Clofarabine by Meitheal Pharmaceuticals Inc. / Kindos Pharmaceuticals Co., Ltd. CLOFARABINE injection
Clofarabine by
Drug Labeling and Warnings
Clofarabine by is a Prescription medication manufactured, distributed, or labeled by Meitheal Pharmaceuticals Inc., Kindos Pharmaceuticals Co., Ltd.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use CLOFARABINE INJECTION safely and effectively. See full prescribing information for CLOFARABINE INJECTION.
CLOFARABINE injection, for intravenous use
Initial U.S. Approval: 2004RECENT MAJOR CHANGES
Indications and Usage (1) 7/2022
INDICATIONS AND USAGE
Clofarabine Injection is a nucleoside metabolic inhibitor indicated for the treatment of pediatric patients 1 to 21 years old with relapsed or refractory acute lymphoblastic leukemia after at least two prior regimens. (1)
DOSAGE AND ADMINISTRATION
- Administer the recommended pediatric dose of 52 mg/m2 as an intravenous infusion over 2 hours daily for 5 consecutive days of a 28-day cycle. Repeat cycles every 2-6 weeks. (2.1)
- Provide supportive care, such as intravenous infusion fluids, antihyperuricemic treatment, and alkalinization of urine throughout the 5 days of clofarabine injection administration to reduce the risk of tumor lysis and other adverse reactions. (2.1)
- Discontinue clofarabine injection if hypotension develops during the 5 days of administration. (2.1)
- Reduce the dose in patients with renal impairment. (2.2)
- Use dose modification for toxicity. (2.4)
DOSAGE FORMS AND STRENGTHS
- Injection: 20 mg per 20 mL (1 mg per mL) single-dose vial. (3)
CONTRAINDICATIONS
- None. (4)
WARNINGS AND PRECAUTIONS
- Myelosuppression: May be severe and prolonged. Monitor complete blood counts and platelet counts during clofarabine therapy. (5.1)
- Hemorrhage: Serious and fatal cerebral, gastrointestinal and pulmonary hemorrhage. Monitor platelets and coagulation parameters and treat accordingly. (5.2)
- Infections: Severe and fatal sepsis as a result of bone marrow suppression. Monitor for signs and symptoms of infection; discontinue clofarabine and treat promptly. (5.3)
- Tumor Lysis syndrome: Anticipate, monitor for signs and symptoms and treat promptly. (5.4)
- Systemic Inflammatory Response Syndrome (SIRS) or Capillary Leak Syndrome: Monitor for and discontinue clofarabine immediately if suspected. (5.5)
- Venous Occlusive Disease of the Liver: Monitor for and discontinue clofarabine if suspected. (5.6)
- Hepatotoxicity: Severe and fatal hepatotoxicity. Monitor liver function, for signs and symptoms of hepatitis and hepatic failure. Discontinue clofarabine immediately for Grade 3 or greater liver enzyme and/or bilirubin elevations. (5.7)
- Renal Toxicity: Increased creatinine and acute renal failure; monitor renal function and interrupt or discontinue clofarabine. (5.8)
- Enterocolitis: Serious and fatal enterocolitis, occurring more frequently within 30 days of treatment and with combination chemotherapy. Monitor patients for signs and symptoms of enterocolitis and treat promptly. (5.9)
- Skin Reactions: Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN), including fatal cases. Discontinue for exfoliative or bullous rash, or if SJS or TEN is suspected. (5.10)
- Embryo-Fetal Toxicity: Can cause fetal harm. Advise females of reproductive potential and males with female partners of reproductive potential of the potential risk to a fetus and to use effective contraception. (5.11)
ADVERSE REACTIONS
Most common adverse reactions (≥25%): vomiting, nausea, diarrhea, febrile neutropenia, pruritus, headache, bacteremia, pyrexia, rash, tachycardia, abdominal pain, chills, fatigue, anorexia, pain in extremity, hypotension, epistaxis, and petechiae. (6)
To report SUSPECTED ADVERSE REACTIONS, contact Meitheal Pharmaceuticals Inc. at 1-844-824-8426 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 9/2022
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
2.2 Recommended Dosage Reduction for Renal Impairment
2.3 Potential Concomitant Medications and Medications to Avoid
2.4 Dose Modifications and Reinitiation of Therapy after Adverse Reactions
2.5 Reconstitution/Preparation
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Myelosuppression
5.2 Hemorrhage
5.3 Infections
5.4 Tumor Lysis Syndrome
5.5 Systemic Inflammatory Response Syndrome (SIRS) and Capillary Leak Syndrome
5.6 Venous Occlusive Disease of the Liver
5.7 Hepatotoxicity
5.8 Renal Toxicity
5.9 Enterocolitis
5.10 Skin Reactions
5.11 Embryo-Fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
15 REFERENCES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
Administer the recommended pediatric dose of 52 mg/m2 as an intravenous infusion over 2 hours daily for 5 consecutive days.
