EXTAVIA- interferon beta-1b kit
EXTAVIA by
Drug Labeling and Warnings
EXTAVIA by is a Prescription medication manufactured, distributed, or labeled by Novartis Pharmaceuticals Corporation. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use EXTAVIA safely and effectively. See full prescribing information for EXTAVIA.
EXTAVIA® (interferon beta-1b) for injection, for subcutaneous use
Initial U.S. Approval: 1993
RECENT MAJOR CHANGES
Indications and Usage (1)
8/2019
INDICATIONS AND USAGE
EXTAVIA is an interferon beta indicated for the treatment of relapsing forms of multiple sclerosis (MS), to include clinically isolated syndrome, relapsing-remitting disease, and active secondary progressive disease, in adults. (1)
DOSAGE AND ADMINISTRATION
- For subcutaneous use only. (2.1)
- The recommended dose is 0.25 mg every other day. Generally, start at 0.0625 mg (0.25 mL) every other day, and increase over a six-week period to 0.25 mg (1 mL) every other day. (2.1)
- Reconstitute lyophilized powder with supplied diluent; the removable diluent cap contains natural rubber latex. (2.2)
DOSAGE FORMS AND STRENGTHS
For injection: 0.3 mg of lyophilized powder in a single-dose vial for reconstitution. (3)
CONTRAINDICATIONS
History of hypersensitivity to natural or recombinant interferon beta, albumin or mannitol. (4)
WARNINGS AND PRECAUTIONS
- Hepatic Injury: Monitor liver function tests and signs and symptoms of hepatic injury; consider discontinuing EXTAVIA if serious hepatic injury occurs. (5.1, 5.11)
- Anaphylaxis and Other Allergic Reactions: Discontinue if anaphylaxis occurs. (5.2)
- Depression and Suicide: Advise patients to immediately report any symptom of depression and/or suicidal ideation; consider discontinuation of EXTAVIA if depression occurs. (5.3)
- Congestive Heart Failure (CHF): Monitor patients with CHF for worsening of cardiac symptoms; consider discontinuation of EXTAVIA if worsening of CHF occurs. (5.4)
- Injection Site Necrosis and Reactions: Do not administer EXTAVIA into affected area until fully healed; if multiple lesions occur, discontinue EXTAVIA until healing of skin lesions. (5.5)
- Leukopenia: Monitor complete blood count. (5.6, 5.11)
- Thrombotic Microangiopathy (TMA): Cases of thrombotic microangiopathy have been reported. Discontinue EXTAVIA if clinical symptoms and laboratory findings consistent with TMA occur. (5.7)
- Flu-like Symptom Complex: Consider analgesics and/or antipyretics on injection days. (5.8)
- Drug-induced Lupus Erythematosus: Cases of drug-induced lupus erythematosus have been reported. Discontinue EXTAVIA if patients develop new characteristic signs and symptoms. (5.10)
ADVERSE REACTIONS
In controlled clinical trials, the most common adverse reactions (at least 5% more frequent on interferon beta-1b than on placebo) were: injection-site reaction, lymphopenia, flu-like symptoms, myalgia, leukopenia, neutropenia, increased liver enzymes, headache, hypertonia, pain, rash, insomnia, abdominal pain, and asthenia. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Novartis Pharmaceuticals Corporation at 1-888-669-6682 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.USE IN SPECIFIC POPULATIONS
Pregnancy: Based on animal data, may cause fetal harm (8.1)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 8/2019
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Dosing Information
2.2 Reconstitution of the Lyophilized Powder
2.3 Important Administration Instructions
2.4 Premedication for Flu-like Symptoms
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hepatic Injury
5.2 Anaphylaxis and Other Allergic Reactions
5.3 Depression and Suicide
5.4 Congestive Heart Failure
5.5 Injection-Site Necrosis and Reactions
5.6 Leukopenia
5.7 Thrombotic Microangiopathy
5.8 Flu-like Symptom Complex
5.9 Seizures
5.10 Drug-induced Lupus Erythematosus
5.11 Monitoring for Laboratory Abnormalities
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Immunogenicity
6.3 Postmarketing Experience
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Stability and Storage
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Dosing Information
The recommended starting dose is 0.0625 mg (0.25 mL) subcutaneously every other day, with dose increases over a six-week period to the recommended dose of 0.25 mg (1 mL) every other day (see Table 1).
Table 1: Schedule for Dose Titration aDosed every other day, subcutaneously. EXTAVIA
Dosea
Percentage of
Recommended DoseVolume
Weeks 1-2
0.0625 mg
25%
0.25 mL
Weeks 3-4
0.125 mg
50%
0.5 mL
Weeks 5-6
0.1875 mg
75%
0.75 mL
Week 7 and thereafter
0.25 mg
100%
1 mL
If a dose of EXTAVIA is missed, then it should be taken as soon as the patient remembers or is able to take it. The patient should not take EXTAVIA on two consecutive days. The next injection should be taken about 48 hours (two days) after that dose. If the patient accidentally takes more than their prescribed dose, or takes it on two consecutive days, they should be instructed to call their healthcare provider immediately.
2.2 Reconstitution of the Lyophilized Powder
(a) Prior to reconstitution, verify that the vial containing lyophilized EXTAVIA is not cracked or damaged. Do not use cracked or damaged vials.
(b) To reconstitute lyophilized EXTAVIA for injection, attach the pre-filled syringe containing the diluent (0.54% Sodium Chloride Solution, USP) to the EXTAVIA vial using the vial adapter.
The removable rubber cap of the diluent (0.54% Sodium Chloride Solution, USP) pre-filled syringe contains natural rubber latex, which may cause allergic reactions and should not be handled by latex-sensitive individuals.
(c) Slowly inject 1.2 mL of diluent into the EXTAVIA vial.
(d) Gently swirl the vial to dissolve the lyophilized powder completely; do not shake. Foaming may occur during reconstitution or if the vial is swirled or shaken too vigorously. If foaming occurs, allow the vial to sit undisturbed until the foam settles.
(e) 1 mL of reconstituted EXTAVIA solution contains 0.25 mg of interferon beta-1b.
(f) After reconstitution, if not used immediately, refrigerate the reconstituted EXTAVIA solution at 35°F to 46°F (2°C to 8°C) and use within three hours. Do not freeze.
2.3 Important Administration Instructions
(a) Perform the first EXTAVIA injection under the supervision of an appropriately qualified healthcare professional. If patients or caregivers are to administer EXTAVIA, train them in the proper subcutaneous injection technique and assess their ability to inject subcutaneously to ensure the proper administration of EXTAVIA.
(b) Visually inspect the reconstituted EXTAVIA solution before use; discard if it contains particulate matter or is discolored.
(c) Keeping the syringe and vial adapter in place, turn the assembly over so that the vial is on top. Withdraw the appropriate dose of EXTAVIA solution. Remove the vial from the vial adapter before injecting EXTAVIA.
(d) Use safe disposal procedures for needles and syringes.
(e) Do not re-use needles or syringes.
(f) Advise patients and caregivers to rotate sites for subcutaneous injections to minimize the likelihood of severe injection site reactions, including necrosis or localized infection.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Hepatic Injury
Severe hepatic injury including cases of hepatic failure, some of which have been due to autoimmune hepatitis, has been rarely reported in patients taking EXTAVIA. In some cases, these events have occurred in the presence of other drugs or comorbid medical conditions that have been associated with hepatic injury. Consider the potential risk of EXTAVIA used in combination with known hepatotoxic drugs or other products (e.g., alcohol) prior to EXTAVIA administration, or when adding new agents to the regimen of patients already on EXTAVIA. Monitor patients for signs and symptoms of hepatic injury. Consider discontinuing EXTAVIA if serum transaminase levels significantly increase, or if they are associated with clinical symptoms such as jaundice.
Asymptomatic elevation of serum transaminases is common in patients treated with EXTAVIA. In controlled clinical trials, elevations of serum glutamic-pyruvic transaminase (SGPT) to greater than five times baseline value were reported in 12% of patients receiving interferon beta-1b (compared to 4% on placebo), and increases of serum glutamic-oxaloacetic transaminase (SGOT) to greater than five times baseline value were reported in 4% of patients receiving interferon beta-1b (compared to 1% on placebo), leading to dose-reduction or discontinuation of treatment in some patients [see Adverse Reactions (6.1)]. Monitor liver function tests [see Warnings and Precautions (5.11)].
