HELIXATE FS- antihemophilic factor, recombinant kit
Helixate FS by
Drug Labeling and Warnings
Helixate FS by is a Other medication manufactured, distributed, or labeled by CSL Behring LLC, Bayer HealthCare LLC. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use Helixate FS safely and effectively. See full prescribing information for Helixate FS.
Helixate FS
[Antihemophilic Factor (Recombinant), Formulated with Sucrose]
For Intravenous Use, Lyophilized Powder for Reconstitution
Initial U.S. Approval: 1993INDICATIONS AND USAGE
Helixate FS is an Antihemophilic Factor (Recombinant) indicated for: (1)
- On-demand treatment and control of bleeding episodes in adults and children with hemophilia A.
- Perioperative management of bleeding in adults and children with hemophilia A.
- Routine prophylaxis to reduce the frequency of bleeding episodes in children with hemophilia A and to reduce the risk of joint damage in children without pre-existing joint damage.
- Routine prophylaxis to reduce the frequency of bleeding episodes in adults with hemophilia A.
Helixate FS is not indicated for the treatment of von Willebrand disease. (1)
DOSAGE AND ADMINISTRATION
For intravenous use only.
Each vial of Helixate FS contains the labeled amount of recombinant factor VIII in international units (IU, unit).
Control of bleeding episodes and perioperative management (2.1):
- Dose (units) = body weight (kg) × desired factor VIII rise (IU/dL or % of normal) × 0.5 (IU/kg per IU/dL)
- Titrate doses to patient's clinical response.
- Determine treatment frequency based on type of bleeding episode.
For routine prophylaxis in adults: 25 units per kg three times a week (2.1).
For routine prophylaxis in children: 25 units per kg every other day (2.1).
DOSAGE FORMS AND STRENGTHS
Available as lyophilized powder in single use vials containing nominally 250, 500, 1000, 2000, and 3000 IU (3).
CONTRAINDICATIONS
Do not use in patients who have life-threatening hypersensitivity reactions, including anaphylaxis to mouse or hamster protein or other constituents of the product (4).
WARNINGS AND PRECAUTIONS
- Hypersensitivity reactions, including anaphylaxis, are possible. Should symptoms occur, discontinue treatment with Helixate FS and administer appropriate treatment (5.1).
- Development of activity-neutralizing antibodies can occur in patients receiving factor VIII-containing products, including Helixate FS. If expected plasma factor VIII activity levels are not attained, or if bleeding is not controlled with an expected dose, perform an assay that measures factor VIII inhibitor concentration (5.2).
- When clotting is normalized by treatment with factor VIII, development of cardiovascular risk factors may be the same as the risk for non-hemophilic patients (5.3).
- Monitor plasma factor VIII levels during infusions when indicated (5.4).
ADVERSE REACTIONS
The most common adverse reactions (≥4%) in clinical trials are inhibitor formation (neutralizing antibodies) in previously untreated and minimally treated patients (PUPs and MTPs), skin-associated hypersensitivity reactions (e.g., rash, pruritus, urticaria), infusion site reactions (e.g., inflammation, pain), and central venous access device (CVAD) associated infections. (6)
To report SUSPECTED ADVERSE REACTIONS, contact CSL Behring Pharmacovigilance Department at 1-866-915-6958 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch. (6)
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 5/2016
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Dose
2.2 Preparation and Reconstitution
2.3 Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity Reactions
5.2 Neutralizing Antibodies
5.3 Cardiovascular Risk Factors
5.4 Monitoring Laboratory Tests
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Labor and Delivery
8.3 Nursing Mothers
8.4 Pediatric Use
8.5 Geriatric Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
15 REFERENCES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
Helixate® FS is a recombinant antihemophilic factor indicated for:
- On-demand treatment and control of bleeding episodes in adults and children with hemophilia A.
- Perioperative management of bleeding in adults and children with hemophilia A.
- Routine prophylaxis to reduce the frequency of bleeding episodes in children with hemophilia A and to reduce the risk of joint damage in children without pre-existing joint damage.
- Routine prophylaxis to reduce the frequency of bleeding episodes in adults with hemophilia A.
Helixate FS is not indicated for the treatment of von Willebrand disease.
-
2 DOSAGE AND ADMINISTRATION
For intravenous use after reconstitution only.
2.1 Dose
Dosage and duration of treatment depend on the severity of the factor VIII deficiency, the location and extent of bleeding, and the patient's clinical condition.1 Careful control of replacement therapy is especially important in cases of major surgery or life-threatening bleeding episodes.
Each vial of Helixate FS has the recombinant factor VIII (rFVIII) potency in international units (IU, unit) stated on the label. One IU (unit), as defined by the World Health Organization standard for blood coagulation factor VIII, human, is approximately equal to the level of factor VIII activity found in 1 mL of fresh pooled human plasma.
The expected in vivo peak increase in factor VIII level expressed as IU/dL (or % normal) can be estimated using the following formulas:
Dosage (units) = body weight (kg) × desired factor VIII rise (IU/dL or % of normal) × 0.5 (IU/kg per IU/dL)
or
IU/dL (or % normal) = Total Dose (IU)/body weight (kg) × 2 [IU/dL]/[IU/kg]
Titrate dose to the patient's clinical response. Patients may vary in their pharmacokinetic (e.g., half-life, in vivo recovery) and clinical responses to Helixate FS.2,3,4 Although the dose can be estimated by the calculations above, it is highly recommended that appropriate laboratory tests, including serial factor VIII activity assays, are performed [see Warnings and Precautions (5.3) and Clinical Pharmacology (12.3)].
On-Demand Treatment and Control of Bleeding Episodes
A guide for dosing Helixate FS for on-demand treatment and control of bleeding episodes is provided in Table 1. The goal of treatment is to maintain a plasma factor VIII activity level at or above the plasma levels (in % of normal or in IU/dL) outlined in Table 1.
Table 1 Dosing for On-Demand Treatment and Control of Bleeding Episodes Type of Bleeding Episodes Factor VIII Level Required (IU/dL or % of normal) Dose (IU/kg) Frequency of Doses (hours) Duration of Therapy (days) Minor
Early hemarthrosis, minor muscle or oral bleeds.20 – 40
10 – 20
Repeat dose if there is evidence of further bleeding.
Until bleeding is resolved
Moderate
Bleeding into muscles, bleeding into the oral cavity, definite hemarthroses, and known trauma.30 – 60
15 – 30
12 – 24
Until bleeding is resolved
Major
Gastrointestinal bleeding. Intracranial, intra-abdominal or intrathoracic bleeding, central nervous system bleeding, bleeding in the retropharyngeal or retroperitoneal spaces, or iliopsoas sheath.
Fractures.
Head trauma.80 – 100
Initial: 40 – 50
Repeat: 20 – 258 – 12
Until bleeding is resolved
Perioperative Management of Bleeding
A guide for dosing Helixate FS during surgery (perioperative management of bleeding) is provided in Table 2. The goal of treatment is to maintain a plasma factor VIII activity level at or above the plasma level (in % of normal or in IU/dL) outlined in Table 2.