- Repeat treatment cycles following recovery or return to baseline organ function, approximately every 2 to 6 weeks. Base dosage on the patient's body surface area (BSA), calculated using the actual height and weight before the start of each cycle. To prevent drug incompatibilities, do not administer other medications through the same intravenous line. Administer subsequent cycles no sooner than 14 days from the starting day of the previous cycle and provided the patient's ANC is ≥0.75 × 109/L.
- Provide supportive care, such as intravenous fluids, antihyperuricemic treatment, and alkalinize urine throughout the 5 days of clofarabine injection administration to reduce the effects of tumor lysis and other adverse reactions.
- Discontinue clofarabine injection if hypotension develops during the 5 days of administration.
- Monitor renal and hepatic function during the 5 days of clofarabine injection administration [see Warnings and Precautions (5.7, 5.8)].
- Monitor patients taking medications known to affect blood pressure. Monitor cardiac function during administration of clofarabine injection.
2.2 Recommended Dosage Reduction for Renal Impairment
- Reduce the dose by 50% in patients with creatinine clearance (CrCL) between 30 and 60 mL/min. There is insufficient information to make a dosage recommendation in patients with CrCL less than 30 mL/min [see Use in Specific Populations (8.6)].
2.3 Potential Concomitant Medications and Medications to Avoid
- Consider prophylactic antiemetic medications as clofarabine injection is moderately emetogenic.
- Consider the use of prophylactic steroids to mitigate Systemic Inflammatory Response Syndrome (SIRS) or capillary leak syndrome (e.g., hypotension, tachycardia, tachypnea, and pulmonary edema).
- Minimize exposure to drugs with known renal toxicity during the 5 days of clofarabine injection administration since the risk of renal toxicity may be increased.
- Avoid concomitant use of medications known to induce hepatic toxicity.
2.4 Dose Modifications and Reinitiation of Therapy after Adverse Reactions
Hematologic Toxicity
- If a patient experiences a Grade 4 neutropenia (ANC <0.5 × 109/L) lasting ≥4 weeks, reduce dose by 25% for the next cycle.
Non-hematologic Toxicity
- Withhold clofarabine injection if a patient develops a clinically significant infection, until the infection is controlled, then restart at the full dose.
- Withhold clofarabine injection for a Grade 3 non-infectious non-hematologic toxicity (excluding transient elevations in serum transaminases and/or serum bilirubin and/or nausea/vomiting controlled by antiemetic therapy). Re-institute clofarabine injection administration at a 25% dose reduction when resolution or return to baseline.
- Discontinue clofarabine injection administration for a Grade 4 non-infectious non-hematologic toxicity.
- Discontinue clofarabine injection administration if a patient shows early signs or symptoms of SIRS or capillary leak syndrome (e.g., hypotension, tachycardia, tachypnea, and pulmonary edema) occur and provide appropriate supportive measures.
- Discontinue clofarabine injection administration if Grade 3 or higher increases in creatinine or bilirubin are noted. Re-institute clofarabine injection with a 25% dose reduction, when the patient is stable and organ function has returned to baseline. If hyperuricemia is anticipated (tumor lysis), initiate measures to control uric acid.
