YUFLYMA- adalimumab-aaty kit
Yuflyma by
Drug Labeling and Warnings
Yuflyma by is a Prescription medication manufactured, distributed, or labeled by CELLTRION USA, Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use YUFLYMA safely and effectively. See full prescribing information for YUFLYMA.
YUFLYMA (adalimumab-aaty) injection, for subcutaneous use
Initial U.S. Approval: 2023
YUFLYMA (adalimumab-aaty) is biosimilar* to HUMIRA (adalimumab).WARNING: SERIOUS INFECTIONS and MALIGNANCY
See full prescribing information for complete boxed warning.
SERIOUS INFECTIONS (5.1, 6.1):
- Increased risk of serious infections leading to hospitalization or death, including tuberculosis (TB), bacterial sepsis, invasive fungal infections (such as histoplasmosis), and infections due to other opportunistic pathogens.
- Discontinue YUFLYMA if a patient develops a serious infection or sepsis during treatment.
- Perform test for latent TB; if positive, start treatment for TB prior to starting YUFLYMA.
- Monitor all patients for active TB during treatment, even if initial latent TB test is negative.
MALIGNANCY (5.2):
- Lymphoma and other malignancies, some fatal, have been reported in children and adolescent patients treated with TNF blockers including adalimumab products.
- Post-marketing cases of hepatosplenic T-cell lymphoma (HSTCL), a rare type of T-cell lymphoma, have occurred in adolescent and young adults with inflammatory bowel disease treated with TNF blockers including adalimumab products.
RECENT MAJOR CHANGES
Indications and Usage, Hidradenitis Suppurativa (1.8) 10/2025 Indications and Usage, Uveitis (1.9) 10/2025 Dosage and Administration, Recommended Dosage in Juvenile Idiopathic Arthritis or Pediatric Uveitis (2.3) 10/2025 Dosage and Administration, Recommended Dosage in Hidradenitis Suppurativa (2.7) 10/2025 Warnings and Precautions, Autoimmunity (5.9) 10/2025 INDICATIONS AND USAGE
YUFLYMA is a tumor necrosis factor (TNF) blocker indicated for:
- Reducing signs and symptoms, inducing major clinical response, inhibiting the progression of structural damage, and improving physical function in adult patients with moderately to severely active rheumatoid arthritis. (1.1)
- Reducing signs and symptoms of moderately to severely active polyarticular juvenile idiopathic arthritis in patients 2 years of age and older. (1.2)
- Reducing signs and symptoms, inhibiting the progression of structural damage, and improving physical function in adult patients with active psoriatic arthritis. (1.3)
- Reducing signs and symptoms in adult patients with active ankylosing spondylitis. (1.4)
- Treatment of moderately to severely active Crohn's disease in adults and pediatric patients 6 years of age and older. (1.5)
- Treatment of moderately to severely active ulcerative colitis in adult patients. (1.6)
Limitations of Use: Effectiveness has not been established in patients who have lost response to or were intolerant to TNF blockers. - Treatment of adult patients with moderate to severe chronic plaque psoriasis who are candidates for systemic therapy or phototherapy, and when other systemic therapies are medically less appropriate. (1.7)
- Treatment of moderate to severe hidradenitis suppurativa in patients 12 years of age and older. (1.8)
- Treatment of non-infectious intermediate, posterior, and panuveitis in adults and pediatric patients 2 years of age and older. (1.9)
DOSAGE AND ADMINISTRATION
- Administer by subcutaneous injection (2)
Rheumatoid Arthritis, Psoriatic Arthritis, Ankylosing Spondylitis (2.2):
-
Adults:40 mg every other week.
- Some patients with RA not receiving methotrexate may benefit from increasing the dosage to 40 mg every week or 80 mg every other week.
Juvenile Idiopathic Arthritis or Pediatric Uveitis (2.3):
Pediatric Weight
2 Years of Age and OlderRecommended Dosage 10 kg (22 lbs) to less than 15 kg (33 lbs) 10 mg every other week 15 kg (33 lbs) to less than 30 kg (66 lbs) 20 mg every other week 30 kg (66 lbs) and greater 40 mg every other week Crohn's Disease (2.4):
- Adults: 160 mg on Day 1 (given in one day or split over two consecutive days); 80 mg on Day 15; and 40 mg every other week starting on Day 29.
- Pediatric Patients 6 Years of Age and Older:
Pediatric Weight Recommended Dosage Days 1 and 15 Starting on Day 29 17 kg (37 lbs) to less than 40 kg (88 lbs) Day 1: 80 mg
Day 15: 40 mg20 mg every other week 40 kg (88 lbs) and greater Day 1: 160 mg (single dose or split over two consecutive days)
Day 15: 80 mg40 mg every other week Ulcerative Colitis (2.5):
- Adults: 160 mg on Day 1 (given in one day or split over two consecutive days), 80 mg on Day 15 and 40 mg every other week starting on Day 29. Discontinue in patients without evidence of clinical remission by eight weeks (Day 57).
Plaque Psoriasis or Adult Uveitis (2.6):
- Adults: 80 mg initial dose followed by 40 mg every other week starting one week after initial dose.
Hidradenitis Suppurativa (2.7):
-
Adults:
- Day 1: 160 mg (given in one day or split over two consecutive days)
- Day 15: 80 mg
- Day 29 and subsequent doses: 40 mg every week or 80 mg every other week
- Adolescents 12 years of age and older:
Adolescent Weight Recommended Dosage 30 kg (66 lbs) to less than 60 kg (132 lbs) Day 1: 80 mg
Day 8 and subsequent doses: 40 mg every other week60 kg (132 lbs) and greater Day 1: 160 mg (given in one day or split over two consecutive days)
Day 15: 80 mg
Day 29 and subsequent doses: 40 mg every week or 80 mg every other weekDOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONS
None (4)
WARNINGS AND PRECAUTIONS
- Serious infections: Do not start YUFLYMA during an active infection. If an infection develops, monitor carefully, and stop YUFLYMA if infection becomes serious (5.1)
- Invasive fungal infections: For patients who develop a systemic illness on YUFLYMA, consider empiric antifungal therapy for those who reside or travel to regions where mycoses are endemic (5.1)
- Malignancies: Incidence of malignancies was greater in adalimumab-treated patients than in controls (5.2)
- Anaphylaxis or serious hypersensitivity reactions may occur (5.3)
- Hepatitis B virus reactivation: Monitor HBV carriers during and several months after therapy. If reactivation occurs, stop YUFLYMA and begin anti-viral therapy (5.4)
- Demyelinating disease: Exacerbation or new onset, may occur (5.5)
- Cytopenias, pancytopenia: Advise patients to seek immediate medical attention if symptoms develop, and consider stopping YUFLYMA (5.6)
- Heart failure: Worsening or new onset, may occur (5.8)
- Autoimmunity: Stop YUFLYMA if lupus-like syndrome or autoimmune hepatitis develop (5.9)
ADVERSE REACTIONS
Most common adverse reactions (> 10%) are: infections (e.g. upper respiratory, sinusitis), injection site reactions, headache and rash (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact CELLTRION USA Inc., at 1-800-560-9414 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
- * Biosimilar means that the biological product is approved basetd on data demonstrating that it is highly similar to an FDA-approved biological product, known as a reference product, and that there are no clinically meaningful differences between the biosimilar product and the reference product. Biosimilarity of YUFLYMA has been demonstrated for the condition(s) of use (e.g. indication(s), dosing regimen(s)), strength(s), dosage form(s), and route(s) of administration described in its Full Prescribing Information.
Revised: 11/2025
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: SERIOUS INFECTIONS and MALIGNANCY
1 INDICATIONS AND USAGE
1.1 Rheumatoid Arthritis
1.2 Juvenile Idiopathic Arthritis
1.3 Psoriatic Arthritis
1.4 Ankylosing Spondylitis
1.5 Crohn's Disease
1.6 Ulcerative Colitis
1.7 Plaque Psoriasis
1.8 Hidradenitis Suppurativa
1.9 Uveitis
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Tuberculosis Evaluation
2.2 Recommended Dosage in Rheumatoid Arthritis, Psoriatic Arthritis, and Ankylosing Spondylitis
2.3 Recommended Dosage in Juvenile Idiopathic Arthritis or Pediatric Patients with Uveitis
2.4 Recommended Dosage in Crohn's Disease
2.5 Recommended Dosage in Ulcerative Colitis
2.6 Recommended Dosage in Plaque Psoriasis or Adults with Uveitis
2.7 Recommended Dosage in Hidradenitis Suppurativa
2.8 General Considerations for Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Serious Infections
5.2 Malignancies
5.3 Hypersensitivity Reactions
5.4 Hepatitis B Virus Reactivation
5.5 Neurologic Reactions
5.6 Hematological Reactions
5.7 Increased Risk of Infection when Used with Anakinra
5.8 Heart Failure
5.9 Autoimmunity
5.10 Immunizations
5.11 Increased Risk of Infection When Used with Abatacept
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Immunogenicity
6.3 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Methotrexate
7.2 Biological Products
7.3 Live Vaccines
7.4 Cytochrome P450 Substrates
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Clinical Studies in Rheumatoid Arthritis
14.2 Clinical Studies in Juvenile Idiopathic Arthritis
14.3 Clinical Studies in Psoriatic Arthritis
14.4 Clinical Studies in Ankylosing Spondylitis
14.5 Clinical Studies in Adults with Crohn's Disease
14.6 Clinical Studies in Pediatric Subjects with Crohn's Disease
14.7 Clinical Studies in Adults with Ulcerative Colitis
14.8 Clinical Studies in Plaque Psoriasis
14.9 Clinical Studies in Hidradenitis Suppurativa
14.10 Clinical Studies in Adults with Uveitis
14.11 Clinical Studies in Pediatric Subjects with Uveitis
15 REFERENCES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: SERIOUS INFECTIONS and MALIGNANCY
SERIOUS INFECTIONS
Patients treated with adalimumab products, including YUFLYMA, are at increased risk for developing serious infections that may lead to hospitalization or death [see Warnings and Precautions (5.1)]. Most patients who developed these infections were taking concomitant immunosuppressants such as methotrexate or corticosteroids.
Discontinue YUFLYMA if a patient develops a serious infection or sepsis.
Reported infections include:
- Active tuberculosis (TB), including reactivation of latent TB. Patients with TB have frequently presented with disseminated or extrapulmonary disease. Test patients for latent TB before YUFLYMA use and during therapy. Initiate treatment for latent TB prior to YUFLYMA use.
- Invasive fungal infections, including histoplasmosis, coccidioidomycosis, candidiasis, aspergillosis, blastomycosis, and pneumocystosis. Patients with histoplasmosis or other invasive fungal infections may present with disseminated, rather than localized, disease. Antigen and antibody testing for histoplasmosis may be negative in some patients with active infection. Consider empiric anti-fungal therapy in patients at risk for invasive fungal infections who develop severe systemic illness.
- Bacterial, viral and other infections due to opportunistic pathogens, including Legionella and Listeria.
Carefully consider the risks and benefits of treatment with YUFLYMA prior to initiating therapy in patients with chronic or recurrent infection.
Monitor patients closely for the development of signs and symptoms of infection during and after treatment with YUFLYMA, including the possible development of TB in patients who tested negative for latent TB infection prior to initiating therapy [see Warnings and Precautions (5.1) and Adverse Reactions (6.1)].
MALIGNANCY
Lymphoma and other malignancies, some fatal, have been reported in children and adolescent patients treated with TNF blockers including adalimumab products [see Warnings and Precautions (5.2)]. Post-marketing cases of hepatosplenic T-cell lymphoma (HSTCL), a rare type of T-cell lymphoma, have been reported in patients treated with TNF blockers including adalimumab products. These cases have had a very aggressive disease course and have been fatal. The majority of reported TNF blocker cases have occurred in patients with Crohn's disease or ulcerative colitis and the majority were in adolescent and young adult males. Almost all these patients had received treatment with azathioprine or 6-mercaptopurine (6–MP) concomitantly with a TNF blocker at or prior to diagnosis. It is uncertain whether the occurrence of HSTCL is related to use of a TNF blocker or a TNF blocker in combination with these other immunosuppressants [see Warnings and Precautions (5.2)].
-
1 INDICATIONS AND USAGE
1.1 Rheumatoid Arthritis
YUFLYMA is indicated for reducing signs and symptoms, inducing major clinical response, inhibiting the progression of structural damage, and improving physical function in adult patients with moderately to severely active rheumatoid arthritis. YUFLYMA can be used alone or in combination with methotrexate or other non-biologic disease-modifying anti-rheumatic drugs (DMARDs).
1.2 Juvenile Idiopathic Arthritis
YUFLYMA is indicated for reducing signs and symptoms of moderately to severely active polyarticular juvenile idiopathic arthritis in patients 2 years of age and older. YUFLYMA can be used alone or in combination with methotrexate.
1.3 Psoriatic Arthritis
YUFLYMA is indicated for reducing signs and symptoms, inhibiting the progression of structural damage, and improving physical function in adult patients with active psoriatic arthritis. YUFLYMA can be used alone or in combination with non-biologic DMARDs.
1.4 Ankylosing Spondylitis
YUFLYMA is indicated for reducing signs and symptoms in adult patients with active ankylosing spondylitis.
1.5 Crohn's Disease
YUFLYMA is indicated for the treatment of moderately to severely active Crohn's disease in adults and pediatric patients 6 years of age and older.
1.6 Ulcerative Colitis
YUFLYMA is indicated for the treatment of moderately to severely active ulcerative colitis in adult patients.
Limitations of Use
The effectiveness of adalimumab products has not been established in patients who have lost response to or were intolerant to TNF blockers [see Clinical Studies (14.7)].
1.7 Plaque Psoriasis
YUFLYMA is indicated for the treatment of adult patients with moderate to severe chronic plaque psoriasis who are candidates for systemic therapy or phototherapy, and when other systemic therapies are medically less appropriate. YUFLYMA should only be administered to patients who will be closely monitored and have regular follow-up visits with a physician [see Warnings and Precautions (5)].
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Tuberculosis Evaluation
Prior to initiating YUFLYMA and periodically during therapy, evaluate patients for active tuberculosis and test for latent infection [see Warnings and Precautions (5.1)].
2.2 Recommended Dosage in Rheumatoid Arthritis, Psoriatic Arthritis, and Ankylosing Spondylitis
The recommended subcutaneous dosage of YUFLYMA for adult patients with rheumatoid arthritis (RA), psoriatic arthritis (PsA), or ankylosing spondylitis (AS) [see Indications and Usage (1.1, 1.3, 1.4)] is 40 mg administered every other week. Methotrexate (MTX), other non-biologic DMARDS, glucocorticoids, nonsteroidal anti-inflammatory drugs (NSAIDs), and/or analgesics may be continued during treatment with YUFLYMA. In the treatment of RA, some patients not taking concomitant MTX may derive additional benefit from increasing the dosage of YUFLYMA to 40 mg every week or 80 mg every other week.
2.3 Recommended Dosage in Juvenile Idiopathic Arthritis or Pediatric Patients with Uveitis
The recommended subcutaneous dosage of YUFLYMA for pediatric patients 2 years of age and older with polyarticular juvenile idiopathic arthritis (JIA) or pediatric uveitis [see Indications and Usage (1.2, 1.9)], based on weight, is shown below. MTX, glucocorticoids, NSAIDs, and/or analgesics may be continued during treatment with YUFLYMA.
Pediatric Weight
(2 years of age and older)Recommended Dosage 10 kg (22 lbs) to less than 15 kg (33 lbs) 10 mg every other week 15 kg (33 lbs) to less than 30 kg (66 lbs) 20 mg every other week 30 kg (66 lbs) and greater 40 mg every other week Adalimumab products have not been studied in patients with polyarticular JIA or pediatric uveitis less than 2 years of age or in patients with a weight below 10 kg.
2.4 Recommended Dosage in Crohn's Disease
Subcutaneous Adult Dosage Regimen
The recommended subcutaneous dosage of YUFLYMA for adult patients with moderately to severely active Crohn's disease (CD) is 160 mg initially on Day 1 (given in one day or split over two consecutive days), followed by 80 mg two weeks later (Day 15). Two weeks later (Day 29) begin a dosage of 40 mg every other week. Aminosalicylates and/or corticosteroids may be continued during treatment with YUFLYMA. Azathioprine, 6-mercaptopurine (6-MP) [see Warnings and Precautions (5.2)] or MTX may be continued during treatment with YUFLYMA if necessary.
Subcutaneous Pediatric Dosage Regimen
The recommended subcutaneous dosage of YUFLYMA for pediatric patients 6 years of age and older with moderately to severely active Crohn's disease (CD), based on body weight, is shown below:
Pediatric Weight Recommended Dosage Days 1 through 15 Starting on Day 29 17 kg (37 lbs) to less than 40 kg (88 lbs) Day 1: 80 mg
Day 15: 40 mg20 mg every other week 40 kg (88 lbs) and greater Day 1: 160 mg (single dose or split over two consecutive days)
Day 15: 80 mg40 mg every other week 2.5 Recommended Dosage in Ulcerative Colitis
Subcutaneous Adult Dosage Regimen
The recommended subcutaneous dosage of YUFLYMA for adult patients with moderately to severely active ulcerative colitis is 160 mg initially on Day 1 (given in one day or split over two consecutive days), followed by 80 mg two weeks later (Day 15). Two weeks later (Day 29) continue with a dosage of 40 mg every other week.
Discontinue YUFLYMA in adult patients without evidence of clinical remission by eight weeks (Day 57) of therapy. Aminosalicylates and/or corticosteroids may be continued during treatment with YUFLYMA. Azathioprine and 6-mercaptopurine (6-MP) [see Warnings and Precautions (5.2)] may be continued during treatment with YUFLYMA if necessary.
2.6 Recommended Dosage in Plaque Psoriasis or Adults with Uveitis
The recommended subcutaneous dosage of YUFLYMA for adult patients with plaque psoriasis (Ps) or uveitis (UV) [see Indications and Usage (1.7, 1.9)] is an initial dose of 80 mg, followed by 40 mg given every other week starting one week after the initial dose. The use of adalimumab products in moderate to severe chronic Ps beyond one year has not been evaluated in controlled clinical studies.
2.7 Recommended Dosage in Hidradenitis Suppurativa
Subcutaneous Adult Dosage Regimen
The recommended subcutaneous dosage of YUFLYMA for adult patients with moderate to severe hidradenitis suppurativa (HS) is an initial dose of 160 mg (given in one day or split over two consecutive days), followed by 80 mg two weeks later (Day 15). Begin 40 mg weekly or 80 mg every other week dosing two weeks later (Day 29).
Subcutaneous Pediatric Dosage Regimen
The recommended subcutaneous dosage of YUFLYMA for pediatric patients 12 years of age and older weighing at least 30 kg with moderate to severe hidradenitis suppurativa (HS), based on body weight, is shown below [see Use in Specific Populations (8.4) and Clinical Pharmacology (12.3)]:
Body Weight of Pediatric Patients
(12 years of age and older)Recommended Dosage 30 kg (66 lbs) to less than 60 kg (132 lbs) Day 1: 80 mg
Day 8 and subsequent doses: 40 mg every other week60 kg (132 lbs) and greater Day 1: 160 mg (given in one day or split over two consecutive days)
Day 15: 80 mg
Day 29 and subsequent doses: 40 mg every week or 80 mg every other week2.8 General Considerations for Administration
YUFLYMA is intended for use under the guidance and supervision of a physician. A patient may self-inject YUFLYMA or a caregiver may inject YUFLYMA using either the YUFLYMA AI or YUFLYMA prefilled syringe if a physician determines that it is appropriate, and with medical follow-up, as necessary, after proper training in subcutaneous injection technique.
YUFLYMA can be taken out of the refrigerator for about 15 to 30 minutes before injecting to allow the liquid to come to room temperature. Do not remove the cap or cover while allowing it to reach room temperature. Do not shake. Carefully inspect the solution in the YUFLYMA AI or YUFLYMA prefilled syringe for particulate matter and discoloration prior to subcutaneous administration. If particulates and discolorations are noted, do not use the product. YUFLYMA does not contain preservatives; therefore, discard unused portions of drug remaining from the syringe.
Instruct patients using the YUFLYMA AI or YUFLYMA prefilled syringe to inject the full amount in the syringe, according to the directions provided in the Instructions for Use [see Instructions for Use].
Injections should occur at separate sites in the thigh or abdomen. Rotate injection sites and do not give injections into areas where the skin is tender, bruised, red or hard.
If a dose is missed, administer the dose as soon as possible. Thereafter, resume dosing at the regular scheduled time.
-
3 DOSAGE FORMS AND STRENGTHS
YUFLYMA is a clear to opalescent, and colorless to pale brown solution available as:
-
Auto-injector (YUFLYMA AI)
Injection: 80 mg/0.8 mL in a single-dose auto-injector.
Injection: 40 mg/0.4 mL in a single-dose auto-injector. -
Prefilled Syringe with Safety Guard
Injection: 80 mg/0.8 mL in a single-dose prefilled syringe with safety guard.
Injection: 40 mg/0.4 mL in a single-dose prefilled syringe with safety guard. -
Prefilled Syringe
Injection: 80 mg/0.8 mL in a single-dose prefilled syringe.
Injection: 40 mg/0.4 mL in a single-dose prefilled syringe.
Injection: 20 mg/0.2 mL in a single-dose prefilled syringe.
Injection: 10 mg/0.1 mL in a single-dose prefilled syringe.
-
Auto-injector (YUFLYMA AI)
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Serious Infections
Patients treated with adalimumab products, including YUFLYMA, are at increased risk for developing serious infections involving various organ systems and sites that may lead to hospitalization or death. Opportunistic infections due to bacterial, mycobacterial, invasive fungal, viral, parasitic, or other opportunistic pathogens including aspergillosis, blastomycosis, candidiasis, coccidioidomycosis, histoplasmosis, legionellosis, listeriosis, pneumocystosis and tuberculosis have been reported with TNF blockers. Patients have frequently presented with disseminated rather than localized disease.
The concomitant use of a TNF blocker and abatacept or anakinra was associated with a higher risk of serious infections in patients with rheumatoid arthritis (RA); therefore, the concomitant use of YUFLYMA and these biologic products is not recommended in the treatment of patients with RA [see Warnings and Precautions (5.7, 5.11) and Drug Interactions (7.2)].
Treatment with YUFLYMA should not be initiated in patients with an active infection, including localized infections. Patients 65 years of age and older, patients with co-morbid conditions and/or patients taking concomitant immunosuppressants (such as corticosteroids or methotrexate), may be at greater risk of infection. Consider the risks and benefits of treatment prior to initiating therapy in patients:
- with chronic or recurrent infection;
- who have been exposed to tuberculosis;
- with a history of an opportunistic infection;
- who have resided or traveled in areas of endemic tuberculosis or endemic mycoses, such as histoplasmosis, coccidioidomycosis, or blastomycosis; or
- with underlying conditions that may predispose them to infection.
Tuberculosis
Cases of reactivation of tuberculosis and new onset tuberculosis infections have been reported in patients receiving adalimumab products, including patients who have previously received treatment for latent or active tuberculosis. Reports included cases of pulmonary and extrapulmonary (i.e., disseminated) tuberculosis. Evaluate patients for tuberculosis risk factors and test for latent infection prior to initiating YUFLYMA and periodically during therapy.
Treatment of latent tuberculosis infection prior to therapy with TNF blocking agents has been shown to reduce the risk of tuberculosis reactivation during therapy. Prior to initiating YUFLYMA, assess if treatment for latent tuberculosis is needed; and consider an induration of ≥ 5 mm a positive tuberculin skin test result, even for patients previously vaccinated with Bacille Calmette-Guerin (BCG).
Consider anti-tuberculosis therapy prior to initiation of YUFLYMA in patients with a past history of latent or active tuberculosis in whom an adequate course of treatment cannot be confirmed, and for patients with a negative test for latent tuberculosis but having risk factors for tuberculosis infection. Despite prophylactic treatment for tuberculosis, cases of reactivated tuberculosis have occurred in patients treated with adalimumab products. Consultation with a physician with expertise in the treatment of tuberculosis is recommended to aid in the decision whether initiating anti-tuberculosis therapy is appropriate for an individual patient.
Strongly consider tuberculosis in the differential diagnosis in patients who develop a new infection during YUFLYMA treatment, especially in patients who have previously or recently traveled to countries with a high prevalence of tuberculosis, or who have had close contact with a person with active tuberculosis.
Monitoring
Closely monitor patients for the development of signs and symptoms of infection during and after treatment with YUFLYMA, including the development of tuberculosis in patients who tested negative for latent tuberculosis infection prior to initiating therapy. Tests for latent tuberculosis infection may also be falsely negative while on therapy with YUFLYMA.
Discontinue YUFLYMA if a patient develops a serious infection or sepsis. For a patient who develops a new infection during treatment with YUFLYMA, closely monitor them, perform a prompt and complete diagnostic workup appropriate for an immunocompromised patient, and initiate appropriate antimicrobial therapy.
Invasive Fungal Infections
If patients develop a serious systemic illness and they reside or travel in regions where mycoses are endemic, consider invasive fungal infection in the differential diagnosis. Antigen and antibody testing for histoplasmosis may be negative in some patients with active infection. Consider appropriate empiric antifungal therapy, taking into account both the risk for severe fungal infection and the risks of antifungal therapy, while a diagnostic workup is being performed. To aid in the management of such patients, consider consultation with a physician with expertise in the diagnosis and treatment of invasive fungal infections.
5.2 Malignancies
Consider the risks and benefits of TNF-blocker treatment including YUFLYMA prior to initiating therapy in patients with a known malignancy other than a successfully treated non-melanoma skin cancer (NMSC) or when considering continuing a TNF blocker in patients who develop a malignancy.
Malignancies in Adults
In the controlled portions of clinical trials of some TNF-blockers, including adalimumab products, more cases of malignancies have been observed among TNF-blocker-treated adult subjects compared to control-treated adult subjects. During the controlled portions of 39 global adalimumab clinical trials in adult patients with rheumatoid arthritis (RA), psoriatic arthritis (PsA), ankylosing spondylitis (AS), Crohn's disease (CD), ulcerative colitis (UC), plaque psoriasis (Ps), hidradenitis suppurativa (HS) and uveitis (UV), malignancies, other than non-melanoma (basal cell and squamous cell) skin cancer, were observed at a rate (95% confidence interval) of 0.7 (0.48, 1.03) per 100 patient-years among 7973 adalimumab-treated subjects versus a rate of 0.7 (0.41, 1.17) per 100 patient-years among 4848 control-treated subjects (median duration of treatment of 4 months for adalimumab-treated subjects and 4 months for control-treated subjects). In 52 global controlled and uncontrolled clinical trials of adalimumab in adult patients with RA, PsA, AS, CD, UC, Ps, HS and UV, the most frequently observed malignancies, other than lymphoma and NMSC, were breast, colon, prostate, lung, and melanoma. The malignancies in adalimumab-treated subjects in the controlled and uncontrolled portions of the studies were similar in type and number to what would be expected in the general U.S. population according to the SEER database (adjusted for age, gender, and race).1
In controlled trials of other TNF blockers in adult patients at higher risk for malignancies (i.e., subjects with COPD with a significant smoking history and cyclophosphamide-treated patients with Wegener's granulomatosis), a greater portion of malignancies occurred in the TNF blocker group compared to the control group.
Non-Melanoma Skin Cancer
During the controlled portions of 39 global adalimumab clinical trials in adult patients with RA, PsA, AS, CD, UC, Ps, HS and UV, the rate (95% confidence interval) of NMSC was 0.8 (0.52, 1.09) per 100 patient-years among adalimumab-treated subjects and 0.2 (0.10, 0.59) per 100 patient-years among control-treated subjects. Examine all patients, and in particular patients with a medical history of prior prolonged immunosuppressant therapy or psoriasis patients with a history of PUVA treatment for the presence of NMSC prior to and during treatment with YUFLYMA.
Lymphoma and Leukemia
In the controlled portions of clinical trials of all the TNF-blockers in adults, more cases of lymphoma have been observed among TNF-blocker-treated subjects compared to control-treated subjects. In the controlled portions of 39 global adalimumab clinical trials in adult patients with RA, PsA, AS, CD, UC, Ps, HS and UV, 2 lymphomas occurred among 7973 adalimumab-treated subjects versus 1 among 4848 control-treated subjects. In 52 global controlled and uncontrolled clinical trials of adalimumab in adult patients with RA, PsA, AS, CD, UC, Ps, HS and UV with a median duration of approximately 0.7 years, including 24,605 subjects and over 40,215 patient-years of adalimumab, the observed rate of lymphomas was approximately 0.11 per 100 patient-years. This is approximately 3-fold higher than expected in the general U.S. population according to the SEER database (adjusted for age, gender, and race).1 Rates of lymphoma in clinical trials of adalimumab cannot be compared to rates of lymphoma in clinical trials of other TNF blockers and may not predict the rates observed in a broader patient population. Patients with RA and other chronic inflammatory diseases, particularly those with highly active disease and/or chronic exposure to immunosuppressant therapies, may be at a higher risk (up to several fold) than the general population for the development of lymphoma, even in the absence of TNF blockers. Post-marketing cases of acute and chronic leukemia have been reported in association with TNF-blocker use in RA and other indications. Even in the absence of TNF-blocker therapy, patients with RA may be at a higher risk (approximately 2-fold) than the general population for the development of leukemia.
Malignancies in Pediatric Patients and Young Adults
Malignancies, some fatal, have been reported among children, adolescents, and young adults who received treatment with TNF-blockers (initiation of therapy ≤ 18 years of age), of which YUFLYMA is a member. Approximately half the cases were lymphomas, including Hodgkin's and non-Hodgkin's lymphoma. The other cases represented a variety of different malignancies and included rare malignancies usually associated with immunosuppression and malignancies that are not usually observed in children and adolescents. The malignancies occurred after a median of 30 months of therapy (range 1 to 84 months). Most of the patients were receiving concomitant immunosuppressants. These cases were reported post-marketing and are derived from a variety of sources including registries and spontaneous postmarketing reports.
Postmarketing cases of hepatosplenic T-cell lymphoma (HSTCL), a rare type of T-cell lymphoma, have been reported in patients treated with TNF blockers including adalimumab products. These cases have had a very aggressive disease course and have been fatal. The majority of reported TNF blocker cases have occurred in patients with Crohn's disease or ulcerative colitis and the majority were in adolescent and young adult males. Almost all of these patients had received treatment with the immunosuppressants azathioprine or 6-mercaptopurine (6–MP) concomitantly with a TNF blocker at or prior to diagnosis. It is uncertain whether the occurrence of HSTCL is related to use of a TNF blocker or a TNF blocker in combination with these other immunosuppressants. The potential risk with the combination of azathioprine or 6-mercaptopurine and YUFLYMA should be carefully considered.
5.3 Hypersensitivity Reactions
Anaphylaxis and angioneurotic edema have been reported following administration of adalimumab products. If an anaphylactic or other serious allergic reaction occurs, immediately discontinue administration of YUFLYMA and institute appropriate therapy. In clinical trials of adalimumab, hypersensitivity reactions (e.g., rash, anaphylactoid reaction, fixed drug reaction, non-specified drug reaction, urticaria) have been observed.
5.4 Hepatitis B Virus Reactivation
Use of TNF blockers, including YUFLYMA, may increase the risk of reactivation of hepatitis B virus (HBV) in patients who are chronic carriers of this virus. In some instances, HBV reactivation occurring in conjunction with TNF blocker therapy has been fatal. The majority of these reports have occurred in patients concomitantly receiving other medications that suppress the immune system, which may also contribute to HBV reactivation. Evaluate patients at risk for HBV infection for prior evidence of HBV infection before initiating TNF blocker therapy. Exercise caution in prescribing TNF blockers for patients identified as carriers of HBV. Adequate data are not available on the safety or efficacy of treating patients who are carriers of HBV with anti-viral therapy in conjunction with TNF blocker therapy to prevent HBV reactivation. For patients who are carriers of HBV and require treatment with TNF blockers, closely monitor such patients for clinical and laboratory signs of active HBV infection throughout therapy and for several months following termination of therapy. In patients who develop HBV reactivation, stop YUFLYMA and initiate effective anti-viral therapy with appropriate supportive treatment. The safety of resuming TNF blocker therapy after HBV reactivation is controlled is not known. Therefore, exercise caution when considering resumption of YUFLYMA therapy in this situation and monitor patients closely.
5.5 Neurologic Reactions
Use of TNF blocking agents, including adalimumab products, has been associated with rare cases of new onset or exacerbation of clinical symptoms and/or radiographic evidence of central nervous system demyelinating disease, including multiple sclerosis (MS) and optic neuritis, and peripheral demyelinating disease, including Guillain-Barré syndrome. Exercise caution in considering the use of YUFLYMA in patients with preexisting or recent-onset central or peripheral nervous system demyelinating disorders; discontinuation of YUFLYMA should be considered if any of these disorders develop. There is a known association between intermediate uveitis and central demyelinating disorders.
5.6 Hematological Reactions
Rare reports of pancytopenia including aplastic anemia have been reported with TNF blocking agents. Adverse reactions of the hematologic system, including medically significant cytopenia (e.g., thrombocytopenia, leukopenia) have been infrequently reported with adalimumab products. The causal relationship of these reports to adalimumab products remains unclear. Advise all patients to seek immediate medical attention if they develop signs and symptoms suggestive of blood dyscrasias or infection (e.g., persistent fever, bruising, bleeding, pallor) while on YUFLYMA. Consider discontinuation of YUFLYMA therapy in patients with confirmed significant hematologic abnormalities.
5.7 Increased Risk of Infection when Used with Anakinra
Concurrent use of anakinra (an interleukin-1 antagonist) and another TNF-blocker, was associated with a greater proportion of serious infections and neutropenia and no added benefit compared with the TNF-blocker alone in patients with RA. Therefore, the combination of YUFLYMA and anakinra is not recommended [see Drug Interactions (7.2)].
5.8 Heart Failure
Cases of worsening congestive heart failure (CHF) and new onset CHF have been reported with TNF blockers. Cases of worsening CHF have also been observed with adalimumab products. Adalimumab products have not been formally studied in patients with CHF; however, in clinical trials of another TNF blocker, a higher rate of serious CHF-related adverse reactions was observed. Exercise caution when using YUFLYMA in patients who have heart failure and monitor them carefully.
5.9 Autoimmunity
Treatment with adalimumab products may result in the formation of autoantibodies and, rarely, in the development of a lupus-like syndrome or autoimmune hepatitis [see Adverse Reactions (6.1, 6.3)]. If a patient develops symptoms and findings suggestive of a lupus-like syndrome or autoimmune hepatitis following treatment with YUFLYMA, discontinue treatment and evaluate the patient.
5.10 Immunizations
In a placebo-controlled clinical trial of patients with RA, no difference was detected in anti-pneumococcal antibody response between adalimumab and placebo treatment groups when the pneumococcal polysaccharide vaccine and influenza vaccine were administered concurrently with adalimumab. Similar proportions of subjects developed protective levels of anti-influenza antibodies between adalimumab and placebo treatment groups; however, titers in aggregate to influenza antigens were moderately lower in patients receiving adalimumab. The clinical significance of this is unknown. Patients on YUFLYMA may receive concurrent vaccinations, except for live vaccines. No data are available on the secondary transmission of infection by live vaccines in patients receiving adalimumab products.
It is recommended that pediatric patients, if possible, be brought up to date with all immunizations in agreement with current immunization guidelines prior to initiating YUFLYMA therapy. Patients on YUFLYMA may receive concurrent vaccinations, except for live vaccines.
The safety of administering live or live-attenuated vaccines in infants exposed to adalimumab products in utero is unknown. Risks and benefits should be considered prior to vaccinating (live or live-attenuated) exposed infants [see Use in Specific Populations (8.1, 8.4)].
5.11 Increased Risk of Infection When Used with Abatacept
In controlled trials, the concurrent administration of TNF-blockers and abatacept was associated with a greater proportion of serious infections than the use of a TNF-blocker alone; the combination therapy, compared to the use of a TNF-blocker alone, has not demonstrated improved clinical benefit in the treatment of RA. Therefore, the combination of abatacept with TNF-blockers including YUFLYMA is not recommended [see Drug Interactions (7.2)].
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Serious Infections [see Warnings and Precautions (5.1)]
- Malignancies [see Warnings and Precautions (5.2)]
- Hypersensitivity Reactions [see Warnings and Precautions (5.3)]
- Hepatitis B Virus Reactivation [see Warnings and Precautions (5.4)]
- Neurologic Reactions [see Warnings and Precautions (5.5)]
- Hematological Reactions [see Warnings and Precautions (5.6)]
- Heart Failure [see Warnings and Precautions (5.8)]
- Autoimmunity [see Warnings and Precautions (5.9)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The most common adverse reaction with adalimumab was injection site reactions. In placebo-controlled trials, 20% of subjects treated with adalimumab developed injection site reactions (erythema and/or itching, hemorrhage, pain or swelling), compared to 14% of subjects receiving placebo. Most injection site reactions were described as mild and generally did not necessitate drug discontinuation.
The proportion of subjects who discontinued treatment due to adverse reactions during the double-blind, placebo-controlled portion of studies in subjects with RA (i.e., Studies RA-I, RA-II, RA-III and RA-IV) was 7% for subjects taking adalimumab and 4% for placebo-treated subjects. The most common adverse reactions leading to discontinuation of adalimumab in these RA studies were clinical flare reaction (0.7%), rash (0.3%) and pneumonia (0.3%).
Infections
In the controlled portions of the 39 global adalimumab clinical trials in adult subjects with RA, PsA, AS, CD, UC, Ps, HS and UV, the rate of serious infections was 4.3 per 100 patient-years in 7973 adalimumab-treated subjects versus a rate of 2.9 per 100 patient-years in 4848 control-treated subjects. Serious infections observed included pneumonia, septic arthritis, prosthetic and post-surgical infections, erysipelas, cellulitis, diverticulitis, and pyelonephritis [see Warnings and Precautions (5.1)].
Tuberculosis and Opportunistic Infections
In 52 global controlled and uncontrolled clinical trials in RA, PsA, AS, CD, UC, Ps, HS and UV that included 24,605 adalimumab-treated subjects, the rate of reported active tuberculosis was 0.20 per 100 patient-years and the rate of positive PPD conversion was 0.09 per 100 patient-years. In a subgroup of 10,113 U.S. and Canadian adalimumab-treated subjects, the rate of reported active TB was 0.05 per 100 patient-years and the rate of positive PPD conversion was 0.07 per 100 patient-years. These trials included reports of miliary, lymphatic, peritoneal, and pulmonary TB. Most of the TB cases occurred within the first eight months after initiation of therapy and may reflect recrudescence of latent disease. In these global clinical trials, cases of serious opportunistic infections have been reported at an overall rate of 0.05 per 100 patient-years. Some cases of serious opportunistic infections and TB have been fatal [see Warnings and Precautions (5.1)].
Autoantibodies
In the rheumatoid arthritis-controlled trials, 12% of subjects treated with adalimumab and 7% of placebo-treated subjects that had negative baseline ANA titers developed positive titers at week 24. Two subjects out of 3046 treated with adalimumab developed clinical signs suggestive of new-onset lupus-like syndrome. The subjects improved following discontinuation of therapy. No subjects developed lupus nephritis or central nervous system symptoms. The impact of long-term treatment with adalimumab products on the development of autoimmune diseases is unknown.
Liver Enzyme Elevations
There have been reports of severe hepatic reactions including acute liver failure in subjects receiving TNF-blockers. In controlled Phase 3 trials of adalimumab (40 mg SC every other week) in subjects with RA, PsA, and AS with control period duration ranging from 4 to 104 weeks, ALT elevations ≥ 3 × ULN occurred in 3.5% of adalimumab-treated subjects and 1.5% of control-treated subjects. Since many of these subjects in these trials were also taking medications that cause liver enzyme elevations (e.g., NSAIDS, MTX), the relationship between adalimumab and the liver enzyme elevations is not clear. In a controlled Phase 3 trial of adalimumab in subjects with polyarticular JIA who were 4 to 17 years, ALT elevations ≥ 3 × ULN occurred in 4.4% of adalimumab-treated subjects and 1.5% of control-treated subjects (ALT more common than AST); liver enzyme test elevations were more frequent among those treated with the combination of adalimumab and MTX than those treated with adalimumab alone. In general, these elevations did not lead to discontinuation of adalimumab treatment. No ALT elevations ≥ 3 × ULN occurred in the open-label study of adalimumab in subjects with polyarticular JIA who were 2 to < 4 years.
In controlled Phase 3 trials of adalimumab (initial doses of 160 mg and 80 mg, or 80 mg and 40 mg on Days 1 and 15, respectively, followed by 40 mg every other week) in adult subjects with Crohn's disease with a control period duration ranging from 4 to 52 weeks, ALT elevations ≥ 3 × ULN occurred in 0.9% of adalimumab-treated subjects and 0.9% of control-treated subjects. In the Phase 3 trial of adalimumab in pediatric subjects with Crohn's disease which evaluated efficacy and safety of two body weight based maintenance dose regimens following body weight based induction therapy up to 52 weeks of treatment, ALT elevations ≥ 3 × ULN occurred in 2.6% (5/192) of subjects, of whom 4 were receiving concomitant immunosuppressants at baseline; none of these subjects discontinued due to abnormalities in ALT tests. In controlled Phase 3 trials of adalimumab (initial doses of 160 mg and 80 mg on Days 1 and 15 respectively, followed by 40 mg every other week) in adult subjects with UC with control period duration ranging from 1 to 52 weeks, ALT elevations ≥ 3 × ULN occurred in 1.5% of adalimumab-treated subjects and 1.0% of control-treated subjects. In controlled Phase 3 trials of adalimumab (initial dose of 80 mg then 40 mg every other week) in subjects with Ps with control period duration ranging from 12 to 24 weeks, ALT elevations ≥ 3 × ULN occurred in 1.8% of adalimumab-treated subjects and 1.8% of control-treated subjects. In controlled trials of adalimumab (initial doses of 160 mg at Week 0 and 80 mg at Week 2, followed by 40 mg every week starting at Week 4), in subjects with HS with a control period duration ranging from 12 to 16 weeks, ALT elevations ≥ 3 × ULN occurred in 0.3% of adalimumab-treated subjects and 0.6% of control-treated subjects. In controlled trials of adalimumab (initial doses of 80 mg at Week 0 followed by 40 mg every other week starting at Week 1) in adult subjects with uveitis with an exposure of 165.4 PYs and 119.8 PYs in adalimumab-treated and control-treated subjects, respectively, ALT elevations ≥ 3 x ULN occurred in 2.4% of adalimumab-treated subjects and 2.4% of control-treated subjects.