Table 2 Dosing for Perioperative Management of Bleeding Type of Surgery Factor VIII Level Required (IU/dL or % of normal) Dose (IU/kg) Frequency of Doses (hours) Duration of Therapy (days) Minor
Including tooth extraction30 – 60
15 – 30
12 – 24
Until bleeding is resolved.
Major
Examples include tonsillectomy, inguinal herniotomy, synovectomy, total knee replacement, craniotomy, osteosynthesis, trauma.100
50
Pre-operatively to achieve 100% activity.6 – 12 to keep FVIII activity in desired range
Until healing is complete.
2.2 Preparation and Reconstitution
Helixate FS is administered by intravenous injection after reconstitution. Patients should follow the specific reconstitution and administration procedures provided by their physicians.
Reconstitute and administer Helixate FS with the components provided with each package. If any component of the package is opened or damaged, do not use this component.
Product reconstitution, administration, and handling of the administration set and needles must be done with caution because percutaneous puncture with a needle contaminated with blood can transmit infectious viruses, including HIV (AIDS) and hepatitis. Place needles in a sharps container after single use. Discard all equipment, including any reconstituted Helixate FS product, in an appropriate container. Obtain immediate medical attention if injury occurs.
For any questions about the handling, reconstitution and administration of Helixate FS, contact CSL Behring Medical Affairs at 1-800-504-5434.
For instructions, patients should follow the recommendations in the FDA-Approved Patient Labeling.
The procedures below are provided as general guidelines for the reconstitution of Helixate FS.
- Work on a clean flat surface and wash hands thoroughly using soap and warm water before performing the procedures.
- Reconstitute the product with the components provided with each package. If any component of the package is opened or damaged, do not use this component.
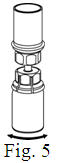
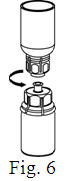
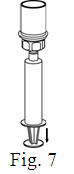
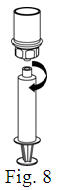
- Filter the reconstituted product prior to administration to remove potential particulate matter in the solution. Filtering can be achieved by using the Mix2Vial® vial adapter.
Vacuum Transfer and Reconstitution
2.3 Administration
For intravenous use after reconstitution only.
- Inspect Helixate FS visually for particulate matter and discoloration prior to administration, whenever solution and container permit. Turbid or discolored solution should be discarded.
- Store the reconstituted Helixate FS at room temperature prior to administration, but administer it within 3 hours.
- Administer Helixate FS over a period of 1 to 15 minutes. Adapt the rate of administration to the response of each individual patient. Determine the pulse rate before and during administration of Helixate FS. If there is a significant increase in pulse rate, reduce the rate of administration or temporarily halt the infusion allowing the symptoms to disappear promptly.
-
3 DOSAGE FORMS AND STRENGTHS
Helixate FS is available as a lyophilized powder in single use glass vials containing nominally 250, 500, 1000, 2000, and 3000 International Units (IU, unit).
Each vial of Helixate FS is labeled with the recombinant antihemophilic factor activity expressed in International Units per vial. This potency assignment employs a factor VIII concentrate standard that is referenced to a WHO International Standard for factor VIII concentrates, and is evaluated by appropriate methodology to ensure accuracy of the results.
-
4 CONTRAINDICATIONS
Helixate FS is contraindicated in patients who have life-threatening hypersensitivity reactions, including anaphylaxis to mouse or hamster protein or other constituents of the product (sucrose, glycine, histidine, sodium, calcium chloride, polysorbate 80, imidazole, tri-n-butyl phosphate, and copper).
-
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity Reactions
Hypersensitivity reactions, including anaphylaxis have been reported with Helixate FS. Reported symptoms included facial swelling, flushing, hives, decrease in blood pressure, nausea, rash, restlessness, shortness of breath, tachycardia, tightness of the chest, tingling, urticaria, and vomiting.
Helixate FS contains trace amounts of mouse immunoglobulin G (MuIgG) and hamster (BHK) proteins [see Description (11)]. Patients treated with this product may develop hypersensitivity to these non-human mammalian proteins.
Discontinue Helixate FS if symptoms occur and seek immediate emergency treatment.
5.2 Neutralizing Antibodies
Neutralizing antibodies (inhibitors) have been reported following administration of Helixate FS, predominantly in previously untreated patients (PUPs) [see Adverse Reactions (6)]. Carefully monitor patients for the development of factor VIII inhibitors, using appropriate clinical observations and laboratory tests.6 If expected plasma factor VIII activity levels are not attained, or if bleeding is not controlled with an expected dose, perform an assay that measures factor VIII inhibitor concentration [see Warnings and Precautions (5.4)].
5.3 Cardiovascular Risk Factors
Hemophilic patients with cardiovascular risk factors or diseases may be at the same risk to develop cardiovascular events as non-hemophilic patients when clotting has been normalized by treatment with factor VIII.
5.4 Monitoring Laboratory Tests
- Monitor plasma factor VIII activity levels by the one-stage clotting assay to confirm the adequate factor VIII levels have been achieved and maintained, when clinically indicated [see Dosage and Administration (2)].
- Monitor for development of factor VIII inhibitors. Perform assay to determine if factor VIII inhibitor is present. If expected factor VIII activity plasma levels are not attained or if bleeding is not controlled with the expected dose of Helixate FS, use Bethesda Units (BU) to titer inhibitors.
- If the inhibitor is less than 10 BU per mL, the administration of additional Helixate FS concentrate may neutralize the inhibitor and may permit an appropriate hemostatic response.
- If inhibitor titers are above 10 BU per mL, adequate hemostasis may not be achieved. The inhibitor titer may rise following Helixate FS infusion as a result of an anamnestic response to factor VIII. The on-demand treatment and control of bleeding in such patients requires the use of alternative therapeutic approaches and agents.
-
6 ADVERSE REACTIONS
Serious adverse reactions seen with Helixate FS are systemic hypersensitivity reactions, including bronchospastic reactions and/or hypotension and anaphylaxis, and the development of high-titer inhibitors necessitating alternative treatments to factor VIII.
The most common adverse reactions (≥ 4%) observed in clinical trials were inhibitor formation in previously untreated patients (PUPs) and minimally treated patients (MTPs), skin-related hypersensitivity reactions (e.g., rash, pruritus), infusion site reactions (e.g., inflammation, pain), and central venous access device (CVAD) associated infections.
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in clinical trials of another drug and may not reflect the rates observed in clinical practice.
Previously Treated Patients (PTPs)
During the open-label clinical studies conducted in 73 PTPs, there were 24 adverse reactions reported in the course of 24,936 infusions.
Adverse reactions reported by ≥ 4% of the patients are listed in Table 3 below.
Table 3 Adverse Reactions (AR) in Previously Treated Patients with Frequency of ≥ 4% (Age Range 12–59 years) MedDRA Primary SOC Preferred Term N = 73 AR (%) SOC = System Organ Class Skin and Subcutaneous Tissue Disorders
Rash, pruritus
6 (8.2%)
General Disorders and Administration Site Conditions
Infusion site reactions
3 (4.1%)
Previously Untreated Patients (PUPs) and Minimally Treated Patients (MTPs)
In clinical studies with pediatric PUPs and MTPs, there were 29 adverse reactions reported in the course of 9,389 infusions.
