deferasirox [see Drug Interactions (7.2)].
Repaglinide: The concomitant administration of deferasirox tablets for oral suspension (30 mg per kg/day for 4 days) and the CYP2C8 probe substrate repaglinide (single dose of 0.5 mg) increased repaglinide AUCinfto 2.3 fold and Cmaxof 1.6-fold [see Drug Interactions (7.3)].
Theophylline: The concomitant administration of deferasirox tablets for oral suspension (repeated dose of 30 mg per kg/day) and the CYP1A2 substrate theophylline (single dose of 120 mg) resulted in an approximate doubling of the theophylline AUCinfand elimination half-life. The single dose Cmaxwas not affected, but an increase in theophylline Cmaxis expected to occur with chronic dosing [see Drug Interactions (7.4)].
Rifampicin: The concomitant administration of deferasirox tablets for oral suspension (single dose of 30 mg per kg) and the strong uridine diphosphate glucuronosyltransferase (UGT) inducer rifampicin (600 mg per day for 9 days) decreased deferasirox AUCinfby 44% [see Drug Interactions (7.5)].
Cholestyramine: The concomitant administration of cholestyramine after a single dose of deferasirox tablets for oral suspension decreased deferasirox AUCinfby 45% [see Drug Interactions (7.6)].
Busulfan: Concomitant administration of deferasirox and busulfan resulted in an increase of busulfan exposure (AUC).
In vitro Studies: Deferasirox inhibited human CYP2A6, CYP2D6, and CYP2C19 in vitro.
Deferasirox is not a substrate of P-glycoprotein, MRP1 or MRP2.
Pharmacokinetics in Specific Populations
Pediatric: Following oral administration of single or multiple doses, systemic exposure of adolescents and children to deferasirox was less than in adult patients. In children less than 6 years of age, systemic exposure was about 50% lower than in adults.
Sex: The apparent clearance is 17.5% lower in females compared to males.
Renal Impairment: Compared to patients with MDS and eGFR greater than 60 mL/min/1.73 m2, patients with MDS and eGFR 40 to 60 mL/min/1.73 m2 (n = 34) had approximately 50% higher mean deferasirox trough plasma concentrations.
Hepatic Impairment: In a single dose (20 mg/kg) study in patients with varying degrees of hepatic impairment, deferasirox exposure was increased compared to patients with normal hepatic function. The average total (free and bound) AUCinfof deferasirox increased 16% in 6 patients with mild (Child-Pugh A) hepatic impairment, and 76% in 6 patients with moderate (Child-Pugh B) hepatic impairment compared to 6 patients with normal hepatic function. The impact of severe (Child-Pugh C) hepatic impairment was assessed in only 1 patient.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
A 104-week oral carcinogenicity study in Wistar rats showed no evidence of carcinogenicity from deferasirox at doses up to 60 mg/kg/day (0.7 times the MRHD on an mg/m2basis). A 26-week oral carcinogenicity study in p53 (+/-) transgenic mice has shown no evidence of carcinogenicity from deferasirox at doses up to 200 mg/kg/day (1.2 times the MRHD on a mg/m2basis) in males and 300 mg/kg/day (1.7 times the MRHD on a mg/m2 basis) in females.
Deferasirox was negative in the Ames test and chromosome aberration test with human peripheral blood lymphocytes. It was positive in 1 of 3 in vivo oral rat micronucleus tests.
Deferasirox at oral doses up to 75 mg/kg/day (0.9 times the MRHD on an mg/m2basis) was found to have no adverse effect on fertility and reproductive performance of male and female rats.
-
14 CLINICAL STUDIES
Deferasirox was evaluated in healthy subjects. There are no clinical data in patients with deferasirox. Deferasirox contains the same active ingredient as Exjade® (deferasirox) tablets for oral suspension. The following information is based on clinical trials conducted with Exjade® tablets for oral suspension.
TransfusionalIronOverload
The primary efficacy study, Study 1 (NCT00061750), was a multicenter, open-label, randomized, active-comparator control study to compare deferasirox tablets for oral suspension and deferoxamine in patients with beta-thalassemia and transfusional hemosiderosis. Patients greater than or equal to 2 years of age were randomized in a 1:1 ratio to receive either oral deferasirox tablets for oral suspension at starting doses of 5, 10, 20, or 30 mg per kg once daily or subcutaneous deferoxamine at starting doses of 20 to 60 mg per kg for at least 5 days per week based on LIC at baseline (2 to 3, greater than 3 to 7, greater than 7 to 14, and greater than 14 mg Fe/g dry weight). Patients randomized to deferoxamine who had LIC values less than 7 mg Fe/g dry weight were permitted to continue on their prior deferoxamine dose, even though the dose may have been higher than specified in the protocol.