5.2 Anaphylaxis and Other Allergic Reactions
Anaphylaxis has been reported as a rare complication of interferon beta-1b use. Other allergic reactions have included dyspnea, bronchospasm, tongue edema, skin rash, and urticaria [see Adverse Reactions (6.1)]. Discontinue EXTAVIA if anaphylaxis occurs.
The removable rubber cap of the diluent (0.54% Sodium Chloride Solution, USP) pre-filled syringe contains natural rubber latex, which may cause allergic reactions and should not be handled by latex-sensitive individuals. The safe use of EXTAVIA pre-filled syringe in latex-sensitive individuals has not been studied.
5.3 Depression and Suicide
Depression and suicide have been reported to occur with increased frequency in patients receiving interferon beta products, including interferon beta-1b. Advise patients to report any symptom of depression and/or suicidal ideation to their healthcare provider. If a patient develops depression, discontinuation of EXTAVIA therapy should be considered.
In randomized controlled clinical trials, there were three suicides and eight suicide attempts among the 1532 patients on interferon beta-1b compared to one suicide and four suicide attempts among 965 patients on placebo.
5.4 Congestive Heart Failure
Monitor patients with preexisting congestive heart failure (CHF) for worsening of their cardiac condition during initiation of and continued treatment with EXTAVIA. While beta interferons do not have any known direct-acting cardiac toxicity, cases of CHF, cardiomyopathy, and cardiomyopathy with CHF have been reported in patients without known predisposition to these events, and without other known etiologies being established. In some cases, these events have been temporally related to the administration of interferon beta-1b. Recurrence upon rechallenge was observed in some patients. Consider discontinuation of EXTAVIA if worsening of CHF occurs with no other etiology.
5.5 Injection-Site Necrosis and Reactions
Injection-site necrosis (ISN) was reported in 4% of interferon beta-1b-treated patients in controlled clinical trials (compared to 0% on placebo) [see Adverse Reactions (6.1)]. Typically, ISN occurs within the first four months of therapy, although postmarketing reports have been received of ISN occurring over one year after initiation of therapy. The necrotic lesions are typically 3 cm or less in diameter, but larger areas have been reported. Generally the necrosis has extended only to subcutaneous fat, but has extended to the fascia overlying muscle. In some lesions where biopsy results are available, vasculitis has been reported. For some lesions, debridement, and/or skin grafting have been required. In most cases healing was associated with scarring.
Whether to discontinue therapy following a single site of necrosis is dependent on the extent of necrosis. For patients who continue therapy with EXTAVIA after injection-site necrosis has occurred, avoid administration of EXTAVIA into the affected area until it is fully healed. If multiple lesions occur, discontinue therapy until healing occurs.
Periodically evaluate patient understanding and use of aseptic self-injection techniques and procedures, particularly if injection site necrosis has occurred.
In controlled clinical trials, injection-site reactions occurred in 78% of patients receiving interferon beta-1b with injection-site necrosis in 4%. Injection-site inflammation (42%), injection-site pain (16%), injection-site hypersensitivity (4%), injection-site necrosis (4%), injection-site mass (2%), injection-site edema (2%), and nonspecific reactions were significantly associated with interferon beta-1b treatment. The incidence of injection-site reactions tended to decrease over time. Approximately 69% of patients experienced injection-site reactions during the first three months of treatment, compared to approximately 40% at the end of the studies.
5.6 Leukopenia
In controlled clinical trials, leukopenia was reported in 18% of patients receiving interferon beta-1b (compared to 6% on placebo), leading to a reduction of the dose of interferon beta-1b in some patients [see Adverse Reactions (6.1)]. Monitoring of complete blood and differential white blood cell counts is recommended. Patients with myelosuppression may require more intensive monitoring of complete blood cell counts, with differential and platelet counts.
5.7 Thrombotic Microangiopathy
Cases of thrombotic microangiopathy (TMA), including thrombotic thrombocytopenic purpura and hemolytic uremic syndrome, some fatal, have been reported with interferon beta products, including EXTAVIA. Cases have been reported several weeks to years after starting interferon beta products. Discontinue EXTAVIA if clinical symptoms and laboratory findings consistent with TMA occur, and manage as clinically indicated.
5.8 Flu-like Symptom Complex
In controlled clinical trials, the rate of flu-like symptom complex for patients on interferon beta-1b was 57% [see Adverse Reactions (6.1)]. The incidence decreased over time, with 10% of patients reporting flu-like symptom complex at the end of the studies. The median duration of flu-like symptom complex in Study 1 was 7.5 days [see Clinical Studies (14)]. Analgesics and/or antipyretics on treatment days may help ameliorate flu-like symptoms associated with EXTAVIA use.
5.9 Seizures
Seizures have been temporally associated with the use of beta interferons in clinical trials and postmarketing safety surveillance. It is not known whether these events were related to a primary seizure disorder, the effects of MS alone, the use of beta interferons, other potential precipitants of seizures (e.g., fever), or to some combination of these.
5.10 Drug-induced Lupus Erythematosus
Cases of drug-induced lupus erythematosus have been reported with some interferon beta products, including EXTAVIA. Signs and symptoms of drug-induced lupus reported in EXTAVIA-treated patients have included rash, serositis, polyarthritis, nephritis, and Raynaud’s phenomenon. Cases have occurred with positive serologic testing (including positive anti-nuclear and/or anti-double-stranded DNA antibody testing). If EXTAVIA-treated patients develop new signs and symptoms characteristic of this syndrome, EXTAVIA therapy should be stopped.
5.11 Monitoring for Laboratory Abnormalities
In addition to those laboratory tests normally required for monitoring patients with MS, complete blood and differential white blood cell counts, platelet counts and blood chemistries, including liver function tests, are recommended at regular intervals (one, three, and six months) following introduction of EXTAVIA therapy, and then periodically thereafter in the absence of clinical symptoms.
-
6 ADVERSE REACTIONS
The following serious adverse reactions are discussed in more details in other sections of labeling:
- Hepatic Injury [see Warnings and Precautions (5.1)]
- Anaphylaxis and Other Allergic Reactions [see Warnings and Precautions (5.2)]
- Depression and Suicide [see Warnings and Precautions (5.3)]
- Congestive Heart Failure [see Warnings and Precautions (5.4)]
- Injection-Site Necrosis and Reactions [see Warnings and Precautions (5.5)]
- Leukopenia [see Warnings and Precautions (5.6)]
- Thrombotic Microangiopathy [see Warnings and Precautions (5.7)]
- Flu-like Symptom Complex [see Warnings and Precautions (5.8)]
- Seizures [see Warnings and Precautions (5.9)]
- Drug-induced Lupus Erythematosus [see Warnings and Precautions (5.10)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions and over varying lengths of time, adverse reaction rates observed in the clinical trials of interferon beta-1b cannot be directly compared to rates in clinical trials of other drugs, and may not reflect the rates observed in practice.
Among 1407 patients with MS treated with interferon beta-1b 0.25 mg every other day (including 1261 patients treated for greater than one year), the most commonly reported adverse reactions (at least 5% more frequent on interferon beta-1b than on placebo) were injection-site reaction, lymphopenia, flu-like symptoms, myalgia, leukopenia, neutropenia, increased liver enzymes, headache, hypertonia, pain, rash, insomnia, abdominal pain, and asthenia. The most frequently reported adverse reactions resulting in clinical intervention (for example, discontinuation of interferon beta-1b, adjustment in dosage, or the need for concomitant medication to treat an adverse reaction symptom) were depression, flu-like symptom complex, injection-site reactions, leukopenia, increased liver enzymes, asthenia, hypertonia, and myasthenia.
Table 2 enumerates adverse reactions and laboratory abnormalities that occurred among patients treated with 0.25 mg of interferon beta-1b every other day by subcutaneous injection in the pooled placebo-controlled trials (Study 1-4) at an incidence that was at least 2% more than that observed in the placebo-treated patients [see Clinical Studies (14)].