Adverse reactions reported by ≥ 4% of the patients are listed in Table 4 below.
Table 4 Adverse Reactions (AR) in Previously Untreated Patients and Minimally Treated Patients with Frequency of ≥ 4% (Age Range 2–27 months) MedDRA Primary SOC Preferred Term N = 61 AR (%) SOC = System Organ Class - * Denominator for de novo inhibitors is N = 60, since one patient had a pre-existing inhibitor.
Skin and Subcutaneous Tissue Disorders
Rash, pruritus, urticaria
10 (16%)
Blood and Lymphatic System Disorders
Factor VIII inhibition (neutralizing antibodies)
9 (15%)*
General Disorders and Administration Site Conditions
Infusion site reactions
4 (7%)
Minimally Treated Patients (MTPs) in the Joint Outcome Study
In the Joint Outcome Study with pediatric MTPs treated with routine prophylaxis or episodic enhanced treatment for 5.5 years, 46 of the 65 randomized patients experienced adverse events over the study duration.
Table 5 Adverse Reactions in Minimally Treated Patients in the Joint Outcome Study (Age Range 0–6 years) MedDRA Primary SOC Preferred Term Prophylaxis Arm N = 32 AR (%) Enhanced Episodic Arm N = 33 AR (%) SOC = System Organ Class - * Three patients from the enhanced episodic arm had catheter removal.
Surgical and Medical Procedures
Central venous catheterization, Catheter removal
19 (59%)
18 (55%)*
Infections and Infestations
Central line infection
6 (19%)
6 (18%)
General Disorders and Administration Site Conditions
Pyrexia
1 (3%)
4 (12%)
Immunogenicity
In clinical studies with 73 PTPs (defined as having more than 100 exposure days), one patient had a pre-existing inhibitor. In the other 72 patients, followed over 4 years, no de novo inhibitors were observed.
In clinical studies with pediatric PUPs and MTPs, inhibitor development was observed in 9 out of 60 patients (15%), 6 were high titer1 (> 5 BU) and 3 were low-titer inhibitors. Inhibitors were detected at a median number of 7 exposure days (range 2 to 16 exposure days).
In the Joint Outcome Study with Helixate FS,5 de novo inhibitor development was observed in 8 of 64 patients with negative baseline values (12.5%), 2 patients developed high titer1 (> 5 BU) and were withdrawn from the study. Six patients developed low-titer inhibitors. Inhibitors were detected at a median number of 44 exposure days (range 5 to 151 exposure days).
Inhibitor data in PUPs have been collected in several postmarketing registries [see Postmarketing Experience (6.2)].
The detection of antibody formation is dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to Helixate FS with the incidence of antibodies to other products may be misleading.
6.2 Postmarketing Experience
Because adverse reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure. The following adverse reaction has been identified during post approval use of Helixate FS:
Sensory System – Dysgeusia
Immunogenicity – Postmarketing Registries
Data from the Research of Determinants of Inhibitor Development (RODIN) study7, French National Registry (FranceCoag)8 and United Kingdom Haemophilia Centre Doctors' Organisation (UKHCDO)9 registry reported an inhibitor development rate in PUPs for Hexliate FS of 38%, 50% and 35%, respectively, which is comparable to previously-reported inhibitor rates10 for FVIII products. These registry studies show a trend towards an increased risk of inhibitor development in PUPs, as compared to the reference rFVIII product. A survey of Canadian hemophilia centers11 (2005 to 2012) and available data from the European Haemophilia Safety Surveillance (EUHASS) 12 registry from 2009 to 2013, reported an inhibitor development rate in PUPs for Helixate FS of 42% and 31%, respectively, with no statistically significant differences observed across FVIII products.
-
8 USE IN SPECIFIC POPULATIONS
8.2 Labor and Delivery
There is no information available on the effect of factor VIII replacement therapy on labor and delivery. Helixate FS should be used only if clinically needed.
8.3 Nursing Mothers
It is not known whether this drug is excreted into human milk. Because many drugs are excreted into human milk, caution should be exercised if Helixate FS is administered to a nursing woman.
8.4 Pediatric Use
Safety and efficacy studies have been performed in previously untreated and minimally treated pediatric patients. Children, in comparison to adults, present higher factor VIII clearance values and, thus, lower half-life and recovery of factor VIII. This may be due to differences in body composition.13 Account for this difference in clearance when dosing or following factor VIII levels in the pediatric population [see Clinical Pharmacology (12.3)].
Routine prophylactic treatment in children ages 0–2.5 years with no pre-existing joint damage has been shown to reduce spontaneous joint bleeding and the risk of joint damage. This data can be extrapolated to ages >2.5–16 years for children who have no existing joint damage [see Clinical Studies (14)].
-
11 DESCRIPTION
Helixate FS Antihemophilic Factor (Recombinant) is a coagulation factor VIII produced by recombinant DNA technology. It is produced by Baby Hamster Kidney (BHK) cells into which the human factor VIII gene has been introduced.14 The cell culture medium contains Human Plasma Protein Solution (HPPS) and recombinant insulin, but does not contain any proteins derived from animal sources. Helixate FS is a purified glycoprotein consisting of multiple peptides including an 80 kD and various extensions of the 90 kD subunit. It has the same biological activity as factor VIII derived from human plasma. No human or animal proteins, such as albumin, are added during the purification and formulation processes of Helixate FS.
The purification process includes a solvent/detergent virus inactivation step in addition to the use of the purification methods of ion exchange chromatography, monoclonal antibody immunoaffinity chromatography, along with other chromatographic steps designed to purify recombinant factor VIII and remove contaminating substances.
Additionally, the manufacturing process was investigated for its capacity to decrease the infectivity of an experimental agent of transmissible spongiform encephalopathy (TSE), considered as a model for the vCJD and CJD agents.15-19 Several of the individual production and raw material preparation steps in the Helixate FS manufacturing process have been shown to decrease TSE infectivity of that experimental model agent. These TSE reduction steps include the Fraction II+III separation step for HPPS (6.0 log10) and an anion exchange chromatography step (3.6 log10).
Helixate FS is formulated with the following as stabilizers (see Table 6) in the final container and is then lyophilized. The final product does not contain any preservative. It is a sterile, nonpyrogenic powder preparation for intravenous injection. Intravenous administration of sucrose contained in Helixate FS will not affect blood glucose levels.
Table 6 Stabilizers Contained in Helixate FS Final Container Stabilizer 250 IU, 500 IU, 1000 IU
Nominal Vial Sizes2000 IU, 3000 IU
Nominal Vial SizesSucrose
0.9–1.3%
0.9–1.2%
Glycine
21–25 mg/mL
20–24 mg/mL
Histidine
18–23 mmol/L
17–22 mmol/L
Table 7 lists the inactive ingredients/excipients also contained in the final product.
Table 7 Inactive Ingredients/Excipients Inactive Ingredient/Excipient 250 IU, 500 IU, 1000 IU 2000 IU, 3000 IU Sodium
27–36 mEq/L
26–34 mEq/L
Calcium
2.0–3.0 mmol/L
1.9–2.9 mmol/L
Chloride
32–40 mEq/L
31–38 mEq/L
Polysorbate 80
64–96 μg/mL
64–96 μg/mL
Sucrose
28 mg/vial
52 mg/vial
Imidazole, tri-n-butyl phosphate, and copper
Trace amounts
Trace amounts
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Helixate FS temporarily replaces the missing clotting factor VIII that is needed for effective hemostasis.