Patients were to have a liver biopsy at baseline and end of study (after 12 months) for LIC. The primary efficacy endpoint was defined as a reduction in LIC of greater than or equal to 3 mg Fe/g dry weight for baseline values greater than or equal to 10 mg Fe/g dry weight, reduction of baseline values between 7 and less than 10 to less than 7 mg Fe/g dry weight, or maintenance or reduction for baseline values less than 7 mg Fe/g dry weight.
A total of 586 patients were randomized and treated, 296 with deferasirox tablets for oral suspension and 290 with deferoxamine. The mean age was 17.1 years (range, 2 to 53 years); 52% were females and 88% were Caucasian. The primary efficacy population consisted of 553 patients (deferasirox tablets for oral suspension n = 276; deferoxamine n = 277) who had LIC evaluated at baseline and 12 months or discontinued due to an adverse event. The percentage of patients achieving the primary endpoint was 52.9% for deferasirox tablets for oral suspension and 66.4% for deferoxamine. The relative efficacy of deferasirox to deferoxamine cannot be determined from this study.
In patients who had an LIC at baseline and at end of study, the mean change in LIC was -2.4 mg Fe/g dry weight in patients treated with deferasirox tablets for oral suspension and -2.9 mg Fe/g dry weight in patients treated with deferoxamine.
Reduction of LIC and serum ferritin was observed with deferasirox tablet for oral suspension doses of 20 to 30 mg per kg per day. Deferasirox tablets for oral suspension doses below 20 mg per kg per day failed to provide consistent lowering of LIC and serum ferritin levels (Figure 1). Therefore, a starting dose of 20 mg per kg per day is recommended [see Dosage and Administration (2.1)].
Figure 1. Changes in Liver Iron Concentration and Serum Ferritin Following Deferasirox Tablets for Oral Suspension (5 to 30 mg per kg per day) in Study 1
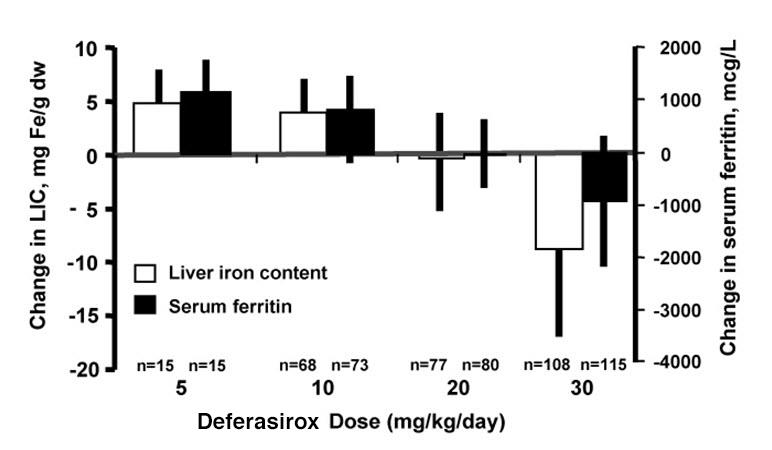
Study 2 (NCT00061763) was an open-label, noncomparative trial of efficacy and safety of deferasirox tablets for oral suspension given for 1 year to patients with chronic anemias and transfusional hemosiderosis. Similar to Study 1, patients received 5, 10, 20, or 30 mg per kg per day of deferasirox tablets for oral suspension based on baseline LIC.
A total of 184 patients were treated in this study: 85 patients with beta-thalassemia and 99 patients with other congenital or acquired anemias (myelodysplastic syndromes, n = 47; Diamond-Blackfan syndrome, n = 30; other, n = 22). Nineteen percent of patients were less than 16 years of age and 16% were greater than or equal to 65 years of age. There was a reduction in the absolute LIC from baseline to end of study (-4.2 mg Fe/g dry weight).
Study 3 (NCT00067080) was a multicenter, open-label, randomized trial of the safety and efficacy of deferasirox tablets for oral suspension relative to deferoxamine given for 1 year in patients with sickle cell disease and transfusional hemosiderosis. Patients were randomized to deferasirox tablets for oral suspension at doses of 5, 10, 20, or 30 mg per kg per day or subcutaneous deferoxamine at doses of 20-60 mg per kg per day for 5 days per week according to baseline LIC.
