LOVENOX- enoxaparin sodium injection
Lovenox by
Drug Labeling and Warnings
Lovenox by is a Prescription medication manufactured, distributed, or labeled by Sanofi-Aventis U.S. LLC, Aventis Pharma Manufacturing Pte. Ltd., Genzyme Corporation, Sanofi-Aventis Deutschland GmbH, Sanofi Winthrop Industrie. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use Lovenox safely and effectively. See full prescribing information for Lovenox.
Lovenox (enoxaparin sodium injection), for subcutaneous and intravenous use
Initial U.S. Approval: 1993WARNING: SPINAL/EPIDURAL HEMATOMAS
See full prescribing information for complete boxed warning.
Epidural or spinal hematomas may occur in patients who are anticoagulated with low molecular weight heparins (LMWH) or heparinoids and are receiving neuraxial anesthesia or undergoing spinal puncture. These hematomas may result in long-term or permanent paralysis. Consider these risks when scheduling patients for spinal procedures. Factors that can increase the risk of developing epidural or spinal hematomas in these patients include:
- Use of indwelling epidural catheters
- Concomitant use of other drugs that affect hemostasis, such as non-steroidal anti-inflammatory drugs (NSAIDs), platelet inhibitors, and other anticoagulants
- A history of traumatic or repeated epidural or spinal punctures
- A history of spinal deformity or spinal surgery
- Optimal timing between the administration of Lovenox and neuraxial procedures is not known
Monitor patients frequently for signs and symptoms of neurological impairment. If neurological compromise is noted, urgent treatment is necessary. (5.1, 7)
INDICATIONS AND USAGE
Lovenox is a low molecular weight heparin (LMWH) indicated for:
- Prophylaxis of deep vein thrombosis (DVT) in abdominal surgery, hip replacement surgery, knee replacement surgery, or medical patients with severely restricted mobility during acute illness (1.1)
- Inpatient treatment of acute DVT with or without pulmonary embolism (1.2)
- Outpatient treatment of acute DVT without pulmonary embolism (1.2)
- Prophylaxis of ischemic complications of unstable angina and non–Q-wave myocardial infarction (MI) (1.3)
- Treatment of acute ST-segment elevation myocardial infarction (STEMI) managed medically or with subsequent percutaneous coronary intervention (PCI) (1.4)
DOSAGE AND ADMINISTRATION
See full prescribing information for dosing and administration information. (2)
DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONS
- Active major bleeding (4)
- History of heparin-induced thrombocytopenia (HIT) within the past 100 days or in the presence of circulating antibodies (4)
- Hypersensitivity to enoxaparin sodium (4)
- Hypersensitivity to heparin or pork products (4)
- Hypersensitivity to benzyl alcohol (for multiple-dose formulation only) (4)
WARNINGS AND PRECAUTIONS
- Increased Risk of Hemorrhage: Monitor for signs of bleeding. (5.1, 5.2, 5.3)
- Risk of Heparin-Induced Thrombocytopenia with or without Thrombosis. (5.4)
- Thrombocytopenia: Monitor platelet count closely. (5.5)
- Interchangeability with other heparins: Do not exchange with heparin or other LMWHs. (5.6)
- Increased Risk of Thrombosis in Pregnant Women with Mechanical Prosthetic Heart Valves: Women and their fetuses may be at increased risk. Monitor more frequently and adjust dosage as needed. (5.7)
ADVERSE REACTIONS
Most common adverse reactions (>1%) were bleeding, anemia, thrombocytopenia, elevation of serum aminotransferase, diarrhea, nausea, ecchymosis, fever, edema, peripheral edema, dyspnea, confusion, and injection site pain. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact sanofi-aventis at 1-800-633-1610 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 12/2018
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: SPINAL/EPIDURAL HEMATOMAS
1 INDICATIONS AND USAGE
1.1 Prophylaxis of Deep Vein Thrombosis
1.2 Treatment of Acute Deep Vein Thrombosis
1.3 Prophylaxis of Ischemic Complications of Unstable Angina and Non–Q-Wave Myocardial Infarction
1.4 Treatment of Acute ST-Segment Elevation Myocardial Infarction
2 DOSAGE AND ADMINISTRATION
2.1 Pretreatment Evaluation
2.2 Adult Dosage
2.3 Dose Reduction for Patients with Severe Renal Impairment
2.4 Recommended Dosage for Geriatric Patients with Acute ST-Segment Elevation Myocardial Infarction
2.5 Administration
2.6 Monitoring for Safety
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Increased Risk of Hemorrhage
5.2 Increased Risk of Bleeding following Percutaneous Coronary Revascularization Procedures
5.3 Increased Risk of Bleeding in Patients with Concomitant Medical Conditions
5.4 Risk of Heparin-Induced Thrombocytopenia with or without Thrombosis
5.5 Thrombocytopenia
5.6 Interchangeability with other Heparins
5.7 Increased Risk of Thrombosis in Pregnant Women with Mechanical Prosthetic Heart Valves
5.8 Risk of Serious Adverse Reactions in Infants due to Benzyl Alcohol Preservative
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Patients with Mechanical Prosthetic Heart Valves
8.7 Renal Impairment
8.8 Low-Weight Patients
8.9 Obese Patients
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
13.3 Reproductive and Developmental Toxicology
14 CLINICAL STUDIES
14.1 Prophylaxis of Deep Vein Thrombosis following Abdominal Surgery in Patients at Risk for Thromboembolic Complications
14.2 Prophylaxis of Deep Vein Thrombosis following Hip or Knee Replacement Surgery
14.3 Prophylaxis of Deep Vein Thrombosis in Medical Patients with Severely Restricted Mobility during Acute Illness
14.4 Treatment of Deep Vein Thrombosis with or without Pulmonary Embolism
14.5 Prophylaxis of Ischemic Complications in Unstable Angina and Non–Q-Wave Myocardial Infarction
14.6 Treatment of Acute ST-Segment Elevation Myocardial Infarction
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: SPINAL/EPIDURAL HEMATOMAS
Epidural or spinal hematomas may occur in patients who are anticoagulated with low molecular weight heparins (LMWH) or heparinoids and are receiving neuraxial anesthesia or undergoing spinal puncture. These hematomas may result in long-term or permanent paralysis. Consider these risks when scheduling patients for spinal procedures. Factors that can increase the risk of developing epidural or spinal hematomas in these patients include:
- Use of indwelling epidural catheters
- Concomitant use of other drugs that affect hemostasis, such as non-steroidal anti-inflammatory drugs (NSAIDs), platelet inhibitors, and other anticoagulants
- A history of traumatic or repeated epidural or spinal punctures
- A history of spinal deformity or spinal surgery
- Optimal timing between the administration of Lovenox and neuraxial procedures is not known
Monitor patients frequently for signs and symptoms of neurological impairment. If neurological compromise is noted, urgent treatment is necessary.
Consider the benefits and risks before neuraxial intervention in patients anticoagulated or to be anticoagulated for thromboprophylaxis [see Warnings and Precautions (5.1) and Drug Interactions (7)].
-
1 INDICATIONS AND USAGE
1.1 Prophylaxis of Deep Vein Thrombosis
Lovenox® is indicated for the prophylaxis of deep vein thrombosis (DVT), which may lead to pulmonary embolism (PE):
- in patients undergoing abdominal surgery who are at risk for thromboembolic complications [see Clinical Studies (14.1)]
- in patients undergoing hip replacement surgery, during and following hospitalization
- in patients undergoing knee replacement surgery
- in medical patients who are at risk for thromboembolic complications due to severely restricted mobility during acute illness
1.2 Treatment of Acute Deep Vein Thrombosis
Lovenox is indicated for:
- the inpatient treatment of acute deep vein thrombosis with or without pulmonary embolism, when administered in conjunction with warfarin sodium
- the outpatient treatment of acute deep vein thrombosis without pulmonary embolism when administered in conjunction with warfarin sodium
1.3 Prophylaxis of Ischemic Complications of Unstable Angina and Non–Q-Wave Myocardial Infarction
Lovenox is indicated for the prophylaxis of ischemic complications of unstable angina and non–Q-wave myocardial infarction, when concurrently administered with aspirin.
1.4 Treatment of Acute ST-Segment Elevation Myocardial Infarction
Lovenox, when administered concurrently with aspirin, has been shown to reduce the rate of the combined endpoint of recurrent myocardial infarction or death in patients with acute ST-segment elevation myocardial infarction (STEMI) receiving thrombolysis and being managed medically or with percutaneous coronary intervention (PCI).
-
2 DOSAGE AND ADMINISTRATION
2.1 Pretreatment Evaluation
Evaluate all patients for a bleeding disorder before starting Lovenox treatment, unless treatment is urgently needed.
2.2 Adult Dosage
Abdominal Surgery
The recommended dose of Lovenox is 40 mg by subcutaneous injection once a day (with the initial dose given 2 hours prior to surgery) in patients undergoing abdominal surgery who are at risk for thromboembolic complications. The usual duration of administration is 7 to 10 days [see Clinical Studies (14.1)].
Hip or Knee Replacement Surgery
The recommended dose of Lovenox is 30 mg every 12 hours administered by subcutaneous injection in patients undergoing hip or knee replacement surgery. Administer the initial dose 12 to 24 hours after surgery, provided that hemostasis has been established. The usual duration of administration is 7 to 10 days [see Clinical Studies (14.2)].
A dose of Lovenox of 40 mg once a day subcutaneously may be considered for hip replacement surgery for up to 3 weeks. Administer the initial dose 12 (±3) hours prior to surgery.
Medical Patients During Acute Illness
The recommended dose of Lovenox is 40 mg once a day administered by subcutaneous injection for medical patients at risk for thromboembolic complications due to severely restricted mobility during acute illness. The usual duration of administration is 6 to 11 days [see Clinical Studies (14.3)].
Treatment of Deep Vein Thrombosis with or without Pulmonary Embolism
The recommended dose of Lovenox is 1 mg/kg every 12 hours administered subcutaneously in patients with acute deep vein thrombosis without pulmonary embolism, who can be treated at home in an outpatient setting.
The recommended dose of Lovenox is 1 mg/kg every 12 hours administered subcutaneously or 1.5 mg/kg once a day administered subcutaneously at the same time every day for inpatient (hospital) treatment of patients with acute deep vein thrombosis with pulmonary embolism or patients with acute deep vein thrombosis without pulmonary embolism (who are not candidates for outpatient treatment).
In both outpatient and inpatient (hospital) treatments, initiate warfarin sodium therapy when appropriate (usually within 72 hours of Lovenox). Continue Lovenox for a minimum of 5 days and until a therapeutic oral anticoagulant effect has been achieved (International Normalization Ratio 2 to 3). The average duration of administration is 7 days [see Clinical Studies (14.4)].
Unstable Angina and Non–Q-Wave Myocardial Infarction
The recommended dose of Lovenox is 1 mg/kg administered subcutaneously every 12 hours in conjunction with oral aspirin therapy (100 to 325 mg once daily) in patients with unstable angina or non–Q-wave myocardial infarction. Treat with Lovenox for a minimum of 2 days and continue until clinical stabilization. The usual duration of treatment is 2 to 8 days [see Warnings and Precautions (5.2) and Clinical Studies (14.5)].
Treatment of Acute ST-Segment Elevation Myocardial Infarction
The recommended dose of Lovenox is a single intravenous bolus of 30 mg plus a 1 mg/kg subcutaneous dose followed by 1 mg/kg administered subcutaneously every 12 hours (maximum 100 mg for the first two doses only, followed by 1 mg/kg dosing for the remaining doses) in patients with acute ST-segment elevation myocardial infarction. Reduce the dosage in patients ≥75 years of age [see Dosage and Administration (2.4)]. Unless contraindicated, administer aspirin to all patients as soon as they are identified as having STEMI and continue dosing with 75 to 325 mg once daily.
When administered in conjunction with a thrombolytic (fibrin specific or non–fibrin specific), administer Lovenox between 15 minutes before and 30 minutes after the start of fibrinolytic therapy. The usual duration of Lovenox therapy is 8 days or until hospital discharge.
For patients managed with percutaneous coronary intervention (PCI), if the last Lovenox subcutaneous administration was given less than 8 hours before balloon inflation, no additional dosing is needed. If the last Lovenox subcutaneous administration was given more than 8 hours before balloon inflation, administer an intravenous bolus of 0.3 mg/kg of Lovenox [see Warnings and Precautions (5.2)].
2.3 Dose Reduction for Patients with Severe Renal Impairment
The recommended prophylaxis and treatment dosage regimens for patients with severe renal impairment (creatinine clearance <30 mL/min) are described in Table 1 [see Use in Specific Populations (8.7) and Clinical Pharmacology (12.3)].