12.2 Pharmacodynamics
The activated partial thromboplastin time (aPTT) is prolonged in patients with hemophilia. Determination of aPTT is a conventional in vitro assay for biological activity of factor VIII. Treatment with Helixate FS normalizes the aPTT over the effective dosing period.
12.3 Pharmacokinetics
The pharmacokinetic properties of Helixate FS were investigated in two separate studies in adult and pediatric previously treated patients (PTPs).
Pharmacokinetic studies with Helixate FS were conducted in 20 PTPs (ages 12 to 33 years) with severe hemophilia A. The pharmacokinetic parameters for Helixate FS were measured in a randomized, crossover clinical trial with the predecessor HELIXATE product using a single dose administration of 50 IU per kg. After 24 weeks, the same dose of Helixate FS was administered to the same patients. The recovery and half-life data for Helixate FS were unchanged after 24 weeks of continued treatment with sustained efficacy and no evidence of factor VIII inhibition (see Table 8).
Table 8 Pharmacokinetic Parameters for Helixate FS Compared to HELIXATE Parameter Helixate FS HELIXATE Initial PK (Mean±SD) PK at week 24 (Mean±SD) Reference (Mean±SD) AUC (IU∙h/dL)
1588.05 ± 344.32
1487.08 ± 381.73
1879.02 ± 412.32
Cmax (IU/dL)
114.95 ± 20.19
109.42 ± 20.09
127.40 ± 33.21
Half-life (hr)
13.74 ± 1.82
14.60 ± 4.38
14.07 ± 2.62
In Vivo Recovery (IU/dL / IU/kg)
2.20 ± 0.34
2.11 ± 0.37
2.43 ± 0.60
The pharmacokinetics of Helixate FS were investigated in pediatric PTPs (4.4–18.1 years of age, average age 12).13 The pharmacokinetic parameters in children compared to adults show differences in higher clearance, lower incremental in vivo factor VIII recovery and a shorter factor VIII half-life. The pharmacokinetic parameters are depicted in Table 9.
Table 9 Pharmacokinetic Parameters for Helixate FS in Children Parameter Mean (range) AUC (IU∙h/dL)
1320.0
Clearance (mL/h∙kg)
4.1
Half-life (hr)
10.7 (7.8–15.3)
In Vivo Recovery (IU/dL / IU/kg)
1.9 (1.25–2.76)
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No studies have been conducted with Helixate FS to assess its mutagenic or carcinogenic potential and impairment of fertility. By inference, the predecessor product HELIXATE and Helixate FS would be expected to have equivalent mutagenic and carcinogenic potential.
The predecessor product did not demonstrate reverse mutation or chromosomal aberrations at doses substantially greater than the maximum expected clinical dose. In vivo evaluation with the predecessor product in animals using doses ranging between 10 and 40 times the expected clinical maximum also indicated that the predecessor product did not possess a mutagenic potential. Long-term investigations of carcinogenic potential in animals have not been performed due to the immune response to heterologous proteins in all non-human mammalian species.
13.2 Animal Toxicology and/or Pharmacology
Preclinical studies evaluating Helixate FS in hemophilia A with mice, rats, rabbits, and dogs demonstrated safe and effective restoration of hemostasis. Doses several fold higher than the recommended clinical dose (related to body weight) did not demonstrate any acute or subacute toxic effect for Helixate FS in laboratory animals.
Helixate FS has been shown to be comparable to the predecessor product HELIXATE with respect to its biochemical and physiochemical properties, as well as its non-clinical in vivo pharmacology and toxicology.
-
14 CLINICAL STUDIES
Previously Treated Patients (PTPs) Clinical Studies
A total of 73 patients with severe (≤ 2% FVIII) hemophilia A, ages 12–59, who had been previously treated with other recombinant or with plasma-derived AHF products, were treated up to 54 months in open label studies with Helixate FS. A total of 5,684 bleeding episodes were treated during the studies; 92.7% of the bleeds were treated with one (79.7%) or two (13.0%) infusions. Patients could be treated with on-demand or prophylaxis. Regularly scheduled prophylaxis treatment represented 76% of all infusions (treatment regimens of 2–3 infusions per week).
A total of 30 patients received Helixate FS for 41 surgical procedures during the PTP studies. There were both minor and major surgery types, 16 and 25 respectively. Efficacy was measured by the attending surgeon based on a comparison of estimated blood loss from experience with non-hemophilic patients undergoing similar procedures. The surgeon or treating physician assigned a rating to the hemostatic outcome according to 4 categories: "excellent (blood loss less than expected)," "good (blood loss as expected)," "moderate (blood loss more than expected)," or "none (uncontrolled bleeding)." Hemostasis was rated as satisfactory ("excellent" or "good") in all cases.
Previously Untreated Patients (PUPs) and Minimally Treated Patients (MTPs) Clinical Study
Helixate FS has been used in the treatment of bleeding episodes in pediatric PUPs and MTPs with severe (<2% FVIII) hemophilia A. There were 37 PUPs and 24 MTPs (defined as having equal to or less than 4 exposure days) treated with a total of 9,419 infusions of Helixate FS for a follow up duration up to 3.1 years. A total of 1047 bleeding episodes were treated; the bleeds were treated with one (73%) or two (15%) infusions.
A total of 27 surgical procedures were performed in 22 patients during the PUPs and MTPs study. There were both minor and major surgery types, 21 and 6 respectively. The attending surgeon measured efficacy and assigned a rating to the hemostatic outcome according to 4 categories as described above for PTPs. Hemostasis was rated as satisfactory ("excellent" or "good") in all cases.
Adult Prophylaxis for Bleeding Frequency Reduction
A 3-year, multicenter, open-label, parallel-group, prospective, randomized, controlled clinical study of the effect of routine prophylaxis with Helixate FS versus on-demand use on bleeding frequency in adults and adolescents included 84 PTPs with severe Hemophilia A (FVIII level < 1 IU/dL), age 15 to 50 years. Patients were matched at baseline on demographic and disease characteristics. The median number of bleeds in the year before enrollment was 18.
Patients were randomized 1:1 to prophylaxis (25 units per kg three times a week) or on-demand use of Helixate FS. Escalation of the prophylaxis dose by 5 units per kg/infusion after years 1 and 2, up to a maximum of 35 units per kg/infusion, was allowed.
Bleeding frequency was analyzed in the intent-to-treat population after a median follow-up period of 1.4 years. Patients who received prophylaxis experienced statistically significantly fewer bleeds (p<0.0001) compared to patients treated on-demand regardless of baseline subgroups examined including age, bleeding history, and presence or absence of target joints. The ratio of the mean bleeding frequency was 15.2 (95% CI: 8.5, 27.2; p<0.0001) for on-demand versus prophylaxis, indicating that patients who received on-demand treatment experienced on average 15.2 times as many bleeds compared to patients treated with prophylaxis. The mean annualized bleed rates (bleeds/subject/year) were 37 in the on-demand group versus 2 in the prophylaxis group. The median annualized bleed rate (bleeds/subject/year) in the on-demand group was 33 versus zero in the prophylaxis group. Most of the bleeding occurred in joints: the median joint bleed rate (joint bleeds/subject/year) was 24 in the on-demand group versus zero in the prophylaxis group. The mean annualized joint bleed rate was 29 in the on-demand group versus 2 in the prophylaxis group.