A total of 195 patients were treated in this study: 132 with deferasirox tablets for oral suspension and 63 with deferoxamine. Forty-four percent (44%) of patients were less than 16 years of age and 91% were black. At end of study, the mean change in LIC (as measured by magnetic susceptometry by a superconducting quantum interference device) in the per protocol-1 (PP-1) population, which consisted of patients who had at least 1 post- baseline LIC assessment, was -1.3 mg Fe/g dry weight for patients receiving deferasirox tablets for oral suspension (n = 113) and -0.7 mg Fe/g dry weight for patients receiving deferoxamine (n = 54).
One-hundred five (105) patients with thalassemia major and cardiac iron overload were enrolled in a study assessing the change in cardiac MRI T2* value [(measured in milliseconds, (ms)] before and after treatment with deferasirox. Cardiac T2* values at baseline ranged from 5 to less than 20 ms. The geometric mean of cardiac T2* in the 68 patients who completed 3 years of deferasirox tablets for oral suspension therapy increased from 11.98 ms at baseline to 17.12 ms at 3 years. Cardiac T2* values improved in patients with severe cardiac iron overload (less than 10 ms) and in those with mild to moderate cardiac iron overload (greater than or equal to 10 to less than 20 ms). The clinical significance of these observations is unknown.
Six hundred twenty-seven patients with MDS were enrolled across 5 uncontrolled trials. Two hundred thirty- nine of the 627 patients were enrolled in trials that limited enrollment to patients with IPSS Low or Intermediate 1 risk MDS, and the remaining 388 patients were enrolled in trials that did not specify MDS risk stratification but required a life expectancy of greater than 1 year. Planned duration of treatment in these trials ranged from 1 year (365 patients) to 5 years (47 patients). These trials evaluated the effects of deferasirox tablets for oral suspension therapy on parameters of iron overload, including LIC (125 patients) and serum ferritin (627 patients). The percent of patients completing planned duration of treatment was 51% in the largest 1-year study, 52% in the 3-year study and 22% in the 5-year study. The major causes for treatment discontinuation were withdrawal of consent, adverse reaction, and death. Over 1 year of follow-up across these pooled studies, mean change in serum ferritin was -332.8 (±2615.59) mcg/L (n = 593) and mean change in LIC was -5.9 (±8.32) mg Fe/g dw (n = 68). Results of these pooled studies in 627 patients with MDS suggest a progressive decrease in serum ferritin and LIC beyond 1 year in those patients who are able to continue deferasirox tablets for oral suspension.
Non-Transfusion-Dependent Thalassemia
Study 5 (NCT00873041) was a randomized, double-blind, placebo-controlled trial of treatment with deferasirox tablets for oral suspension for patients 10 years of age or older with NTDT syndromes and iron overload. Eligible patients had an LIC of at least 5 mg Fe/g dw measured by R2 MRI and a serum ferritin exceeding 300 mcg/L at screening (2 consecutive values at least 14 days apart from each other). A total of 166 patients were randomized, 55 to the deferasirox tablets for oral suspension 5 mg/kg/day dose group, 55 to the deferasirox tablets for oral suspension 10 mg/kg/day dose group, and 56 to placebo (28 to each matching placebo group). Doses could be increased after 6 months if the LIC exceeded 7 mg Fe/g dw and the LIC reduction from baseline was less than 15%. The patients enrolled included 89 males and 77 females. The underlying disease was betathalassemia intermedia in 95 (57%) patients, HbE beta-thalassemia in 49 (30%) patients, and alpha-thalassemia in 22 (13%) patients. There were 17 pediatric patients in the study. Caucasians comprised 57% of the study population and Asians comprised 42%. The median baseline LIC (range) for all patients was 12.1 (2.6 to 49.1) mg Fe/g dw. Follow-up was for 1 year. The primary efficacy endpoint of change in LIC from baseline to Week 52 was statistically significant in favor of both deferasirox dose groups compared with placebo (p less than or equal to 0.001) (Table 5). Furthermore, a statistically significant dose effect of deferasirox was observed in favor of the 10 mg/kg/day dose group (10 versus 5 mg/kg/day, p = 0.009). In a descriptive analysis, the target LIC (less than 5 mg Fe/g dw) was reached by 15 (27%) of 55 patients in the 10 mg/kg/day arm, 8 (15%) of 55 patients in the 5 mg/kg/day arm and 2 (4%) of 56 patients in the combined placebo groups.