Table 1: Dosage Regimens for Patients with Severe Renal Impairment (creatinine clearance <30 mL/minute) Indication Dosage Regimen Prophylaxis in abdominal surgery 30 mg administered subcutaneously once daily Prophylaxis in hip or knee replacement surgery 30 mg administered subcutaneously once daily Prophylaxis in medical patients during acute illness 30 mg administered subcutaneously once daily Inpatient treatment of acute deep vein thrombosis with or without pulmonary embolism, when administered in conjunction with warfarin sodium 1 mg/kg administered subcutaneously once daily Outpatient treatment of acute deep vein thrombosis without pulmonary embolism, when administered in conjunction with warfarin sodium 1 mg/kg administered subcutaneously once daily Prophylaxis of ischemic complications of unstable angina and non–Q-wave myocardial infarction, when concurrently administered with aspirin 1 mg/kg administered subcutaneously once daily Treatment of acute ST-segment elevation myocardial infarction in patients <75 years of age, when administered in conjunction with aspirin 30 mg single intravenous bolus plus a 1 mg/kg subcutaneous dose followed by 1 mg/kg administered subcutaneously once daily Treatment of acute ST-segment elevation myocardial infarction in geriatric patients ≥75 years of age, when administered in conjunction with aspirin 1 mg/kg administered subcutaneously once daily
(no initial bolus)Although no dose adjustment is recommended in patients with creatinine clearance 30 to 50 mL/min and creatinine clearance 50 to 80 mL/min, observe these patients frequently for signs and symptoms of bleeding.
2.4 Recommended Dosage for Geriatric Patients with Acute ST-Segment Elevation Myocardial Infarction
For treatment of acute ST-segment elevation myocardial infarction in geriatric patients ≥75 years of age, do not use an initial intravenous bolus. Initiate dosing with 0.75 mg/kg subcutaneously every 12 hours (maximum 75 mg for the first two doses only, followed by 0.75 mg/kg dosing for the remaining doses) [see Use in Specific Populations (8.5) and Clinical Pharmacology (12.3)].
No dose adjustment is necessary for other indications in geriatric patients unless kidney function is impaired [see Dosage and Administration (2.2)].
2.5 Administration
Do not administer Lovenox by intramuscular injection.
Administer Lovenox by intravenous or subcutaneous injection only.
Lovenox is a clear, colorless to pale yellow sterile solution, and as with other parenteral drug products, should be inspected visually for particulate matter and discoloration prior to administration.
Use a tuberculin syringe or equivalent when using Lovenox multiple-dose vials to assure withdrawal of the appropriate volume of drug.
Patients may self-inject by the subcutaneous route of administration only after their physicians determine that it is appropriate and with medical follow-up, as necessary. Provide proper training in subcutaneous injection technique before allowing self-injection (with or without the assistance of an injection device).
Subcutaneous Injection Technique
- Position patients in a supine position for Lovenox administration by deep subcutaneous injection.
- Do not expel the air bubble from the prefilled syringes before the injection, to avoid the loss of drug.
- Alternate injection sites between the left and right anterolateral and left and right posterolateral abdominal wall.
- Introduce the whole length of the needle into a skin fold held between the thumb and forefinger; hold the skin fold throughout the injection. To minimize bruising, do not rub the injection site after completion of the injection.
Lovenox prefilled syringes and graduated prefilled syringes are for single, one-time use only and are available with a system that shields the needle after injection.
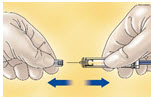
Remove the prefilled syringe from the blister packaging by peeling at the arrow as directed on the blister. Do not remove by pulling on the plunger as this may damage the syringe.
- Remove the needle shield by pulling it straight off the syringe (see Figure A). If less than the full syringe volume is needed to administer the prescribed dose, eject syringe contents until the prescribed dose is left in the syringe.
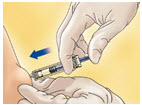
- Inject using standard technique, pushing the plunger to the bottom of the syringe (see Figure B).
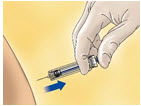
- Remove the syringe from the injection site keeping your finger on the plunger rod (see Figure C).
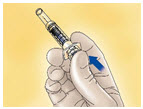
- Orient the needle away from you and others, and activate the safety system by firmly pushing the plunger rod. The protective sleeve will automatically cover the needle and an audible "click" will be heard to confirm shield activation (see Figure D).
- Immediately dispose of the syringe in the nearest sharps container (see Figure E).

NOTE:
- The safety system can only be activated once the syringe has been emptied.
- Activation of the safety system must be done only after removing the needle from the patient's skin.
- Do not replace the needle shield after injection.
- The safety system should not be sterilized.
Activation of the safety system may cause minimal splatter of fluid. For optimal safety, activate the system while orienting it downwards away from yourself and others.
Intravenous (Bolus) Injection Technique
Use the multiple-dose vial for intravenous injections. Administer Lovenox through an intravenous line. Do not mix or coadminister Lovenox with other medications. Flush the intravenous access device with a sufficient volume of saline or dextrose solution prior to and following the intravenous bolus administration of Lovenox, to prevent mixing of drugs. Lovenox is compatible with normal saline solution (0.9%) or 5% dextrose in water.
2.6 Monitoring for Safety
During therapy monitor complete blood counts including platelets and stool occult blood.
Assess for signs and symptoms of bleeding.
In patients with renal impairment anti-Factor Xa levels may be used to monitor the anticoagulant effects of Lovenox.
If during Lovenox therapy abnormal coagulation parameters or bleeding should occur, anti-Factor Xa levels may be used to monitor the anticoagulant effects of Lovenox [see Clinical Pharmacology (12.3)].
Prothrombin Time (PT) and Activated Partial Thromboplastin Time (aPTT) are not adequate for monitoring the anticoagulant effects of Lovenox.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
Lovenox is contraindicated in patients with:
- Active major bleeding
- History of immune-mediated heparin-induced thrombocytopenia (HIT) within the past 100 days or in the presence of circulating antibodies [see Warnings and Precautions (5.4)]
- Known hypersensitivity to enoxaparin sodium (e.g., pruritus, urticaria, anaphylactic/anaphylactoid reactions) [see Adverse Reactions (6.2)]
- Known hypersensitivity to heparin or pork products
- Known hypersensitivity to benzyl alcohol (which is in only the multiple-dose formulation of Lovenox) [see Warnings and Precautions (5.8)]
-
5 WARNINGS AND PRECAUTIONS
5.1 Increased Risk of Hemorrhage
Cases of epidural or spinal hemorrhage and subsequent hematomas have been reported with the use of Lovenox and epidural or spinal anesthesia/analgesia or spinal puncture procedures, resulting in long-term or permanent paralysis. The risk of these events is higher with the use of postoperative indwelling epidural catheters, with the concomitant use of additional drugs affecting hemostasis such as NSAIDs, with traumatic or repeated epidural or spinal puncture, or in patients with a history of spinal surgery or spinal deformity [see Boxed Warning, Adverse Reactions (6.2) and Drug Interactions (7)].
To reduce the potential risk of bleeding associated with the concurrent use of Lovenox and epidural or spinal anesthesia/analgesia or spinal puncture, consider the pharmacokinetic profile of Lovenox [see Clinical Pharmacology (12.3)]. Placement or removal of an epidural catheter or lumbar puncture is best performed when the anticoagulant effect of Lovenox is low; however, the exact timing to reach a sufficiently low anticoagulant effect in each patient is not known.
Placement or removal of a catheter should be delayed for at least 12 hours after administration of lower doses (30 mg once or twice daily or 40 mg once daily) of Lovenox, and at least 24 hours after the administration of higher doses (0.75 mg/kg twice daily, 1 mg/kg twice daily, or 1.5 mg/kg once daily) of Lovenox. Anti-Xa levels are still detectable at these time points, and these delays are not a guarantee that neuraxial hematoma will be avoided. Patients receiving the 0.75 mg/kg twice-daily dose or the 1 mg/kg twice-daily dose should not receive the second Lovenox dose in the twice-daily regimen to allow a longer delay before catheter placement or removal. Likewise, although a specific recommendation for timing of a subsequent Lovenox dose after catheter removal cannot be made, consider delaying this next dose for at least four hours, based on a benefit-risk assessment considering both the risk for thrombosis and the risk for bleeding in the context of the procedure and patient risk factors. For patients with creatinine clearance <30 mL/minute, additional considerations are necessary because elimination of Lovenox is more prolonged; consider doubling the timing of removal of a catheter, at least 24 hours for the lower prescribed dose of Lovenox (30 mg once daily) and at least 48 hours for the higher dose (1 mg/kg/day) [see Clinical Pharmacology (12.3)].
Should the physician decide to administer anticoagulation in the context of epidural or spinal anesthesia/analgesia or lumbar puncture, frequent monitoring must be exercised to detect any signs and symptoms of neurological impairment such as midline back pain, sensory and motor deficits (numbness or weakness in lower limbs), and bowel and/or bladder dysfunction. Instruct patients to report immediately if they experience any of the above signs or symptoms. If signs or symptoms of spinal hematoma are suspected, initiate urgent diagnosis and treatment including consideration for spinal cord decompression even though such treatment may not prevent or reverse neurological sequelae.
Use Lovenox with extreme caution in conditions with increased risk of hemorrhage, such as bacterial endocarditis, congenital or acquired bleeding disorders, active ulcerative and angiodysplastic gastrointestinal disease, hemorrhagic stroke, or shortly after brain, spinal, or ophthalmological surgery, or in patients treated concomitantly with platelet inhibitors.
Major hemorrhages including retroperitoneal and intracranial bleeding have been reported. Some of these cases have been fatal.
Bleeding can occur at any site during therapy with Lovenox. An unexplained fall in hematocrit or blood pressure should lead to a search for a bleeding site.
5.2 Increased Risk of Bleeding following Percutaneous Coronary Revascularization Procedures
To minimize the risk of bleeding following the vascular instrumentation during the treatment of unstable angina, non–Q-wave myocardial infarction and acute ST-segment elevation myocardial infarction, adhere precisely to the intervals recommended between Lovenox doses. It is important to achieve hemostasis at the puncture site after PCI. In case a closure device is used, the sheath can be removed immediately. If a manual compression method is used, sheath should be removed 6 hours after the last intravenous/subcutaneous Lovenox. If the treatment with Lovenox is to be continued, the next scheduled dose should be given no sooner than 6 to 8 hours after sheath removal. The site of the procedure should be observed for signs of bleeding or hematoma formation [see Dosage and Administration (2.1)].
5.3 Increased Risk of Bleeding in Patients with Concomitant Medical Conditions
Lovenox should be used with care in patients with a bleeding diathesis, uncontrolled arterial hypertension or a history of recent gastrointestinal ulceration, diabetic retinopathy, renal dysfunction and hemorrhage.
5.4 Risk of Heparin-Induced Thrombocytopenia with or without Thrombosis
Lovenox may cause heparin-induced thrombocytopenia (HIT) or heparin-induced thrombocytopenia with thrombosis (HITTS). HITTS may lead to organ infarction, limb ischemia, or death. Monitor thrombocytopenia of any degree closely.
Use of Lovenox in patients with a history of immune-mediated HIT within the past 100 days or in the presence of circulating antibodies is contraindicated [see Contraindications (4)]. Circulating antibodies may persist for several years.
Only use Lovenox in patients with a history of HIT if more than 100 days have elapsed since the prior HIT episode and no circulating antibodies are present. Because HIT may still occur in these circumstances, the decision to use Lovenox in such a case must be made only after a careful benefit-risk assessment and after non-heparin alternative treatments are considered.
5.5 Thrombocytopenia
Thrombocytopenia can occur with the administration of Lovenox.
Moderate thrombocytopenia (platelet counts between 100,000/mm3 and 50,000/mm3) occurred at a rate of 1.3% in patients given Lovenox, 1.2% in patients given heparin, and 0.7% in patients given placebo in clinical trials.
Platelet counts less than 50,000/mm3 occurred at a rate of 0.1% in patients given Lovenox, in 0.2% of patients given heparin, and 0.4% of patients given placebo in the same trials.
Thrombocytopenia of any degree should be monitored closely. If the platelet count falls below 100,000/mm3, Lovenox should be discontinued.
5.6 Interchangeability with other Heparins
Lovenox cannot be used interchangeably (unit for unit) with heparin or other low molecular weight heparins as they differ in manufacturing process, molecular weight distribution, anti-Xa and anti-IIa activities, units, and dosage. Each of these medicines has its own instructions for use.
5.7 Increased Risk of Thrombosis in Pregnant Women with Mechanical Prosthetic Heart Valves
Use of Lovenox for thromboprophylaxis in pregnant women with mechanical prosthetic heart valves may result in valve thrombosis. In a clinical study of pregnant women with mechanical prosthetic heart valves given Lovenox (1 mg/kg twice daily) to reduce the risk of thromboembolism, 2 of 8 women developed clots resulting in blockage of the valve and leading to maternal and fetal death. No patients in the heparin/warfarin group (0 of 4 women) died. There also have been isolated postmarketing reports of valve thrombosis in pregnant women with mechanical prosthetic heart valves while receiving Lovenox for thromboprophylaxis. Women with mechanical prosthetic heart valves may be at higher risk for thromboembolism during pregnancy and, when pregnant, have a higher rate of fetal loss from stillbirth, spontaneous abortion, and premature delivery. Therefore, frequent monitoring of peak and trough anti-Factor Xa levels, and adjusting of dosage may be needed [see Use in Specific Populations (8.6)].
5.8 Risk of Serious Adverse Reactions in Infants due to Benzyl Alcohol Preservative
Lovenox multiple-dose vials are not approved for use in neonates or infants.