Twenty-two of 42 (52%) prophylaxis subjects experienced no bleeding, and 12 of 42 (29%) prophylaxis subjects experienced only 1–2 bleeds during the follow-up period. Among prophylaxis patients the mean number of infusions/week was 2.8, and the median dose per prophylaxis infusion was 26 units per kg.
Pediatric Prophylaxis for Joint Damage Risk Reduction
A total of 65 boys less than 30 months of age with severe hemophilia A (FVIII level ≤ 2 IU/dL) and with ≤ 2 bleeds into each index joint and normal baseline joint imaging, were observed for up to 5.5 years in a multicenter, open-label, prospective, randomized, controlled clinical study.5 Patients received either 25 IU per kg every other day (primary prophylaxis; n = 32) or at least 3 doses totaling a minimum of 80 IU per kg at the time of a bleeding episode (enhanced episodic; n = 33). Joint damage was evaluated by magnetic resonance imaging (MRI) or radiography, as well as the frequency of bleeding episodes. Joint damage detected by MRI or radiography in the ankles, knees, and elbows (i.e., index joints) was statistically significantly lower (p = 0.002) for subjects receiving prophylactic therapy (7%) than for subjects receiving episodic therapy (42%). This corresponds to a 6.29-fold relative risk of joint damage for subjects treated with enhanced episodic therapy compared to prophylaxis. The mean rate of index joint hemorrhages for subjects on episodic therapy was 4.89 bleeds per year, versus 0.63 bleeds per year observed in the prophylaxis arm. Three of 33 (9.1%) subjects in the episodic arm experienced recurrent life threatening bleeds (intracranial, gastrointestinal) compared to no subjects in the prophylaxis arm. On a per joint basis, joints in the regular prophylaxis arm were 8-fold more likely to remain damage-free than those in the episodic arm. Joint damage was most frequently observed in ankle joints and was detected at higher rates by MRI than by radiography. Ankles were also the index joint that demonstrated the highest frequency of bleeding events in this study (left ankle, mean 2.7 hemorrhages; right ankle, mean 2.6 hemorrhages).
As shown in Table 10 below, the incidence of joint damage was statistically significantly lower in the prophylactic group as compared to the episodic treatment group when assessed by MRI, or either MRI or radiography, using predefined criteria (described below) for establishing joint damage. However, there was no statistically significant difference between the two groups when joint damage was assessed by radiography alone.
To evaluate joint damage, MRIs were scored using a scale developed by Nuss et al.,20 and X-rays were scored using the method of Pettersson et al.21 Both scales have been validated in various clinical trials and are routinely used for joint damage evaluation in hemophiliacs. Joint damage was defined as bone and/or cartilage damage including subchondral cysts, erosions and cartilage loss with narrowing of joint space. This corresponded to a total MRI score of ≥ 7 or an X-ray score of ≥1 in any of the following categories: subchondral cysts, erosions of joint surfaces or narrowing of joint spaces. Images were read separately by two independent radiologists centrally. Any discrepant reading was read by an independent third radiologist who was not aware of the initial reading results. The concordant reading of two out of three readers was used for analysis purposes.
Table 10 Subjects with Joint Damage (Subjects with Available Baseline and Endpoint Data) Endpoint Assessment Prophylaxis Episodic Therapy p-value Incidence (%) Relative Risk (95% CI) Incidence (%) Relative Risk (95% CI) Relative Risk is the risk of damage to one or more index joints on the given therapy as compared to the other therapy.
P-value is from the 2-sided Fisher Exact Test comparing the incidence of joint damage between treatment groups.MRI
2/27 (7%)
0.17 (0.04, 0.67)
13/29 (45%)
6.05 (1.50, 24.38)
0.002
Radiography
1/28 (4%)
0.19 (0.02, 1.55)
5/27 (19%)
5.19 (0.65, 41.54)
0.101
MRI or Radiography
2/30 (7%)
0.16 (0.04, 0.65)
13/31 (42%)
6.29 (1.55, 25.55)
0.002
-
15 REFERENCES
- White GC, Rosendaal F, Aledort LM, Lusher JM, Rothschild C, Ingerslev J, for the Factor VIII and Factor IX Subcommittee of the Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis. Definitions in hemophilia. Thromb Haemost 85:560-75, 2001.
- Abildgaard CF, Simone JV, Corrigan JJ, et al: Treatment of hemophilia with glycine-precipitated Factor VIII. N Engl J Med 275(9):471–5, 1966.
- Schwartz RS, Abildgaard CF, Aledort LM, et al: Human recombinant DNA-derived antihemophilic factor (factor VIII) in the treatment of hemophilia A. Recombinant Factor VIII Study Group. N Engl J Med 323(26):1800-5, 1990.
- White GC 2nd, Courter S, Bray GL, et al: A multicenter study of recombinant factor VIII (Recombinate) in previously treated patients with hemophilia A. The Recombinate Previously Treated Patient Study Group. Thromb Haemost 77(4):660-667, 1997.
- Manco-Johnson MJ, Abshire TC, Shapiro AD, Riske B, Hacker MR, Kilcoyne R, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med 2007;357(6):535-44.
- Kasper CK: Complications of hemophilia A treatment: factor VIII inhibitors. Ann NY Acad Sci 614:97-105, 1991.
- Gouw SC, van den Berg HM, et al: Intensity of factor VIII treatment and inhibitor development in children with severe hemophilia A: the RODIN study. Blood 121(20): 4046-4055, 2013.
- Calvez T, Chambost H, et al: Recombinant factor VIII products and inhibitor development in previously untreated boys with severe hemophilia A. Blood 124(23): 3398-3408, 2014.
- Collins PW, Palmer BP, et al: Factor VIII brand and the incidence of factor VIII inhibitors in previously untreated UK children with severe hemophilia A, 2000-2011. Blood 124(23): 3389-3397, 2014.
- Franchini M, Coppola A, et al: Systematic Review of the Role of FVIII Concentrates in Inhibitor Development in Previously Untreated Patients with Severe Hemophilia A: A 2013 Update. Semin Throm Hemost (39): 752-766, 2013.
- Vezina C, Carcao M, et al: Incidence and risk factors for inhibitor development in previously untreated severe haemophilia A patients born between 2005 and 2010. Haemophilia 20(6): 771-776, 2014.
- Fisher K, Lassila, R, et al. Inhibitor development in haemophilia according to concentrate: Four-year results from the European HAemophilia Safety Surveillance (EUHASS) project. Thromb Haemost 113.4, 2015.
- Barnes C, Lillicrap D, Pazmino-Canizares J, et al: Pharmacokinetics of recombinant factor VIII (Helixate-FS®) in children and causes of inter-patient pharmacokinetic variability. Haemophilia 12 (Suppl. 4): 40-49, 2006.