Table 5. Absolute Change in LIC at Week 52 in NTDT Patients
Deferasirox tablets for oral suspension Starting Dose1
Placebo
5 mg/kg/day
10 mg/kg/day
20 mg/kg/day
Study 52
Number of Patients
n = 54
n = 51
n = 54
-
Mean LIC at Baseline (mg Fe/g dw)
16.1
13.4
14.4
-
Mean Change (mg Fe/g dw)
+0.4
-2.0
-3.8
-
(95% Confidence Interval)
(-0.6, +1.3)
(-2.9, -1.0)
(-4.8, -2.9)
-
Study 6
Number of Patients
-
n = 8
n = 77
n = 43
Mean LIC at Baseline (mg Fe/g dw)
-
5.6
8.8
23.5
Mean Change (mg Fe/g dw)
-
-1.5
-2.8
-9.1
(95% Confidence Interval)
-
(-3.7, +0.7)
(-3.4, -2.2)
(-11.0, -7.3)
1Randomized dose in Study 5 or assigned starting dose in Study 6
2Least square mean change for Study 5
Study 6 (NCT00873041) was an open-label trial of deferasirox tablets for oral suspension for the treatment of patients previously enrolled on Study 5, including cross-over to active treatment for those previously treated with placebo. The starting dose of deferasirox tablets for oral suspension in Study 6 was assigned based on the patient’s LIC at completion of Study 5, being 20 mg/kg/day for an LIC exceeding 15 mg Fe/g dw, 10 mg/kg/day for LIC 3 to 15 mg Fe/g dw, and observation if the LIC was less than 3 mg Fe/g dw. Patients could continue on 5 mg/kg/day if they had previously exhibited at least a 30% reduction in LIC. Doses could be increased to a maximum of 20 mg/kg/day after 6 months if the LIC was more than 7 mg Fe/g dw and the LIC reduction from baseline was less than 15%. The primary efficacy endpoint in Study 6 was the proportion of patients achieving an LIC less than 5 mg Fe/g dw. A total of 133 patients were enrolled. Twenty patients began Study 6 with an LIC less than 5 mg Fe/g dw. Of the 113 patients with a baseline LIC of at least 5 mg Fe/g dw in Study 6, the target LIC (less than 5 mg Fe/g dw) was reached by 39 (35%). The responders included 4 (10%) of 39 patients treated at 20 mg/kg/day for a baseline LIC exceeding 15 mg Fe/g dw, and 31 (51%) of 61 patients treated at 10 mg/kg/day for a baseline LIC between 5 and 15 mg Fe/g dw. The absolute change in LIC at Week 52 by starting dose is shown in Table 5.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
Deferasirox 90 mg tablets are light blue, oval shaped, biconvex, beveled edges, film coated tablet, debossed with “90” on one side and plain on other side. They are available in bottles of 30 tablets (NDC: 70700-269-30).
Deferasirox 360 mg tablets are dark blue, oval shaped, biconvex, beveled edges, film coated tablet, debossed with “360” on one side and plain on other side. They are available in bottles of 30 tablets (NDC: 70700-271-30).
Store deferasirox tablets at 25°C (77°F); excursions are permitted to 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature]. Protect from moisture.
-
17 PATIENT COUNSELING INFORMATION
Dosing Instructions
Advise patients to take deferasirox tablets with water or other liquids. Advise patients to swallow deferasirox tablets once daily with water or other liquids, preferably at the same time each day. Advise patients to take deferasirox tablets on an empty stomach or with a light meal (contains less than 7% fat content and approximately 250 calories). Examples of light meals include 1 whole wheat English muffin, 1 packet jelly (0.5 ounces), and skim milk (8 fluid ounces) or a turkey sandwich (2 oz. turkey on whole wheat bread w/lettuce, tomato, and 1 packet mustard). For patients who have difficulty swallowing whole tablets, deferasirox tablets may be crushed and mixed with soft foods (e.g., yogurt or applesauce) immediately prior to use and administered orally. Advise against the use of commercial crushers with serrated surfaces for crushing a single 90 mg tablet. Advise patients to immediately and completely consume the dose and not store it for future use [see Dosage and Administration (2.3)].
Blood Testing
Advise patients that blood tests will be performed frequently to check for damage to kidneys, liver, or blood cells [see Warnings and Precautions (5.1, 5.2, 5.3, 5.4, 5.5)].
Acute Kidney Injury, Including Acute Renal Failure
Caution patients about the potential for kidney toxicity when taking deferasirox tablets. Inform patients of the signs and symptoms of kidney injury. Advise patients to contact their healthcare provider immediately if they experience any of these symptoms [see Warnings and Precautions (5.1)].
Hepatic Toxicity and Failure
Caution patients about the potential for hepatic toxicity when taking deferasirox tablets. Inform patients of the signs and symptoms of hepatic toxicity. Advise patients to contact their healthcare provider immediately if they experience any of these symptoms [see Warnings and Precautions (5.2)].
