IBTROZI- taletrectinib capsule
IBTROZI by
Drug Labeling and Warnings
IBTROZI by is a Prescription medication manufactured, distributed, or labeled by Nuvation Bio Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use IBTROZI safely and effectively. See full prescribing information for IBTROZI.
IBTROZI® (taletrectinib) capsules, for oral use
Initial U.S. Approval: 2025INDICATIONS AND USAGE
IBTROZI is a kinase inhibitor indicated for the treatment of adult patients with locally advanced or metastatic ROS1-positive non-small cell lung cancer (NSCLC). (1)
DOSAGE AND ADMINISTRATION
- Select patients for the treatment of locally advanced or metastatic NSCLC based on the presence of ROS1 rearrangement(s). (2.1)
- Recommended Dosage: 600 mg orally once daily on an empty stomach (no food intake at least 2 hours before and 2 hours after taking IBTROZI). (2.3)
- Continue treatment until disease progression or unacceptable toxicity. (2.3)
DOSAGE FORMS AND STRENGTHS
Capsules: 200 mg of taletrectinib (3)
CONTRAINDICATIONS
- None (4)
WARNINGS AND PRECAUTIONS
- Hepatotoxicity: Monitor liver function tests prior to initiating, every 2 weeks during the first 2 months of treatment, then monthly thereafter as clinically indicated, with more frequent testing in patients who develop transaminase elevations. Based on severity and resolution, withhold and then resume at reduced dose, or permanently discontinue. (2.4, 5.1)
- Interstitial Lung Disease (ILD)/Pneumonitis: Monitor patients for new or worsening pulmonary symptoms indicative of ILD/pneumonitis. Immediately withhold in patients with suspected ILD/pneumonitis. Based on severity and resolution, resume at the same or reduced dose, or permanently discontinue. (2.4, 5.2)
- QTc Interval Prolongation: Monitor ECG and electrolytes prior to initiating and periodically during treatment. Based on severity and resolution, withhold and then resume at same or reduced dose, or permanently discontinue. (2.4, 5.3)
- Hyperuricemia: Monitor serum uric acid levels prior to initiating and periodically during treatment. Initiate treatment with urate-lowering medications as clinically indicated. Withhold and resume at same or reduced dose or permanently discontinue based on severity. (2.4, 5.4)
- Myalgia with Creatine Phosphokinase (CPK) Elevation: Monitor serum CPK levels during treatment in patients reporting unexplained muscle pain, tenderness, or weakness. Based on severity, withhold and resume at same or reduced dose upon improvement. (2.4, 5.5)
- Skeletal Fractures: Promptly evaluate patients with signs or symptoms (e.g., pain, changes in mobility, deformity) of fractures. (5.6)
- Embryo-Fetal Toxicity: Can cause fetal harm. Advise patients of the potential risk to a fetus and to use effective contraception. (5.7)
ADVERSE REACTIONS
The most frequently reported adverse reactions (≥20%) were: diarrhea, nausea, vomiting, dizziness, rash, constipation, and fatigue. (6.1)
The most frequently reported Grade 3 or 4 laboratory abnormalities (≥5%) were: increased ALT, increased AST, decreased neutrophils, and increased creatine phosphokinase. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Nuvation Bio Inc. at 1-844-688-4550 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- Strong and Moderate CYP3A inhibitors: Avoid concomitant use. (7.1)
- Strong and Moderate CYP3A inducers: Avoid concomitant use. (7.1)
- Gastric Acid Reducing Agents: Avoid concomitant use with proton pump inhibitors (PPIs) and H2 receptor antagonists. If an acid-reducing agent cannot be avoided, administer IBTROZI 2 hours before or 2 hours after taking a locally acting antacid. (7.1)
- Drugs That Prolong the QTc Interval: Avoid concomitant use. (7.2)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 3/2026
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection
2.2 Recommended Testing and Evaluation Before Initiating IBTROZI
2.3 Recommended Dosage and Administration
2.4 Dosage Modifications for Adverse Reactions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hepatotoxicity
5.2 Interstitial Lung Disease/Pneumonitis
5.3 QTc Interval Prolongation
5.4 Hyperuricemia
5.5 Myalgia with Creatine Phosphokinase Elevation
5.6 Skeletal Fractures
5.7 Embryo-Fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 Effects of Other Drugs on IBTROZI
7.2. Drugs That Prolong the QTc Interval
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Locally Advanced or Metastatic ROS1-Positive NSCLC
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
IBTROZI® (taletrectinib) is indicated for the treatment of adult patients with locally advanced or metastatic ROS1-positive non-small cell lung cancer (NSCLC) [see Dosage and Administration (2.1)].
-
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection
Select patients for the treatment of locally advanced or metastatic NSCLC with IBTROZI based on the presence of ROS1 rearrangement(s) in tumor specimens [see Clinical Studies (14.1)].
An FDA-approved test to detect ROS1 rearrangement(s) for selecting patients for treatment with IBTROZI is not currently available.
2.2 Recommended Testing and Evaluation Before Initiating IBTROZI
Before initiating IBTROZI, evaluate liver function tests (including ALT, AST, and bilirubin), electrolytes, ECG, and uric acid [see Warnings and Precautions (5.1, 5.3, 5.4)].
2.3 Recommended Dosage and Administration
- The recommended dosage of IBTROZI is 600 mg orally once daily on an empty stomach (no food intake at least 2 hours before and 2 hours after taking IBTROZI) [see Clinical Pharmacology (12.2, 12.3)] until disease progression or unacceptable toxicity.
- Take IBTROZI at approximately the same time each day. Swallow IBTROZI capsules whole. Do not open, chew, crush, or dissolve the capsule prior to swallowing.
- Avoid food or drink containing grapefruit during treatment with IBTROZI.
- Minimize sun exposure and use sun protection, including broad-spectrum sunscreen, during treatment with IBTROZI and for at least 5 days after discontinuation [see Adverse Reactions (6.1)].
Missed Dose
- If a dose is missed, take the next dose at its scheduled time on the following day.
Vomiting
- If vomiting occurs at any time after taking a dose, take the next dose at its scheduled time on the following day.
2.4 Dosage Modifications for Adverse Reactions
The recommended dosage reductions for the management of adverse reactions are provided in Table 1.
Table 1: Recommended Dose Reductions for IBTROZI Adverse Reactions Dosage Reduction Recommended Dosage First Dose Reduction 400 mg once daily Second Dose Reduction 200 mg once daily Permanently discontinue IBTROZI capsules in patients unable to tolerate 200 mg once daily. The recommended dosage modifications of IBTROZI for the management of adverse reactions are provided in Table 2.
Table 2: Recommended Dosage Modifications for IBTROZI Adverse Reactions Adverse Reaction Severity* Dosage Modification Hepatotoxicity (Elevation of ALT or AST) [see Warnings and
Precautions (5.1)]Grade 3 (>5 - 20 × ULN) Withhold IBTROZI until recovery to Grade ≤1 or baseline.
- If resolved within 6 weeks, resume IBTROZI at a reduced dose level.
- If unresolved after 6 weeks, permanently discontinue IBTROZI.