- Lawn RM, Vehar GA: The molecular genetics of hemophilia. Sci Am 254(3):48–54, 1986.
- Stenland CJ, et al: Partitioning of human and sheep forms of the pathogenic prion protein during the purification of therapeutic proteins from human plasma. Transfusion 42(11):1497-500, 2002.
- Lee DC, Miller JL, Petteway SR: Pathogen safety of manufacturing processes for biological products: special emphasis on HELIXATE® Bayer. Haemophilia 8(Suppl. 2):6-9, 2002.
- Lee DC, Stenland CJ, Miller JL, et al: A direct relationship between the partitioning of the pathogenic prion protein and transmissible spongiform encephalopathy infectivity during the purification of plasma proteins. Transfusion 41(4):449-55, 2001.
- Cai K, Miller JL, Stenland CJ, et al: Solvent-dependent precipitation of prion protein. Biochim Biophys Acta 1597(1):28-35, 2002.
- Trejo SR, Hotta JA, Lebing W, et al: Evaluation of virus and prion reduction in a new intravenous immunoglobulin manufacturing process. Vox Sang 84(3):176-87, 2003.
- Nuss R, Kilcoyne RF, Geraghty S, et al: MRI findings in haemophilic joints treated with radiosynoviorthesis with development of an MRI scale of joint damage. Haemophilia 6:162-169, 2000.
- Pettersson H, Ahlberg A, Nilsson IM: A radiologic classification of hemophilia arthropathy. Clin Orthop Relat Res 149:153-159, 1980.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
Helixate FS is available as a kit in the following single-use glass vial sizes. A suitable volume of Sterile Water for Injection, USP and Mix2Vial® filter transfer device are provided in the kit.
Kit NDC Number Approximate FVIII Activity (IU) Diluent (mL) 0053-8131-02
250
2.5
0053-8132-02
500
2.5
0053-8133-02
1000
2.5
0053-8134-02
2000
5.0
0053-8135-02
3000
5.0
Actual factor VIII activity in IU is stated on the label of each Helixate FS vial. Use the actual potency as indicated on the vial label to calculate the dose.
Storage and Handling
The product vial and diluent vial are not made with natural rubber latex.
Product as Packaged for Sale
- Store Helixate FS at +2°C to +8°C (36°F to 46°F) for up to 30 months from the date of manufacture. Within this period, Helixate FS may be stored for a period of up to 12 months at temperatures up to +25°C or 77°F.
- Record the starting date of room temperature storage on the unopened product carton. Once stored at room temperature, do not return the product to the refrigerator. The shelf-life then expires after storage at room temperature, or after the expiration date on the product vial, whichever is earlier.
- Do not use Helixate FS after the expiration date indicated on the vial.
- Do not freeze.
- Protect from extreme exposure to light and store the lyophilized powder in the carton prior to use.
-
17 PATIENT COUNSELING INFORMATION
- Advise the patient to read the FDA-approved patient labeling (Patient Information and Instructions for Use).
- Advise patients to report any adverse reactions or problems following Helixate FS administration to their physician or healthcare provider.
- Allergic-type hypersensitivity reactions have been reported with Helixate FS. Warn patients of the early signs of hypersensitivity reactions [including hives (rash with itching), generalized urticaria, tightness of the chest, wheezing, hypotension] and anaphylaxis. Advise patients to discontinue use of the product if these symptoms occur and seek immediate emergency treatment with resuscitative measures such as the administration of epinephrine and oxygen.
- Inhibitor formation may occur at any time in the treatment of a patient with hemophilia A. Advise patients to contact their physician or treatment center for further treatment and/or assessment, if they experience a lack of clinical response to factor VIII replacement therapy, as this may be a manifestation of an inhibitor.
- Advise patients to consult with their healthcare provider prior to travel. While traveling advise patients to bring an adequate supply of Helixate FS based on their current regimen of treatment.
-
PATIENT PACKAGE INSERT
FDA-APPROVED PATIENT LABELING
PATIENT INFORMATION
Helixate FS (he-liks-āt)
Antihemophilic Factor (Recombinant)
Formulated with Sucrose
This leaflet summarizes important information about Helixate FS. Please read it carefully before using this medicine. This information does not take the place of talking with your healthcare provider, and it does not include all of the important information about Helixate FS. If you have any questions after reading this, ask your healthcare provider.
Do not attempt to self-infuse unless you have been taught how by your healthcare provider or hemophilia center.
What is Helixate FS?
Helixate FS is a medicine used to replace clotting factor (factor VIII or antihemophilic factor) that is missing in people with hemophilia A (also called "classic" hemophilia). Hemophilia A is an inherited bleeding disorder that prevents blood from clotting normally.
Helixate FS is used to treat and control bleeding in adults and children with hemophilia A. Your healthcare provider may give you Helixate FS when you have surgery. Helixate FS can reduce the number of bleeding episodes when used regularly (prophylaxis). Helixate FS can reduce the risk of joint damage in children.
Helixate FS is not used to treat von Willebrand Disease.
Who should not use Helixate FS?
You should not use Helixate FS if you
- are allergic to rodents (like mice and hamsters).
- are allergic to any ingredients in Helixate FS.
Tell your healthcare provider if you are pregnant or breast-feeding because Helixate FS may not be right for you.
What should I tell my healthcare provider before I use Helixate FS?
Tell your healthcare provider about all of your medical conditions.
Tell your healthcare provider and pharmacist about all of the medicines you take, including all prescription and non-prescription medicines, such as over-the-counter medicines, supplements, or herbal remedies.
Tell your healthcare provider if you have been told you have heart disease or are at risk for heart disease.
Tell your healthcare provider if you have been told that you have inhibitors to factor VIII (because Helixate FS may not work for you).
What are the possible side effects of Helixate FS?
You could have an allergic reaction to Helixate FS. Call your healthcare provider right away and stop treatment if you get
- rash or hives
- itching
- tightness of the chest or throat
- difficulty breathing
- light-headed, dizziness
- nausea
- decrease in blood pressure
Your body can also make antibodies, called "inhibitors," against Helixate FS, which may stop Helixate FS from working properly. Consult with your healthcare provider to make sure you are carefully monitored with blood tests for the development of inhibitors to factor VIII.
Other common side effects of Helixate FS are
- Local injection site reactions (pain, swelling, irritation at infusion site)
- Infections from implanted injection device
Tell your healthcare provider about any side effect that bothers you or that does not go away.
Finding veins for injections may be difficult in young children. When frequent injections are required your child's healthcare provider may propose to have a device surgically placed under the skin to facilitate access to the bloodstream. These devices may result in infections.
These are not all the possible side effects with Helixate FS.
You can ask your healthcare provider for information that is written for healthcare professionals.
How do I store Helixate FS?
Do not freeze Helixate FS.
Store Helixate FS at +2°C to +8°C (36°F to 46°F) for up to 30 months from the date of manufacture. Within this period, Helixate FS may be stored for a period of up to 12 months at temperatures up to +25°C or 77°F.
Record the starting date of room temperature storage on the unopened product carton. Once stored at room temperature, do not return the product to the refrigerator. The product then expires after storage at room temperature, or after the expiration date on the product vial, whichever is earlier. Store vials in their original carton and protect them from extreme exposure to light.