Gastrointestinal Ulceration and Hemorrhage
Caution patients about the potential for the development of GI ulcers or bleeding when taking deferasirox in combination with drugs that have ulcerogenic or hemorrhagic potential, such as NSAIDs, corticosteroids, oral bisphosphonates, or anticoagulants.Inform patients of the signs and symptoms of GI ulcers or bleeding.Advise patients to contact their healthcare provider for symptoms of heartburn but to seek immediate medical attention for symptoms of gastrointestinal hemorrhage [see Warnings and Precautions (5.3)].
Allergic Reactions
Serious allergic reactions (which include swelling of the throat) have been reported in patients taking deferasirox, usually within the first month of treatment. If reactions are severe, advise patients to stop taking deferasirox immediately and seek immediate medical attention [see Warnings and Precautions (5.7)].
Severe Skin Reactions
Severe skin reactions have been reported in patients taking deferasirox tablets. Inform patients of the signs and symptoms of severe skin reactions. If reactions are severe, advise patients to stop taking deferasirox tablets immediately and seek immediate medical attention [see Warnings and Precautions (5.8)].
Skin Rash
Skin rashes may occur during deferasirox treatment. If the skin rash is severe, advise patients to stop taking deferasirox and seek medical attention [see Warnings and Precautions (5.9)].
Pediatric Patients with Acute IllnessInstruct pediatric patients and their caregivers to contact their healthcare provider during episodes of acute illness, especially if the patient has not been drinking fluids or the patient has volume depletion due to fever, vomiting, or diarrhea [see Warnings and Precautions (5.1)].
Auditory and Ocular Testing
Because auditory and ocular disturbances have been reported with deferasirox, conduct auditory testing and ophthalmic testing before starting deferasirox treatment and thereafter at regular intervals. Advise patients to contact their healthcare provider if they develop visual or auditory changes during treatment [see Warnings and Precautions (5.10)].
Drug Interactions
Caution patients not to take aluminum containing antacids and deferasirox tablets simultaneously [see Drug Interactions (7.1)].
Caution patients about potential loss of effectiveness of drugs metabolized by CYP3A4 (e.g., cyclosporine, simvastatin, hormonal contraceptive agents) when deferasirox is administered with these drugs [see Drug Interactions (7.2)].
Caution patients about potential loss of effectiveness of deferasirox when administered with drugs that are potent UGT inducers (e.g., rifampicin, phenytoin, phenobarbital, ritonavir). Based on serum ferritin levels and clinical response, consider increases in the dose of deferasirox when concomitantly used with potent UGT inducers [see Drug Interactions (7.5)].
Caution patients about potential loss of effectiveness of deferasirox when administered with drugs that are bile acid sequestrants (e.g., cholestyramine, colesevelam, colestipol). Based on serum ferritin levels and clinical response, consider increases in the dose of deferasirox when concomitantly used with bile acid sequestrants [see Drug Interactions (7.6)].
Caution patients with diabetes to monitor their glucose levels more frequently when repaglinide is used concomitantly with deferasirox [see Drug Interactions (7.3)].
Handling Instructions
Advise patients to store deferasirox in a dry, room-temperature environment [see How Supplied/Storage and Handling (16)].
Driving and Using Machines
Caution patients experiencing dizziness to avoid driving or operating machinery [see Adverse Reactions (6.1)].
The brands listed are trademarks of their respective owners and are not trademarks of Xiromed, LLC. The makers of these brands are not affiliated with and do not endorse Xiromed, LLC. or its products.
Manufactured for:

Xiromed, LLC.
Florham Park, NJ, 07932
Made in India
April 2020
PI269-03
MEDICATION GUIDE
Deferasirox (de FER a sir ox) Tablets
What is the most important information I should know about deferasirox tablets?
Deferasirox tablets can cause serious side effects, including:
Kidney problems: Deferasirox tablets can cause sudden (acute) kidney problems, including kidney failure that may require treatment with dialysis, and may cause death. Deaths have happened mostly in people who also have other health problems and had a blood disorder that was in an advanced stage. Adults and children who already have kidney problems and are taking certain medicines with deferasirox tablets may also have an increased risk of sudden kidney problems. Be sure to tell your healthcare provider about all the medicines you take during treatment with deferasirox tablets.
Your healthcare provider should do blood and urine tests to check your or your child’s kidney function before and during treatment with deferasirox tablets. Call your healthcare provider right away if:
- your child becomes sick with fever, vomiting, or diarrhea and cannot drink fluids normally during treatment with deferasirox tablets. Your child may be dehydrated. Your healthcare provider may need to temporarily stop treatment with deferasirox tablets and treat your child for dehydration to help prevent kidney problems. Your healthcare provider may monitor your child’s kidney function more closely.