Serious and fatal adverse reactions including "gasping syndrome" can occur in neonates and low birth weight infants treated with benzyl alcohol-preserved drugs, including Lovenox multiple-dose vials. The "gasping syndrome" is characterized by central nervous system depression, metabolic acidosis, and gasping respirations. The minimum amount of benzyl alcohol at which serious adverse reactions may occur is not known (Lovenox multiple-dose vials contain 15 mg of benzyl alcohol per mL) [see Use in Specific Populations (8.4)].
Because benzyl alcohol may cross the placenta, if anticoagulation with Lovenox is needed during pregnancy, use the preservative-free formulations where possible [see Use in Specific Populations (8.1)].
-
6 ADVERSE REACTIONS
The following serious adverse reactions are also discussed in other sections of the labeling:
- Spinal/epidural hematomas [see Boxed Warning and Warnings and Precautions (5.1)]
- Increased Risk of Hemorrhage [see Warnings and Precautions (5.1)]
- Thrombocytopenia [see Warnings and Precautions (5.5)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
During clinical development for the approved indications, 15,918 patients were exposed to Lovenox. These included 1,228 for prophylaxis of deep vein thrombosis following abdominal surgery in patients at risk for thromboembolic complications, 1,368 for prophylaxis of deep vein thrombosis following hip or knee replacement surgery, 711 for prophylaxis of deep vein thrombosis in medical patients with severely restricted mobility during acute illness, 1,578 for prophylaxis of ischemic complications in unstable angina and non–Q-wave myocardial infarction, 10,176 for treatment of acute ST-elevation myocardial infarction, and 857 for treatment of deep vein thrombosis with or without pulmonary embolism. Lovenox doses in the clinical trials for prophylaxis of deep vein thrombosis following abdominal or hip or knee replacement surgery or in medical patients with severely restricted mobility during acute illness ranged from 40 mg subcutaneously once daily to 30 mg subcutaneously twice daily. In the clinical studies for prophylaxis of ischemic complications of unstable angina and non–Q-wave myocardial infarction doses were 1 mg/kg every 12 hours and in the clinical studies for treatment of acute ST-segment elevation myocardial infarction Lovenox doses were a 30 mg intravenous bolus followed by 1 mg/kg every 12 hours subcutaneously.
Hemorrhage
The following rates of major bleeding events have been reported during clinical trials with Lovenox (see Tables 2 to 7).
Table 2: Major Bleeding Episodes Following Abdominal and Colorectal Surgery* Dosing Regimen Indications Lovenox
40 mg daily subcutaneouslyHeparin
5000 U q8h subcutaneously- * Bleeding complications were considered major: (1) if the hemorrhage caused a significant clinical event, or (2) if accompanied by a hemoglobin decrease ≥2 g/dL or transfusion of 2 or more units of blood products. Retroperitoneal, intraocular, and intracranial hemorrhages were always considered major.
Abdominal Surgery n=555
23 (4%)n=560
16 (3%)Colorectal Surgery n=673
28 (4%)n=674
21 (3%)Table 3: Major Bleeding Episodes Following Hip or Knee Replacement Surgery* Dosing Regimen Indications Lovenox
40 mg daily subcutaneouslyLovenox
30 mg q12h subcutaneouslyHeparin
15,000 U/24h subcutaneously- * Bleeding complications were considered major: (1) if the hemorrhage caused a significant clinical event, or (2) if accompanied by a hemoglobin decrease ≥2 g/dL or transfusion of 2 or more units of blood products. Retroperitoneal and intracranial hemorrhages were always considered major. In the knee replacement surgery trials, intraocular hemorrhages were also considered major hemorrhages.
- † Lovenox 30 mg every 12 hours subcutaneously initiated 12 to 24 hours after surgery and continued for up to 14 days after surgery
- ‡ Lovenox 40 mg subcutaneously once a day initiated up to 12 hours prior to surgery and continued for up to 7 days after surgery
- § Lovenox 40 mg subcutaneously once a day for up to 21 days after discharge
Hip Replacement Surgery without Extended Prophylaxis† n=786
31 (4%)n=541
32 (6%)Hip Replacement Surgery with Extended Prophylaxis Peri-operative Period‡ n=288
4 (2%)Extended Prophylaxis Period§ n=221
0 (0%)Knee Replacement Surgery without Extended Prophylaxis† n=294
3 (1%)n=225
3 (1%)NOTE: At no time point were the 40 mg once a day pre-operative and the 30 mg every 12 hours postoperative hip replacement surgery prophylactic regimens compared in clinical trials. Injection site hematomas during the extended prophylaxis period after hip replacement surgery occurred in 9% of the Lovenox patients versus 1.8% of the placebo patients.
Table 4: Major Bleeding Episodes in Medical Patients with Severely Restricted Mobility During Acute Illness* Dosing Regimen Indication Lovenox†
20 mg daily subcutaneouslyLovenox†
40 mg daily subcutaneouslyPlacebo† - * Bleeding complications were considered major: (1) if the hemorrhage caused a significant clinical event, (2) if the hemorrhage caused a decrease in hemoglobin of ≥2 g/dL or transfusion of 2 or more units of blood products. Retroperitoneal and intracranial hemorrhages were always considered major although none were reported during the trial.
- † The rates represent major bleeding on study medication up to 24 hours after last dose.
Medical Patients During Acute Illness n=351
1 (<1%)n=360
3 (<1%)n=362
2 (<1%)Table 5: Major Bleeding Episodes in Deep Vein Thrombosis with or without Pulmonary Embolism Treatment* Dosing Regimen† Indication Lovenox
1.5 mg/kg daily subcutaneouslyLovenox
1 mg/kg q12h subcutaneouslyHeparin
aPTT Adjusted Intravenous Therapy- * Bleeding complications were considered major: (1) if the hemorrhage caused a significant clinical event, or (2) if accompanied by a hemoglobin decrease ≥2 g/dL or transfusion of 2 or more units of blood products. Retroperitoneal, intraocular, and intracranial hemorrhages were always considered major.
- † All patients also received warfarin sodium (dose-adjusted according to PT to achieve an INR of 2.0 to 3.0) commencing within 72 hours of Lovenox or standard heparin therapy and continuing for up to 90 days.
Treatment of DVT and PE n=298
5 (2%)n=559
9 (2%)n=554
9 (2%)Table 6: Major Bleeding Episodes in Unstable Angina and Non–Q-Wave Myocardial Infarction Dosing Regimen Indication Lovenox*
1 mg/kg q12h subcutaneouslyHeparin*
aPTT Adjusted Intravenous Therapy- * The rates represent major bleeding on study medication up to 12 hours after dose.
- † Aspirin therapy was administered concurrently (100 to 325 mg per day).
- ‡ Bleeding complications were considered major: (1) if the hemorrhage caused a significant clinical event, or (2) if accompanied by a hemoglobin decrease by ≥3 g/dL or transfusion of 2 or more units of blood products. Intraocular, retroperitoneal, and intracranial hemorrhages were always considered major.
Unstable Angina and Non–Q-Wave MI†,‡ n=1578
17 (1%)n=1529
18 (1%)Table 7: Major Bleeding Episodes in Acute ST-Segment Elevation Myocardial Infarction Dosing Regimen Indication Lovenox*
Initial 30 mg intravenous bolus followed by 1 mg/kg q12h subcutaneouslyHeparin*
aPTT Adjusted Intravenous Therapy- * The rates represent major bleeding (including ICH) up to 30 days
- † Bleedings were considered major if the hemorrhage caused a significant clinical event associated with a hemoglobin decrease by ≥5 g/dL. ICH were always considered major.
Acute ST-Segment Elevation Myocardial Infarction n=10176
n (%)n=10151
n (%)Major bleeding (including ICH)† 211 (2.1) 138 (1.4) Intracranial hemorrhages (ICH) 84 (0.8) 66 (0.7) Elevations of Serum Aminotransferases
Asymptomatic increases in aspartate (AST [SGOT]) and alanine (ALT [SGPT]) aminotransferase levels greater than three times the upper limit of normal of the laboratory reference range have been reported in up to 6.1% and 5.9% of patients, respectively, during treatment with Lovenox.
Since aminotransferase determinations are important in the differential diagnosis of myocardial infarction, liver disease, and pulmonary emboli, elevations that might be caused by drugs like Lovenox should be interpreted with caution.
Local Reactions
Local irritation, pain, hematoma, ecchymosis, and erythema may follow subcutaneous injection of Lovenox.
Adverse Reactions in Patients Receiving Lovenox for Prophylaxis or Treatment of DVT, PE
Other adverse reactions that were thought to be possibly or probably related to treatment with Lovenox, heparin, or placebo in clinical trials with patients undergoing hip or knee replacement surgery, abdominal or colorectal surgery, or treatment for DVT and that occurred at a rate of at least 2% in the Lovenox group, are provided below (see Tables 8 to 11).
Table 8: Adverse Reactions Occurring at ≥2% Incidence in Lovenox-Treated Patients Undergoing Abdominal or Colorectal Surgery Dosing Regimen Lovenox
40 mg daily subcutaneously
n=1228
%Heparin
5000 U q8h subcutaneously
n=1234
%Adverse Reaction Severe Total Severe Total Hemorrhage <1 7 <1 6 Anemia <1 3 <1 3 Ecchymosis 0 3 0 3 Table 9: Adverse Reactions Occurring at ≥2% Incidence in Lovenox-Treated Patients Undergoing Hip or Knee Replacement Surgery Dosing Regimen Lovenox
40 mg daily subcutaneouslyLovenox
30 mg q12h subcutaneouslyHeparin
15,000 U/24h subcutaneouslyPlacebo
q12h subcutaneouslyPeri-operative Period Extended Prophylaxis Period n=288*
%n=131†
%n=1080
%n=766
%n=115
%Adverse Reaction Severe Total Severe Total Severe Total Severe Total Severe Total - * Data represent Lovenox 40 mg subcutaneously once a day initiated up to 12 hours prior to surgery in 288 hip replacement surgery patients who received Lovenox peri-operatively in an unblinded fashion in one clinical trial.
- † Data represent Lovenox 40 mg subcutaneously once a day given in a blinded fashion as extended prophylaxis at the end of the peri-operative period in 131 of the original 288 hip replacement surgery patients for up to 21 days in one clinical trial.
Fever 0 8 0 0 <1 5 <1 4 0 3 Hemorrhage <1 13 0 5 <1 4 1 4 0 3 Nausea <1 3 <1 2 0 2 Anemia 0 16 0 <2 <1 2 2 5 <1 7 Edema <1 2 <1 2 0 2 Peripheral edema 0 6 0 0 <1 3 <1 4 0 3 Table 10: Adverse Reactions Occurring at ≥2% Incidence in Lovenox-Treated Medical Patients with Severely Restricted Mobility During Acute Illness Dosing Regimen Adverse Reaction Lovenox
40 mg daily subcutaneously
n=360
%Placebo
daily subcutaneously
n=362
%Dyspnea 3.3 5.2 Thrombocytopenia 2.8 2.8 Confusion 2.2 1.1 Diarrhea 2.2 1.7 Nausea 2.5 1.7 Table 11: Adverse Reactions Occurring at ≥2% Incidence in Lovenox-Treated Patients Undergoing Treatment of Deep Vein Thrombosis with or without Pulmonary Embolism Dosing Regimen Lovenox
1.5 mg/kg daily subcutaneously
n=298
%Lovenox
1 mg/kg q12h subcutaneously
n=559
%Heparin
aPTT Adjusted Intravenous Therapy
n=544
%Adverse Reaction Severe Total Severe Total Severe Total Injection Site Hemorrhage 0 5 0 3 <1 <1 Injection Site Pain 0 2 0 2 0 0 Hematuria 0 2 0 <1 <1 2 Adverse Events in Lovenox-Treated Patients with Unstable Angina or Non–Q-Wave Myocardial Infarction
Non-hemorrhagic clinical events reported to be related to Lovenox therapy occurred at an incidence of ≤1%.
Non-major hemorrhagic events, primarily injection site ecchymosis and hematomas, were more frequently reported in patients treated with subcutaneous Lovenox than in patients treated with intravenous heparin.
Serious adverse events with Lovenox or heparin in a clinical trial in patients with unstable angina or non–Q-wave myocardial infarction that occurred at a rate of at least 0.5% in the Lovenox group are provided below (see Table 12).
Table 12: Serious Adverse Events Occurring at ≥0.5% Incidence in Lovenox-Treated Patients with Unstable Angina or Non–Q-Wave Myocardial Infarction Dosing Regimen Lovenox
1 mg/kg q12h subcutaneouslyHeparin
aPTT Adjusted Intravenous TherapyAdverse Event n=1578
n (%)n=1529
n (%)Atrial fibrillation 11 (0.70) 3 (0.20) Heart failure 15 (0.95) 11 (0.72) Lung edema 11 (0.70) 11 (0.72) Pneumonia 13 (0.82) 9 (0.59) 6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of Lovenox. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
There have been reports of epidural or spinal hematoma formation with concurrent use of Lovenox and spinal/epidural anesthesia or spinal puncture. The majority of patients had a postoperative indwelling epidural catheter placed for analgesia or received additional drugs affecting hemostasis such as NSAIDs. Many of the epidural or spinal hematomas caused neurologic injury, including long-term or permanent paralysis.
