LAMOTRIGINE tablet, chewable
Lamotrigine by
Drug Labeling and Warnings
Lamotrigine by is a Prescription medication manufactured, distributed, or labeled by Glenmark Pharmaceuticals Inc., USA, Glenmark Pharmaceuticals Limited. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use LAMOTRIGINE TABLETS FOR ORAL SUSPENSION safely and effectively. See full prescribing information for LAMOTRIGINE TABLETS FOR ORAL SUSPENSION.
LAMOTRIGINE tablets for oral suspension (chewable, dispersible tablets), for oral use
Initial U.S. Approval: 1994WARNING: SERIOUS SKIN RASHES
See full prescribing information for complete boxed warning.
-
Cases of life-threatening serious rashes, including Stevens-Johnson syndrome and toxic epidermal necrolysis, and/or rash-related death have been caused by lamotrigine. The rate of serious rash is greater in pediatric patients than in adults. Additional factors that may increase the risk of rash include:
- coadministration with valproate.
- exceeding recommended initial dose of lamotrigine.
- exceeding recommended dose escalation for lamotrigine. (5.1)
- Benign rashes are also caused by lamotrigine; however, it is not possible to predict which rashes will prove to be serious or life threatening. Lamotrigine should be discontinued at the first sign of rash, unless the rash is clearly not drug related. (5.1)
RECENT MAJOR CHANGES
Warnings and Precautions, Hemophagocytic Lymphohistiocytosis (5.2)
8/2019
INDICATIONS AND USAGE
Lamotrigine tablets for oral suspension (chewable, dispersible tablets) are indicated for:
Epilepsy – adjunctive therapy in patients aged 2 years and older:
- partial-onset seizures.
- primary generalized tonic-clonic seizures.
- generalized seizures of Lennox-Gastaut syndrome. (1.1)
Epilepsy – monotherapy in patients aged 16 years and older: Conversion to monotherapy in patients with partial-onset seizures who are receiving treatment with carbamazepine, phenytoin, phenobarbital, primidone, or valproate as the single antiepileptic drug. (1.1)
Bipolar disorder: Maintenance treatment of bipolar I disorder to delay the time to occurrence of mood episodes in patients treated for acute mood episodes with standard therapy. (1.2)
Limitations of Use: Treatment of acute manic or mixed episodes is not recommended. Effectiveness of lamotrigine tablets for oral suspension (chewable dispersible tablets) in the acute treatment of mood episodes has not been established.
DOSAGE AND ADMINISTRATION
- Dosing is based on concomitant medications, indication, and patient age. (2.1, 2.2, 2.3, 2.4)
- To avoid an increased risk of rash, the recommended initial dose and subsequent dose escalations should not be exceeded. (2.1, 16)
- Do not restart lamotrigine tablets for oral suspension (chewable, dispersible tablets) in patients who discontinued due to rash unless the potential benefits clearly outweigh the risks. (2.1, 5.1)
- Adjustments to maintenance doses will be necessary in most patients starting or stopping estrogen-containing oral contraceptives. (2.1, 5.8)
- Discontinuation: Taper over a period of at least 2 weeks (approximately 50% dose reduction per week). (2.1, 5.9)
Epilepsy:
- Adjunctive therapy – See Table 1 for patients older than 12 years and Tables 2 and 3 for patients aged 2 to 12 years. (2.2)
- Conversion to monotherapy – See Table 4. (2.3)
Bipolar disorder: See Tables 5 and 6. (2.4)
DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONS
Hypersensitivity to the drug or its ingredients. (Boxed Warning, 4)
WARNINGS AND PRECAUTIONS
- Life-threatening serious rash and/or rash-related death: Discontinue at the first sign of rash, unless the rash is clearly not drug related. (Boxed Warning, 5.1)
- Hemophagocytic lymphohistiocytosis: Consider this diagnosis and evaluate patients immediately if they develop signs or symptoms of systemic inflammation. Discontinue lamotrigine if an alternative etiology is not established. (5.2)
- Fatal or life-threatening hypersensitivity reaction: Multiorgan hypersensitivity reactions, also known as drug reaction with eosinophilia and systemic symptoms, may be fatal or life threatening. Early signs may include rash, fever, and lymphadenopathy. These reactions may be associated with other organ involvement, such as hepatitis, hepatic failure, blood dyscrasias, or acute multiorgan failure. Lamotrigine should be discontinued if alternate etiology for this reaction is not found. (5.3)
- Blood dyscrasias (e.g., neutropenia, thrombocytopenia, pancytopenia): May occur, either with or without an associated hypersensitivity syndrome. Monitor for signs of anemia, unexpected infection, or bleeding. (5.4)
- Suicidal behavior and ideation: Monitor for suicidal thoughts or behaviors. (5.5)
- Aseptic meningitis: Monitor for signs of meningitis. (5.6)
- Medication errors due to product name confusion: Strongly advise patients to visually inspect tablets to verify the received drug is correct. (5.7, 16, 17)
- Risk in Patients with Phenylketonuria: Phenylalanine can be harmful to patients with phenylketonuria (PKU). Lamotrigine tablets for oral suspension (chewable, dispersible tablets), 5 mg and 25 mg, each contain 1.123 mg phenylalanine. (5.15)
ADVERSE REACTIONS
- Epilepsy: Most common adverse reactions (incidence ≥10%) in adults were dizziness, headache, diplopia, ataxia, nausea, blurred vision, somnolence, rhinitis, pharyngitis, and rash. Additional adverse reactions (incidence ≥10%) reported in children included vomiting, infection, fever, accidental injury, diarrhea, abdominal pain, and tremor. (6.1)
- Bipolar disorder: Most common adverse reactions (incidence >5%) in adults were nausea, insomnia, somnolence, back pain, fatigue, rash, rhinitis, abdominal pain, and xerostomia. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Glenmark Pharmaceuticals Inc., USA at 1 (888) 721-7115 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- Valproate increases lamotrigine concentrations more than 2-fold. (7, 12.3)
- Carbamazepine, phenytoin, phenobarbital, primidone, and rifampin decrease lamotrigine concentrations by approximately 40%. (7, 12.3)
- Estrogen-containing oral contraceptives decrease lamotrigine concentrations by approximately 50%. (7, 12.3)
- Protease inhibitors lopinavir/ritonavir and atazanavir/lopinavir decrease lamotrigine exposure by approximately 50% and 32%, respectively. (7, 12.3)
- Coadministration with organic cationic transporter 2 substrates with narrow therapeutic index is not recommended (7, 12.3)
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 2/2020
-
Cases of life-threatening serious rashes, including Stevens-Johnson syndrome and toxic epidermal necrolysis, and/or rash-related death have been caused by lamotrigine. The rate of serious rash is greater in pediatric patients than in adults. Additional factors that may increase the risk of rash include:
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: SERIOUS SKIN RASHES
1 INDICATIONS AND USAGE
1.1 Epilepsy
1.2 Bipolar Disorder
2 DOSAGE AND ADMINISTRATION
2.1 General Dosing Considerations
2.2 Epilepsy-Adjunctive Therapy
2.3 Epilepsy-Conversion from Adjunctive Therapy to Monotherapy
2.4 Bipolar Disorder
2.5 Administration of Lamotrigine Tablets For Oral Suspension (Chewable, Dispersible Tablets)
3 DOSAGE FORMS AND STRENGTHS
3.2 Tablets For Oral Suspension (Chewable, Dispersible Tablets)
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Serious Skin Rashes [see Boxed Warning]
5.2 Hemophagocytic Lymphohistiocytosis
5.3 Multiorgan Hypersensitivity Reactions and Organ Failure
5.4 Blood Dyscrasias
5.5 Suicidal Behavior and Ideation
5.6 Aseptic Meningitis
5.7 Potential Medication Errors
5.8 Concomitant Use with Oral Contraceptives
5.9 Withdrawal Seizures
5.10 Status Epilepticus
5.11 Sudden Unexplained Death in Epilepsy (SUDEP)
5.12 Addition of Lamotrigine to a Multidrug Regimen that Includes Valproate
5.13 Binding in the Eye and Other Melanin-Containing Tissues
5.14 Laboratory Tests
5.15 Risk in Patients with Phenylketonuria
6 ADVERSE REACTIONS
6.1 Clinical Trial Experience
6.2 Other Adverse Reactions Observed in All Clinical Trials
6.3 Postmarketing Experience
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
8.7 Renal Impairment
10 OVERDOSAGE
10.1 Human Overdose Experience
10.2 Management of Overdose
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Epilepsy
14.2 Bipolar Disorder
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: SERIOUS SKIN RASHES
Lamotrigine can cause serious rashes requiring hospitalization and discontinuation of treatment. The incidence of these rashes, which have included Stevens-Johnson syndrome, is approximately 0.3% to 0.8% in pediatric patients (aged 2 to 17 years) and 0.08% to 0.3% in adults receiving lamotrigine. One rash-related death was reported in a prospectively followed cohort of 1,983 pediatric patients (aged 2 to 16 years) with epilepsy taking lamotrigine as adjunctive therapy. In worldwide postmarketing experience, rare cases of toxic epidermal necrolysis and/or rash-related death have been reported in adult and pediatric patients, but their numbers are too few to permit a precise estimate of the rate.
Other than age, there are as yet no factors identified that are known to predict the risk of occurrence or the severity of rash caused by lamotrigine. There are suggestions, yet to be proven, that the risk of rash may also be increased by (1) coadministration of lamotrigine with valproate (includes valproic acid and divalproex sodium), (2) exceeding the recommended initial dose of lamotrigine, or (3) exceeding the recommended dose escalation for lamotrigine. However, cases have occurred in the absence of these factors.
Nearly all cases of life-threatening rashes caused by lamotrigine have occurred within 2 to 8 weeks of treatment initiation. However, isolated cases have occurred after prolonged treatment (e.g., 6 months). Accordingly, duration of therapy cannot be relied upon as means to predict the potential risk heralded by the first appearance of a rash.
Although benign rashes are also caused by lamotrigine, it is not possible to predict reliably which rashes will prove to be serious or life threatening. Accordingly, lamotrigine should ordinarily be discontinued at the first sign of rash, unless the rash is clearly not drug related. Discontinuation of treatment may not prevent a rash from becoming life threatening or permanently disabling or disfiguring [see Warnings and Precautions (5.1)].
-
1 INDICATIONS AND USAGE
1.1 Epilepsy
Adjunctive Therapy
Lamotrigine tablets for oral suspension (chewable, dispersible tablets) are indicated as adjunctive therapy for the following seizure types in patients aged 2 years and older:
- partial-onset seizures.
- primary generalized tonic-clonic (PGTC) seizures.
- generalized seizures of Lennox-Gastaut syndrome.
Monotherapy
Lamotrigine tablets for oral suspension (chewable, dispersible tablets) are indicated for conversion to monotherapy in adults (aged 16 years and older) with partial-onset seizures who are receiving treatment with carbamazepine, phenytoin, phenobarbital, primidone, or valproate as the single antiepileptic drug (AED).
Safety and effectiveness of lamotrigine tablets for oral suspension (chewable, dispersible tablets) have not been established (1) as initial monotherapy; (2) for conversion to monotherapy from AEDs other than carbamazepine, phenytoin, phenobarbital, primidone, or valproate; or (3) for simultaneous conversion to monotherapy from 2 or more concomitant AEDs.
1.2 Bipolar Disorder
Lamotrigine tablets for oral suspension (chewable, dispersible tablets) are indicated for the maintenance treatment of bipolar I disorder to delay the time to occurrence of mood episodes (depression, mania, hypomania, mixed episodes) in patients treated for acute mood episodes with standard therapy [see Clinical Studies (14.1)].
Limitations of Use
Treatment of acute manic or mixed episodes is not recommended. Effectiveness of lamotrigine tablets for oral suspension (chewable, dispersible tablets) in the acute treatment of mood episodes has not been established.
-
2 DOSAGE AND ADMINISTRATION
2.1 General Dosing Considerations
Rash
There are suggestions, yet to be proven, that the risk of severe, potentially life-threatening rash may be increased by (1) coadministration of lamotrigine with valproate, (2) exceeding the recommended initial dose of lamotrigine, or (3) exceeding the recommended dose escalation for lamotrigine. However, cases have occurred in the absence of these factors [see Boxed Warning]. Therefore, it is important that the dosing recommendations be followed closely.
The risk of nonserious rash may be increased when the recommended initial dose and/or the rate of dose escalation for lamotrigine is exceeded and in patients with a history of allergy or rash to other AEDs.
It is recommended that lamotrigine tablets for oral suspension (chewable, dispersible tablets) not be restarted in patients who discontinued due to rash associated with prior treatment with lamotrigine unless the potential benefits clearly outweigh the risks. If the decision is made to restart a patient who has discontinued lamotrigine tablets for oral suspension (chewable, dispersible tablets), the need to restart with the initial dosing recommendations should be assessed. The greater the interval of time since the previous dose, the greater consideration should be given to restarting with the initial dosing recommendations. If a patient has discontinued lamotrigine for a period of more than 5 half-lives, it is recommended that initial dosing recommendations and guidelines be followed. The half-life of lamotrigine is affected by other concomitant medications [see Clinical Pharmacology (12.3)].
Lamotrigine Tablets For Oral Suspension (Chewable, Dispersible Tablets) Added to Drugs Known to Induce or Inhibit Glucuronidation
Because lamotrigine is metabolized predominantly by glucuronic acid conjugation, drugs that are known to induce or inhibit glucuronidation may affect the apparent clearance of lamotrigine. Drugs that induce glucuronidation include carbamazepine, phenytoin, phenobarbital, primidone, rifampin, estrogen-containing oral contraceptives, and the protease inhibitors lopinavir/ritonavir and atazanavir/ritonavir. Valproate inhibits glucuronidation. For dosing considerations for lamotrigine tablets for oral suspension (chewable, dispersible tablets) in patients on estrogen-containing contraceptives and atazanavir/ritonavir, see below and Table 13. For dosing considerations for lamotrigine tablets for oral suspension (chewable, dispersible tablets) in patients on other drugs known to induce or inhibit glucuronidation, see Tables 1, 2, 5 to 6, and 13.
Target Plasma Levels for Patients with Epilepsy or Bipolar Disorder
A therapeutic plasma concentration range has not been established for lamotrigine. Dosing of lamotrigine should be based on therapeutic response [see Clinical Pharmacology (12.3)].
Women Taking Estrogen-Containing Oral Contraceptives
Starting Lamotrigine Tablets For Oral Suspension (Chewable, Dispersible Tablets) in Women Taking Estrogen-Containing Oral Contraceptives: Although estrogen-containing oral contraceptives have been shown to increase the clearance of lamotrigine [see Clinical Pharmacology (12.3)], no adjustments to the recommended dose-escalation guidelines for lamotrigine tablets for oral suspension (chewable, dispersible tablets) should be necessary solely based on the use of estrogen-containing oral contraceptives. Therefore, dose escalation should follow the recommended guidelines for initiating adjunctive therapy with lamotrigine tablets for oral suspension (chewable, dispersible tablets) based on the concomitant AED or other concomitant medications (see Tables 1, 5, and 7). See below for adjustments to maintenance doses of lamotrigine tablets for oral suspension (chewable, dispersible tablets) in women taking estrogen-containing oral contraceptives.
Adjustments to the Maintenance Dose of Lamotrigine Tablets For Oral Suspension (Chewable, Dispersible Tablets) in Women Taking Estrogen-Containing Oral Contraceptives:
(1) Taking Estrogen-Containing Oral Contraceptives: In women not taking carbamazepine, phenytoin, phenobarbital, primidone, or other drugs such as rifampin and the protease inhibitors lopinavir/ritonavir and atazanavir/ritonavir that induce lamotrigine glucuronidation [see Drug Interactions (7), Clinical Pharmacology (12.3)], the maintenance dose of lamotrigine tablets for oral suspension (chewable, dispersible tablets) will in most cases need to be increased by as much as 2-fold over the recommended target maintenance dose to maintain a consistent lamotrigine plasma level.
(2) Starting Estrogen-Containing Oral Contraceptives: In women taking a stable dose of lamotrigine tablets for oral suspension (chewable, dispersible tablets) and not taking carbamazepine, phenytoin, phenobarbital, primidone, or other drugs such as rifampin and the protease inhibitors lopinavir/ritonavir and atazanavir/ritonavir that induce lamotrigine glucuronidation [see Drug Interactions (7), Clinical Pharmacology (12.3)], the maintenance dose will in most cases need to be increased by as much as 2-fold to maintain a consistent lamotrigine plasma level. The dose increases should begin at the same time that the oral contraceptive is introduced and continue, based on clinical response, no more rapidly than 50 to 100 mg/day every week. Dose increases should not exceed the recommended rate (see Tables 1 and 5) unless lamotrigine plasma levels or clinical response support larger increases. Gradual transient increases in lamotrigine plasma levels may occur during the week of inactive hormonal preparation (pill-free week), and these increases will be greater if dose increases are made in the days before or during the week of inactive hormonal preparation. Increased lamotrigine plasma levels could result in additional adverse reactions, such as dizziness, ataxia, and diplopia. If adverse reactions attributable to lamotrigine tablets for oral suspension (chewable, dispersible tablets) consistently occur during the pill-free week, dose adjustments to the overall maintenance dose may be necessary. Dose adjustments limited to the pill-free week are not recommended. For women taking lamotrigine tablets for oral suspension (chewable, dispersible tablets) in addition to carbamazepine, phenytoin, phenobarbital, primidone, or other drugs such as rifampin and the protease inhibitors lopinavir/ritonavir and atazanavir/ritonavir that induce lamotrigine glucuronidation [see Drug Interactions (7), Clinical Pharmacology (12.3)], no adjustment to the dose of lamotrigine tablets for oral suspension (chewable, dispersible tablets) should be necessary.
(3) Stopping Estrogen-Containing Oral Contraceptives: In women not taking carbamazepine, phenytoin, phenobarbital, primidone, or other drugs such as rifampin and the protease inhibitors lopinavir/ritonavir and atazanavir/ritonavir that induce lamotrigine glucuronidation [see Drug Interactions (7), Clinical Pharmacology (12.3)], the maintenance dose of lamotrigine tablets for oral suspension (chewable, dispersible tablets) will in most cases need to be decreased by as much as 50% in order to maintain a consistent lamotrigine plasma level. The decrease in dose of lamotrigine tablets for oral suspension (chewable, dispersible tablets) should not exceed 25% of the total daily dose per week over a 2-week period, unless clinical response or lamotrigine plasma levels indicate otherwise [see Clinical Pharmacology (12.3)]. In women taking lamotrigine tablets for oral suspension (chewable, dispersible tablets) in addition to carbamazepine, phenytoin, phenobarbital, primidone, or other drugs such as rifampin and the protease inhibitors lopinavir/ritonavir and atazanavir/ritonavir that induce lamotrigine glucuronidation [see Drug Interactions (7), Clinical Pharmacology (12.3)], no adjustment to the dose of lamotrigine tablets for oral suspension (chewable, dispersible tablets) should be necessary.
Women and Other Hormonal Contraceptive Preparations or Hormone Replacement Therapy
The effect of other hormonal contraceptive preparations or hormone replacement therapy on the pharmacokinetics of lamotrigine has not been systematically evaluated. It has been reported that ethinylestradiol, not progestogens, increased the clearance of lamotrigine up to 2-fold, and the progestin-only pills had no effect on lamotrigine plasma levels. Therefore, adjustments to the dosage of lamotrigine tablets for oral suspension (chewable, dispersible tablets) in the presence of progestogens alone will likely not be needed.