- you notice that you or your child are passing less urine than usual during treatment with deferasirox tablets.
Liver problems. Deferasirox tablets can cause liver problems, including liver failure that can sometimes cause death. Liver problems with deferasirox tablets may be more common in people who are over 55 years of age but can also happen in children. Liver failure has happened more often in people with cirrhosis of the liver and failure of other organs. Liver failure has also happened along with kidney problems in certain children who become dehydrated. See “Kidney problems” above.
Your healthcare provider should do blood tests to check your liver function before you start and regularly during treatment with deferasirox tablets. Call your healthcare provider right away, if you develop any of the following signs and symptoms:
- drowsiness
- upper right stomach-area (abdomen) pain
- yellowing or increased yellowing of your skin or eyes
- dark urine
Bleeding, ulcers and tears of the stomach or intestine. Severe stomach and intestine bleeding (hemorrhage) that have caused death have happened in some people treated with deferasirox tablets, especially in elderly people who have advanced blood cancers or low platelet counts. Some people have also had ulcers of the stomach or intestine, sometimes with tears (perforation) that have caused death. In some people who have taken deferasirox tablets, including children and adolescents, irritation of the upper gastrointestinal tract, ulcers, and bleeding have happened, but did not cause death.
Your risk of severe bleeding (hemorrhage) may be increased if you take deferasirox tablets along with other medicines that can cause ulcers or bleeding, such as:
- nonsteroidal anti-inflammatory drugs (NSAIDs)
- corticosteroids,
- certain osteoporosis medicines called oral bisphosphonates
- blood thinner medicines
Before you start taking deferasirox tablets, tell your healthcare provider if you are taking one of these medicines. Ask your healthcare provider if you are not sure. If you develop an ulcer of the stomach or intestine, or severe bleeding, your healthcare provider may stop deferasirox tablets.
Elderly people may be at a higher risk of developing serious side effects and death due to serious side effects with deferasirox tablets. Your healthcare provider may need to monitor you more closely during treatment with deferasirox tablets.
- Tell your healthcare provider if you get heartburn during treatment with deferasirox tablets.
- Get emergency medical help right away if you vomit blood or pass black or bloody stools, or if you have severe stomach-area (abdomen) pain during treatment with deferasirox tablets.
See “What are possible side effects of deferasirox tablets?” for more information about side effects.
What are deferasirox tablets?
Deferasirox tablets are a prescription medicine that is used to treat:
- people 2 years of age and older who have an increased amount of iron in their blood for a long period of time (chronic), caused by repeated blood transfusions
- certain people 10 years of age or older with thalassemia who have an increased amount of iron in their blood but who are not receiving regular blood transfusions
It is not known if deferasirox tablets are safe and effective when used with other medicines to treat an increased amount of iron in the blood.
It is not known if deferasirox tablets are safe and effective for treating children under 2 years of age who have an increased amount of iron in their blood for a long period of time (chronic) caused by repeated blood transfusions.
It is not known if deferasirox tablets are safe and effective for treating children under 10 years of age with thalassemia who have an increased amount of iron in their blood, but who are not receiving regular blood transfusions.
Do not take deferasirox tablets if you:
- have certain kidney problems
- have high-risk myelodysplastic syndrome (MDS)
- have advanced cancer
- have a low platelet count
- are allergic to deferasirox or any of the ingredients in deferasirox tablets. See the end of this leaflet for a list of the ingredients in deferasirox tablets.
Ask your healthcare provider if you are not sure if you have any of the medical conditions listed above.
Before taking deferasirox tablets tell your healthcare provider about all of your medical conditions, including if you:
- have kidney problems
- have liver problems
- have advanced cancer. See “Do not take deferasirox tablets if you?”
- have a blood disorder that may increase your risk for bleeding
- are pregnant or plan to become pregnant. It is not known if deferasirox tablets can harm your unborn baby. Hormonal forms of birth control may not be as effective if used during treatment with deferasirox tablets. You could become pregnant. Talk to your healthcare provider about other birth control options that you can use during this time. Tell your healthcare provider right away if you become pregnant during treatment with deferasirox tablets.
- are breastfeeding or plan to breastfeed. It is not known if deferasirox passes into your breast milk and can harm your baby. You and your healthcare provider should decide if you will take deferasirox tablets or breastfeed. You should not do both.