Local reactions at the injection site (e.g. nodules, inflammation, oozing), systemic allergic reactions (e.g. pruritus, urticaria, anaphylactic/anaphylactoid reactions including shock), vesiculobullous rash, cases of hypersensitivity cutaneous vasculitis, purpura, skin necrosis (occurring at either the injection site or distant from the injection site), thrombocytosis, and thrombocytopenia with thrombosis [see Warnings and Precautions (5.5)] have been reported.
Cases of hyperkalemia have been reported. Most of these reports occurred in patients who also had conditions that tend toward the development of hyperkalemia (e.g., renal dysfunction, concomitant potassium-sparing drugs, administration of potassium, hematoma in body tissues). Very rare cases of hyperlipidemia have also been reported, with one case of hyperlipidemia, with marked hypertriglyceridemia, reported in a diabetic pregnant woman; causality has not been determined.
Cases of headache, hemorrhagic anemia, eosinophilia, alopecia, hepatocellular and cholestatic liver injury have been reported.
Osteoporosis has also been reported following long-term therapy.
-
7 DRUG INTERACTIONS
Whenever possible, agents which may enhance the risk of hemorrhage should be discontinued prior to initiation of Lovenox therapy. These agents include medications such as: anticoagulants, platelet inhibitors including acetylsalicylic acid, salicylates, NSAIDs (including ketorolac tromethamine), dipyridamole, or sulfinpyrazone. If coadministration is essential, conduct close clinical and laboratory monitoring [see Warnings and Precautions (5.1)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Placental transfer of enoxaparin was observed in the animal studies. Human data from a retrospective cohort study, which included 693 live births, suggest that enoxaparin does not increase the risk of major developmental abnormalities [see Data]. Based on animal data, Lovenox is not predicted to increase the risk of major developmental abnormalities (see Data).
Adverse outcomes in pregnancy occur regardless of the health of the mother or the use of medications. The estimated background risk of major birth defects and miscarriage for the indicated populations is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Clinical Considerations
Pregnancy alone confers an increased risk for thromboembolism that is even higher for women with thromboembolic disease and certain high risk pregnancy conditions. While not adequately studied, pregnant women with mechanical prosthetic heart valves may be at even higher risk for thrombosis [see Warnings and Precautions (5.7) and Use in Specific Populations (8.6)]. Pregnant women with thromboembolic disease, including those with mechanical prosthetic heart valves and those with inherited or acquired thrombophilias, have an increased risk of other maternal complications and fetal loss regardless of the type of anticoagulant used.
All patients receiving anticoagulants, including pregnant women, are at risk for bleeding. Pregnant women receiving Lovenox should be carefully monitored for evidence of bleeding or excessive anticoagulation. Consideration for use of a shorter acting anticoagulant should be specifically addressed as delivery approaches [see Boxed Warning]. Hemorrhage can occur at any site and may lead to death of mother and/or fetus. Pregnant women should be apprised of the potential hazard to the fetus and the mother if Lovenox is administered during pregnancy.
It is not known if monitoring of anti-Factor Xa activity and dose adjustment (by weight or anti-Factor Xa activity) of Lovenox affect the safety and the efficacy of the drug during pregnancy.
Cases of "gasping syndrome" have occurred in premature infants when large amounts of benzyl alcohol have been administered (99–405 mg/kg/day). The multiple-dose vial of Lovenox contains 15 mg benzyl alcohol per 1 mL as a preservative [see Warnings and Precautions (5.8)].
Data
Human data
There are no adequate and well-controlled studies in pregnant women. A retrospective study reviewed the records of 604 women who used Lovenox during pregnancy. A total of 624 pregnancies resulted in 693 live births. There were 72 hemorrhagic events (11 serious) in 63 women. There were 14 cases of neonatal hemorrhage. Major congenital anomalies in live births occurred at rates (2.5%) similar to background rates.
There have been postmarketing reports of fetal death when pregnant women received Lovenox. Causality for these cases has not been determined. Insufficient data, the underlying disease, and the possibility of inadequate anticoagulation complicate the evaluation of these cases.
A clinical study using Lovenox in pregnant women with mechanical prosthetic heart valves has been conducted [see Warnings and Precautions (5.7)].
Animal data
Teratology studies have been conducted in pregnant rats and rabbits at subcutaneous doses of enoxaparin up to 15 times the recommended human dose (by comparison with 2 mg/kg as the maximum recommended daily dose). There was no evidence of teratogenic effects or fetotoxicity due to enoxaparin. Because animal reproduction studies are not always predictive of human response, this drug should be used during pregnancy only if clearly needed.
8.2 Lactation
Risk Summary
It is unknown whether Lovenox is excreted in human milk. In lactating rats, the passage of enoxaparin or its metabolites in the milk is very limited. There is no information available on the effect of enoxaparin or its metabolites on the breastfed child, or on the milk production. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for Lovenox and any potential adverse effects on the breastfed child from Lovenox or from the underlying maternal condition.
8.4 Pediatric Use
Safety and effectiveness of Lovenox in pediatric patients have not been established.
Lovenox is not approved for use in neonates or infants.
Serious adverse reactions including fatal reactions and the "gasping syndrome" occurred in premature neonates and low birth weight infants in the neonatal intensive care unit who received drugs containing benzyl alcohol as a preservative. In these cases, benzyl alcohol dosages of 99 to 234 mg/kg/day produced high levels of benzyl alcohol and its metabolites in the blood and urine (blood levels of benzyl alcohol were 0.61 to 1.378 mmol/L). Additional adverse reactions included gradual neurological deterioration, seizures, intracranial hemorrhage, hematologic abnormalities, skin breakdown, hepatic and renal failure, hypotension, bradycardia, and cardiovascular collapse. Preterm, low-birth weight infants may be more likely to develop these reactions because they may be less able to metabolize benzyl alcohol. The minimum amount of benzyl alcohol at which serious adverse reactions may occur is not known.
Lovenox multiple-dose vials contain 15 mg/mL of benzyl alcohol (at the dose of 1.5 mg/kg twice a day, benzyl alcohol exposure in patients is 0.45 mg/kg daily) [see Warnings and Precautions (5.8)].
8.5 Geriatric Use
Prevention of Deep Vein Thrombosis in Hip, Knee and Abdominal Surgery; Treatment of Deep Vein Thrombosis, Prevention of Ischemic Complications of Unstable Angina and Non–Q-Wave Myocardial Infarction
Over 2800 patients, 65 years and older, have received Lovenox in clinical trials. The efficacy of Lovenox in the geriatric (≥65 years) was similar to that seen in younger patients (<65 years). The incidence of bleeding complications was similar between geriatric and younger patients when 30 mg every 12 hours or 40 mg once a day doses of Lovenox were employed. The incidence of bleeding complications was higher in geriatric patients as compared to younger patients when Lovenox was administered at doses of 1.5 mg/kg once a day or 1 mg/kg every 12 hours. The risk of Lovenox-associated bleeding increased with age. Serious adverse events increased with age for patients receiving Lovenox. Other clinical experience (including postmarketing surveillance and literature reports) has not revealed additional differences in the safety of Lovenox between geriatric and younger patients. Careful attention to dosing intervals and concomitant medications (especially antiplatelet medications) is advised. Lovenox should be used with care in geriatric patients who may show delayed elimination of enoxaparin. Monitoring of geriatric patients with low body weight (<45 kg) and those predisposed to decreased renal function should be considered [see Warnings and Precautions (2.6) and Clinical Pharmacology (12.3)].
Treatment of Acute ST-Segment Elevation Myocardial Infarction
In the clinical study for treatment of acute ST-segment elevation myocardial infarction, there was no evidence of difference in efficacy between patients ≥75 years of age (n=1241) and patients less than 75 years of age (n=9015). Patients ≥75 years of age did not receive a 30 mg intravenous bolus prior to the normal dosage regimen and had their subcutaneous dose adjusted to 0.75 mg/kg every 12 hours [see Dosage and Administration (2.4)]. The incidence of bleeding complications was higher in patients ≥65 years of age as compared to younger patients (<65 years).
8.6 Patients with Mechanical Prosthetic Heart Valves
The use of Lovenox has not been adequately studied for thromboprophylaxis in patients with mechanical prosthetic heart valves and has not been adequately studied for long-term use in this patient population. Isolated cases of prosthetic heart valve thrombosis have been reported in patients with mechanical prosthetic heart valves who have received Lovenox for thromboprophylaxis. Some of these cases were pregnant women in whom thrombosis led to maternal and fetal deaths. Insufficient data, the underlying disease and the possibility of inadequate anticoagulation complicate the evaluation of these cases. Pregnant women with mechanical prosthetic heart valves may be at higher risk for thromboembolism [see Warnings and Precautions (5.7)].
8.7 Renal Impairment
In patients with renal impairment, there is an increase in exposure of enoxaparin sodium. All such patients should be observed carefully for signs and symptoms of bleeding. Because exposure of enoxaparin sodium is significantly increased in patients with severe renal impairment (creatinine clearance <30 mL/min), a dosage adjustment is recommended for therapeutic and prophylactic dosage ranges. No dosage adjustment is recommended in patients with creatinine clearance 30 to <50 mL/min and creatinine clearance 50 to 80 mL/min [see Dosage and Administration (2.3) and Clinical Pharmacology (12.3)]. In patients with renal failure, treatment with Lovenox has been associated with the development of hyperkalemia [see Adverse Reactions (6.2)].
8.8 Low-Weight Patients
An increase in exposure of enoxaparin sodium with prophylactic dosages (non-weight adjusted) has been observed in low-weight women (<45 kg) and low-weight men (<57 kg). Observe low-weight patients frequently for signs and symptoms of bleeding [see Clinical Pharmacology (12.3)].
8.9 Obese Patients
Obese patients are at higher risk for thromboembolism. The safety and efficacy of prophylactic doses of Lovenox in obese patients (BMI >30 kg/m2) has not been fully determined and there is no consensus for dose adjustment. Observe these patients carefully for signs and symptoms of thromboembolism.
-
10 OVERDOSAGE
Accidental overdosage following administration of Lovenox may lead to hemorrhagic complications. Injected Lovenox may be largely neutralized by the slow intravenous injection of protamine sulfate (1% solution). The dose of protamine sulfate should be equal to the dose of Lovenox injected: 1 mg protamine sulfate should be administered to neutralize 1 mg Lovenox, if Lovenox was administered in the previous 8 hours. An infusion of 0.5 mg protamine per 1 mg of Lovenox may be administered if Lovenox was administered greater than 8 hours previous to the protamine administration, or if it has been determined that a second dose of protamine is required. The second infusion of 0.5 mg protamine sulfate per 1 mg of Lovenox may be administered if the aPTT measured 2 to 4 hours after the first infusion remains prolonged.
If at least 12 hours have elapsed since the last Lovenox injection, protamine administration may not be required; however, even with higher doses of protamine, the aPTT may remain more prolonged than following administration of heparin. In all cases, the anti-Factor Xa activity is never completely neutralized (maximum about 60%). Particular care should be taken to avoid overdosage with protamine sulfate. Administration of protamine sulfate can cause severe hypotensive and anaphylactoid reactions. Because fatal reactions, often resembling anaphylaxis, have been reported with protamine sulfate, it should be given only when resuscitation techniques and treatment of anaphylactic shock are readily available. For additional information consult the labeling of protamine sulfate injection products.
-
11 DESCRIPTION
Lovenox is a sterile aqueous solution containing enoxaparin sodium, a low molecular weight heparin. The pH of the injection is 5.5 to 7.5.
Enoxaparin sodium is obtained by alkaline depolymerization of heparin benzyl ester derived from porcine intestinal mucosa. Its structure is characterized by a 2-O-sulfo-4-enepyranosuronic acid group at the non-reducing end and a 2-N,6-O-disulfo-D-glucosamine at the reducing end of the chain. About 20% (ranging between 15% and 25%) of the enoxaparin structure contains a 1,6 anhydro derivative on the reducing end of the polysaccharide chain. The drug substance is the sodium salt. The average molecular weight is about 4500 daltons. The molecular weight distribution is:
<2000 daltons ≤20% 2000 to 8000 daltons ≥68% >8000 daltons ≤18% STRUCTURAL FORMULA
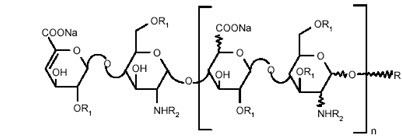
R1 = H or SO3Na and R2 = SO3Na or COCH3
- * X = Percent of polysaccharide chain containing 1,6 anhydro derivative on the reducing end
R X*=15 to 25% 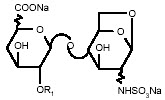
n=0 to 20 100-X H n=1 to 21 Lovenox 100 mg/mL Concentration contains 10 mg enoxaparin sodium (approximate anti-Factor Xa activity of 1000 IU [with reference to the W.H.O. First International Low Molecular Weight Heparin Reference Standard]) per 0.1 mL Water for Injection.
Lovenox 150 mg/mL Concentration contains 15 mg enoxaparin sodium (approximate anti-Factor Xa activity of 1500 IU [with reference to the W.H.O. First International Low Molecular Weight Heparin Reference Standard]) per 0.1 mL Water for Injection.
The Lovenox prefilled syringes and graduated prefilled syringes are preservative-free and intended for use only as a single-dose injection. The multiple-dose vial contains 15 mg benzyl alcohol per 1 mL as a preservative [see Dosage and Administration (2) and How Supplied/Storage and Handling (16)].