Patients Taking Atazanavir/Ritonavir
While atazanavir/ritonavir does reduce the lamotrigine plasma concentration, no adjustments to the recommended dose-escalation guidelines for lamotrigine tablets for oral suspension (chewable, dispersible tablets) should be necessary solely based on the use of atazanavir/ritonavir. Dose escalation should follow the recommended guidelines for initiating adjunctive therapy with lamotrigine tablets for oral suspension (chewable, dispersible tablets) based on concomitant AED or other concomitant medications (see Tables 1, 2, and 5). In patients already taking maintenance doses of lamotrigine tablets for oral suspension (chewable, dispersible tablets) and not taking glucuronidation inducers, the dose of lamotrigine tablets for oral suspension (chewable, dispersible tablets) may need to be increased if atazanavir/ritonavir is added or decreased if atazanavir/ritonavir is discontinued [see Clinical Pharmacology(12.3)].
Patients with Hepatic Impairment
Experience in patients with hepatic impairment is limited. Based on a clinical pharmacology study in 24 subjects with mild, moderate, and severe liver impairment [see Use in Specific Populations (8.6), Clinical Pharmacology (12.3)], the following general recommendations can be made. No dosage adjustment is needed in patients with mild liver impairment. Initial, escalation, and maintenance doses should generally be reduced by approximately 25% in patients with moderate and severe liver impairment without ascites and 50% in patients with severe liver impairment with ascites. Escalation and maintenance doses may be adjusted according to clinical response.
Patients with Renal Impairment
Initial doses of lamotrigine tablets for oral suspension (chewable, dispersible tablets) should be based on patients’ concomitant medications (see Tables 1 to 3 and 5); reduced maintenance doses may be effective for patients with significant renal impairment [see Use in Specific Populations (8.7), Clinical Pharmacology (12.3)]. Few patients with severe renal impairment have been evaluated during chronic treatment with lamotrigine tablets for oral suspension (chewable, dispersible tablets). Because there is inadequate experience in this population, lamotrigine tablets for oral suspension (chewable, dispersible tablets) should be used with caution in these patients.
Discontinuation Strategy
Epilepsy: For patients receiving lamotrigine tablets for oral suspension (chewable, dispersible tablets) in combination with other AEDs, a re-evaluation of all AEDs in the regimen should be considered if a change in seizure control or an appearance or worsening of adverse reactions is observed.
If a decision is made to discontinue therapy with lamotrigine tablets for oral suspension (chewable, dispersible tablets), a step-wise reduction of dose over at least 2 weeks (approximately 50% per week) is recommended unless safety concerns require a more rapid withdrawal [see Warnings and Precautions (5.9)].
Discontinuing carbamazepine, phenytoin, phenobarbital, primidone, or other drugs such as rifampin and the protease inhibitors lopinavir/ritonavir and atazanavir/ritonavir that induce lamotrigine glucuronidation should prolong the half-life of lamotrigine; discontinuing valproate should shorten the half-life of lamotrigine.
Bipolar Disorder: In the controlled clinical trials, there was no increase in the incidence, type, or severity of adverse reactions following abrupt termination of lamotrigine tablets for oral suspension (chewable, dispersible tablets). In the clinical development program in adults with bipolar disorder, 2 patients experienced seizures shortly after abrupt withdrawal of lamotrigine tablets for oral suspension (chewable, dispersible tablets). Discontinuation of lamotrigine tablets for oral suspension (chewable, dispersible tablets) should involve a step-wise reduction of dose over at least 2 weeks (approximately 50% per week) unless safety concerns require a more rapid withdrawal [see Warnings and Precautions (5.9)].
2.2 Epilepsy-Adjunctive Therapy
This section provides specific dosing recommendations for patients older than 12 years and patients aged 2 to 12 years. Within each of these age-groups, specific dosing recommendations are provided depending upon concomitant AEDs or other concomitant medications (see Table 1 for patients older than 12 years and Table 2 for patients aged 2 to 12 years). A weight-based dosing guide for patients aged 2 to 12 years on concomitant valproate is provided in Table 3.
Patients Older than 12 Years
Recommended dosing guidelines are summarized in Table 1.
Table 1. Escalation Regimen for Lamotrigine Tablets For Oral Suspension (Chewable, Dispersible Tablets) in Patients Older than 12 Years with Epilepsy In Patients
TAKING Valproatea
In Patients
NOT TAKING
Carbamazepine, Phenytoin,
Phenobarbital, Primidone,b or
Valproatea
In Patients TAKING Carbamazepine,
Phenytoin,
Phenobarbital, or Primidoneb and NOT TAKING Valproatea
Weeks 1 and 2
25 mg every other day
25 mg every day
50 mg/day
Weeks 3 and 4
25 mg every day
50 mg/day
100 mg/day
(in 2 divided doses)
Week 5 onward to maintenance
Increase by
25 to 50 mg/day
every 1 to 2 weeks.
Increase by
50 mg/day
every 1 to 2 weeks.
Increase by
100 mg/day
every 1 to 2 weeks.
Usual maintenance dose
100 to 200 mg/day with valproate alone
100 to 400 mg/day with valproate and other drugs that induce glucuronidation
(in 1 or 2 divided doses)
225 to 375 mg/day
(in 2 divided doses)
300 to 500 mg/day
(in 2 divided doses)
- a Valproate has been shown to inhibit glucuronidation and decrease the apparent clearance of lamotrigine [see Drug Interactions (7), Clinical Pharmacology (12.3)].
- b Drugs that induce lamotrigine glucuronidation and increase clearance, other than the specified antiepileptic drugs, include estrogen-containing oral contraceptives, rifampin, and the protease inhibitors lopinavir/ritonavir and atazanavir/ritonavir. Dosing recommendations for oral contraceptives and the protease inhibitor atazanavir/ritonavir can be found in General Dosing Considerations [see Dosage and Administration (2.1)]. Patients on rifampin and the protease inhibitor lopinavir/ritonavir should follow the same dosing titration/maintenance regimen used with antiepileptic drugs that induce glucuronidation and increase clearance [see Dosage and Administration (2.1), Drug Interactions (7), Clinical Pharmacology (12.3)].
Patients Aged 2 to 12 Years
Recommended dosing guidelines are summarized in Table 2.
Lower starting doses and slower dose escalations than those used in clinical trials are recommended because of the suggestion that the risk of rash may be decreased by lower starting doses and slower dose escalations. Therefore, maintenance doses will take longer to reach in clinical practice than in clinical trials. It may take several weeks to months to achieve an individualized maintenance dose. Maintenance doses in patients weighing ˂30 kg, regardless of age or concomitant AED, may need to be increased as much as 50%, based on clinical response.
The smallest available strength of lamotrigine tablets for oral suspension (chewable, dispersible tablets) is 2 mg, and only whole tablets should be administered. If the calculated dose cannot be achieved using whole tablets, the dose should be rounded down to the nearest whole tablet [see How Supplied/Storage and Handling (16), Medication Guide].
Table 2. Escalation Regimen for Lamotrigine Tablets For Oral Suspension (Chewable, Dispersible Tablets) in Patients Aged 2 to 12 Years with Epilepsy In Patients TAKING Valproatea
In Patients
NOT TAKING Carbamazepine,
Phenytoin,
Phenobarbital,
Primidone,b or
Valproatea
In Patients TAKING Carbamazepine,
Phenytoin,
Phenobarbital, or
Primidoneb and NOT
TAKING Valproatea
Weeks 1 and 2
0.15 mg/kg/day
in 1 or 2 divided doses, rounded down to the nearest whole tablet
(see Table 3 for weight-based dosing guide)
0.3 mg/kg/day
in 1 or 2 divided doses, rounded down to the nearest whole tablet
0.6 mg/kg/day
in 2 divided doses, rounded down to the nearest whole tablet
Weeks 3 and 4
0.3 mg/kg/day
in 1 or 2 divided doses, rounded down to the nearest whole tablet
(see Table 3 for weight-based dosing guide)
0.6 mg/kg/day
in 2 divided doses, rounded down to the nearest whole tablet
1.2 mg/kg/day
in 2 divided doses, rounded down to the nearest whole tablet
Week 5 onward to maintenance
The dose should be increased every 1 to 2 weeks as follows: calculate 0.3 mg/kg/day, round this amount down to the nearest whole tablet, and add this amount to the previously administered daily dose.
The dose should be increased every 1 to 2 weeks as follows: calculate 0.6 mg/kg/day, round this amount down to the nearest whole tablet, and add this amount to the previously administered daily dose.
The dose should be increased every 1 to 2 weeks as follows: calculate 1.2 mg/kg/day, round this amount down to the nearest whole tablet, and add this amount to the previously administered daily dose.
Usual maintenance dose
1 to 5 mg/kg/day
(maximum 200 mg/day in 1 or 2 divided doses)
1 to 3 mg/kg/day with valproate alone
4.5 to 7.5 mg/kg/day
(maximum 300 mg/day in 2 divided doses)
5 to 15 mg/kg/day
(maximum 400 mg/day in 2 divided doses)
Maintenance dose in patients <30 kg
May need to be increased by as much as 50%, based on clinical response.
May need to be increased by as much as 50%, based on clinical response.
May need to be increased by as much as 50%, based on clinical response.
Note: Only whole tablets should be used for dosing.
- a Valproate has been shown to inhibit glucuronidation and decrease the apparent clearance of lamotrigine [see Drug Interactions (7), Clinical Pharmacology (12.3)].
- b Drugs that induce lamotrigine glucuronidation and increase clearance, other than the specified antiepileptic drugs, include estrogen-containing oral contraceptives, rifampin, and the protease inhibitors lopinavir/ritonavir and atazanavir/ritonavir. Dosing recommendations for oral contraceptives and the protease inhibitor atazanavir/ritonavir can be found in General Dosing Considerations [see Dosage and Administration (2.1)]. Patients on rifampin and the protease inhibitor lopinavir/ritonavir should follow the same dosing titration/maintenance regimen used with antiepileptic drugs that induce glucuronidation and increase clearance [see Dosage and Administration (2.1), Drug Interactions (7), Clinical Pharmacology (12.3)].
Table 3. The Initial Weight-Based Dosing Guide for Patients Aged 2 to 12 Years Taking Valproate (Weeks 1 to 4) with Epilepsy If the patient’s weight is
Give this daily dose, using the most appropriate combination of lamotrigine 2- and 5-mg tablets for oral suspension (chewable, dispersible tablets)
Greater than
And less than
Weeks 1 and 2
Weeks 3 and 4
6.7 kg
14 kg
2 mg every other day
2 mg every day
14.1 kg
27 kg
2 mg every day
4 mg every day
27.1 kg
34 kg
4 mg every day
8 mg every day
34.1 kg
40 kg
5 mg every day
10 mg every day
Usual Adjunctive Maintenance Dose for Epilepsy
The usual maintenance doses identified in Tables 1 and 2 are derived from dosing regimens employed in the placebo-controlled adjunctive trials in which the efficacy of lamotrigine tablets for oral suspension (chewable, dispersible tablets) was established. In patients receiving multidrug regimens employing carbamazepine, phenytoin, phenobarbital, or primidone without valproate, maintenance doses of adjunctive lamotrigine tablets for oral suspension (chewable, dispersible tablets) as high as 700 mg/day have been used. In patients receiving valproate alone, maintenance doses of adjunctive lamotrigine tablets for oral suspension (chewable, dispersible tablets) as high as 200 mg/day have been used. The advantage of using doses above those recommended in Tables 1 to 4 has not been established in controlled trials.
2.3 Epilepsy-Conversion from Adjunctive Therapy to Monotherapy
The goal of the transition regimen is to attempt to maintain seizure control while mitigating the risk of serious rash associated with the rapid titration of lamotrigine tablets for oral suspension (chewable, dispersible tablets).
The recommended maintenance dose of lamotrigine tablets for oral suspension (chewable, dispersible tablets) as monotherapy is 500 mg/day given in 2 divided doses.
To avoid an increased risk of rash, the recommended initial dose and subsequent dose escalations for lamotrigine tablets for oral suspension (chewable, dispersible tablets) should not be exceeded [see Boxed Warning].
Conversion from Adjunctive Therapy with Carbamazepine, Phenytoin, Phenobarbital, or Primidone to Monotherapy with Lamotrigine Tablets for oral suspension (Chewable, Dispersible tablets)
After achieving a dose of 500 mg/day of lamotrigine tablets for oral suspension (chewable, dispersible tablets) using the guidelines in Table 1, the concomitant enzyme-inducing AED should be withdrawn by 20% decrements each week over a 4-week period. The regimen for the withdrawal of the concomitant AED is based on experience gained in the controlled monotherapy clinical trial.
Conversion from Adjunctive Therapy with Valproate to Monotherapy with Lamotrigine Tablets For Oral Suspension (Chewable, Dispersible Tablets)
The conversion regimen involves the 4 steps outlined in Table 4.
Table 4. Conversion from Adjunctive Therapy with Valproate to Monotherapy with Lamotrigine Tablets For Oral Suspension (Chewable, Dispersible Tablets) in Patients Aged 16 Years and Older with Epilepsy Lamotrigine Tablets For Oral Suspension (Chewable, Dispersible Tablets)
Valproate
Step 1
Achieve a dose of 200 mg/day according to guidelines in Table 1.
Maintain established stable dose.
Step 2
Maintain at 200 mg/day.
Decrease dose by decrements no greater than 500 mg/day/week to 500 mg/day and then maintain for 1 week.
Step 3
Increase to 300 mg/day and maintain for 1 week.
Simultaneously decrease to 250 mg/day and maintain for 1 week.
Step 4
Increase by 100 mg/day every week to achieve maintenance dose of 500 mg/day.
Discontinue.
Conversion from Adjunctive Therapy with Antiepileptic Drugs other than Carbamazepine, Phenytoin, Phenobarbital, Primidone, or Valproate to Monotherapy with Lamotrigine Tablets for oral suspension (Chewable, Dispersible tablets)
No specific dosing guidelines can be provided for conversion to monotherapy with lamotrigine tablets for oral suspension (chewable, dispersible tablets) with AEDs other than carbamazepine, phenytoin, phenobarbital, primidone, or valproate.
2.4 Bipolar Disorder
The goal of maintenance treatment with lamotrigine tablets for oral suspension (chewable, dispersible tablets) is to delay the time to occurrence of mood episodes (depression, mania, hypomania, mixed episodes) in patients treated for acute mood episodes with standard therapy [see Indications and Usage (1.2)].
Patients taking lamotrigine tablets for oral suspension (chewable, dispersible tablets) for more than 16 weeks should be periodically reassessed to determine the need for maintenance treatment.
Adults
The target dose of lamotrigine tablets for oral suspension (chewable, dispersible tablets) is 200 mg/day (100 mg/day in patients taking valproate, which decreases the apparent clearance of lamotrigine, and 400 mg/day in patients not taking valproate and taking either carbamazepine, phenytoin, phenobarbital, primidone, or other drugs such as rifampin and the protease inhibitor lopinavir/ritonavir that increase the apparent clearance of lamotrigine). In the clinical trials, doses up to 400 mg/day as monotherapy were evaluated; however, no additional benefit was seen at 400 mg/day compared with 200 mg/day [see Clinical Studies (14.2)]. Accordingly, doses above 200 mg/day are not recommended.
Treatment with lamotrigine tablets for oral suspension (chewable, dispersible tablets) are introduced, based on concurrent medications, according to the regimen outlined in Table 5. If other psychotropic medications are withdrawn following stabilization, the dose of lamotrigine tablets for oral suspension (chewable, dispersible tablets) should be adjusted. In patients discontinuing valproate, the dose of lamotrigine tablets for oral suspension (chewable, dispersible tablets) should be doubled over a 2-week period in equal weekly increments (see Table 6). In patients discontinuing carbamazepine, phenytoin, phenobarbital, primidone, or other drugs such as rifampin and the protease inhibitors lopinavir/ritonavir and atazanavir/ritonavir that induce lamotrigine glucuronidation, the dose of lamotrigine tablets for oral suspension (chewable, dispersible tablets) should remain constant for the first week and then should be decreased by half over a 2-week period in equal weekly decrements (see Table 6). The dose of lamotrigine tablets for oral suspension (chewable, dispersible tablets) may then be further adjusted to the target dose (200 mg) as clinically indicated.
If other drugs are subsequently introduced, the dose of lamotrigine tablets for oral suspension (chewable, dispersible tablets) may need to be adjusted. In particular, the introduction of valproate requires reduction in the dose of lamotrigine tablets for oral suspension (chewable, dispersible tablets) [see Drug Interactions (7), Clinical Pharmacology (12.3)].
To avoid an increased risk of rash, the recommended initial dose and subsequent dose escalations of lamotrigine tablets for oral suspension (chewable, dispersible tablets) should not be exceeded [see Boxed Warning].
Table 5. Escalation Regimen for Lamotrigine Tablets For Oral Suspension (Chewable, Dispersible Tablets) in Adults with Bipolar Disorder In Patients TAKING Valproatea
In Patients
NOT TAKING Carbamazepine, Phenytoin, Phenobarbital, Primidone,b or ValproateaIn Patients TAKING Carbamazepine, Phenytoin, Phenobarbital, or Primidoneb and NOT TAKING Valproatea
Weeks 1 and 2
25 mg every other day
25 mg daily
50 mg daily
Weeks 3 and 4
25 mg daily
50 mg daily
100 mg daily,
in divided dosesWeek 5
50 mg daily
100 mg daily
200 mg daily,
in divided dosesWeek 6
100 mg daily
200 mg daily
300 mg daily,
in divided dosesWeek 7
100 mg daily
200 mg daily
up to 400 mg daily,
in divided doses- a Valproate has been shown to inhibit glucuronidation and decrease the apparent clearance of lamotrigine [see Drug Interactions (7), Clinical Pharmacology (12.3)].
- b Drugs that induce lamotrigine glucuronidation and increase clearance, other than the specified antiepileptic drugs, include estrogen-containing oral contraceptives, rifampin, and the protease inhibitors lopinavir/ritonavir and atazanavir/ritonavir. Dosing recommendations for oral contraceptives and the protease inhibitor atazanavir/ritonavir can be found in General Dosing Considerations [see Dosage and Administration (2.1)]. Patients on rifampin and the protease inhibitor lopinavir/ritonavir should follow the same dosing titration/maintenance regimen used with antiepileptic drugs that induce glucuronidation and increase clearance [see Dosage and Administration (2.1), Drug Interactions (7), Clinical Pharmacology (12.3)].