Table 2: Adverse Reactions and Laboratory Abnormalities in Patients with MS in Pooled Studies 1, 2, 3, and 4 a"Injection-site reaction" comprises all adverse reactions occurring at the injection site (except injection-site necrosis), that is, the following terms: injection-site reaction, injection-site hemorrhage, injection-site hypersensitivity, injection-site inflammation, injection-site mass, injection-site pain, injection-site edema and injection-site atrophy.
b"Flu-like symptom (complex)" denotes flu syndrome and/or a combination of at least two adverse reactions from fever, chills, myalgia, malaise, sweating.Adverse Reaction Placebo
(N = 965)Interferon beta-1b
(N = 1407)Blood and lymphatic system disorders Lymphocytes count decreased (< 1500/mm3) 66% 86% Absolute neutrophil count decreased (< 1500/mm3) 5% 13% White blood cell count decreased (< 3000/mm3) 4% 13% Lymphadenopathy 3% 6% Nervous system disorders Headache 43% 50% Insomnia 16% 21% Incoordination 15% 17% Vascular disorders Hypertension 4% 6% Respiratory, thoracic, and mediastinal disorders Dyspnea 3% 6% Gastrointestinal disorders Abdominal pain 11% 16% Hepatobiliary disorders Alanine aminotransferase increased
(SGPT > 5 times baseline)4% 12% Aspartate aminotransferase increased
(SGOT > 5 times baseline)1% 4% Skin and subcutaneous tissue disorders Rash 15% 21% Skin disorder 8% 10% Musculoskeletal and
connective tissue disordersHypertonia 33% 40% Myalgia 14% 23% Renal and urinary disorders Urinary urgency 8% 11% Reproductive system and breast disorders Metrorrhagia 7% 9% Impotence 6% 8% General disorders and administration-site conditions Injection-site reactiona 26% 78% Asthenia 48% 53% Flu-like symptoms (complex)b 37% 57% Pain 35% 42% Fever 19% 31% Chills 9% 21% Peripheral edema 10% 12% Chest pain 6% 9% Malaise 3% 6% Injection-site necrosis 0% 4% In addition to the adverse reactions listed in Table 2, the following adverse reactions occurred more frequently on interferon beta-1b than on placebo, but with a difference smaller than 2%: alopecia, anxiety, arthralgia, constipation, diarrhea, dizziness, dyspepsia, dysmenorrhea, leg cramps, menorrhagia, myasthenia, nausea, nervousness, palpitations, peripheral vascular disorder, prostatic disorder, tachycardia, urinary frequency, vasodilatation, and weight increase.
Laboratory Abnormalities
In the four clinical trials (Studies 1, 2, 3, and 4), leukopenia was reported in 18% and 6% of patients in interferon beta-1b- and placebo-treated groups, respectively. No patients were withdrawn or dose reduced for neutropenia in Study 1. Three percent (3%) of patients in Studies 2 and 3 experienced leukopenia and were dose-reduced. Other abnormalities included increase of SGPT to greater than five times baseline value (12%), and increase of SGOT to greater than five times baseline value (4%). In Study 1, two patients were dose reduced for increased hepatic enzymes; one continued on treatment and one was ultimately withdrawn. In Studies 2 and 3, 1.5% of interferon beta-1b patients were dose-reduced or interrupted treatment for increased hepatic enzymes. In Study 4, 1.7% of patients were withdrawn from treatment due to increased hepatic enzymes, two of them after a dose reduction. In Studies 1-4, nine (0.6%) patients were withdrawn from treatment with interferon beta-1b for any laboratory abnormality, including four (0.3%) patients following dose reduction.
6.2 Immunogenicity
As with all therapeutic proteins, there is a potential for immunogenicity. Serum samples were monitored for the development of antibodies to interferon beta-1b during Study 1. In patients receiving 0.25 mg every other day 56/124 (45%) were found to have serum neutralizing activity at one or more of the time points tested. In Study 4, neutralizing activity was measured every 6 months and at end of study. At individual visits after start of therapy, activity was observed in 17% up to 25% of the interferon beta-1b-treated patients. Such neutralizing activity was measured at least once in 75 (30%) out of 251 interferon beta-1b patients who provided samples during treatment phase; of these, 17 (23%) converted to negative status later in the study. Based on all the available evidence, the relationship between antibody formation and clinical safety or efficacy is not known.
These data reflect the percentage of patients whose test results were considered positive for antibodies to interferon beta-1b using a biological neutralization assay that measures the ability of immune sera to inhibit the production of the interferon-inducible protein, MxA. Neutralization assays are highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of neutralizing activity in an assay may be influenced by several factors including sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to interferon beta-1b with the incidence of antibodies to other products may be misleading.
Anaphylactic reactions have been reported with the use of interferon beta-1b [see Warnings and Precautions (5.2)].
6.3 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of interferon beta-1b. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Blood and lymphatic system disorders: Anemia, Thrombocytopenia
Endocrine disorders: Hypothyroidism, Hyperthyroidism, Thyroid dysfunction
Metabolism and nutrition disorders: Triglyceride increased, Anorexia, Weight decrease, Weight increase
Psychiatric disorders: Anxiety, Confusion, Emotional lability
Nervous system disorders: Convulsion, Dizziness, Psychotic symptoms
Cardiac disorders: Cardiomyopathy, Palpitations, Tachycardia
Vascular disorders: Vasodilatation
Respiratory, thoracic, and mediastinal disorders: Bronchospasm
Gastrointestinal disorders: Diarrhea, Nausea, Pancreatitis, Vomiting
Hepatobiliary disorders: Hepatitis, Gamma GT increased
Skin and subcutaneous tissue disorders: Alopecia, Pruritus, Skin discoloration, Urticaria
Musculoskeletal and connective tissue disorders: Arthralgia; drug-induced lupus erythematosus
Reproductive system and breast disorder: Menorrhagia
General disorders and administration site conditions: Fatal capillary leak syndrome*
*The administration of cytokines to patients with a preexisting monoclonal gammopathy has been associated with the development of this syndrome.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Although there have been no well-controlled studies in pregnant women, available data, which include prospective observational studies, have not generally indicated a drug-associated risk of major birth defects with interferon beta-1b during pregnancy.
Administration of interferon beta-1b to monkeys during gestation resulted in increased embryo-fetal death at or above exposures greater than 3 times the human therapeutic dose (see Animal Data).
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2%-4% and 15%-20%, respectively. The background risk of major birth defects and miscarriage for the indicated population is unknown.
Data
Human Data
The majority of the observational studies reporting on pregnancies exposed to interferon beta-1b did not identify an association between the use of interferon beta-1b during pregnancy and an increased risk of major birth defects.
Animal Data
When interferon beta-1b (doses ranging from 0.028 to 0.42 mg/kg/day) was administered to pregnant rhesus monkeys throughout the period of organogenesis (gestation days 20 to 70), a dose-related increase in the incidence of abortion was observed. The low-effect dose is approximately 3 times the recommended human dose of 0.25 mg on a body surface area (mg/m2) basis. A no-effect dose for embryo-fetal developmental toxicity in rhesus monkeys was not established.
8.2 Lactation
Risk Summary
There are no data on the presence of interferon beta-1b in human milk, the effects on the breastfed infant, or the effects of the drug on milk production.
The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for EXTAVIA and any potential adverse effects on the breastfed child from EXTAVIA or from the underlying maternal condition.
-
11 DESCRIPTION
EXTAVIA (interferon beta-lb) is a purified, sterile, lyophilized protein product produced by recombinant DNA techniques. Interferon beta-1b is manufactured by bacterial fermentation of a strain of Escherichia coli that bears a genetically engineered plasmid containing the gene for human interferon betaser17. The native gene was obtained from human fibroblasts and altered in a way that substitutes serine for the cysteine residue found at position 17. Interferon beta-1b has 165 amino acids and an approximate molecular weight of 18,500 daltons. It does not include the carbohydrate side chains found in the natural material.
The specific activity of EXTAVIA is approximately 32 million international units (IU)/mg interferon beta-lb. Each vial contains 0.3 mg of interferon beta-lb. The unit measurement is derived by comparing the antiviral activity of the product to the World Health Organization (WHO) reference standard of recombinant human interferon beta. Mannitol, USP and Albumin (Human), USP (15 mg each/vial) are added as stabilizers.