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Enoxaparin is a low molecular weight heparin which has antithrombotic properties.
12.2 Pharmacodynamics
In humans, enoxaparin given at a dose of 1.5 mg/kg subcutaneously is characterized by a higher ratio of anti-Factor Xa to anti-Factor IIa activity (mean ±SD, 14.0±3.1) (based on areas under anti-Factor activity versus time curves) compared to the ratios observed for heparin (mean ±SD, 1.22±0.13). Increases of up to 1.8 times the control values were seen in the thrombin time (TT) and the activated partial thromboplastin time (aPTT). Enoxaparin at a 1 mg/kg dose (100 mg/mL concentration), administered subcutaneously every 12 hours to patients in a large clinical trial resulted in aPTT values of 45 seconds or less in the majority of patients (n=1607). A 30 mg intravenous bolus immediately followed by a 1 mg/kg subcutaneous administration resulted in aPTT postinjection values of 50 seconds. The average aPTT prolongation value on Day 1 was about 16% higher than on Day 4.
12.3 Pharmacokinetics
Absorption
Pharmacokinetic trials were conducted using the 100 mg/mL formulation. Maximum anti-Factor Xa and anti-thrombin (anti-Factor IIa) activities occur 3 to 5 hours after subcutaneous injection of enoxaparin. Mean peak anti-Factor Xa activity was 0.16 IU/mL (1.58 mcg/mL) and 0.38 IU/mL (3.83 mcg/mL) after the 20 mg and the 40 mg clinically tested subcutaneous doses, respectively. Mean (n=46) peak anti-Factor Xa activity was 1.1 IU/mL at steady state in patients with unstable angina receiving 1 mg/kg subcutaneously every 12 hours for 14 days. Mean absolute bioavailability of enoxaparin, after 1.5 mg/kg given subcutaneously, based on anti-Factor Xa activity is approximately 100% in healthy subjects.
A 30 mg intravenous bolus immediately followed by 1 mg/kg subcutaneously every 12 hours provided initial peak anti-Factor Xa levels of 1.16 IU/mL (n=16) and average exposure corresponding to 84% of steady-state levels. Steady state is achieved on the second day of treatment.
Enoxaparin pharmacokinetics appears to be linear over the recommended dosage ranges [see Dosage and Administration (2)]. After repeated subcutaneous administration of 40 mg once daily and 1.5 mg/kg once-daily regimens in healthy volunteers, the steady state is reached on day 2 with an average exposure ratio about 15% higher than after a single dose. Steady-state enoxaparin activity levels are well predicted by single-dose pharmacokinetics. After repeated subcutaneous administration of the 1 mg/kg twice-daily regimen, the steady state is reached from day 4 with mean exposure about 65% higher than after a single dose and mean peak and trough levels of about 1.2 and 0.52 IU/mL, respectively. Based on enoxaparin sodium pharmacokinetics, this difference in steady state is expected and within the therapeutic range.
Although not studied clinically, the 150 mg/mL concentration of enoxaparin sodium is projected to result in anticoagulant activities similar to those of 100 mg/mL and 200 mg/mL concentrations at the same enoxaparin dose. When a daily 1.5 mg/kg subcutaneous injection of enoxaparin sodium was given to 25 healthy male and female subjects using a 100 mg/mL or a 200 mg/mL concentration the following pharmacokinetic profiles were obtained (see Table 13).
Table 13: Pharmacokinetic Parameters* After 5 Days of 1.5 mg/kg Subcutaneous Once-Daily Doses of Enoxaparin Sodium Using 100 mg/mL or 200 mg/mL Concentrations Concentration Anti-Xa Anti-IIa Heptest aPTT - * Means ±SD at Day 5 and 90% Confidence Interval (CI) of the ratio
- † Median (range)
Amax
(IU/mL or Δ sec)100 mg/mL 1.37 (±0.23) 0.23 (±0.05) 105 (±17) 19 (±5) 200 mg/mL 1.45 (±0.22) 0.26 (±0.05) 111 (±17) 22 (±7) 90% CI 102%–110% 102%–111% tmax† (h) 100 mg/mL 3 (2–6) 4 (2–5) 2.5 (2–4.5) 3 (2–4.5) 200 mg/mL 3.5 (2–6) 4.5 (2.5–6) 3.3 (2–5) 3 (2–5) AUC (ss)
(h*IU/mL or h* Δ sec)100 mg/mL 14.26 (±2.93) 1.54 (±0.61) 1321 (±219) 200 mg/mL 15.43 (±2.96) 1.77 (±0.67) 1401 (±227) 90% CI 105%–112% 103%–109% Elimination
Following intravenous dosing, the total body clearance of enoxaparin is 26 mL/min. After intravenous dosing of enoxaparin labeled with the gamma-emitter, 99mTc, 40% of radioactivity and 8 to 20% of anti-Factor Xa activity were recovered in urine in 24 hours. Elimination half-life based on anti-Factor Xa activity was 4.5 hours after a single subcutaneous dose to about 7 hours after repeated dosing. Significant anti-Factor Xa activity persists in plasma for about 12 hours following a 40 mg subcutaneous once a day dose.
Following subcutaneous dosing, the apparent clearance (CL/F) of enoxaparin is approximately 15 mL/min.
Metabolism
Enoxaparin sodium is primarily metabolized in the liver by desulfation and/or depolymerization to lower molecular weight species with much reduced biological potency. Renal clearance of active fragments represents about 10% of the administered dose and total renal excretion of active and non-active fragments 40% of the dose.
Special Populations
Gender
Apparent clearance and Amax derived from anti-Factor Xa values following single subcutaneous dosing (40 mg and 60 mg) were slightly higher in males than in females. The source of the gender difference in these parameters has not been conclusively identified; however, body weight may be a contributing factor.
Geriatric
Apparent clearance and Amax derived from anti-Factor Xa values following single and multiple subcutaneous dosing in geriatric subjects were close to those observed in young subjects. Following once a day subcutaneous dosing of 40 mg enoxaparin, the Day 10 mean area under anti-Factor Xa activity versus time curve (AUC) was approximately 15% greater than the mean Day 1 AUC value [see Dosage and Administration (2.4) and Use in Specific Populations (8.5)].
Renal impairment
A linear relationship between anti-Factor Xa plasma clearance and creatinine clearance at steady state has been observed, which indicates decreased clearance of enoxaparin sodium in patients with reduced renal function. Anti-Factor Xa exposure represented by AUC, at steady state, is marginally increased in patients with creatinine clearance 50 to 80 mL/min and patients with creatinine clearance 30 to <50 mL/min renal impairment after repeated subcutaneous 40 mg once-daily doses. In patients with severe renal impairment (creatinine clearance <30 mL/min), the AUC at steady state is significantly increased on average by 65% after repeated subcutaneous 40 mg once-daily doses [see Dosage and Administration (2.3) and Use in Specific Populations (8.7)].
Hemodialysis
In a single study, elimination rate appeared similar but AUC was two-fold higher than control population, after a single 0.25 or 0.5 mg/kg intravenous dose.
Hepatic impairment
Studies with Lovenox in patients with hepatic impairment have not been conducted and the impact of hepatic impairment on the exposure to enoxaparin is unknown.
Weight
After repeated subcutaneous 1.5 mg/kg once-daily dosing, mean AUC of anti-Factor Xa activity is marginally higher at steady state in obese healthy volunteers (BMI 30–48 kg/m2) compared to non-obese control subjects, while Amax is not increased.
When non–weight-adjusted dosing was administered, it was found after a single-subcutaneous 40 mg dose, that anti-Factor Xa exposure is 52% higher in low-weight women (<45 kg) and 27% higher in low-weight men (<57 kg) when compared to normal weight control subjects [see Use in Specific Populations (8.8)].
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No long-term studies in animals have been performed to evaluate the carcinogenic potential of enoxaparin. Enoxaparin was not mutagenic in in vitro tests, including the Ames test, mouse lymphoma cell forward mutation test, and human lymphocyte chromosomal aberration test, and the in vivo rat bone marrow chromosomal aberration test. Enoxaparin was found to have no effect on fertility or reproductive performance of male and female rats at subcutaneous doses up to 20 mg/kg/day or 141 mg/m2/day. The maximum human dose in clinical trials was 2.0 mg/kg/day or 78 mg/m2/day (for an average body weight of 70 kg, height of 170 cm, and body surface area of 1.8 m2).
13.2 Animal Toxicology and/or Pharmacology
A single subcutaneous dose of 46.4 mg/kg enoxaparin was lethal to rats. The symptoms of acute toxicity were ataxia, decreased motility, dyspnea, cyanosis, and coma.
13.3 Reproductive and Developmental Toxicology
Teratology studies have been conducted in pregnant rats and rabbits at subcutaneous doses of enoxaparin up to 30 mg/kg/day corresponding to 211 mg/m2/day and 410 mg/m2/day in rats and rabbits respectively. There was no evidence of teratogenic effects or fetotoxicity due to enoxaparin.
-
14 CLINICAL STUDIES
14.1 Prophylaxis of Deep Vein Thrombosis following Abdominal Surgery in Patients at Risk for Thromboembolic Complications
Abdominal surgery patients at risk include those who are over 40 years of age, obese, undergoing surgery under general anesthesia lasting longer than 30 minutes or who have additional risk factors such as malignancy or a history of deep vein thrombosis (DVT) or pulmonary embolism (PE).
In a double-blind, parallel group study of patients undergoing elective cancer surgery of the gastrointestinal, urological, or gynecological tract, a total of 1116 patients were enrolled in the study, and 1115 patients were treated. Patients ranged in age from 32 to 97 years (mean age 67 years) with 52.7% men and 47.3% women. Patients were 98% Caucasian, 1.1% Black, 0.4% Asian and 0.4% others. Lovenox 40 mg subcutaneously, administered once a day, beginning 2 hours prior to surgery and continuing for a maximum of 12 days after surgery, was comparable to heparin 5000 U every 8 hours subcutaneously in reducing the risk of DVT. The efficacy data are provided below (see Table 14).
Table 14: Efficacy of Lovenox in the Prophylaxis of Deep Vein Thrombosis Following Abdominal Surgery Dosing Regimen Indication Lovenox
40 mg daily subcutaneously
n (%)Heparin
5000 U q8h subcutaneously
n (%)- * VTE = Venous thromboembolic events which included DVT, PE, and death considered to be thromboembolic in origin
- † CI = Confidence Interval
All Treated Abdominal Surgery Patients 555 (100) 560 (100) Treatment Failures Total VTE* (%) 56 (10.1)
(95% CI†: 8 to 13)63 (11.3)
(95% CI: 9 to 14)DVT Only (%) 54 (9.7)
(95% CI: 7 to 12)61 (10.9)
(95% CI: 8 to 13)In a second double-blind, parallel group study, Lovenox 40 mg subcutaneously once a day was compared to heparin 5000 U every 8 hours subcutaneously in patients undergoing colorectal surgery (one-third with cancer). A total of 1347 patients were randomized in the study and all patients were treated. Patients ranged in age from 18 to 92 years (mean age 50.1 years) with 54.2% men and 45.8% women. Treatment was initiated approximately 2 hours prior to surgery and continued for approximately 7 to 10 days after surgery. The efficacy data are provided below (see Table 15).
Table 15: Efficacy of Lovenox in the Prophylaxis of Deep Vein Thrombosis Following Colorectal Surgery Dosing Regimen Indication Lovenox
40 mg daily subcutaneously
n (%)Heparin
5000 U q8h subcutaneously
n (%)- * VTE = Venous thromboembolic events which included DVT, PE, and death considered to be thromboembolic in origin
- † CI = Confidence Interval
All Treated Colorectal Surgery Patients 673 (100) 674 (100) Treatment Failures Total VTE* (%) 48 (7.1)
(95% CI†: 5 to 9)45 (6.7)
(95% CI: 5 to 9)DVT Only (%) 47 (7.0)
(95% CI: 5 to 9)44 (6.5)
(95% CI: 5 to 8)14.2 Prophylaxis of Deep Vein Thrombosis following Hip or Knee Replacement Surgery
Lovenox has been shown to reduce the risk of postoperative deep vein thrombosis (DVT) following hip or knee replacement surgery.
In a double-blind study, Lovenox 30 mg every 12 hours subcutaneously was compared to placebo in patients with hip replacement. A total of 100 patients were randomized in the study and all patients were treated. Patients ranged in age from 41 to 84 years (mean age 67.1 years) with 45% men and 55% women. After hemostasis was established, treatment was initiated 12 to 24 hours after surgery and was continued for 10 to 14 days after surgery. The efficacy data are provided below (see Table 16).
Table 16: Efficacy of Lovenox in the Prophylaxis of Deep Vein Thrombosis Following Hip Replacement Surgery Dosing Regimen Indication Lovenox
30 mg q12h subcutaneously
n (%)Placebo
q12h subcutaneously
n (%)- * p value versus placebo = 0.0002
- † p value versus placebo = 0.0134
All Treated Hip Replacement Patients 50 (100) 50 (100) Treatment Failures Total DVT (%) 5 (10)* 23 (46) Proximal DVT (%) 1 (2)† 11 (22) A double-blind, multicenter study compared three dosing regimens of Lovenox in patients with hip replacement. A total of 572 patients were randomized in the study and 568 patients were treated. Patients ranged in age from 31 to 88 years (mean age 64.7 years) with 63% men and 37% women. Patients were 93% Caucasian, 6% Black, <1% Asian, and 1% others. Treatment was initiated within two days after surgery and was continued for 7 to 11 days after surgery. The efficacy data are provided below (see Table 17).