Reconstituted product (after mixing dry products with wet diluent) must be used within 3 hours and cannot be stored.
Throw away any unused Helixate FS after the expiration date.
Do not use reconstituted Helixate FS if it is not clear to slightly cloudy and colorless.
What else should I know about Helixate FS and hemophilia A?
Medicines are sometimes prescribed for purposes other than those listed here. Do not use Helixate FS for a condition for which it is not prescribed. Do not share Helixate FS with other people, even if they have the same symptoms that you have.
This leaflet summarizes the most important information about Helixate FS. If you would like more information, talk to your healthcare provider. You can ask your healthcare provider or pharmacist for information about Helixate FS that was written for healthcare professionals.
Instructions for use
How should I take Helixate FS?
Do not attempt to self-infuse unless you have been taught how by your healthcare provider or hemophilia center.
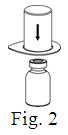
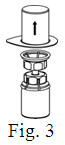
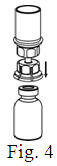
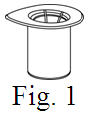
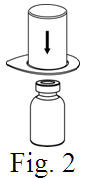
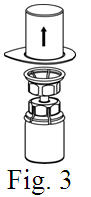
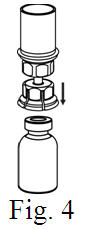
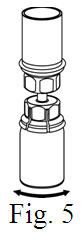
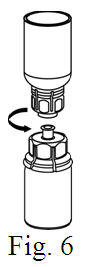
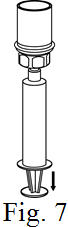
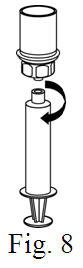
See the step-by-step instructions for reconstituting Helixate FS at the end of this leaflet and the Mix2Vial® filter transfer device instruction leaflet provided.
You should always follow the specific instructions given by your healthcare provider. The steps listed below are general guidelines for using Helixate FS. If you are unsure of the procedures, please call your healthcare provider before using.
Call your healthcare provider right away if bleeding is not controlled after using Helixate FS.
Your healthcare provider will prescribe the dose that you should take.
Your healthcare provider may need to take blood tests from time to time.
Talk to your healthcare provider before traveling. You should plan to bring enough Helixate FS for your treatment during this time.
Carefully handle Helixate FS. Dispose of all materials, including any leftover reconstituted Helixate FS product, in an appropriate container.
Reconstitution and use of Helixate FS
Always work on a clean flat surface and wash your hands before performing the following procedure. Use only the components for reconstitution and administration that are provided with each package of Helixate FS. If a package is opened or damaged, do not use this component. If these components cannot be used, please contact your healthcare provider. If you have any questions about Helixate FS contact CSL Behring Customer Support 1-800-683-1288.
Rate of administration
The entire dose of Helixate FS can usually be infused within 1 to 15 minutes. However, your healthcare provider will determine the rate of administration that is best for you.
Resources at CSL Behring available to the patient:
For Adverse Reaction Reporting contact:
CSL Behring Pharmacovigilance Department at 1-866-915-6958Contact CSL Behring to receive more product information:
Consumer Affairs 1-888-508-6978
Customer Support 1-800-683-1288
Reimbursement Services 1-800-676-4266For more information, visit www.HelixateFS.com
Manufactured by:
Bayer HealthCare LLC Whippany, NJ 07981 USA
U.S. License No. 8 (License Holder: Bayer Corporation)
Distributed by:
CSL Behring LLC Kankakee, IL 60901 USA
Mix2Vial® is a trademark of West Pharmaceutical Services, Inc. in the United States.
-
PRINCIPAL DISPLAY PANEL - 250 IU Range Carton
250 IU Range
NDC: 0053-8131-02
Helixate® FS
Antihemophilic Factor
(Recombinant)Formulated with Sucrose
Recombinant Factor VIII with
2.5 mL diluentCSL Behring
-
PRINCIPAL DISPLAY PANEL - 500 IU Range Carton
500 IU Range
NDC: 0053-8132-02
Helixate® FS
Antihemophilic Factor
(Recombinant)Formulated with Sucrose
Recombinant Factor VIII with
2.5 mL diluentCSL Behring
-
PRINCIPAL DISPLAY PANEL - 1000 IU Range Carton
1000 IU Range
NDC: 0053-8133-02
Helixate® FS
Antihemophilic Factor
(Recombinant)Formulated with Sucrose
Recombinant Factor VIII with
2.5 mL diluentCSL Behring
-
PRINCIPAL DISPLAY PANEL - 2000 IU Range Carton
2000 IU Range
NDC: 0053-8134-02
Helixate® FS
Antihemophilic Factor
(Recombinant)Formulated with Sucrose
Recombinant Factor VIII with
5 mL diluentCSL Behring
-
PRINCIPAL DISPLAY PANEL - 3000 IU Range Carton
3000 IU Range
NDC: 0053-8135-02
Helixate® FS
Antihemophilic Factor
(Recombinant)Formulated with Sucrose
Recombinant Factor VIII with
5 mL diluentCSL Behring
-
INGREDIENTS AND APPEARANCE
HELIXATE FS
antihemophilic factor, recombinant kitProduct Information Product Type PLASMA DERIVATIVE Item Code (Source) NDC: 0053-8131 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0053-8131-02 1 in 1 BOX; Type 0: Not a Combination Product Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 1 VIAL, SINGLE-USE 2.5 mL Part 2 1 VIAL 2.5 mL Part 1 of 2 HELIXATE FS
antihemophilic factor, recombinant injection, powder, lyophilized, for solutionProduct Information Item Code (Source) NDC: 0053-8141 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ANTIHEMOPHILIC FACTOR, HUMAN RECOMBINANT (UNII: P89DR4NY54) (Antihemophilic Factor, Human Recombinant - UNII:P89DR4NY54) ANTIHEMOPHILIC FACTOR, HUMAN RECOMBINANT 250 [iU] in 2.5 mL Inactive Ingredients Ingredient Name Strength GLYCINE (UNII: TE7660XO1C) HISTIDINE (UNII: 4QD397987E) CALCIUM (UNII: SY7Q814VUP) POLYSORBATE 80 (UNII: 6OZP39ZG8H) SUCROSE (UNII: C151H8M554) TRI-N-BUTYL PHOSPHATE (UNII: 95UAS8YAF5) COPPER (UNII: 789U1901C5) CHLORIDE ION (UNII: Q32ZN48698) SODIUM POLYMETAPHOSPHATE (UNII: P1BM4ZH95L) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0053-8141-01 2.5 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA103332 08/18/2000 Part 2 of 2 DILUENT
water injectionProduct Information Item Code (Source) NDC: 0053-7653 Route of Administration INTRAVENOUS Inactive Ingredients Ingredient Name Strength WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0053-7653-52 2.5 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA103332 08/18/2000 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA103332 08/18/2000 HELIXATE FS