EXTAVIA (interferon beta-1b) for injection is a sterile, preservative-free, white to off-white lyophilized powder, for subcutaneous injection after reconstitution with the diluent supplied (0.54% Sodium Chloride Solution, USP). Each vial contains Albumin (Human) USP (15 mg) and Mannitol, USP (15 mg).
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
The mechanism of action of EXTAVIA (interferon beta-1b) in patients with MS is unknown.
12.2 Pharmacodynamics
Interferons (IFNs) are a family of naturally occurring proteins, produced by eukaryotic cells in response to viral infection and other biologic agents. Three major types of interferons have been defined: type 1 (IFN-alpha, beta, epsilon, kappa, and omega), type II (IFN-gamma) and type III (IFN-lambda). Interferon-beta is a member of the type I subset of interferons. The type I interferons have considerably overlapping but also distinct biologic activities. The bioactivities of all IFNs, including IFN-beta, are induced via their binding to specific receptors on the membranes of human cells. Differences in the bioactivities induced by the three major subtypes of IFNs likely reflect differences in the signal transduction pathways induced by signaling through their cognate receptors.
Interferon beta-1b receptor binding induces the expression of proteins that are responsible for the pleiotropic bioactivities of interferon beta-1b. A number of these proteins (including neopterin, β2-microglobulin, MxA protein, and IL-10) have been measured in blood fractions from interferon beta-1b-treated patients and interferon beta-1b-treated healthy volunteers. Immunomodulatory effects of interferon beta-1b include the enhancement of suppressor T-cell activity, reduction of pro-inflammatory cytokine production, down-regulation of antigen presentation, and inhibition of lymphocyte trafficking into the central nervous system. It is not known if these effects play an important role in the observed clinical activity of interferon beta-1b in MS.
12.3 Pharmacokinetics
Because serum concentrations of interferon beta-1b are low or not detectable following subcutaneous administration of 0.25 mg or less of interferon beta-1b, pharmacokinetic information in patients with MS receiving the recommended dose of interferon beta-1b is not available.
Following single and multiple daily subcutaneous administrations of 0.5 mg interferon beta-1b to healthy volunteers (N = 12), serum interferon beta-1b concentrations were generally below 100 units/mL. Peak serum interferon beta-1b concentrations occurred between one to eight hours, with a mean peak serum interferon concentration of 40 units/mL. Bioavailability, based on a total dose of 0.5 mg interferon beta-1b given as two subcutaneous injections at different sites, was approximately 50%.
After intravenous administration of interferon beta-1b (0.006 mg to 2 mg), similar pharmacokinetic profiles were obtained from healthy volunteers (N = 12) and from patients with diseases other than MS (N = 142). In patients receiving single intravenous doses up to 2 mg, increases in serum concentrations were dose proportional. Mean serum clearance values ranged from 9.4 mL/minkg-1 to 28.9 mL/minkg-1 and were independent of dose. Mean terminal elimination half-life values ranged from 8 minutes to 4.3 hours, and mean steady-state volume of distribution values ranged from 0.25 L/kg to 2.88 L/kg. Three-times-a-week intravenous dosing for two weeks resulted in no accumulation of interferon beta-1b in sera of patients. Pharmacokinetic parameters after single and multiple intravenous doses of interferon beta-1b were comparable.
Following every other day subcutaneous administration of 0.25 mg interferon beta-1b in healthy volunteers, biologic response marker levels (neopterin, β2-microglobulin, MxA protein, and the immunosuppressive cytokine, IL-10) increased significantly above baseline six-twelve hours after the first interferon beta-1b dose. Biologic response marker levels peaked between 40 and 124 hours and remained elevated above baseline throughout the seven-day (168-hour) study. The relationship between serum interferon beta-1b levels or induced biologic response marker levels and the clinical effects of interferon beta-1b in MS is unknown.
Drug Interaction Studies
No formal drug interaction studies have been conducted with interferon beta-1b.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Interferon beta-1b has not been tested for its carcinogenic potential in animals.
Mutagenesis
Interferon beta-1b was not genotoxic in the in vitro bacterial reverse mutation assay or the in vitro chromosomal aberration assay in human peripheral blood lymphocytes. Interferon beta-1b treatment of mouse BALBc-3T3 cells did not result in increased transformation frequency in an in vitro model of tumor transformation.
Impairment of Fertility
Administration of interferon beta-1b (doses of up to 0.33 mg/kg/day) to normally cycling female rhesus monkeys had no apparent adverse effects on either menstrual cycle duration or associated hormonal profiles (progesterone and estradiol) when administered over three consecutive menstrual cycles. The highest dose tested is approximately 30 times the recommended human dose of 0.25 mg on a body surface area (mg/m2) basis. The potential for other effects on fertility or reproductive performance was not evaluated.
-
14 CLINICAL STUDIES
The clinical effects of interferon beta-1b were studied in four randomized, multicenter, double-blind, placebo-controlled studies in patients with MS (Studies 1, 2, 3, and 4).
Patients with Relapsing-Remitting MS
The effectiveness of interferon beta-1b in relapsing-remitting MS (RRMS) was evaluated in a double blind, multiclinic, randomized, parallel, placebo controlled clinical study of two years duration (Study 1). The study enrolled MS patients, aged 18 to 50, who were ambulatory [Kurtzke Expanded Disability Status Scale (EDSS) of ≤ 5.5-score 5.5 is ambulatory for 100 meters, disability precludes full daily activities], exhibited a relapsing-remitting clinical course, met Poser’s criteria for clinically definite and/or laboratory-supported definite MS and had experienced at least two exacerbations over two years preceding the trial without exacerbation in the preceding month. The EDSS score is a method of quantifying disability in patients with MS and ranges from 0 (normal neurologic exam) to 10 (death due to MS). Patients who had received prior immunosuppressant therapy were excluded.
An exacerbation was defined as the appearance of a new clinical sign/symptom or the clinical worsening of a previous sign/symptom (one that had been stable for at least 30 days) that persisted for a minimum of 24 hours.
Patients selected for study were randomized to treatment with either placebo (N = 123), 0.05 mg of interferon beta-1b (N = 125), or 0.25 mg of interferon beta-1b (N = 124), self-administered subcutaneously every other day. Outcome based on the 372 randomized patients was evaluated after two years.
Patients who required more than three 28-day courses of corticosteroids were removed from the study. Minor analgesics (acetaminophen, codeine), antidepressants, and oral baclofen were allowed ad libitum, but chronic nonsteroidal anti-inflammatory drug (NSAID) use was not allowed.
The primary protocol-defined outcome measures were 1) frequency of exacerbations per patient and 2) proportion of exacerbation-free patients. A number of secondary clinical and magnetic resonance imaging (MRI) measures were also employed. All patients underwent annual T2 MRI imaging, and a subset of 52 patients at one site had MRIs performed every six weeks for assessment of new or expanding lesions.
The study results are shown in Table 3.
Table 3: Two-Year RRMS Study Results of Primary and Secondary Clinical Outcomes (Study 1) a14 exacerbation-free patients (0 from placebo, six from 0.05 mg, and eight from 0.25 mg) dropped out of the study before completing six months of therapy. These patients are excluded from this analysis.
bSequelae and Functional Neurologic Status, both required by protocol, were not analyzed individually but are included as a function of the EDSS.
cEDSS scores range from 1-10, with higher scores reflecting greater disability.
dScripps neurologic rating scores range from 0-100, with smaller scores reflecting greater disability.
eND =Not done.Efficacy Parameters Treatment Groups Statistical Comparisons p-value Primary End Points Placebo
(N = 123)Interferon beta-1b
0.05 mg
(N = 125)Interferon beta-1b
0.25 mg
(N = 124)Placebo
vs
0.05 mg0.05 mg
vs
0.25 mgPlacebo
vs
0.25 mgAnnual exacerbation rate 1.31 1.14 0.9 0.005 0.113 0.0001 Proportion of exacerbation-free patientsa 16% 18% 25% 0.609 0.288 0.094 Exacerbation frequency per patient 0a
1
2
3
4
> 520%
32%
20%
15%
15%
21%22%
31%
28%
15%
7%
16%29%
39%
17%
14%
9%
8%0.151 0.077 0.001 Secondary Endpointsb Median number of months to first on-study exacerbation 5 6 9 0.299 0.097 0.01 Rate of moderate or severe exacerbations per year 0.47 0.29 0.23 0.02 0.257 0.001 Mean number of moderate or severe exacerbation days per patient 44 33 20 0.229 0.064 0.001 Mean change in EDSS scorec at endpoint 0.21 0.21 -0.07 0.995 0.108 0.144 Mean change in Scripps scored at endpoint -0.53 -0.5 0.66 0.641 0.051 0.126 Median duration in days per exacerbation 36 33 36 NDe NDe NDe % change in mean MRI lesion area at endpoint 21.4% 9.8% -0.9% 0.015 0.019 0.0001 Of the 372 RRMS patients randomized, 72 (19%) failed to complete two full years on their assigned treatments.