Table 17: Efficacy of Lovenox in the Prophylaxis of Deep Vein Thrombosis Following Hip Replacement Surgery Dosing Regimen Indication 10 mg daily subcutaneously
n (%)30 mg q12h subcutaneously
n (%)40 mg daily subcutaneously
n (%)- * p value versus Lovenox 10 mg once a day = 0.0008
- † p value versus Lovenox 10 mg once a day = 0.0168
All Treated Hip Replacement Patients 161 (100) 208 (100) 199 (100) Treatment Failures Total DVT (%) 40 (25) 22 (11)* 27 (14) Proximal DVT (%) 17 (11) 8 (4)† 9 (5) There was no significant difference between the 30 mg every 12 hours and 40 mg once a day regimens. In a double-blind study, Lovenox 30 mg every 12 hours subcutaneously was compared to placebo in patients undergoing knee replacement surgery. A total of 132 patients were randomized in the study and 131 patients were treated, of which 99 had total knee replacement and 32 had either unicompartmental knee replacement or tibial osteotomy. The 99 patients with total knee replacement ranged in age from 42 to 85 years (mean age 70.2 years) with 36.4% men and 63.6% women. After hemostasis was established, treatment was initiated 12 to 24 hours after surgery and was continued up to 15 days after surgery. The incidence of proximal and total DVT after surgery was significantly lower for Lovenox compared to placebo. The efficacy data are provided below (see Table 18).
Table 18: Efficacy of Lovenox in the Prophylaxis of Deep Vein Thrombosis Following Total Knee Replacement Surgery Dosing Regimen Indication Lovenox
30 mg q12h subcutaneously
n (%)Placebo
q12h subcutaneously
n (%)- * p value versus placebo = 0.0001
- † CI = Confidence Interval
- ‡ p value versus placebo = 0.013
- § CL = Confidence Limit
All Treated Total Knee Replacement Patients 47 (100) 52 (100) Treatment Failures Total DVT (%) 5 (11)*
(95% CI†: 1 to 21)32 (62)
(95% CI: 47 to 76)Proximal DVT (%) 0 (0)‡
(95% Upper CL§: 5)7 (13)
(95% CI: 3 to 24)Additionally, in an open-label, parallel group, randomized clinical study, Lovenox 30 mg every 12 hours subcutaneously in patients undergoing elective knee replacement surgery was compared to heparin 5000 U every 8 hours subcutaneously. A total of 453 patients were randomized in the study and all were treated. Patients ranged in age from 38 to 90 years (mean age 68.5 years) with 43.7% men and 56.3% women. Patients were 92.5% Caucasian, 5.3% Black, and 0.6% others. Treatment was initiated after surgery and continued up to 14 days. The incidence of deep vein thrombosis was lower for Lovenox compared to heparin.
Extended Prophylaxis of Deep Vein Thrombosis Following Hip Replacement Surgery: In a study of extended prophylaxis for patients undergoing hip replacement surgery, patients were treated, while hospitalized, with Lovenox 40 mg subcutaneously, initiated up to 12 hours prior to surgery for the prophylaxis of postoperative DVT. At the end of the peri-operative period, all patients underwent bilateral venography. In a double-blind design, those patients with no venous thromboembolic disease were randomized to a post-discharge regimen of either Lovenox 40 mg (n=90) once a day subcutaneously or to placebo (n=89) for 3 weeks. A total of 179 patients were randomized in the double-blind phase of the study and all patients were treated. Patients ranged in age from 47 to 87 years (mean age 69.4 years) with 57% men and 43% women. In this population of patients, the incidence of DVT during extended prophylaxis was significantly lower for Lovenox compared to placebo. The efficacy data are provided below (see Table 19).
Table 19: Efficacy of Lovenox in the Extended Prophylaxis of Deep Vein Thrombosis Following Hip Replacement Surgery Post-discharge Dosing Regimen Indication (Post Discharge) Lovenox
40 mg daily subcutaneously
n (%)Placebo
daily subcutaneously
n (%)- * p value versus placebo = 0.008
- † CI= Confidence Interval
- ‡ p value versus placebo = 0.537
All Treated Extended Prophylaxis Patients 90 (100) 89 (100) Treatment Failures Total DVT (%) 6 (7)*
(95% CI†: 3 to 14)18 (20)
(95% CI: 12 to 30)Proximal DVT (%) 5 (6)‡
(95% CI: 2 to 13)7 (8)
(95% CI: 3 to 16)In a second study, patients undergoing hip replacement surgery were treated, while hospitalized, with Lovenox 40 mg subcutaneously, initiated up to 12 hours prior to surgery. All patients were examined for clinical signs and symptoms of venous thromboembolic (VTE) disease. In a double-blind design, patients without clinical signs and symptoms of VTE disease were randomized to a post-discharge regimen of either Lovenox 40 mg (n=131) once a day subcutaneously or to placebo (n=131) for 3 weeks. A total of 262 patients were randomized in the study double-blind phase and all patients were treated. Patients ranged in age from 44 to 87 years (mean age 68.5 years) with 43.1% men and 56.9% women. Similar to the first study the incidence of DVT during extended prophylaxis was significantly lower for Lovenox compared to placebo, with a statistically significant difference in both total DVT (Lovenox 21 [16%] versus placebo 45 [34%]; p=0.001) and proximal DVT (Lovenox 8 [6%] versus placebo 28 [21%]; p=<0.001).
14.3 Prophylaxis of Deep Vein Thrombosis in Medical Patients with Severely Restricted Mobility during Acute Illness
In a double blind multicenter, parallel group study, Lovenox 20 mg or 40 mg once a day subcutaneously was compared to placebo in the prophylaxis of deep vein thrombosis (DVT) in medical patients with severely restricted mobility during acute illness (defined as walking distance of <10 meters for ≤3 days). This study included patients with heart failure (NYHA Class III or IV); acute respiratory failure or complicated chronic respiratory insufficiency (not requiring ventilatory support): acute infection (excluding septic shock); or acute rheumatic disorder (acute lumbar or sciatic pain, vertebral compression [due to osteoporosis or tumor], acute arthritic episodes of the lower extremities). A total of 1102 patients were enrolled in the study, and 1073 patients were treated. Patients ranged in age from 40 to 97 years (mean age 73 years) with equal proportions of men and women. Treatment continued for a maximum of 14 days (median duration 7 days). When given at a dose of 40 mg once a day subcutaneously, Lovenox significantly reduced the incidence of DVT as compared to placebo. The efficacy data are provided below (see Table 20).
Table 20: Efficacy of Lovenox in the Prophylaxis of Deep Vein Thrombosis in Medical Patients with Severely Restricted Mobility during Acute Illness Dosing Regimen Lovenox
20 mg daily subcutaneouslyLovenox
40 mg daily subcutaneouslyPlacebo Indication n (%) n (%) n (%) - * Treatment failures during therapy, between Days 1 and 14
- † VTE = Venous thromboembolic events which included DVT, PE, and death considered to be thromboembolic in origin
- ‡ CI = Confidence Interval
All Treated Medical Patients During Acute Illness 351 (100) 360 (100) 362 (100) Treatment Failure* Total VTE† (%) 43 (12.3) 16 (4.4) 43 (11.9) Total DVT (%) 43 (12.3)
(95% CI‡ 8.8 to 15.7)16 (4.4)
(95% CI‡ 2.3 to 6.6)41 (11.3)
(95% CI‡ 8.1 to 14.6)Proximal DVT (%) 13 (3.7) 5 (1.4) 14 (3.9) At approximately 3 months following enrollment, the incidence of venous thromboembolism remained lower in the Lovenox 40 mg treatment group versus the placebo treatment group.
14.4 Treatment of Deep Vein Thrombosis with or without Pulmonary Embolism
In a multicenter, parallel group study, 900 patients with acute lower extremity deep vein thrombosis (DVT) with or without pulmonary embolism (PE) were randomized to an inpatient (hospital) treatment of either (i) Lovenox 1.5 mg/kg once a day subcutaneously, (ii) Lovenox 1 mg/kg every 12 hours subcutaneously, or (iii) heparin intravenous bolus (5000 IU) followed by a continuous infusion (administered to achieve an aPTT of 55 to 85 seconds). A total of 900 patients were randomized in the study and all patients were treated. Patients ranged in age from 18 to 92 years (mean age 60.7 years) with 54.7% men and 45.3% women. All patients also received warfarin sodium (dose adjusted according to PT to achieve an International Normalization Ratio [INR] of 2.0 to 3.0), commencing within 72 hours of initiation of Lovenox or standard heparin therapy, and continuing for 90 days. Lovenox or standard heparin therapy was administered for a minimum of 5 days and until the targeted warfarin sodium INR was achieved. Both Lovenox regimens were equivalent to standard heparin therapy in reducing the risk of recurrent venous thromboembolism (DVT and/or PE). The efficacy data are provided below (see Table 21).
Table 21: Efficacy of Lovenox in Treatment of Deep Vein Thrombosis with or without Pulmonary Embolism Dosing Regimen* Lovenox
1.5 mg/kg daily subcutaneouslyLovenox
1 mg/kg q12h subcutaneouslyHeparin
aPTT Adjusted Intravenous TherapyIndication n (%) n (%) n (%) - * All patients were also treated with warfarin sodium commencing within 72 hours of Lovenox or standard heparin therapy.
- † VTE = venous thromboembolic event (DVT and/or PE)
- ‡ The 95% Confidence Intervals for the treatment differences for total VTE were:
Lovenox once a day versus heparin (-3.0 to 3.5)
Lovenox every 12 hours versus heparin (-4.2 to 1.7)All Treated DVT Patients with or without PE 298 (100) 312 (100) 290 (100) Patient Outcome Total VTE† (%) 13 (4.4)‡ 9 (2.9)‡ 12 (4.1) DVT Only (%) 11 (3.7) 7 (2.2) 8 (2.8) Proximal DVT (%) 9 (3.0) 6 (1.9) 7 (2.4) PE (%) 2 (0.7) 2 (0.6) 4 (1.4) Similarly, in a multicenter, open-label, parallel group study, patients with acute proximal DVT were randomized to Lovenox or heparin. Patients who could not receive outpatient therapy were excluded from entering the study. Outpatient exclusion criteria included the following: inability to receive outpatient heparin therapy because of associated comorbid conditions or potential for non-compliance and inability to attend follow-up visits as an outpatient because of geographic inaccessibility. Eligible patients could be treated in the hospital, but ONLY Lovenox patients were permitted to go home on therapy (72%). A total of 501 patients were randomized in the study and all patients were treated. Patients ranged in age from 19 to 96 years (mean age 57.8 years) with 60.5% men and 39.5% women. Patients were randomized to either Lovenox 1 mg/kg every 12 hours subcutaneously or heparin intravenous bolus (5000 IU) followed by a continuous infusion administered to achieve an aPTT of 60 to 85 seconds (in-patient treatment). All patients also received warfarin sodium as described in the previous study. Lovenox or standard heparin therapy was administered for a minimum of 5 days. Lovenox was equivalent to standard heparin therapy in reducing the risk of recurrent venous thromboembolism. The efficacy data are provided below (see Table 22).
Table 22: Efficacy of Lovenox in Treatment of Deep Vein Thrombosis Dosing Regimen* Lovenox
1 mg/kg q12h subcutaneouslyHeparin
aPTT Adjusted Intravenous TherapyIndication n (%) n (%) - * All patients were also treated with warfarin sodium commencing on the evening of the second day of Lovenox or standard heparin therapy.
- † VTE = venous thromboembolic event (deep vein thrombosis [DVT] and/or pulmonary embolism [PE]).
- ‡ The 95% Confidence Intervals for the treatment difference for total VTE was: Lovenox versus heparin (-5.6 to 2.7).
All Treated DVT Patients 247 (100) 254 (100) Patient Outcome Total VTE† (%) 13 (5.3)‡ 17 (6.7) DVT Only (%) 11 (4.5) 14 (5.5) Proximal DVT (%) 10 (4.0) 12 (4.7) PE (%) 2 (0.8) 3 (1.2) 14.5 Prophylaxis of Ischemic Complications in Unstable Angina and Non–Q-Wave Myocardial Infarction
In a multicenter, double-blind, parallel group study, patients who recently experienced unstable angina or non–Q-wave myocardial infarction were randomized to either Lovenox 1 mg/kg every 12 hours subcutaneously or heparin intravenous bolus (5000 U) followed by a continuous infusion (adjusted to achieve an aPTT of 55 to 85 seconds). A total of 3171 patients were enrolled in the study, and 3107 patients were treated. Patients ranged in age from 25 to 94 years (median age 64 years), with 33.4% of patients female and 66.6% male. Race was distributed as follows: 89.8% Caucasian, 4.8% Black, 2.0% Asian, and 3.5% other. All patients were also treated with aspirin 100 to 325 mg per day. Treatment was initiated within 24 hours of the event and continued until clinical stabilization, revascularization procedures, or hospital discharge, with a maximal duration of 8 days of therapy. The combined incidence of the triple endpoint of death, myocardial infarction, or recurrent angina was lower for Lovenox compared with heparin therapy at 14 days after initiation of treatment. The lower incidence of the triple endpoint was sustained up to 30 days after initiation of treatment. These results were observed in an analysis of both all-randomized and all-treated patients. The efficacy data are provided below (see Table 23).