Table 6. Dosage Adjustments to Lamotrigine Tablets For Oral Suspension (Chewable, Dispersible Tablets) in Adults with Bipolar Disorder following Discontinuation of Psychotropic Medications Discontinuation of Psychotropic Drugs (excluding Valproate,a Carbamazepine, Phenytoin, Phenobarbital, or Primidoneb)
After Discontinuation of Valproatea
After Discontinuation of Carbamazepine,
Phenytoin, Phenobarbital,
or Primidoneb
Current Dose of Lamotrigine Tablets For Oral Suspension (Chewable, Dispersible Tablets) (mg/day) 100
Current Dose of Lamotrigine Tablets For Oral Suspension (Chewable, Dispersible Tablets) (mg/day) 400
Week 1
Maintain current dose of lamotrigine tablets for oral suspension (chewable, dispersible tablets)
150
400
Week 2
Maintain current dose of lamotrigine tablets for oral suspension (chewable, dispersible tablets)
200
300
Week 3 onward
Maintain current dose of lamotrigine tablets for oral suspension (chewable, dispersible tablets)
200
200
- a Valproate has been shown to inhibit glucuronidation and decrease the apparent clearance of lamotrigine [see Drug Interactions (7), Clinical Pharmacology (12.3)].
b Drugs that induce lamotrigine glucuronidation and increase clearance, other than the specified antiepileptic drugs, include estrogen-containing oral contraceptives, rifampin, and the protease inhibitors lopinavir/ritonavir and atazanavir/ritonavir. Dosing recommendations for oral contraceptives and the protease inhibitor atazanavir/ritonavir can be found in General Dosing Considerations [see Dosage and Administration (2.1)]. Patients on rifampin and the protease inhibitor lopinavir/ritonavir should follow the same dosing titration/maintenance regimen used with antiepileptic drugs that induce glucuronidation and increase clearance [see Dosage and Administration (2.1), Drug Interactions (7), Clinical Pharmacology (12.3)].
2.5 Administration of Lamotrigine Tablets For Oral Suspension (Chewable, Dispersible Tablets)
Lamotrigine tablets for oral suspension (chewable, dispersible tablets) may be swallowed whole, chewed, or dispersed in water or diluted fruit juice. If the tablets are chewed, consume a small amount of water or diluted fruit juice to aid in swallowing.
To disperse lamotrigine tablets for oral suspension (chewable, dispersible tablets), add the tablets to a small amount of liquid (1 teaspoon, or enough to cover the medication). Approximately 1 minute later, when the tablets are completely dispersed, swirl the solution and consume the entire quantity immediately. No attempt should be made to administer partial quantities of the dispersed tablets.
-
3 DOSAGE FORMS AND STRENGTHS
3.2 Tablets For Oral Suspension (Chewable, Dispersible Tablets)
5 mg: White to off-white, caplet shaped, flat faced beveled edged, uncoated tablets with a score line on one side and “228” debossed on the other side.
25 mg: White to off-white, circular, flat faced beveled edged, uncoated tablets with “G” debossed on one side and “229” debossed on the other side.
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Serious Skin Rashes [see Boxed Warning]
Pediatric Population
The incidence of serious rash associated with hospitalization and discontinuation of lamotrigine in a prospectively followed cohort of pediatric patients (aged 2 to 17 years) is approximately 0.3% to 0.8%. One rash-related death was reported in a prospectively followed cohort of 1,983 pediatric patients (aged 2 to 16 years) with epilepsy taking lamotrigine as adjunctive therapy. Additionally, there have been rare cases of toxic epidermal necrolysis with and without permanent sequelae and/or death in U.S. and foreign postmarketing experience.
There is evidence that the inclusion of valproate in a multidrug regimen increases the risk of serious, potentially life-threatening rash in pediatric patients. In pediatric patients who used valproate concomitantly for epilepsy, 1.2% (6 of 482) experienced a serious rash compared with 0.6% (6 of 952) patients not taking valproate.
Adult Population
Serious rash associated with hospitalization and discontinuation of lamotrigine occurred in 0.3% (11 of 3,348) of adult patients who received lamotrigine in premarketing clinical trials of epilepsy. In the bipolar and other mood disorders clinical trials, the rate of serious rash was 0.08% (1 of 1,233) of adult patients who received lamotrigine as initial monotherapy and 0.13% (2 of 1,538) of adult patients who received lamotrigine as adjunctive therapy. No fatalities occurred among these individuals. However, in worldwide postmarketing experience, rare cases of rash-related death have been reported, but their numbers are too few to permit a precise estimate of the rate.
Among the rashes leading to hospitalization were Stevens-Johnson syndrome, toxic epidermal necrolysis, angioedema, and those associated with multiorgan hypersensitivity [see Warnings and Precautions (5.3)].
There is evidence that the inclusion of valproate in a multidrug regimen increases the risk of serious, potentially life-threatening rash in adults. Specifically, of 584 patients administered lamotrigine with valproate in epilepsy clinical trials, 6 (1%) were hospitalized in association with rash; in contrast, 4 (0.16%) of 2,398 clinical trial patients and volunteers administered lamotrigine in the absence of valproate were hospitalized.
Patients with History of Allergy or Rash to Other Antiepileptic Drugs
The risk of nonserious rash may be increased when the recommended initial dose and/or the rate of dose escalation for lamotrigine is exceeded and in patients with a history of allergy or rash to other AEDs.
5.2 Hemophagocytic Lymphohistiocytosis
Hemophagocytic lymphohistiocytosis (HLH) has occurred in pediatric and adult patients taking lamotrigine for various indications. HLH is a life-threatening syndrome of pathologic immune activation characterized by clinical signs and symptoms of extreme systemic inflammation. It is associated with high mortality rates if not recognized early and treated. Common findings include fever, hepatosplenomegaly, rash, lymphadenopathy, neurologic symptoms, cytopenias, high serum ferritin, hypertriglyceridemia, and liver function and coagulation abnormalities. In cases of HLH reported with lamotrigine, patients have presented with signs of systemic inflammation (fever, rash, hepatosplenomegaly, and organ system dysfunction) and blood dyscrasias. Symptoms have been reported to occur within 8 to 24 days following the initiation of treatment. Patients who develop early manifestations of pathologic immune activation should be evaluated immediately, and a diagnosis of HLH should be considered. Lamotrigine should be discontinued if an alternative etiology for the signs or symptoms cannot be established.
5.3 Multiorgan Hypersensitivity Reactions and Organ Failure
Multiorgan hypersensitivity reactions, also known as drug reaction with eosinophilia and systemic symptoms (DRESS), have occurred with lamotrigine. Some have been fatal or life threatening. DRESS typically, although not exclusively, presents with fever, rash, and/or lymphadenopathy in association with other organ system involvement, such as hepatitis, nephritis, hematologic abnormalities, myocarditis, or myositis, sometimes resembling an acute viral infection. Eosinophilia is often present. This disorder is variable in its expression, and other organ systems not noted here may be involved.
Fatalities associated with acute multiorgan failure and various degrees of hepatic failure have been reported in 2 of 3,796 adult patients and 4 of 2,435 pediatric patients who received lamotrigine in epilepsy clinical trials. Rare fatalities from multiorgan failure have also been reported in postmarketing use.
Isolated liver failure without rash or involvement of other organs has also been reported with lamotrigine.
It is important to note that early manifestations of hypersensitivity (e.g., fever, lymphadenopathy) may be present even though a rash is not evident. If such signs or symptoms are present, the patient should be evaluated immediately. Lamotrigine should be discontinued if an alternative etiology for the signs or symptoms cannot be established.
Prior to initiation of treatment with lamotrigine, the patient should be instructed that a rash or other signs or symptoms of hypersensitivity (e.g., fever, lymphadenopathy) may herald a serious medical event and that the patient should report any such occurrence to a healthcare provider immediately.
5.4 Blood Dyscrasias
There have been reports of blood dyscrasias that may or may not be associated with multiorgan hypersensitivity (also known as DRESS) [see Warnings and Precautions (5.3)]. These have included neutropenia, leukopenia, anemia, thrombocytopenia, pancytopenia, and, rarely, aplastic anemia and pure red cell aplasia.
5.5 Suicidal Behavior and Ideation
AEDs, including lamotrigine, increase the risk of suicidal thoughts or behavior in patients taking these drugs for any indication. Patients treated with any AED for any indication should be monitored for the emergence or worsening of depression, suicidal thoughts or behavior, and/or any unusual changes in mood or behavior.
Pooled analyses of 199 placebo-controlled clinical trials (monotherapy and adjunctive therapy) of 11 different AEDs showed that patients randomized to 1 of the AEDs had approximately twice the risk (adjusted Relative Risk 1.8, 95% CI: 1.2, 2.7) of suicidal thinking or behavior compared with patients randomized to placebo. In these trials, which had a median treatment duration of 12 weeks, the estimated incidence of suicidal behavior or ideation among 27,863 AED-treated patients was 0.43%, compared with 0.24% among 16,029 placebo-treated patients, representing an increase of approximately 1 case of suicidal thinking or behavior for every 530 patients treated. There were 4 suicides in drug-treated patients in the trials and none in placebo-treated patients, but the number of events is too small to allow any conclusion about drug effect on suicide.
The increased risk of suicidal thoughts or behavior with AEDs was observed as early as 1 week after starting treatment with AEDs and persisted for the duration of treatment assessed. Because most trials included in the analysis did not extend beyond 24 weeks, the risk of suicidal thoughts or behavior beyond 24 weeks could not be assessed.
The risk of suicidal thoughts or behavior was generally consistent among drugs in the data analyzed. The finding of increased risk with AEDs of varying mechanism of action and across a range of indications suggests that the risk applies to all AEDs used for any indication. The risk did not vary substantially by age (5 to 100 years) in the clinical trials analyzed.
Table 7 shows absolute and relative risk by indication for all evaluated AEDs.
Table 7. Risk by Indication for Antiepileptic Drugs in the Pooled Analysis Indication
Placebo Patients with Events per 1,000 Patients
Drug Patients with Events per 1,000 Patients
Relative Risk:
Incidence of Events in Drug Patients/Incidence in Placebo Patients
Risk Difference:
Additional Drug Patients with Events per 1,000 Patients
Epilepsy
1
3.4
3.5
2.4
Psychiatric
5.7
8.5
1.5
2.9
Other
1
1.8
1.9
0.9
Total
2.4
4.3
1.8
1.9
The relative risk for suicidal thoughts or behavior was higher in clinical trials for epilepsy than in clinical trials for psychiatric or other conditions, but the absolute risk differences were similar for the epilepsy and psychiatric indications.
Anyone considering prescribing lamotrigine or any other AED must balance the risk of suicidal thoughts or behavior with the risk of untreated illness. Epilepsy and many other illnesses for which AEDs are prescribed are themselves associated with morbidity and mortality and an increased risk of suicidal thoughts and behavior. Should suicidal thoughts and behavior emerge during treatment, the prescriber needs to consider whether the emergence of these symptoms in any given patient may be related to the illness being treated.
Patients, their caregivers, and families should be informed that AEDs increase the risk of suicidal thoughts and behavior and should be advised of the need to be alert for the emergence or worsening of the signs and symptoms of depression, any unusual changes in mood or behavior, the emergence of suicidal thoughts or suicidal behavior, or thoughts about self-harm. Behaviors of concern should be reported immediately to healthcare providers.
5.6 Aseptic Meningitis
Therapy with lamotrigine increases the risk of developing aseptic meningitis. Because of the potential for serious outcomes of untreated meningitis due to other causes, patients should also be evaluated for other causes of meningitis and treated as appropriate.
Postmarketing cases of aseptic meningitis have been reported in pediatric and adult patients taking lamotrigine for various indications. Symptoms upon presentation have included headache, fever, nausea, vomiting, and nuchal rigidity. Rash, photophobia, myalgia, chills, altered consciousness, and somnolence were also noted in some cases. Symptoms have been reported to occur within 1 day to one and a half months following the initiation of treatment. In most cases, symptoms were reported to resolve after discontinuation of lamotrigine. Re-exposure resulted in a rapid return of symptoms (from within 30 minutes to 1 day following re-initiation of treatment) that were frequently more severe. Some of the patients treated with lamotrigine who developed aseptic meningitis had underlying diagnoses of systemic lupus erythematosus or other autoimmune diseases.
Cerebrospinal fluid (CSF) analyzed at the time of clinical presentation in reported cases was characterized by a mild to moderate pleocytosis, normal glucose levels, and mild to moderate increase in protein. CSF white blood cell count differentials showed a predominance of neutrophils in a majority of the cases, although a predominance of lymphocytes was reported in approximately one third of the cases. Some patients also had new onset of signs and symptoms of involvement of other organs (predominantly hepatic and renal involvement), which may suggest that in these cases the aseptic meningitis observed was part of a hypersensitivity reaction [see Warnings and Precautions (5.3)].
5.7 Potential Medication Errors
Medication errors involving lamotrigine have occurred. In particular, the name lamotrigine can be confused with the names of other commonly used medications. Medication errors may also occur between the different formulations of lamotrigine. To reduce the potential of medication errors, write and say lamotrigine clearly. Depictions of the lamotrigine tablets for oral suspension (chewable, dispersible tablets) can be found in the Medication Guide that accompanies the product to highlight the distinctive markings, colors, and shapes that serve to identify the different presentations of the drug and thus may help reduce the risk of medication errors. To avoid the medication error of using the wrong drug or formulation, patients should be strongly advised to visually inspect their tablets to verify that they are lamotrigine tablets for oral suspension (chewable, dispersible tablets), as well as the correct formulation of lamotrigine, each time they fill their prescription.
5.8 Concomitant Use with Oral Contraceptives
Some estrogen-containing oral contraceptives have been shown to decrease serum concentrations of lamotrigine [see Clinical Pharmacology (12.3)]. Dosage adjustments will be necessary in most patients who start or stop estrogen-containing oral contraceptives while taking lamotrigine [see Dosage and Administration (2.1)]. During the week of inactive hormone preparation (pill-free week) of oral contraceptive therapy, plasma lamotrigine levels are expected to rise, as much as doubling at the end of the week. Adverse reactions consistent with elevated levels of lamotrigine, such as dizziness, ataxia, and diplopia, could occur.
5.9 Withdrawal Seizures
As with other AEDs, lamotrigine should not be abruptly discontinued. In patients with epilepsy there is a possibility of increasing seizure frequency. In clinical trials in adults with bipolar disorder, 2 patients experienced seizures shortly after abrupt withdrawal of lamotrigine. Unless safety concerns require a more rapid withdrawal, the dose of lamotrigine should be tapered over a period of at least 2 weeks (approximately 50% reduction per week) [see Dosage and Administration (2.1)].
5.10 Status Epilepticus
Valid estimates of the incidence of treatment-emergent status epilepticus among patients treated with lamotrigine are difficult to obtain because reporters participating in clinical trials did not all employ identical rules for identifying cases. At a minimum, 7 of 2,343 adult patients had episodes that could unequivocally be described as status epilepticus. In addition, a number of reports of variably defined episodes of seizure exacerbation (e.g., seizure clusters, seizure flurries) were made.
5.11 Sudden Unexplained Death in Epilepsy (SUDEP)
During the premarketing development of lamotrigine, 20 sudden and unexplained deaths were recorded among a cohort of 4,700 patients with epilepsy (5,747 patient-years of exposure).
Some of these could represent seizure-related deaths in which the seizure was not observed, e.g., at night. This represents an incidence of 0.0035 deaths per patient-year. Although this rate exceeds that expected in a healthy population matched for age and sex, it is within the range of estimates for the incidence of sudden unexplained death in epilepsy (SUDEP) in patients not receiving lamotrigine (ranging from 0.0005 for the general population of patients with epilepsy, to 0.004 for a recently studied clinical trial population similar to that in the clinical development program for lamotrigine, to 0.005 for patients with refractory epilepsy). Consequently, whether these figures are reassuring or suggest concern depends on the comparability of the populations reported upon with the cohort receiving lamotrigine and the accuracy of the estimates provided. Probably most reassuring is the similarity of estimated SUDEP rates in patients receiving lamotrigine and those receiving other AEDs, chemically unrelated to each other, that underwent clinical testing in similar populations. This evidence suggests, although it certainly does not prove, that the high SUDEP rates reflect population rates, not a drug effect.
5.12 Addition of Lamotrigine to a Multidrug Regimen that Includes Valproate
Because valproate reduces the clearance of lamotrigine, the dosage of lamotrigine in the presence of valproate is less than half of that required in its absence [see Dosage and Administration (2.2, 2.3, 2.4), Drug Interactions (7)].
5.13 Binding in the Eye and Other Melanin-Containing Tissues
Because lamotrigine binds to melanin, it could accumulate in melanin-rich tissues over time. This raises the possibility that lamotrigine may cause toxicity in these tissues after extended use. Although ophthalmological testing was performed in 1 controlled clinical trial, the testing was inadequate to exclude subtle effects or injury occurring after long-term exposure. Moreover, the capacity of available tests to detect potentially adverse consequences, if any, of lamotrigine's binding to melanin is unknown [see Clinical Pharmacology (12.2)].
Accordingly, although there are no specific recommendations for periodic ophthalmological monitoring, prescribers should be aware of the possibility of long-term ophthalmologic effects.
5.14 Laboratory Tests
False-Positive Drug Test Results
Lamotrigine has been reported to interfere with the assay used in some rapid urine drug screens, which can result in false-positive readings, particularly for phencyclidine (PCP). A more specific analytical method should be used to confirm a positive result.
Plasma Concentrations of Lamotrigine
The value of monitoring plasma concentrations of lamotrigine in patients treated with lamotrigine has not been established. Because of the possible pharmacokinetic interactions between lamotrigine and other drugs, including AEDs (see Table 13), monitoring of the plasma levels of lamotrigine and concomitant drugs may be indicated, particularly during dosage adjustments. In general, clinical judgment should be exercised regarding monitoring of plasma levels of lamotrigine and other drugs and whether or not dosage adjustments are necessary.
-
6 ADVERSE REACTIONS
The following serious adverse reactions are described in more detail in the Warnings and Precautions section of the labeling:
- Serious Skin Rashes [see Warnings and Precautions (5.1)]
- Hemophagocytic Lymphohistiocytosis [see Warnings and Precautions (5.2)]
- Multiorgan Hypersensitivity Reactions and Organ Failure [see Warnings and Precautions (5.3)]
- Blood Dyscrasias [see Warnings and Precautions (5.4)]
- Suicidal Behavior and Ideation [see Warnings and Precautions (5.5)]
- Aseptic Meningitis [see Warnings and Precautions (5.6)]
- Withdrawal Seizures [see Warnings and Precautions (5.9)]
- Status Epilepticus [see Warnings and Precautions (5.10)]
- Sudden Unexplained Death in Epilepsy [see Warnings and Precautions (5.11)]
6.1 Clinical Trial Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Epilepsy
Most Common Adverse Reactions in All Clinical Trials: Adjunctive Therapy in Adults with Epilepsy: The most commonly observed (≥5% for lamotrigine and more common on drug than placebo) adverse reactions seen in association with lamotrigine during adjunctive therapy in adults and not seen at an equivalent frequency among placebo-treated patients were: dizziness, ataxia, somnolence, headache, diplopia, blurred vision, nausea, vomiting, and rash. Dizziness, diplopia, ataxia, blurred vision, nausea, and vomiting were dose related. Dizziness, diplopia, ataxia, and blurred vision occurred more commonly in patients receiving carbamazepine with lamotrigine than in patients receiving other AEDs with lamotrigine. Clinical data suggest a higher incidence of rash, including serious rash, in patients receiving concomitant valproate than in patients not receiving valproate [see Warnings and Precautions (5.1)].
Approximately 11% of the 3,378 adult patients who received lamotrigine as adjunctive therapy in premarketing clinical trials discontinued treatment because of an adverse reaction. The adverse reactions most commonly associated with discontinuation were rash (3%), dizziness (2.8%), and headache (2.5%).
In a dose-response trial in adults, the rate of discontinuation of lamotrigine for dizziness, ataxia, diplopia, blurred vision, nausea, and vomiting was dose related.