Over the two-year period in Study 1, there were 25 MS-related hospitalizations in the 0.25 mg interferon beta-1b-treated group compared to 48 hospitalizations in the placebo group. In comparison, non-MS hospitalizations were evenly distributed among the groups, with 16 in the 0.25 mg interferon beta-1b group and 15 in the placebo group. The average number of days of MS-related steroid use was 41 days in the 0.25 mg interferon beta-1b group and 55 days in the placebo group (p = 0.004).
MRI data were also analyzed for patients in this study. A frequency distribution of the observed percent changes in MRI area at the end of two years was obtained by grouping the percentages in successive intervals of equal width. Figure 1 displays a histogram of the proportions of patients, which fell into each of these intervals. The median percent change in MRI area for the 0.25 mg group was -1.1%, which was significantly smaller than the 16.5% observed for the placebo group (p = 0.0001).
Figure 1: Distribution of Change in MRI Area in Patients with RRMS in Study 1
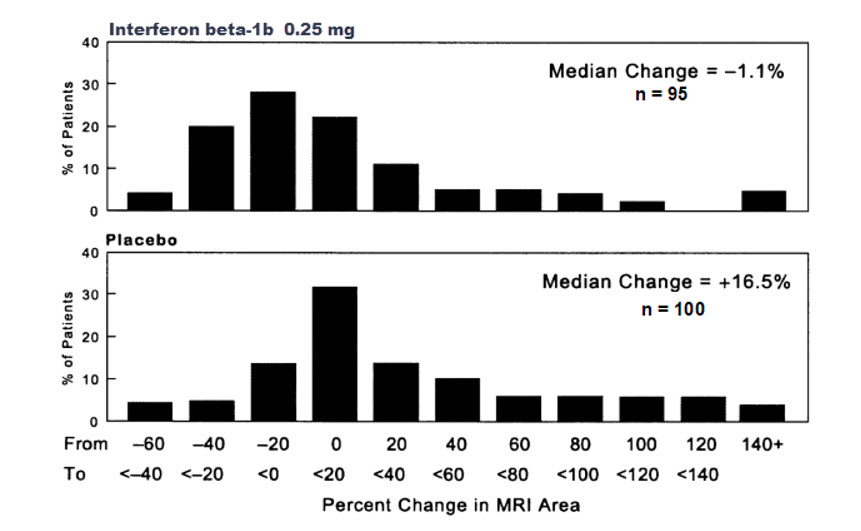
In an evaluation of frequent MRI scans (every six weeks) on 52 patients at one site in Study 1, the percent of scans with new or expanding lesions was 29% in the placebo group and 6% in the 0.25 mg treatment group (p = 0.006).
Patients with Secondary Progressive MS
Studies 2 and 3 were multicenter, randomized, double-blind, placebo-controlled trials conducted to assess the effect of interferon beta-1b in patients with secondary progressive MS (SPMS). Study 2 was conducted in Europe, and Study 3 was conducted in North America. Both studies enrolled patients with clinically definite or laboratory-supported MS in the secondary progressive phase, and who had evidence of disability progression (both Study 2 and 3) or two relapses (Study 2 only) within the previous two years. Baseline Kurtzke EDSS scores ranged from 3.0 to 6.5. Patients in Study 2 were randomized to receive interferon beta-1b 0.25 mg (N = 360) or placebo (N = 358). Patients in Study 3 were randomized to interferon beta-1b 0.25 mg (N = 317), interferon beta-1b 0.16 mg/m2 of body surface area (N = 314, mean assigned dose 0.3 mg), or placebo (N = 308). Test agents were administered subcutaneously, every other day for three years.
The primary outcome measure was progression of disability, defined as a 1.0 point increase in the EDSS score, or a 0.5 point increase for patients with baseline EDSS ≥ 6.0. In Study 2, time to progression in EDSS was longer in the interferon beta-1b treatment group (p = 0.005), with estimated annualized rates of progression of 16% and 19% in the interferon beta-1b and placebo groups, respectively. In Study 3, the rates of progression did not differ significantly between treatment groups, with estimated annualized rates of progression of 12%, 14%, and 12% in the interferon beta-1b fixed dose, surface area-adjusted dose, and placebo groups, respectively.
Multiple analyses, including covariate and subset analyses based on sex, age, disease duration, clinical disease activity prior to study enrollment, MRI measures at baseline, and early changes in MRI following treatment were evaluated in order to interpret the discordant study results. No demographic or disease-related factors enabled identification of a patient subset where interferon beta-1b treatment was predictably associated with delayed progression of disability.
In Studies 2 and 3, like Study 1, a statistically significant decrease in the incidence of relapses associated with interferon beta-1b treatment was demonstrated. In Study 2, the mean annual relapse rates were 0.42 and 0.63 in the interferon beta-1b and placebo groups, respectively (p < 0.001). In Study 3, the mean annual relapse rates were 0.16, 0.20, and 0.28, for the fixed dose, surface area-adjusted dose, and placebo groups, respectively (p < 0.02).
MRI endpoints in both Study 2 and Study 3 showed smaller increases in T2 MRI lesion area and decreased number of active MRI lesions in patients in the interferon beta-1b groups compared to the placebo group.
Patients with an Isolated Demyelinating Event and Typical MS Lesions on Brain MRI
In Study 4, 468 patients who had recently (within 60 days) experienced an isolated demyelinating event, and who had lesions typical of MS on brain MRI were randomized to receive either 0.25 mg interferon beta-1b (N = 292) or placebo (N = 176) subcutaneously every other day (ratio 5:3). The primary outcome measure was time to development of a second exacerbation with involvement of at least two distinct anatomical regions. Secondary outcomes were brain MRI measures, including the cumulative number of newly active lesions, and the absolute change in T2 lesion volume. Patients were followed for up to two years or until they fulfilled the primary endpoint. Eight percent of subjects on interferon beta-1b and 6% of subjects on placebo withdrew from the study for a reason other than the development of a second exacerbation. Time to development of a second exacerbation was significantly delayed in patients treated with interferon beta-1b compared to patients treated with placebo (p < 0.0001). The Kaplan-Meier estimates of the percentage of patients developing an exacerbation within 24 months were 45% in the placebo group and 28% of the interferon beta-1b group (Figure 2). The risk for developing a second exacerbation in the interferon beta-1b group was 53% of the risk in the placebo group (Hazard ratio = 0.53; 95% confidence interval 0.39 to 0.73).
Figure 2: Onset of Second Exacerbation by Time in Patients with Isolated Demyelinating Event with Typical MS Lesions on Brain MRI in Study 4*
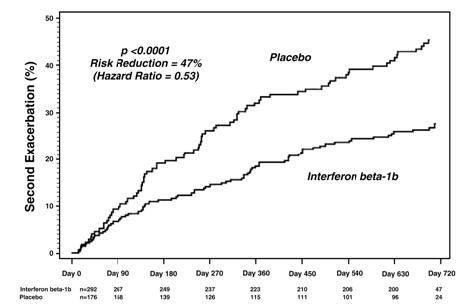
*Kaplan-Meier Methodology
In Study 4, patients treated with interferon beta-1b demonstrated a lower number of newly active lesions during the course of the study. A significant difference between interferon beta-1b and placebo was not seen in the absolute change in T2 lesion volume during the course of the study.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
EXTAVIA (interferon beta-1b) for injection is supplied as a white to off-white lyophilized powder in a clear glass, single-dose vial (3 mL capacity). Each carton contains 15 blister units: NDC: 0078-0569-12.