Tell your healthcare provider about all the medicines you take, including prescription medicines, over-the-counter medicines, vitamins, and herbal supplements. Some medicines may affect how deferasirox tablets works, and deferasirox tablets may affect how other medicines work. Also, your risk of sudden kidney problems or severe bleeding may be increased if you take deferasirox tablets with certain medicines. See “What is the most important information I should know about deferasirox tablets?”
- Avoid taking the following medicines during treatment with deferasirox tablets:
- antacid products (medicines used to treat heartburn) that contain aluminum
- theophylline
- certain medicines to lower your cholesterol, called bile acid sequestrants.
Ask your healthcare provider if you are not sure if you take one of these medicines.
Know the medicines you take. Keep a list of them to show your healthcare provider or pharmacist when you get a new medicine.
How should I take deferasirox tablets?
- Take deferasirox tablets exactly as your healthcare provider tells you.
- Do not change your dose of deferasirox tablets or stop taking unless your healthcare provider tells you to.
- Deferasirox comes as tablets.
- You may take deferasirox tablets on an empty stomach or with a light meal. Examples of a light meal include:
- 1 whole wheat English muffin, 1 packet of jelly (0.5 ounce), and skim milk (8 fluid ounces), or
- A turkey sandwich (2 ounces of turkey on whole wheat bread with lettuce, tomato, and 1 packet of mustard)
- You may take deferasirox tablets on an empty stomach or with a light meal. Examples of a light meal include:
Taking deferasirox tablets:
- Take deferasirox tablets by mouth with water or other liquids 1 time every day, preferably at the same time every day.
- If you have trouble swallowing the tablets whole, you may crush deferasirox tablets and mix them with soft foods such as yogurt or apple sauce right before taking your dose.
- Take the dose right away. Do not save any of the deferasirox tablets and soft food mixture for later use.
- Do not use store-bought pill crushers with serrated surfaces for crushing deferasirox 90 mg tablets.
- Do not take deferasirox tablets with aluminum-containing antacid products. See “Before taking deferasirox tablets”
- Tell your healthcare provider if you or your child gain or lose any weight. Your or your child’s dose of deferasirox tablets may need to be adjusted.
- If you take the diabetes medicine repaglinide during treatment with deferasirox tablets you may need to test your blood sugar (glucose) levels more often. Follow your healthcare provider’s instructions about how often to test your blood sugar during this time.
- Your healthcare provider should do blood and urine tests before, and during treatment to check how you respond to deferasirox tablets, and to monitor you for side effects. Your healthcare provider may change your dose, temporarily or permanently stop deferasirox tablets if you have certain side effects.
- In people who have thalassemia, your healthcare provider will check the amount of iron in your liver before and during treatment with deferasirox tablets.
- If you or your child take too much deferasirox tablets, call your healthcare provider right away or go to the nearest hospital emergency room.
What should I avoid while taking deferasirox tablets?
- Deferasirox tablets may cause dizziness. Avoid driving or operating machinery until you know how deferasirox tablets affect you. Do not drive or operate machinery if deferasirox tablets make you dizzy.
What are the possible side effects of deferasirox tablets?
Deferasirox tablets can cause serious side effects, including:
- See “What is the most important information I should know about deferasirox tablets?”
- Effects on your bone marrow. Deferasirox tablets can affect your bone marrow and cause you to have low white blood cell count which can be serious, decreased platelets, or worsening of your anemia, and may lead to death. Your risk for effects on your bone marrow may be increased if you already have other blood disorders. Your healthcare provider will do blood tests to monitor your blood cell counts for these problems.
-
Serious allergic reactions. Deferasirox tablets may cause serious allergic reactions, which usually start within the first month of treatment. Get medical help right away if you develop any of the following symptoms of a serious allergic reaction including:
- difficulty in breathing or swallowing
- chest pain
- rapid heartbeat
- feeling faint
- swelling of the face, lips, mouth, tongue or throat
- severe itching of the skin with a red rash or raised bumps
- hives
-
Skin rash and severe skin reactions. Skin rashes are common with deferasirox tablets. If you get a more severe rash, your healthcare provider may temporarily stop deferasirox tablets. Severe skin reactions can also happen with deferasirox tablets and can be life-threatening or lead to death. Get medical help right away if you develop any one or more of the following signs and symptoms of a severe skin reaction, including:
- rash or red skin
- blisters on your lips, or around your mouth or eyes
- mouth sores
- skin peeling
- high fever or flu-like symptoms
- enlarged lymph nodes
- Hearing and vision problems. Deferasirox tablets can cause decreased hearing and changes in your vision including cataracts, increased pressure in your eye, and problems with your retinas. Your healthcare provider should do hearing and vision tests before you start and then regularly during treatment. Your healthcare provider may decrease your dose or stop deferasirox tablets if you develop hearing or vision problems.