- Monotherapy in Adults with Epilepsy: The most commonly observed (≥5% for lamotrigine and more common on drug than placebo) adverse reactions seen in association with the use of lamotrigine during the monotherapy phase of the controlled trial in adults not seen at an equivalent rate in the control group were vomiting, coordination abnormality, dyspepsia, nausea, dizziness, rhinitis, anxiety, insomnia, infection, pain, weight decrease, chest pain, and dysmenorrhea. The most commonly observed (≥5% for lamotrigine and more common on drug than placebo) adverse reactions associated with the use of lamotrigine during the conversion to monotherapy (add-on) period, not seen at an equivalent frequency among low-dose valproate-treated patients, were dizziness, headache, nausea, asthenia, coordination abnormality, vomiting, rash, somnolence, diplopia, ataxia, accidental injury, tremor, blurred vision, insomnia, nystagmus, diarrhea, lymphadenopathy, pruritus, and sinusitis.
Approximately 10% of the 420 adult patients who received lamotrigine as monotherapy in premarketing clinical trials discontinued treatment because of an adverse reaction. The adverse reactions most commonly associated with discontinuation were rash (4.5%), headache (3.1%), and asthenia (2.4%).
- Adjunctive Therapy in Pediatric Patients with Epilepsy: The most commonly observed (≥5% for lamotrigine and more common on drug than placebo) adverse reactions seen in association with the use of lamotrigine as adjunctive treatment in pediatric patients aged 2 to 16 years and not seen at an equivalent rate in the control group were infection, vomiting, rash, fever, somnolence, accidental injury, dizziness, diarrhea, abdominal pain, nausea, ataxia, tremor, asthenia, bronchitis, flu syndrome, and diplopia.
In 339 patients aged 2 to 16 years with partial-onset seizures or generalized seizures of Lennox‑Gastaut syndrome, 4.2% of patients on lamotrigine and 2.9% of patients on placebo discontinued due to adverse reactions. The most commonly reported adverse reaction that led to discontinuation of lamotrigine was rash.
Approximately 11.5% of the 1,081 pediatric patients aged 2 to 16 years who received lamotrigine as adjunctive therapy in premarketing clinical trials discontinued treatment because of an adverse reaction. The adverse reactions most commonly associated with discontinuation were rash (4.4%), reaction aggravated (1.7%), and ataxia (0.6%).
- Controlled Adjunctive Clinical Trials in Adults with Epilepsy: Table 8 lists adverse reactions that occurred in adult patients with epilepsy treated with lamotrigine in placebo-controlled trials. In these trials, either lamotrigine or placebo was added to the patient’s current AED therapy.
Table 8. Adverse Reactions in Pooled, Placebo-Controlled Adjunctive Trials in Adult Patients with Epilepsya,b Body System/
Adverse Reaction
Percent of Patients
Receiving Adjunctive
Lamotrigine
(n = 711)
Percent of Patients
Receiving Adjunctive
Placebo
(n = 419)
Body as a whole
Headache
29
19
Flu syndrome
7
6
Fever
6
4
Abdominal pain
5
4
Neck pain
2
1
Reaction aggravated
(seizure exacerbation)
2
1
Digestive
Nausea
19
10
Vomiting
9
4
Diarrhea
6
4
Dyspepsia
5
2
Constipation
4
3
Anorexia
2
1
Musculoskeletal
Arthralgia
2
0
Nervous
Dizziness
38
13
Ataxia
22
6
Somnolence
14
7
Incoordination
6
2
Insomnia
6
2
Tremor
4
1
Depression
4
3
Anxiety
4
3
Convulsion
3
1
Irritability
3
2
Speech disorder
3
0
Concentration disturbance
2
1
Respiratory
Rhinitis
14
9
Pharyngitis
10
9
Cough increased
8
6
Skin and appendages
Rash
10
5
Pruritus
3
2
Special senses
Diplopia
28
7
Blurred vision
16
5
Vision abnormality
3
1
Urogenital
Female patients only
(n = 365)
(n = 207)
Dysmenorrhea
7
6
Vaginitis
4
1
Amenorrhea
2
1
- a Adverse reactions that occurred in at least 2% of patients treated with lamotrigine and at a greater incidence than placebo.
- b Patients in these adjunctive trials were receiving 1 to 3 of the concomitant antiepileptic drugs carbamazepine, phenytoin, phenobarbital, or primidone in addition to lamotrigine or placebo. Patients may have reported multiple adverse reactions during the trial or at discontinuation; thus, patients may be included in more than 1 category.
In a randomized, parallel trial comparing placebo with 300 and 500 mg/day of lamotrigine, some of the more common drug-related adverse reactions were dose related (see Table 9).
Table 9. Dose-Related Adverse Reactions from a Randomized, Placebo-Controlled, Adjunctive Trial in Adults with Epilepsy Adverse Reaction
Percent of Patients Experiencing Adverse Reactions
Placebo
(n = 73)
Lamotrigine
300 mg
(n = 71)
Lamotrigine
500 mg
(n = 72)
Ataxia
10
10
28a,b
Blurred vision
10
11
25a,b
Diplopia
8
24a
49a,b
Dizziness
27
31
54a,b
Nausea
11
18
25a
Vomiting
4
11
18a
a Significantly greater than placebo group (P<0.05).
b Significantly greater than group receiving lamotrigine 300 mg (P<0.05).
The overall adverse reaction profile for lamotrigine was similar between females and males and was independent of age. Because the largest non-Caucasian racial subgroup was only 6% of patients exposed to lamotrigine in placebo-controlled trials, there are insufficient data to support a statement regarding the distribution of adverse reaction reports by race. Generally, females receiving either lamotrigine as adjunctive therapy or placebo were more likely to report adverse reactions than males. The only adverse reaction for which the reports on lamotrigine were >10% more frequent in females than males (without a corresponding difference by gender on placebo) was dizziness (difference = 16.5%). There was little difference between females and males in the rates of discontinuation of lamotrigine for individual adverse reactions.
- Controlled Monotherapy Trial in Adults with Partial-Onset Seizures: Table 10 lists adverse reactions that occurred in patients with epilepsy treated with monotherapy with lamotrigine in a double-blind trial following discontinuation of either concomitant carbamazepine or phenytoin not seen at an equivalent frequency in the control group.
Table 10. Adverse Reactions in a Controlled Monotherapy Trial in Adult Patients with Partial-Onset Seizuresa,b Body System/
Adverse Reaction
Percent of Patients
Receiving Lamotriginec
as Monotherapy
(n = 43)
Percent of Patients
Receiving Low-Dose
Valproated Monotherapy
(n = 44)
Body as a whole
Pain
5
0
Infection
5
2
Chest pain
5
2
Digestive
Vomiting
9
0
Dyspepsia
7
2
Nausea
7
2
Metabolic and nutritional
Weight decrease
5
2
Nervous
Coordination
abnormality
7
0
Dizziness
7
0
Anxiety
5
0
Insomnia
5
2
Respiratory
Rhinitis
7
2
Urogenital (female patients only)
(n = 21)
(n = 28)
Dysmenorrhea
5
0
a Adverse reactions that occurred in at least 5% of patients treated with lamotrigine and at a greater incidence than valproate-treated patients.
- b Patients in this trial were converted to lamotrigine or valproate monotherapy from adjunctive therapy with carbamazepine or phenytoin. Patients may have reported multiple adverse reactions during the trial; thus, patients may be included in more than 1 category.
c Up to 500 mg/day.
d 1,000 mg/day.
Adverse reactions that occurred with a frequency of < 5% and > 2% of patients receiving lamotrigine and numerically more frequent than placebo were:
Body as a Whole: Asthenia, fever.
Digestive: Anorexia, dry mouth, rectal hemorrhage, peptic ulcer.
Metabolic and Nutritional: Peripheral edema.
Nervous System: Amnesia, ataxia, depression, hypesthesia, libido increase, decreased reflexes, increased reflexes, nystagmus, irritability, suicidal ideation.
Respiratory: Epistaxis, bronchitis, dyspnea.
Skin and Appendages: Contact dermatitis, dry skin, sweating.
Special Senses: Vision abnormality.
- Incidence in Controlled Adjunctive Trials in Pediatric Patients with Epilepsy: Table 11 lists adverse reactions that occurred in 339 pediatric patients with partial-onset seizures or generalized seizures of Lennox-Gastaut syndrome who received lamotrigine up to 15 mg/kg/day or a maximum of 750 mg/day.
Table 11. Adverse Reactions in Pooled, Placebo-Controlled, Adjunctive Trials in Pediatric Patients with Epilepsya Body System/Adverse Reaction
Percent of Patients
Receiving Lamotrigine
(n = 168)
Percent of Patients
Receiving Placebo
(n = 171)
Body as a whole
Infection
20
17
Fever
15
14
Accidental injury
14
12
Abdominal pain
10
5
Asthenia
8
4
Flu syndrome
7
6
Pain
5
4
Facial edema
2
1
Photosensitivity
2
0
Cardiovascular
Hemorrhage
2
1
Digestive
Vomiting
20
16
Diarrhea
11
9
Nausea
10
2
Constipation
4
2
Dyspepsia
2
1
Hemic and lymphatic
Lymphadenopathy
2
1
Metabolic and nutritional
Edema
2
0
Nervous system
Somnolence
17
15
Dizziness
14
4
Ataxia
11
3
Tremor
10
1
Emotional lability
4
2
Gait abnormality
4
2
Thinking abnormality
3
2
Convulsions
2
1
Nervousness
2
1
Vertigo
2
1
Respiratory
Pharyngitis
14
11
Bronchitis
7
5
Increased cough
7
6
Sinusitis
2
1
Bronchospasm
2
1
Skin
Rash
14
12
Eczema
2
1
Pruritus
2
1
Special senses
Diplopia
5
1
Blurred vision
4
1
Visual abnormality
2
0
Urogenital
Male and female patients
Urinary tract infection
3
0
- a Adverse reactions that occurred in at least 2% of patients treated with lamotrigine and at a greater incidence than placebo.
Bipolar Disorder in Adults
The most common adverse reactions seen in association with the use of lamotrigine as monotherapy (100 to 400 mg/day) in adult patients (aged 18 to 82 years) with bipolar disorder in the 2 double-blind, placebo-controlled trials of 18 months’ duration are included in Table 12. Adverse reactions that occurred in at least 5% of patients and were numerically more frequent during the dose-escalation phase of lamotrigine in these trials (when patients may have been receiving concomitant medications) compared with the monotherapy phase were: headache (25%), rash (11%), dizziness (10%), diarrhea (8%), dream abnormality (6%), and pruritus (6%).
During the monotherapy phase of the double-blind, placebo-controlled trials of 18 months’ duration, 13% of 227 patients who received lamotrigine (100 to 400 mg/day), 16% of 190 patients who received placebo, and 23% of 166 patients who received lithium discontinued therapy because of an adverse reaction. The adverse reactions that most commonly led to discontinuation of lamotrigine were rash (3%) and mania/hypomania/mixed mood adverse reactions (2%). Approximately 16% of 2,401 patients who received lamotrigine (50 to 500 mg/day) for bipolar disorder in premarketing trials discontinued therapy because of an adverse reaction, most commonly due to rash (5%) and mania/hypomania/mixed mood adverse reactions (2%).
The overall adverse reaction profile for lamotrigine was similar between females and males, between elderly and nonelderly patients, and among racial groups.
Table 12. Adverse Reactions in 2 Placebo-Controlled Trials in Adult Patients with Bipolar I Disordera,b Body System/Adverse Reaction
Percent of Patients
Receiving Lamotrigine
(n = 227)
Percent of Patients
Receiving Placebo
(n = 190)
General
Back pain
8
6
Fatigue
8
5
Abdominal pain
6
3
Digestive
Nausea
14
11
Constipation
5
2
Vomiting
5
2
Nervous System
Insomnia
10
6
Somnolence
9
7
Xerostomia (dry mouth)
6
4
Respiratory
Rhinitis
7
4
Exacerbation of cough
5
3
Pharyngitis
5
4
Skin
Rash (nonserious)c
7
5
- a Adverse reactions that occurred in at least 5% of patients treated with lamotrigine and at a greater incidence than placebo.
- b Patients in these trials were converted to lamotrigine (100 to 400 mg/day) or placebo monotherapy from add-on therapy with other psychotropic medications. Patients may have reported multiple adverse reactions during the trial; thus, patients may be included in more than 1 category.
- c In the overall bipolar and other mood disorders clinical trials, the rate of serious rash was 0.08% (1 of 1,233) of adult patients who received lamotrigine as initial monotherapy and 0.13% (2 of 1,538) of adult patients who received lamotrigine as adjunctive therapy [see Warnings and Precautions (5.1)].
Other reactions that occurred in 5% or more patients but equally or more frequently in the placebo group included: dizziness, mania, headache, infection, influenza, pain, accidental injury, diarrhea, and dyspepsia.
Adverse reactions that occurred with a frequency of < 5% and > 1% of patients receiving lamotrigine and numerically more frequent than placebo were:
General: Fever, neck pain.
Cardiovascular: Migraine.
Digestive: Flatulence.
Metabolic and Nutritional: Weight gain, edema.
Musculoskeletal: Arthralgia, myalgia.
Nervous System: Amnesia, depression, agitation, emotional lability, dyspraxia, abnormal thoughts, dream abnormality, hypoesthesia.
Respiratory: Sinusitis.
Urogenital: Urinary frequency.
Adverse Reactions following Abrupt Discontinuation: In the 2 controlled clinical trials, there was no increase in the incidence, severity, or type of adverse reactions in patients with bipolar disorder after abruptly terminating therapy with lamotrigine. In the clinical development program in adults with bipolar disorder, 2 patients experienced seizures shortly after abrupt withdrawal of lamotrigine [see Warnings and Precautions (5.9)].
Mania/Hypomania/Mixed Episodes: During the double-blind, placebo-controlled clinical trials in bipolar I disorder in which adults were converted to monotherapy with lamotrigine (100 to 400 mg/day) from other psychotropic medications and followed for up to 18 months, the rates of manic or hypomanic or mixed mood episodes reported as adverse reactions were 5% for patients treated with lamotrigine (n = 227), 4% for patients treated with lithium (n = 166), and 7% for patients treated with placebo (n = 190). In all bipolar controlled trials combined, adverse reactions of mania (including hypomania and mixed mood episodes) were reported in 5% of patients treated with lamotrigine (n = 956), 3% of patients treated with lithium (n = 280), and 4% of patients treated with placebo (n = 803).
6.2 Other Adverse Reactions Observed in All Clinical Trials
Lamotrigine has been administered to 6,694 individuals for whom complete adverse reaction data was captured during all clinical trials, only some of which were placebo controlled. During these trials, all adverse reactions were recorded by the clinical investigators using terminology of their own choosing. To provide a meaningful estimate of the proportion of individuals having adverse reactions, similar types of adverse reactions were grouped into a smaller number of standardized categories using modified COSTART dictionary terminology. The frequencies presented represent the proportion of the 6,694 individuals exposed to lamotrigine who experienced an event of the type cited on at least 1 occasion while receiving lamotrigine. All reported adverse reactions are included except those already listed in the previous tables or elsewhere in the labeling, those too general to be informative, and those not reasonably associated with the use of the drug.
Adverse reactions are further classified within body system categories and enumerated in order of decreasing frequency using the following definitions:
frequent adverse reactions are defined as those occurring in at least 1/100 patients;
infrequent adverse reactions are those occurring in 1/100 to 1/1,000 patients;
rare adverse reactions are those occurring in fewer than 1/1,000 patients.
Body as a Whole
Infrequent: Allergic reaction, chills, malaise.
Cardiovascular System
Infrequent: Flushing, hot flashes, hypertension, palpitations, postural hypotension, syncope, tachycardia, vasodilation.
Dermatological
Infrequent: Acne, alopecia, hirsutism, maculopapular rash, skin discoloration, urticaria.
Rare: Angioedema, erythema, exfoliative dermatitis, fungal dermatitis, herpes zoster, leukoderma, multiforme erythema, petechial rash, pustular rash, Stevens-Johnson syndrome, vesiculobullous rash.
Digestive System
Infrequent: Dysphagia, eructation, gastritis, gingivitis, increased appetite, increased salivation, liver function tests abnormal, mouth ulceration.
Rare: Gastrointestinal hemorrhage, glossitis, gum hemorrhage, gum hyperplasia, hematemesis, hemorrhagic colitis, hepatitis, melena, stomach ulcer, stomatitis, tongue edema.
Endocrine System
Rare: Goiter, hypothyroidism.
Hematologic and Lymphatic System
Infrequent: Ecchymosis, leukopenia.
Rare: Anemia, eosinophilia, fibrin decrease, fibrinogen decrease, iron deficiency anemia, leukocytosis, lymphocytosis, macrocytic anemia, petechia, thrombocytopenia.
Metabolic and Nutritional Disorders
Infrequent: Aspartate transaminase increased.
Rare: Alcohol intolerance, alkaline phosphatase increase, alanine transaminase increase, bilirubinemia, general edema, gamma glutamyl transpeptidase increase, hyperglycemia.
Musculoskeletal System
Infrequent: Arthritis, leg cramps, myasthenia, twitching.
Rare: Bursitis, muscle atrophy, pathological fracture, tendinous contracture.
Nervous System
Frequent: Confusion, paresthesia.
Infrequent: Akathisia, apathy, aphasia, central nervous system depression, depersonalization, dysarthria, dyskinesia, euphoria, hallucinations, hostility, hyperkinesia, hypertonia, libido decreased, memory decrease, mind racing, movement disorder, myoclonus, panic attack, paranoid reaction, personality disorder, psychosis, sleep disorder, stupor, suicidal ideation. Rare: Choreoathetosis, delirium, delusions, dysphoria, dystonia, extrapyramidal syndrome, faintness, grand mal convulsions, hemiplegia, hyperalgesia, hyperesthesia, hypokinesia, hypotonia, manic depression reaction, muscle spasm, neuralgia, neurosis, paralysis, peripheral neuritis.
Respiratory System
Infrequent: Yawn.
Rare: Hiccup, hyperventilation.
Special Senses
Frequent: Amblyopia. Infrequent: Abnormality of accommodation, conjunctivitis, dry eyes, ear pain, photophobia, taste perversion, tinnitus.
Rare: Deafness, lacrimation disorder, oscillopsia, parosmia, ptosis, strabismus, taste loss, uveitis, visual field defect.
Urogenital System
Infrequent: Abnormal ejaculation, hematuria, impotence, menorrhagia, polyuria, urinary incontinence.
Rare: Acute kidney failure, anorgasmia, breast abscess, breast neoplasm, creatinine increase, cystitis, dysuria, epididymitis, female lactation, kidney failure, kidney pain, nocturia, urinary retention, urinary urgency.
6.3 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of for oral suspension (chewable, dispersible tablets). Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Blood and Lymphatic
Agranulocytosis, hemolytic anemia, lymphadenopathy not associated with hypersensitivity disorder.
Gastrointestinal
Esophagitis.
Hepatobiliary Tract and Pancreas
Pancreatitis.
Immunologic
Hypogammaglobulinemia, Lupus-like reaction, vasculitis.
Lower Respiratory
Apnea.
Musculoskeletal
Rhabdomyolysis has been observed in patients experiencing hypersensitivity reactions.
Nervous System
Aggression, exacerbation of Parkinsonian symptoms in patients with pre-existing Parkinson’s disease, tics.
Non-site Specific
Progressive immunosuppression.
-
7 DRUG INTERACTIONS
Significant drug interactions with lamotrigine are summarized in this section.
Uridine 5´-diphospho-glucuronyl transferases (UGT) have been identified as the enzymes responsible for metabolism of lamotrigine. Drugs that induce or inhibit glucuronidation may, therefore, affect the apparent clearance of lamotrigine. Strong or moderate inducers of the cytochrome P450 3A4 (CYP3A4) enzyme, which are also known to induce UGT, may also enhance the metabolism of lamotrigine.