2.5 Reconstitution/Preparation
Filter clofarabine injection through a sterile 0.2 micron syringe filter and then dilute with 5% Dextrose Injection, USP or 0.9% Sodium Chloride Injection, USP prior to intravenous infusion to a final concentration between 0.15 mg per mL and 0.4 mg per mL. Use within 24 hours of preparation. Parenteral drug products should be visually inspected for particulate matter and discoloration prior to administration, whenever solution and container permit. Store diluted clofarabine injection at room temperature (15° to 30ºC).
Discard unused portion in vial. - 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Myelosuppression
Clofarabine causes myelosuppression which may be severe and prolonged. Febrile neutropenia occurred in 55% and non-febrile neutropenia in an additional 10% of pediatric patients in clinical trials. At initiation of treatment, most patients in the clinical studies had hematological impairment as a manifestation of leukemia. Myelosuppression is usually reversible with interruption of clofarabine treatment and appears to be dose-dependent. Monitor complete blood counts [see Dosage and Administration (2.4)].
5.2 Hemorrhage
Serious and fatal hemorrhage, including cerebral, gastrointestinal and pulmonary hemorrhage, has occurred. The majority of the cases were associated with thrombocytopenia. Monitor platelets and coagulation parameters and treat accordingly [see Adverse Reactions (6.2)].
5.3 Infections
Clofarabine increases the risk of infection, including severe and fatal sepsis, and opportunistic infections. At baseline, 48% of the pediatric patients had one or more concurrent infections. A total of 83% of patients experienced at least one infection after clofarabine treatment, including fungal, viral and bacterial infections. Monitor patients for signs and symptoms of infection, discontinue clofarabine, and treat promptly.
5.4 Tumor Lysis Syndrome
Administration of clofarabine may result in tumor lysis syndrome associated with the break-down metabolic products from peripheral leukemia cell death. Monitor patients undergoing treatment for signs and symptoms of tumor lysis syndrome and initiate preventive measures including adequate intravenous fluids and measures to control uric acid.
5.5 Systemic Inflammatory Response Syndrome (SIRS) and Capillary Leak Syndrome
Clofarabine may cause a cytokine release syndrome (e.g., tachypnea, tachycardia, hypotension, pulmonary edema) that may progress to the systemic inflammatory response syndrome (SIRS) with capillary leak syndrome and organ impairment which may be fatal. Monitor patients frequently for these conditions. In clinical trials, SIRS was reported in two patients (2%); capillary leak syndrome was reported in four patients (4%). Symptoms included rapid onset of respiratory distress, hypotension, pleural and pericardial effusion, and multiorgan failure. Close monitoring for this syndrome and early intervention may reduce the risk. Immediately discontinue clofarabine and provide appropriate supportive measures. The use of prophylactic steroids (e.g., 100 mg/m2 hydrocortisone on Days 1 through 3) may be of benefit in preventing signs or symptoms of SIRS or capillary leak syndrome. Consider use of diuretics and/or albumin. After the patient is stabilized and organ function has returned to baseline, retreatment with clofarabine can be considered with a 25% dose reduction.
5.6 Venous Occlusive Disease of the Liver
Patients who have previously received a hematopoietic stem cell transplant (HSCT) are at higher risk for veno-occlusive disease (VOD) of the liver following treatment with clofarabine (40 mg/m2) when used in combination with etoposide (100 mg/m2) and cyclophosphamide (440 mg/m2). Severe hepatotoxic events have been reported in a combination study of clofarabine in pediatric patients with relapsed or refractory acute leukemia. Two cases (2%) of VOD in the monotherapy studies were considered related to study drug. Monitor for and discontinue clofarabine if VOD is suspected.