Other Adverse Reactions
Rheumatoid Arthritis Clinical Studies
The data described below reflect exposure to adalimumab in 2468 subjects, including 2073 exposed for 6 months, 1497 exposed for greater than one year and 1380 in adequate and well-controlled studies (Studies RA-I, RA-II, RA-III, and RA-IV). Adalimumab was studied primarily in placebo-controlled trials and in long-term follow up studies for up to 36 months duration. The population had a mean age of 54 years, 77% were female, 91% were Caucasian and had moderately to severely active rheumatoid arthritis. Most subjects received 40 mg adalimumab every other week [see Clinical Studies (14.1)].
Table 1 summarizes reactions reported at a rate of at least 5% in subjects treated with adalimumab 40 mg every other week compared to placebo and with an incidence higher than placebo. In Study RA-III, the types and frequencies of adverse reactions in the second year open-label extension were similar to those observed in the one-year double-blind portion.
Table 1. Adverse Reactions Reported by ≥ 5% of Subjects with Adalimumab During Placebo-Controlled Period of Pooled RA Studies (Studies RA-I, RA-II, RA-III, and RA-IV) Adalimumab
40 mg subcutaneous
Every Other WeekPlacebo (N=705) (N=690) Adverse Reaction (Preferred Term) Respiratory Upper respiratory infection 17% 13% Sinusitis 11% 9% Flu syndrome 7% 6% Gastrointestinal Nausea 9% 8% Abdominal pain 7% 4% Laboratory Tests* Laboratory test abnormal 8% 7% Hypercholesterolemia 6% 4% Hyperlipidemia 7% 5% Hematuria 5% 4% Alkaline phosphatase increased 5% 3% Other Headache 12% 8% Rash 12% 6% Accidental injury 10% 8% Injection site reaction ** 8% 1% Back pain 6% 4% Urinary tract infection 8% 5% Hypertension 5% 3% * Laboratory test abnormalities were reported as adverse reactions in European trials
** Does not include injection site erythema, itching, hemorrhage, pain or swellingLess Common Adverse Reactions in Rheumatoid Arthritis Clinical Studies
Other infrequent serious adverse reactions that do not appear in the Warnings and Precautions or Adverse Reaction sections that occurred at an incidence of less than 5% in adalimumab-treated subjects in RA studies (RA-I, RA-II, RA-III, and RA-IV) were:
Body As A Whole: Pain in extremity, pelvic pain, surgery, thorax pain
Cardiovascular System: Arrhythmia, atrial fibrillation, chest pain, coronary artery disorder, heart arrest, hypertensive encephalopathy, myocardial infarct, palpitation, pericardial effusion, pericarditis, syncope, tachycardia
Digestive System: Cholecystitis, cholelithiasis, esophagitis, gastroenteritis, gastrointestinal hemorrhage, hepatic necrosis, vomiting
Endocrine System: Parathyroid disorder
Hemic And Lymphatic System: Agranulocytosis, polycythemia
Metabolic And Nutritional Disorders: Dehydration, healing abnormal, ketosis, paraproteinemia, peripheral edema
Musculo-Skeletal System: Arthritis, bone disorder, bone fracture (not spontaneous), bone necrosis, joint disorder, muscle cramps, myasthenia, pyogenic arthritis, synovitis, tendon disorder
Neoplasia: Adenoma
Nervous System: Confusion, paresthesia, subdural hematoma, tremor
Respiratory System: Asthma, bronchospasm, dyspnea, lung function decreased, pleural effusion
Special Senses: Cataract
Thrombosis: Thrombosis leg
Urogenital System: Cystitis, kidney calculus, menstrual disorder
Juvenile Idiopathic Arthritis Clinical Studies
In general, the adverse reactions in the adalimumab-treated subjects in the polyarticular juvenile idiopathic arthritis (JIA) trials (Studies JIA-I and JIA-II) [see Clinical Studies (14.2)] were similar in frequency and type to those seen in adult subjects [see Warnings and Precautions (5), Adverse Reactions (6)]. Important findings and differences from adults are discussed in the following paragraphs.
In Study JIA-I, adalimumab was studied in 171 subjects who were 4 to 17 years of age, with polyarticular JIA. Severe adverse reactions reported in the study included neutropenia, streptococcal pharyngitis, increased aminotransferases, herpes zoster, myositis, metrorrhagia, and appendicitis. Serious infections were observed in 4% of subjects within approximately 2 years of initiation of treatment with adalimumab and included cases of herpes simplex, pneumonia, urinary tract infection, pharyngitis, and herpes zoster.
In Study JIA-I, 45% of subjects experienced an infection while receiving adalimumab with or without concomitant MTX in the first 16 weeks of treatment. The types of infections reported in adalimumab-treated subjects were generally similar to those commonly seen in polyarticular JIA subjects who are not treated with TNF blockers. Upon initiation of treatment, the most common adverse reactions occurring in this population treated with adalimumab were injection site pain and injection site reaction (19% and 16%, respectively). A less commonly reported adverse event in subjects receiving adalimumab was granuloma annulare which did not lead to discontinuation of adalimumab treatment.
In the first 48 weeks of treatment in Study JIA-I, non-serious hypersensitivity reactions were seen in approximately 6% of subjects and included primarily localized allergic hypersensitivity reactions and allergic rash.
In Study JIA-I, 10% of subjects treated with adalimumab who had negative baseline anti-dsDNA antibodies developed positive titers after 48 weeks of treatment. No subject developed clinical signs of autoimmunity during the clinical trial.
Approximately 15% of subjects treated with adalimumab developed mild-to-moderate elevations of creatine phosphokinase (CPK) in Study JIA-I. Elevations exceeding 5 times the upper limit of normal were observed in several subjects. CPK concentrations decreased or returned to normal in all subjects. Most subjects were able to continue adalimumab without interruption.
In Study JIA-II, adalimumab was studied in 32 subjects who were 2 to < 4 years of age or 4 years of age and older weighing < 15 kg with polyarticular JIA. The safety profile for this population was similar to the safety profile seen in subjects 4 to 17 years of age with polyarticular JIA.
In Study JIA-II, 78% of subjects experienced an infection while receiving adalimumab. These included nasopharyngitis, bronchitis, upper respiratory tract infection, otitis media, and were mostly mild to moderate in severity. Serious infections were observed in 9% of subjects receiving adalimumab in the study and included dental caries, rotavirus gastroenteritis, and varicella.
In Study JIA-II, non-serious allergic reactions were observed in 6% of subjects and included intermittent urticaria and rash, which were all mild in severity.
Psoriatic Arthritis and Ankylosing Spondylitis Clinical Studies
Adalimumab has been studied in 395 subjects with psoriatic arthritis (PsA) in two placebo-controlled trials and in an open label study and in 393 subjects with ankylosing spondylitis (AS) in two placebo-controlled studies [see Clinical Studies (14.3, 14.4)]. The safety profile for subjects with PsA and AS treated with adalimumab 40 mg every other week was similar to the safety profile seen in subjects with RA, adalimumab Studies RA-I through IV.
Crohn's Disease Clinical Studies
Adults: The safety profile of adalimumab in 1478 adult subjects with Crohn's disease from four placebo-controlled and two open-label extension studies [see Clinical Studies (14.5)] was similar to the safety profile seen in subjects with RA.
Pediatric Patients 6 Years to 17 Years: The safety profile of adalimumab in 192 pediatric subjects from one double-blind study (Study PCD-I) and one open-label extension study [see Clinical Studies (14.6)] was similar to the safety profile seen in adult subjects with Crohn's disease.
During the 4-week open label induction phase of Study PCD-I, the most common adverse reactions occurring in the pediatric population treated with adalimumab were injection site pain and injection site reaction (6% and 5%, respectively).
A total of 67% of children experienced an infection while receiving adalimumab in Study PCD-I. These included upper respiratory tract infection and nasopharyngitis.
A total of 5% of children experienced a serious infection while receiving adalimumab in Study PCD-I. These included viral infection, device related sepsis (catheter), gastroenteritis, H1N1 influenza, and disseminated histoplasmosis.
In Study PCD-I, allergic reactions were observed in 5% of children which were all non-serious and were primarily localized reactions.
Ulcerative Colitis Clinical Studies
Adults: The safety profile of adalimumab in 1010 adult subjects with ulcerative colitis (UC) from two placebo-controlled studies and one open-label extension study [see Clinical Studies (14.7)] was similar to the safety profile seen in subjects with RA.
Plaque Psoriasis Clinical Studies
Adalimumab has been studied in 1696 subjects with plaque psoriasis (Ps) in placebo-controlled and open-label extension studies [see Clinical Studies (14.8)]. The safety profile for subjects with Ps treated with adalimumab was similar to the safety profile seen in subjects with RA with the following exceptions. In the placebo-controlled portions of the clinical trials in Ps subjects, adalimumab-treated subjects had a higher incidence of arthralgia when compared to controls (3% vs. 1%).
Hidradenitis Suppurativa Clinical Studies
Adalimumab has been studied in 727 subjects with hidradenitis suppurativa (HS) in three placebo-controlled studies and one open-label extension study [see Clinical Studies (14.9)]. The safety profile for subjects with HS treated with adalimumab weekly was consistent with the known safety profile of adalimumab.
Flare of HS, defined as ≥ 25% increase from baseline in abscesses and inflammatory nodule counts and with a minimum of 2 additional lesions, was documented in 22 (22%) of the 100 subjects who were withdrawn from adalimumab treatment following the primary efficacy timepoint in two studies.
Uveitis Clinical Studies
Adalimumab has been studied in 464 adult subjects with uveitis (UV) in placebo-controlled and open-label extension studies and in 90 pediatric subjects with uveitis (Study PUV-I) [see Clinical Studies (14.10, 14.11)]. The safety profile for subjects with UV treated with adalimumab was similar to the safety profile seen in subjects with RA.
6.2 Immunogenicity
The observed incidence of anti-drug antibodies is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of anti-drug antibodies in the studies described below with the incidence of anti-drug antibodies in other studies, including those of adalimumab or of other adalimumab products.
There are two assays that have been used to measure anti-adalimumab antibodies. With the ELISA, antibodies to adalimumab could be detected only when serum adalimumab concentrations were < 2 mcg/mL. The ECL assay can detect anti-adalimumab antibody titers independent of adalimumab concentrations in the serum samples. The incidence of anti-adalimumab antibody (AAA) development in patients treated with adalimumab are presented in Table 2.
Table 2: Anti-Adalimumab Antibody Development Determined by ELISA and ECL Assay in Patients Treated with Adalimumab Indications Study Duration Anti-Adalimumab Antibody Incidence by ELISA (n/N) Anti-Adalimumab Antibody Incidence by ECL Assay (n/N) In all patients who received adalimumab In patients with serum adalimumab concentrations < 2 mcg/mL Rheumatoid Arthritisa 6 to 12 months 5% (58/1062) NR NA Juvenile Idiopathic Arthritis (JIA) 4 to 17 years of ageb 48 weeks 16% (27/171) NR NA 2 to 4 years of age or ≥ 4 years of age and weighing < 15 kg 24 weeks 7% (1/15)c NR NA Psoriatic Arthritisd 48 weekse 13% (24/178) NR NA Ankylosing Spondylitis 24 weeks 9% (16/185) NR NA Adult Crohn’s Disease 56 weeks 3% (7/269) 8% (7/86) NA Pediatric Crohn’s Disease 52 weeks 3% (6/182) 10% (6/58) NA Adult Ulcerative Colitis 52 weeks 5% (19/360) 21% (19/92) NA Plaque Psoriasisf Up to 52 weeksg 8% (77/920) 21% (77/372) NA Hidradenitis Suppurativa 36 weeks 7% (30/461) 28% (58/207)h 61% (272/445)i Non-infectious Uveitis 52 weeks 5% (12/249) 21% (12/57) 40% (99/249)j n: number of patients with anti-adalimumab antibody; NR: not reported; NA: Not applicable (not performed)
a In patients receiving concomitant methotrexate (MTX), the incidence of anti-adalimumab antibody was 1% compared to 12% with adalimumab monotherapy
b In patients receiving concomitant MTX, the incidence of anti-adalimumab antibody was 6% compared to 26% with adalimumab monotherapy
c This patient received concomitant MTX
d In patients receiving concomitant MTX, the incidence of antibody development was 7% compared to 1% in RA
e Subjects enrolled after completing 2 previous studies of 24 weeks or 12 weeks of treatments.
f In plaque psoriasis patients who were on adalimumab monotherapy and subsequently withdrawn from the treatment, the rate of antibodies to adalimumab after retreatment was similar to the rate observed prior to withdrawal
g One 12-week Phase 2 study and one 52-week Phase 3 study
h Among subjects in the 2 Phase 3 studies who stopped adalimumab treatment for up to 24 weeks and in whom adalimumab serum levels subsequently declined to <2 mcg/mL (approximately 22% of total subjects studied)
i No apparent association between antibody development and safety was observed
j No correlation of antibody development to safety or efficacy outcomes was observedRheumatoid Arthritis and Psoriatic Arthritis: Subjects in Studies RA-I, RA-II, and RA-III were tested at multiple time points for antibodies to adalimumab using the ELISA during the 6- to 12-month period. No apparent correlation of antibody development to adverse reactions was observed. With monotherapy, subjects receiving every other week dosing may develop antibodies more frequently than those receiving weekly dosing. In subjects receiving the recommended dosage of 40 mg every other week as monotherapy, the ACR 20 response was lower among antibody-positive subjects than among antibody-negative subjects. The long-term immunogenicity of adalimumab products is unknown.
6.3 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of adalimumab products. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to adalimumab products exposure.
Gastrointestinal disorders: Diverticulitis, large bowel perforations including perforations associated with diverticulitis and appendiceal perforations associated with appendicitis, pancreatitis
General disorders and administration site conditions: Pyrexia
Hepato-biliary disorders: Liver failure, hepatitis, autoimmune hepatitis
Immune system disorders: Sarcoidosis
Neoplasms benign, malignant and unspecified (including cysts and polyps): Merkel Cell Carcinoma (neuroendocrine carcinoma of the skin)
Nervous system disorders: Demyelinating disorders (e.g., optic neuritis, Guillain-Barré syndrome), cerebrovascular accident
Respiratory disorders: Interstitial lung disease, including pulmonary fibrosis, pulmonary embolism
Skin reactions: Stevens Johnson Syndrome, cutaneous vasculitis, erythema multiforme, new or worsening psoriasis (all sub-types including pustular and palmoplantar), alopecia, lichenoid skin reaction
Vascular disorders: Systemic vasculitis, deep vein thrombosis
-
7 DRUG INTERACTIONS
7.1 Methotrexate
Adalimumab has been studied in rheumatoid arthritis (RA) patients taking concomitant methotrexate (MTX). Although MTX reduced the apparent clearance of adalimumab, the data do not suggest the need for dose adjustment of either YUFLYMA or MTX [see Clinical Pharmacology (12.3)].
7.2 Biological Products
In clinical studies in patients with RA, an increased risk of serious infections has been observed with the combination of TNF blockers with anakinra or abatacept, with no added benefit; therefore, use of YUFLYMA with abatacept or anakinra is not recommended in patients with RA [see Warnings and Precautions (5.7, 5.11)]. A higher rate of serious infections has also been observed in patients with RA treated with rituximab who received subsequent treatment with a TNF blocker. There is insufficient information regarding the concomitant use of YUFLYMA and other biologic products for the treatment of RA, PsA, AS, CD, UC, Ps, HS and UV. Concomitant administration of YUFLYMA with other biologic DMARDS (e.g., anakinra and abatacept) or other TNF blockers is not recommended based upon the possible increased risk for infections and other potential pharmacological interactions.
7.3 Live Vaccines
Avoid the use of live vaccines with YUFLYMA [see Warnings and Precautions (5.10)].
7.4 Cytochrome P450 Substrates
The formation of CYP450 enzymes may be suppressed by increased concentrations of cytokines (e.g., TNFα, IL-6) during chronic inflammation. It is possible for products that antagonize cytokine activity, such as adalimumab products, to influence the formation of CYP450 enzymes. Upon initiation or discontinuation of YUFLYMA in patients being treated with CYP450 substrates with a narrow therapeutic index, monitoring of the effect (e.g., warfarin) or drug concentration (e.g., cyclosporine or theophylline) is recommended and the individual dose of the drug product may be adjusted as needed.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Available studies with use of adalimumab during pregnancy do not reliably establish an association between adalimumab and major birth defects. Clinical data are available from the Organization of Teratology Information Specialists (OTIS)/MotherToBaby Pregnancy Registry in pregnant women with rheumatoid arthritis (RA) or Crohn's disease (CD) treated with adalimumab. Registry results showed a rate of 10% for major birth defects with first trimester use of adalimumab in pregnant women with RA or CD and a rate of 7.5% for major birth defects in the disease-matched comparison cohort. The lack of pattern of major birth defects is reassuring and differences between exposure groups may have impacted the occurrence of birth defects (see Data).
Adalimumab is actively transferred across the placenta during the third trimester of pregnancy and may affect immune response in the in-utero exposed infant (see Clinical Considerations). In an embryo-fetal perinatal development study conducted in cynomolgus monkeys, no fetal harm or malformations were observed with intravenous administration of adalimumab during organogenesis and later in gestation, at doses that produced exposures up to approximately 373 times the maximum recommended human dose (MRHD) of 40 mg subcutaneous without methotrexate (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated populations is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Clinical Considerations
Disease-associated maternal and embryo/fetal risk
Published data suggest that the risk of adverse pregnancy outcomes in women with RA or inflammatory bowel disease (IBD) is associated with increased disease activity. Adverse pregnancy outcomes include preterm delivery (before 37 weeks of gestation), low birth weight (less than 2500 g) infants, and small for gestational age at birth.
Fetal/Neonatal Adverse Reactions
Monoclonal antibodies are increasingly transported across the placenta as pregnancy progresses, with the largest amount transferred during the third trimester [see Data]. Risks and benefits should be considered prior to administering live or live-attenuated vaccines to infants exposed to adalimumab products in utero [see Use in Specific Populations (8.4)].
Data
Human Data
A prospective cohort pregnancy exposure registry conducted by OTIS/MotherToBaby in the U.S. and Canada between 2004 and 2016 compared the risk of major birth defects in live-born infants of 221 women (69 RA, 152 CD) treated with adalimumab during the first trimester and 106 women (74 RA, 32 CD) not treated with adalimumab.
The proportion of major birth defects among live-born infants in the adalimumab-treated and untreated cohorts was 10% (8.7% RA, 10.5% CD) and 7.5% (6.8% RA, 9.4% CD), respectively. The lack of pattern of major birth defects is reassuring and differences between exposure groups may have impacted the occurrence of birth defects. This study cannot reliably establish whether there is an association between adalimumab and major birth defects because of methodological limitations of the registry, including small sample size, the voluntary nature of the study, and the non-randomized design.
In an independent clinical study conducted in ten pregnant women with IBD treated with adalimumab, adalimumab concentrations were measured in maternal serum as well as in cord blood (n=10) and infant serum (n=8) on the day of birth. The last dose of adalimumab was given between 1 and 56 days prior to delivery. Adalimumab concentrations were 0.16-19.7 mcg/mL in cord blood, 4.28-17.7 mcg/mL in infant serum, and 0-16.1 mcg/mL in maternal serum. In all but one case, the cord blood concentration of adalimumab was higher than the maternal serum concentration, suggesting adalimumab actively crosses the placenta. In addition, one infant had serum concentrations at each of the following: 6 weeks (1.94 mcg/mL), 7 weeks (1.31 mcg/mL), 8 weeks (0.93 mcg/mL), and 11 weeks (0.53 mcg/mL), suggesting adalimumab can be detected in the serum of infants exposed in utero for at least 3 months from birth.
Animal Data
In an embryo-fetal perinatal development study, pregnant cynomolgus monkeys received adalimumab from gestation days 20 to 97 at doses that produced exposures up to 373 times that achieved with the MRHD without methotrexate (on an AUC basis with maternal IV doses up to 100 mg/kg/week). Adalimumab did not elicit harm to the fetuses or malformations.
8.2 Lactation
Risk Summary
Limited data from case reports in the published literature describe the presence of adalimumab in human milk at infant doses of 0.1% to 1% of the maternal serum concentration. Published data suggest that the systemic exposure to a breastfed infant is expected to be low because adalimumab is a large molecule and is degraded in the gastrointestinal tract. However, the effects of local exposure in the gastrointestinal tract are unknown. There are no reports of adverse effects of adalimumab products on the breastfed infant and no effects on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for YUFLYMA and any potential adverse effects on the breastfed child from YUFLYMA or from the underlying maternal condition.
8.4 Pediatric Use
The safety and effectiveness of YUFLYMA have not been established in pediatric patients with psoriatic arthritis, ankylosing spondylitis, or plaque psoriasis.
The safety and effectiveness of YUFLYMA have been established for:
- reducing signs and symptoms of moderately to severely active polyarticular JIA in pediatric patients 2 years of age and older.
- the treatment of moderately to severely active Crohn's disease in pediatric patients 6 years of age and older.
- the treatment of moderate to severe hidradenitis suppurativa in patients 12 years of age and older.
- the treatment of non-infectious intermediate, posterior, and panuveitis in pediatric patients 2 years of age and older.
A pediatric assessment for YUFLYMA demonstrates that YUFLYMA is safe and effective for pediatric patients in an indication for which Humira (adalimumab) is approved. However, YUFLYMA is not approved for such indication due to marketing exclusivity for Humira (adalimumab).
Due to their inhibition of TNFα, adalimumab products administered during pregnancy could affect immune response in the in utero-exposed newborn and infant. Data from eight infants exposed to adalimumab in utero suggest adalimumab crosses the placenta [see Use in Specific Populations (8.1)]. The clinical significance of elevated adalimumab concentrations in infants is unknown. The safety of administering live or live-attenuated vaccines in exposed infants is unknown. Risks and benefits should be considered prior to vaccinating (live or live-attenuated) exposed infants.
Post-marketing cases of lymphoma, including hepatosplenic T-cell lymphoma and other malignancies, some fatal, have been reported among children, adolescents, and young adults who received treatment with TNF-blockers including adalimumab products [see Warnings and Precautions (5.2)].
Juvenile Idiopathic Arthritis
The safety and effectiveness of YUFLYMA for the treatment of moderately to severely active polyarticular JIA have been established in pediatric patients 2 years of age and older. Use for this indication is supported by evidence from an adequate and well-controlled study (Study JIA) of adalimumab in patients 4 to 17 years of age [see Clinical Studies (14.2)] and a safety study (Study JIA-II) of adalimumab in patients 2 to < 4 years of age where the safety profile was similar to patients 4 to 17 years of age [see Adverse Reactions (6.1)]. Adalimumab products have not been studied in patients with polyarticular JIA less than 2 years of age or in patients with a weight below 10 kg.
The safety of adalimumab in pediatric patients in the polyarticular JIA trials was generally similar to that observed in adults with certain exceptions [see Adverse Reactions (6.1)].
The safety and effectiveness of YUFLYMA have not been established in pediatric patients with JIA less than 2 years of age.
Pediatric Crohn's Disease
The safety and effectiveness of YUFLYMA for the treatment of moderately to severely active Crohn's disease have been established in pediatric patients 6 years of age and older. Use of YUFLYMA for this indication is supported by evidence from adequate and well-controlled studies in adults with additional data from a randomized, double-blind, 52-week clinical study of two dose concentrations of adalimumab in 192 pediatric patients (6 years to 17 years of age) [see Adverse Reactions (6.1), Clinical Pharmacology (12.2, 12.3), Clinical Studies (14.6)]. The adverse reaction profile in patients 6 years to 17 years of age was similar to adults.
The safety and effectiveness of YUFLYMA have not been established in pediatric patients with Crohn's disease less than 6 years of age.
Pediatric Uveitis
The safety and effectiveness of YUFLYMA for the treatment of non-infectious, intermediate, posterior, and panuveitis have been established in pediatric patients 2 years of age and older. Use of YUFLYMA for this indication is supported by evidence from adequate and well-controlled studies of adalimumab in adults and a 2:1 randomized, controlled clinical study of adalimumab in 90 pediatric patients [see Clinical Studies (14.11)]. The safety and effectiveness of YUFLYMA have not been established in pediatric patients with uveitis less than 2 years of age.
Hidradenitis Suppurativa
Use of YUFLYMA in pediatric patients 12 years of age and older for the treatment of moderate to severe HS is supported by evidence from adequate and well-controlled studies of adalimumab in adult HS patients. Additional population pharmacokinetic modeling and simulation predicted that weight-based dosing of adalimumab in pediatric patients 12 years of age and older can provide generally similar exposure to adult HS patients. The course of HS is sufficiently similar in adult and adolescent patients to allow extrapolation of data from adult to adolescent patients. The recommended dosage in pediatric patients 12 years of age or older is based on body weight [see Dosage and Administration (2.7), Clinical Pharmacology (12.3), and Clinical Studies (14.9)].
The safety and effectiveness of YUFLYMA have not been established in patients less than 12 years of age with HS.
8.5 Geriatric Use
In clinical studies of RA (Studies RA-I, RA-II, RA-III, and RA-IV), a total of 519 subjects 65 years of age and older, including 107 subjects 75 years of age and older, received adalimumab. No overall difference in effectiveness was observed between these subjects and younger adult subjects.
The frequency of serious infection and malignancy among adalimumab treated subjects 65 years of age and older was higher than for those less than 65 years of age. Consider the benefits and risks of YUFLYMA in patients 65 years of age and older. In patients treated with YUFLYMA, closely monitor for the development of infection or malignancy [see Warnings and Precautions (5.1, 5.2)].
-
10 OVERDOSAGE
Doses up to 10 mg/kg have been administered to patients in clinical trials without evidence of dose-limiting toxicities. In case of overdosage, it is recommended that the patient be monitored for any signs or symptoms of adverse reactions or effects and appropriate symptomatic treatment instituted immediately.
Consider contacting the Poison Help line (1-800-222-1222) or medical toxicologist for additional overdose management recommendations.
-
11 DESCRIPTION
Adalimumab-aaty is a tumor necrosis factor blocker. Adalimumab-aaty is a recombinant human IgG1 monoclonal antibody created using phage display technology resulting in an antibody with human derived heavy and light chain variable regions and human IgG1:k constant regions. Adalimumab-aaty is produced by recombinant DNA technology in a mammalian cell (Chinese Hamster Ovary (CHO)) expression system and is purified by a process that includes specific viral inactivation and removal steps. It consists of 1330 amino acids and has a molecular weight of approximately 148 kilodaltons.
YUFLYMA (adalimumab-aaty) injection is supplied as a sterile, preservative-free solution for subcutaneous administration. The drug product is supplied as either a single-dose, auto-injector (YUFLYMA AI), as a single-dose, 1 mL prefilled syringe with safety guard, or a single-dose, 1 mL prefilled syringe. Enclosed within the auto-injector is a single-dose, 1 mL prefilled syringe. The solution of YUFLYMA is clear to opalescent, and colorless to pale brown, with a pH of about 5.2.
Each 80 mg/0.8 mL prefilled syringe or prefilled syringe with safety guard or auto-injector delivers 0.8 mL (80 mg) of drug product. Each 0.8 mL of YUFLYMA contains adalimumab-aaty (80 mg), acetic acid (0.13 mg), glycine (15.02 mg), polysorbate 80 (0.8 mg), sodium acetate (0.48 mg) and Water for Injection, USP.
Each 40 mg/0.4 mL prefilled syringe or prefilled syringe with safety guard or auto-injector delivers 0.4 mL (40 mg) of drug product. Each 0.4 mL of YUFLYMA contains adalimumab-aaty (40 mg), acetic acid (0.06 mg), glycine (7.51 mg), polysorbate 80 (0.4 mg), sodium acetate (0.24 mg) and Water for Injection, USP.
Each 20 mg/0.2 mL prefilled syringe delivers 0.2 mL (20 mg) of drug product. Each 0.2 mL of YUFLYMA contains adalimumab-aaty (20 mg), acetic acid (0.03 mg), glycine (3.75 mg), polysorbate 80 (0.2 mg), sodium acetate (0.12 mg) and Water for Injection, USP.
Each 10 mg/0.1 mL prefilled syringe delivers 0.1 mL (10 mg) of drug product. Each 0.1 mL of YUFLYMA contains adalimumab-aaty (10 mg), acetic acid (0.02 mg), glycine (1.88 mg), polysorbate 80 (0.1 mg), sodium acetate (0.06 mg) and Water for Injection, USP.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Adalimumab products bind specifically to TNF-alpha and block its interaction with the p55 and p75 cell surface TNF receptors. Adalimumab products also lyse surface TNF expressing cells in vitro in the presence of complement. Adalimumab products do not bind or inactivate lymphotoxin (TNF-beta). TNF is a naturally occurring cytokine that is involved in normal inflammatory and immune responses. Elevated concentrations of TNF are found in the synovial fluid of patients with RA, JIA, PsA, and AS and play an important role in both the pathologic inflammation and the joint destruction that are hallmarks of these diseases. Increased concentrations of TNF are also found in psoriasis plaques. In Ps, treatment with YUFLYMA may reduce the epidermal thickness and infiltration of inflammatory cells. The relationship between these pharmacodynamic activities and the mechanism(s) by which adalimumab products exert their clinical effects is unknown.
Adalimumab products also modulate biological responses that are induced or regulated by TNF, including changes in the concentrations of adhesion molecules responsible for leukocyte migration (ELAM-1, VCAM-1, and ICAM-1 with an IC50 of 1-2 × 10-10 M).
12.2 Pharmacodynamics
After treatment with adalimumab, a decrease in concentrations of acute phase reactants of inflammation (C-reactive protein [CRP] and erythrocyte sedimentation rate [ESR]) and serum cytokines (IL-6) was observed compared to baseline in patients with rheumatoid arthritis. A decrease in CRP concentrations was also observed in patients with Crohn's disease, ulcerative colitis and hidradenitis suppurativa. Serum concentrations of matrix metalloproteinases (MMP-1 and MMP-3) that produce tissue remodeling responsible for cartilage destruction were also decreased after adalimumab administration.
12.3 Pharmacokinetics
The pharmacokinetics of adalimumab were linear over the dose range of 0.5 to 10 mg/kg following administration of a single intravenous dose (adalimumab products are not approved for intravenous use). Following 20, 40, and 80 mg every other week and every week subcutaneous administration, adalimumab mean serum trough concentrations at steady state increased approximately proportionally with dose in RA patients. The mean terminal half-life was approximately 2 weeks, ranging from 10 to 20 days across studies. Healthy subjects and patients with RA displayed similar adalimumab pharmacokinetics.
Adalimumab exposure in patients treated with 80 mg every other week is estimated to be comparable with that in patients treated with 40 mg every week.
Absorption
The average absolute bioavailability of adalimumab following a single 40 mg subcutaneous dose was 64%. The mean time to reach the maximum concentration was 5.5 days (131 ± 56 hours) and the maximum serum concentration was 4.7 ± 1.6 mcg/mL in healthy subjects following a single 40 mg subcutaneous administration of adalimumab.
Distribution
The distribution volume (Vss) ranged from 4.7 to 6.0 L following intravenous administration of doses ranging from 0.25 to 10 mg/kg in RA patients.
Elimination
The single dose pharmacokinetics of adalimumab in RA patients were determined in several studies with intravenous doses ranging from 0.25 to 10 mg/kg. The systemic clearance of adalimumab is approximately 12 mL/hr. In long-term studies with dosing more than two years, there was no evidence of changes in clearance over time in RA patients.
Patient Population
Rheumatoid Arthritis and Ankylosing Spondylitis: In patients receiving 40 mg adalimumab every other week, adalimumab mean steady-state trough concentrations were approximately 5 mcg/mL and 8 to 9 mcg/mL, without and with MTX concomitant treatment, respectively. Adalimumab concentrations in the synovial fluid from five rheumatoid arthritis patients ranged from 31 to 96% of those in serum. The pharmacokinetics of adalimumab in patients with AS were similar to those in patients with RA.
Psoriatic Arthritis: In patients receiving 40 mg every other week, adalimumab mean steady-state trough concentrations were 6 to 10 mcg/mL and 8.5 to 12 mcg/mL, without and with MTX concomitant treatment, respectively.
Plaque Psoriasis: Adalimumab mean steady-state trough concentration was approximately 5 to 6 mcg/mL during adalimumab 40 mg every other week treatment.
Adult Uveitis: Adalimumab mean steady concentration was approximately 8 to 10 mcg/mL during adalimumab 40 mg every other week treatment.
Adult Hidradenitis Suppurativa: Adalimumab trough concentrations were approximately 7 to 8 mcg/mL at Week 2 and Week 4, respectively, after receiving 160 mg on Week 0 followed by 80 mg on Week 2. Mean steady-state trough concentrations at Week 12 through Week 36 were approximately 7 to 11 mcg/mL during adalimumab 40 mg every week treatment.
Adult Crohn's Disease: Adalimumab mean trough concentrations were approximately 12 mcg/mL at Week 2 and Week 4 after receiving 160 mg on Week 0 followed by 80 mg on Week 2. Mean steady-state trough concentrations were 7 mcg/mL at Week 24 and Week 56 during adalimumab 40 mg every other week treatment.
Adult Ulcerative Colitis: Adalimumab mean trough concentrations were approximately 12 mcg/mL at Week 2 and Week 4 after receiving 160 mg on Week 0 followed by 80 mg on Week 2. Mean steady-state trough concentrations were approximately 8 mcg/mL and 15 mcg/mL at Week 52 after receiving a dose of adalimumab 40 mg every other week and 40 mg every week, respectively.
Anti-Drug Antibody Effects on Pharmacokinetics
Specific Populations
Geriatric Patients: A lower clearance with increasing age was observed in patients with RA aged 40 to > 75 years.
Pediatric Patients:
Juvenile Idiopathic Arthritis:
- 4 years to 17 years of age: The adalimumab mean steady-state trough concentrations were 6.8 mcg/mL and 10.9 mcg/mL in patients weighing < 30 kg receiving 20 mg adalimumab subcutaneously every other week as monotherapy or with concomitant MTX, respectively. The adalimumab mean steady-state trough concentrations were 6.6 mcg/mL and 8.1 mcg/mL in patients weighing ≥ 30 kg receiving 40 mg adalimumab subcutaneously every other week as monotherapy or with MTX concomitant treatment, respectively.
- 2 years to < 4 years of age or 4 years of age and older weighing < 15 kg: The adalimumab mean steady-state trough adalimumab concentrations were 6.0 mcg/mL and 7.9 mcg/mL in patients receiving adalimumab subcutaneously every other week as monotherapy or with MTX concomitant treatment, respectively.
Pediatric Hidradenitis Suppurativa: Adalimumab concentrations in adolescent patients with HS receiving the recommended dosage regimens are predicted to be similar to those observed in adult subjects with HS based on population pharmacokinetic modeling and simulation.
Pediatric Crohn's Disease: Adalimumab mean ± SD concentrations were 15.7±6.5 mcg/mL at Week 4 following 160 mg at Week 0 and 80 mg at Week 2, and 10.5±6.0 mcg/mL at Week 52 following 40 mg every other week dosing in patients weighing ≥ 40 kg. Adalimumab mean ± SD concentrations were 10.6±6.1 mcg/mL at Week 4 following dosing 80 mg at Week 0 and 40 mg at Week 2, and 6.9±3.6 mcg/mL at Week 52 following 20 mg every other week dosing in patients weighing < 40 kg.
Male and Female Patients: No gender-related pharmacokinetic differences were observed after correction for a patient's body weight. Healthy subjects and patients with rheumatoid arthritis displayed similar adalimumab pharmacokinetics.
Drug Interaction Studies:
Methotrexate: MTX reduced adalimumab apparent clearance after single and multiple dosing by 29% and 44% respectively, in patients with RA [see Drug Interactions (7.1)].
- 13 NONCLINICAL TOXICOLOGY
-
14 CLINICAL STUDIES
14.1 Clinical Studies in Rheumatoid Arthritis
The efficacy and safety of adalimumab were assessed in five randomized, double-blind studies in subjects ≥ 18 years of age with active rheumatoid arthritis (RA) diagnosed according to American College of Rheumatology (ACR) criteria. Subjects had at least 6 swollen and 9 tender joints. Adalimumab was administered subcutaneously in combination with methotrexate (MTX) (12.5 to 25 mg, Studies RA-I, RA-III and RA-V) or as monotherapy (Studies RA-II and RA-V) or with other disease-modifying anti-rheumatic drugs (DMARDs) (Study RA-IV).
Study RA-I evaluated 271 subjects who had failed therapy with at least one but no more than four DMARDs and had inadequate response to MTX. Doses of 20, 40 or 80 mg of adalimumab or placebo were given every other week for 24 weeks.
Study RA-II evaluated 544 subjects who had failed therapy with at least one DMARD. Doses of placebo, 20 or 40 mg of adalimumab were given as monotherapy every other week or weekly for 26 weeks.
Study RA-III evaluated 619 subjects who had an inadequate response to MTX. Subjects received placebo, 40 mg of adalimumab every other week with placebo injections on alternate weeks, or 20 mg of adalimumab weekly for up to 52 weeks. Study RA-III had an additional primary endpoint at 52 weeks of inhibition of disease progression (as detected by X-ray results). Upon completion of the first 52 weeks, 457 subjects enrolled in an open-label extension phase in which 40 mg of adalimumab was administered every other week for up to 5 years.
Study RA-IV assessed safety in 636 subjects who were either DMARD-naive or were permitted to remain on their pre-existing rheumatologic therapy provided that therapy was stable for a minimum of 28 days. Subjects were randomized to 40 mg of adalimumab or placebo every other week for 24 weeks.
Study RA-V evaluated 799 subjects with moderately to severely active RA of less than 3 years duration who were ≥ 18 years old and MTX naïve. Subjects were randomized to receive either MTX (optimized to 20 mg/week by week 8), adalimumab 40 mg every other week or adalimumab/MTX combination therapy for 104 weeks. Subjects were evaluated for signs and symptoms, and for radiographic progression of joint damage. The median disease duration among subjects enrolled in the study was 5 months. The median MTX dose achieved was 20 mg.
Clinical Response
The percent of adalimumab treated subjects achieving ACR 20, 50 and 70 responses in Studies RA-II and III are shown in Table 3.
Table 3. ACR Responses in Studies RA-II and RA-III (Percent of Subjects) Study RA-II Monotherapy
(26 weeks)Study RA-III Methotrexate Combination
(24 and 52 weeks)Response Placebo Adalimumab 40 mg every other week Adalimumab 40 mg weekly Placebo/MTX Adalimumab/MTX 40 mg every other week N=110 N=113 N=103 N=200 N=207 ACR20 Month 6 19% 46%* 53%* 30% 63%* Month 12 NA NA NA 24% 59%* ACR50 Month 6 8% 22%* 35%* 10% 39%* Month 12 NA NA NA 10% 42%* ACR70 Month 6 2% 12%* 18%* 3% 21%* Month 12 NA NA NA 5% 23%* * p<0.01, adalimumab vs. placebo The results of Study RA-I were similar to Study RA-III; subjects receiving adalimumab 40 mg every other week in Study RA-I also achieved ACR 20, 50 and 70 response rates of 65%, 52% and 24%, respectively, compared to placebo responses of 13%, 7% and 3% respectively, at 6 months (p<0.01).
The results of the components of the ACR response criteria for Studies RA-II and RA-III are shown in Table 4. ACR response rates and improvement in all components of ACR response were maintained to week 104. Over the 2 years in Study RA-III, 20% of adalimumab subjects receiving 40 mg every other week achieved a major clinical response, defined as maintenance of an ACR 70 response over a 6-month period. ACR responses were maintained in similar proportions of subjects for up to 5 years with continuous adalimumab treatment in the open-label portion of Study RA-III.
Table 4. Components of ACR Response in Studies RA-II and RA-III Study RA-II Study RA-III Parameter (median) Placebo
N=110Adalimumaba
N=113Placebo/MTX
N=200Adalimumaba/MTX
N=207Baseline Wk 26 Baseline Wk 26 Baseline Wk 24 Baseline Wk 24 Number of tender joints (0-68) 35 26 31 16* 26 15 24 8* Number of swollen joints (0-66) 19 16 18 10* 17 11 18 5* Physician global assessmentb 7.0 6.1 6.6 3.7* 6.3 3.5 6.5 2.0* Patient global assessmentb 7.5 6.3 7.5 4.5* 5.4 3.9 5.2 2.0* Painb 7.3 6.1 7.3 4.1* 6.0 3.8 5.8 2.1* Disability index (HAQ)c 2.0 1.9 1.9 1.5* 1.5 1.3 1.5 0.8* CRP (mg/dL) 3.9 4.3 4.6 1.8* 1.0 0.9 1.0 0.4* a 40 mg adalimumab administered every other week
b Visual analogue scale; 0 = best, 10 = worst
c Disability Index of the Health Assessment Questionnaire; 0 = best, 3 = worst, measures the patient’s ability to perform the following: dress/groom, arise, eat, walk, reach, grip, maintain hygiene, and maintain daily activity
* p<0.001, adalimumab vs. placebo, based on mean change from baselineThe time course of ACR 20 response for Study RA-III is shown in Figure 1.
In Study RA-III, 85% of subjects with ACR 20 responses at week 24 maintained the response at 52 weeks. The time course of ACR 20 response for Study RA-I and Study RA-II were similar.
Figure 1. Study RA-III ACR 20 Responses over 52 Weeks 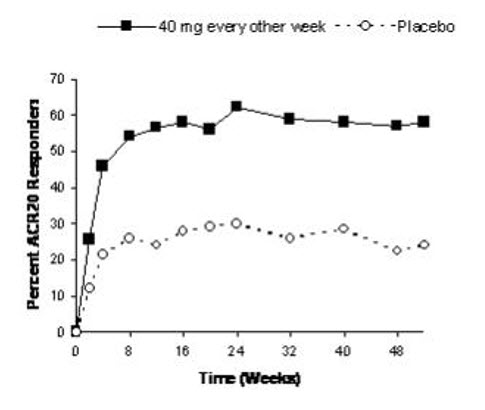
In Study RA-IV, 53% of subjects treated with adalimumab 40 mg every other week plus standard of care had an ACR 20 response at week 24 compared to 35% on placebo plus standard of care (p<0.001). No unique adverse reactions related to the combination of adalimumab and other DMARDs were observed.
In Study RA-V with MTX naïve subjects with recent onset RA, the combination treatment with adalimumab plus MTX led to greater percentages of subjects achieving ACR responses than either MTX monotherapy or adalimumab monotherapy at Week 52 and responses were sustained at Week 104 (see Table 5).
Table 5. ACR Response in Study RA-V (Percent of Subjects) Response MTXb
N=257Adalimumabc
N=274Adalimumab/MTX