Those drugs that have been demonstrated to have a clinically significant impact on lamotrigine metabolism are outlined in Table 13. Specific dosing guidance for these drugs is provided in the Dosage and Administration section [see Dosage and Administration (2.1)].
Additional details of these drug interaction studies are provided in the Clinical Pharmacology section [see Clinical Pharmacology (12.3)].
Table 13. Established and Other Potentially Significant Drug Interactions
Concomitant Drug
Effect on Concentration of Lamotrigine or Concomitant Drug
Clinical Comment
Estrogen-containing oral contraceptive preparations containing 30 mcg ethinylestradiol and 150 mcg levonorgestrel
↓ lamotrigine
↓ levonorgestrel
Decreased lamotrigine concentrations approximately 50%.
Decrease in levonorgestrel component by 19%.
Carbamazepine and carbamazepine epoxide
↓ lamotrigine
? carbamazepine epoxide
Addition of carbamazepine decreases lamotrigine concentration approximately 40%.
May increase carbamazepine epoxide levels.
Lopinavir/ritonavir
↓ lamotrigine
Decreased lamotrigine concentration approximately 50%.
Atazanavir/ritonavir
↓ lamotrigine
Decreased lamotrigine AUC approximately 32%.
Phenobarbital/primidone
↓ lamotrigine
Decreased lamotrigine concentration approximately 40%.
Phenytoin
↓ lamotrigine
Decreased lamotrigine concentration approximately 40%.
Rifampin
↓ lamotrigine
Decreased lamotrigine AUC approximately 40%.
Valproate
↑ lamotrigine
? valproate
Increased lamotrigine concentrations slightly more than 2-fold.
There are conflicting study results regarding effect of lamotrigine on valproate concentrations: 1) a mean 25% decrease in valproate concentrations in healthy volunteers, 2) no change in valproate concentrations in controlled clinical trials in patients with epilepsy.
↓ = Decreased (induces lamotrigine glucuronidation).
↑ = Increased (inhibits lamotrigine glucuronidation).
? = Conflicting data.
Effect of Lamotrigine on Organic Cationic Transporter 2 Substrates
Lamotrigine is an inhibitor of renal tubular secretion via organic cationic transporter 2 (OCT2) proteins [see Clinical Pharmacology (12.3)]. This may result in increased plasma levels of certain drugs that are substantially excreted via this route. Coadministration of lamotrigine with OCT2 substrates with a narrow therapeutic index (e.g., dofetilide) is not recommended.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to AEDs, including lamotrigine, during pregnancy. Encourage women who are taking lamotrigine during pregnancy to enroll in the North American Antiepileptic Drug (NAAED) Pregnancy Registry by calling 1-888-233-2334 or visiting http://www.aedpregnancyregistry.org/.
Risk Summary
Data from several prospective pregnancy exposure registries and epidemiological studies of pregnant women have not detected an increased frequency of major congenital malformations or a consistent pattern of malformations among women exposed to lamotrigine compared with the general population (see Data). The majority of lamotrigine pregnancy exposure data are from women with epilepsy. In animal studies, administration of lamotrigine during pregnancy resulted in developmental toxicity (increased mortality, decreased body weight, increased structural variation, neurobehavioral abnormalities) at doses lower than those administered clinically.
Lamotrigine decreased fetal folate concentrations in rats, an effect known to be associated with adverse pregnancy outcomes in animals and humans (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Clinical Considerations
As with other AEDs, physiological changes during pregnancy may affect lamotrigine concentrations and/or therapeutic effect. There have been reports of decreased lamotrigine concentrations during pregnancy and restoration of pre-pregnancy concentrations after delivery. Dose adjustments may be necessary to maintain clinical response.
Data
Human Data: Data from several international pregnancy registries have not shown an increased risk for malformations overall. The International Lamotrigine Pregnancy Registry reported major congenital malformations in 2.2% (95% CI: 1.6%, 3.1%) of 1,558 infants exposed to lamotrigine monotherapy in the first trimester of pregnancy. The NAAED Pregnancy Registry reported major congenital malformations among 2.0% of 1,562 infants exposed to lamotrigine monotherapy in the first trimester. EURAP, a large international pregnancy registry focused outside of North America, reported major birth defects in 2.9% (95% CI: 2.3%, 3.7%) of 2,514 exposures to lamotrigine monotherapy in the first trimester. The frequency of major congenital malformations was similar to estimates from the general population.
The NAAED Pregnancy Registry observed an increased risk of isolated oral clefts: among 2,200 infants exposed to lamotrigine early in pregnancy, the risk of oral clefts was 3.2 per 1,000 (95% CI: 1.4, 6.3), a 3-fold increased risk versus unexposed healthy controls. This finding has not been observed in other large international pregnancy registries. Furthermore, a case-control study based on 21 congenital anomaly registries covering over 10 million births in Europe reported an adjusted odds ratio for isolated oral clefts with lamotrigine exposure of 1.45 (95% CI: 0.8, 2.63).
Several meta-analyses have not reported an increased risk of major congenital malformations following lamotrigine exposure in pregnancy compared with healthy and disease-matched controls. No patterns of specific malformation types were observed.
The same meta-analyses evaluated the risk of additional maternal and infant outcomes including fetal death, stillbirth, preterm birth, small for gestational age, and neurodevelopmental delay. Although there are no data suggesting an increased risk of these outcomes with lamotrigine monotherapy exposure, differences in outcome definition, ascertainment methods, and comparator groups limit the conclusions that can be drawn.
Animal Data: When lamotrigine was administered to pregnant mice, rats, or rabbits during the period of organogenesis (oral doses of up to 125, 25, and 30 mg/kg, respectively), reduced fetal body weight and increased incidences of fetal skeletal variations were seen in mice and rats at doses that were also maternally toxic. The no-effect doses for embryofetal developmental toxicity in mice, rats, and rabbits (75, 6.25, and 30 mg/kg, respectively) are similar to (mice and rabbits) or less than (rats) the human dose of 400 mg/day on a body surface area (mg/m2) basis.
In a study in which pregnant rats were administered lamotrigine (oral doses of 0, 5, or 25 mg/kg) during the period of organogenesis and offspring were evaluated postnatally, neurobehavioral abnormalities were observed in exposed offspring at both doses. The lowest effect dose for developmental neurotoxicity in rats is less than the human dose of 400 mg/day on a mg/m2 basis. Maternal toxicity was observed at the higher dose tested.
When pregnant rats were administered lamotrigine (oral doses of 0, 5, 10, or 20 mg/kg) during the latter part of gestation and throughout lactation, increased offspring mortality (including stillbirths) was seen at all doses. The lowest effect dose for pre- and post-natal developmental toxicity in rats is less than the human dose of 400 mg/day on a mg/m2 basis. Maternal toxicity was observed at the 2 highest doses tested.
When administered to pregnant rats, lamotrigine decreased fetal folate concentrations at doses greater than or equal to 5 mg/kg/day, which is less than the human dose of 400 mg/day on a mg/m2 basis.
8.2 Lactation
Risk Summary
Lamotrigine is present in milk from lactating women taking lamotrigine tablets (see Data). Neonates and young infants are at risk for high serum levels because maternal serum and milk levels can rise to high levels postpartum if lamotrigine dosage has been increased during pregnancy but is not reduced after delivery to the pre-pregnancy dosage. Glucuronidation is required for drug clearance. Glucuronidation capacity is immature in the infant and this may also contribute to the level of lamotrigine exposure. Events including rash, apnea, drowsiness, poor sucking, and poor weight gain (requiring hospitalization in some cases) have been reported in infants who have been human milk-fed by mothers using lamotrigine; whether or not these events were caused by lamotrigine is unknown. No data are available on the effects of the drug on milk production.
The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for lamotrigine and any potential adverse effects on the breastfed infant from lamotrigine or from the underlying maternal condition.
Clinical Considerations
Human milk-fed infants should be closely monitored for adverse events resulting from lamotrigine. Measurement of infant serum levels should be performed to rule out toxicity if concerns arise. Human milk-feeding should be discontinued in infants with lamotrigine toxicity.
Data
Data from multiple small studies indicate that lamotrigine plasma levels in nursing infants have been reported to be as high as 50% of maternal plasma concentrations.
8.4 Pediatric Use
Epilepsy
Lamotrigine is indicated as adjunctive therapy in patients aged 2 years and older for partial-onset seizures, the generalized seizures of Lennox-Gastaut syndrome, and PGTC seizures.
Safety and efficacy of lamotrigine used as adjunctive treatment for partial-onset seizures were not demonstrated in a small, randomized, double-blind, placebo-controlled withdrawal trial in very young pediatric patients (aged 1 to 24 months). Lamotrigine was associated with an increased risk for infectious adverse reactions (lamotrigine 37%, placebo 5%), and respiratory adverse reactions (lamotrigine 26%, placebo 5%). Infectious adverse reactions included bronchiolitis, bronchitis, ear infection, eye infection, otitis externa, pharyngitis, urinary tract infection, and viral infection. Respiratory adverse reactions included nasal congestion, cough, and apnea.
Bipolar Disorder
Safety and efficacy of lamotrigine for the maintenance treatment of bipolar disorder were not established in a double-blind, randomized withdrawal, placebo-controlled trial that evaluated 301 pediatric patients aged 10 to 17 years with a current manic/hypomanic, depressed, or mixed mood episode as defined by DSM-IV-TR. In the randomized phase of the trial, adverse reactions that occurred in at least 5% of patients taking lamotrigine (n = 87) and were twice as common compared with patients taking placebo (n = 86) were influenza (lamotrigine 8%, placebo 2%), oropharyngeal pain (lamotrigine 8%, placebo 2%), vomiting (lamotrigine 6%, placebo 2%), contact dermatitis (lamotrigine 5%, placebo 2%), upper abdominal pain (lamotrigine 5%, placebo 1%), and suicidal ideation (lamotrigine 5%, placebo 0%).
Juvenile Animal Data
In a juvenile animal study in which lamotrigine (oral doses of 0, 5, 15, or 30 mg/kg) was administered to young rats from postnatal day 7 to 62, decreased viability and growth were seen at the highest dose tested and long-term neurobehavioral abnormalities (decreased locomotor activity, increased reactivity, and learning deficits in animals tested as adults) were observed at the 2 highest doses. The no-effect dose for adverse developmental effects in juvenile animals is less than the human dose of 400 mg/day on a mg/m2 basis.
8.5 Geriatric Use
Clinical trials of lamotrigine for epilepsy and bipolar disorder did not include sufficient numbers of patients aged 65 years and older to determine whether they respond differently from younger patients or exhibit a different safety profile than that of younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function and of concomitant disease or other drug therapy.
8.6 Hepatic Impairment
Experience in patients with hepatic impairment is limited. Based on a clinical pharmacology study in 24 subjects with mild, moderate, and severe liver impairment [see Clinical Pharmacology (12.3)], the following general recommendations can be made. No dosage adjustment is needed in patients with mild liver impairment. Initial, escalation, and maintenance doses should generally be reduced by approximately 25% in patients with moderate and severe liver impairment without ascites and 50% in patients with severe liver impairment with ascites. Escalation and maintenance doses may be adjusted according to clinical response [see Dosage and Administration (2.1)].
8.7 Renal Impairment
Lamotrigine is metabolized mainly by glucuronic acid conjugation, with the majority of the metabolites being recovered in the urine. In a small study comparing a single dose of lamotrigine in subjects with varying degrees of renal impairment with healthy volunteers, the plasma half-life of lamotrigine was approximately twice as long in the subjects with chronic renal failure [see Clinical Pharmacology (12.3)].
Initial doses of lamotrigine should be based on patients’ AED regimens; reduced maintenance doses may be effective for patients with significant renal impairment. Few patients with severe renal impairment have been evaluated during chronic treatment with lamotrigine. Because there is inadequate experience in this population, lamotrigine should be used with caution in these patients [see Dosage and Administration (2.1)].
-
10 OVERDOSAGE
10.1 Human Overdose Experience
Overdoses involving quantities up to 15 g have been reported for lamotrigine, some of which have been fatal. Overdose has resulted in ataxia, nystagmus, seizures (including tonic-clonic seizures), decreased level of consciousness, coma, and intraventricular conduction delay.
10.2 Management of Overdose
There are no specific antidotes for lamotrigine. Following a suspected overdose, hospitalization of the patient is advised. General supportive care is indicated, including frequent monitoring of vital signs and close observation of the patient. If indicated, emesis should be induced; usual precautions should be taken to protect the airway. It should be kept in mind that immediate-release lamotrigine is rapidly absorbed [see Clinical Pharmacology (12.3)]. It is uncertain whether hemodialysis is an effective means of removing lamotrigine from the blood. In 6 renal failure patients, about 20% of the amount of lamotrigine in the body was removed by hemodialysis during a 4-hour session. A Poison Control Center should be contacted for information on the management of overdosage of lamotrigine.
-
11 DESCRIPTION
Lamotrigine, USP, an AED of the phenyltriazine class, is chemically unrelated to existing AEDs. Lamotrigine’s chemical name is 3,5-diamino-6-(2,3-dichlorophenyl)-as-triazine, its molecular formula is C9H7N5Cl2, and its molecular weight is 256.09 g/mol. Lamotrigine, USP is a white powder and has a pKa of 5.7. Lamotrigine, USP is slightly soluble in 0.1 N hydrochloric acid, in acetone, in methanol, and in water. The structural formula is:
Lamotrigine Tablets for oral suspension (Chewable, Dispersible Tablets) 5 mg and 25 mg are supplied for oral administration. The tablets contain 5 mg (white to off-white) or 25 mg (white to off-white) of lamotrigine, USP and the following inactive ingredients: aspartame, colloidal silicon dioxide, magnesium stearate, microcrystalline cellulose, povidone K-25, sodium starch glycolate and strawberry guarana 586997 AP 0551.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
The precise mechanism(s) by which lamotrigine exerts its anticonvulsant action are unknown. In animal models designed to detect anticonvulsant activity, lamotrigine was effective in preventing seizure spread in the maximum electroshock (MES) and pentylenetetrazol (scMet) tests, and prevented seizures in the visually and electrically evoked after-discharge (EEAD) tests for antiepileptic activity. Lamotrigine also displayed inhibitory properties in the kindling model in rats both during kindling development and in the fully kindled state. The relevance of these models to human epilepsy, however, is not known.
One proposed mechanism of action of lamotrigine, the relevance of which remains to be established in humans, involves an effect on sodium channels. In vitro pharmacological studies suggest that lamotrigine inhibits voltage-sensitive sodium channels, thereby stabilizing neuronal membranes and consequently modulating presynaptic transmitter release of excitatory amino acids (e.g., glutamate and aspartate).
Effect of Lamotrigine on N-Methyl d-Aspartate-Receptor-Mediated Activity
Lamotrigine did not inhibit N-methyl d-aspartate (NMDA)-induced depolarizations in rat cortical slices or NMDA-induced cyclic GMP formation in immature rat cerebellum, nor did lamotrigine displace compounds that are either competitive or noncompetitive ligands at this glutamate receptor complex (CNQX, CGS, TCHP). The IC50 for lamotrigine effects on NMDA-induced currents (in the presence of 3 mcM of glycine) in cultured hippocampal neurons exceeded 100 mcM.
The mechanisms by which lamotrigine exerts its therapeutic action in bipolar disorder have not been established.
12.2 Pharmacodynamics
Folate Metabolism
In vitro, lamotrigine inhibited dihydrofolate reductase, the enzyme that catalyzes the reduction of dihydrofolate to tetrahydrofolate. Inhibition of this enzyme may interfere with the biosynthesis of nucleic acids and proteins. When oral daily doses of lamotrigine were given to pregnant rats during organogenesis, fetal, placental, and maternal folate concentrations were reduced. Significantly reduced concentrations of folate are associated with teratogenesis [see Use in Specific Populations (8.1)]. Folate concentrations were also reduced in male rats given repeated oral doses of lamotrigine. Reduced concentrations were partially returned to normal when supplemented with folinic acid.
Accumulation in Kidneys
Lamotrigine accumulated in the kidney of the male rat, causing chronic progressive nephrosis, necrosis, and mineralization. These findings are attributed to α-2 microglobulin, a species- and sex-specific protein that has not been detected in humans or other animal species.
Melanin Binding
Lamotrigine binds to melanin-containing tissues, e.g., in the eye and pigmented skin. It has been found in the uveal tract up to 52 weeks after a single dose in rodents.
Cardiovascular
In dogs, lamotrigine is extensively metabolized to a 2-N-methyl metabolite. This metabolite causes dose-dependent prolongation of the PR interval, widening of the QRS complex, and, at higher doses, complete AV conduction block. Similar cardiovascular effects are not anticipated in humans because only trace amounts of the 2-N-methyl metabolite (<0.6% of lamotrigine dose) have been found in human urine [see Clinical Pharmacology (12.3)]. However, it is conceivable that plasma concentrations of this metabolite could be increased in patients with a reduced capacity to glucuronidate lamotrigine (e.g., in patients with liver disease, patients taking concomitant medications that inhibit glucuronidation).
12.3 Pharmacokinetics
The pharmacokinetics of lamotrigine have been studied in subjects with epilepsy, healthy young and elderly volunteers, and volunteers with chronic renal failure. Lamotrigine pharmacokinetic parameters for adult and pediatric subjects and healthy normal volunteers are summarized in Tables 14 and 16.
Table 14. Mean Pharmacokinetic Parametersa in Healthy Volunteers and Adult Subjects with Epilepsy Adult Study Population
Number of Subjects
Tmax: Time of Maximum Plasma Concentration (h)
t1/2: Elimination Half-life (h)
CL/F: Apparent Plasma Clearance (mL/min/kg)
Healthy volunteers taking no other medications:
- Single-dose lamotrigine
- Multiple-dose lamotrigine
179
36
2.2
(0.25 to 12)
1.7
(0.5 to 4)
32.8
(14 to 103)
25.4
(11.6 to 61.6)
0.44
(0.12 to 1.1)
0.58
(0.24 to 1.15)
Healthy volunteers taking valproate:
- Single-dose lamotrigine
- Multiple-dose lamotrigine
6
18
1.8
(1 to 4)
1.9
(0.5 to 3.5)
48.3
(31.5 to 88.6)
70.3
(41.9 to 113.5)
0.3
(0.14 to 0.42)
0.18
(0.12 to 0.33)
Subjects with epilepsy taking valproate only:
- Single-dose lamotrigine
4
4.8
(1.8 to 8.4)
58.8
(30.5 to 88.8)
0.28
(0.16 to 0.4)
Subjects with epilepsy taking carbamazepine, phenytoin, phenobarbital, or primidoneb plus valproate:
- Single-dose lamotrigine
25
3.8
(1 to 10)
27.2
(11.2 to 51.6)
0.53
(0.27 to 1.04)
Subjects with epilepsy taking carbamazepine, phenytoin, phenobarbital, or primidone:b
- Single-dose lamotrigine
- Multiple-dose lamotrigine
24
17
2.3
(0.5 to 5)
2
(0.75 to 5.93)
14.4
(6.4 to 30.4)
12.6
(7.5 to 23.1)
1.1
(0.51 to 2.22)
1.21
(0.66 to 1.82)
- a The majority of parameter means determined in each study had coefficients of variation between 20% and 40% for half-life and CL/F and between 30% and 70% for Tmax. The overall mean values were calculated from individual study means that were weighted based on the number of volunteers/subjects in each study. The numbers in parentheses below each parameter mean represent the range of individual volunteer/subject values across studies.