5.7 Hepatotoxicity
Severe and fatal hepatotoxicity, including hepatitis and hepatic failure, has occurred with the use of clofarabine. In clinical studies, Grade 3-4 liver enzyme elevations were observed in pediatric patients during treatment with clofarabine at the following rates: elevated aspartate aminotransferase (AST) occurred in 36% of patients; elevated alanine aminotransferase (ALT) occurred in 44% of patients. AST and ALT elevations typically occurred within 10 days of clofarabine administration and returned to Grade 2 or less within 15 days. Grade 3 or 4 elevated bilirubin occurred in 13% of patients, with 2 events reported as Grade 4 hyperbilirubinemia (2%), one of which resulted in treatment discontinuation and one patient had multiorgan failure and died. Eight patients (7%) had Grade 3 or 4 elevations in serum bilirubin at the last time point measured; these patients died due to sepsis and/or multiorgan failure. Monitor hepatic function, and for signs and symptoms of hepatitis and hepatic failure. Discontinue clofarabine immediately for Grade 3 or greater liver enzyme and/or bilirubin elevations [see Dosage and Administration (2.4)].
5.8 Renal Toxicity
Clofarabine may cause acute renal failure. In clofarabine treated patients in clinical studies, Grade 3 or 4 elevated creatinine occurred in 8% of patients and acute renal failure was reported as Grade 3 in three patients (3%) and Grade 4 in two patients (2%). Patients with infection, sepsis, or tumor lysis syndrome may be at increased risk of renal toxicity when treated with clofarabine. Hematuria occurred in 13% of clofarabine treated patients overall. Monitor patients for renal toxicity and interrupt or discontinue clofarabine as necessary [see Dosage and Administration (2.4)].
5.9 Enterocolitis
Fatal and serious cases of enterocolitis, including neutropenic colitis, cecitis, and C. difficile colitis, have occurred during treatment with clofarabine. This has occurred more frequently within 30 days of treatment, and in the setting of combination chemotherapy. Enterocolitis may lead to necrosis, perforation, hemorrhage or sepsis complications. Monitor patients for signs and symptoms of enterocolitis and treat promptly.
5.10 Skin Reactions
Serious and fatal cases of Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN) have been reported. Discontinue clofarabine for exfoliative or bullous rash, or if SJS or TEN is suspected [see Adverse Reactions (6.2)].
5.11 Embryo-Fetal Toxicity
Based on findings from animal reproductive studies and the drug's mechanism of action, clofarabine can cause fetal harm when administered to a pregnant woman. Intravenous doses of clofarabine in rats and rabbits administered during organogenesis at doses that were below the maximum recommended human dose of 52 mg/m2 based on body surface area (mg/m2) caused an increase in resorptions, malformations, and variations. Advise females of reproductive potential of the potential risk to a fetus and to use an effective method of contraception during treatment with clofarabine and for 6 months after the last dose. Advise males with female partners of reproductive potential to use effective contraception during treatment with clofarabine and for 3 months after the last dose [see Use in Specific Populations (8.1)].
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are discussed in greater detail in other sections of the label:
- Myelosuppression [see Warnings and Precautions (5.1)]
- Hemorrhage [see Warnings and Precautions (5.2)]
- Serious Infections [see Warnings and Precautions (5.3)]
- Hyperuricemia (tumor lysis syndrome) [see Warnings and Precautions (5.4)]
- Systemic Inflammatory Response Syndrome (SIRS) and Capillary Leak Syndrome [see Warnings and Precautions (5.5)]
- Venous Occlusive Disease of the Liver [see Warnings and Precautions (5.6)]
- Hepatotoxicity [see Warnings and Precautions (5.7)]
- Renal Toxicity [see Warnings and Precautions (5.8)]
- Enterocolitis [see Warnings and Precautions (5.9)]
- Skin Reactions [see Warnings and Precautions (5.10)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data described below reflect exposure to clofarabine in 115 pediatric patients with relapsed or refractory Acute Lymphoblastic Leukemia (ALL) (70 patients) or Acute Myelogenous Leukemia (AML) (45 patients).
In total, 115 pediatric patients treated in clinical trials received the recommended dose of clofarabine 52 mg/m2 daily × 5. The median number of cycles was 2. The median cumulative amount of clofarabine received by pediatric patients during all cycles was 540 mg.