N=268ACR20 Week 52 63% 54% 73% Week 104 56% 49% 69% ACR50 Week 52 46% 41% 62% Week 104 43% 37% 59% ACR70 Week 52 27% 26% 46% Week 104 28% 28% 47% Major Clinical Responsea 28% 25% 49% a Major clinical response is defined as achieving an ACR70 response for a continuous six month period
b p<0.05, adalimumab/MTX vs. MTX for ACR 20
p<0.001, adalimumab/MTX vs. MTX for ACR 50 and 70, and Major Clinical Response
c p<0.001, adalimumab/MTX vs. adalimumabAt Week 52, all individual components of the ACR response criteria for Study RA-V improved in the adalimumab/MTX group and improvements were maintained to Week 104.
Radiographic Response
In Study RA-III, structural joint damage was assessed radiographically and expressed as change in Total Sharp Score (TSS) and its components, the erosion score and Joint Space Narrowing (JSN) score, at month 12 compared to baseline. At baseline, the median TSS was approximately 55 in the placebo and 40 mg every other week groups. The results are shown in Table 6. Adalimumab/MTX treated subjects demonstrated less radiographic progression than subjects receiving MTX alone at 52 weeks.
Table 6. Radiographic Mean Changes Over 12 Months in Study RA-III Placebo/MTX Adalimumab/MTX 40 mg every other week Placebo/MTX-Adalimumab/MTX (95% Confidence Interval*) P-value** Total Sharp score 2.7 0.1 2.6 (1.4, 3.8) <0.001 Erosion score 1.6 0.0 1.6 (0.9, 2.2) <0.001 JSN score 1.0 0.1 0.9 (0.3, 1.4) 0.002 * 95% confidence intervals for the differences in change scores between MTX and adalimumab.
** Based on rank analysisIn the open-label extension of Study RA-III, 77% of the original subjects treated with any dose of adalimumab were evaluated radiographically at 2 years. Subjects maintained inhibition of structural damage, as measured by the TSS. Fifty-four percent had no progression of structural damage as defined by a change in the TSS of zero or less. Fifty-five percent (55%) of subjects originally treated with 40 mg adalimumab every other week have been evaluated radiographically at 5 years. Subjects had continued inhibition of structural damage with 50% showing no progression of structural damage defined by a change in the TSS of zero or less.
In Study RA-V, structural joint damage was assessed as in Study RA-III. Greater inhibition of radiographic progression, as assessed by changes in TSS, erosion score and JSN was observed in the adalimumab/MTX combination group as compared to either the MTX or adalimumab monotherapy group at Week 52 as well as at Week 104 (see Table 7).
Table 7. Radiographic Mean Change* in Study RA-V MTXa
N=257Adalimumaba,b
N=274Adalimumab/MTX
N=26852 Weeks Total Sharp score 5.7 (4.2, 7.3) 3.0 (1.7, 4.3) 1.3 (0.5, 2.1) Erosion score 3.7 (2.7, 4.8) 1.7 (1.0, 2.4) 0.8 (0.4, 1.2) JSN score 2.0 (1.2, 2.8) 1.3 (0.5, 2.1) 0.5 (0.0, 1.0) 104 Weeks Total Sharp score 10.4 (7.7, 13.2) 5.5 (3.6, 7.4) 1.9 (0.9, 2.9) Erosion score 6.4 (4.6, 8.2) 3.0 (2.0, 4.0) 1.0 (0.4, 1.6) JSN score 4.1 (2.7, 5.4) 2.6 (1.5, 3.7) 0.9 (0.3, 1.5) * mean (95% confidence interval)
a p<0.001, adalimumab/MTX vs. MTX at 52 and 104 weeks and for adalimumab/MTX vs. adalimumab at 104 weeks
b p<0.01, for adalimumab/MTX vs. adalimumab at 52 weeksPhysical Function Response
In studies RA-I through IV, adalimumab showed significantly greater improvement than placebo in the disability index of Health Assessment Questionnaire (HAQ-DI) from baseline to the end of study, and significantly greater improvement than placebo in the health-outcomes as assessed by The Short Form Health Survey (SF 36). Improvement was seen in both the Physical Component Summary (PCS) and the Mental Component Summary (MCS).
In Study RA-III, the mean (95% CI) improvement in HAQ-DI from baseline at week 52 was 0.60 (0.55, 0.65) for the adalimumab subjects and 0.25 (0.17, 0.33) for placebo/MTX (p<0.001) subjects. Sixty-three percent of adalimumab-treated subjects achieved a 0.5 or greater improvement in HAQ-DI at week 52 in the double-blind portion of the study. Eighty-two percent of these subjects maintained that improvement through week 104 and a similar proportion of subjects maintained this response through week 260 (5 years) of open-label treatment. Mean improvement in the SF-36 was maintained through the end of measurement at week 156 (3 years).
In Study RA-V, the HAQ-DI and the physical component of the SF-36 showed greater improvement (p<0.001) for the adalimumab/MTX combination therapy group versus either the MTX monotherapy or the adalimumab monotherapy group at Week 52, which was maintained through Week 104.
14.2 Clinical Studies in Juvenile Idiopathic Arthritis
The safety and efficacy of adalimumab was assessed in two studies (Studies JIA-I and JIA-II) in subjects with active polyarticular juvenile idiopathic arthritis (JIA).
Study JIA-I
The safety and efficacy of adalimumab were assessed in a multicenter, randomized, withdrawal, double-blind, parallel-group study in 171 subjects who were 4 to 17 years of age with polyarticular JIA. In the study, the subjects were stratified into two groups: MTX-treated or non-MTX-treated. All subjects had to show signs of active moderate or severe disease despite previous treatment with NSAIDs, analgesics, corticosteroids, or DMARDS. Subjects who received prior treatment with any biologic DMARDS were excluded from the study.
The study included four phases: an open-label lead in phase (OL-LI; 16 weeks), a double-blind randomized withdrawal phase (DB; 32 weeks), an open-label extension phase (OLE-BSA; up to 136 weeks), and an open-label fixed dose phase (OLE-FD; 16 weeks). In the first three phases of the study, adalimumab was administered based on body surface area at a dose of 24 mg/m2 up to a maximum total body dose of 40 mg subcutaneously (SC) every other week. In the OLE-FD phase, the subjects were treated with 20 mg of adalimumab SC every other week if their weight was less than 30 kg and with 40 mg of adalimumab SC every other week if their weight was 30 kg or greater. Subjects remained on stable doses of NSAIDs and or prednisone (≤ 0.2 mg/kg/day or 10 mg/day maximum).
Subjects demonstrating a Pediatric ACR 30 response at the end of OL-LI phase were randomized into the double blind (DB) phase of the study and received either adalimumab or placebo every other week for 32 weeks or until disease flare. Disease flare was defined as a worsening of ≥ 30% from baseline in ≥ 3 of 6 Pediatric ACR core criteria, ≥ 2 active joints, and improvement of > 30% in no more than 1 of the 6 criteria. After 32 weeks or at the time of disease flare during the DB phase, subjects were treated in the open-label extension phase based on the BSA regimen (OLE-BSA), before converting to a fixed dose regimen based on body weight (OLE-FD phase).
Study JIA-I Clinical Response
At the end of the 16-week OL-LI phase, 94% of the subjects in the MTX stratum and 74% of the subjects in the non-MTX stratum were Pediatric ACR 30 responders. In the DB phase significantly fewer subjects who received adalimumab experienced disease flare compared to placebo, both without MTX (43% vs. 71%) and with MTX (37% vs. 65%). More subjects treated with adalimumab continued to show pediatric ACR 30/50/70 responses at Week 48 compared to subjects treated with placebo. Pediatric ACR responses were maintained for up to two years in the OLE phase in subjects who received adalimumab throughout the study.
Study JIA-II
Adalimumab was assessed in an open-label, multicenter study in 32 subjects who were 2 to < 4 years of age or 4 years of age and older weighing < 15 kg with moderately to severely active polyarticular JIA. Most subjects (97%) received at least 24 weeks of adalimumab treatment dosed 24 mg/m2 up to a maximum of 20 mg every other week as a single SC injection up to a maximum of 120 weeks duration. During the study, most subjects used concomitant MTX, with fewer reporting use of corticosteroids or NSAIDs. The primary objective of the study was evaluation of safety [see Adverse Reactions (6.1)].
14.3 Clinical Studies in Psoriatic Arthritis
The safety and efficacy of adalimumab was assessed in two randomized, double-blind, placebo-controlled studies in 413 subjects with psoriatic arthritis (PsA). Upon completion of both studies, 383 subjects enrolled in an open-label extension study, in which 40 mg adalimumab was administered every other week.
Study PsA-I enrolled 313 adult subjects with moderately to severely active PsA (> 3 swollen and > 3 tender joints) who had an inadequate response to NSAID therapy in one of the following forms: (1) distal interphalangeal (DIP) involvement (N=23); (2) polyarticular arthritis (absence of rheumatoid nodules and presence of plaque psoriasis) (N=210); (3) arthritis mutilans (N=1); (4) asymmetric PsA (N=77); or (5) AS-like (N=2). Subjects on MTX therapy (158 of 313 subjects) at enrollment (stable dose of ≤ 30 mg/week for > 1 month) could continue MTX at the same dose. Doses of adalimumab 40 mg or placebo every other week were administered during the 24-week double-blind period of the study.
Compared to placebo, treatment with adalimumab resulted in improvements in the measures of disease activity (see Tables 8 and 9). Among subjects with PsA who received adalimumab, the clinical responses were apparent in some subjects at the time of the first visit (two weeks) and were maintained up to 88 weeks in the ongoing open-label study. Similar responses were seen in subjects with each of the subtypes of psoriatic arthritis, although few subjects were enrolled with the arthritis mutilans and ankylosing spondylitis-like subtypes. Responses were similar in subjects who were or were not receiving concomitant MTX therapy at baseline.
Subjects with psoriatic involvement of at least three percent body surface area (BSA) were evaluated for Psoriatic Area and Severity Index (PASI) responses. At 24 weeks, the proportions of subjects achieving a 75% or 90% improvement in the PASI were 59% and 42% respectively, in the adalimumab group (N=69), compared to 1% and 0% respectively, in the placebo group (N=69) (p<0.001). PASI responses were apparent in some subjects at the time of the first visit (two weeks). Responses were similar in subjects who were or were not receiving concomitant MTX therapy at baseline.
Table 8. ACR Response in Study PsA-I (Percent of Subjects) Placebo
N=162Adalimumab*
N=151ACR20 Week 12 14% 58% Week 24 15% 57% ACR50 Week 12 4% 36% Week 24 6% 39% ACR70 Week 12 1% 20% Week 24 1% 23% * p<0.001 for all comparisons between adalimumab and placebo Table 9. Components of Disease Activity in Study PsA-I Placebo
N= 162Adalimumab*
N=151Parameter: median Baseline 24 weeks Baseline 24 weeks Number of tender jointsa 23.0 17.0 20.0 5.0 Number of swollen jointsb 11.0 9.0 11.0 3.0 Physician global assessmentc 53.0 49.0 55.0 16.0 Patient global assessmentc 49.5 49.0 48.0 20.0 Painc 49.0 49.0 54.0 20.0 Disability index (HAQ)d 1.0 0.9 1.0 0.4 CRP (mg/dL)e 0.8 0.7 0.8 0.2 * p<0.001 for adalimumab vs. placebo comparisons based on median changes
a Scale 0-78
b Scale 0-76
c Visual analog scale; 0=best, 100=worst
d Disability Index of the Health Assessment Questionnaire; 0=best, 3=worst; measures the patient’s ability to perform the following: dress/groom, arise, eat, walk, reach, grip, maintain hygiene, and maintain daily activity.
e Normal range: 0-0.287 mg/dLSimilar results were seen in an additional, 12-week study in 100 subjects with moderate to severe psoriatic arthritis who had suboptimal response to DMARD therapy as manifested by ≥ 3 tender joints and ≥ 3 swollen joints at enrollment.
Radiographic Response
Radiographic changes were assessed in the PsA studies. Radiographs of hands, wrists, and feet were obtained at baseline and Week 24 during the double-blind period when subjects were on adalimumab or placebo and at Week 48 when all subjects were on open-label adalimumab. A modified Total Sharp Score (mTSS), which included distal interphalangeal joints (i.e., not identical to the TSS used for rheumatoid arthritis), was used by readers blinded to treatment group to assess the radiographs.
Adalimumab-treated subjects demonstrated greater inhibition of radiographic progression compared to placebo-treated subjects and this effect was maintained at 48 weeks (see Table 10).
Table 10. Change in Modified Total Sharp Score in Psoriatic Arthritis Placebo
N=141Adalimumab
N=133Week 24 Week 24 Week 48 Baseline mean 22.1 23.4 23.4 Mean Change ± SD 0.9 ± 3.1 -0.1 ± 1.7 -0.2 ± 4.9* * <0.001 for the difference between adalimumab, Week 48 and Placebo, Week 24 (primary analysis) Physical Function Response
In Study PsA-I, physical function and disability were assessed using the HAQ Disability Index (HAQ-DI) and the SF-36 Health Survey. Subjects treated with 40 mg of adalimumab every other week showed greater improvement from baseline in the HAQ-DI score (mean decreases of 47% and 49% at Weeks 12 and 24 respectively) in comparison to placebo (mean decreases of 1% and 3% at Weeks 12 and 24 respectively). At Weeks 12 and 24, subjects treated with adalimumab showed greater improvement from baseline in the SF-36 Physical Component Summary score compared to subjects treated with placebo, and no worsening in the SF-36 Mental Component Summary score. Improvement in physical function based on the HAQ-DI was maintained for up to 84 weeks through the open-label portion of the study.
14.4 Clinical Studies in Ankylosing Spondylitis
The safety and efficacy of adalimumab 40 mg every other week was assessed in 315 adult subjects in a randomized, 24 week double-blind, placebo-controlled study in subjects with active ankylosing spondylitis (AS) who had an inadequate response to glucocorticoids, NSAIDs, analgesics, methotrexate or sulfasalazine. Active AS was defined as subjects who fulfilled at least two of the following three criteria: (1) a Bath AS disease activity index (BASDAI) score 4 cm, (2) a visual analog score (VAS) for total back pain 40 mm, and (3) morning stiffness 1 hour. The blinded period was followed by an open-label period during which subjects received adalimumab 40 mg every other week subcutaneously for up to an additional 28 weeks.
Improvement in measures of disease activity was first observed at Week 2 and maintained through 24 weeks as shown in Figure 2 and Table 11.
Responses of subjects with total spinal ankylosis (n=11) were similar to those without total ankylosis.
Figure 2. ASAS 20 Response By Visit, Study AS-I 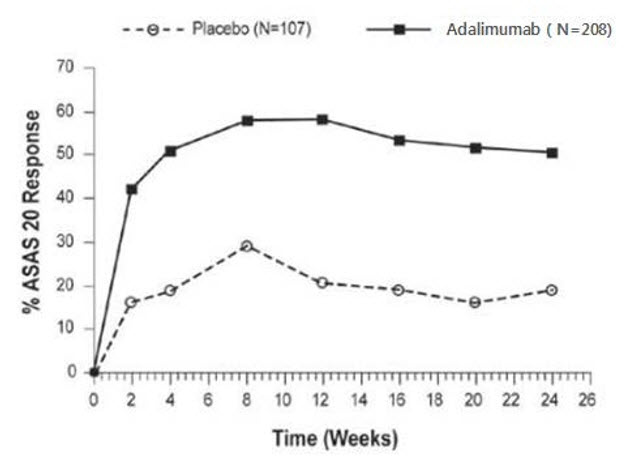
At 12 weeks, the ASAS 20/50/70 responses were achieved by 58%, 38%, and 23%, respectively, of subjects receiving adalimumab, compared to 21%, 10%, and 5% respectively, of subjects receiving placebo (p<0.001). Similar responses were seen at Week 24 and were sustained in subjects receiving open-label adalimumab for up to 52 weeks.
A greater proportion of subjects treated with adalimumab (22%) achieved a low level of disease activity at 24 weeks (defined as a value < 20 [on a scale of 0 to 100 mm] in each of the four ASAS response parameters) compared to subjects treated with placebo (6%).
Table 11. Components of Ankylosing Spondylitis Disease Activity Placebo
N=107Adalimumab
N=208Baseline
meanWeek 24
meanBaseline
meanWeek 24
meanASAS 20 Response Criteria* Patient's Global Assessment of Disease Activitya* 65 60 63 38 Total back pain* 67 58 65 37 Inflammationb* 6.7 5.6 6.7 3.6 BASFIc* 56 51 52 34 BASDAId score* 6.3 5.5 6.3 3.7 BASMIe score* 4.2 4.1 3.8 3.3 Tragus to wall (cm) 15.9 15.8 15.8 15.4 Lumbar flexion (cm) 4.1 4.0 4.2 4.4 Cervical rotation (degrees) 42.2 42.1 48.4 51.6 Lumbar side flexion (cm) 8.9 9.0 9.7 11.7 Intermalleolar distance (cm) 92.9 94.0 93.5 100.8 CRPf* 2.2 2.0 1.8 0.6 a Percent of subjects with at least a 20% and 10-unit improvement measured on a Visual Analog Scale (VAS) with 0 = “none” and 100 = “severe”
b mean of questions 5 and 6 of BASDAI (defined in ‘d’)
c Bath Ankylosing Spondylitis Functional Index
d Bath Ankylosing Spondylitis Disease Activity Index
e Bath Ankylosing Spondylitis Metrology Index
f C-Reactive Protein (mg/dL)
* statistically significant for comparisons between adalimumab and placebo at Week 24A second randomized, multicenter, double-blind, placebo-controlled study of 82 subjects with ankylosing spondylitis showed similar results.
Subjects treated with adalimumab achieved improvement from baseline in the Ankylosing Spondylitis Quality of Life Questionnaire (ASQoL) score (-3.6 vs. -1.1) and in the Short Form Health Survey (SF-36) Physical Component Summary (PCS) score (7.4 vs. 1.9) compared to placebo-treated subjects at Week 24.
14.5 Clinical Studies in Adults with Crohn's Disease
The safety and efficacy of multiple doses of adalimumab were assessed in adult subjects with moderately to severely active Crohn's disease, CD, (Crohn's Disease Activity Index (CDAI) ≥ 220 and ≤ 450) in randomized, double-blind, placebo-controlled studies. Concomitant stable doses of aminosalicylates, corticosteroids, and/or immunomodulatory agents were permitted, and 79% of subjects continued to receive at least one of these medications.
Induction of clinical remission (defined as CDAI < 150) was evaluated in two studies. In Study CD-I, 299 TNF-blocker naïve subjects were randomized to one of four treatment groups: the placebo group received placebo at Weeks 0 and 2, the 160/80 group received 160 mg adalimumab at Week 0 and 80 mg at Week 2, the 80/40 group received 80 mg at Week 0 and 40 mg at Week 2, and the 40/20 group received 40 mg at Week 0 and 20 mg at Week 2. Clinical results were assessed at Week 4.
In the second induction study, Study CD-II, 325 subjects who had lost response to, or were intolerant to, previous infliximab therapy were randomized to receive either 160 mg adalimumab at Week 0 and 80 mg at Week 2, or placebo at Weeks 0 and 2. Clinical results were assessed at Week 4.
Maintenance of clinical remission was evaluated in Study CD-III. In this study, 854 subjects with active disease received open-label adalimumab, 80 mg at week 0 and 40 mg at Week 2. Subjects were then randomized at Week 4 to 40 mg adalimumab every other week, 40 mg adalimumab every week, or placebo. The total study duration was 56 weeks. Subjects in clinical response (decrease in CDAI ≥ 70) at Week 4 were stratified and analyzed separately from those not in clinical response at Week 4.
Induction of Clinical Remission
A greater percentage of the subjects treated with 160/80 mg adalimumab achieved induction of clinical remission versus placebo at Week 4 regardless of whether the subjects were TNF blocker naïve (CD-I), or had lost response to or were intolerant to infliximab (CD-II) (see Table 12).
Table 12. Induction of Clinical Remission in Studies CD-I and CD-II (Percent of Subjects) CD-I CD-II Placebo
N=74Adalimumab 160/80 mg
N=76Placebo
N=166Adalimumab 160/80 mg
N=159Week 4 Clinical remission 12% 36%* 7% 21%* Clinical response 34% 58%** 34% 52%** Clinical remission is CDAI score < 150; clinical response is decrease in CDAI of at least 70 points.
* p<0.001 for adalimumab vs. placebo pairwise comparison of proportions
** p<0.01 for adalimumab vs. placebo pairwise comparison of proportionsMaintenance of Clinical Remission
In Study CD-III at Week 4, 58% (499/854) of subjects were in clinical response and were assessed in the primary analysis. At Weeks 26 and 56, greater proportions of subjects who were in clinical response at Week 4 achieved clinical remission in the adalimumab 40 mg every other week maintenance group compared to subjects in the placebo maintenance group (see Table 13). The group that received adalimumab therapy every week did not demonstrate significantly higher remission rates compared to the group that received adalimumab every other week.
Table 13. Maintenance of Clinical Remission in CD-III (Percent of Subjects) Placebo 40 mg Adalimumab every other week N=170 N=172 Week 26 Clinical remission 17% 40%* Clinical response 28% 54%* Week 56 Clinical remission 12% 36%* Clinical response 18% 43%* Clinical remission is CDAI score < 150; clinical response is decrease in CDAI of at least 70 points.
* p<0.001 for adalimumab vs. placebo pairwise comparisons of proportionsOf those in response at Week 4 who attained remission during the study, subjects in the adalimumab every other week group maintained remission for a longer time than subjects in the placebo maintenance group. Among subjects who were not in response by Week 12, therapy continued beyond 12 weeks did not result in significantly more responses.
14.6 Clinical Studies in Pediatric Subjects with Crohn's Disease
A randomized, double-blind, 52-week clinical study of 2 dose concentrations of adalimumab (Study PCD-I) was conducted in 192 pediatric subjects (6 to 17 years of age) with moderately to severely active Crohn's disease (defined as Pediatric Crohn's Disease Activity Index (PCDAI) score > 30). Enrolled subjects had over the previous two-year period an inadequate response to corticosteroids or an immunomodulator (i.e., azathioprine, 6-mercaptopurine, or methotrexate). Subjects who had previously received a TNF blocker were allowed to enroll if they had previously had loss of response or intolerance to that TNF blocker.
Subjects received open-label induction therapy at a dose based on their body weight (≥ 40 kg and < 40 kg). Subjects weighing ≥ 40 kg received 160 mg (at Week 0) and 80 mg (at Week 2). Subjects weighing < 40 kg received 80 mg (at Week 0) and 40 mg (at Week 2). At Week 4, subjects within each body weight category (≥ 40 kg and < 40 kg) were randomized 1:1 to one of two maintenance dose regimens (high dose and low dose). The high dose was 40 mg every other week for subjects weighing ≥ 40 kg and 20 mg every other week for subjects weighing < 40 kg. The low dose was 20 mg every other week for subjects weighing ≥ 40 kg and 10 mg every other week for subjects weighing < 40 kg.
Concomitant stable dosages of corticosteroids (prednisone dosage ≤ 40 mg/day or equivalent) and immunomodulators (azathioprine, 6-mercaptopurine, or methotrexate) were permitted throughout the study.
At Week 12, subjects who experienced a disease flare (increase in PCDAI of ≥ 15 from Week 4 and absolute PCDAI > 30) or who were non-responders (did not achieve a decrease in the PCDAI of ≥ 15 from baseline for 2 consecutive visits at least 2 weeks apart) were allowed to dose-escalate (i.e., switch from blinded every other week dosing to blinded every week dosing); subjects who dose-escalated were considered treatment failures.
At baseline, 38% of subjects were receiving corticosteroids, and 62% of subjects were receiving an immunomodulator. Forty-four percent (44%) of subjects had previously lost response or were intolerant to a TNF blocker. The median baseline PCDAI score was 40.
Of the 192 subjects total, 188 subjects completed the 4 week induction period, 152 subjects completed 26 weeks of treatment, and 124 subjects completed 52 weeks of treatment. Fifty-one percent (51%) (48/95) of subjects in the low maintenance dose group dose-escalated, and 38% (35/93) of subjects in the high maintenance dose group dose-escalated.
At Week 4, 28% (52/188) of subjects were in clinical remission (defined as PCDAI ≤ 10).
The proportions of subjects in clinical remission (defined as PCDAI ≤ 10) and clinical response (defined as reduction in PCDAI of at least 15 points from baseline) were assessed at Weeks 26 and 52.
At both Weeks 26 and 52, the proportion of subjects in clinical remission and clinical response was numerically higher in the high dose group compared to the low dose group (Table 14). The recommended maintenance regimen is 20 mg every other week for subjects weighing < 40 kg and 40 mg every other week for subjects weighing ≥ 40 kg. Every week dosing is not the recommended maintenance dosing regimen [see Dosage and Administration (2.3)].
Table 14. Clinical Remission and Clinical Response in Study PCD-I Low Maintenance Dose†
(20 or 10 mg every other week)
N = 95High Maintenance Dose#
(40 or 20 mg every other week)
N = 93Week 26 Clinical Remission‡ 28% 39% Clinical Response§ 48% 59% Week 52 Clinical Remission‡ 23% 33% Clinical Response§ 28% 42% † The low maintenance dose was 20 mg every other week for subjects weighing ≥ 40 kg and 10 mg every other week for subjects weighing < 40 kg.
# The high maintenance dose was 40 mg every other week for subjects weighing ≥ 40 kg and 20 mg every other week for subjects weighing < 40 kg.
‡ Clinical remission defined as PCDAI ≤ 10.
§ Clinical response defined as reduction in PCDAI of at least 15 points from baseline.14.7 Clinical Studies in Adults with Ulcerative Colitis
The safety and efficacy of adalimumab were assessed in adult subjects with moderately to severely active ulcerative colitis (Mayo score 6 to 12 on a 12-point scale, with an endoscopy subscore of 2 to 3 on a scale of 0 to 3) despite concurrent or prior treatment with immunosuppressants such as corticosteroids, azathioprine, or 6-MP in two randomized, double-blind, placebo-controlled clinical studies (Studies UC-I and UC-II). Both studies enrolled TNF-blocker naïve subjects, but Study UC-II also allowed entry of subjects who lost response to or were intolerant to TNF-blockers. Forty percent (40%) of subjects enrolled in Study UC-II had previously used another TNF-blocker.
Concomitant stable doses of aminosalicylates and immunosuppressants were permitted. In Studies UC-I and II, subjects were receiving aminosalicylates (69%), corticosteroids (59%) and/or azathioprine or 6-MP (37%) at baseline. In both studies, 92% of subjects received at least one of these medications.
Induction of clinical remission (defined as Mayo score ≤ 2 with no individual subscores > 1) at Week 8 was evaluated in both studies. Clinical remission at Week 52 and sustained clinical remission (defined as clinical remission at both Weeks 8 and 52) were evaluated in Study UC-II.
In Study UC-I, 390 TNF-blocker naïve subjects were randomized to one of three treatment groups for the primary efficacy analysis. The placebo group received placebo at Weeks 0, 2, 4 and 6. The 160/80 group received 160 mg adalimumab at Week 0 and 80 mg at Week 2, and the 80/40 group received 80 mg adalimumab at Week 0 and 40 mg at Week 2. After Week 2, subjects in both adalimumab treatment groups received 40 mg every other week.
In Study UC-II, 518 subjects were randomized to receive either adalimumab 160 mg at Week 0, 80 mg at Week 2, and 40 mg every other week starting at Week 4 through Week 50, or placebo starting at Week 0 and every other week through Week 50. Corticosteroid taper was permitted starting at Week 8.
In both Studies UC-I and UC-II, a greater percentage of the subjects treated with 160/80 mg of adalimumab compared to subjects treated with placebo achieved induction of clinical remission. In Study UC-II, a greater percentage of the subjects treated with 160/80 mg of adalimumab compared to subjects treated with placebo achieved sustained clinical remission (clinical remission at both Weeks 8 and 52) (Table 15).
Table 15. Induction of Clinical Remission in Studies UC-I and UC-II and Sustained Clinical Remission in Study UC-II (Percent of Subjects) Study UC-I Study UC-II Placebo
N=130Adalimumab 160/80 mg
N=130Treatment Difference
(95% CI)Placebo
N=246Adalimumab 160/80 mg
N=248Treatment Difference
(95% CI)Induction of Clinical Remission (Clinical Remission at Week 8) 9.2% 18.5% 9.3%*
(0.9%,17.6%)9.3% 16.5% 7.2%*
(1.2%,12.9%)Sustained Clinical Remission (Clinical Remission at both Weeks 8 and 52) N/A N/A N/A 4.1% 8.5% 4.4%*
(0.1%, 8.6%)Clinical remission is defined as Mayo score ≤ 2 with no individual subscores > 1.
CI=Confidence interval
* p<0.05 for adalimumab vs. placebo pairwise comparison of proportionsIn Study UC-I, there was no statistically significant difference in clinical remission observed between the adalimumab 80/40 mg group and the placebo group at Week 8.
In Study UC-II, 17.3% (43/248) in the adalimumab group were in clinical remission at Week 52 compared to 8.5% (21/246) in the placebo group (treatment difference: 8.8%; 95% confidence interval (CI): [2.8%, 14.5%]; p<0.05).
In the subgroup of subjects in Study UC-II with prior TNF-blocker use, the treatment difference for induction of clinical remission appeared to be lower than that seen in the whole study population, and the treatment differences for sustained clinical remission and clinical remission at Week 52 appeared to be similar to those seen in the whole study population. The subgroup of subjects with prior TNF-blocker use achieved induction of clinical remission at 9% (9/98) in the adalimumab group versus 7% (7/101) in the placebo group and sustained clinical remission at 5% (5/98) in the adalimumab group versus 1% (1/101) in the placebo group. In the subgroup of subjects with prior TNF-blocker use, 10% (10/98) were in clinical remission at Week 52 in the adalimumab group versus 3% (3/101) in the placebo group.
14.8 Clinical Studies in Plaque Psoriasis
The safety and efficacy of adalimumab were assessed in randomized, double-blind, placebo-controlled studies in 1696 adult subjects with moderate to severe chronic plaque psoriasis (Ps) who were candidates for systemic therapy or phototherapy.
Study Ps-I evaluated 1212 subjects with chronic Ps with ≥ 10% body surface area (BSA) involvement, Physician's Global Assessment (PGA) of at least moderate disease severity, and Psoriasis Area and Severity Index (PASI) ≥ 12 within three treatment periods. In period A, subjects received placebo or adalimumab at an initial dose of 80 mg at Week 0 followed by a dose of 40 mg every other week starting at Week 1. After 16 weeks of therapy, subjects who achieved at least a PASI 75 response at Week 16, defined as a PASI score improvement of at least 75% relative to baseline, entered period B and received open-label 40 mg adalimumab every other week. After 17 weeks of open label therapy, subjects who maintained at least a PASI 75 response at Week 33 and were originally randomized to active therapy in period A were re-randomized in period C to receive 40 mg adalimumab every other week or placebo for an additional 19 weeks. Across all treatment groups the mean baseline PASI score was 19 and the baseline Physician's Global Assessment score ranged from "moderate" (53%) to "severe" (41%) to "very severe" (6%).
Study Ps-II evaluated 99 subjects randomized to adalimumab and 48 subjects randomized to placebo with chronic plaque psoriasis with ≥ 10% BSA involvement and PASI ≥ 12. Subjects received placebo, or an initial dose of 80 mg adalimumab at Week 0 followed by 40 mg every other week starting at Week 1 for 16 weeks. Across all treatment groups the mean baseline PASI score was 21 and the baseline PGA score ranged from "moderate" (41%) to "severe" (51%) to "very severe" (8%).
Studies Ps-I and II evaluated the proportion of subjects who achieved "clear" or "minimal" disease on the 6-point PGA scale and the proportion of subjects who achieved a reduction in PASI score of at least 75% (PASI 75) from baseline at Week 16 (see Table 16 and 17).
Additionally, Study Ps-I evaluated the proportion of subjects who maintained a PGA of "clear" or "minimal" disease or a PASI 75 response after Week 33 and on or before Week 52.
Table 16. Efficacy Results at 16 Weeks in Study Ps-I Number of Subjects (%) Adalimumab 40 mg every other week Placebo N = 814 N = 398 PGA: Clear or minimal* 506 (62%) 17 (4%) PASI 75 578 (71%) 26 (7%) * Clear = no plaque elevation, no scale, plus or minus hyperpigmentation or diffuse pink or red coloration
Minimal = possible but difficult to ascertain whether there is slight elevation of plaque above normal skin, plus or minus surface dryness with some white coloration, plus or minus up to red colorationTable 17. Efficacy Results at 16 Weeks in Study Ps-II Number of Subjects (%) Adalimumab 40 mg every other week Placebo N = 99 N = 48 PGA: Clear or minimal* 70 (71%) 5 (10%) PASI 75 77 (78%) 9 (19%) * Clear = no plaque elevation, no scale, plus or minus hyperpigmentation or diffuse pink or red coloration
Minimal = possible but difficult to ascertain whether there is slight elevation of plaque above normal skin, plus or minus surface dryness with some white coloration, plus or minus up to red colorationAdditionally, in Study Ps-I, subjects on adalimumab who maintained a PASI 75 were re-randomized to adalimumab (N = 250) or placebo (N = 240) at Week 33. After 52 weeks of treatment with adalimumab, more subjects on adalimumab maintained efficacy when compared to subjects who were re-randomized to placebo based on maintenance of PGA of "clear" or "minimal" disease (68% vs. 28%) or a PASI 75 (79% vs. 43%).
A total of 347 stable responders participated in a withdrawal and retreatment evaluation in an open-label extension study. Median time to relapse (decline to PGA "moderate" or worse) was approximately 5 months. During the withdrawal period, no subject experienced transformation to either pustular or erythrodermic psoriasis. A total of 178 subjects who relapsed re-initiated treatment with 80 mg of adalimumab, then 40 mg every other week beginning at week 1. At week 16, 69% (123/178) of subjects had a response of PGA "clear" or "minimal".
A randomized, double-blind study (Study Ps-III) compared the efficacy and safety of adalimumab versus placebo in 217 adult subjects. Subjects in the study had to have chronic plaque psoriasis of at least moderate severity on the PGA scale, fingernail involvement of at least moderate severity on a 5-point Physician's Global Assessment of Fingernail Psoriasis (PGA-F) scale, a Modified Nail Psoriasis Severity Index (mNAPSI) score for the target-fingernail of ≥ 8, and either a BSA involvement of at least 10% or a BSA involvement of at least 5% with a total mNAPSI score for all fingernails of ≥ 20. Subjects received an initial dose of 80 mg adalimumab followed by 40 mg every other week (starting one week after the initial dose) or placebo for 26 weeks followed by open-label adalimumab treatment for an additional 26 weeks. This study evaluated the proportion of subjects who achieved "clear" or "minimal" assessment with at least a 2-grade improvement on the PGA-F scale and the proportion of subjects who achieved at least a 75% improvement from baseline in the mNAPSI score (mNAPSI 75) at Week 26.
At Week 26, a higher proportion of subjects in the adalimumab group than in the placebo group achieved the PGA-F endpoint. Furthermore, a higher proportion of subjects in the adalimumab group than in the placebo group achieved mNAPSI 75 at Week 26 (see Table 18).
Table 18. Efficacy Results at 26 Weeks Endpoint Adalimumab 40 mg every other week*
N=109Placebo
N=108PGA-F: ≥ 2-grade improvement and clear or minimal 49% 7% mNAPSI 75 47% 3% * Subjects received 80 mg of adalimumab at Week 0, followed by 40 mg every other week starting at Week 1. Nail pain was also evaluated and improvement in nail pain was observed in Study Ps-III.
14.9 Clinical Studies in Hidradenitis Suppurativa
Two randomized, double-blind, placebo-controlled studies (Studies HS-I and II) evaluated the safety and efficacy of adalimumab in a total of 633 adult subjects with moderate to severe hidradenitis suppurativa (HS) with Hurley Stage II or III disease and with at least 3 abscesses or inflammatory nodules. In both studies, subjects received placebo or adalimumab at an initial dose of 160 mg at Week 0, 80 mg at Week 2, and 40 mg every week starting at Week 4 and continued through Week 11. Subjects used topical antiseptic wash daily. Concomitant oral antibiotic use was allowed in Study HS-II.
Both studies evaluated Hidradenitis Suppurativa Clinical Response (HiSCR) at Week 12. HiSCR was defined as at least a 50% reduction in total abscess and inflammatory nodule count with no increase in abscess count and no increase in draining fistula count relative to baseline (see Table 19). Reduction in HS-related skin pain was assessed using a Numeric Rating Scale in subjects who entered the study with an initial baseline score of 3 or greater on a 11 point scale.
In both studies, a higher proportion of adalimumab- than placebo-treated subjects achieved HiSCR (see Table 19).
Table 19. Efficacy Results at 12 Weeks in Subjects with Moderate to Severe Hidradenitis Suppurativa HS Study I HS Study II* Placebo Adalimumab
40 mg WeeklyPlacebo Adalimumab
40 mg WeeklyHidradenitis Suppurativa Clinical Response (HiSCR) N = 154
40 (26%)N = 153
64 (42%)N = 163
45 (28%)N = 163
96 (59%)* 19.3% of subjects in Study HS-II continued baseline oral antibiotic therapy during the study. In both studies, from Week 12 to Week 35 (Period B), subjects who had received adalimumab were re-randomized to 1 of 3 treatment groups (adalimumab 40 mg every week, adalimumab 40 mg every other week, or placebo). Subjects who had been randomized to placebo were assigned to receive adalimumab 40 mg every week (Study HS-I) or placebo (Study HS-II).
During Period B, flare of HS, defined as ≥ 25% increase from baseline in abscesses and inflammatory nodule counts and with a minimum of 2 additional lesions, was documented in 22 (22%) of the 100 subjects who were withdrawn from adalimumab treatment following the primary efficacy timepoint in two studies.
14.10 Clinical Studies in Adults with Uveitis
The safety and efficacy of adalimumab were assessed in adult subjects with non-infectious intermediate, posterior and panuveitis excluding subjects with isolated anterior uveitis, in two randomized, double-masked, placebo-controlled studies (UV I and II). Subjects received placebo or adalimumab at an initial dose of 80 mg followed by 40 mg every other week starting one week after the initial dose. The primary efficacy endpoint in both studies was ´time to treatment failure´.
Treatment failure was a multi-component outcome defined as the development of new inflammatory chorioretinal and/or inflammatory retinal vascular lesions, an increase in anterior chamber (AC) cell grade or vitreous haze (VH) grade or a decrease in best corrected visual acuity (BCVA).
Study UV I evaluated 217 subjects with active uveitis while being treated with corticosteroids (oral prednisone at a dose of 10 to 60 mg/day). All subjects received a standardized dose of prednisone 60 mg/day at study entry followed by a mandatory taper schedule, with complete corticosteroid discontinuation by Week 15.
Study UV II evaluated 226 subjects with inactive uveitis while being treated with corticosteroids (oral prednisone 10 to 35 mg/day) at baseline to control their disease. Subjects subsequently underwent a mandatory taper schedule, with complete corticosteroid discontinuation by Week 19.
Clinical Response
Results from both studies demonstrated statistically significant reduction of the risk of treatment failure in subjects treated with adalimumab versus subjects receiving placebo. In both studies, all components of the primary endpoint contributed cumulatively to the overall difference between adalimumab and placebo groups (Table 20).
Table 20. Time to Treatment Failure in Studies UV I and UV II UV I UV II Placebo
(N = 107)Adalimumab
(N = 110)HR
[95% CI]aPlacebo
(N = 111)Adalimumab
(N = 115)HR
[95% CI]aFailureb n (%) 84 (78.5) 60 (54.5) 0.50
[0.36, 0.70]61 (55.0) 45 (39.1) 0.57
[0.39, 0.84]Median Time to Failure (Months)
[95% CI]3.0
[2.7, 3.7]5.6
[3.9, 9.2]N/A 8.3
[4.8, 12.0]NEc N/A b HR of adalimumab versus placebo from proportional hazards regression with treatment as factor.
b Treatment failure at or after Week 6 in Study UV I, or at or after Week 2 in Study UV II, was counted as event. Subjects who discontinued the study were censored at the time of dropping out.
c NE = not estimable. Fewer than half of at-risk subjects had an event.Figure 3: Kaplan-Meier Curves Summarizing Time to Treatment Failure on or after Week 6 (Study UV I) or Week 2 (Study UV II) 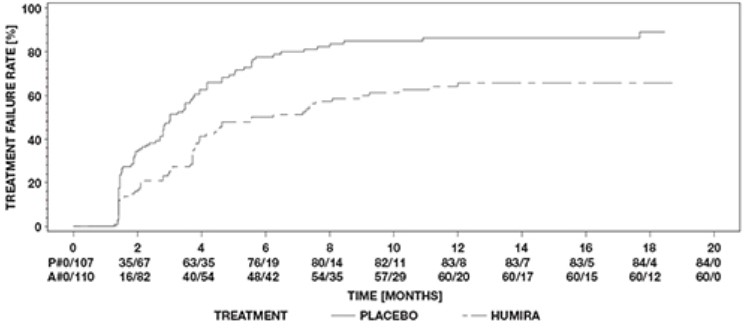
Study UV I 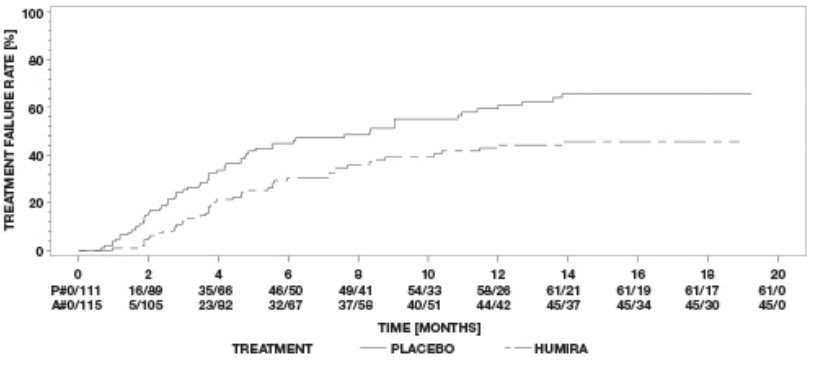
Study UV II Note: P# = Placebo (Number of Events/Number at Risk); A# = Adalimumab (Number of Events/Number at Risk).
14.11 Clinical Studies in Pediatric Subjects with Uveitis
The safety and efficacy of adalimumab were assessed in a randomized, double-masked, placebo-controlled study of 90 pediatric subjects from 2 to < 18 years of age with active JIA-associated non-infectious uveitis (PUV-I). Subjects received either placebo or 20 mg adalimumab (if < 30 kg) or 40 mg adalimumab (if 30 kg) every other week in combination with a dose of methotrexate. Concomitant dosages of corticosteroids were permitted at study entry followed by a mandatory reduction in topical corticosteroids within 3 months.
The primary endpoint was time to treatment failure. The criteria determining treatment failure were worsening or sustained non-improvement in ocular inflammation, or worsening of ocular co-morbidities.
Clinical Response
Adalimumab significantly decreased the risk of treatment failure by 75% relative to placebo (HR = 0.25 [95% CI: 0.12, 0.49]) (Table 21).
Table 21. Analysis Results of Time to Treatment Failure (Study PUV-I) Placebo
(N=30)Adalimumab
(N=60)HR (95% CI)a Failure (n[%]) 18 (60%) 16 (26.7%) 0.25
(0.12, 0.49)Median Time to
Failure (Weeks)
(95% CI)b24.1
(12.4, 81.0)NEc a HR of adalimumab versus placebo from proportional hazards regression with treatment as factor.
b Estimated based on Kaplan-Meier curve.
c NE = not estimable. Fewer than half of at-risk subjects had an event.Figure 4: Kaplan-Meier Curves Summarizing Time to Treatment Failure (Study PUV-I) 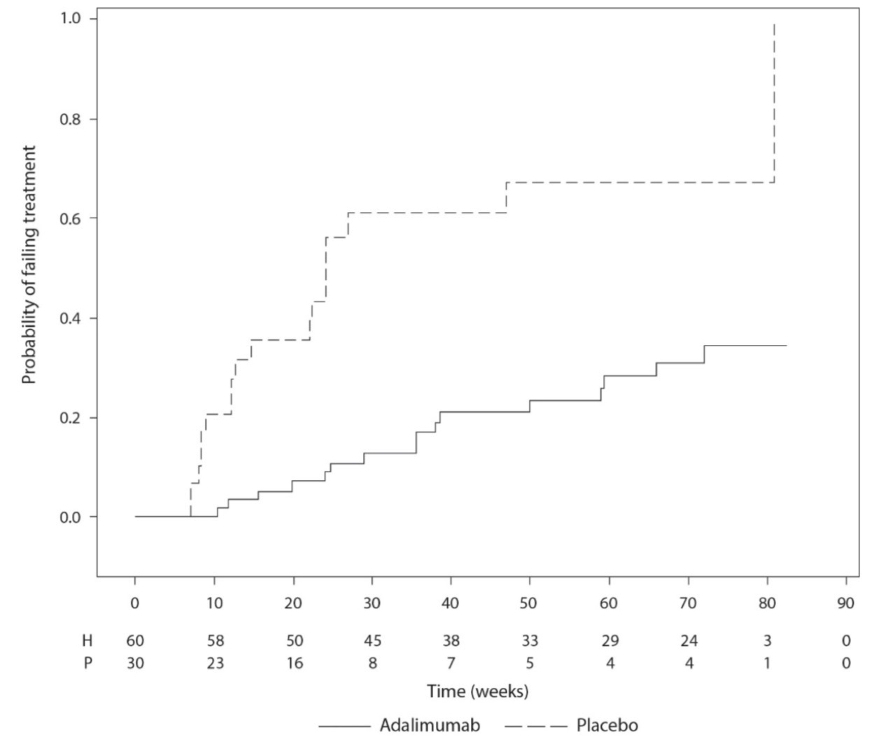
Study PUV-I
Note: P# = Placebo (Number of Events/Number at Risk); A# = Adalimumab (Number of Events/Number at Risk).
- 15 REFERENCES
-
16 HOW SUPPLIED/STORAGE AND HANDLING
YUFLYMA (adalimumab-aaty) is supplied as a preservative-free, sterile, clear to opalescent, and colorless to pale brown solution for subcutaneous administration. The following packaging configurations are available.
-
YUFLYMA AI Carton - 40 mg/0.4 mL (1 Count)
YUFLYMA is supplied in a carton containing two alcohol preps and one dose tray. The dose tray consists of a single-dose prefilled auto-injector, containing a 1 mL prefilled syringe with a fixed thin wall, ½ inch needle, providing 40 mg/0.4 mL of YUFLYMA. The NDC number is 72606-030-09. -
YUFLYMA AI Carton - 40 mg/0.4 mL (2 Count)
YUFLYMA is supplied in a carton containing two alcohol preps and two dose trays. Each dose tray consists of a single-dose prefilled auto-injector, containing a 1 mL prefilled syringe with a fixed thin wall, ½ inch needle, providing 40 mg/0.4 mL of YUFLYMA. The NDC number is 72606-030-10. -
YUFLYMA AI Carton - 40 mg/0.4 mL (4 Count)
YUFLYMA is supplied in a carton containing four alcohol preps and four dose trays. Each dose tray consists of a single-dose prefilled auto-injector, containing a 1 mL prefilled syringe with a fixed thin wall, ½ inch needle, providing 40 mg/0.4 mL of YUFLYMA. The NDC number is 72606-030-11. -
YUFLYMA AI Carton - 40 mg/0.4 mL (6 Count)
YUFLYMA is supplied in a carton containing six alcohol preps and six dose trays. Each dose tray consists of a single-dose prefilled auto-injector, containing a 1 mL prefilled syringe with a fixed thin wall, ½ inch needle, providing 40 mg/0.4 mL of YUFLYMA. The NDC number is 72606-030-12. -
Prefilled Syringe with Safety Guard Carton - 40 mg/0.4 mL (1 Count)
YUFLYMA is supplied in a carton containing two alcohol preps and one dose tray. The dose tray consists of a single-dose prefilled syringe with safety guard, containing a 1 mL prefilled syringe with a fixed thin wall, ½ inch needle, providing 40 mg/0.4 mL of YUFLYMA. The NDC number is 72606-030-05. -
Prefilled Syringe with Safety Guard Carton - 40 mg/0.4 mL (2 Count)
YUFLYMA is supplied in a carton containing two alcohol preps and two dose trays. Each dose tray consists of a single-dose prefilled syringe with safety guard, containing a 1 mL prefilled syringe with a fixed thin wall, ½ inch needle, providing 40 mg/0.4 mL of YUFLYMA. The NDC number is 72606-030-06. -
Prefilled Syringe with Safety Guard Carton - 40 mg/0.4 mL (4 Count)
YUFLYMA is supplied in a carton containing four alcohol preps and four dose trays. Each dose tray consists of a single-dose prefilled syringe with safety guard, containing a 1 mL prefilled syringe with a fixed thin wall, ½ inch needle, providing 40 mg/0.4 mL of YUFLYMA. The NDC number is 72606-030-07. -
Prefilled Syringe with Safety Guard Carton - 40 mg/0.4 mL (6 Count)
YUFLYMA is supplied in a carton containing six alcohol preps and six dose trays. Each dose tray consists of a single-dose prefilled syringe with safety guard, containing a 1 mL prefilled syringe with a fixed thin wall, ½ inch needle, providing 40 mg/0.4 mL of YUFLYMA. The NDC number is 72606-030-08. -
Prefilled Syringe Carton - 40 mg/0.4 mL (1 Count)
YUFLYMA is supplied in a carton containing two alcohol preps and one dose tray. The dose tray consists of a single-dose prefilled syringe, containing a 1 mL prefilled syringe with a fixed thin wall, ½ inch needle, providing 40 mg/0.4 mL of YUFLYMA. The NDC number is 72606-030-01. -
Prefilled Syringe Carton - 40 mg/0.4 mL (2 Count)
YUFLYMA is supplied in a carton containing two alcohol preps and two dose trays. Each dose tray consists of a single-dose prefilled syringe, containing a 1 mL prefilled syringe with a fixed thin wall, ½ inch needle, providing 40 mg/0.4 mL of YUFLYMA. The NDC number is 72606-030-02. -
Prefilled Syringe Carton - 40 mg/0.4 mL (4 Count)