- b Carbamazepine, phenytoin, phenobarbital, and primidone have been shown to increase the apparent clearance of lamotrigine. Estrogen-containing oral contraceptives and other drugs, such as rifampin and protease inhibitors lopinavir/ritonavir and atazanavir/ritonavir, that induce lamotrigine glucuronidation have also been shown to increase the apparent clearance of lamotrigine [see Drug Interactions (7)].
Absorption
Lamotrigine is rapidly and completely absorbed after oral administration with negligible first-pass metabolism (absolute bioavailability is 98%). The bioavailability is not affected by food. Peak plasma concentrations occur anywhere from 1.4 to 4.8 hours following drug administration. The lamotrigine tablets for oral suspension (chewable, dispersible tablets) were found to be equivalent, whether administered as dispersed in water, chewed and swallowed, or swallowed whole, to the lamotrigine compressed tablets in terms of rate and extent of absorption. In terms of rate and extent of absorption, lamotrigine orally disintegrating tablets, whether disintegrated in the mouth or swallowed whole with water, were equivalent to the lamotrigine compressed tablets swallowed with water.
Dose Proportionality
In healthy volunteers not receiving any other medications and given single doses, the plasma concentrations of lamotrigine increased in direct proportion to the dose administered over the range of 50 to 400 mg. In 2 small studies (n = 7 and 8) of patients with epilepsy who were maintained on other AEDs, there also was a linear relationship between dose and lamotrigine plasma concentrations at steady state following doses of 50 to 350 mg twice daily.
Distribution
Estimates of the mean apparent volume of distribution (Vd/F) of lamotrigine following oral administration ranged from 0.9 to 1.3 L/kg. Vd/F is independent of dose and is similar following single and multiple doses in both patients with epilepsy and in healthy volunteers.
Protein Binding
Data from in vitro studies indicate that lamotrigine is approximately 55% bound to human plasma proteins at plasma lamotrigine concentrations from 1 to 10 mcg/mL (10 mcg/mL is 4 to 6 times the trough plasma concentration observed in the controlled efficacy trials). Because lamotrigine is not highly bound to plasma proteins, clinically significant interactions with other drugs through competition for protein binding sites are unlikely. The binding of lamotrigine to plasma proteins did not change in the presence of therapeutic concentrations of phenytoin, phenobarbital, or valproate. Lamotrigine did not displace other AEDs (carbamazepine, phenytoin, phenobarbital) from protein-binding sites.
Metabolism
Lamotrigine is metabolized predominantly by glucuronic acid conjugation; the major metabolite is an inactive 2-N-glucuronide conjugate. After oral administration of 240 mg of 14C-lamotrigine (15 μCi) to 6 healthy volunteers, 94% was recovered in the urine and 2% was recovered in the feces. The radioactivity in the urine consisted of unchanged lamotrigine (10%), the 2-N-glucuronide (76%), a 5-N-glucuronide (10%), a 2-N-methyl metabolite (0.14%), and other unidentified minor metabolites (4%).
Enzyme Induction
The effects of lamotrigine on the induction of specific families of mixed-function oxidase isozymes have not been systematically evaluated.
Following multiple administrations (150 mg twice daily) to normal volunteers taking no other medications, lamotrigine induced its own metabolism, resulting in a 25% decrease in t½ and a 37% increase in CL/F at steady state compared with values obtained in the same volunteers following a single dose. Evidence gathered from other sources suggests that self-induction by lamotrigine may not occur when lamotrigine is given as adjunctive therapy in patients receiving enzyme-inducing drugs such as carbamazepine, phenytoin, phenobarbital, primidone, or other drugs such as rifampin and the protease inhibitors lopinavir/ritonavir and atazanavir/ritonavir that induce lamotrigine glucuronidation [see Drug Interactions (7)].
Elimination
The elimination half-life and apparent clearance of lamotrigine following oral administration of lamotrigine to adult subjects with epilepsy and healthy volunteers is summarized in Table 14. Half-life and apparent oral clearance vary depending on concomitant AEDs.
Drug Interactions
The apparent clearance of lamotrigine is affected by the coadministration of certain medications [see Warnings and Precautions (5.8, 5.12), Drug Interactions (7)].
The net effects of drug interactions with lamotrigine are summarized in Tables 13 and 15, followed by details of the drug interaction studies below.
Table 15. Summary of Drug Interactions with Lamotrigine Drug
Drug Plasma Concentration with Adjunctive Lamotriginea
Lamotrigine Plasma Concentration with Adjunctive Drugsb
Oral contraceptives (e.g., ethinylestradiol/levonorgestrel)c
↔d
↓
Aripiprazole
Not assessed
↔e
Atazanavir/ritonavir
↔f
↓
Bupropion
Not assessed
↔
Carbamazepine
↔
↓
Carbamazepine epoxideg
?
Felbamate
Not assessed
↔
Gabapentin
Not assessed
↔
Lacosamide
Not assessed
↔
Levetiracetam
↔
↔
Lithium
↔
Not assessed
Lopinavir/ritonavir
↔e
↓
Olanzapine
↔
↔e
Oxcarbazepine
↔
↔
10-Monohydroxy oxcarbazepine metaboliteh
↔
Perampanel
Not assessed
↔e
Phenobarbital/primidone
↔
↓
Phenytoin
↔
↓
Pregabalin
↔
↔
Rifampin
Not assessed
↓
Risperidone
↔
Not assessed
9-Hydroxyrisperidonei
↔
Topiramate
↔j
↔
Valproate
↓
↑
Valproate + phenytoin and/or carbamazepine
Not assessed
↔
Zonisamide
Not assessed
↔
a From adjunctive clinical trials and volunteer trials.
- b Net effects were estimated by comparing the mean clearance values obtained in adjunctive clinical trials and volunteer trials.
- c The effect of other hormonal contraceptive preparations or hormone replacement therapy on the pharmacokinetics of lamotrigine has not been systematically evaluated in clinical trials, although the effect may be similar to that seen with the ethinylestradiol/levonorgestrel combinations.
d Modest decrease in levonorgestrel.
e Slight decrease, not expected to be clinically meaningful.
f Compared with historical controls.
g Not administered, but an active metabolite of carbamazepine.
h Not administered, but an active metabolite of oxcarbazepine.
i Not administered, but an active metabolite of risperidone.
j Slight increase, not expected to be clinically meaningful.
↔ = No significant effect.
? = Conflicting data.
Estrogen-Containing Oral Contraceptives
In 16 female volunteers, an oral contraceptive preparation containing 30 mcg ethinylestradiol and 150 mcg levonorgestrel increased the apparent clearance of lamotrigine (300 mg/day) by approximately 2-fold with mean decreases in AUC of 52% and in Cmax of 39%. In this study, trough serum lamotrigine concentrations gradually increased and were approximately 2-fold higher on average at the end of the week of the inactive hormone preparation compared with trough lamotrigine concentrations at the end of the active hormone cycle.
Gradual transient increases in lamotrigine plasma levels (approximate 2 -fold increase) occurred during the week of inactive hormone preparation (pill-free week) for women not also taking a drug that increased the clearance of lamotrigine (carbamazepine, phenytoin, phenobarbital, primidone, or other drugs such as rifampin and the protease inhibitors lopinavir/ritonavir and atazanavir/ritonavir that induce lamotrigine glucuronidation) [see Drug Interactions (7)]. The increase in lamotrigine plasma levels will be greater if the dose of lamotrigine is increased in the few days before or during the pill-free week. Increases in lamotrigine plasma levels could result in dose-dependent adverse reactions.
In the same study, coadministration of lamotrigine (300 mg/day) in 16 female volunteers did not affect the pharmacokinetics of the ethinylestradiol component of the oral contraceptive preparation. There were mean decreases in the AUC and Cmax of the levonorgestrel component of 19% and 12%, respectively. Measurement of serum progesterone indicated that there was no hormonal evidence of ovulation in any of the 16 volunteers, although measurement of serum FSH, LH, and estradiol indicated that there was some loss of suppression of the hypothalamic-pituitary-ovarian axis.
The effects of doses of lamotrigine other than 300 mg/day have not been systematically evaluated in controlled clinical trials.
The clinical significance of the observed hormonal changes on ovulatory activity is unknown. However, the possibility of decreased contraceptive efficacy in some patients cannot be excluded. Therefore, patients should be instructed to promptly report changes in their menstrual pattern (e.g., break-through bleeding).
Dosage adjustments may be necessary for women receiving estrogen-containing oral contraceptive preparations [see Dosage and Administration (2.1)].
Other Hormonal Contraceptives or Hormone Replacement Therapy
The effect of other hormonal contraceptive preparations or hormone replacement therapy on the pharmacokinetics of lamotrigine has not been systematically evaluated. It has been reported that ethinylestradiol, not progestogens, increased the clearance of lamotrigine up to 2-fold, and the progestin-only pills had no effect on lamotrigine plasma levels. Therefore, adjustments to the dosage of lamotrigine in the presence of progestogens alone will likely not be needed.
Aripiprazole
In 18 patients with bipolar disorder on a stable regimen of 100 to 400 mg/day of lamotrigine, the lamotrigine AUC and Cmax were reduced by approximately 10% in patients who received aripiprazole 10 to 30 mg/day for 7 days, followed by 30 mg/day for an additional 7 days. This reduction in lamotrigine exposure is not considered clinically meaningful.
Atazanavir/Ritonavir
In a study in healthy volunteers, daily doses of atazanavir/ritonavir (300 mg/100 mg) reduced the plasma AUC and Cmax of lamotrigine (single 100-mg dose) by an average of 32% and 6%, respectively, and shortened the elimination half-lives by 27%. In the presence of atazanavir/ritonavir (300 mg/100 mg), the metabolite-to-lamotrigine ratio was increased from 0.45 to 0.71 consistent with induction of glucuronidation. The pharmacokinetics of atazanavir/ritonavir were similar in the presence of concomitant lamotrigine to the historical data of the pharmacokinetics in the absence of lamotrigine.
Bupropion
The pharmacokinetics of a 100-mg single dose of lamotrigine in healthy volunteers (n = 12) were not changed by coadministration of bupropion sustained-release formulation (150 mg twice daily) starting 11 days before lamotrigine.
Carbamazepine
Lamotrigine has no appreciable effect on steady-state carbamazepine plasma concentration. Limited clinical data suggest there is a higher incidence of dizziness, diplopia, ataxia, and blurred vision in patients receiving carbamazepine with lamotrigine than in patients receiving other AEDs with lamotrigine [see Adverse Reactions (6.1)]. The mechanism of this interaction is unclear. The effect of lamotrigine on plasma concentrations of carbamazepine-epoxide is unclear. In a small subset of patients (n = 7) studied in a placebo-controlled trial, lamotrigine had no effect on carbamazepine-epoxide plasma concentrations, but in a small, uncontrolled study (n = 9), carbamazepine-epoxide levels increased.
The addition of carbamazepine decreases lamotrigine steady-state concentrations by approximately 40%.
Felbamate
In a trial in 21 healthy volunteers, coadministration of felbamate (1,200 mg twice daily) with lamotrigine (100 mg twice daily for 10 days) appeared to have no clinically relevant effects on the pharmacokinetics of lamotrigine.
Folate Inhibitors
Lamotrigine is a weak inhibitor of dihydrofolate reductase. Prescribers should be aware of this action when prescribing other medications that inhibit folate metabolism.
Gabapentin
Based on a retrospective analysis of plasma levels in 34 subjects who received lamotrigine both with and without gabapentin, gabapentin does not appear to change the apparent clearance of lamotrigine.
Lacosamide
Plasma concentrations of lamotrigine were not affected by concomitant lacosamide (200, 400, or 600 mg/day) in placebo-controlled clinical trials in patients with partial-onset seizures.
Levetiracetam
Potential drug interactions between levetiracetam and lamotrigine were assessed by evaluating serum concentrations of both agents during placebo-controlled clinical trials. These data indicate that lamotrigine does not influence the pharmacokinetics of levetiracetam and that levetiracetam does not influence the pharmacokinetics of lamotrigine.
Lithium
The pharmacokinetics of lithium were not altered in healthy subjects (n = 20) by coadministration of lamotrigine (100 mg/day) for 6 days.
Lopinavir/Ritonavir
The addition of lopinavir (400 mg twice daily)/ritonavir (100 mg twice daily) decreased the AUC, Cmax, and elimination half-life of lamotrigine by approximately 50% to 55.4% in 18 healthy subjects. The pharmacokinetics of lopinavir/ritonavir were similar with concomitant lamotrigine, compared with that in historical controls.
Olanzapine
The AUC and Cmax of olanzapine were similar following the addition of olanzapine (15 mg once daily) to lamotrigine (200 mg once daily) in healthy male volunteers (n = 16) compared with the AUC and Cmax in healthy male volunteers receiving olanzapine alone (n = 16).
In the same trial, the AUC and Cmax of lamotrigine were reduced on average by 24% and 20%, respectively, following the addition of olanzapine to lamotrigine in healthy male volunteers compared with those receiving lamotrigine alone. This reduction in lamotrigine plasma concentrations is not expected to be clinically meaningful.
Oxcarbazepine
The AUC and Cmax of oxcarbazepine and its active 10-monohydroxy oxcarbazepine metabolite were not significantly different following the addition of oxcarbazepine (600 mg twice daily) to lamotrigine (200 mg once daily) in healthy male volunteers (n = 13) compared with healthy male volunteers receiving oxcarbazepine alone (n = 13).
In the same trial, the AUC and Cmax of lamotrigine were similar following the addition of oxcarbazepine (600 mg twice daily) to lamotrigine in healthy male volunteers compared with those receiving lamotrigine alone. Limited clinical data suggest a higher incidence of headache, dizziness, nausea, and somnolence with coadministration of lamotrigine and oxcarbazepine compared with lamotrigine alone or oxcarbazepine alone.
Perampanel
In a pooled analysis of data from 3 placebo-controlled clinical trials investigating adjunctive perampanel in patients with partial-onset and primary generalized tonic-clonic seizures, the highest perampanel dose evaluated (12 mg/day) increased lamotrigine clearance by <10%. An effect of this magnitude is not considered to be clinically relevant.
Phenobarbital, Primidone
The addition of phenobarbital or primidone decreases lamotrigine steady-state concentrations by approximately 40%.
Phenytoin
Lamotrigine has no appreciable effect on steady-state phenytoin plasma concentrations in patients with epilepsy. The addition of phenytoin decreases lamotrigine steady-state concentrations by approximately 40%.
Pregabalin
Steady-state trough plasma concentrations of lamotrigine were not affected by concomitant pregabalin (200 mg 3 times daily) administration. There are no pharmacokinetic interactions between lamotrigine and pregabalin.
Rifampin
In 10 male volunteers, rifampin (600 mg/day for 5 days) significantly increased the apparent clearance of a single 25-mg dose of lamotrigine by approximately 2-fold (AUC decreased by approximately 40%).
Risperidone
In a 14 healthy volunteers study, multiple oral doses of lamotrigine 400 mg daily had no clinically significant effect on the single-dose pharmacokinetics of risperidone 2 mg and its active metabolite 9-OH risperidone. Following the coadministration of risperidone 2 mg with lamotrigine, 12 of the 14 volunteers reported somnolence compared with 1 out of 20 when risperidone was given alone, and none when lamotrigine was administered alone.
Topiramate
Topiramate resulted in no change in plasma concentrations of lamotrigine. Administration of lamotrigine resulted in a 15% increase in topiramate concentrations.
Valproate
When lamotrigine was administered to healthy volunteers (n = 18) receiving valproate, the trough steady-state valproate plasma concentrations decreased by an average of 25% over a 3-week period, and then stabilized. However, adding lamotrigine to the existing therapy did not cause a change in valproate plasma concentrations in either adult or pediatric patients in controlled clinical trials.
The addition of valproate increased lamotrigine steady-state concentrations in normal volunteers by slightly more than 2-fold. In 1 trial, maximal inhibition of lamotrigine clearance was reached at valproate doses between 250 and 500 mg/day and did not increase as the valproate dose was further increased.
Zonisamide
In a study in 18 patients with epilepsy, coadministration of zonisamide (200 to 400 mg/day) with lamotrigine (150 to 500 mg/day for 35 days) had no significant effect on the pharmacokinetics of lamotrigine.
Known Inducers or Inhibitors of Glucuronidation
Drugs other than those listed above have not been systematically evaluated in combination with lamotrigine. Since lamotrigine is metabolized predominately by glucuronic acid conjugation, drugs that are known to induce or inhibit glucuronidation may affect the apparent clearance of lamotrigine and doses of lamotrigine may require adjustment based on clinical response.
Other
In vitro assessment of the inhibitory effect of lamotrigine at OCT2 demonstrate that lamotrigine, but not the N(2)-glucuronide metabolite, is an inhibitor of OCT2 at potentially clinically relevant concentrations, with IC50 value of 53.8 μM [see Drug Interactions (7)].
Results of in vitro experiments suggest that clearance of lamotrigine is unlikely to be reduced by concomitant administration of amitriptyline, clonazepam, clozapine, fluoxetine, haloperidol, lorazepam, phenelzine, sertraline, or trazodone.
Results of in vitro experiments suggest that lamotrigine does not reduce the clearance of drugs eliminated predominantly by CYP2D6.
Specific Populations
Patients with Renal Impairment: Twelve volunteers with chronic renal failure (mean creatinine clearance: 13 mL/min, range: 6 to 23) and another 6 individuals undergoing hemodialysis were each given a single 100-mg dose of lamotrigine. The mean plasma half-lives determined in the study were 42.9 hours (chronic renal failure), 13 hours (during hemodialysis), and 57.4 hours (between hemodialysis) compared with 26.2 hours in healthy volunteers. On average, approximately 20% (range: 5.6 to 35.1) of the amount of lamotrigine present in the body was eliminated by hemodialysis during a 4-hour session [see Dosage and Administration (2.1)].
Patients with Hepatic Impairment: The pharmacokinetics of lamotrigine following a single 100-mg dose of lamotrigine were evaluated in 24 subjects with mild, moderate, and severe hepatic impairment (Child-Pugh classification system) and compared with 12 subjects without hepatic impairment. The subjects with severe hepatic impairment were without ascites (n = 2) or with ascites (n = 5). The mean apparent clearances of lamotrigine in subjects with mild (n = 12), moderate (n = 5), severe without ascites (n = 2), and severe with ascites (n = 5) liver impairment were 0.30 ± 0.09, 0.24 ± 0.1, 0.21 ± 0.04, and 0.15 ± 0.09 mL/min/kg, respectively, as compared with 0.37 ± 0.1 mL/min/kg in the healthy controls. Mean half-lives of lamotrigine in subjects with mild, moderate, severe without ascites, and severe with ascites hepatic impairment were 46 ± 20, 72 ± 44, 67 ± 11, and 100 ± 48 hours, respectively, as compared with 33 ± 7 hours in healthy controls [see Dosage and Administration (2.1)].
Pediatric Patients: The pharmacokinetics of lamotrigine following a single 2-mg/kg dose were evaluated in 2 studies in pediatric subjects (n = 29 for subjects aged 10 months to 5.9 years and n = 26 for subjects aged 5 to 11 years). Forty-three subjects received concomitant therapy with other AEDs and 12 subjects received lamotrigine as monotherapy. Lamotrigine pharmacokinetic parameters for pediatric patients are summarized in Table 16.