Table 23: Efficacy of Lovenox in the Prophylaxis of Ischemic Complications in Unstable Angina and Non–Q-Wave Myocardial Infarction (combined endpoint of death, myocardial infarction, or recurrent angina) Dosing Regimen* Lovenox
1 mg/kg q12h subcutaneousHeparin
aPTT Adjusted Intravenous TherapyReduction
(%)p Value Indication n (%) n (%) - * All patients were also treated with aspirin 100 to 325 mg per day.
- † Evaluation time points are after initiation of treatment. Therapy continued for up to 8 days (median duration of 2.6 days).
All Treated Unstable Angina and Non–Q-Wave MI Patients 1578 (100) 1529 (100) Time point† 48 Hours 96 (6.1) 112 (7.3) 1.2 0.120 14 Days 261 (16.5) 303 (19.8) 3.3 0.017 30 Days 313 (19.8) 358 (23.4) 3.6 0.014 The combined incidence of death or myocardial infarction at all time points was lower for Lovenox compared to standard heparin therapy, but did not achieve statistical significance. The efficacy data are provided below (see Table 24).
Table 24: Efficacy of Lovenox in the Prophylaxis of Ischemic Complications in Unstable Angina and Non–Q-Wave Myocardial Infarction (Combined endpoint of death or myocardial infarction) Dosing Regimen* Lovenox
1 mg/kg q12h subcutaneouslyHeparin
aPTT Adjusted Intravenous TherapyReduction
(%)p Value Indication n (%) n (%) - * All patients were also treated with aspirin 100 to 325 mg per day.
- † Evaluation time points are after initiation of treatment. Therapy continued for up to 8 days (median duration of 2.6 days).
All Treated Unstable Angina and Non–Q-Wave MI Patients 1578 (100) 1529 (100) Time point† 48 Hours 16 (1.0) 20 (1.3) 0.3 0.126 14 Days 76 (4.8) 93 (6.1) 1.3 0.115 30 Days 96 (6.1) 118 (7.7) 1.6 0.069 In a survey one year following treatment, with information available for 92% of enrolled patients, the combined incidence of death, myocardial infarction, or recurrent angina remained lower for Lovenox versus heparin (32.0% vs 35.7%).
Urgent revascularization procedures were performed less frequently in the Lovenox group as compared to the heparin group, 6.3% compared to 8.2% at 30 days (p=0.047).
14.6 Treatment of Acute ST-Segment Elevation Myocardial Infarction
In a multicenter, double-blind, double-dummy, parallel-group study, patients with acute ST-segment elevation myocardial infarction (STEMI) who were to be hospitalized within 6 hours of onset and were eligible to receive fibrinolytic therapy were randomized in a 1:1 ratio to receive either Lovenox or unfractionated heparin.
Study medication was initiated between 15 minutes before and 30 minutes after the initiation of fibrinolytic therapy. Unfractionated heparin was administered beginning with an intravenous bolus of 60 U/kg (maximum 4000 U) and followed with an infusion of 12 U/kg per hour (initial maximum 1000 U per hour) that was adjusted to maintain an aPTT of 1.5 to 2 times the control value. The intravenous infusion was to be given for at least 48 hours. The Lovenox dosing strategy was adjusted according to the patient's age and renal function. For patients younger than 75 years of age, Lovenox was given as a single 30 mg intravenous bolus plus a 1 mg/kg subcutaneous dose followed by a subcutaneous injection of 1 mg/kg every 12 hours. For patients at least 75 years of age, the intravenous bolus was not given and the subcutaneous dose was reduced to 0.75 mg/kg every 12 hours. For patients with severe renal insufficiency (estimated creatinine clearance of less than 30 mL per minute), the dose was to be modified to 1 mg/kg every 24 hours. The subcutaneous injections of Lovenox were given until hospital discharge or for a maximum of eight days (whichever came first). The mean treatment duration for Lovenox was 6.6 days. The mean treatment duration of unfractionated heparin was 54 hours.
When percutaneous coronary intervention was performed during study medication period, patients received antithrombotic support with blinded study drug. For patients on Lovenox, the PCI was to be performed on Lovenox (no switch) using the regimen established in previous studies, i.e. no additional dosing, if the last subcutaneous administration was less than 8 hours before balloon inflation, intravenous bolus of 0.3 mg/kg Lovenox if the last subcutaneous administration was more than 8 hours before balloon inflation.
All patients were treated with aspirin for a minimum of 30 days. Eighty percent of patients received a fibrin-specific agent (19% tenecteplase, 5% reteplase and 55% alteplase) and 20% received streptokinase.
Among 20,479 patients in the ITT population, the mean age was 60 years, and 76% were male. Racial distribution was: 87% Caucasian, 9.8% Asian, 0.2% Black, and 2.8% other. Medical history included previous MI (13%), hypertension (44%), diabetes (15%) and angiographic evidence of CAD (5%). Concomitant medication included aspirin (95%), beta-blockers (86%), ACE inhibitors (78%), statins (70%) and clopidogrel (27%). The MI at entry was anterior in 43%, non-anterior in 56%, and both in 1%.
The primary efficacy endpoint was the composite of death from any cause or myocardial re-infarction in the first 30 days after randomization. Total follow-up was one year.
The rate of the primary efficacy endpoint (death or myocardial re-infarction) was 9.9% in the Lovenox group, and 12% in the unfractionated heparin group, a 17% reduction in the relative risk, (P=0.000003) (see Table 25).
Table 25: Efficacy of Lovenox in the Treatment of Acute ST-Segment Elevation Myocardial Infarction Lovenox
(N=10,256)UFH
(N=10,223)Relative Risk
(95% CI)P Value Note: Urgent revascularization denotes episodes of recurrent myocardial ischemia (without infarction) leading to the clinical decision to perform coronary revascularization during the same hospitalization. CI denotes confidence intervals. Outcome at 48 hours n (%) n (%) Death or Myocardial Re-infarction 478 (4.7) 531 (5.2) 0.90 (0.80 to 1.01) 0.08 Death 383 (3.7) 390 (3.8) 0.98 (0.85 to 1.12) 0.76 Myocardial Re-infarction 102 (1.0) 156 (1.5) 0.65 (0.51 to 0.84) <0.001 Urgent Revascularization 74 (0.7) 96 (0.9) 0.77 (0.57 to 1.04) 0.09 Death or Myocardial Re-infarction or Urgent Revascularization 548 (5.3) 622 (6.1) 0.88 (0.79 to 0.98) 0.02 Outcome at 8 Days Death or Myocardial Re-infarction 740 (7.2) 954 (9.3) 0.77 (0.71 to 0.85) <0.001 Death 559 (5.5) 605 (5.9) 0.92 (0.82 to 1.03) 0.15 Myocardial Re-infarction 204 (2.0) 379 (3.7) 0.54 (0.45 to 0.63) <0.001 Urgent Revascularization 145 (1.4) 247 (2.4) 0.59 (0.48 to 0.72) <0.001 Death or Myocardial Re-infarction or Urgent Revascularization 874 (8.5) 1181 (11.6) 0.74 (0.68 to 0.80) <0.001 Outcome at 30 Days Primary efficacy endpoint
(Death or Myocardial Re-infarction)1017 (9.9) 1223 (12.0) 0.83 (0.77 to 0.90) 0.000003 Death 708 (6.9) 765 (7.5) 0.92 (0.84 to 1.02) 0.11 Myocardial Re-infarction 352 (3.4) 508 (5.0) 0.69 (0.60 to 0.79) <0.001 Urgent Revascularization 213 (2.1) 286 (2.8) 0.74 (0.62 to 0.88) <0.001 Death or Myocardial Re-infarction or Urgent Revascularization 1199 (11.7) 1479 (14.5) 0.81 (0.75 to 0.87) <0.001 The beneficial effect of Lovenox on the primary endpoint was consistent across key subgroups including age, gender, infarct location, history of diabetes, history of prior myocardial infarction, fibrinolytic agent administered, and time to treatment with study drug (see Figure 1); however, it is necessary to interpret such subgroup analyses with caution.
- * The primary efficacy endpoint was the composite of death from any cause or myocardial re-infarction in the first 30 days. The overall treatment effect of Lovenox as compared to the unfractionated heparin is shown at the bottom of the figure. For each subgroup, the circle is proportional to the number and represents the point estimate of the treatment effect and the horizontal lines represent the 95% confidence intervals. Fibrin-specific fibrinolytic agents included alteplase, tenecteplase, and reteplase. Time to treatment indicates the time from the onset of symptoms to the administration of study drug (median: 3.2 hours).
Figure 1: Relative Risks of and Absolute Event Rates for the Primary Endpoint at 30 Days in Various Subgroups* 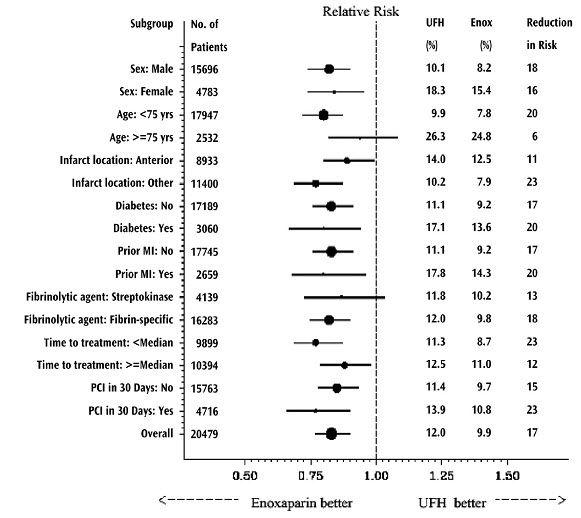
The beneficial effect of Lovenox on the primary endpoint observed during the first 30 days was maintained over a 12 month follow-up period (see Figure 2).
Figure 2: Kaplan-Meier Plot – Death or Myocardial Re-infarction at 30 Days – ITT Population
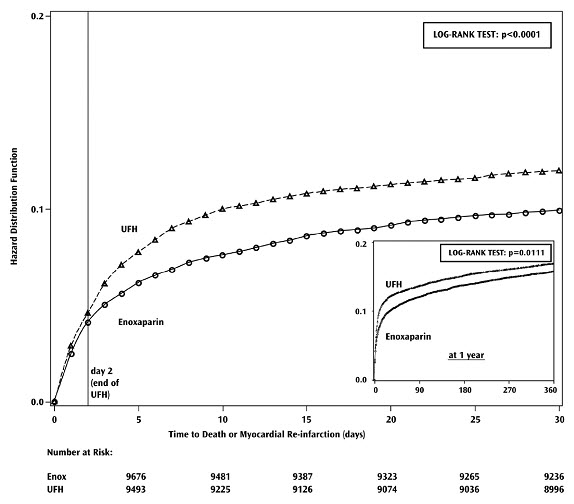
There is a trend in favor of Lovenox during the first 48 hours, but most of the treatment difference is attributed to a step increase in the event rate in the UFH group at 48 hours (seen in Figure 2), an effect that is more striking when comparing the event rates just prior to and just subsequent to actual times of discontinuation. These results provide evidence that UFH was effective and that it would be better if used longer than 48 hours. There is a similar increase in endpoint event rate when Lovenox was discontinued, suggesting that it too was discontinued too soon in this study.
The rates of major hemorrhages (defined as requiring 5 or more units of blood for transfusion, or 15% drop in hematocrit or clinically overt bleeding, including intracranial hemorrhage) at 30 days were 2.1% in the Lovenox group and 1.4% in the unfractionated heparin group. The rates of intracranial hemorrhage at 30 days were 0.8% in the Lovenox group and 0.7% in the unfractionated heparin group. The 30-day rate of the composite endpoint of death, myocardial re-infarction or ICH (a measure of net clinical benefit) was significantly lower in the Lovenox group (10.1%) as compared to the heparin group (12.2%).
-
16 HOW SUPPLIED/STORAGE AND HANDLING
Lovenox is available in two concentrations (see Tables 26 and 27).
Table 26: 100 mg/mL Concentration Dosage Unit/Strength* Anti-Xa Activity† Package Size
(per carton)Label Color NDC # 0075- - * Strength represents the number of milligrams of enoxaparin sodium in Water for Injection. Lovenox 30 and 40 mg prefilled syringes, and 60, 80, and 100 mg graduated prefilled syringes each contain 10 mg enoxaparin sodium per 0.1 mL Water for Injection.
- † Approximate anti-Factor Xa activity based on reference to the W.H.O. First International Low Molecular Weight Heparin Reference Standard.
- ‡ Each Lovenox prefilled syringe is for single, one-time use only and is affixed with a 27 gauge × 1/2-inch needle.
- § Each Lovenox multiple-dose vial contains 15 mg benzyl alcohol per 1 mL as a preservative.