Each blister unit contains:
- A single-dose vial containing 0.3 mg EXTAVIA (interferon beta-1b)
- A pre-filled single-use syringe containing 1.2 mL diluent (0.54% Sodium Chloride Solution, USP). The rubber cap of the pre-filled syringe contains natural rubber latex.
- A vial adapter with a 27-gauge needle attached
- 2 alcohol prep pads
16.2 Stability and Storage
EXTAVIA and the diluent are for single-use only. Discard unused portions. The reconstituted product contains no preservative. Store EXTAVIA vials at room temperature 68°F to 77°F (20°C to 25°C). Excursions of 59°F to 86°F (15°C to 30°C) are permitted for up to 3 months. After reconstitution, if not used immediately, refrigerate the reconstituted solution and use within three hours. Do not freeze.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide and Instructions for Use).
Instruction on Self-injection Technique and Procedures
Provide appropriate instruction for reconstitution of EXTAVIA and methods of self-injection, including careful review of the EXTAVIA Medication Guide. Instruct patients in the use of aseptic technique when administering EXTAVIA.
Tell patients not to re-use needles or syringes, and instruct patients on safe disposal procedures. Advise patients of the importance of rotating areas of injection with each dose, to minimize the likelihood of severe injection-site reactions, including necrosis or localized infection (see Medication Guide).
Hepatic Injury
Advise patients that severe hepatic injury, including hepatic failure, has been reported during the use of EXTAVIA.
Inform patients of symptoms of hepatic dysfunction, and instruct patients to report them immediately to their healthcare provider [see Warnings and Precautions (5.1)].
Anaphylaxis and Other Allergic Reactions
Advise patients of the symptoms of allergic reactions and anaphylaxis, and instruct patients to seek immediate medical attention if these symptoms occur. Inform latex-sensitive patients that the removable rubber cap of the diluent pre-filled syringe contains natural rubber latex [see Warnings and Precautions (5.2)].
Depression and Suicide
Advise patients that depression and suicidal ideation have been reported during the use of EXTAVIA. Inform patients of the symptoms of depression or suicidal ideation, and instruct patients to report them immediately to their healthcare provider [see Warnings and Precautions (5.3)].
Congestive Heart Failure
Advise patients that worsening of preexisting congestive heart failure have been reported in patients using EXTAVIA.
Advise patients of symptoms of worsening cardiac condition, and instruct patients to report them immediately to their healthcare provider [see Warnings and Precautions (5.4)].
Injection-Site Necrosis and Reactions
Advise patients that injection-site reactions occur in most patients treated with EXTAVIA, and that injection-site necrosis may occur at one or multiple sites. Instruct patients to promptly report any break in the skin, which may be associated with blue-black discoloration, swelling, or drainage of fluid from the injection site, prior to continuing their EXTAVIA therapy [see Warnings and Precautions (5.5)].
Flu-like Symptom Complex
Inform patients that flu-like symptoms are common following initiation of therapy with EXTAVIA, and that concurrent use of analgesics and/or antipyretics on treatment days may help ameliorate flu-like symptoms associated with EXTAVIA use [see Warnings and Precautions (5.8), Dosage and Administration (2.4)]
Seizures
Instruct patients to report seizures immediately to their healthcare provider [see Warnings and Precautions (5.9)].
Drug-induced Lupus Erythematosus
Advise patients that drug-induced lupus erythematosus has been reported during the use of EXTAVIA. Inform patients of the symptoms of rash, redness of the skin on the face, joint pain, fever and weakness, and instruct patients to report them immediately to their healthcare provider [see Warnings and Precautions (5.10)]
Pregnancy
Advise patients to notify their healthcare provider if they are pregnant or plan to become pregnant [see Use in Specific Population (8.1)].
Manufactured by:
Novartis Pharmaceuticals Corporation
East Hanover, New Jersey 07936
U.S. License No. 1244© Novartis
T2019-94
-
MEDICATION GUIDE
This Medication Guide has been approved by the U.S. Food and Drug Administration. Revised: August 2019 Medication Guide
EXTAVIA
(ex tā vee uh)
interferon beta-1b
(in-ter-feer-on beta-one-be)
for injection, for subcutaneous useRead this Medication Guide before you start taking EXTAVIA and each time you get a refill. There may be new information. This information does not take the place of talking with your healthcare provider about your medical condition or your treatment.
What is the most important information I should know about EXTAVIA?
EXTAVIA can cause serious side effects, including:liver problems including liver failure. Symptoms of liver problems may include:
yellowing of your eyes
flu-like symptoms
itchy skin
nausea or vomiting
feeling very tired
bruising easily or bleeding problemsYour healthcare provider will do blood tests to check for these problems while you take EXTAVIA. serious allergic reactions. Serious allergic reactions can happen quickly and may happen after your first dose of EXTAVIA or after you have taken EXTAVIA many times. Symptoms may include: difficulty breathing or swallowing or swelling of the mouth or tongue, rash, itching, or skin bumps
depression or suicidal thoughts. Call your healthcare provider right away if you have any of the following symptoms, especially if they are new, worse, or worry you:thoughts about suicide or dying
trouble sleeping (insomnia)
hallucinationsnew or worse depression
acting aggressive, being angry, or violent
other unusual changes in behavior or moodnew or worse anxiety
acting on dangerous impulsesWhat is EXTAVIA?
EXTAVIA is a prescription medicine used to treat relapsing forms of multiple sclerosis (MS), to include clinically isolated syndrome, relapsing-remitting disease, and active secondary progressive disease, in adults. EXTAVIA is similar to certain interferon proteins that are produced in the body. It is not known if EXTAVIA is safe and effective in children.Who should not take EXTAVIA?
Do not take EXTAVIA if you are allergic to interferon beta-1b, to another interferon beta, to human albumin, or mannitol. See the end of this leaflet for a complete list of ingredients in EXTAVIA.What should I tell my healthcare provider before taking EXTAVIA?
Before you take EXTAVIA, tell your healthcare provider if you:- have or have had depression (sinking feeling or sadness), anxiety (feeling uneasy, nervous, or fearful for no reason) or trouble sleeping
- have or have had liver problems
- have or have had blood problems such as bleeding or bruising easily, low red blood cells (anemia) or low white blood cells
- have or have had seizures
- have or have had heart problems
- have or have had an allergic reaction to rubber or latex. The rubber cap of the diluent pre-filled syringe contains natural rubber latex.
- are pregnant or plan to become pregnant.
- are breastfeeding or plan to breastfeed. It is not known if EXTAVIA passes into your breast milk.
Tell your healthcare provider about all the medicines you take, including prescription and nonprescription medicines, vitamins, and herbal supplements.
Know the medicines you take. Keep a list of them to show your healthcare provider and pharmacist when you get a new medicine.
How should I take EXTAVIA? - See the Instructions for Use at the end of this Medication Guide for complete information on how to use EXTAVIA.
- EXTAVIA is given by injection under your skin (subcutaneous injection) every other day.
- Take EXTAVIA exactly as your healthcare provider tells you to take it.
- If your healthcare provider feels that you or someone else may give you the injections, then you or the other person should be trained by your healthcare provider in how to give an injection.
- Do not try to give yourself or have another person give you injections until you or both of you understand and are comfortable with how to prepare your dose and give the injection.
- You may be started on a lower dose when you first start taking EXTAVIA. Your healthcare provider will tell you what dose of EXTAVIA to use.
- Your healthcare provider may change your dose of EXTAVIA. You should not change your dose without talking to your healthcare provider.
- If you miss a dose, you should take your next dose as soon as you remember or are able to take it. Your next injection should be taken about 48 hours (2 days) after that dose. Do not take EXTAVIA on 2 consecutive days. If you accidentally take more than your prescribed dose, or take it on 2 consecutive days, call your healthcare provider right away.
- Always use a new, unopened vial of EXTAVIA and pre-filled diluent syringe for each injection. Throw away any unused medicine. Do not re-use any vials, syringes, or needles.