antihemophilic factor, recombinant kitProduct Information Product Type PLASMA DERIVATIVE Item Code (Source) NDC: 0053-8132 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0053-8132-02 1 in 1 BOX; Type 0: Not a Combination Product Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 1 VIAL, SINGLE-USE 2.5 mL Part 2 1 VIAL 2.5 mL Part 1 of 2 HELIXATE FS
antihemophilic factor, recombinant injection, powder, lyophilized, for solutionProduct Information Item Code (Source) NDC: 0053-8142 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ANTIHEMOPHILIC FACTOR, HUMAN RECOMBINANT (UNII: P89DR4NY54) (ANTIHEMOPHILIC FACTOR, HUMAN RECOMBINANT - UNII:P89DR4NY54) ANTIHEMOPHILIC FACTOR, HUMAN RECOMBINANT 500 [iU] in 2.5 mL Inactive Ingredients Ingredient Name Strength GLYCINE (UNII: TE7660XO1C) HISTIDINE (UNII: 4QD397987E) CALCIUM (UNII: SY7Q814VUP) POLYSORBATE 80 (UNII: 6OZP39ZG8H) SUCROSE (UNII: C151H8M554) TRI-N-BUTYL PHOSPHATE (UNII: 95UAS8YAF5) COPPER (UNII: 789U1901C5) CHLORIDE ION (UNII: Q32ZN48698) SODIUM POLYMETAPHOSPHATE (UNII: P1BM4ZH95L) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0053-8142-01 2.5 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA103332 08/18/2000 Part 2 of 2 DILUENT
water injectionProduct Information Item Code (Source) NDC: 0053-7653 Route of Administration INTRAVENOUS Inactive Ingredients Ingredient Name Strength WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0053-7653-52 2.5 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA103332 08/18/2000 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA103332 08/18/2000 HELIXATE FS
antihemophilic factor, recombinant kitProduct Information Product Type PLASMA DERIVATIVE Item Code (Source) NDC: 0053-8133 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0053-8133-02 1 in 1 BOX; Type 0: Not a Combination Product Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 1 VIAL, SINGLE-USE 2.5 mL Part 2 1 VIAL 2.5 mL Part 1 of 2 HELIXATE FS
antihemophilic factor, recombinant injection, powder, lyophilized, for solutionProduct Information Item Code (Source) NDC: 0053-8143 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ANTIHEMOPHILIC FACTOR, HUMAN RECOMBINANT (UNII: P89DR4NY54) (ANTIHEMOPHILIC FACTOR, HUMAN RECOMBINANT - UNII:P89DR4NY54) ANTIHEMOPHILIC FACTOR, HUMAN RECOMBINANT 1000 [iU] in 2.5 mL Inactive Ingredients Ingredient Name Strength GLYCINE (UNII: TE7660XO1C) HISTIDINE (UNII: 4QD397987E) CALCIUM (UNII: SY7Q814VUP) POLYSORBATE 80 (UNII: 6OZP39ZG8H) SUCROSE (UNII: C151H8M554) TRI-N-BUTYL PHOSPHATE (UNII: 95UAS8YAF5) COPPER (UNII: 789U1901C5) CHLORIDE ION (UNII: Q32ZN48698) SODIUM POLYMETAPHOSPHATE (UNII: P1BM4ZH95L) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0053-8143-01 2.5 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA103332 08/18/2000 Part 2 of 2 DILUENT
water injectionProduct Information Item Code (Source) NDC: 0053-7653 Route of Administration INTRAVENOUS Inactive Ingredients Ingredient Name Strength WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0053-7653-52 2.5 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA103332 08/18/2000 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA103332 08/18/2000 HELIXATE FS
antihemophilic factor, recombinant kitProduct Information Product Type PLASMA DERIVATIVE Item Code (Source) NDC: 0053-8134 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0053-8134-02 1 in 1 BOX; Type 0: Not a Combination Product Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 1 VIAL, SINGLE-USE 5 mL Part 2 1 VIAL 5 mL Part 1 of 2 HELIXATE FS
antihemophilic factor, recombinant injection, powder, lyophilized, for solutionProduct Information Item Code (Source) NDC: 0053-8144 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ANTIHEMOPHILIC FACTOR, HUMAN RECOMBINANT (UNII: P89DR4NY54) (ANTIHEMOPHILIC FACTOR, HUMAN RECOMBINANT - UNII:P89DR4NY54) ANTIHEMOPHILIC FACTOR, HUMAN RECOMBINANT 2000 [iU] in 5 mL Inactive Ingredients Ingredient Name Strength GLYCINE (UNII: TE7660XO1C) HISTIDINE (UNII: 4QD397987E) CALCIUM (UNII: SY7Q814VUP) POLYSORBATE 80 (UNII: 6OZP39ZG8H) SUCROSE (UNII: C151H8M554) TRI-N-BUTYL PHOSPHATE (UNII: 95UAS8YAF5) COPPER (UNII: 789U1901C5) CHLORIDE ION (UNII: Q32ZN48698) SODIUM POLYMETAPHOSPHATE (UNII: P1BM4ZH95L) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0053-8144-01 5 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA103332 01/16/2008 Part 2 of 2 DILUENT
water injectionProduct Information Item Code (Source) NDC: 0053-7653 Route of Administration INTRAVENOUS Inactive Ingredients Ingredient Name Strength WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0053-7653-07 5 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA103332 01/16/2008 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA103332 01/16/2008 HELIXATE FS
antihemophilic factor, recombinant kitProduct Information Product Type PLASMA DERIVATIVE Item Code (Source) NDC: 0053-8135 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0053-8135-02 1 in 1 BOX; Type 0: Not a Combination Product Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 1 VIAL, SINGLE-USE 5 mL Part 2 1 VIAL 5 mL Part 1 of 2 HELIXATE FS
antihemophilic factor, recombinant injection, powder, lyophilized, for solutionProduct Information Item Code (Source) NDC: 0053-8145 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ANTIHEMOPHILIC FACTOR, HUMAN RECOMBINANT (UNII: P89DR4NY54) (ANTIHEMOPHILIC FACTOR, HUMAN RECOMBINANT - UNII:P89DR4NY54) ANTIHEMOPHILIC FACTOR, HUMAN RECOMBINANT 3000 [iU] in 5 mL Inactive Ingredients Ingredient Name Strength GLYCINE (UNII: TE7660XO1C) HISTIDINE (UNII: 4QD397987E) CALCIUM (UNII: SY7Q814VUP) POLYSORBATE 80 (UNII: 6OZP39ZG8H) SUCROSE (UNII: C151H8M554) TRI-N-BUTYL PHOSPHATE (UNII: 95UAS8YAF5) COPPER (UNII: 789U1901C5) CHLORIDE ION (UNII: Q32ZN48698) SODIUM POLYMETAPHOSPHATE (UNII: P1BM4ZH95L) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0053-8145-01 5 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA103332 07/31/2009 Part 2 of 2 DILUENT
water injectionProduct Information Item Code (Source) NDC: 0053-7653 Route of Administration INTRAVENOUS Inactive Ingredients Ingredient Name Strength WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0053-7653-07 5 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA103332 07/31/2009 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA103332 07/31/2009 Labeler - CSL Behring LLC (058268293) Registrant - Bayer HealthCare LLC (127769128) Establishment Name Address ID/FEI Business Operations Bayer HealthCare LLC 127769128 MANUFACTURE(0053-8131, 0053-8132, 0053-8133, 0053-8134, 0053-8135)





© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.