Recurrence:
- If resolved within 6 weeks, resume IBTROZI at a reduced dose level.
- If unresolved after 6 weeks, permanently discontinue IBTROZI.
Grade 4 (>20 × ULN) Withhold IBTROZI until recovery to Grade ≤1 or baseline.
- If resolved within 6 weeks, resume IBTROZI at a reduced dose level.
- If unresolved after 6 weeks, permanently discontinue IBTROZI.
Recurrence:
- Permanently discontinue IBTROZI.
ALT or AST ≥3 × ULN with concurrent total bilirubin ≥2 × ULN (in the absence of cholestasis or hemolysis) Permanently discontinue IBTROZI. ILD/pneumonitis
[see Warnings and
Precautions (5.2)]Grade 1 Withhold IBTROZI if ILD/pneumonitis occurs or is suspected until recovery to Grade 0 or baseline.
- If resolved within 6 weeks, resume IBTROZI at the same dose level.
- If unresolved after 6 weeks, permanently discontinue IBTROZI.
Recurrence:
- Permanently discontinue IBTROZI.
Grade 2 Withhold IBTROZI if ILD/pneumonitis occurs or is suspected until recovery to Grade 0 or baseline.
- If resolved within 6 weeks, resume IBTROZI at a reduced dose level.
- If unresolved after 6 weeks, permanently discontinue IBTROZI.
Recurrence:
- Permanently discontinue IBTROZI.
Grade 3 or 4 Permanently discontinue IBTROZI. QTc Interval Prolongation [see Warnings and Precautions (5.3)] Grade 2 (QTc interval 481-500 msec) Withhold IBTROZI until recovery to Grade ≤1 or baseline.
- Correct electrolytes and/or change concomitant medications.
- Resume IBTROZI at same dose.
Abbreviations: ALT = alanine aminotransferase; AST = aspartate aminotransferase; ULN = Upper limit of normal
*Graded per National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE), version 5.0.
Adverse Reaction Severity* Dosage Modification Grade 3 (QTc interval ≥501 msec or QTc interval increase of >60 msec from baseline) Withhold IBTROZI until recovery to Grade ≤1 or baseline.
- Correct electrolytes and/or change concomitant medications.
- Resume IBTROZI at a reduced dose.
Grade 4 (Torsade de pointes; polymorphic ventricular tachycardia; signs/symptoms of serious arrhythmia) Permanently discontinue IBTROZI. Hyperuricemia [see
Warnings and
Precautions (5.4)]Grade 3 or 4 Withhold IBTROZI until improvement of signs or symptoms.
- Resume IBTROZI at same or reduced dose level or permanently discontinue.
Creatine
Phosphokinase Elevation [see
Warnings and
Precautions (5.5)]CPK elevation >5 times
ULNWithhold until recovery to baseline or ≤2.5 times ULN, then resume at same dose. CPK elevation >10 times ULN or second occurrence of CPK elevation of >5 times ULN Withhold until recovery to baseline or ≤2.5 times ULN, then resume at a reduced dose. Other Adverse
Reactions [see
Adverse Reactions
(6.1)]Grade 3 Withhold IBTROZI until recovery to Grade ≤1 or baseline.
- If resolved within 6 weeks, resume IBTROZI at a reduced dose level.
- If unresolved after 6 weeks, permanently discontinue IBTROZI.
Recurrence:
- Resume treatment at a reduced dose or permanently discontinue IBTROZI.
Grade 4 Withhold IBTROZI until recovery to Grade ≤1 or baseline. Resume IBTROZI at reduced dose or permanently discontinue as clinically indicated.
Recurrence:
- Permanently discontinue IBTROZI.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Hepatotoxicity
IBTROZI can cause hepatotoxicity, including drug-induced liver injury and fatal adverse reactions.
In the pooled safety population [see Adverse Reactions (6.1)], based on laboratory values, 88% of patients treated with IBTROZI experienced increased aspartate aminotransferase (AST), including 10% Grade 3 or 4. Increased alanine aminotransferase (ALT) occurred in 85% of patients treated with IBTROZI, including 13% Grade 3 or 4. The median time to first onset of AST or ALT elevation was 15 days (range: 3 days to 20.8 months).
Increased AST or ALT each led to dose interruption in 7% of patients. Increased AST and ALT leading to dose reduction occurred in 5% and 9% of patients, respectively. Increased AST and ALT each led to permanent discontinuation of IBTROZI in 0.3% of patients. Other liver-related adverse reactions leading to permanent discontinuation of IBTROZI were hepatotoxicity (0.6% of patients) and increased bilirubin (0.3% of patients).
Concurrent elevations in AST or ALT ≥3 times the upper limit of normal (ULN) and total bilirubin ≥2 times the ULN, with normal alkaline phosphatase, occurred in 2 (0.6%) patients treated with IBTROZI. Fatal liver events occurred in 2 (0.6%) patients.
Monitor liver function tests (AST, ALT, and bilirubin) prior to administration of IBTROZI, every 2 weeks during the first 2 months of treatment, and then monthly thereafter as clinically indicated with more frequent testing in patients who develop transaminase elevations. Withhold, then resume at reduced dose upon improvement, or permanently discontinue IBTROZI based on severity [see Dosage and Administration (2.4)].
5.2 Interstitial Lung Disease/Pneumonitis
IBTROZI can cause severe, life-threatening, or fatal interstitial lung disease (ILD) or pneumonitis.
In the pooled safety population [see Adverse Reactions (6.1)], interstitial lung disease (ILD)/pneumonitis occurred in 2.3% of patients treated with IBTROZI, including Grade 3 or 4 in 1.1% of patients. The median time to first onset of ILD/pneumonitis was 3.8 months (range: 12 days to 11.8 months).
ILD/pneumonitis led to dose interruption of IBTROZI in 1.1% of patients. ILD/pneumonitis required dose reduction in 0.6% of patients and permanent discontinuation of IBTROZI in 0.6% of patients. One fatal ILD case occurred in a patient who received 400 mg once daily dose of IBTROZI.
Monitor patients for new or worsening pulmonary symptoms indicative of ILD/pneumonitis. Immediately withhold IBTROZI in patients with suspected ILD/pneumonitis. Withhold, then reduce the dose or permanently discontinue IBTROZI if Grade ≥2 ILD/pneumonitis is confirmed [see Dosage and Administration (2.4)].
5.3 QTc Interval Prolongation
IBTROZI can cause QTc interval prolongation, which can increase the risk for ventricular tachyarrhythmias (e.g., torsades de pointes) or sudden death. IBTROZI prolongs the QTc interval in a concentration-dependent manner [see Clinical Pharmacology (12.2)].
In the pooled safety population [see Adverse Reactions (6.1)], of the 351 patients who underwent at least one post baseline ECG assessment, 13% experienced an increase in QTcF of >60 msec compared to baseline after receiving IBTROZI and 2.6% had an increase in QTcF to >500 msec. Overall, 3.4% of patients had Grade ≥3 QTc interval prolongation. The median time from the first dose of IBTROZI to the onset of ECG QT prolongation was 22 days (range: 1 day to 38.7 months). QTc prolongation led to dose interruption and dose reduction, each in 2.8% of patients treated with IBTROZI.