YUFLYMA is supplied in a carton containing four alcohol preps and four dose trays. Each dose tray consists of a single-dose prefilled syringe, containing a 1 mL prefilled syringe with a fixed thin wall, ½ inch needle, providing 40 mg/0.4 mL of YUFLYMA. The NDC number is 72606-030-03. -
Prefilled Syringe Carton - 40 mg/0.4 mL (6 Count)
YUFLYMA is supplied in a carton containing six alcohol preps and six dose trays. Each dose tray consists of a single-dose prefilled syringe, containing a 1 mL prefilled syringe with a fixed thin wall, ½ inch needle, providing 40 mg/0.4 mL of YUFLYMA. The NDC number is 72606-030-04. -
YUFLYMA AI 40 mg/0.4 mL - Plaque Psoriasis Starter Package (4 Count)
YUFLYMA is supplied in a carton containing four alcohol preps and four dose trays (Plaque Psoriasis Starter Package). Each dose tray consists of a single-dose prefilled auto-injector, containing a 1 mL prefilled syringe with a fixed thin wall, ½ inch needle, providing 40 mg/0.4 mL of YUFLYMA. The NDC number is 72606-030-13. -
YUFLYMA AI 40 mg/0.4 mL - Crohn's Disease, Pediatric Crohn's Disease, Ulcerative Colitis or Hidradenitis Suppurativa Starter Package (6 Count)
YUFLYMA is supplied in a carton containing six alcohol preps and six dose trays (Crohn's Disease, Pediatric Crohn’s Disease, Ulcerative Colitis or Hidradenitis Suppurativa Starter Package). Each dose tray consists of a single-dose prefilled auto-injector, containing a 1 mL prefilled syringe with a fixed thin wall, ½ inch needle, providing 40 mg/0.4 mL of YUFLYMA. The NDC number is 72606-030-14. -
YUFLYMA AI Carton - 80 mg/0.8 mL (1 Count)
YUFLYMA is supplied in a carton containing two alcohol preps and one dose tray. The dose tray consists of a single-dose prefilled auto-injector, containing a 1 mL prefilled syringe with a fixed thin wall, ½ inch needle, providing 80 mg/0.8 mL of YUFLYMA. The NDC number is 72606-023-04. -
YUFLYMA AI Carton - 80 mg/0.8 mL (2 Count)
YUFLYMA is supplied in a carton containing two alcohol preps and two dose trays. Each dose tray consists of a single-dose prefilled auto-injector, containing a 1 mL prefilled syringe with a fixed thin wall, ½ inch needle, providing 80 mg/0.8 mL of YUFLYMA. The NDC number is 72606-023-05. -
Prefilled Syringe with Safety Guard Carton - 80 mg/0.8 mL (1 Count)
YUFLYMA is supplied in a carton containing two alcohol preps and one dose tray. The dose tray consists of a single-dose prefilled syringe with safety guard, containing a 1 mL prefilled syringe with a fixed thin wall, ½ inch needle, providing 80 mg/0.8 mL of YUFLYMA. The NDC number is 72606-023-02. -
Prefilled Syringe with Safety Guard Carton - 80 mg/0.8 mL (2 Count)
YUFLYMA is supplied in a carton containing two alcohol preps and two dose trays. Each dose tray consists of a single-dose prefilled syringe with safety guard, containing a 1 mL prefilled syringe with a fixed thin wall, ½ inch needle, providing 80 mg/0.8 mL of YUFLYMA. The NDC number is 72606-023-03. -
Prefilled Syringe Carton - 80 mg/0.8 mL (1 Count)
YUFLYMA is supplied in a carton containing two alcohol preps and one dose tray. The dose tray consists of a single-dose prefilled syringe, containing a 1 mL prefilled syringe with a fixed thin wall, ½ inch needle, providing 80 mg/0.8 mL of YUFLYMA. The NDC number is 72606-023-01. -
Prefilled Syringe Carton - 20 mg/0.2 mL (2 Count)
YUFLYMA is supplied in a carton containing two alcohol preps and two dose trays. Each dose tray consists of a single-dose prefilled syringe, containing a 1 mL prefilled syringe with a fixed thin wall, ½ inch needle, providing 20 mg/0.2 mL of YUFLYMA. The NDC number is 72606-024-01. -
Prefilled Syringe Carton - 10 mg/0.1 mL (2 Count)
YUFLYMA is supplied in a carton containing two alcohol preps and two dose trays. Each dose tray consists of a single-dose prefilled syringe, containing a 1 mL prefilled syringe with a fixed thin wall, ½ inch needle, providing 10 mg/0.1 mL of YUFLYMA. The NDC number is 72606-063-01. -
YUFLYMA AI 80 mg/0.8 mL and 40 mg/0.4 mL - Plaque Psoriasis Starter Package (3 Count)
YUFLYMA is supplied in a carton containing four alcohol preps and three dose trays (Plaque Psoriasis Starter Package). One dose tray consists of a single-dose prefilled auto-injector, containing a 1 mL prefilled syringe with a fixed thin wall, ½ inch needle, providing 80 mg/0.8 mL of YUFLYMA. Each of two other dose trays consists of a single-dose prefilled auto-injector, containing a 1 mL prefilled syringe with a fixed thin wall, ½ inch needle, providing 40 mg/0.4 mL of YUFLYMA. The NDC number is 72606-046-01. -
YUFLYMA AI 80 mg/0.8 mL - Crohn's Disease, Ulcerative Colitis or Hidradenitis Suppurativa Starter Package (3 Count)
YUFLYMA is supplied in a carton containing four alcohol preps and three dose trays (Crohn’s Disease, Ulcerative Colitis or Hidradenitis Suppurativa Starter Package). Each dose tray consists of a single-dose prefilled auto-injector, containing a 1 mL prefilled syringe with a fixed thin wall, ½ inch needle, providing 80 mg/0.8 mL of YUFLYMA. The NDC number is 72606-023-07. -
YUFLYMA Prefilled Syringe with Safety Guard 80 mg/0.8 mL and 40 mg/0.4 mL - Pediatric Crohn's Disease Starter Package (2 Count)
YUFLYMA is supplied in a carton containing two alcohol preps and two dose trays (Pediatric Crohn’s Disease Starter Package). One dose tray consists of a single-dose prefilled syringe with safety guard, containing a 1 mL prefilled syringe with a fixed thin wall, ½ inch needle, providing 80 mg/0.8 mL of YUFLYMA. The other dose tray consists of a single-dose prefilled syringe with safety guard, containing a 1 mL prefilled syringe with a fixed thin wall, ½ inch needle, providing 40 mg/0.4 mL of YUFLYMA. The NDC number is 72606-039-01. -
YUFLYMA Prefilled Syringe with Safety Guard 80 mg/0.8 mL - Pediatric Crohn's Disease Starter Package (3 Count)
YUFLYMA is supplied in a carton containing four alcohol preps and three dose trays (Pediatric Crohn’s Disease Starter Package). Each dose tray consists of a single-dose prefilled syringe with safety guard, containing a 1 mL prefilled syringe with a fixed thin wall, ½ inch needle, providing 80 mg/0.8 mL of YUFLYMA. The NDC number is 72606-023-08.
Storage and Stability
Do not use beyond the expiration date on the container. YUFLYMA must be refrigerated at 36°F to 46°F (2°C to 8°C). DO NOT FREEZE. Do not use if frozen even if it has been thawed.
Store in original carton until time of administration to protect from light. Do not shake.
If needed, for example when traveling, YUFLYMA may be stored at room temperature up to a maximum of 77°F (25°C) for a period of up to 31 days, with protection from light. YUFLYMA should be discarded if not used within the 31-day period. Record the date when YUFLYMA is first removed from the refrigerator in the spaces provided on the carton and dose pack.
Do not store YUFLYMA in extreme heat or cold.
-
YUFLYMA AI Carton - 40 mg/0.4 mL (1 Count)
-
17 PATIENT COUNSELING INFORMATION
Advise the patient or caregiver to read the FDA-approved patient labeling (Medication Guide and Instructions for Use).
Infections
Inform patients that YUFLYMA may lower the ability of their immune system to fight infections. Instruct patients of the importance of contacting their doctor if they develop any symptoms of infection, including tuberculosis, invasive fungal infections, and reactivation of hepatitis B virus infections [see Warnings and Precautions (5.1, 5.2, 5.4)].
Malignancies
Counsel patients about the risk of malignancies while receiving YUFLYMA [see Warnings and Precautions (5.2)].
Hypersensitivity Reactions
Advise patients to seek immediate medical attention if they experience any symptoms of severe hypersensitivity reactions. [see Warnings and Precautions (5.3)].
Other Medical Conditions
Advise patients to report any signs of new or worsening medical conditions such as congestive heart failure, neurological disease, autoimmune disorders, or cytopenias. Advise patients to report any symptoms suggestive of a cytopenia such as bruising, bleeding, or persistent fever [see Warnings and Precautions (5.5, 5.6, 5.8, 5.9)].
Instructions on Injection Technique
Inform patients that the first injection is to be performed under the supervision of a qualified health care professional. If a patient or caregiver is to administer YUFLYMA, instruct them in injection techniques and assess their ability to inject subcutaneously to ensure the proper administration of YUFLYMA [see Instructions for Use].
For patients who will use the YUFLYMA AI, tell them that they:
- May hear a 1st loud "click" when they place the auto-injector straight on their skin and push the entire device down firmly. The click means the start of the injection.
- Must keep holding the YUFLYMA AI against their skin until all of the medicine is injected.
- Will know that the injection has finished when the blue plunger rod fills the medication window and stops moving. Also they may hear a 2nd loud "click" several seconds after starting the injection. Then, count slowly to 5 to ensure full dose has been given.
Instruct patients to dispose of their used needles and syringes or used auto-injector in a FDA-cleared sharps disposal container immediately after use. Instruct patients not to dispose of loose needles and syringes or auto-injector in their household trash. Instruct patients that if they do not have a FDA-cleared sharps disposal container, they may use a household container that is made of a heavy-duty plastic, can be closed with a tight-fitting and puncture-resistant lid without sharps being able to come out, upright and stable during use, leak-resistant, and properly labeled to warn of hazardous waste inside the container.
Instruct patients that when their sharps disposal container is almost full, they will need to follow their community guidelines for the correct way to dispose of their sharps disposal container. Instruct patients that there may be state or local laws regarding disposal of used needles and syringes. Refer patients to the FDA's website at http://www.fda.gov/safesharpsdisposal for more information about safe sharps disposal, and for specific information about sharps disposal in the state that they live in.
Instruct patients not to dispose of their used sharps disposal container in their household trash unless their community guidelines permit this. Instruct patients not to recycle their used sharps disposal container.
- SPL UNCLASSIFIED SECTION
-
MEDICATION GUIDE
MEDICATION GUIDE
YUFLYMA® (yoo-fly'-mah)
(adalimumab-aaty)
injection, for subcutaneous useThis Medication Guide has been approved by the U.S. Food and Drug Administration. Revised: 10/2025 Read the Medication Guide that comes with YUFLYMA before you start taking it and each time you get a refill. There may be new information. This Medication Guide does not take the place of talking with your doctor about your medical condition or treatment. What is the most important information I should know about YUFLYMA?
YUFLYMA is a medicine that affects your immune system. YUFLYMA can lower the ability of your immune system to fight infections. Serious infections have happened in people taking adalimumab products. These serious infections include tuberculosis (TB) and infections caused by viruses, fungi or bacteria that have spread throughout the body. Some people have died from these infections.- Your doctor should test you for TB before starting YUFLYMA.
- Your doctor should check you closely for signs and symptoms of TB during treatment with YUFLYMA.
Before starting YUFLYMA, tell your doctor if you:- think you have an infection or have symptoms of infection such as:
- fever, sweats, or chills
- muscle aches
- cough
- shortness of breath
- blood in phlegm
- warm, red, or painful skin or sores on your body
- diarrhea or stomach pain
- burning when you urinate or urinate more often than normal
- feel very tired
- weight loss
- are being treated for an infection.
- get a lot of infections or have infections that keep coming back.
- have diabetes.
- have TB, or have been in close contact with someone with TB.
- were born in, lived in, or traveled to countries where there is more risk for getting TB. Ask your doctor if you are not sure.
- live or have lived in certain parts of the country (such as the Ohio and Mississippi River valleys) where there is an increased risk for getting certain kinds of fungal infections (histoplasmosis, coccidioidomycosis, or blastomycosis). These infections may happen or become more severe if you use YUFLYMA. Ask your doctor if you do not know if you have lived in an area where these infections are common.
- have or have had hepatitis B.
- use the medicine ORENCIA® (abatacept), KINERET® (anakinra), RITUXAN® (rituximab), IMURAN® (azathioprine), or PURINETHOL® (6–mercaptopurine, 6-MP).
- are scheduled to have major surgery.
After starting YUFLYMA, call your doctor right away if you have an infection, or any sign of an infection.
YUFLYMA can make you more likely to get infections or make any infection that you may have worse.
Cancer- For children and adults taking Tumor Necrosis Factor (TNF)-blockers, including YUFLYMA, the chances of getting cancer may increase.
- There have been cases of unusual cancers in children, teenagers, and young adults using TNF-blockers.
- People with rheumatoid arthritis (RA), especially more serious RA, may have a higher chance for getting a kind of cancer called lymphoma.
- If you use TNF blockers including YUFLYMA, your chance of getting two types of skin cancer may increase (basal cell cancer and squamous cell cancer of the skin). These types of cancer are generally not life-threatening if treated. Tell your doctor if you have a bump or open sore that does not heal.
- Some people receiving TNF blockers including YUFLYMA developed a rare type of cancer called hepatosplenic T-cell lymphoma. This type of cancer often results in death. Most of these people were male teenagers or young men. Also, most people were being treated for Crohn's disease or ulcerative colitis with another medicine called IMURAN® (azathioprine) or PURINETHOL® (6-mercaptopurine, 6–MP).
What is YUFLYMA?
YUFLYMA is a medicine called a Tumor Necrosis Factor (TNF) blocker. YUFLYMA is used:- To reduce the signs and symptoms of:
- moderate to severe RA in adults. YUFLYMA can be used alone, with methotrexate, or with certain other medicines.
- moderate to severe polyarticular juvenile idiopathic arthritis (JIA) in children 2 years and older. YUFLYMA can be used alone or with methotrexate.
- psoriatic arthritis (PsA) in adults. YUFLYMA can be used alone or with certain other medicines.
- ankylosing spondylitis (AS) in adults.
- moderate to severe hidradenitis suppurativa (HS) in people 12 years and older.
- To treat moderate to severe Crohn's disease (CD) in adults and children 6 years of age and older.
- To treat moderate to severe ulcerative colitis (UC) in adults. It is not known if adalimumab products are effective in people who stopped responding to or could not tolerate TNF-blocker medicines.
- To treat moderate to severe chronic (lasting a long time) plaque psoriasis (Ps) in adults who have the condition in many areas of their body and who may benefit from taking injections or pills (systemic therapy) or phototherapy (treatment using ultraviolet light alone or with pills).
- To treat non-infectious intermediate, posterior, and panuveitis in adults and children 2 years of age and older.
What should I tell my doctor before taking YUFLYMA?
YUFLYMA may not be right for you. Before starting YUFLYMA, tell your doctor about all of your medical conditions, including if you:- have an infection. See "What is the most important information I should know about YUFLYMA?"
- have or have had cancer.
- have any numbness or tingling or have a disease that affects your nervous system such as multiple sclerosis or Guillain-Barré syndrome.
- have or had heart failure.
- have recently received or are scheduled to receive a vaccine. You may receive vaccines, except for live vaccines while using YUFLYMA. Children should be brought up to date with all vaccines before starting YUFLYMA.
- are allergic to YUFLYMA or to any of its ingredients. See the end of this Medication Guide for a list of ingredients in YUFLYMA.
- are pregnant or plan to become pregnant, breastfeeding or plan to breastfeed. You and your doctor should decide if you should take YUFLYMA while you are pregnant or breastfeeding.
- have a baby and you were using YUFLYMA during your pregnancy. Tell your baby's doctor before your baby receives any vaccines.
Especially tell your doctor if you use:- ORENCIA® (abatacept), KINERET® (anakinra), REMICADE® (infliximab), ENBREL® (etanercept), CIMZIA® (certolizumab pegol) or SIMPONI® (golimumab), because you should not use YUFLYMA while you are also using one of these medicines.
- RITUXAN® (rituximab). Your doctor may not want to give you YUFLYMA if you have received RITUXAN® (rituximab) recently.
- IMURAN® (azathioprine) or PURINETHOL® (6–mercaptopurine, 6-MP).
How should I take YUFLYMA? - YUFLYMA is given by an injection under the skin. Your doctor will tell you how often to take an injection of YUFLYMA. This is based on your condition to be treated. Do not inject YUFLYMA more often than you were prescribed.
- See the Instructions for Use inside the carton for complete instructions for the right way to prepare and inject YUFLYMA.
- Make sure you have been shown how to inject YUFLYMA before you do it yourself. You can call your doctor or 1-877-888-4231 if you have any questions about giving yourself an injection. Someone you know can also help you with your injection after they have been shown how to prepare and inject YUFLYMA.
- Do not try to inject YUFLYMA yourself until you have been shown the right way to give the injections. If your doctor decides that you or a caregiver may be able to give your injections of YUFLYMA at home, you should receive training on the right way to prepare and inject YUFLYMA.
- Do not miss any doses of YUFLYMA unless your doctor says it is okay. If you forget to take YUFLYMA, inject a dose as soon as you remember. Then, take your next dose at your regular scheduled time. This will put you back on schedule. In case you are not sure when to inject YUFLYMA, call your doctor or pharmacist.
- If you take more YUFLYMA than you were told to take, call your doctor.
What are the possible side effects of YUFLYMA?
YUFLYMA can cause serious side effects, including:
See "What is the most important information I should know about YUFLYMA?"-
Serious Infections.
Your doctor will examine you for TB and perform a test to see if you have TB. If your doctor feels that you are at risk for TB, you may be treated with medicine for TB before you begin treatment with YUFLYMA and during treatment with YUFLYMA. Even if your TB test is negative your doctor should carefully monitor you for TB infections while you are taking YUFLYMA. People who had a negative TB skin test before receiving adalimumab products have developed active TB. Tell your doctor if you have any of the following symptoms while taking or after taking YUFLYMA:
- cough that does not go away
- low grade fever
- weight loss
- loss of body fat and muscle (wasting)
-
Hepatitis B infection in people who carry the virus in their blood.
If you are a carrier of the hepatitis B virus (a virus that affects the liver), the virus can become active while you use YUFLYMA. Your doctor should do blood tests before you start treatment, while you are using YUFLYMA, and for several months after you stop treatment with YUFLYMA. Tell your doctor if you have any of the following symptoms of a possible hepatitis B infection:
- muscle aches
- feel very tired
- dark urine
- skin or eyes look yellow
- little or no appetite
- vomiting
- clay-colored bowel movements
- fever
- chills
- stomach discomfort
- skin rash
- Allergic reactions. Allergic reactions can happen in people who use YUFLYMA. Call your doctor or get medical help right away if you have any of these symptoms of a serious allergic reaction:
- hives
- trouble breathing
- swelling of your face, eyes, lips or mouth
- Nervous system problems. Signs and symptoms of a nervous system problem include: numbness or tingling, problems with your vision, weakness in your arms or legs, and dizziness.
- Blood problems. Your body may not make enough of the blood cells that help fight infections or help to stop bleeding. Symptoms include a fever that does not go away, bruising or bleeding very easily, or looking very pale.
- New heart failure or worsening of heart failure you already have. Call your doctor right away if you get new worsening symptoms of heart failure while taking YUFLYMA, including:
- shortness of breath
- sudden weight gain
- swelling of your ankles or feet
- Immune reactions including a lupus-like syndrome. Symptoms include chest discomfort or pain that does not go away, shortness of breath, joint pain, or a rash on your cheeks or arms that gets worse in the sun. Symptoms may improve when you stop YUFLYMA.
- Liver Problems. Liver problems can happen in people who use TNF-blocker medicines. These problems can lead to liver failure and death. Call your doctor right away if you have any of these symptoms:
- feel very tired
- poor appetite or vomiting
- skin or eyes look yellow
- pain on the right side of your stomach (abdomen)
- Psoriasis. Some people using adalimumab products had new psoriasis or worsening of psoriasis they already had. Tell your doctor if you develop red scaly patches or raised bumps that are filled with pus. Your doctor may decide to stop your treatment with YUFLYMA.
The most common side effects of YUFLYMA include:- injection site reactions: redness, rash, swelling, itching, or bruising. These symptoms usually will go away within a few days. Call your doctor right away if you have pain, redness or swelling around the injection site that does not go away within a few days or gets worse.
- upper respiratory infections (including sinus infections)
- headaches
- rash
Call your doctor for medical advice about side effects. You may report side effects to the FDA at 1-800-FDA-1088.How should I store YUFLYMA? - Store YUFLYMA in the refrigerator at 36°F to 46°F (2°C to 8°C). Store YUFLYMA in the original carton until use to protect it from light. Do not shake.
- Do not freeze YUFLYMA. Do not use YUFLYMA if frozen, even if it has been thawed.
- Refrigerated YUFLYMA may be used until the expiration date printed on the YUFLYMA carton, dose pack, auto-injector or prefilled syringe. Do not use YUFLYMA after the expiration date.
- If needed, for example when you are traveling, you may also store YUFLYMA at room temperature up to 77°F (25°C) for up to 31 days. Store YUFLYMA in the original carton until use to protect it from light.
- Throw away YUFLYMA if it has been kept at room temperature and not been used within 31 days.
- Record the date you first remove YUFLYMA from the refrigerator in the spaces provided on the carton and dose pack.
- Do not store YUFLYMA in extreme heat or cold.
- Do not use an auto-injector or prefilled syringe if the liquid is cloudy, discolored, or has flakes or particles in it.
- Do not drop or crush YUFLYMA. The prefilled syringe is glass.
General information about the safe and effective use of YUFLYMA
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use YUFLYMA for a condition for which it was not prescribed. Do not give YUFLYMA to other people, even if they have the same condition. It may harm them.
This Medication Guide summarizes the most important information about YUFLYMA. If you would like more information, talk with your doctor. You can ask your doctor or pharmacist for information about YUFLYMA that is written for health professionals.What are the ingredients in YUFLYMA?
Active ingredient: adalimumab-aaty
YUFLYMA AI 40 mg/0.4 mL, YUFLYMA 40 mg/0.4 mL prefilled syringe with safety guard, and YUFLYMA 40 mg/0.4 mL prefilled syringe, YUFLYMA AI 80 mg/0.8 mL, YUFLYMA 80 mg/0.8 mL prefilled syringe with safety guard, YUFLYMA 80 mg/0.8 mL prefilled syringe, YUFLYMA 20 mg/0.2 mL prefilled syringe and YUFLYMA 10 mg/0.1 mL prefilled syringe:
Inactive ingredients: acetic acid, glycine, polysorbate 80, sodium acetate, and Water for Injection, USP.
Manufactured by: CELLTRION, Inc., 23, Academy-ro, Yeonsu-gu, Incheon, 22014, Republic of Korea
US License Number 1996
Distributed by: CELLTRION USA, Inc., One Evertrust Plaza, Suite 1207, Jersey City, NJ 07302, USA -
INSTRUCTIONS FOR USE YUFLYMA® (yoo-fly'-mah) (adalimumab-aaty) injection, for subcutaneous use 80 mg/0.8 mL, 40 mg/0.4 mLSingle-Dose Auto-injector
For subcutaneous use only
Read and follow the Instructions for Use that come with your YUFLYMA Auto-injector before you start using it and each time you get a refill. There may be new information. This information does not take the place of talking to your doctor about your medical condition or treatment.
Figure A: Parts of YUFLYMA Auto-injector 
Important Information
- Use the Auto-injector only if your doctor has trained you on the right way to prepare for and to give an injection.
- Ask your doctor how often you will need to give an injection.
- Do not shake the Auto-injector at any time.
- Do not remove the Cap until you are ready to inject.
- Do not share the Auto-injector with anyone.
How to store the Auto-injector
- Store the Auto-injector in a refrigerator between 36°F to 46°F (2°C to 8°C).
- Keep the Auto-injector in the original carton until use to protect it from light.
- Do not use an Auto-injector that has been left in direct sunlight.
- Do not freeze the Auto-injector. If the Auto-injector has been frozen, do not use the Auto-injector even if it is thawed.
- If needed, you may store the Auto-injector at room temperature up to 77°F (25°C) for up to 31 days.
- After the Auto-injector has reached room temperature, do not put it back in the refrigerator.
- Keep the Auto-injector and all medicines out of the reach of children.
Read Instructions on All Pages Before Using the YUFLYMA Auto-injector
Prepare for Injection
This Instructions for Use has been approved by the U.S. Food and Drug Administration. Issued: 9/2023 1.Gather the supplies for the injection.1a. Prepare a clean, flat surface, such as a table or countertop, in a well-lit area.1b. Remove 1 Auto-injector from the carton stored in your refrigerator.1c. Make sure you have the following supplies:-Auto-injector-1 Alcohol swabNot included in the carton:-1 Cotton ball or gauze-1 Adhesive bandage-FDA-cleared sharps disposal container 2.Inspect the Auto-injector.2a. Make sure you have the correct medicine (YUFLYMA) and dosage.2b. Check the expiration date on the label of the Auto-injector (see Figure B).2c. Look at the Auto-injector and make sure it is not cracked or damaged. - Do not use the Auto-injector if;-it is cracked or damaged.-the expiration date has passed.
Figure B
3.Inspect the Medicine.3a. Look through the Window and make sure that the liquid is clear, colorless to pale brown, and free of particles (see Figure C). - Do not use the Auto-injector if the liquid is the discolored (yellow or dark brown), cloudy, or contains particles in it.
- You may see air bubbles in the liquid. This is normal.
Figure C
4.Wait 15 to 30 minutes.4a. Leave the Auto-injector at room temperature 68°F to 77°F (20°C to 25°C) for 15 to 30 minutes to allow it to warm up (see Figure D). - Do not warm the Auto-injector using heat sources such as hot water or a microwave.
Figure D
5.Choose an injection site (see Figure E).5a. You may inject into:-the front of your thighs.-your abdomen except for the 2 in (5 cm) around the belly button (navel). - Do not inject into skin that is within 2 in (5 cm) of your belly button (navel), or is red, hard, tender, damaged, bruised, or scarred.
- If you have psoriasis, do not inject directly into any raised, thick, red or scaly skin patches or lesions on your skin.
- Do not inject through your clothes.
- Do not inject the same injection site each time you give an injection.
- Each new injection site should be at least 1.2 in (3 cm) away from the injection site you used before.
Figure E
6.Wash your hands.6a. Wash your hands with soap and water and dry them thoroughly (see Figure F). Figure F
7.Clean the injection site.7a. Clean the injection site with an alcohol swab using a circular motion (see Figure G).7b. Let the skin dry before injecting. - Do not blow on or touch the injection site again before giving the injection.
Figure G
Give the Injection8.Remove the Cap.8a. Hold the Auto-injector by the injector body with the Cap on top using one hand. Gently pull the Cap straight off with the other hand. - Do not recap the Auto-injector.
- Do not remove the Cap until you are ready to inject.
- Do not touch the Needle or Needle Cover. Doing so may result in a needle stick injury because the Needle is inside the Needle Cover.
Figure H
8b. Dispose of the Cap in an FDA cleared sharps container (see Step 12 and Figure H). 9.Place the Auto-injector on the injection site.9a. Hold the Auto-injector so that you can see the Window.9b. Without pinching or stretching the skin, place the Auto-injector over the injection site at a 90-degree angle (see Figure I). Figure I
10.Give the injection (see Figure J).10a. Press the Auto-injector firmly against the skin. - When the injection starts you will hear the 1st loud "click" and the blue Plunger Rod will begin to fill the Window.
- Do not change the position of the Auto-injector after the injection has started.
Figure J
11.Remove the Auto-injector from your skin.11a. Remove the Auto-injector from your skin (see Figure K). - After you remove the Auto-injector from the injection site, the needle will be automatically covered (see Figure L).
- If the Window has not turned completely blue or if the medicine is still injecting, this means you have not received a full dose. Call your doctor immediately.
- You may see grey stopper in the Window. This is normal.
- Some bleeding may occur.
- Do not reuse the Auto-injector.
- Do not rub the injection site.
Figure L
After the Injection 12.Dispose of the Auto-injector.12a. Put the used Auto-injector in a FDA-cleared sharps disposal container right away after use (see Figure M). - Do not throw away (dispose of) the Auto-injector in your household trash.
- If you do not have an FDA-cleared sharps disposal container, you may use a household container that is:-made of a heavy-duty plastic,-can be closed with a tight-fitting, puncture-resistant lid, without sharps being able to come out,-upright and stable during use,-leak-resistant, and-properly labeled to warn of hazardous waste inside the container.
Figure M
- When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of it. There may be state or local laws about how you should throw away used needles and syringes. For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA's website at: http://www.fda.gov/safesharpsdisposal.
- Do not dispose of your used sharps disposal container in your household trash unless your community guidelines permit this. Do not recycle your used sharps disposal container.
13.Care for the injection site.13a. Treat the injection site by gently pressing, not rubbing, a cotton ball or gauze to the site and apply an adhesive bandage, if necessary. Manufactured by: CELLTRION, Inc., 23, Academy-ro, Yeonsu-gu, Incheon, 22014, Republic of Korea
US License Number 1996Distributed by: CELLTRION USA, Inc., One Evertrust Plaza Suite 1207, Jersey City, NJ 07302 -
INSTRUCTIONS FOR USE YUFLYMA® (yoo-fly'-mah) (adalimumab-aaty) injection for subcutaneous use 80 mg/0.8 mL, 40 mg/0.4 mLSingle-Dose Prefilled Syringe with Safety Guard
For subcutaneous use only
Read and follow the Instructions for Use that come with your YUFLYMA Prefilled Syringe before you start using it and each time you get a refill. There may be new information. This information does not take the place of talking to your doctor about your medical condition or treatment.
Figure A: Parts of YUFLYMA Prefilled Syringe with Safety Guard 
Important Information
- Use the Prefilled Syringe with Needle Guard only if your doctor has trained you on the right way to prepare for and to give an injection.
- Ask your doctor how often you will need to give an injection.
- Do not shake the Prefilled Syringe at any time.
- Do not remove the Cap until you are ready to inject.
- Do not share the Prefilled Syringe with anyone.
- Only use each Prefilled Syringe for one injection.
- Do not pull back on the plunger rod at any time.
How to store the Prefilled Syringe
- Store the Prefilled Syringe in a refrigerator between 36°F to 46°F (2°C to 8°C).
- Keep the Prefilled Syringe in the original carton to protect it from light.
- Do not use a Prefilled Syringe that has been left in direct sunlight.
- Do not freeze the Prefilled Syringe. If the Prefilled Syringe has been frozen, do not use the Prefilled Syringe even if it is thawed.
- If needed, you may store the Prefilled Syringe at room temperature up to 77°F (25°C) for up to 31 days.
- After the Prefilled Syringe has reached room temperature, do not put it back in the refrigerator.
- Keep the Prefilled Syringe and all medicines out of the reach of children.
Read Instructions on All Pages Before Using the YUFLYMA Prefilled Syringe
Prepare for Injection
This Instructions for Use has been approved by the U.S. Food and Drug Administration. Issued: 9/2023 1.Gather the supplies for the injection.1a.Prepare a clean, flat surface, such as a table or countertop, in a well-lit area.1b. Remove 1 Prefilled Syringe from the carton stored in your refrigerator. - Hold the Prefilled Syringe body when removing it from the carton. Do not touch the plunger rod.
2.Inspect the Prefilled Syringe.2a. Make sure you have the correct medicine (YUFLYMA) and dosage.2b. Check the expiration date on the label of the Prefilled Syringe (see Figure B).2c. Look at the Prefilled Syringe and make sure it is not cracked or damaged. - Do not use the Prefilled Syringe if;-it is cracked or damaged.-the expiration date has passed.
Figure B
3.Inspect the Medicine.3a. Look at the Medicine and confirm that the liquid is clear, colorless to pale brown, and free of particles
(see Figure C).- Do not use the Prefilled Syringe if the liquid is the discolored (yellow or dark brown), cloudy, or contains particles in it.
- You may see air bubbles in the liquid. This is normal.
4.Wait 15 to 30 minutes.4a. Leave the Prefilled Syringe at room temperature 68°F to 77°F (20°C to 25°C) for 15 to 30 minutes to allow it to warm up (see Figure D). - Do not warm the Prefilled Syringe using heat sources such as hot water or a microwave.
5.Choose an injection site (see Figure E).5a. You may inject into:-the front of your thighs.-your abdomen except for the 2 in (5 cm) around the belly button (navel). - Do not inject into skin that is within 2 in (5 cm) of your belly button (navel), or is red, hard, tender, damaged, bruised, or scarred.
- If you have psoriasis, do not inject directly into any raised, thick, red or scaly skin patches or lesions on your skin.
- Do not inject through your clothes.
6.Wash your hands.6a. Wash your hands with soap and water and dry them thoroughly (see Figure F). 7.Clean the injection site.7a. Clean the injection site with an alcohol swab using a circular motion (see Figure G).7b. Let the skin dry before injecting. - Do not blow on or touch the injection site again before giving the injection.
Give the Injection8.Remove the Cap.8a. Hold the Prefilled Syringe by the injector body with the Cap on top using one hand. Gently pull the Cap straight off with the other hand. - Do not pull back on the plunger rod at any time.
- Do not recap the Prefilled Syringe.
- Do not remove the Cap until you are ready to inject.
- Do not touch the Needle. Doing so may result in a needle stick injury.
9.Insert the Prefilled Syringe into the injection site.9a. Gently pinch a fold of skin at the injection site with one hand.9b. With a quick and "dart-like" motion, insert the Needle completely into the fold of the skin at a 45-degree angle (see Figure I). - Do not change the position of the Prefilled Syringe after the injection has started.
10.Give the injection.10a. After the Needle is inserted, release the pinch.10b. Slowly push the Plunger all the way down until all of the liquid is injected and the Syringe is empty
(see Figure J).11.Remove the Prefilled Syringe from the injection site.11a. After the Prefilled Syringe is empty, slowly lift your thumb from the Plunger until Needle is completely covered by the Needle Guard (see Figure K). - Some bleeding may occur.
- Do not reuse the Prefilled Syringe.
- Do not touch or recap the needle.
- Do not rub the injection site.
After the Injection 12.Dispose of the Prefilled Syringe.12a. Put the used Prefilled Syringe in a FDA-cleared sharps disposal container right away after use (see Figure L). - Do not throw away (dispose of) the Prefilled Syringe in your household trash.
- If you do not have an FDA-cleared sharps disposal container, you may use a household container that is:-made of a heavy-duty plastic,-can be closed with a tight-fitting, puncture-resistant lid, without sharps being able to come out,-upright and stable during use,-leak-resistant, and-properly labeled to warn of hazardous waste inside the container.
- When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of it. There may be state or local laws about how you should throw away used needles and syringes. For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA's website at: http://www.fda.gov/safesharpsdisposal.
- Do not dispose of your used sharps disposal container in your household trash unless your community guidelines permit this. Do not recycle your used sharps disposal container.
13.Care for the injection site.13a. Treat the injection site by gently pressing, not rubbing, a cotton ball or gauze to the site and apply an adhesive bandage, if necessary. Manufactured by: CELLTRION, Inc., 23, Academy-ro, Yeonsu-gu, Incheon, 22014, Republic of Korea
US License Number 1996Distributed by: CELLTRION USA, Inc., One Evertrust Plaza Suite 1207, Jersey City, NJ 07302 -
INSTRUCTIONS FOR USE YUFLYMA® (yoo-fly'-mah) (adalimumab-aaty) injection, for subcutaneous use 80 mg/0.8 mL, 40 mg/0.4 mL, 20 mg/0.2 mL, 10 mg/0.1 mLSingle-Dose Prefilled Syringe
For subcutaneous use only
Read and follow the Instructions for Use that come with your YUFLYMA Prefilled Syringe before you start using it and each time you get a refill. There may be new information. This information does not take the place of talking to your doctor about your medical condition or treatment.
Figure A: Parts of YUFLYMA Prefilled Syringe 
Important Information
- Use the Prefilled Syringe only if your doctor has trained you on the right way to prepare for and to give an injection.
- Ask your doctor how often you will need to give an injection.
- Do not shake the Prefilled Syringe at any time.
- Do not remove the Cap until you are ready to inject.
- Do not share the Prefilled Syringe with anyone.
- Do not pull back on the plunger rod at any time.
How to store the Prefilled Syringe
- Store the Prefilled Syringe in a refrigerator between 36°F to 46°F (2°C to 8°C).
- Keep the Prefilled Syringe in the original carton to protect it from light.
- Do not use a Prefilled Syringe that has been left in direct sunlight.
- Do not freeze the Prefilled Syringe. If the Prefilled Syringe has been frozen, do not use the Prefilled Syringe even if it is thawed.
- If needed, you may store the Prefilled Syringe at room temperature up to 77°F (25°C) for up to 31 days.
- After the Prefilled Syringe has reached room temperature, do not put it back in the refrigerator.
- Keep the Prefilled Syringe and all medicines out of the reach of children.
Read Instructions on All Pages Before Using the YUFLYMA Prefilled Syringe
Prepare for Injection
This Instructions for Use has been approved by the U.S. Food and Drug Administration. Issued: 10/2025 1.Gather the supplies for the injection.1a. Prepare a clean, flat surface, such as a table or countertop, in a well-lit area.1b. Remove 1 Prefilled Syringe from the carton stored in your refrigerator. - Hold the Prefilled Syringe body when removing it from the carton. Do not touch the plunger rod.
2.Inspect the Prefilled Syringe.2a. Make sure you have the correct medicine (YUFLYMA) and dosage.2b. Check the expiration date on the label of the Prefilled Syringe (see Figure B)2c. Look at the Prefilled Syringe and make sure it is not cracked or damaged. - Do not use the Prefilled Syringe if;-it is cracked or damaged.-the expiration date has passed.
Figure B
3.Inspect the Medicine.3a. Look at the Medicine and confirm that the liquid is clear, colorless to pale brown, and free of particles (see Figure C). - Do not use the Prefilled Syringe if the liquid is the discolored (yellow or dark brown), cloudy, or contains particles in it.
- You may see air bubbles in the liquid. This is normal.
Figure C
4.Wait 15 to 30 minutes.4a. Leave the Prefilled Syringe at room temperature 68°F to 77°F (20°C to 25°C) for 15 to 30 minutes to allow it to warm up (see Figure D). - Do not warm the Prefilled Syringe using heat sources such as hot water or a microwave.
Figure D
5.Choose an injection site (see Figure E).5a. You may inject into:-the front of your thighs.-your abdomen except for the 2 in (5 cm) around the belly button (navel). - Do not inject into skin that is within 2 in (5 cm) of your belly button (navel), or is red, hard, tender, damaged, bruised, or scarred.
- If you have psoriasis, do not inject directly into any raised, thick, red or scaly skin patches or lesions on your skin.
- Do not inject through your clothes.
Figure E
6.Wash your hands.6a. Wash your hands with soap and water and dry them thoroughly (see Figure F). Figure F
7.Clean the injection site.7a. Clean the injection site with an alcohol swab using a circular motion (see Figure G).7b. Let the skin dry before injecting. - Do not blow on or touch the injection site again before giving the injection.
Figure G
Give the Injection8.Remove the Cap.8a. Hold the Prefilled Syringe by the injector body with the Cap on top using one hand. Gently pull the Cap straight off with the other hand. - Do not pull back on the plunger rod at any time.
- Do not recap the Prefilled Syringe.
- Do not remove the Cap until you are ready to inject.
- Do not touch the Needle. Doing so may result in a needle stick injury.
Figure H
9.Insert the Prefilled Syringe into the injection site.9a. Gently pinch a fold of skin at the injection site with one hand.9b. With a quick and "dart-like" motion, insert the Needle completely into the fold of the skin at a 45-degree angle (see Figure I). - Do not change the position of the Prefilled Syringe after the injection has started.
Figure I
10.Give the injection.10a. After the Needle is inserted, release the pinch.10b. Slowly push the Plunger all the way down until all of the liquid is injected and the Syringe is empty (see Figure J). Figure J
11.Remove the Prefilled Syringe from the injection site.11a. After the Prefilled Syringe is empty, remove the Prefilled Syringe from your skin (see Figure K). - Some bleeding may occur.
- Do not reuse the Prefilled Syringe.
- Do not touch the Needle.
- Do not rub the injection site.
Figure K
After the Injection 12.Dispose of the Prefilled Syringe.12a. Put the used Prefilled Syringe in a FDA-cleared sharps disposal container right away after use (see Figure L). - Do not throw away (dispose of) the Prefilled Syringe in your household trash.
- If you do not have an FDA-cleared sharps disposal container, you may use a household container that is:-made of a heavy-duty plastic,-can be closed with a tight-fitting, puncture-resistant lid, without sharps being able to come out,-upright and stable during use,-leak-resistant, and-properly labeled to warn of hazardous waste inside the container.
Figure L
- When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of it. There may be state or local laws about how you should throw away used needles and syringes. For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA's website at: http://www.fda.gov/safesharpsdisposal.
- Do not dispose of your used sharps disposal container in your household trash unless your community guidelines permit this. Do not recycle your used sharps disposal container.
13.Care for the injection site.13a. Treat the injection site by gently pressing, not rubbing, a cotton ball or gauze to the site and apply an adhesive bandage, if necessary. Manufactured by: CELLTRION, Inc., 23, Academy-ro, Yeonsu-gu, Incheon, 22014, Republic of Korea
US License Number 1996Distributed by: CELLTRION USA, Inc., One Evertrust Plaza Suite 1207, Jersey City, NJ 07302 - PRINCIPAL DISPLAY PANEL - 40 mg/0.4 mL Auto-injector Label
-
PRINCIPAL DISPLAY PANEL - 40 mg/0.4 mL Auto-injector Carton
- PRINCIPAL DISPLAY PANEL - 40 mg/0.4 mL Auto-injector Carton 1PK
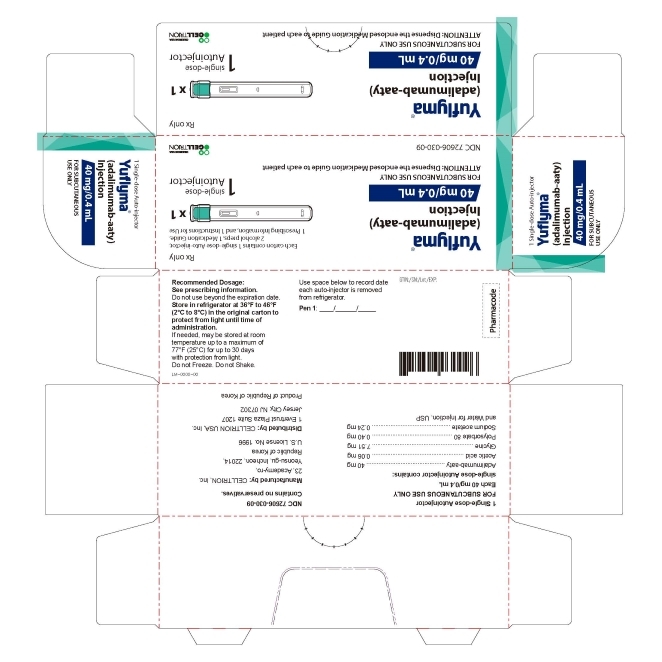
1 Single-dose Auto-injector
Rx only
Yuflyma 40 mg/0.4 mL
adalimumab-aatyFOR SUBCUTANEOUS USE ONLY
1 auto-injector + 2 alcohol preps
NDC: 72606-030-09
CELLTRION Inc.
- PRINCIPAL DISPLAY PANEL - 40 mg/0.4 mL Auto-injector Carton 2PK
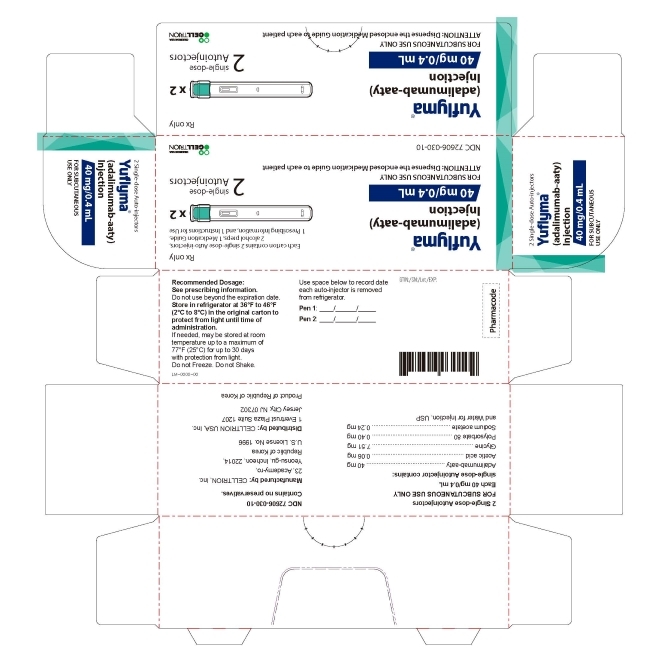
2 Single-dose Auto-injector
Rx only
Yuflyma 40 mg/0.4 mL
adalimumab-aaty
FOR SUBCUTANEOUS USE ONLY
2 auto-injector + 2 alcohol preps
NDC: 72606-030-10
CELLTRION Inc.
- PRINCIPAL DISPLAY PANEL - 40 mg/0.4 mL Auto-injector Carton 4PK