- It is important for you to change your injection site each time you inject EXTAVIA. This will lessen the chance of you having a serious skin reaction at the site where you inject EXTAVIA. Avoid injecting EXTAVIA into an area of skin that is sore, reddened, infected, or has other problems.
What are the possible side effects of EXTAVIA?
EXTAVIA may cause serious side effects. Call your healthcare provider right away if you have any of the serious side effects of EXTAVIA, including:- See “What is the most important information I should know about EXTAVIA?”
- heart problems. EXTAVIA may worsen heart problems, including congestive heart failure. Symptoms of heart problems may include:
swollen ankles
tightness in chestshortness of breath
increased need to urinate at nightdecreased ability to exercise
not being able to lay flat in bedfast heartbeat Injection-site problems. Serious skin reactions can happen in some people including areas of severe damage to skin and the tissue below the skin (necrosis). These reactions can happen anywhere you inject EXTAVIA. Symptoms of injection site problems may include swelling, redness, or pain at the injection site, fluid drainage from the injection site, and breaks in your skin or blue-black skin discoloration
flu-like symptoms. EXTAVIA can cause flu-like symptoms including:fever chills tiredness sweating muscle aches when you first start to use it These symptoms may decrease over time. Taking medicines for fever and pain relief on the days you are using EXTAVIA may help decrease these symptoms. seizures. Some people have had seizures while taking EXTAVIA, including people who have never had seizures before. It is not known if the seizures were related to their MS, to EXTAVIA, or to a combination of both. If you have a seizure after taking EXTAVIA, call your healthcare provider right away.
The most common side effects of EXTAVIA include:
low white blood cell count
increases in your muscle tension
problems sleepingincreases in your liver enzymes
pain
stomach painheadache
rash
weaknessTell your healthcare provider if you have any side effect that bothers you or that does not go away.
These are not all the possible side effects of EXTAVIA. For more information, ask your healthcare provider or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.How should I store EXTAVIA? - Before mixing, store EXTAVIA at room temperature between 68°F to 77°F (20°C to 25°C).
- Before mixing, EXTAVIA may be stored for up to 3 months between 59°F to 86°F (15°C to 30°C).
- After mixing, you can refrigerate EXTAVIA for up to 3 hours before using. Your EXTAVIA must be used within 3 hours of mixing even if refrigerated.
- Do not freeze.
Keep EXTAVIA and all medicines out of the reach of children.
General information about the safe and effective use of EXTAVIA.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use EXTAVIA for a condition for which it was not prescribed. Do not give EXTAVIA to other people, even if they have the same symptoms that you have. It may harm them.
This Medication Guide summarizes the most important information about EXTAVIA. If you would like more information, talk with your healthcare provider. You can ask your pharmacist or healthcare provider for information about EXTAVIA that is written for health professionals.
For more information, go to www.EXTAVIA.com or call the EXTAVIA toll-free medical information line at 1-888-669-6682.What are the ingredients in EXTAVIA?
Active ingredient: interferon beta-1b
Inactive ingredients: albumin (human), mannitol
Diluent contains sodium chloride solution.Manufactured by:
Novartis Pharmaceuticals Corporation
East Hanover, New Jersey 07936
U.S. License No. 1244T2019-95
-
INSTRUCTIONS FOR USE
EXTAVIA (interferon beta-1b) Patient Instructions for Use
If your doctor decides that you or a caregiver may be able to give your injections of EXTAVIA at home, your doctor or nurse should instruct you on the right way to prepare and inject EXTAVIA. To lower your risk of infection, it is important that you follow the technique that your doctor or nurse discussed with you to prepare and inject EXTAVIA. Do not try to inject EXTAVIA yourself until you have been shown by your doctor or nurse the right way to prepare and give the injections.
It is important for you to read, understand, and follow these instructions. Call your doctor if you or your caregiver has any questions about the right way to prepare or inject EXTAVIA.
Important safety information
- The rubber cap on the diluent pre-filled syringe is made of natural rubber latex. Tell your doctor if you are allergic to rubber or latex.
- Do not leave the blister pack containing EXTAVIA where others might tamper with it.
- Keep the blister pack containing EXTAVIA out of the reach of children.
- Do not open the blister pack or take out any of the items until right before you are ready to use them.
- Do not use EXTAVIA if the seal on the vial is broken. If the seal is broken, the product may not be safe for you to use.
- Do not use EXTAVIA after the expiration date shown on the blister pack label or box (Figure 1). If it has expired, return the entire pack to the pharmacy.
Figure 1
- Do not use any of the items in the blister pack more than one time. See the section at the end of this leaflet, “Dispose of used syringes, needles, and vials.” Throw away any open and unused medicine.
Gather your supplies.
You will need the following supplies to get ready to give your injection of EXTAVIA:
-
A blister pack containing the following items (Figure 2)
- a vial of EXTAVIA
- a pre-filled syringe of diluent (0.54% Sodium Chloride Solution)
- a vial adapter with a 27-gauge needle attached (in its own container)
- two (2) alcohol wipes
- a vial of EXTAVIA
Figure 2
- a dry cotton ball and gauze
- a sharps disposal container (Figure 3). See the section “Dispose of used syringes, needles, and vials.”
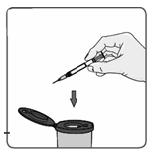
Figure 3
Prepare for self-injection
- Wash your hands well with soap and water.
- Open the blister pack by peeling off the label and take out all the items. Make sure the blister pack containing the vial adapter is sealed. Check to make sure the rubber cap on the diluent syringe is firmly attached.
- Turn the blister pack over, and place the vial in the well (vial holder) and place the pre-filled syringe in the U-shaped trough (Figure 4).
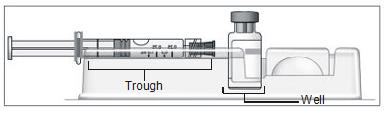
Figure 4
Mix EXTAVIA
4. Remove the EXTAVIA vial from the well and take the cap off the vial (Figure 5).
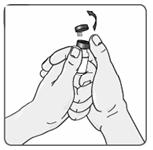
Figure 5
5. Place the vial back in the vial holder.
6. Use an alcohol wipe to clean the top of the vial (Figure 6). Wipe in one direction only.
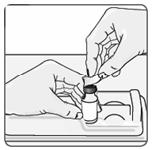
Figure 6
7. Leave the alcohol wipe on top of the vial until step 9 below.
8. Peel the label off the container with the vial adapter in it, but do not remove the vial adapter. The vial adapter is sterile, so do not touch it.
9. Remove the alcohol wipe from the top of the vial. Pick up the container that holds the vial adapter. Turn over the container keeping the vial adaptor inside. Put the adapter on top of the vial. Push down on the adapter until it pierces the rubber top of the vial and snaps in place (Figure 7). Lift the container off the vial adapter.
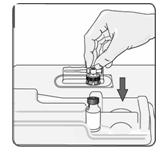
Figure 7
10. Remove the rubber cap from the pre-filled syringe using a twist and pull motion (Figure 8). Throw away the rubber cap.
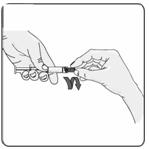
Figure 8
11. Remove the vial from the vial holder by grasping the vial. Do not touch any part of the vial adapter. Be careful not to pull the vial adapter off the top of the vial.
12. Connect the pre-filled syringe of diluent to the vial adapter by turning clockwise and tighten carefully (Figure 9).
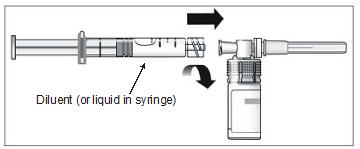
Figure 9
13. Slowly push the plunger of the pre-filled syringe all the way in. This will push all of the liquid from the syringe into the vial (Figure 10). Continue to hold the plunger while you mix EXTAVIA with the liquid from the syringe. If you do not hold the plunger in, it may return to its original position after you let go.
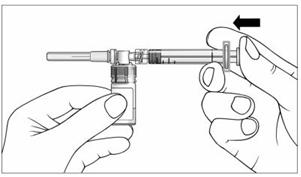
Figure 10
14. Gently swirl the vial to completely dissolve the white powder (EXTAVIA). Do not shake. Shaking and even gentle mixing can cause foaming of the medicine. If there is foam, let the vial sit until the foam settles.