Monitor ECGs and electrolytes prior to administration of IBTROZI, and then periodically thereafter as clinically indicated during treatment. Adjust the frequency of monitoring based on risk factors such as known long QT syndromes, clinically significant bradyarrhythmias, severe or uncontrolled heart failure, and concomitant medications associated with QTc interval prolongation.
Significant prolongation of the QTc interval may occur when IBTROZI is taken with food, strong and moderate CYP3A inhibitors, and/or drugs with a known potential to prolong QTc. Administer IBTROZI on an empty stomach. Avoid coadministration of IBTROZI with strong and moderate CYP3A inhibitors and/or drugs with a known potential to prolong QTc [see Dosage and Administration (2.3), Drug Interactions (7.1, 7.2)].
Withhold, then resume at the same or reduced dose, or permanently discontinue IBTROZI based on severity [see Dosage and Administration (2.4)].
5.4 Hyperuricemia
IBTROZI can cause hyperuricemia.
In the pooled safety population [see Adverse Reactions (6.1)], 14% of patients treated with IBTROZI experienced hyperuricemia reported as an adverse reaction, with 16% of these patients requiring urate-lowering medication without pre-existing gout or hyperuricemia. Hyperuricemia Grade ≥3 occurred in one patient. The median time to first onset of hyperuricemia was 2.1 months (range: 7 days to 35.8 months). Hyperuricemia leading to dose interruption occurred in 0.3% of patients.
Monitor serum uric acid levels prior to administration of IBTROZI and periodically during treatment. Initiate treatment with urate-lowering medications as clinically indicated. Withhold, then resume at the same or reduced dose, or permanently discontinue IBTROZI based on severity [see Dosage and Administration (2.4)].
5.5 Myalgia with Creatine Phosphokinase Elevation
IBTROZI can cause myalgia with or without creatine phosphokinase (CPK) elevation.
In the pooled safety population [see Adverse Reactions (6.1)], myalgia occurred in 10% of patients treated with IBTROZI. The median time to first onset of myalgia was 11 days (range: 2 days to 10 months).
Concurrent myalgia with increased CPK within a 7-day time period was observed in 0.9% of patients. IBTROZI was interrupted in one patient (0.3%) with myalgia, who also presented with concurrent CPK elevation.
Advise patients to report any unexplained muscle pain, tenderness, or weakness. Monitor serum CPK levels during IBTROZI treatment every 2 weeks during the first month of treatment and then as clinically indicated in patients reporting unexplained muscle pain, tenderness, or weakness. Withhold, then resume at the same or reduced dose upon improvement [see Dosage and Administration (2.4)].
5.6 Skeletal Fractures
IBTROZI can increase the risk of fractures. ROS1 inhibitors as a class have been associated with skeletal fractures.
In the pooled safety population [see Adverse Reactions (6.1)], 3.4% of patients experienced fractures including 1.4% Grade 3. Some fractures occurred in the setting of an accidental fall or other predisposing factors such as osteoporosis, bone metastasis, and age-related degenerative conditions. Fractures involved the ribs (1.4%), spine (0.9%), femur (0.6%), humerus (0.3%), and acetabulum (0.3%). The median time to first onset of fracture was 10.7 months (range: 26 days to 29.1 months). Dose interruption occurred in 0.3% of patients.
Promptly evaluate patients with signs or symptoms (e.g., pain, changes in mobility, deformity) of fractures. There are no data on the effects of IBTROZI on healing of known fractures and risk of future fractures.
5.7 Embryo-Fetal Toxicity
Based on literature reports in humans with congenital mutations leading to changes in tropomyosin receptor kinase (TRK) signaling, findings from animal studies and its mechanism of action, IBTROZI can cause fetal harm when administered to a pregnant woman.
In animal reproduction studies, oral administration of taletrectinib to pregnant rats during the period of organogenesis caused structural abnormalities. Oral administration of taletrectinib to pregnant rabbits during the period of organogenesis resulted in embryo-fetal mortalities and structural abnormalities. Findings in both species occurred at doses resulting in exposures below or equal to the human exposure based on AUC at the recommended dose.
Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with IBTROZI and for 3 weeks after the last dose. Advise male patients with female partners of reproductive potential to use effective contraception during treatment with IBTROZI and for 3 weeks after the last dose [see Use in Specific Populations (8.1, 8.3)].
-
6 ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in the WARNINGS AND PRECAUTIONS section:
- Hepatotoxicity [see Warnings and Precautions (5.1)]
- Interstitial Lung Disease/Pneumonitis [see Warnings and Precautions (5.2)]
- QTc Interval Prolongation [see Warnings and Precautions (5.3)]
- Hyperuricemia [see Warnings and Precautions (5.4)]
- Myalgia with Creatine Phosphokinase Elevation [see Warnings and Precautions (5.5)]
- Skeletal fractures [see Warnings and Precautions (5.6)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The pooled safety population described in the WARNINGS AND PRECAUTIONS section and below reflects exposure to IBTROZI as a single agent at 600 mg orally once daily until disease progression or unacceptable toxicity in 352 patients with ROS1-positive NSCLC (N=337) and other solid tumors (N=15). Among the 352 patients who received IBTROZI, 68% were exposed for at least 6 months, and 47% were exposed for greater than 1 year. In this pooled safety population, the most common (≥20%) adverse reactions were diarrhea, nausea, vomiting, dizziness, rash, constipation, and fatigue. The most common (≥2%) Grade 3 or 4 laboratory abnormalities were increased ALT, increased AST, decreased neutrophils, increased creatine phosphokinase, decreased lymphocytes, increased magnesium, decreased hemoglobin, and increased triglycerides.
Locally Advanced or Metastatic ROS1-Positive NSCLC
The safety of IBTROZI was evaluated in the TRUST-I and TRUST-II studies [see Clinical Studies (14.1)]. Key eligibility criteria were histologically confirmed, locally advanced or metastatic, ROS1-positive NSCLC, ECOG performance status ≤1, and measurable disease per RECIST v1.1. Patients received IBTROZI as a single agent at 600 mg orally once daily until disease progression or unacceptable toxicity. Among patients who received IBTROZI, 68% were exposed for 6 months or longer and 47% were exposed for greater than one year.
The median age of patients who received IBTROZI was 56 years (range: 26 to 83); 56% female; 76% Asian, 15% White, 0.6% Black or African American, 8% unknown or other races; and 1.8% were of Hispanic or Latino ethnicity.
Serious adverse reactions occurred in 31% of patients who received IBTROZI. Serious adverse reactions in ≥2% of patients included pneumonia (7%), pleural effusion (4.7%), and hepatotoxicity (2.4%). Fatal adverse reactions occurred in 18 (5%) patients who received IBTROZI, including pneumonia (2.4%), multiple organ dysfunction syndrome (0.6%), hepatotoxicity (0.6%), cardiac arrest (0.6%), cardiac failure (0.3%), cardiopulmonary failure (0.3%), respiratory failure (0.3%), and death not otherwise specified (0.3%).