Population pharmacokinetic analyses involving subjects aged 2 to 18 years demonstrated that lamotrigine clearance was influenced predominantly by total body weight and concurrent AED therapy. The oral clearance of lamotrigine was higher, on a body weight basis, in pediatric patients than in adults. Weight-normalized lamotrigine clearance was higher in those subjects weighing < 30 kg compared with those weighing > 30 kg. Accordingly, patients weighing < 30 kg may need an increase of as much as 50% in maintenance doses, based on clinical response, as compared with subjects weighing ˃ 30 kg being administered the same AEDs [see Dosage and Administration (2.2)]. These analyses also revealed that, after accounting for body weight, lamotrigine clearance was not significantly influenced by age. Thus, the same weight-adjusted doses should be administered to children irrespective of differences in age. Concomitant AEDs which influence lamotrigine clearance in adults were found to have similar effects in children.
Table 16. Mean Pharmacokinetic Parameters in Pediatric Subjects with Epilepsy Pediatric Study Population
Number of Subjects
Tmax
(h)
t1/2
(h)
CL/F
(mL/min/kg)
Ages 10 months to 5.3 years
- Subjects taking carbamazepine, phenytoin, phenobarbital, or primidonea
- Subjects taking antiepileptic drugs with no known effect on the apparent clearance of lamotrigine
- Subjects taking valproate only
10
7
8
3
(1 to 5.9)
5.2
(2.9 to 6.1)
2.9
(1 to 6)
7.7
(5.7 to 11.4)
19
(12.9 to 27.1)
44.9
(29.5 to 52.5)
3.62
(2.44 to 5.28)
1.2
(0.75 to 2.42)
0.47
(0.23 to 0.77)
Ages 5 to 11 years
- Subjects taking carbamazepine, phenytoin, phenobarbital, or primidonea
- Subjects taking carbamazepine, phenytoin, phenobarbital, or primidonea plus valproate
- Subjects taking valproate onlyb
7
8
3
1.6
(1 to 3)
3.3
(1 to 6.4)
4.5
(3 to 6)
7
(3.8 to 9.8)
19.1
(7 to 31.2)
65.8
(50.7 to 73.7)
2.54
(1.35 to 5.58)
0.89
(0.39 to 1.93)
0.24
(0.21 to 0.26)
Ages 13 to 18 years
- Subjects taking carbamazepine, phenytoin, phenobarbital, or primidonea
- Subjects taking carbamazepine, phenytoin, phenobarbital, or primidonea plus valproate
- Subjects taking valproate only
11
8
4
______c
______c
______c
______c
______c
______c
1.3
0.5
0.3
- a Carbamazepine, phenytoin, phenobarbital, and primidone have been shown to increase the apparent clearance of lamotrigine. Estrogen-containing oral contraceptives, rifampin, and the protease inhibitors lopinavir/ritonavir and atazanavir/ritonavir have also been shown to increase the apparent clearance of lamotrigine [see Drug Interactions (7)].
b Two subjects were included in the calculation for mean Tmax.
c Parameter not estimated.
- Geriatric Patients: The pharmacokinetics of lamotrigine following a single 150-mg dose of lamotrigine were evaluated in 12 elderly volunteers between the ages of 65 and 76 years (mean creatinine clearance = 61 mL/min, range: 33 to 108 mL/min). The mean half-life of lamotrigine in these subjects was 31.2 hours (range: 24.5 to 43.4 hours), and the mean clearance was 0.4 mL/min/kg (range: 0.26 to 0.48 mL/min/kg).
Male and Female Patients: The clearance of lamotrigine is not affected by gender. However, during dose escalation of lamotrigine in 1 clinical trial in patients with epilepsy on a stable dose of valproate (n = 77), mean trough lamotrigine concentrations unadjusted for weight were 24% to 45% higher (0.3 to 1.7 mcg/mL) in females than in males.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No evidence of carcinogenicity was seen in mice or rats following oral administration of lamotrigine for up to 2 years at doses up to 30 mg/kg/day and 10 to 15 mg/kg/day, respectively. The highest doses tested are less than the human dose of 400 mg/day on a body surface area (mg/m2) basis.
Lamotrigine was negative in in vitro gene mutation (Ames and mouse lymphoma tk) assays and in clastogenicity (in vitro human lymphocyte and in vivo rat bone marrow) assays.
No evidence of impaired fertility was detected in rats given oral doses of lamotrigine up to 20 mg/kg/day. The highest dose tested is less than the human dose of 400 mg/day on a mg/m2 basis.
-
14 CLINICAL STUDIES
14.1 Epilepsy
Monotherapy with Lamotrigine in Adults with Partial-Onset Seizures Already Receiving Treatment with Carbamazepine, Phenytoin, Phenobarbital, or Primidone as the Single Antiepileptic Drug
The effectiveness of monotherapy with lamotrigine was established in a multicenter, double-blind clinical trial enrolling 156 adult outpatients with partial-onset seizures. The patients experienced at least 4 simple partial-onset, complex partial-onset, and/or secondarily generalized seizures during each of 2 consecutive 4-week periods while receiving carbamazepine or phenytoin monotherapy during baseline. Lamotrigine (target dose of 500 mg/day) or valproate (1,000 mg/day) was added to either carbamazepine or phenytoin monotherapy over a 4-week period. Patients were then converted to monotherapy with lamotrigine or valproate during the next 4 weeks, then continued on monotherapy for an additional 12-week period.
Trial endpoints were completion of all weeks of trial treatment or meeting an escape criterion. Criteria for escape relative to baseline were: (1) doubling of average monthly seizure count, (2) doubling of highest consecutive 2-day seizure frequency, (3) emergence of a new seizure type (defined as a seizure that did not occur during the 8-week baseline) that is more severe than seizure types that occur during study treatment, or (4) clinically significant prolongation of generalized tonic-clonic seizures. The primary efficacy variable was the proportion of patients in each treatment group who met escape criteria.
The percentages of patients who met escape criteria were 42% (32/76) in the group receiving lamotrigine and 69% (55/80) in the valproate group. The difference in the percentage of patients meeting escape criteria was statistically significant (P = 0.0012) in favor of lamotrigine. No differences in efficacy based on age, sex, or race were detected.
Patients in the control group were intentionally treated with a relatively low dose of valproate; as such, the sole objective of this trial was to demonstrate the effectiveness and safety of monotherapy with lamotrigine, and cannot be interpreted to imply the superiority of lamotrigine to an adequate dose of valproate.
Adjunctive Therapy with Lamotrigine in Adults with Partial-Onset Seizures
The effectiveness of lamotrigine as adjunctive therapy (added to other AEDs) was initially established in 3 pivotal, multicenter, placebo-controlled, double-blind clinical trials in 355 adults with refractory partial-onset seizures. The patients had a history of at least 4 partial-onset seizures per month in spite of receiving 1 or more AEDs at therapeutic concentrations and in 2 of the trials were observed on their established AED regimen during baselines that varied between 8 to 12 weeks. In the third trial, patients were not observed in a prospective baseline. In patients continuing to have at least 4 seizures per month during the baseline, lamotrigine or placebo was then added to the existing therapy. In all 3 trials, change from baseline in seizure frequency was the primary measure of effectiveness. The results given below are for all partial-onset seizures in the intent-to-treat population (all patients who received at least 1 dose of treatment) in each trial, unless otherwise indicated. The median seizure frequency at baseline was 3 per week while the mean at baseline was 6.6 per week for all patients enrolled in efficacy trials.
One trial (n = 216) was a double-blind, placebo-controlled, parallel trial consisting of a 24-week treatment period. Patients could not be on more than 2 other anticonvulsants and valproate was not allowed. Patients were randomized to receive placebo, a target dose of 300 mg/day of lamotrigine, or a target dose of 500 mg/day of lamotrigine. The median reductions in the frequency of all partial-onset seizures relative to baseline were 8% in patients receiving placebo, 20% in patients receiving 300 mg/day of lamotrigine, and 36% in patients receiving 500 mg/day of lamotrigine. The seizure frequency reduction was statistically significant in the 500-mg/day group compared with the placebo group, but not in the 300-mg/day group.
A second trial (n = 98) was a double-blind, placebo-controlled, randomized, crossover trial consisting of two 14-week treatment periods (the last 2 weeks of which consisted of dose tapering) separated by a 4-week washout period. Patients could not be on more than 2 other anticonvulsants and valproate was not allowed. The target dose of lamotrigine was 400 mg/day. When the first 12 weeks of the treatment periods were analyzed, the median change in seizure frequency was a 25% reduction on lamotrigine compared with placebo (P<0.001).
The third trial (n = 41) was a double-blind, placebo-controlled, crossover trial consisting of two 12-week treatment periods separated by a 4-week washout period. Patients could not be on more than 2 other anticonvulsants. Thirteen patients were on concomitant valproate; these patients received 150 mg/day of lamotrigine. The 28 other patients had a target dose of 300 mg/day of lamotrigine. The median change in seizure frequency was a 26% reduction on lamotrigine compared with placebo (P<0.01).
No differences in efficacy based on age, sex, or race, as measured by change in seizure frequency, were detected.
Adjunctive Therapy with Lamotrigine in Pediatric Patients with Partial-Onset Seizures
The effectiveness of lamotrigine as adjunctive therapy in pediatric patients with partial-onset seizures was established in a multicenter, double-blind, placebo-controlled trial in 199 patients aged 2 to 16 years (n = 98 on lamotrigine, n = 101 on placebo). Following an 8-week baseline phase, patients were randomized to 18 weeks of treatment with lamotrigine or placebo added to their current AED regimen of up to 2 drugs. Patients were dosed based on body weight and valproate use. Target doses were designed to approximate 5 mg/kg/day for patients taking valproate (maximum dose: 250 mg/day) and 15 mg/kg/day for the patients not taking valproate (maximum dose: 750 mg/day). The primary efficacy endpoint was percentage change from baseline in all partial-onset seizures. For the intent-to-treat population, the median reduction of all partial-onset seizures was 36% in patients treated with lamotrigine and 7% on placebo, a difference that was statistically significant (P<0.01).
Adjunctive Therapy with Lamotrigine in Pediatric and Adult Patients with Lennox-Gastaut Syndrome
The effectiveness of lamotrigine as adjunctive therapy in patients with Lennox-Gastaut syndrome was established in a multicenter, double-blind, placebo-controlled trial in 169 patients aged 3 to 25 years (n = 79 on lamotrigine, n = 90 on placebo). Following a 4-week, single-blind, placebo phase, patients were randomized to 16 weeks of treatment with lamotrigine or placebo added to their current AED regimen of up to 3 drugs. Patients were dosed on a fixed-dose regimen based on body weight and valproate use. Target doses were designed to approximate 5 mg/kg/day for patients taking valproate (maximum dose: 200 mg/day) and 15 mg/kg/day for patients not taking valproate (maximum dose: 400 mg/day). The primary efficacy endpoint was percentage change from baseline in major motor seizures (atonic, tonic, major myoclonic, and tonic-clonic seizures). For the intent-to-treat population, the median reduction of major motor seizures was 32% in patients treated with lamotrigine and 9% on placebo, a difference that was statistically significant (P<0.05). Drop attacks were significantly reduced by lamotrigine (34%) compared with placebo (9%), as were tonic-clonic seizures (36% reduction versus 10% increase for lamotrigine and placebo, respectively).
Adjunctive Therapy with Lamotrigine in Pediatric and Adult Patients with Primary Generalized Tonic-Clonic Seizures
The effectiveness of lamotrigine as adjunctive therapy in patients with PGTC seizures was established in a multicenter, double-blind, placebo-controlled trial in 117 pediatric and adult patients aged 2 years and older (n = 58 on lamotrigine, n = 59 on placebo). Patients with at least 3 PGTC seizures during an 8-week baseline phase were randomized to 19 to 24 weeks of treatment with lamotrigine or placebo added to their current AED regimen of up to 2 drugs. Patients were dosed on a fixed-dose regimen, with target doses ranging from 3 to 12 mg/kg/day for pediatric patients and from 200 to 400 mg/day for adult patients based on concomitant AEDs.
The primary efficacy endpoint was percentage change from baseline in PGTC seizures. For the intent-to-treat population, the median percent reduction in PGTC seizures was 66% in patients treated with lamotrigine and 34% on placebo, a difference that was statistically significant (P = 0.006).
14.2 Bipolar Disorder
Adults
The effectiveness of lamotrigine in the maintenance treatment of bipolar I disorder was established in 2 multicenter, double-blind, placebo-controlled trials in adult patients (aged 18 to 82 years) who met DSM-IV criteria for bipolar I disorder. Trial 1 enrolled patients with a current or recent (within 60 days) depressive episode as defined by DSM-IV and Trial 2 included patients with a current or recent (within 60 days) episode of mania or hypomania as defined by DSM-IV. Both trials included a cohort of patients (30% of 404 subjects in Trial 1 and 28% of 171 patients in Trial 2) with rapid cycling bipolar disorder (4 to 6 episodes per year).
In both trials, patients were titrated to a target dose of 200 mg of lamotrigine, as add-on therapy or as monotherapy with gradual withdrawal of any psychotropic medications during an 8- to 16-week open-label period. Overall 81% of 1,305 patients participating in the open-label period were receiving 1 or more other psychotropic medications, including benzodiazepines, selective serotonin reuptake inhibitors (SSRIs), atypical antipsychotics (including olanzapine), valproate, or lithium, during titration of lamotrigine. Patients with a CGI-severity score of 3 or less maintained for at least 4 continuous weeks, including at least the final week on monotherapy with lamotrigine, were randomized to a placebo-controlled double-blind treatment period for up to 18 months. The primary endpoint was TIME (time to intervention for a mood episode or one that was emerging, time to discontinuation for either an adverse event that was judged to be related to bipolar disorder, or for lack of efficacy). The mood episode could be depression, mania, hypomania, or a mixed episode.
In Trial 1, patients received double-blind monotherapy with lamotrigine 50 mg/day (n = 50), lamotrigine 200 mg/day (n = 124), lamotrigine 400 mg/day (n = 47), or placebo (n = 121). Lamotrigine (200- and 400-mg/day treatment groups combined) was superior to placebo in delaying the time to occurrence of a mood episode (Figure 1). Separate analyses of the 200- and 400-mg/day dose groups revealed no added benefit from the higher dose.
In Trial 2, patients received double-blind monotherapy with lamotrigine (100 to 400 mg/day, n = 59), or placebo (n = 70). Lamotrigine was superior to placebo in delaying time to occurrence of a mood episode (Figure 2). The mean dose of lamotrigine was about 211 mg/day.
Although these trials were not designed to separately evaluate time to the occurrence of depression or mania, a combined analysis for the 2 trials revealed a statistically significant benefit for lamotrigine over placebo in delaying the time to occurrence of both depression and mania, although the finding was more robust for depression.
Figure 1: Kaplan-Meier Estimation of Cumulative Proportion of Patients with Mood Episode (Trial 1)
Figure 2: Kaplan-Meier Estimation of Cumulative Proportion of Patients with Mood Episode (Trial 2)
-
16 HOW SUPPLIED/STORAGE AND HANDLING
Lamotrigine Tablets for oral suspension (Chewable, Dispersible Tablets), 5 mg: White to off-white, caplet shaped, flat faced beveled edged, uncoated tablets with a score line on one side and “228” debossed on the other side.
Bottles of 90 (NDC: 68462-228-90)
Bottles of 100 (NDC: 68462-228-01)
Bottles of 1000 (NDC: 68462-228-10)
Lamotrigine Tablets for oral suspension (Chewable, Dispersible Tablets), 25 mg: White to off-white, circular, flat faced beveled edged, uncoated tablets with ‘G’ debossed on one side and ‘229’ debossed on the other side.
Bottles of 90 (NDC: 68462-229-90)
Bottles of 100 (NDC: 68462-229-01)
Bottles of 1000 (NDC: 68462-229-10)
Store at 20°C to 25°C (68°F to 77°F); excursions permitted to 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature] in a dry place.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Rash
Prior to initiation of treatment with lamotrigine tablets for oral suspension (chewable, dispersible tablets), inform patients that a rash or other signs or symptoms of hypersensitivity (e.g., fever, lymphadenopathy) may herald a serious medical event and instruct them to report any such occurrence to their healthcare providers immediately.
Hemophagocytic Lymphohistiocytosis
Prior to initiation of treatment with lamotrigine tablets for oral suspension (chewable, dispersible tablets), inform patients that excessive immune activation may occur with lamotrigine tablets for oral suspension (chewable, dispersible tablets), and that they should report signs or symptoms such as fever, rash, or lymphadenopathy to a healthcare provider immediately.
Multiorgan Hypersensitivity Reactions, Blood Dyscrasias, and Organ Failure
Inform patients that multiorgan hypersensitivity reactions and acute multiorgan failure may occur with lamotrigine tablets for oral suspension (chewable, dispersible tablets). Isolated organ failure or isolated blood dyscrasias without evidence of multiorgan hypersensitivity may also occur. Instruct patients to contact their healthcare providers immediately if they experience any signs or symptoms of these conditions [see Warnings and Precautions (5.3, 5.4)].
Suicidal Thinking and Behavior
Inform patients, their caregivers, and families that AEDs, including lamotrigine tablets for oral suspension (chewable, dispersible tablets), may increase the risk of suicidal thoughts and behavior. Instruct them to be alert for the emergence or worsening of symptoms of depression, any unusual changes in mood or behavior, or the emergence of suicidal thoughts or behavior or thoughts about self-harm. Instruct them to immediately report behaviors of concern to their healthcare providers.
Worsening of Seizures
Instruct patients to notify their healthcare providers if worsening of seizure control occurs.
Central Nervous System Adverse Effects
Inform patients that lamotrigine tablets for oral suspension (chewable, dispersible tablets) may cause dizziness, somnolence, and other symptoms and signs of central nervous system depression. Accordingly, instruct them neither to drive a car nor to operate other complex machinery until they have gained sufficient experience on lamotrigine tablets for oral suspension (chewable, dispersible tablets) to gauge whether or not it adversely affects their mental and/or motor performance.
Pregnancy and Nursing
Instruct patients to notify their healthcare providers if they become pregnant or intend to become pregnant during therapy and if they intend to breastfeed or are breastfeeding an infant.
Encourage patients to enroll in the NAAED Pregnancy Registry if they become pregnant. This registry is collecting information about the safety of antiepileptic drugs during pregnancy. To enroll, patients can call the toll-free number 1-888-233-2334 [see Use in Specific Populations (8.1)].
Inform patients who intend to breastfeed that lamotrigine is present in breast milk and advise them to monitor their child for potential adverse effects of this drug. Discuss the benefits and risks of continuing breastfeeding.
Oral Contraceptive Use
Instruct women to notify their healthcare providers if they plan to start or stop use of oral contraceptives or other female hormonal preparations. Starting estrogen-containing oral contraceptives may significantly decrease lamotrigine plasma levels and stopping estrogen-containing oral contraceptives (including the pill-free week) may significantly increase lamotrigine plasma levels [see Warnings and Precautions (5.8), Clinical Pharmacology (12.3)]. Also instruct women to promptly notify their healthcare providers if they experience adverse reactions or changes in menstrual pattern (e.g., break-through bleeding) while receiving lamotrigine tablets for oral suspension (chewable, dispersible tablets) in combination with these medications.