4 Single-dose Auto-injector
Rx only
Yuflyma 40 mg/0.4 mL
adalimumab-aaty
FOR SUBCUTANEOUS USE ONLY
4 auto-injector + 4 alcohol preps
NDC: 72606-030-11
CELLTRION Inc.
- PRINCIPAL DISPLAY PANEL - 40 mg/0.4 mL Auto-injector Carton 6PK

6 Single-dose Auto-injector
Rx only
Yuflyma 40 mg/0.4 mL
adalimumab-aatyFOR SUBCUTANEOUS USE ONLY
6 auto-injector + 6 alcohol preps
NDC: 72606-030-12
CELLTRION Inc.
-
PRINCIPAL DISPLAY PANEL - 40 mg/0.4 mL Syringe Label- with Guard
Lot:
EXP:NDC: 72606-030-05
Rx onlyYuflyma 40 mg/0.4 mL
adalimumab-aaty
injectionSubcutaneous use
CELLTRION Inc.
LM000000-00000
-
PRINCIPAL DISPLAY PANEL - 40 mg/0.4 mL Syringe Carton - with Guard
- 40 mg/0.4 mL Syringe Carton - with Guard 1PK
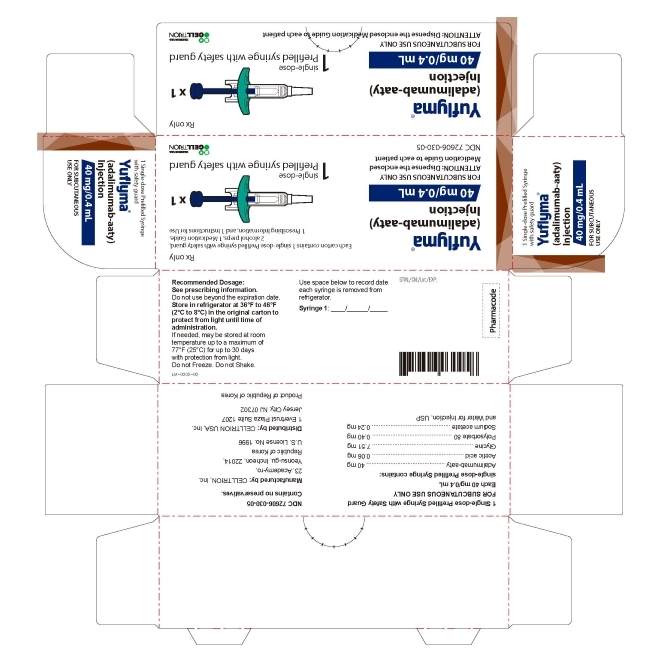
1 Single-dose Prefilled Syringe with Safety Guard
Rx only
Yuflyma 40 mg/0.4 mL
adalimumab-aatyFOR SUBCUTANEOUS USE ONLY
1 prefilled syringe with safety guard + 2 alcohol preps
NDC: 72606-030-05
CELLTRION Inc.
- 40 mg/0.4 mL Syringe Carton - with Guard 2PK