Permanent discontinuation of IBTROZI was required in 7% of patients due to adverse reactions. Adverse reactions resulting in permanent discontinuation of IBTROZI in ≥2 patients were pneumonia, ILD, and hepatotoxicity.
Dosage interruptions of IBTROZI due to an adverse reaction occurred in 41% of patients. Adverse reactions which required dosage interruption in ≥5% of patients included increased AST and increased ALT.
Dose reductions of IBTROZI due to an adverse reaction occurred in 29% of patients. Adverse reactions that required dosage reductions in ≥5% of patients included increased ALT and increased AST.
Table 3 summarizes the adverse reactions in this population.
Table 3: Adverse Reactions (≥15%) in Patients with ROS1-Positive NSCLC Who Received IBTROZI in TRUST-I and TRUST-II 1 Based on NCI CTCAE version 5.0 a Includes enterocolitis
bIncludes vertigo, and vertigo positional
cIncludes dysesthesia, hypoesthesia, neuralgia, paresthesia, and peripheral sensory neuropathy d Includes ageusia
eIncludes dermatitis, dermatitis acneiform, drug eruption, eczema, eyelid rash, palmar-plantar erythrodysesthesia syndrome, rash
maculo-papular, rash papular, skin exfoliation, and drug reaction with eosinophilia and systemic symptoms (DRESS)
fIncludes asthenia
g Includes productive cough
Adverse Reaction1 IBTROZI N=337 All Grades (%) Grade 3 or 4 (%) Gastrointestinal Disorders Diarrheaa 64 2.1 Nausea 47 1.5 Vomiting 43 1.5 Constipation 21 0 Nervous System Disorders Dizzinessb 22 0.3 Peripheral neuropathyc 17 0.3 Dysgeusiad 15 0 Skin and Subcutaneous Tissue Rashe 22 1.8 General Disorders Fatiguef 20 0.9 Cardiac Electrocardiogram QT prolonged 19 3.6 Metabolism and Nutritional Decreased appetite 16 0.3 Respiratory, thoracic and mediastinal disorders Coughg 16 0 Clinically relevant adverse reactions in <15% of patients receiving IBTROZI were pneumonia, eye disorders, myalgia, skeletal fracture, ILD/pneumonitis, dermatologic adverse reactions including drug reaction with eosinophilia and systemic symptoms (DRESS), and photosensitivity reactions.
Table 4 summarizes the laboratory abnormalities.
Table 4: Select Laboratory Abnormalities (≥20%) That Worsened from Baseline in Patients with ROS1-Positive NSCLC Who Received IBTROZI in TRUST-I and TRUST-II Abbreviations: ALT = alanine aminotransferase; AST = aspartate aminotransferase
1Based on NCI CTCAE version 5.0
2The denominator used to calculate the rate varied from 149 to 336 based on the number of patients with a baseline value and at least one post-treatment value.
Laboratory Abnormality1 IBTROZI2
All Grades (%) Grade 3 or 4 (%) Hematology Hemoglobin decreased 48 3.6 Lymphocytes decreased 38 4.8 Neutrophils decreased 25 5 Chemistry AST increased 87 10 ALT increased 85 13 Creatine phosphokinase increased 53 5 Cholesterol increased 41 0 Triglycerides increased 41 2.5 Creatinine increased 39 0.3 Uric acid increased 38 0 Gamma glutamyl transferase increased 36 1.8 Alkaline phosphatase increased 30 0 Calcium decreased 28 1.8 Albumin decreased 25 0.9 Bilirubin increased 24 0.6 Potassium increased 21 1.2 Sodium increased 20 0.9 -
7 DRUG INTERACTIONS
7.1 Effects of Other Drugs on IBTROZI
Strong and Moderate CYP3A Inhibitors
Avoid concomitant use with strong or moderate CYP3A inhibitors.
Taletrectinib is a CYP3A substrate. Concomitant use of IBTROZI with a strong or moderate CYP3A inhibitor increases taletrectinib exposure [see Clinical Pharmacology (12.3)], which may increase the risk of IBTROZI adverse reactions.
Strong and Moderate CYP3A Inducers
Avoid concomitant use with strong or moderate CYP3A inducers.
Concomitant use of IBTROZI with a strong or moderate CYP3A inducer decreases taletrectinib exposure [see Clinical Pharmacology (12.3)], which may reduce the effectiveness of IBTROZI.
Gastric Acid Reducing Agents
Avoid concomitant use with proton pump inhibitors (PPI) and H2 receptor antagonists. Administer locally acting antacids at least 2 hours before or 2 hours after taking IBTROZI [see Clinical Pharmacology (12.3)].
Concomitant use of a proton pump inhibitor decreases taletrectinib exposure [see Clinical Pharmacology (12.3)], which may reduce the effectiveness of IBTROZI.
7.2. Drugs That Prolong the QTc Interval
Avoid concomitant use of IBTROZI with other drug(s) with a known potential to prolong the QTc interval, such as antiarrhythmic drugs. If concomitant use cannot be avoided, adjust the frequency of monitoring as recommended [see Warnings and Precautions (5.3), Clinical Pharmacology (12.2)]. Withhold IBTROZI if the QTc interval is >500 msec or the change from baseline is >60 msec [see Dosage and Administration (2.4)].
IBTROZI causes QTc interval prolongation [see Warnings and Precautions (5.3), Clinical Pharmacology (12.2)]. Concomitant use of IBTROZI with other drugs known to prolong the QTc interval may increase the risk of QTc interval prolongation.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on literature reports in humans with congenital mutations leading to changes in TRK signaling, findings from animal studies, and its mechanism of action [see Clinical Pharmacology (12.1)], IBTROZI can cause fetal harm when administered to a pregnant woman. Limited data from case reports with IBTROZI used in pregnant women are insufficient to inform a drug-associated risk of adverse developmental outcomes. In animal reproduction studies, oral administration of taletrectinib to pregnant rats and rabbits during the period of organogenesis resulted in embryo-fetal mortalities and structural abnormalities at exposures that were below or equal to the human exposure based on AUC at the recommended dose (see Data). Advise pregnant women of the potential risk to a fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Human Data
Published reports of individuals with congenital mutations in TRK pathway proteins suggest that decreases in TRK-mediated signaling are correlated with obesity, developmental delays, cognitive impairment, insensitivity to pain, and anhidrosis.
Animal Data
In an embryo-fetal development study, taletrectinib was administered orally to pregnant rats during the period of organogenesis from gestation day 6 to 17 at doses of 10, 30, and 100 mg/kg/day. Taletrectinib caused fetal abnormalities including abnormal ossification of the pelvis at 100 mg/kg/day (1.3 times the human exposure based on AUC at the recommended dose).
In an embryo-fetal development study, taletrectinib was administered orally to pregnant rabbits during the period of organogenesis from gestation day 6 to 19 at doses of 15, 30, and 90 mg/kg/day. Maternal lethality and increased total pregnancy loss were observed at doses ≥15 mg/kg/day (≥0.04 times the human exposure based on AUC at the recommended dose). Fetal malformations, including undeveloped or no development of eyes, nose, and mouth, ventricular malformations, thoracic vascular malformations, and skull malformations were observed at 30 mg/kg/day (0.1 times the human exposure based on AUC at the recommended dose).