15. After the powder dissolves, look closely at the solution in the vial. Do not use the solution if it is not clear or colorless, or if it contains particles.
The injection should be given right away after you mix EXTAVIA and let any foam in the solution settle. If you must wait for any reason before giving yourself the injection, you may refrigerate the medicine after you mix it. But you should use it within three hours.
16. With your thumb still pushing the plunger, turn the syringe and vial, so that the vial is on top (Figure 11).
17. Slowly pull the plunger back to withdraw the entire contents of the vial into the syringe.
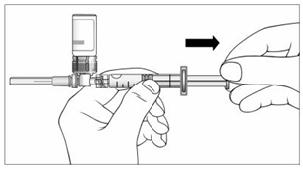
Figure 11
18. Turn the syringe so that the needle end is pointing up. Remove any air bubbles by tapping the outside of the syringe with your fingers (Figure 12). Slowly push the plunger to the 1 mL mark on the syringe or to the mark that matches the amount of EXTAVIA prescribed by your doctor. If too much solution is pushed back into the vial, return to step 16.
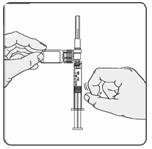
Figure 12
19. Remove the vial adapter and the vial from the syringe by twisting the vial adapter (Figure 13).
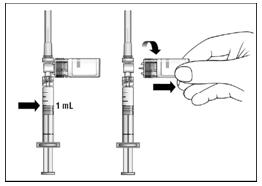
Figure 13
Choose an Injection Site
- EXTAVIA is injected under the skin and into the fat layer between the skin and the muscles (subcutaneous tissue). The best areas for injection are where the skin is loose and soft and away from the joints, nerves, and bones. Do not use the area near your navel (belly button) or waistline. If you are very thin, use only the thigh or outer surface of the arm for injection.
- Choose a different site each time you give yourself an injection. Figure 14 shows different areas for giving injections. Do not inject in the same area for two injections in a row. Keep a record of your injections to help make sure you change (rotate) your injection sites. If there are any sites that are difficult for you to reach, you can ask someone who has been trained to give the injection to you.
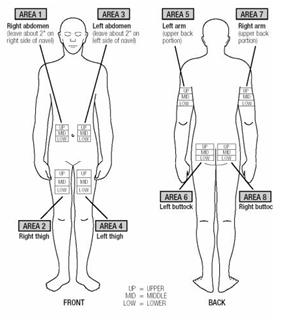
Figure 14
- Do not inject EXTAVIA in a site where the skin is red, bruised, infected, or scabbed, has broken open, or has lumps, bumps, or pain. Tell your doctor if you find skin conditions like the ones mentioned here or any other unusual looking areas where you have been given injections.
Injecting EXTAVIA
20. Using a circular motion, clean the injection site with an alcohol wipe, starting at the injection site and moving outward (Figure 15). Let the skin area air dry.
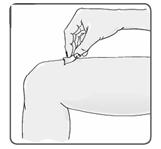
Figure 15
21. Remove the cap from the needle (Figure 16).
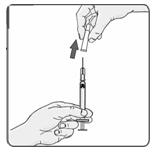
Figure 16
22. Gently pinch the skin around the site with your thumb and forefinger of the other hand (Figure 17). Insert the needle straight up and down into your skin at a 90˚ angle with a quick, dart-like motion.
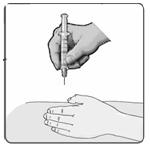
Figure 17
23. Once the needle is in your skin, slowly pull back on the plunger. If blood appears in the syringe, it means that you have entered a blood vessel. Do not inject EXTAVIA. Withdraw the needle. Throw away the syringe and needle in your puncture-proof container. Do not use the same syringe or any of the other supplies that you used for this injection. Repeat the above steps to prepare your dose using a new blister pack. Choose and clean a new injection site.
24. If no blood appears in the syringe, slowly push the plunger all the way in until the syringe is empty (Figure 18). Remove the needle from the skin; then place a dry cotton ball or gauze pad over the injection site. Gently massage the injection site for a few minutes with the dry cotton ball or gauze pad. Throw away the syringe in your puncture-proof disposal container.
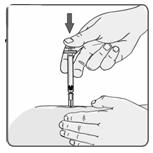
Figure 18
Dispose of used syringes, needles, and vials
- To prevent needle-stick injury and spread of infection, do not try to re-cap the needle.
- Place used needles, syringes, and vials in a closeable, puncture-resistant container. You may use a sharps container (such as a red biohazard container), a hard plastic container (such as a detergent bottle), or a metal container (such as an empty coffee can). Do not use glass or clear plastic containers. Ask your doctor for instructions on the right way to throw away (dispose of) the container. There may be state and local laws about how you should throw away used needles and syringes.
- Do not throw used needles, syringes, or vials in your household trash or recycle.
- Throw away any unused medicine. Do not save any unused EXTAVIA for a future dose.
- Keep the disposal container, needles, syringes, and vials of EXTAVIA out of the reach of children.
Manufactured by:
Novartis Pharmaceuticals Corporation
East Hanover, NJ 07936
U.S. License No. 1244This Instruction for Use has been approved by the U.S. Food and Drug Administration. August 2019
© Novartis
T2019-96
-
PRINCIPAL DISPLAY PANEL
PRINCIPAL DISPLAY PANEL
NDC: 0078-0569-12 NOVARTIS
EXTAVIA®
(interferon beta-1b)
0.3 mg per vial
For subcutaneous use only Rx only
Dispense the enclosed Medication Guide to each patient.
Each blister pack contains:
- one 1.2 mL prefilled single use sodium chloride 0.54% solution diluent syringe
- one single dose vial for reconstitution containing Extavia
- one vial adapter with attached 27-guage needle
- two alcohol prep pads
Dispense in this unit-of-use container. 15 single dose blister packs

- one 1.2 mL prefilled single use sodium chloride 0.54% solution diluent syringe
-
INGREDIENTS AND APPEARANCE
EXTAVIA
interferon beta-1b kitProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0078-0569 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0078-0569-12 15 in 1 CARTON 08/14/2009 1 NDC: 0078-0569-61 1 in 1 BLISTER PACK; Type 0: Not a Combination Product Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 1 VIAL, SINGLE-USE 3 mL Part 2 1 SYRINGE 1.2 mL Part 1 of 2 EXTAVIA
interferon beta 1b injection, powder, lyophilized, for solutionProduct Information Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength INTERFERON BETA-1B (UNII: TTD90R31WZ) (INTERFERON BETA-1B - UNII:TTD90R31WZ) INTERFERON BETA-1B 0.25 mg in 1.0 mL Inactive Ingredients Ingredient Name Strength ALBUMIN HUMAN (UNII: ZIF514RVZR) 15 mg in 1.0 mL MANNITOL (UNII: 3OWL53L36A) 15 mg in 1.0 mL Other Ingredients Ingredient Kind Ingredient Name Quantity May contain NATURAL LATEX RUBBER (UNII: 2LQ0UUW8IN) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 3 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125290 08/14/2009 Part 2 of 2 SODIUM CHLORIDE
sodium chloride solutionProduct Information Route of Administration SUBCUTANEOUS Inactive Ingredients Ingredient Name Strength SODIUM CHLORIDE (UNII: 451W47IQ8X) 0.0054 mL in 1 mL Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 1.2 mL in 1 SYRINGE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125290 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125290 08/14/2009 Labeler - Novartis Pharmaceuticals Corporation (002147023)
Trademark Results [EXTAVIA]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 EXTAVIA 85418581 4543851 Live/Registered |
Novartis AG 2011-09-09 |
 EXTAVIA 85418577 4535484 Live/Registered |
Novartis AG 2011-09-09 |
 EXTAVIA 78848819 3181020 Live/Registered |
Novartis AG 2006-03-29 |
 EXTAVIA 78221165 2813082 Dead/Cancelled |
Novartis AG 2003-03-04 |
 EXTAVIA 78140688 2853387 Dead/Cancelled |
Novartis AG 2002-07-02 |
 EXTAVIA 77384067 not registered Dead/Abandoned |
Novartis AG 2008-01-30 |
 EXTAVIA 75685030 not registered Dead/Abandoned |
Novartis AG 1999-05-07 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.