2 Single-dose Prefilled Syringe with Safety Guard
Rx only
Yuflyma 40 mg/0.4 mL
adalimumab-aatyFOR SUBCUTANEOUS USE ONLY
2 prefilled syringe with safety guard + 2 alcohol preps
NDC: 72606-030-06
CELLTRION Inc.
- 40 mg/0.4 mL Syringe Carton - with Guard 4PK

4 Single-dose Prefilled Syringe with Safety Guard
Rx only
Yuflyma 40 mg/0.4 mL
adalimumab-aatyFOR SUBCUTANEOUS USE ONLY
4 prefilled syringe with safety guard + 4 alcohol preps
NDC: 72606-030-07
CELLTRION Inc.
- 40 mg/0.4 mL Syringe Carton - with Guard 6PK

6 Single-dose Prefilled Syringe with Safety Guard
Rx only
Yuflyma 40 mg/0.4 mL
adalimumab-aatyFOR SUBCUTANEOUS USE ONLY
6 prefilled syringe with safety guard + 6 alcohol preps
NDC: 72606-030-08
CELLTRION Inc.
-
PRINCIPAL DISPLAY PANEL - 40 mg/0.4 mL Syringe Label
Lot:
EXP:NDC: 72606-030-01
Rx onlyYuflyma 40 mg/0.4 mL
adalimumab-aaty
injectionSubcutaneous use
CELLTRION Inc.
LM000000-00000
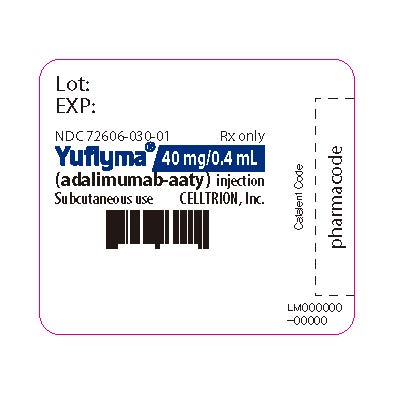
-
PRINCIPAL DISPLAY PANEL - 40 mg/0.4 mL Syringe Carton
- 40 mg/0.4 mL Syringe Carton 1PK