Prefilled Syringes‡ 30 mg/0.3 mL 3000 IU 10 syringes Medium Blue 0624-30 40 mg/0.4 mL 4000 IU 10 syringes Yellow 0620-40 Graduated Prefilled Syringes‡ 60 mg/0.6 mL 6000 IU 10 syringes Orange 0621-60 80 mg/0.8 mL 8000 IU 10 syringes Brown 0622-80 100 mg/1 mL 10,000 IU 10 syringes Black 0623-00 Multiple-Dose Vial§ 300 mg/3 mL 30,000 IU 1 vial Red 0626-03 Table 27: 150 mg/mL Concentration Dosage Unit/Strength* Anti-Xa Activity† Package Size
(per carton)Syringe Label Color NDC # 0075- - * Strength represents the number of milligrams of enoxaparin sodium in Water for Injection. Lovenox 120 and 150 mg graduated prefilled syringes contain 15 mg enoxaparin sodium per 0.1 mL Water for Injection.
- † Approximate anti-Factor Xa activity based on reference to the W.H.O. First International Low Molecular Weight Heparin Reference Standard.
- ‡ Each Lovenox graduated prefilled syringe is for single, one-time use only and is affixed with a 27 gauge × 1/2-inch needle.
Graduated Prefilled Syringes‡ 120 mg/0.8 mL 12,000 IU 10 syringes Purple 2912-01 150 mg/1 mL 15,000 IU 10 syringes Navy Blue 2915-01 -
17 PATIENT COUNSELING INFORMATION
If patients have had neuraxial anesthesia or spinal puncture, and particularly, if they are taking concomitant NSAIDs, platelet inhibitors, or other anticoagulants, advise them to watch for signs and symptoms of spinal or epidural hematoma, such as tingling, numbness (especially in the lower limbs) and muscular weakness. Instruct the patient to seek immediate medical attention if any of these symptoms occur.
Inform patients:
- of the instructions for injecting Lovenox if they continue Lovenox therapy after discharge from the hospital.
- that it may take them longer than usual to stop bleeding.
- that they may bruise and/or bleed more easily when they use Lovenox.
- that they should report any unusual bleeding, bruising, or signs of thrombocytopenia (such as a rash of dark red spots under the skin) to their physician [see Warnings and Precautions (5.1, 5.5)].
- that risks are associated with the use of benzyl alcohol, a preservative in Lovenox multi-dose vials, in neonates, infants, and pregnant women.
- to tell their physicians and dentists they are taking Lovenox and/or any other product known to affect bleeding before any surgery is scheduled and before any new drug is taken [see Warnings and Precautions (5.1, 5.3)].
- to tell their physicians and dentists of all medications they are taking, including those obtained without a prescription, such as aspirin or other NSAIDs [see Drug Interactions (7)].
- SPL UNCLASSIFIED SECTION
-
PRINCIPAL DISPLAY PANEL - 30 mg/0.3 mL Syringe Carton
NDC: 0075-0624-30
LOVENOX®
(enoxaparin sodium injection)30mg/0.3mL
Rx ONLY
SINGLE DOSE SYRINGES WITH AUTOMATIC SAFETY DEVICE
FOR SUBCUTANEOUS INJECTIONTen 0.3mL Syringes
SANOFI
-
PRINCIPAL DISPLAY PANEL - 40 mg/0.4 mL Syringe Carton
NDC: 0075-0620-40
LOVENOX®
(enoxaparin sodium injection)40mg/0.4mL
Rx ONLY
SINGLE DOSE SYRINGES WITH AUTOMATIC SAFETY DEVICE
FOR SUBCUTANEOUS INJECTIONTen 0.4mL Syringes
SANOFI
-
PRINCIPAL DISPLAY PANEL - 60 mg/0.6 mL Syringe Carton
NDC: 0075-0621-60
LOVENOX®
(enoxaparin sodium injection)60mg/0.6mL
Rx ONLY
SINGLE DOSE SYRINGES WITH AUTOMATIC SAFETY DEVICE
FOR SUBCUTANEOUS INJECTIONTen 0.6mL Syringes
SANOFI
-
PRINCIPAL DISPLAY PANEL - 80 mg/0.8 mL Syringe Carton
NDC: 0075-0622-80
LOVENOX®
(enoxaparin sodium injection)80mg/0.8mL
Rx ONLY
SINGLE DOSE SYRINGES WITH AUTOMATIC SAFETY DEVICE
FOR SUBCUTANEOUS INJECTIONTen 0.8mL Syringes
SANOFI
-
PRINCIPAL DISPLAY PANEL - 100 mg/1 mL Syringe Carton
NDC: 0075-0623-00
LOVENOX®
(enoxaparin sodium injection)100mg/1 mL
Rx ONLY
SINGLE DOSE SYRINGES WITH AUTOMATIC SAFETY DEVICE
FOR SUBCUTANEOUS INJECTIONTen 1mL Syringes
SANOFI
-
PRINCIPAL DISPLAY PANEL - 300 mg/3 mL Vial Carton
NDC: 0075-0626-03
LOVENOX®
(enoxaparin sodium injection)300mg/3mL
(100mg/1mL)Rx ONLY
For Subcutaneous or
Intravenous InjectionMultiple Dose Vial
One 3mL Vial
SANOFI

-
PRINCIPAL DISPLAY PANEL - 120 mg/0.8 mL Syringe Carton
NDC: 0075-2912-01
LOVENOX®
(enoxaparin sodium injection)120mg/0.8mL
Rx ONLY
SINGLE DOSE SYRINGES WITH AUTOMATIC SAFETY DEVICE
FOR SUBCUTANEOUS INJECTIONTen 0.8mL Syringes
SANOFI

-
PRINCIPAL DISPLAY PANEL - 150 mg/1 mL Syringe Carton
NDC: 0075-2915-01
LOVENOX®
(enoxaparin sodium injection)150mg/1 mL
Rx ONLY
SINGLE DOSE SYRINGES WITH AUTOMATIC SAFETY DEVICE
FOR SUBCUTANEOUS INJECTIONTen 1mL Syringes
SANOFI

-
INGREDIENTS AND APPEARANCE
LOVENOX
enoxaparin sodium injectionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0075-0624 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength enoxaparin sodium (UNII: 8NZ41MIK1O) (enoxaparin - UNII:E47C0NF7LV) enoxaparin sodium 30 mg in 0.3 mL Inactive Ingredients Ingredient Name Strength water (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0075-0624-30 10 in 1 CARTON 03/29/1993 1 1 in 1 CELLO PACK 1 0.3 mL in 1 SYRINGE; Type 2: Prefilled Drug Delivery Device/System (syringe, patch, etc.) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA020164 03/29/1993 LOVENOX
enoxaparin sodium injectionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0075-0620 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength enoxaparin sodium (UNII: 8NZ41MIK1O) (enoxaparin - UNII:E47C0NF7LV) enoxaparin sodium 40 mg in 0.4 mL Inactive Ingredients Ingredient Name Strength water (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0075-0620-40 10 in 1 CARTON 03/29/1993 1 1 in 1 CELLO PACK 1 0.4 mL in 1 SYRINGE; Type 2: Prefilled Drug Delivery Device/System (syringe, patch, etc.) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA020164 03/29/1993 LOVENOX
enoxaparin sodium injectionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0075-0621 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength enoxaparin sodium (UNII: 8NZ41MIK1O) (enoxaparin - UNII:E47C0NF7LV) enoxaparin sodium 60 mg in 0.6 mL Inactive Ingredients Ingredient Name Strength water (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0075-0621-60 10 in 1 CARTON 03/29/1993 1 1 in 1 CELLO PACK 1 0.6 mL in 1 SYRINGE; Type 2: Prefilled Drug Delivery Device/System (syringe, patch, etc.) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA020164 03/29/1993 LOVENOX
enoxaparin sodium injectionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0075-0622 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength enoxaparin sodium (UNII: 8NZ41MIK1O) (enoxaparin - UNII:E47C0NF7LV) enoxaparin sodium 80 mg in 0.8 mL Inactive Ingredients Ingredient Name Strength water (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0075-0622-80 10 in 1 CARTON 03/29/1993 1 1 in 1 CELLO PACK 1 0.8 mL in 1 SYRINGE; Type 2: Prefilled Drug Delivery Device/System (syringe, patch, etc.) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA020164 03/29/1993 LOVENOX
enoxaparin sodium injectionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0075-0623 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength enoxaparin sodium (UNII: 8NZ41MIK1O) (enoxaparin - UNII:E47C0NF7LV) enoxaparin sodium 100 mg in 1 mL Inactive Ingredients Ingredient Name Strength water (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0075-0623-00 10 in 1 CARTON 03/29/1993 1 1 in 1 CELLO PACK 1 1 mL in 1 SYRINGE; Type 2: Prefilled Drug Delivery Device/System (syringe, patch, etc.) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA020164 03/29/1993 LOVENOX
enoxaparin sodium injectionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0075-0626 Route of Administration SUBCUTANEOUS, INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength enoxaparin sodium (UNII: 8NZ41MIK1O) (enoxaparin - UNII:E47C0NF7LV) enoxaparin sodium 300 mg in 3 mL Inactive Ingredients Ingredient Name Strength water (UNII: 059QF0KO0R) benzyl alcohol (UNII: LKG8494WBH) 45 mg in 3 mL Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0075-0626-03 1 in 1 CARTON 03/29/1993 1 3 mL in 1 VIAL, MULTI-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA020164 03/29/1993 LOVENOX
enoxaparin sodium injectionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0075-2912 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength enoxaparin sodium (UNII: 8NZ41MIK1O) (enoxaparin - UNII:E47C0NF7LV) enoxaparin sodium 120 mg in 0.8 mL Inactive Ingredients Ingredient Name Strength water (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0075-2912-01 10 in 1 CARTON 03/29/1993 1 1 in 1 CELLO PACK 1 0.8 mL in 1 SYRINGE; Type 2: Prefilled Drug Delivery Device/System (syringe, patch, etc.) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA020164 03/29/1993 LOVENOX
enoxaparin sodium injectionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0075-2915 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength enoxaparin sodium (UNII: 8NZ41MIK1O) (enoxaparin - UNII:E47C0NF7LV) enoxaparin sodium 150 mg in 1 mL Inactive Ingredients Ingredient Name Strength water (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0075-2915-01 10 in 1 CARTON 03/29/1993 1 1 in 1 CELLO PACK 1 1 mL in 1 SYRINGE; Type 2: Prefilled Drug Delivery Device/System (syringe, patch, etc.) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA020164 03/29/1993 Labeler - Sanofi-Aventis U.S. LLC (824676584) Establishment Name Address ID/FEI Business Operations Sanofi Winthrop Industries 298045688 ANALYSIS(0075-0621, 0075-0622, 0075-0623, 0075-2912, 0075-2915) , LABEL(0075-0621, 0075-0622, 0075-0623, 0075-2912, 0075-2915) , MANUFACTURE(0075-0621, 0075-0622, 0075-0623, 0075-2912, 0075-2915) , PACK(0075-0621, 0075-0622, 0075-0623, 0075-2912, 0075-2915) Establishment Name Address ID/FEI Business Operations sanofi-aventis Deutschland GmbH 313218430 ANALYSIS(0075-0626) , LABEL(0075-0626) , MANUFACTURE(0075-0626) , PACK(0075-0626) Establishment Name Address ID/FEI Business Operations Genzyme Corporation 098066215 ANALYSIS(0075-0620, 0075-0621, 0075-0622, 0075-0623, 0075-0624, 0075-2912, 0075-2915) , LABEL(0075-0620, 0075-0621, 0075-0622, 0075-0623, 0075-0624, 0075-2912, 0075-2915) , MANUFACTURE(0075-0620, 0075-0621, 0075-0622, 0075-0623, 0075-0624, 0075-2912, 0075-2915) , PACK(0075-0620, 0075-0621, 0075-0622, 0075-0623, 0075-0624, 0075-2912, 0075-2915) Establishment Name Address ID/FEI Business Operations Sanofi Winthrop Industrie 297730488 ANALYSIS(0075-0620, 0075-0621, 0075-0622, 0075-0623, 0075-0624, 0075-2912, 0075-2915) , LABEL(0075-0620, 0075-0621, 0075-0622, 0075-0623, 0075-0624, 0075-2912, 0075-2915) , MANUFACTURE(0075-0620, 0075-0621, 0075-0622, 0075-0623, 0075-0624, 0075-2912, 0075-2915) , PACK(0075-0620, 0075-0621, 0075-0622, 0075-0623, 0075-0624, 0075-2912, 0075-2915) Establishment Name Address ID/FEI Business Operations VLG CHEM 272606430 API MANUFACTURE(0075-0620, 0075-0621, 0075-0622, 0075-0623, 0075-0624, 0075-0626, 0075-2912, 0075-2915) Establishment Name Address ID/FEI Business Operations Aventis Pharma Manufacturing Pte. Ltd. 595299306 ANALYSIS(0075-0620, 0075-0621, 0075-0622, 0075-0623, 0075-0624, 0075-0626, 0075-2912, 0075-2915) , API MANUFACTURE(0075-0620, 0075-0621, 0075-0622, 0075-0623, 0075-0624, 0075-0626, 0075-2912, 0075-2915)
Trademark Results [Lovenox]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 LOVENOX 74219194 1707749 Live/Registered |
AVENTIS PHARMA S.A. 1991-11-05 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.