8.2 Lactation
Risk Summary
There are no data on the presence of taletrectinib or its metabolites in human milk or their effects on a breastfed child or on milk production. Because of the potential for adverse reactions in breastfed children, advise women not to breastfeed during treatment with IBTROZI and for 3 weeks after the last dose.
8.3 Females and Males of Reproductive Potential
IBTROZI can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Pregnancy Testing
Verify the pregnancy status of females of reproductive potential prior to initiating IBTROZI [see Use in Specific Populations (8.1)].
Contraception
Females
Advise females of reproductive potential to use effective contraception during treatment with IBTROZI and for 3 weeks after the last dose [see Nonclinical Toxicology (13.1)].
Males
Advise male patients with female partners of reproductive potential to use effective contraception during treatment with IBTROZI and for 3 weeks after the last dose [see Nonclinical Toxicology (13.1)].
Infertility
Based on findings in animals, IBTROZI may impair fertility in males and females [see Nonclinical Toxicology 13.1]. The effects on animal fertility were reversible.
8.4 Pediatric Use
The safety and effectiveness of IBTROZI in pediatric patients has not been established.
8.5 Geriatric Use
Of the 352 patients who received IBTROZI 600 mg orally once daily, 88 (25%) patients were 65 years of age and older, and 14 (4%) patients were 75 years of age and older. There were no clinically meaningful differences in safety and efficacy between patients younger than 65 years of age and patients 65 years of age or older.
-
11 DESCRIPTION
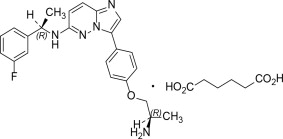
IBTROZI (taletrectinib) capsules contain taletrectinib adipate, a kinase inhibitor for oral use. The molecular formula for taletrectinib adipate is C23H24FN5OC6H10O4 and the molecular weight is 551.61 g/mol. The chemical name is 3-{4-[(2R)-2-aminopropoxy]phenyl}-N-[(1R)-1-(3-fluorophenyl)ethyl]imidazo [1,2b]pyridazin-6-amine monoadipate. The chemical structure of taletrectinib adipate is as follows:
Figure 1: Structure of Taletrectinib Adipate
(R) Indicates chiral center
Taletrectinib adipate is a white to yellowish powder and is practically insoluble or insoluble in water, anhydrous ethanol, isopropanol, acetonitrile, and n-hexane, sparingly soluble in methanol, and slightly soluble in N-methyl pyrrolidone. The aqueous solubility is pH dependent and decreases with increasing pH. The pKa1 is 5.39 and pKa2 is 8.65.
IBTROZI (taletrectinib) capsules for oral use are supplied as immediate-release hard capsules containing 272 mg of taletrectinib adipate (equivalent to 200 mg of taletrectinib free base). Inactive ingredients are colloidal silicon dioxide, low-substituted hydroxypropyl cellulose, mannitol, pregelatinized starch, and sodium stearyl fumarate.
The white opaque capsule shell contains hypromellose and titanium dioxide. The blue printing ink (SB-6008) contains butyl alcohol, dehydrated alcohol, FD&C Blue#1 Aluminum Lake, isopropyl alcohol, propylene glycol, shellac, strong ammonia solution, and titanium dioxide.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Taletrectinib is an inhibitor of tyrosine kinase ROS1, including ROS1 resistance mutations. Taletrectinib also showed inhibitory effects on tropomyosin receptor kinases (TRKs) TRKA, TRKB, and TRKC.
Fusion proteins that include ROS1 domains can drive tumorigenic potential through hyperactivation of downstream signaling pathways leading to unconstrained cell proliferation. Taletrectinib inhibited growth of cancer cells expressing ROS1 fusion genes and mutations.
In mice subcutaneously implanted with tumors harboring ROS1 fusions, including the G2032R mutation, administration of taletrectinib resulted in tumor growth inhibition. Taletrectinib had anticancer activity in an intracranial NSCLC xenograft model harboring a ROS1 fusion.
12.2 Pharmacodynamics
Exposure-response relationship
Higher taletrectinib exposure is associated with an increased risk of Grade ≥3 increased AST/ALT.
Taletrectinib exposure-response relationships for efficacy and the time course of pharmacodynamic response have not been fully characterized.
Cardiac Electrophysiology
The largest mean increase in the QTc interval was 12.8 msec (upper confidence interval of 15.4 msec) at the mean steady-state maximum concentration (Cmax,ss) after administration of IBTROZI 600 mg once daily. The increase in the QTc internal was concentration-dependent [see Warnings and Precautions (5.3)]. At plasma concentrations achieved with administration of IBTROZI 600 mg once daily with high fat food, the predicted increase in the QTc interval is 20.5 (16.3, 24.7) msec.
12.3 Pharmacokinetics
Taletrectinib pharmacokinetics (PK) were characterized at steady state for the approved recommended dosage and the PK parameters presented below are as mean (CV%), unless otherwise specified.
Taletrectinib maximum concentration (Cmax) is 476 (36%) ng/mL and systemic exposure (AUC) is 9,649 (36%) ngh/mL. Taletrectinib Cmax and AUC increase in a dose proportional manner over the dose range of 50 mg to 1200 mg (0.08 to 2 times the approved recommended dosage). Taletrectinib accumulation is approximately 4-fold and steady state reached within 7 days.
Distribution
The estimated apparent (oral) volume of distribution is 9,820 L.
Taletrectinib in vitro plasma protein binding is concentration-dependent and decreases with increasing taletrectinib concentrations (from 96% at 100 ng/mL to 93% at 10,000 ng/mL). The blood-to-plasma ratio is 1.3 to 1.4 in vitro.
Elimination
Taletrectinib effective half-life is approximately 66 hours with an apparent (oral) clearance of 63 L/h (36%).
Specific Populations
No clinically significant differences in the pharmacokinetics of taletrectinib were observed based on age (18 to 80 years), sex, mild to moderate renal impairment (eGFR 30 to 89 mL/min), or mild hepatic impairment (total bilirubin >1 to 1.5 times ULN or AST >ULN). The effect of moderate (total bilirubin >1.5 to 3 times ULN with any AST) or severe (total bilirubin >3 x ULN with any AST) hepatic impairment, severe renal impairment (eGFR<30 mL/min), or dialysis on taletrectinib pharmacokinetics is unknown.
A 30% increase in taletrectinib exposure (AUC) was observed in Asian patients compared to White patients. No clinically significant differences in the AUC were observed between White and Black patients.
Drug Interaction Studies
Clinical Studies and Model-Informed Approaches
Strong CYP3A and P-gp Inhibitors: Taletrectinib Cmax increased by 1.8-fold and AUC by 3.3-fold following concomitant administration of itraconazole (strong CYP3A and P-gp inhibitor) 200 mg daily.
Moderate CYP3A Inhibitors: Concomitant use with moderate CYP3A inhibitors (fluconazole, erythromycin, or verapamil) is predicted to increase taletrectinib AUC up to 2.6-fold and Cmax up to 1.5-fold.