Most common adverse reactions (≥25%): vomiting, nausea, diarrhea, febrile neutropenia, pruritus, headache, bacteremia, pyrexia, rash, tachycardia, abdominal pain, chills, fatigue, anorexia, pain in extremity, hypotension, epistaxis, and petechiae.
Table 1 lists adverse reactions by System Organ Class, including severe or life-threatening (NCI CTCAE Grade 3 or Grade 4), reported in ≥5% of the 115 patients in the 52 mg/m2/day dose group (pooled analysis of pediatric patients with ALL and AML). More detailed information and follow-up of certain events is given below.
Table 1: Most Commonly Reported (≥5% Overall) Adverse Reactions by System Organ Class (N=115 pooled analysis) ¹ Patients with more than one adverse reaction (MedDRA preferred term) within a SOC are counted only once in the SOC totals. Patients with more than one occurrence of the same adverse reaction (MedDRA preferred term) are counted only once within that reaction and at the highest severity grade.
System Organ Class¹
Adverse Reaction
(MedDRA Preferred Term)¹ALL/AML
(All Grades,
N=115)Worst Grade
(NCI Common Terminology Criteria)¹3 4 5 N % N % N % N % Blood and Lymphatic System Disorders Febrile neutropenia 63 55 59 51 3 3 . . Neutropenia 11 10 3 3 8 7 . . Cardiac Disorders Pericardial effusion 9 8 . . 1 1 . . Tachycardia 40 35 6 5 . . . . Gastrointestinal Disorders Abdominal pain 40 35 8 7 . . . . Abdominal pain upper 9 8 1 1 . . . . Diarrhea 64 56 14 12 . . . . Gingival or mouth bleeding 20 17 8 7 1 1 . . Nausea 84 73 16 14 1 1 . . Oral mucosal petechiae 6 5 4 4 . . . . Proctalgia 9 8 2 2 . . . . Stomatitis 8 7 1 1 . . . . Vomiting 90 78 9 8 1 1 . . General Disorders and Administration Site Conditions Asthenia 12 10 1 1 1 1 . . Chills 39 34 3 3 . . . . Fatigue 39 34 3 3 2 2 . . Irritability 11 10 1 1 . . . . Mucosal inflammation 18 16 2 2 . . . . Edema 14 12 2 2 . . . . Pain 17 15 7 6 1 1 . . Pyrexia 45 39 16 14 . . . . Hepatobiliary Disorder Jaundice 9 8 2 2 . . . . Infections and Infestations Bacteremia 10 9 10 9 . . . . Candidiasis 8 7 1 1 . . . . Catheter related infection 14 12 13 11 . . . . Cellulitis 9 8 7 6 . . . . Clostridium colitis 8 7 6 5 . . . . Herpes simplex 11 10 6 5 . . . . Herpes zoster 8 7 6 5 . . . . Oral candidiasis 13 11 2 2 . . . . Pneumonia 11 10 6 5 1 1 1 1 Sepsis, including septic shock 19 17 6 5 4 4 9 8 Staphylococcal bacteremia 7 6 5 4 1 1 . . Staphylococcal sepsis 6 5 5 4 1 1 . . Upper respiratory tract infection 6 5 1 1 . . . . Metabolism and Nutrition
DisordersAnorexia 34 30 6 5 8 7 . . Musculoskeletal and Connective Tissue Disorders Arthralgia 10 9 3 3 . . . . Back pain 12 10 3 3 . . . . Bone pain 11 10 3 3 . . . . Myalgia 16 14 . . . . . . Pain in extremity 34 30 6 5 . . . . Neoplasms Benign,
Malignant and Unspecified (incl. cysts and polyps)
Tumor lysis syndrome
7
6
7
6
.
.
.
.Nervous System Disorders Headache 49 43 6 5 . . . . Lethargy 12 10 1 1 . . . . Somnolence 11 10 1 1 . . . . Psychiatric Disorders Agitation 6 5 1 1 . . . . Anxiety 24 21 2 2 . . . . Renal and Urinary
DisordersHematuria
15
13
2
2
.