The most common side effects in anyone who takes deferasirox tablets include: diarrhea and nausea. Other common side effects in people with too much iron in their blood due to repeated blood transfusions include: vomiting, stomach-area (abdomen) pain, and an abnormal kidney function blood test.
These are not all the possible side effects of deferasirox tablets.
Call your doctor for medical advice about side effects. You may report side effects to Xiromed, LLC. at 1-844-XIROMED (1-844-947-6633) or FDA at 1-800-FDA-1088.
How should I store deferasirox tablets?
- Store deferasirox tablets at room temperature 25°C (77°F); excursion are permitted to 15°C to 30°C (59°F to 86°F).
- Keep the bottle closed tightly and away from moisture.
Keep deferasirox tablets and all medicines out of the reach of children.
General information about the safe and effective use of deferasirox tablets
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use deferasirox tablets for a condition for which it was not prescribed. Do not give it to other people, even if they have the same symptoms you have. It may harm them. You can ask your healthcare provider or pharmacist for more information about deferasirox tablets.
What are the ingredients in deferasirox tablets?
Active ingredient: deferasirox
Inactive ingredients: crospovidone, magnesium stearate, microcrystalline cellulose, poloxamer, povidone and talc. The film coating contains opadry blue which has ingredients FD&C blue, hypromellose, polyethylene glycol, talc and titanium dioxide.
Manufactured for:
Xiromed, LLC.
Florham Park, NJ, 07932
Made in India
For more information, go to www.Xiromed.com or call 1-844-XIROMED (1-844-947-6633).
This Medication Guide has been approved by the U.S. Food and Drug Administration.
April 2020
PIL269-02
- PACKAGE LABEL.PRINCIPAL DISPLAY PANEL - Deferasirox Tablets 90 mg
- PACKAGE LABEL.PRINCIPAL DISPLAY PANEL - Deferasirox Tablets 360 mg
-
INGREDIENTS AND APPEARANCE
DEFERASIROX
deferasirox tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 70700-269 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength DEFERASIROX (UNII: V8G4MOF2V9) (DEFERASIROX - UNII:V8G4MOF2V9) DEFERASIROX 90 mg Inactive Ingredients Ingredient Name Strength CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) CROSPOVIDONE (UNII: 68401960MK) POVIDONE K30 (UNII: U725QWY32X) POLOXAMER 188 (UNII: LQA7B6G8JG) TALC (UNII: 7SEV7J4R1U) MAGNESIUM STEARATE (UNII: 70097M6I30) HYPROMELLOSE 2910 (6 MPA.S) (UNII: 0WZ8WG20P6) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) POLYETHYLENE GLYCOL 6000 (UNII: 30IQX730WE) FD&C BLUE NO. 2--ALUMINUM LAKE (UNII: 4AQJ3LG584) Product Characteristics Color BLUE (Light blue) Score no score Shape OVAL Size 11mm Flavor Imprint Code 90 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 70700-269-30 30 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 01/02/2020 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA212995 01/02/2020 DEFERASIROX
deferasirox tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 70700-271 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength DEFERASIROX (UNII: V8G4MOF2V9) (DEFERASIROX - UNII:V8G4MOF2V9) DEFERASIROX 360 mg Inactive Ingredients Ingredient Name Strength CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) CROSPOVIDONE (UNII: 68401960MK) POVIDONE K30 (UNII: U725QWY32X) POLOXAMER 188 (UNII: LQA7B6G8JG) TALC (UNII: 7SEV7J4R1U) MAGNESIUM STEARATE (UNII: 70097M6I30) HYPROMELLOSE 2910 (6 MPA.S) (UNII: 0WZ8WG20P6) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) POLYETHYLENE GLYCOL 6000 (UNII: 30IQX730WE) FD&C BLUE NO. 2--ALUMINUM LAKE (UNII: 4AQJ3LG584) Product Characteristics Color BLUE (Dark blue) Score no score Shape OVAL Size 17mm Flavor Imprint Code 360 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 70700-271-30 30 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 01/02/2020 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA212995 01/02/2020 Labeler - Xiromed, LLC (080228637) Registrant - Piramal Healthcare UK Limited (345609965) Establishment Name Address ID/FEI Business Operations Piramal Enterprises Limited 862202793 ANALYSIS(70700-269, 70700-271) , MANUFACTURE(70700-269, 70700-271) , PACK(70700-269, 70700-271)


© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.