Discontinuing Lamotrigine Tablets For Oral Suspension (Chewable, Dispersible Tablets)
Instruct patients to notify their healthcare providers if they stop taking lamotrigine tablets for oral suspension (chewable, dispersible tablets) for any reason and not to resume lamotrigine tablets for oral suspension (chewable, dispersible tablets) without consulting their healthcare providers.
Aseptic Meningitis
Inform patients that lamotrigine tablets for oral suspension (chewable, dispersible tablets) may cause aseptic meningitis. Instruct them to notify their healthcare providers immediately if they develop signs and symptoms of meningitis such as headache, fever, nausea, vomiting, stiff neck, rash, abnormal sensitivity to light, myalgia, chills, confusion, or drowsiness while taking lamotrigine tablets for oral suspension (chewable, dispersible tablets).
Potential Medication Errors
To avoid a medication error of using the wrong drug or formulation, strongly advise patients to visually inspect their tablets to verify that they are lamotrigine tablets for oral suspension (chewable, dispersible tablets), as well as the correct formulation of lamotrigine tablets for oral suspension (chewable, dispersible tablets), each time they fill their prescription [see Dosage Forms and Strengths (3.2), How Supplied/Storage and Handling (16)]. Refer the patient to the Medication Guide that provides depictions of the lamotrigine tablets for oral suspension (chewable, dispersible tablets).
Risk in Patients with Phenylketonurics
Inform patients with phenylketonuria (PKU) that lamotrigine tablets for oral suspension (chewable, dispersible tablets), 5 mg and 25 mg, each contain 1.123 mg phenylalanine.
Medication Guide available at www.glenmarkpharma-us.com/medguides
Manufactured by:
Glenmark Pharmaceuticals Ltd.
Colvale-Bardez, Goa 403513, India
Manufactured for:
Glenmark Pharmaceuticals Inc., USA
Mahwah, NJ 07430
Questions? 1 (888) 721-7115
www.glenmarkpharma-us.com
February 2020
-
MEDICATION GUIDE
Lamotrigine (la-moe'-tri-jeen) Tablets For Oral Suspension
(Chewable, Dispersible Tablets)
What is the most important information I should know about lamotrigine tablets for oral suspension (chewable, dispersible tablets)?
1. Lamotrigine tablets for oral suspension (chewable, dispersible tablets) may cause a serious skin rash that may cause you to be hospitalized or even cause death.
There is no way to tell if a mild rash will become more serious. A serious skin rash can happen at any time during your treatment with lamotrigine tablets for oral suspension (chewable, dispersible tablets), but is more likely to happen within the first 2 to 8 weeks of treatment. Children and teenagers aged between 2 and 17 years have a higher chance of getting this serious skin rash while taking lamotrigine tablets for oral suspension (chewable, dispersible tablets).
The risk of getting a serious skin rash is higher if you:
- take lamotrigine tablets for oral suspension (chewable, dispersible tablets) while taking valproate [DEPAKENE (valproic acid) or DEPAKOTE (divalproex sodium)].
- take a higher starting dose of lamotrigine tablets for oral suspension (chewable, dispersible tablets) than your healthcare provider prescribed.
- increase your dose of lamotrigine tablets for oral suspension (chewable, dispersible tablets) faster than prescribed.
Call your healthcare provider right away if you have any of the following:
- a skin rash
- blistering or peeling of your skin
- hives
- painful sores in your mouth or around your eyes
These symptoms may be the first signs of a serious skin reaction. A healthcare provider should examine you to decide if you should continue taking lamotrigine tablets for oral suspension (chewable, dispersible tablets).
2. Other serious reactions, including serious blood problems or liver problems. Lamotrigine tablets for oral suspension (chewable, dispersible tablets) can also cause other types of allergic reactions or serious problems that may affect organs and other parts of your body like your liver or blood cells. You may or may not have a rash with these types of reactions. Call your healthcare provider right away if you have any of these symptoms:
- fever
- frequent infections
- severe muscle pain
- swelling of your face, eyes, lips, or tongue
- swollen lymph glands
- unusual bruising or bleeding
- weakness, fatigue
- yellowing of your skin or the white part of your eyes
3. Like other antiepileptic drugs, lamotrigine tablets for oral suspension (chewable, dispersible tablets) may cause suicidal thoughts or actions in a very small number of people, about 1 in 500.
Call a healthcare provider right away if you have any of these symptoms, especially if they are new, worse, or worry you:
- thoughts about suicide or dying
- attempt to commit suicide
- new or worse depression
- new or worse anxiety
- feeling agitated or restless
- panic attacks
- trouble sleeping (insomnia)
- new or worse irritability
- acting aggressive, being angry, or violent
- acting on dangerous impulses
- an extreme increase in activity and talking (mania)
- other unusual changes in behavior or mood
Do not stop lamotrigine tablets for oral suspension (chewable, dispersible tablets) without first talking to a healthcare provider.
- Stopping lamotrigine tablets for oral suspension (chewable, dispersible tablets) suddenly can cause serious problems.
- Suicidal thoughts or actions can be caused by things other than medicines. If you have suicidal thoughts or actions, your healthcare provider may check for other causes.
How can I watch for early symptoms of suicidal thoughts and actions in myself or a family member?
- Pay attention to any changes, especially sudden changes, in mood, behaviors, thoughts, or feelings.
- Keep all follow-up visits with your healthcare provider as scheduled.
- Call your healthcare provider between visits as needed, especially if you are worried about symptoms.
4. Lamotrigine tablets for oral suspension (chewable, dispersible tablets) may cause aseptic meningitis, a serious inflammation of the protective membrane that covers the brain and spinal cord.
Call your healthcare provider right away if you have any of the following symptoms:
- headache
- fever
- nausea
- vomiting
- stiff neck
- rash
- unusual sensitivity to light
- muscle pains
- chills
- confusion
- drowsiness
Meningitis has many causes other than lamotrigine tablets for oral suspension (chewable, dispersible tablets), which your doctor would check for if you developed meningitis while taking lamotrigine tablets for oral suspension (chewable, dispersible tablets).
Lamotrigine tablets for oral suspension (chewable, dispersible tablets) can cause other serious side effects. For more information ask your healthcare provider or pharmacist. Tell your healthcare provider if you have any side effect that bothers you. Be sure to read the section below entitled “What are the possible side effects of lamotrigine tablets for oral suspension (chewable, dispersible tablets)?”
5. People prescribed lamotrigine tablets for oral suspension (chewable, dispersible tablets) have sometimes been given the wrong medicine because many medicines have names similar to lamotrigine tablets for oral suspension (chewable, dispersible tablets), so always check that you receive lamotrigine tablets for oral suspension (chewable, dispersible tablets).
Taking the wrong medication can cause serious health problems. When your healthcare provider gives you a prescription for lamotrigine tablets for oral suspension (chewable, dispersible tablets):
- Make sure you can read it clearly.
- Talk to your pharmacist to check that you are given the correct medicine.
- Each time you fill your prescription, check the tablets you receive against the pictures of the tablets below.
These pictures show the distinct wording, colors, and shapes of the tablets that help to identify the right strength of lamotrigine tablets for oral suspension (chewable, dispersible tablets). Immediately call your pharmacist if you receive lamotrigine tablets for oral suspension (chewable, dispersible tablets) that do not look like one of the tablets shown below, as you may have received the wrong medication.
Lamotrigine Tablets For Oral Suspension (Chewable, Dispersible Tablets)
What are lamotrigine tablets for oral suspension (chewable, dispersible tablets)?
- Lamotrigine tablets for oral suspension (chewable, dispersible tablets) are a prescription medicine used:
- o together with other medicines to treat certain types of seizures (partial-onset seizures, primary generalized tonic-clonic seizures, generalized seizures of Lennox-Gastaut syndrome) in people aged 2 years and older.
- o alone when changing from 1 other medicine used to treat partial-onset seizures in people aged 16 years and older.
- o for the long-term treatment of bipolar I disorder to lengthen the time between mood episodes in people who have been treated for mood episodes with other medicine.
- It is not known if lamotrigine tablets for oral suspension (chewable, dispersible tablets) are safe or effective in people younger than 18 years with mood episodes such as bipolar disorder or depression.
- It is not known if lamotrigine tablets for oral suspension (chewable, dispersible tablets) are safe or effective when used alone as the first treatment of seizures.
- It is not known if lamotrigine tablets for oral suspension (chewable, dispersible tablets) are safe or effective for people with mood episodes who have not already been treated with other medicines.
- Lamotrigine tablets for oral suspension (chewable, dispersible tablets) should not be used for acute treatment of manic or mixed mood episodes.
Do not take lamotrigine tablets for oral suspension (chewable, dispersible tablets):
- if you have had an allergic reaction to lamotrigine or to any of the inactive ingredients in lamotrigine tablets for oral suspension (chewable, dispersible tablets). See the end of this leaflet for a complete list of ingredients in lamotrigine tablets for oral suspension (chewable, dispersible tablets).
Before taking lamotrigine tablets for oral suspension (chewable, dispersible tablets), tell your healthcare provider about all of your health conditions, including if you:
- have had a rash or allergic reaction to another antiseizure medicine.
- have or have had depression, mood problems, or suicidal thoughts or behavior.
- have had aseptic meningitis after taking lamotrigine tablets for oral suspension (chewable, dispersible tablets) or lamotrigine extended-release tablets.
- are taking oral contraceptives (birth control pills) or other female hormonal medicines. Do not start or stop taking birth control pills or other female hormonal medicine until you have talked with your healthcare provider. Tell your healthcare provider if you have any changes in your menstrual pattern such as breakthrough bleeding. Stopping these medicines while you are taking lamotrigine tablets for oral suspension (chewable, dispersible tablets) may cause side effects (such as dizziness, lack of coordination, or double vision). Starting these medicines may lessen how well lamotrigine tablets for oral suspension (chewable, dispersible tablets) works.
- are pregnant or plan to become pregnant. It is not known if lamotrigine may harm your unborn baby. If you become pregnant while taking lamotrigine tablets for oral suspension (chewable, dispersible tablets), talk to your healthcare provider about registering with the North American Antiepileptic Drug Pregnancy Registry. You can enroll in this registry by calling 1-888-233-2334. The purpose of this registry is to collect information about the safety of antiepileptic drugs during pregnancy.
- are breastfeeding. Lamotrigine passes into breast milk and may cause side effects in a breastfed baby. If you breastfeed while taking lamotrigine tablets for oral suspension (chewable, dispersible tablets), watch your baby closely for trouble breathing, episodes of temporarily stopping breathing, sleepiness, or poor sucking. Call your baby’s healthcare provider right away if you see any of these problems. Talk to your healthcare provider about the best way to feed your baby if you take lamotrigine tablets for oral suspension (chewable, dispersible tablets).
- have phenylketonuria. Lamotrigine tablets for oral suspension (chewable, dispersible tablets) contain aspartame, a source of phenylalanine.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. Lamotrigine tablets for oral suspension (chewable, dispersible tablets) and certain other medicines may interact with each other. This may cause serious side effects.
Know the medicines you take. Keep a list of them to show your healthcare provider and pharmacist when you get a new
medicine.
How should I take lamotrigine tablets for oral suspension (chewable, dispersible tablets)?
- Take lamotrigine tablets for oral suspension (chewable, dispersible tablets) exactly as prescribed.
- Your healthcare provider may change your dose. Do not change your dose without talking to your healthcare provider.
- Do not stop taking lamotrigine tablets for oral suspension (chewable, dispersible tablets) without talking to your healthcare provider. Stopping lamotrigine tablets for oral suspension (chewable, dispersible tablets) suddenly may cause serious problems. For example, if you have epilepsy and you stop taking lamotrigine tablets for oral suspension (chewable, dispersible tablets) suddenly, you may have seizures that do not stop. Talk with your healthcare provider about how to stop lamotrigine tablets for oral suspension (chewable, dispersible tablets) slowly.
- If you miss a dose of lamotrigine tablets for oral suspension (chewable, dispersible tablets), take it as soon as you remember. If it is almost time for your next dose, just skip the missed dose. Take the next dose at your regular time. Do not take 2 doses at the same time.
- If you take too much lamotrigine tablets for oral suspension (chewable, dispersible tablets), call your healthcare provider or your local Poison Control Center or go to the nearest hospital emergency room right away.
- You may not feel the full effect of lamotrigine tablets for oral suspension (chewable, dispersible tablets) for several weeks.
- If you have epilepsy, tell your healthcare provider if your seizures get worse or if you have any new types of seizures.
- Lamotrigine tablets for oral suspension (chewable, dispersible tablets) may be swallowed whole, chewed, or mixed in water or fruit juice mixed with water. If the tablets are chewed, drink a small amount of water or fruit juice mixed with water to help in swallowing. To break up lamotrigine tablets for oral suspension (chewable, dispersible tablets), add the tablets to a small amount of liquid (1 teaspoon, or enough to cover the medicine) in a glass or spoon. Wait at least 1 minute or until the tablets are completely broken up, mix the solution together, and take the whole amount right away.
What should I avoid while taking lamotrigine tablets for oral suspension (chewable, dispersible tablets)?
Do not drive, operate machinery, or do other dangerous activities until you know how lamotrigine tablets for oral suspension (chewable, dispersible tablets) affects you.
What are the possible side effects of lamotrigine tablets for oral suspension (chewable, dispersible tablets)?
Lamotrigine tablets for oral suspension (chewable, dispersible tablets) can cause serious side effects.
See “What is the most important information I should know about lamotrigine tablets for oral suspension (chewable, dispersible tablets)?”
Common side effects of lamotrigine tablets for oral suspension (chewable, dispersible tablets) include:
- dizziness
- tremor
- headache
- rash
- blurred or double vision
- fever
- lack of coordination
- abdominal pain
- infections, including seasonal flu
- sleepiness
- back pain
- nausea, vomiting
- diarrhea
- tiredness
- insomnia
- dry mouth
- stuffy nose
- sore throat
These are not all the possible side effects of lamotrigine tablets for oral suspension (chewable, dispersible tablets).
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store lamotrigine tablets for oral suspension (chewable, dispersible tablets)?
- Store lamotrigine tablets for oral suspension (chewable, dispersible tablets) at room temperature between 68°F and 77°F (20°C and 25°C).
- Keep lamotrigine tablets for oral suspension (chewable, dispersible tablets) and all medicines out of the reach of children.
General information about the safe and effective use of lamotrigine tablets for oral suspension (chewable, dispersible tablets).
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use lamotrigine tablets for oral suspension (chewable, dispersible tablets) for a condition for which it was not prescribed. Do not give lamotrigine tablets for oral suspension (chewable, dispersible tablets) to other people, even if they have the same symptoms that you have. It may harm them.
If you take a urine drug screening test, lamotrigine tablets for oral suspension (chewable, dispersible tablets) may make the test result positive for another drug. If you require a urine drug screening test, tell the healthcare professional administering the test that you are taking lamotrigine tablets for oral suspension (chewable, dispersible tablets).
You can ask your healthcare provider or pharmacist for information about lamotrigine tablets for oral suspension (chewable, dispersible tablets) that is written for health professionals.
What are the ingredients in lamotrigine tablets for oral suspension (chewable, dispersible tablets)?
Active ingredient: lamotrigine
Inactive ingredients: aspartame, colloidal silicon dioxide, magnesium stearate, microcrystalline cellulose, povidone K-25, sodium starch glycolate and strawberry guarana 586997 AP 0551.
People with Phenylketonuria: Lamotrigine tablets for oral suspension (chewable, dispersible tablets), 5 mg and 25 mg, each contain 1.123 mg phenylalanine.
Trademarks are the property of their respective owners.
This Medication Guide has been approved by the U.S. Food and Drug Administration.
Medication Guide available at www.glenmarkpharma-us.com/medguides
Manufactured by:
Glenmark Pharmaceuticals Ltd.
Colvale-Bardez, Goa 403513, India
Manufactured for:
Glenmark Pharmaceuticals Inc., USA
Mahwah, NJ 07430
Questions? 1 (888) 721-7115
February 2020
- Principle Display Panel
- Principle Display Panel
-
INGREDIENTS AND APPEARANCE
LAMOTRIGINE
lamotrigine tablet, chewableProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 68462-228 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength LAMOTRIGINE (UNII: U3H27498KS) (LAMOTRIGINE - UNII:U3H27498KS) LAMOTRIGINE 5 mg Inactive Ingredients Ingredient Name Strength ASPARTAME (UNII: Z0H242BBR1) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) MAGNESIUM STEARATE (UNII: 70097M6I30) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) POVIDONE K25 (UNII: K0KQV10C35) SODIUM STARCH GLYCOLATE TYPE B POTATO (UNII: 27NA468985) Product Characteristics Color WHITE (to off-white) Score 2 pieces Shape CAPSULE (caplet, flat faced beveled edged) Size 8mm Flavor STRAWBERRY (strawberry guarana) Imprint Code 228 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 68462-228-90 90 in 1 BOTTLE; Type 0: Not a Combination Product 02/19/2009 2 NDC: 68462-228-01 100 in 1 BOTTLE; Type 0: Not a Combination Product 02/19/2009 3 NDC: 68462-228-10 1000 in 1 BOTTLE; Type 0: Not a Combination Product 02/19/2009 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA079099 02/19/2009 LAMOTRIGINE
lamotrigine tablet, chewableProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 68462-229 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength LAMOTRIGINE (UNII: U3H27498KS) (LAMOTRIGINE - UNII:U3H27498KS) LAMOTRIGINE 25 mg Inactive Ingredients Ingredient Name Strength ASPARTAME (UNII: Z0H242BBR1) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) MAGNESIUM STEARATE (UNII: 70097M6I30) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) POVIDONE K25 (UNII: K0KQV10C35) SODIUM STARCH GLYCOLATE TYPE B POTATO (UNII: 27NA468985) Product Characteristics Color WHITE (to off-white) Score no score Shape ROUND (flat faced beveled edged) Size 7mm Flavor STRAWBERRY (strawberry guarana) Imprint Code G;229 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 68462-229-90 90 in 1 BOTTLE; Type 0: Not a Combination Product 02/19/2009 2 NDC: 68462-229-01 100 in 1 BOTTLE; Type 0: Not a Combination Product 02/19/2009 3 NDC: 68462-229-10 1000 in 1 BOTTLE; Type 0: Not a Combination Product 02/19/2009 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA079099 02/19/2009 Labeler - Glenmark Pharmaceuticals Inc., USA (130597813) Establishment Name Address ID/FEI Business Operations Glenmark Pharmaceuticals Limited 677318665 ANALYSIS(68462-228, 68462-229) , MANUFACTURE(68462-228, 68462-229)
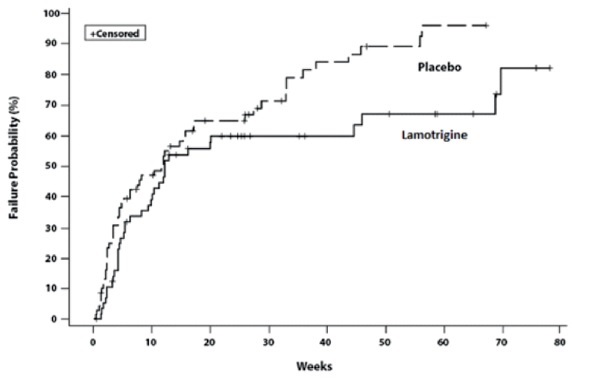
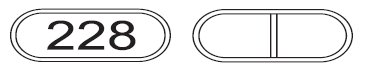
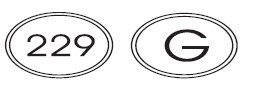


© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.