1 Single-dose Prefilled Syringe
Rx only Yuflyma 40 mg/0.4 mL
adalimumab-aatyFOR SUBCUTANEOUS USE ONLY
1 prefilled syringe + 2 alcohol preps
NDC: 72606-030-01
CELLTRION Inc.
- 40 mg/0.4 mL Syringe Carton 2PK

2 Single-dose Prefilled Syringe
Rx only Yuflyma 40 mg/0.4 mL
adalimumab-aatyFOR SUBCUTANEOUS USE ONLY
2 prefilled syringe + 2 alcohol preps
NDC: 72606-030-02
CELLTRION Inc.
- 40 mg/0.4 mL Syringe Carton 4PK

4 Single-dose Prefilled Syringe
Rx only Yuflyma 40 mg/0.4 mL
adalimumab-aatyFOR SUBCUTANEOUS USE ONLY
4 prefilled syringe + 4 alcohol preps
NDC: 72606-030-03
CELLTRION Inc.
- 40 mg/0.4 mL Syringe Carton 6PK

6 Single-dose Prefilled Syringe
Rx only Yuflyma 40 mg/0.4 mL
adalimumab-aatyFOR SUBCUTANEOUS USE ONLY
6 prefilled syringe + 6 alcohol preps
NDC: 72606-030-04
CELLTRION Inc.
-
PRINCIPAL DISPLAY PANEL - 40 mg/0.4 mL Auto-injector Carton (Starter Package)
- 40 mg/0.4 mL Auto-injector Carton 4PK - Plaque Psoriasis Starter Package

4 Single-dose Auto-injector
Rx only
Yuflyma 40 mg/0.4 mL
adalimumab-aaty
FOR SUBCUTANEOUS USE ONLY
4 auto-injector + 4 alcohol preps
NDC: 72606-030-13
CELLTRION Inc.
- 40 mg/0.4 mL Auto-injector Carton 6PK - Crohn's Disease, Pediatric Crohn's Disease, Ulcerative Colitis or Hidradenitis Suppurativa Starter Package

6 Single-dose Auto-injector
Rx only
Yuflyma 40 mg/0.4 mL
adalimumab-aaty
FOR SUBCUTANEOUS USE ONLY
6 auto-injector + 6 alcohol preps
NDC: 72606-030-14
CELLTRION Inc.
-
PRINCIPAL DISPLAY PANEL - 80 mg/0.8 mL Auto-injector Label
NDC: 72606-023-04
Rx onlyYuflyma 80 mg/0.8 mL
adalimumab-aaty
injectionSubcutaneous use
CELLTRION Inc.
LM-0000-00
Lot: Exp:
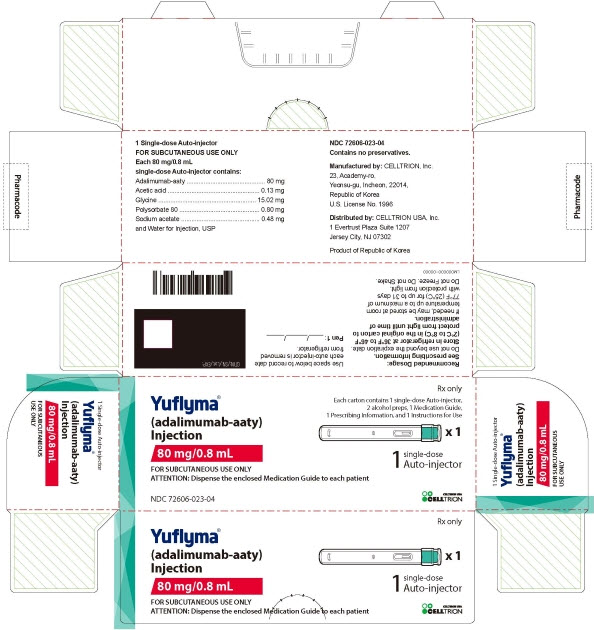
-
PRINCIPAL DISPLAY PANEL - 80 mg/0.8 mL Auto-injector Carton
- PRINCIPAL DISPLAY PANEL - 80 mg/0.8 mL Auto-injector Carton 1PK

1 Single-dose Auto-injector
Rx only
Yuflyma 80 mg/0.8 mL
adalimumab-aatyFOR SUBCUTANEOUS USE ONLY
1 auto-injector + 2 alcohol preps
NDC: 72606-023-04
CELLTRION Inc.
- PRINCIPAL DISPLAY PANEL - 80 mg/0.8 mL Auto-injector Carton 2PK

2 Single-dose Auto-injector
Rx only
Yuflyma 80 mg/0.8 mL
adalimumab-aaty
FOR SUBCUTANEOUS USE ONLY
2 auto-injector + 2 alcohol preps
NDC: 72606-023-05
CELLTRION Inc.
-
PRINCIPAL DISPLAY PANEL - 80 mg/0.8 mL Syringe Label- with Guard
Lot:
EXP:NDC: 72606-023-02
Rx onlyYuflyma 80 mg/0.8 mL
adalimumab-aaty
injectionSubcutaneous use
CELLTRION Inc.
LM000000-00000

-
PRINCIPAL DISPLAY PANEL - 80 mg/0.8 mL Syringe Carton - with Guard
- 80 mg/0.8 mL Syringe Carton - with Guard 1PK

1 Single-dose Prefilled Syringe with Safety Guard
Rx only
Yuflyma 80 mg/0.8 mL
adalimumab-aatyFOR SUBCUTANEOUS USE ONLY
1 prefilled syringe with safety guard + 2 alcohol preps
NDC: 72606-023-02
CELLTRION Inc.
- 80 mg/0.8 mL Syringe Carton - with Guard 2PK

2 Single-dose Prefilled Syringe with Safety Guard
Rx only
Yuflyma 80 mg/0.8 mL
adalimumab-aatyFOR SUBCUTANEOUS USE ONLY
2 prefilled syringe with safety guard + 2 alcohol preps
NDC: 72606-023-03
CELLTRION Inc.
-
PRINCIPAL DISPLAY PANEL - 80 mg/0.8 mL Syringe Label
Lot:
EXP:NDC: 72606-023-01
Rx onlyYuflyma 80 mg/0.8 mL
adalimumab-aaty
injectionSubcutaneous use
CELLTRION Inc.
LM000000-00000

-
PRINCIPAL DISPLAY PANEL - 80 mg/0.8 mL Syringe Carton
- 80 mg/0.8 mL Syringe Carton 1PK

1 Single-dose Prefilled Syringe
Rx only Yuflyma 80 mg/0.8 mL
adalimumab-aatyFOR SUBCUTANEOUS USE ONLY
1 prefilled syringe + 2 alcohol preps
NDC: 72606-023-01
CELLTRION Inc.
-
PRINCIPAL DISPLAY PANEL - Kit Carton (STARTER PACKAGE - 80 mg/0.8 mL Auto-injectors)
NDC: 72606-023-07
Rx onlyYuflyma®
(adalimumab-aaty)
Injection80 mg/0.8 mL
FOR SUBCUTANEOUS USE ONLY
ATTENTION: Dispense the enclosed
Medication Guide to each patientSTARTER PACKAGE
CROHN'S DISEASE, ULCERATIVE COLITIS or HIDRADENITIS SUPPURATIVA

Each carton contains 3 single-dose Auto-injectors.
4 alcohol preps, 1 Medication Guide.
1 Prescribing Information, and 1 Instruction for Use3 single-dose
Auto-injectors
CELLTRION
-
PRINCIPAL DISPLAY PANEL - Kit Carton (STARTER PACKAGE - 80 mg/0.8 mL Pre-filled Syringe with Safety Guard)
NDC: 72606-023-08
Rx onlyYuflyma®
(adalimumab-aaty)
Injection80 mg/0.8 mL
FOR SUBCUTANEOUS USE ONLY
ATTENTION: Dispense the enclosed
Medication Guide to each patientSTARTER PACKAGE
PEDIATRIC CROHN'S DISEASE (≧40 kg (88 lbs))


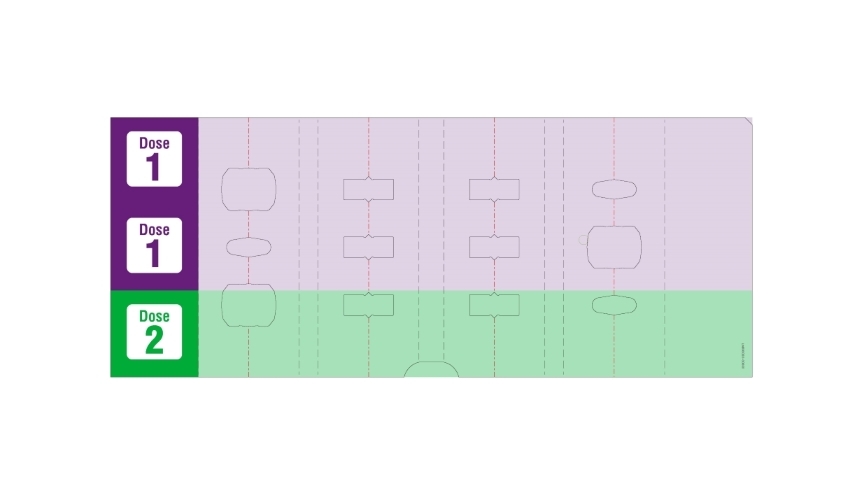
Each carton contains 3 single-dose Prefilled syringes with safety guard.
4 alcohol preps, 1 Medication Guide.
1 Prescribing Information, and 1 Instruction for Use3 single-dose
Prefilled syringes with safety guard
CELLTRION
-
PRINCIPAL DISPLAY PANEL - 20 mg/0.2 mL Syringe Label
Lot:
EXP:NDC: 72606-024-01
Rx onlyYuflyma 20 mg/0.2 mL
adalimumab-aaty
injectionSubcutaneous use
CELLTRION Inc.
LM000000-00000

-
PRINCIPAL DISPLAY PANEL - 20 mg/0.2 mL Syringe Carton
- 20 mg/0.2 mL Syringe Carton 2PK

2 Single-dose Prefilled Syringes
Rx only Yuflyma 20 mg/0.2 mL
adalimumab-aatyFOR SUBCUTANEOUS USE ONLY
2 prefilled syringes + 2 alcohol preps
NDC: 72606-024-01
CELLTRION Inc.
-
PRINCIPAL DISPLAY PANEL - Kit Carton (STARTER PACKAGE - 80 mg/0.8 mL and 40 mg/0.4 mL Auto-injectors)
NDC: 72606-046-01
Rx onlyYuflyma®
(adalimumab-aaty)
Injection80 mg/0.8 mL
40 mg/0.4 mL
FOR SUBCUTANEOUS USE ONLY
ATTENTION: Dispense the enclosed
Medication Guide to each patientSTARTER PACKAGE
PLAQUE PSORIASIS