.
.
.Respiratory, Thoracic and Mediastinal Disorders Dyspnea 15 13 6 5 2 2 . . Epistaxis 31 27 15 13 . . . . Pleural effusion 14 12 4 4 2 2 . . Respiratory distress 12 10 5 4 4 4 1 1 Tachypnea 10 9 4 4 1 1 . . Skin and Subcutaneous Tissue Disorders Erythema 13 11 . . . . . . Palmar-plantar erythrodysesthesia syndrome
18
16
8
7
.
.
.
.Petechiae 30 26 7 6 . . . . Pruritus 49 43 1 1 . . . . Rash 44 38 8 7 . . . . Rash pruritic 9 8 . . . . . . Vascular Disorders Flushing 22 19 . . . . . . Hypertension 15 13 6 5 . . . . Hypotension 33 29 13 11 9 8 . . The following adverse reactions were reported in <5% of the 115 pediatric patients with ALL or AML:
Gastrointestinal Disorders: cecitis, pancreatitis
Hepatobiliary Disorders: hyperbilirubinemia
Immune System Disorders: hypersensitivity
Infections and Infestations: bacterial infection, Enterococcal bacteremia, Escherichia bacteremia, Escherichia sepsis, fungal infection, fungal sepsis, gastroenteritis adenovirus, infection, influenza, parainfluenza virus infection, pneumonia fungal, pneumonia primary atypical, Respiratory syncytial virus infection, sinusitis, staphylococcal infection
Investigations: blood creatinine increased
Psychiatric Disorders: mental status change
Respiratory, Thoracic and Mediastinal Disorder: pulmonary edema
Table 2 lists the incidence of treatment-emergent laboratory abnormalities after clofarabine administration at 52 mg/m2 among pediatric patients with ALL and AML (N=115).
Table 2: Incidence of Treatment-Emergent Laboratory Abnormalities after Clofarabine Administration Parameter Any Grade Grade 3 or higher Anemia (N=114) 83% 75% Leukopenia (N=114) 88% 88% Lymphopenia (N=113) 82% 82% Neutropenia (N=113) 64% 64% Thrombocytopenia (N=114) 81% 80% Elevated Creatinine (N=115) 50% 8% Elevated SGOT (N=100) 74% 36% Elevated SGPT (N=113) 81% 43% Elevated Total Bilirubin (N=114) 45% 13% 6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of clofarabine. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- Gastrointestinal disorders: gastrointestinal hemorrhage including fatalities.
- Metabolism and nutrition disorders: hyponatremia
- Skin and subcutaneous tissue disorders: Stevens-Johnson syndrome (SJS), toxic epidermal necrolysis (TEN) (including fatal cases).
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
In animal reproduction studies, intravenous administration of clofarabine to pregnant rats and rabbits during organogenesis at doses approximately 0.2- to 1-times the maximum recommended human dose of 52 mg/m2 based on body surface area (BSA) resulted in embryo-fetal mortality, alterations to growth, and structural abnormalities (see Data). Advise pregnant women of the potential risk to a fetus. There are no available data on clofarabine use in pregnant women to evaluate for a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes. Clofarabine should be used during pregnancy only if the potential benefits to the mother outweigh the potential risks, including those to the fetus.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal data
Intravenous administration of clofarabine to pregnant rats during organogenesis (gestation days [GD] 7-17) at doses of 1, 3 or 9 mg/kg/day (equivalent to 6, 18, 54 mg/m2/day) resulted in maternal toxicities at the 9 mg/kg dose, as indicated by reduced body weights and food consumption. Developmental toxicity (i.e., reduced fetal body weights and increased postimplantation loss) and increased incidences of external, soft tissue, and skeletal malformations and variations (including retarded ossification) were observed at 9 mg/kg/day (54 mg/m2; approximately equivalent to the recommended human dose based on BSA). Altered ossification patterns (extra metacarpal or metatarsal ossification) were observed in single fetuses at lower doses of clofarabine (1 and 3 mg/kg/day; 0.1- and 0.3-times the recommended human dose based on BSA).