Strong CYP3A Inducers and P-gp Inducers: Taletrectinib Cmax decreased by 42% and AUC by 86% following concomitant administration of rifampin (strong CYP3A and P-gp inducer) 600 mg daily.
Moderate CYP3A Inducers: Concomitant use with a moderate CYP3A inducer (efavirenz) is predicted to reduce taletrectinib AUC by 66% and its Cmax by 40%.
Proton Pump Inhibitors (PPIs): Taletrectinib displays a pH-dependent aqueous solubility [see Description (11)]. Taletrectinib Cmax decreased by 65% and AUC by 40% following concomitant administration of omeprazole (PPI) 40 mg daily.
P-gp substrates: No clinically significant difference in the exposure of digoxin (P-gp substrate) was observed when used concomitantly with taletrectinib.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Mutagenesis
Taletrectinib was not mutagenic in the in vitro bacterial reverse mutation (Ames) assay and additional mutagenicity tests. Taletrectinib was not clastogenic or aneugenic in the in vivo micronucleus study in rats in bone marrow and liver.
Impairment of Fertility
In a fertility and early embryonic development study in rats, male animals were treated with taletrectinib at doses of 4, 25, or 60 mg/kg/day orally for 70 days before mating and during 13 days of the mating period. Female rats, paired with treated males, were administered taletrectinib at doses of 4, 25, or 100 mg/kg/day orally for 14 to 21 days before mating, during the mating period and up to gestational Day 6. No effects on mating or fertility in males or females were observed at doses below or equal to the human exposure based on AUC at the recommended dose. Sperm morphological abnormalities were observed in males at exposures as low as 0.03 times the human exposure based on AUC at the recommended dose.
In repeat-dose toxicity studies up to 12 weeks in duration with oral administration of taletrectinib in rats, adverse effects in reproductive organs included epithelial vacuolation of the seminal vesicle in male rats and endometrial glandular epithelium vacuolation in the uterus with cervix in female rats at exposures 4 times the human exposure based on AUC at the recommended dose for both male and female rats. Findings in the reproductive organs were reversible.
-
14 CLINICAL STUDIES
14.1 Locally Advanced or Metastatic ROS1-Positive NSCLC
The efficacy of IBTROZI was evaluated in 270 patients with ROS1-positive locally advanced or metastatic NSCLC who received IBTROZI at a dose of 600 mg orally once daily, enrolled in two multicenter, single-arm, open-label clinical trials: TRUST-I (NCT04395677) or TRUST-II (NCT04919811). In both trials, patients were required to have histologically confirmed, locally advanced or metastatic, ROS1-positive NSCLC, ECOG performance status of 0 or 1, and measurable disease per Response Evaluation Criteria in Solid Tumors (RECIST) v1.1. Identification of ROS1 gene fusions in tumor specimens was determined in local laboratories using next-generation sequencing (NGS), polymerase chain reaction (PCR), fluorescence in situ (FISH), or immunohistochemistry (IHC).
The major efficacy outcome measures were confirmed overall response rate (ORR) and duration of response (DOR) according to RECIST v1.1 as assessed by a blinded independent central review (BICR). Intracranial response according to modified RECIST v1.1 was assessed by BICR. Tumor assessments with imaging were performed every 6 weeks for the first 24 weeks, every 9 weeks for the following year, and every 12 weeks thereafter. The efficacy populations included 157 patients naïve to treatment with a ROS1 TKI and 113 patients who received one prior ROS1 TKI. Patients could be chemotherapy-naïve or have received prior chemotherapy for locally advanced disease.
ROS1 TKI-Naïve
TRUST-I: Among 103 patients with ROS1 TKI-naïve NSCLC treated in TRUST-I, the median age was 56 years (range: 26 to 78); 55% were female; 100% were Asian; 73% never smoked; and 81% had ECOG performance status of 1. At baseline, 91% of patients had metastatic disease; 17% of patients had CNS metastases by BICR; 96% had adenocarcinoma; and 19% of patients had prior platinum-based chemotherapy for advanced disease.
TRUST-II: Among 54 patients with ROS1 TKI-naïve NSCLC treated in TRUST-II, the median age was 57 years (range: 27 to 82); 56% were female; 65% were Asian; 22% were White; 1.9% were Black or African American; 11% were of unknown race; 1.9% were of Hispanic or Latino ethnicity; 50% never smoked; and 61% had ECOG performance status of 1. At baseline, 91% of patients had metastatic disease, 35% of patients had CNS metastases by BICR; 98% had adenocarcinoma; and 19% of patients had prior platinum-based chemotherapy for advanced disease.
Efficacy results are summarized in Table 5.
Table 5: Efficacy Results in ROS1-Positive TKI-Naïve NSCLC Patients per BICR Assessment Abbreviations: CI = Confidence interval; NR = Not reached; “+” indicating ongoing response
a Median DOR not included for TRUST-II given the shorter duration of follow-up.
b Based on observed duration of response
Efficacy Parameters ROS1 TKI-Naïve N=157 TRUST-I TRUST-II n=103 n=54 Response Rate, % (95% CI) 90%
(83, 95)85%
(73, 93)Complete Response 5% 7% Partial Response 85% 78% Duration of Response (DOR) n= 93 n= 46 Median DOR (95% CI) NR (30.4, NR) --a Range (months) 1.1, 46.9+ 1.4+, 30.4+ % with DORb ≥12 months 72 63 Among 157 ROS1 TKI-naïve patients across TRUST-I and TRUST-II, 15 had measurable CNS metastases at baseline as assessed by BICR and had not received radiation therapy to the brain within 2 months prior to study entry; responses in intracranial lesions were observed in 11 patients.
ROS1 TKI-Pretreated
TRUST-I: Among the 66 patients with ROS1 TKI-pretreated NSCLC, the median age was 51 years (range: 31 to 77); 61% were female; 100% were Asian; 74% never smoked; and 71% had ECOG performance status of 1. At baseline, 97% of patients had metastatic disease, 42% of patients had CNS metastases by BICR; 92% had adenocarcinoma; 35% of patients had prior platinum-based chemotherapy for advanced disease and 100% had prior treatment with crizotinib.
TRUST-II: Among 47 patients with ROS1 TKI-pretreated NSCLC, the median age was 55 years (range: 27 to 79); 57% were female; 47% were Asian; 34% were White; 2.1% were Black or African American, 17% were of unknown or other races; 2.1% were Hispanic or Latino; 62% never smoked; and 55% had ECOG performance status of 1. At baseline, 98% of patients had metastatic disease; 57% of patients had CNS metastases by BICR; 98% had adenocarcinoma; 40% of patients had prior platinum-based chemotherapy for advanced disease, 79% had prior treatment with crizotinib, and 21% had prior treatment with entrectinib.
Efficacy results are summarized in Table 6.
Table 6: Efficacy Results in ROS1-Positive TKI-Pretreated NSCLC Patients per BICR Assessment Abbreviations: CI = Confidence interval; “+” indicating ongoing response
a Median DOR not included for TRUST-II given the shorter duration of follow-up.
b Based on observed duration of response.