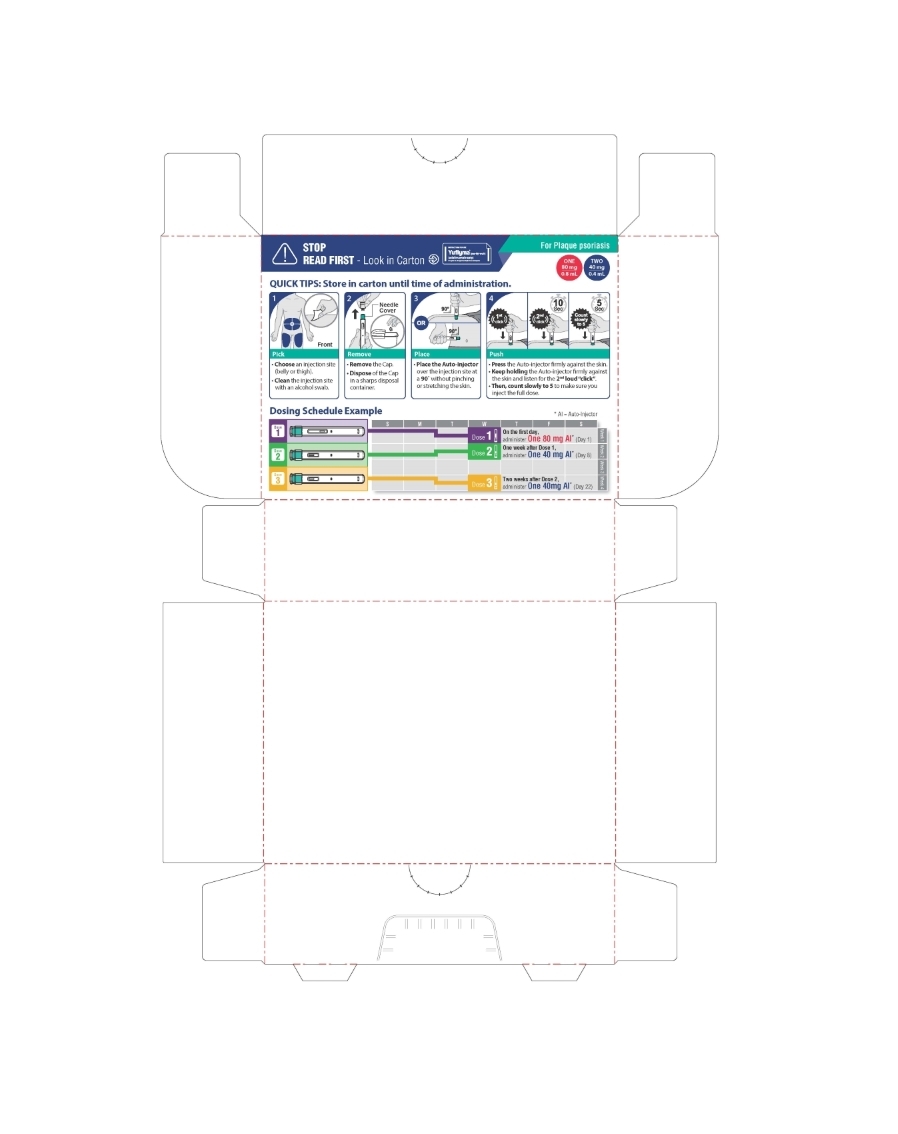
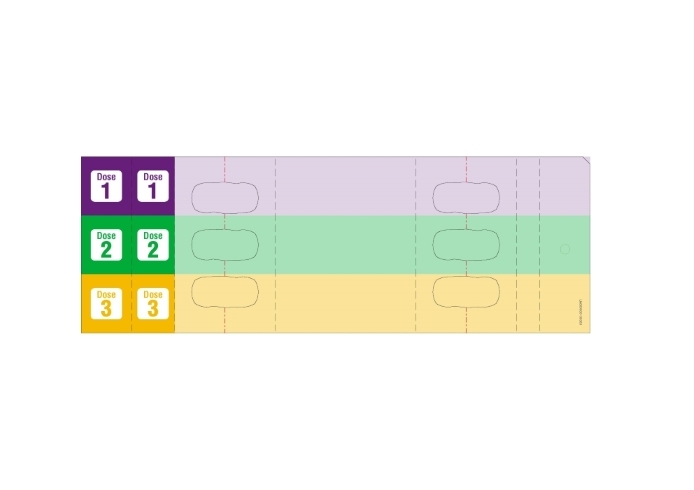
Each carton contains 3 single-dose Auto-injectors.
4 alcohol preps, 1 Medication Guide.
1 Prescribing Information, and 1 Instruction for UseONE 80 mg/0.8 mL
TWO 40 mg/0.4 mL
3 single-dose
Auto-injectors
CELLTRION
-
PRINCIPAL DISPLAY PANEL - Kit Carton (STARTER PACKAGE - 80 mg/0.8 mL and 40 mg/0.4 mL Pre-filled Syringe with Safety Guard)
NDC: 72606-039-01
Rx onlyYuflyma®
(adalimumab-aaty)
Injection80 mg/0.8 mL
40 mg/0.4 mL
FOR SUBCUTANEOUS USE ONLY
ATTENTION: Dispense the enclosed
Medication Guide to each patientSTARTER PACKAGE
PEDIATRIC CROHN'S DISEASE (17 kg (37 lbs) to <40 kg (88 lbs))

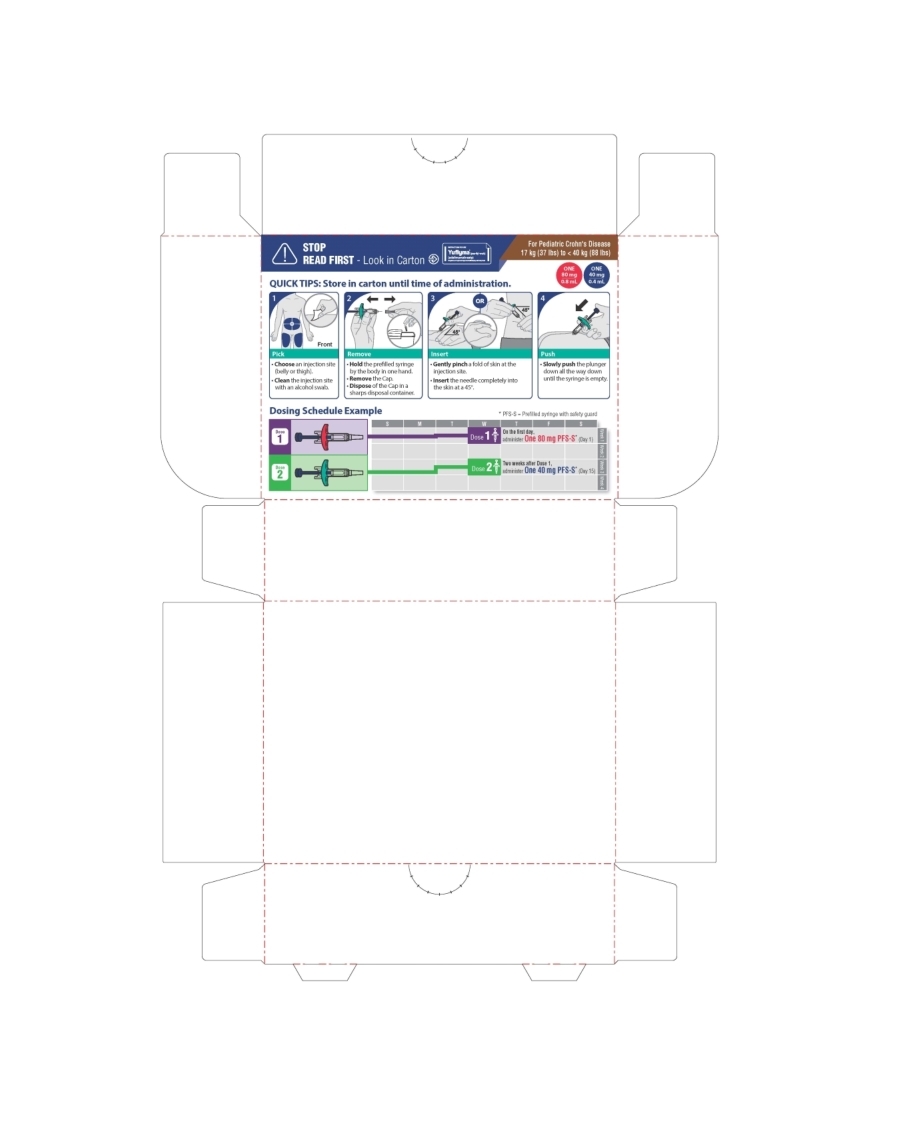
Each carton contains 2 single-dose Pre-filled Syringe with Safety Guard.
2 alcohol preps, 1 Medication Guide.
1 Prescribing Information, and 1 Instruction for UseONE 80 mg/0.8 mL
ONE 40 mg/0.4 mL
2 single-dose
Pre-filled Syringe with Safety Guard
CELLTRION
-
PRINCIPAL DISPLAY PANEL - 10 mg/0.1 mL Syringe Label
Lot:
EXP:NDC: 72606-063-01
Rx onlyYuflyma 10 mg/0.1 mL
adalimumab-aaty
injectionSubcutaneous use
CELLTRION Inc.
LM000000-00000

-
PRINCIPAL DISPLAY PANEL - 10 mg/0.1 mL Syringe Carton
- 10 mg/0.1 mL Syringe Carton 2PK
2 Single-dose Prefilled Syringes
Rx only Yuflyma 10 mg/0.1 mL
adalimumab-aatyFOR SUBCUTANEOUS USE ONLY
2 prefilled syringes + 2 alcohol preps
NDC: 72606-063-01
CELLTRION Inc.
-
INGREDIENTS AND APPEARANCE
YUFLYMA
adalimumab-aaty kitProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 72606-030 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 72606-030-01 1 in 1 CARTON 05/23/2023 1 1 in 1 KIT; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) 2 NDC: 72606-030-02 2 in 1 CARTON 05/23/2023 2 1 in 1 KIT; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) 3 NDC: 72606-030-03 4 in 1 CARTON 05/23/2023 3 1 in 1 KIT; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) 4 NDC: 72606-030-04 6 in 1 CARTON 05/23/2023 4 1 in 1 KIT; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) 5 NDC: 72606-030-05 1 in 1 CARTON 05/23/2023 5 1 in 1 KIT; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) 6 NDC: 72606-030-06 2 in 1 CARTON 05/23/2023 6 1 in 1 KIT; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) 7 NDC: 72606-030-07 4 in 1 CARTON 05/23/2023 7 1 in 1 KIT; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) 8 NDC: 72606-030-08 6 in 1 CARTON 05/23/2023 8 1 in 1 KIT; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) 9 NDC: 72606-030-09 1 in 1 CARTON 05/23/2023 9 1 in 1 KIT; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) 10 NDC: 72606-030-10 2 in 1 CARTON 05/23/2023 10 1 in 1 KIT; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) 11 NDC: 72606-030-11 4 in 1 CARTON 05/23/2023 11 1 in 1 KIT; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) 12 NDC: 72606-030-12 6 in 1 CARTON 05/23/2023 12 1 in 1 KIT; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) 13 NDC: 72606-030-13 4 in 1 CARTON 05/23/2023 13 1 in 1 KIT; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) 14 NDC: 72606-030-14 6 in 1 CARTON 05/23/2023 14 1 in 1 KIT; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 1 SYRINGE 0.4 mL Part 2 1 PACKET 1 mL Part 1 of 2 YUFLYMA
adalimumab-aaty injectionProduct Information Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength adalimumab-aaty (UNII: FYS6T7F842) (ADALIMUMAB - UNII:FYS6T7F842) adalimumab-aaty 40 mg in 0.4 mL Inactive Ingredients Ingredient Name Strength acetic acid (UNII: Q40Q9N063P) 0.06 mg in 0.4 mL sodium acetate (UNII: 4550K0SC9B) 0.24 mg in 0.4 mL glycine (UNII: TE7660XO1C) 7.51 mg in 0.4 mL polysorbate 80 (UNII: 6OZP39ZG8H) 0.4 mg in 0.4 mL water (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 1 in 1 TRAY 1 0.4 mL in 1 SYRINGE; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761219 05/23/2023 Part 2 of 2 ALCOHOL
isopropyl alcohol swabProduct Information Route of Administration TOPICAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ISOPROPYL ALCOHOL (UNII: ND2M416302) (ISOPROPYL ALCOHOL - UNII:ND2M416302) ISOPROPYL ALCOHOL 0.70 mL in 1 mL Inactive Ingredients Ingredient Name Strength water (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 1 mL in 1 PACKET; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date OTC Monograph Drug M003 10/31/2020 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761219 05/23/2023 YUFLYMA
adalimumab-aaty kitProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 72606-023 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 72606-023-01 1 in 1 CARTON 09/29/2023 1 1 in 1 KIT; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) 2 NDC: 72606-023-02 1 in 1 CARTON 09/29/2023 2 1 in 1 KIT; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) 3 NDC: 72606-023-03 2 in 1 CARTON 09/29/2023 3 1 in 1 KIT; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) 4 NDC: 72606-023-04 1 in 1 CARTON 09/29/2023 4 1 in 1 KIT; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) 5 NDC: 72606-023-05 2 in 1 CARTON 09/29/2023 5 1 in 1 KIT; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) 6 NDC: 72606-023-07 3 in 1 CARTON 09/29/2023 6 1 in 1 KIT; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) 7 NDC: 72606-023-08 3 in 1 CARTON 09/29/2023 7 1 in 1 KIT; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 1 SYRINGE 0.8 mL Part 2 1 PACKET 1 mL Part 1 of 2 YUFLYMA
adalimumab-aaty injectionProduct Information Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength adalimumab-aaty (UNII: FYS6T7F842) (ADALIMUMAB - UNII:FYS6T7F842) adalimumab-aaty 80 mg in 0.8 mL Inactive Ingredients Ingredient Name Strength acetic acid (UNII: Q40Q9N063P) 0.13 mg in 0.8 mL sodium acetate (UNII: 4550K0SC9B) 0.48 mg in 0.8 mL glycine (UNII: TE7660XO1C) 15.02 mg in 0.8 mL polysorbate 80 (UNII: 6OZP39ZG8H) 0.8 mg in 0.8 mL water (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 1 in 1 TRAY 1 0.8 mL in 1 SYRINGE; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761219 09/29/2023 Part 2 of 2 ALCOHOL
isopropyl alcohol swabProduct Information Route of Administration TOPICAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength isopropyl alcohol (UNII: ND2M416302) (ISOPROPYL ALCOHOL - UNII:ND2M416302) isopropyl alcohol 0.70 mL in 1 mL Inactive Ingredients Ingredient Name Strength water (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 1 mL in 1 PACKET; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date OTC Monograph Drug M003 10/31/2020 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761219 09/29/2023 YUFLYMA
adalimumab-aaty kitProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 72606-024 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 72606-024-01 2 in 1 CARTON 09/29/2023 1 1 in 1 KIT; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 1 SYRINGE 0.2 mL Part 2 1 PACKET 1 mL Part 1 of 2 YUFLYMA
adalimumab-aaty injectionProduct Information Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength adalimumab-aaty (UNII: FYS6T7F842) (ADALIMUMAB - UNII:FYS6T7F842) adalimumab-aaty 20 mg in 0.2 mL Inactive Ingredients Ingredient Name Strength acetic acid (UNII: Q40Q9N063P) 0.03 mg in 0.2 mL sodium acetate (UNII: 4550K0SC9B) 0.12 mg in 0.2 mL glycine (UNII: TE7660XO1C) 3.75 mg in 0.2 mL polysorbate 80 (UNII: 6OZP39ZG8H) 0.2 mg in 0.2 mL water (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 1 in 1 TRAY 1 0.2 mL in 1 SYRINGE; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761219 09/29/2023 Part 2 of 2 ALCOHOL
isopropyl alcohol swabProduct Information Route of Administration TOPICAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength isopropyl alcohol (UNII: ND2M416302) (ISOPROPYL ALCOHOL - UNII:ND2M416302) isopropyl alcohol 0.70 mL in 1 mL Inactive Ingredients Ingredient Name Strength water (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 1 mL in 1 PACKET; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date OTC Monograph Drug M003 10/31/2020 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761219 09/29/2023 YUFLYMA
adalimumab-aaty kitProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 72606-046 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 72606-046-01 3 in 1 CARTON 09/29/2023 1 1 in 1 KIT; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 2 SYRINGE 0.8 mL Part 2 1 SYRINGE 0.8 mL Part 3 1 PACKET 1 mL Part 1 of 3 YUFLYMA
adalimumab-aaty injectionProduct Information Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength adalimumab-aaty (UNII: FYS6T7F842) (ADALIMUMAB - UNII:FYS6T7F842) adalimumab-aaty 40 mg in 0.4 mL Inactive Ingredients Ingredient Name Strength acetic acid (UNII: Q40Q9N063P) 0.06 mg in 0.4 mL sodium acetate (UNII: 4550K0SC9B) 0.24 mg in 0.4 mL glycine (UNII: TE7660XO1C) 7.51 mg in 0.4 mL polysorbate 80 (UNII: 6OZP39ZG8H) 0.4 mg in 0.4 mL water (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 1 in 1 TRAY 1 0.4 mL in 1 SYRINGE; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761219 09/29/2023 Part 2 of 3 YUFLYMA
adalimumab-aaty injectionProduct Information Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength adalimumab-aaty (UNII: FYS6T7F842) (ADALIMUMAB - UNII:FYS6T7F842) adalimumab-aaty 80 mg in 0.8 mL Inactive Ingredients Ingredient Name Strength acetic acid (UNII: Q40Q9N063P) 0.13 mg in 0.8 mL sodium acetate (UNII: 4550K0SC9B) 0.48 mg in 0.8 mL glycine (UNII: TE7660XO1C) 15.02 mg in 0.8 mL polysorbate 80 (UNII: 6OZP39ZG8H) 0.8 mg in 0.8 mL water (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 1 in 1 TRAY 1 0.8 mL in 1 SYRINGE; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761219 09/29/2023 Part 3 of 3 ALCOHOL
isopropyl alcohol swabProduct Information Route of Administration TOPICAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength isopropyl alcohol (UNII: ND2M416302) (ISOPROPYL ALCOHOL - UNII:ND2M416302) isopropyl alcohol 0.70 mL in 1 mL Inactive Ingredients Ingredient Name Strength water (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 1 mL in 1 PACKET; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date OTC Monograph Drug M003 10/31/2020 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761219 09/29/2023 YUFLYMA
adalimumab-aaty kitProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 72606-039 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 72606-039-01 2 in 1 CARTON 09/29/2023 1 1 in 1 KIT; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 1 SYRINGE 0.4 mL Part 2 1 SYRINGE 0.8 mL Part 3 1 PACKET 1 mL Part 1 of 3 YUFLYMA
adalimumab-aaty injectionProduct Information Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength adalimumab-aaty (UNII: FYS6T7F842) (ADALIMUMAB - UNII:FYS6T7F842) adalimumab-aaty 40 mg in 0.4 mL Inactive Ingredients Ingredient Name Strength acetic acid (UNII: Q40Q9N063P) 0.06 mg in 0.4 mL sodium acetate (UNII: 4550K0SC9B) 0.24 mg in 0.4 mL glycine (UNII: TE7660XO1C) 7.51 mg in 0.4 mL polysorbate 80 (UNII: 6OZP39ZG8H) 0.4 mg in 0.4 mL water (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 1 in 1 TRAY 1 0.4 mL in 1 SYRINGE; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761219 09/29/2023 Part 2 of 3 YUFLYMA
adalimumab-aaty injectionProduct Information Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength adalimumab-aaty (UNII: FYS6T7F842) (ADALIMUMAB - UNII:FYS6T7F842) adalimumab-aaty 80 mg in 0.8 mL Inactive Ingredients Ingredient Name Strength acetic acid (UNII: Q40Q9N063P) 0.13 mg in 0.8 mL sodium acetate (UNII: 4550K0SC9B) 0.48 mg in 0.8 mL glycine (UNII: TE7660XO1C) 15.02 mg in 0.8 mL polysorbate 80 (UNII: 6OZP39ZG8H) 0.8 mg in 0.8 mL water (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 1 in 1 TRAY 1 0.8 mL in 1 SYRINGE; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761219 09/29/2023 Part 3 of 3 ALCOHOL
isopropyl alcohol swabProduct Information Route of Administration TOPICAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength isopropyl alcohol (UNII: ND2M416302) (ISOPROPYL ALCOHOL - UNII:ND2M416302) isopropyl alcohol 0.70 mL in 1 mL Inactive Ingredients Ingredient Name Strength water (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 1 mL in 1 PACKET; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date OTC Monograph Drug M003 10/31/2020 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761219 09/29/2023 YUFLYMA
adalimumab-aaty kitProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 72606-063 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 72606-063-01 2 in 1 CARTON 10/31/2025 1 1 in 1 KIT; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 1 SYRINGE 0.1 mL Part 2 1 PACKET 1 mL Part 1 of 2 YUFLYMA
adalimumab-aaty injectionProduct Information Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength adalimumab-aaty (UNII: FYS6T7F842) (ADALIMUMAB - UNII:FYS6T7F842) adalimumab-aaty 10 mg in 0.1 mL Inactive Ingredients Ingredient Name Strength acetic acid (UNII: Q40Q9N063P) 0.02 mg in 0.1 mL sodium acetate (UNII: 4550K0SC9B) 0.06 mg in 0.1 mL glycine (UNII: TE7660XO1C) 1.88 mg in 0.1 mL polysorbate 80 (UNII: 6OZP39ZG8H) 0.1 mg in 0.1 mL water (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 1 in 1 TRAY 1 0.1 mL in 1 SYRINGE; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761219 10/31/2025 Part 2 of 2 ALCOHOL
isopropyl alcohol swabProduct Information Route of Administration TOPICAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength isopropyl alcohol (UNII: ND2M416302) (ISOPROPYL ALCOHOL - UNII:ND2M416302) isopropyl alcohol 0.70 mL in 1 mL Inactive Ingredients Ingredient Name Strength water (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 1 mL in 1 PACKET; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date OTC Monograph Drug M003 10/31/2020 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761219 10/31/2025 Labeler - CELLTRION USA, Inc. (116587378)
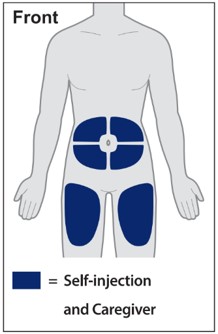
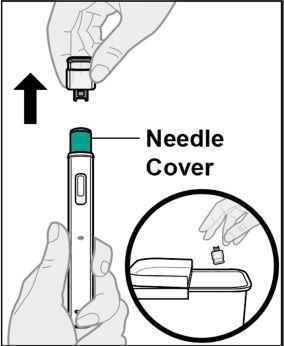
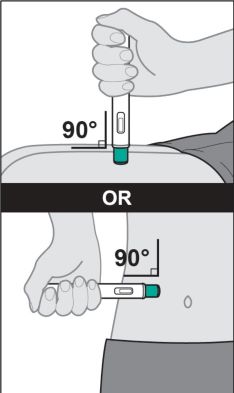
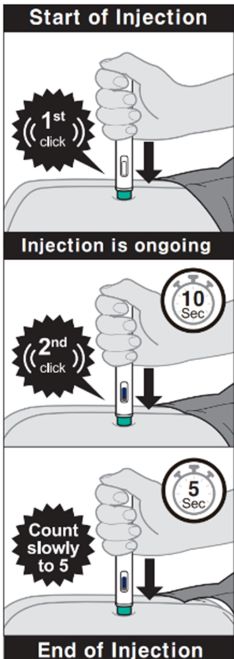
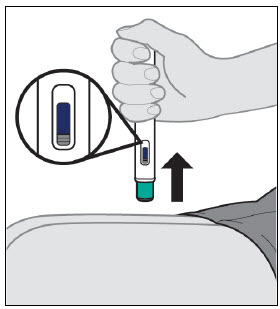
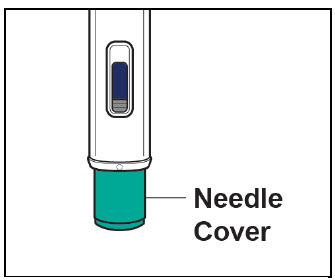
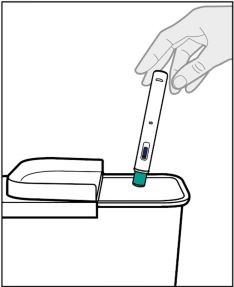
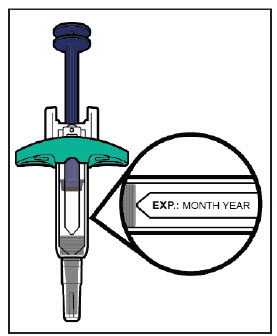
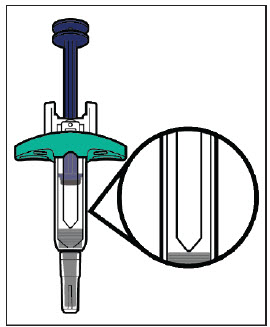
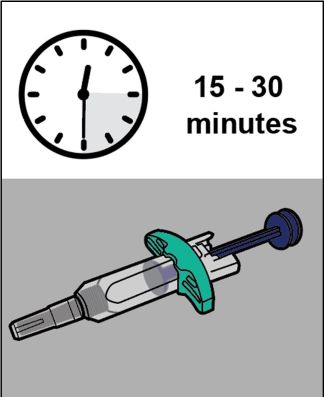
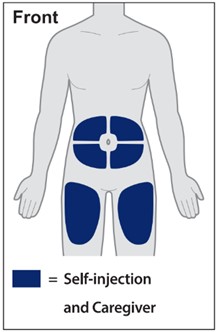
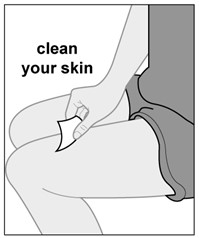
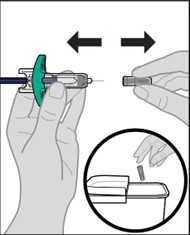
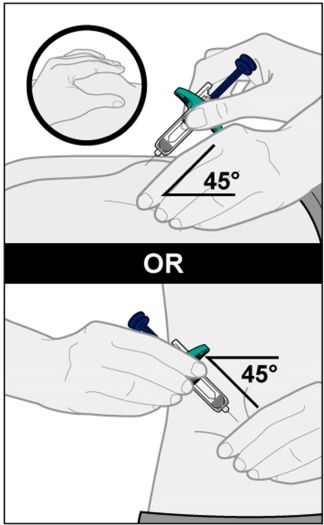
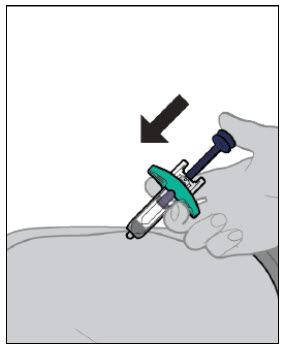
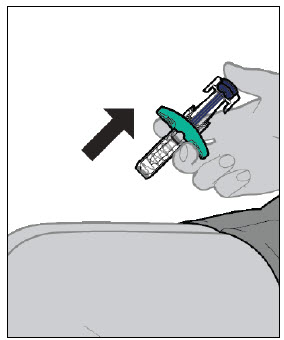
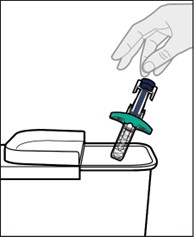
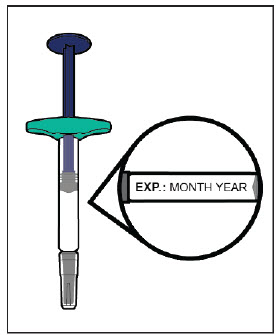
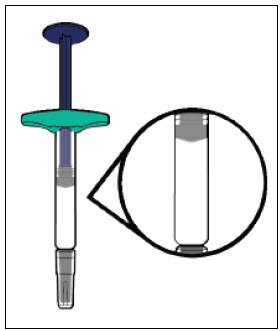
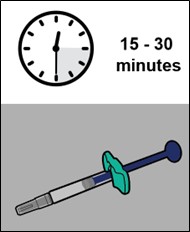
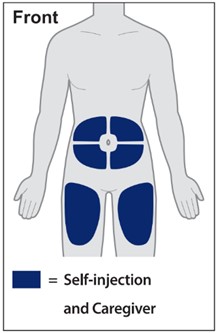
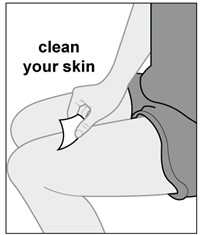
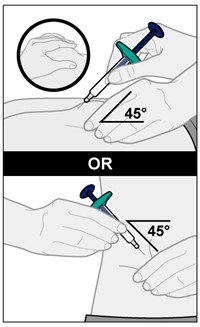
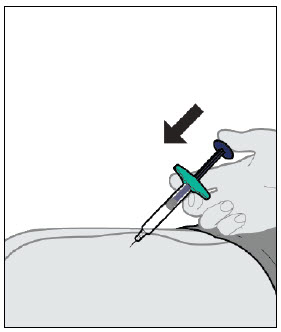
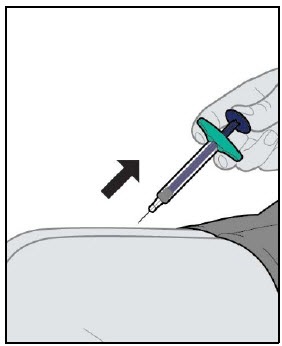
Trademark Results [Yuflyma]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 YUFLYMA 79268175 not registered Live/Pending |
CELLTRION, INC. 2019-08-07 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.