Efficacy Parameters ROS1 TKI-Pretreated N=113 TRUST-I TRUST-II n=66 n=47 Response Rate, % (95% CI) 52%
(39, 64)62%
(46, 75)Complete Response 0% 11% Partial Response 52% 51% Duration of Response (DOR) n= 34 n= 29 Median DOR (95% CI) 13.2 (7.7, 24.9) --a Range (months) 1.4, 38.7+ 1.7+, 30.4+ % with DORb ≥6 months 74 83 % with DORb ≥12 months 44 45 Among 113 ROS1 TKI-pretreated patients, across TRUST-I and TRUST-II, 24 had measurable CNS metastases at baseline as assessed by BICR and had not received radiation therapy to the brain within 2 months prior to study entry; responses in intracranial lesions were observed in 15 patients.
Among 32 patients who had re-biopsied samples tested by next-generation sequencing after failure of a prior ROS1 TKI, 15 had resistance mutations. Responses were observed in 8 of these 15 patients; all responding patients had tumors with solvent front mutation G2032R.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
IBTROZI is supplied as 200 mg, Size 0 white opaque hard capsules imprinted with “TAL” and “200” in blue ink as follows:
Packaging Configuration NDC Carton of three bottles (90 capsules total) – each bottle contains 30 capsules 84651-200-93
– each bottle 84651-200-30Carton of one bottle containing 90 capsules 84651-200-90 -
17 PATIENT COUNSELING INFORMATION
Advise patients to read the FDA-approved patient labeling (Patient Information).
Hepatotoxicity
- Advise patients of the risk of hepatoxicity and of the need for laboratory tests to monitor liver function during treatment with IBTROZI and to immediately contact their healthcare providers if they experience symptoms of hepatotoxicity [see Warnings and Precautions (5.1)].
Interstitial Lung Disease (ILD)/Pneumonitis
- Inform patients of the risk of ILD/pneumonitis during treatment with IBTROZI and to contact their healthcare provider immediately if they experience new or worsening pulmonary symptoms indicative of ILD/pneumonitis [see Warnings and Precautions (5.2)].
QTc Interval Prolongation
- Inform patients of the risk of QT interval prolongation during treatment with IBTROZI and to advise patients to contact their healthcare provider immediately for any symptoms of QT interval prolongation [see Warnings and Precautions (5.3)].
Hyperuricemia
- Advise patients of the risk of hyperuricemia during treatment with IBTROZI and to contact their healthcare provider if they experience signs or symptoms associated with hyperuricemia [see Warnings and Precautions (5.4)].
Myalgia with Creatine Phosphokinase Elevation
- Advise patients of the risk of myalgia with creatine phosphokinase elevation during treatment with IBTROZI and to contact their healthcare provider if they experience unexplained muscle pain, tenderness, or weakness [see Warnings and Precautions (5.5)].
Skeletal Fractures
- Inform patients of the risk of bone fractures during treatment with IBTROZI and advise patients to contact their healthcare provider immediately if they experience signs or symptoms of fracture [see Warnings and Precautions (5.6)].
Embryo-Fetal Toxicity
- Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.7), Use in Specific Populations (8.1, 8.3)].
- Advise females of reproductive potential to use effective contraception during treatment with IBTROZI and for 3 weeks after the last dose [see Use in Specific Populations (8.1, 8.3)].
- Advise male patients with female partners of reproductive potential to use effective contraception during treatment with IBTROZI and for 3 weeks after the last dose [see Use in Specific Populations (8.3)].
Photosensitivity
- Inform patients that IBTROZI can cause photosensitivity. Advise patients to minimize sun exposure while taking IBTROZI and to use sun protection, including broad-spectrum sunscreen, during treatment with IBTROZI and for at least 5 days after discontinuation [see Adverse Reactions (6.1)].
Lactation
- Advise women not to breastfeed during treatment with IBTROZI and for 3 weeks after the last dose [see Use in Specific Populations (8.2)].
Drug Interactions
- Advise patients to inform their healthcare providers of all concomitant medications, including prescription medicines, over-the-counter drugs, vitamins, and herbal products [see Drug Interactions (7)].
- Advise patients to avoid grapefruit or grapefruit juice while taking IBTROZI [see Dosage and Administration (2.3)].
Administration
- Advise patients to swallow IBTROZI capsules on an empty stomach, at least 2 hours before or 2 hours after food intake [see Dosage and Administration (2.3), Clinical Pharmacology (12.3)].
- Advise patients that if a dose of IBTROZI is missed or if vomiting occurs, resume IBTROZI at its regularly scheduled time the next day [see Dosage and Administration (2.3)].
Distributed by: Nuvation Bio Inc., Burlington, MA 01803
IBTROZI® is a trademark of Nuvation Bio Inc.
© 2026 Nuvation Bio Inc. All rights reserved.
Version: 2, 3/2026 -
PATIENT PACKAGE INSERT
PATIENT INFORMATION
IBTROZI® [ib-TRO-zee]
(taletrectinib)
capsules, for oral useThis Patient Information has been approved by the U.S. Food and Drug Administration.
Version 1
Issued: 06/2025
What is IBTROZI?
IBTROZI is a prescription medicine used to treat adults with non-small cell lung cancer (NSCLC) that has spread within your chest or to other parts of the body and is caused by an abnormal ROS1 gene.
It is not known if IBTROZI is safe and effective in children.Before taking IBTROZI tell your healthcare provider about all of your medical conditions, including if you:
- have liver problems.
- have lung or breathing problems other than lung cancer.
- have any heart problems, including a condition called long QT syndrome.
- have gout.
- are pregnant or plan to become pregnant. IBTROZI can harm your unborn baby. Tell your healthcare provider right away if you become pregnant or think you may be pregnant during treatment with IBTROZI.
Females who are able to become pregnant:- Your healthcare provider should do a pregnancy test before you start treatment with IBTROZI.
- You should use effective birth control (contraception) during treatment and for 3 weeks after the last dose of IBTROZI.
- Talk to your healthcare provider about birth control methods that may be right for you.
Males with female partners who are able to become pregnant:
- You should use effective birth control (contraception) during treatment and for 3 weeks after the last dose of IBTROZI.
- are breastfeeding or plan to breastfeed. It is not known if IBTROZI passes into your breast milk. Do not breastfeed during treatment and for 3 weeks after the last dose of IBTROZI. Talk to your healthcare provider about the best way to feed your baby during this time.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, or herbal supplements.
IBTROZI may affect the way other medicines work, and other medicines may affect how IBTROZI works.
You should not start or stop any medicine before you talk with your healthcare provider that prescribed IBTROZI.How should I take IBTROZI?
- Take IBTROZI exactly as your healthcare provider tells you to take it. Do not change your dose or stop taking IBTROZI unless your healthcare provider tells you to.
- Take IBTROZI on an empty stomach. Do not eat food for at least 2 hours before or 2 hours after taking IBTROZI.
- Take IBTROZI at about the same time each day.
- Swallow IBTROZI capsules whole. Do not open, crush, chew or dissolve the capsule before swallowing.
- Avoid taking proton pump inhibitor (PPI), or H2 blocker medicine. If you take an antacid, take it at least 2 hours before or 2 hours after taking IBTROZI.
- If you miss a dose or vomit at any time after taking a dose of IBTROZI, take your next dose at your regularly scheduled time the next day.
What should I avoid while taking IBTROZI?
- You should limit time in the sun during treatment with IBTROZI. IBTROZI may make your skin sensitive to sunlight. Wear a hat and clothes that cover your skin and use sunscreen with sun protective factor (SPF) if you are in the sun during treatment with IBTROZI and for at least 5 days after your last dose of IBTROZI.
- You should avoid grapefruit, grapefruit juice, or products that contain grapefruit during your treatment with IBTROZI. Grapefruit may increase the amount of IBTROZI in your blood, which may increase the risk of IBTROZI side effects.
What are the possible side effects of IBTROZI?
IBTROZI can cause serious side effects, including:
-
Liver problems. Changes in liver function can happen during treatment with IBTROZI and can lead to liver injury and death. Your healthcare provider will do blood tests to check your liver function before starting, every 2 weeks during the first 2 months of treatment, then monthly as needed during your treatment with IBTROZI. Tell your healthcare provider right away if you develop signs and symptoms of liver problems including:
- yellowing of your skin or the white part of your eyes (jaundice)
- dark or “tea-colored” urine
- light-colored stools (bowel movements)
- loss of appetite
- nausea or vomiting
- pain on the upper right side of your stomach
- feeling tired or weak
- Lung problems. IBTROZI can cause lung problems that are severe, life-threatening or lead to death. Tell your healthcare provider right away if you have any new or worsening symptoms of lung problems, including trouble breathing, shortness of breath, cough (with or without mucus), or fever.
- Changes in the electrical activity of your heart called QT prolongation. QT prolongation can cause irregular heartbeats that can be life-threatening. Your healthcare provider will do tests before and during your treatment with IBTROZI to check the electrical activity of your heart and your body salts (electrolytes). Tell your healthcare provider right away if you feel faint, lightheaded, dizzy, or feel your heart beating irregularly or fast during your treatment with IBTROZI. These may be symptoms related to QT prolongation.
-
Increased uric acid level in your blood (hyperuricemia). Your healthcare provider will check your uric acid blood level before and during your treatment with IBTROZI. Your healthcare provider may prescribe medicine to lower uric acid if needed. Tell your healthcare provider if you develop any of the following symptoms of hyperuricemia:
- red, hot, tender or swollen joints, especially in your big toe
- pain in your stomach-area
- nausea or vomiting
- pink or brown urine
- Muscle pain, tenderness, and weakness (myalgia). IBTROZI can cause myalgia with or without an increase in the level of an enzyme in your blood called creatine phosphokinase (CPK), which may be a sign of muscle damage. Your healthcare provider will do blood tests to check your CPK blood levels every 2 weeks during the first month and as needed if you experience unexplained muscle pain, tenderness, or weakness during your treatment with IBTROZI. Tell your healthcare provider if you develop unexplained muscle pain, tenderness, or weakness.
-
Bone fractures. IBTROZI can increase your risk of bone fractures. Bone fractures may happen with or without a fall or other injury. Tell your healthcare provider if you develop pain, changes in movement, or bone abnormalities.
The most common side effects of IBTROZI include:
-
- diarrhea
- nausea
- vomiting
- dizziness
- rash
- constipation
- tiredness
- changes in your liver function tests
- decreased white blood cell levels
IBTROZI may cause fertility problems in males and females. Talk to your healthcare provider if this is a concern for you. Your healthcare provider may decrease your dose, temporarily stop, or completely stop your treatment with IBTROZI if you have serious side effects.
These are not all the possible side effects of IBTROZI.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.How should I store IBTROZI?
- Store IBTROZI at room temperature between 68°F to 77°F (20°C to 25°C).
Keep IBTROZI and all medicines out of the reach of children. General information about the safe and effective use of IBTROZI.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use IBTROZI for a condition for which it was not prescribed. Do not give IBTROZI to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about IBTROZI that is written for health professionals.What are the ingredients of IBTROZI?
Active ingredient: taletrectinib adipate
Inactive ingredients: colloidal silicon dioxide, low-substituted hydroxypropyl cellulose, mannitol, pregelatinized starch, sodium stearyl fumarate. Capsule shell contains hypromellose and titanium dioxide. Blue printing ink contains butyl alcohol, dehydrated alcohol, FD&C Blue#1 Aluminum Lake, isopropyl alcohol, propylene glycol, shellac, strong ammonia solution, and titanium dioxide.
Distributed by: Nuvation Bio Inc., Burlington, MA 01803
IBTROZI® is a trademark of Nuvation Bio Inc.
© 2026 Nuvation Bio Inc. All rights reserved.
For more information, go to www.IBTROZI.com or call 1-844-688-4550. -
PRINCIPAL DISPLAY PANEL
Principal Display Panel – 200 mg Carton Label
NDC: 84651-200-93
90 Capsules
Contains three bottles
of 30 capsules per bottle.IBTROZI™
(taletrectinib) capsules
200 mg
Swallow capsules whole
Rx Only
Nuvation Bio
-
PRINCIPAL DISPLAY PANEL
Principal Display Panel - 200 mg Carton Label
NDC: 84651-200-90
90 capsules
IBTROZI®
(taletrectinib)
capsules200 mg
Swallow capsules whole
Rx Only
Nuvation Bio
-
INGREDIENTS AND APPEARANCE
IBTROZI
taletrectinib capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 84651-200 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength taletrectinib adipate (UNII: 6KLL51GNBG) (taletrectinib - UNII:W4141180YD) taletrectinib adipate 272 mg Inactive Ingredients Ingredient Name Strength mannitol (UNII: 3OWL53L36A) starch, corn (UNII: O8232NY3SJ) low-substituted hydroxypropyl cellulose, unspecified (UNII: 2165RE0K14) silicon dioxide (UNII: ETJ7Z6XBU4) sodium stearyl fumarate (UNII: 7CV7WJK4UI) hypromellose, unspecified (UNII: 3NXW29V3WO) titanium dioxide (UNII: 15FIX9V2JP) shellac (UNII: 46N107B71O) alcohol (UNII: 3K9958V90M) isopropyl alcohol (UNII: ND2M416302) butyl alcohol (UNII: 8PJ61P6TS3) propylene glycol (UNII: 6DC9Q167V3) ammonia (UNII: 5138Q19F1X) FD&C Blue No. 1 Aluminum Lake (UNII: J9EQA3S2JM) Product Characteristics Color white (white) Score no score Shape OVAL (OVAL) Size 22mm Flavor Imprint Code TAL;200 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 84651-200-93 3 in 1 CARTON 06/13/2025 1 NDC: 84651-200-30 30 in 1 BOTTLE; Type 0: Not a Combination Product 2 NDC: 84651-200-90 1 in 1 CARTON 09/01/2026 2 90 in 1 BOTTLE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA219713 06/11/2025 Labeler - Nuvation Bio Inc. (119311388)

Trademark Results [IBTROZI]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 IBTROZI 98258541 not registered Live/Pending |
AnHeart Therapeutics Inc. 2023-11-07 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.