When clofarabine was administered intravenously to pregnant rabbits during organogenesis (GD 6-18) at doses of 0.1, 0.3, or 1 mg/kg/day (equivalent to 1.2, 3.6, 12 mg/m2/day), developmental toxicity (i.e., reduced fetal body weights and increased postimplantation loss) and increased incidences of external, soft tissue, and skeletal malformations and variations (including retarded ossification) were observed at the 1 mg/kg/day dose (12 mg/m2; 0.2-times the recommended human dose based on BSA). Alterations in ossification patterns (increase in the average numbers of ossified thoracic vertebrae and rib pairs, and reduction in the average number of forepaw metacarpals) and abdominal wall defect were observed at 0.3 mg/kg/day (3.6 mg/m2; 0.1-times the recommended human dose based on BSA).
8.2 Lactation
Risk Summary
There are no data on the presence of clofarabine in human milk, the effects on the breastfed child, or the effects on milk production. Because of the potential for serious adverse reactions in the breastfed child including genotoxicity, advise patients not to breastfeed during treatment with clofarabine, and for at least 2 weeks after the last dose.
8.3 Females and Males of Reproductive Potential
Clofarabine can cause embryo-fetal harm when administered to pregnant women [see Use in Specific Populations (8.1)].
Pregnancy Testing
Pregnancy testing is recommended for females of reproductive potential prior to initiating clofarabine.
Contraception
Females
Advise female patients to use effective contraception during treatment with clofarabine and for 6 months after the last dose.
Males
Based on genotoxicity findings, advise males with female partners of reproductive potential to use effective contraception during treatment with clofarabine and for 3 months after the last dose [see Nonclinical Toxicology (13.1)].
Infertility
Females
Based on findings from animal studies, clofarabine may impair female fertility [see Nonclinical Toxicology (13.1)]. The reversibility of the effect on fertility is unknown.
Males
Based on findings from animal studies, clofarabine may impair male fertility [see Nonclinical Toxicology (13.1)]. The reversibility of the effect on fertility is unknown.
8.4 Pediatric Use
Safety and effectiveness have been established in pediatric patients 1 to 21 years old with relapsed or refractory acute lymphoblastic leukemia.
8.5 Geriatric Use
Safety and effectiveness of clofarabine has not been established in geriatric patients aged 65 and older.
8.6 Renal Impairment
Reduce the clofarabine starting dose by 50% in patients with CrCL of 30 to 60 mL/min. There is insufficient information to make a dosage recommendation in patients with CrCL less than 30 mL/min or in patients on dialysis.
The pharmacokinetics of clofarabine in patients with renal impairment and normal renal function were obtained from a population pharmacokinetic analysis of three pediatric and two adult studies. In patients with CrCL 60 to less than 90 mL/min (N=47) and CrCL 30 to less than 60 mL/min (N=30), the average AUC of clofarabine increased by 60% and 140%, respectively, compared to patients with normal (N=66) renal function (CrCL greater than 90 mL/min).
-
10 OVERDOSAGE
There were no known overdoses of clofarabine. The highest daily dose administered to a human to date (on a mg/m2 basis) has been 70 mg/m2/day × 5 days (2 pediatric ALL patients). The toxicities included in these 2 patients included Grade 4 hyperbilirubinemia, Grade 2 and 3 vomiting, and Grade 3 maculopapular rash.
In a Phase 1 study of adults with refractory and/or relapsed hematologic malignancies, the recommended pediatric dose of 52 mg/m2/day was not tolerated.
-
11 DESCRIPTION
Clofarabine Injection contains clofarabine, a purine nucleoside metabolic inhibitor. The chemical name of clofarabine is 2-chloro-9-(2-deoxy-2-fluoro-β-D-arabinofuranosyl)-9H-purin-6-amine. Its molecular formula is C10H11ClFN5O3 with a molecular weight of 303.68 Daltons.
The molecular structure of clofarabine is:
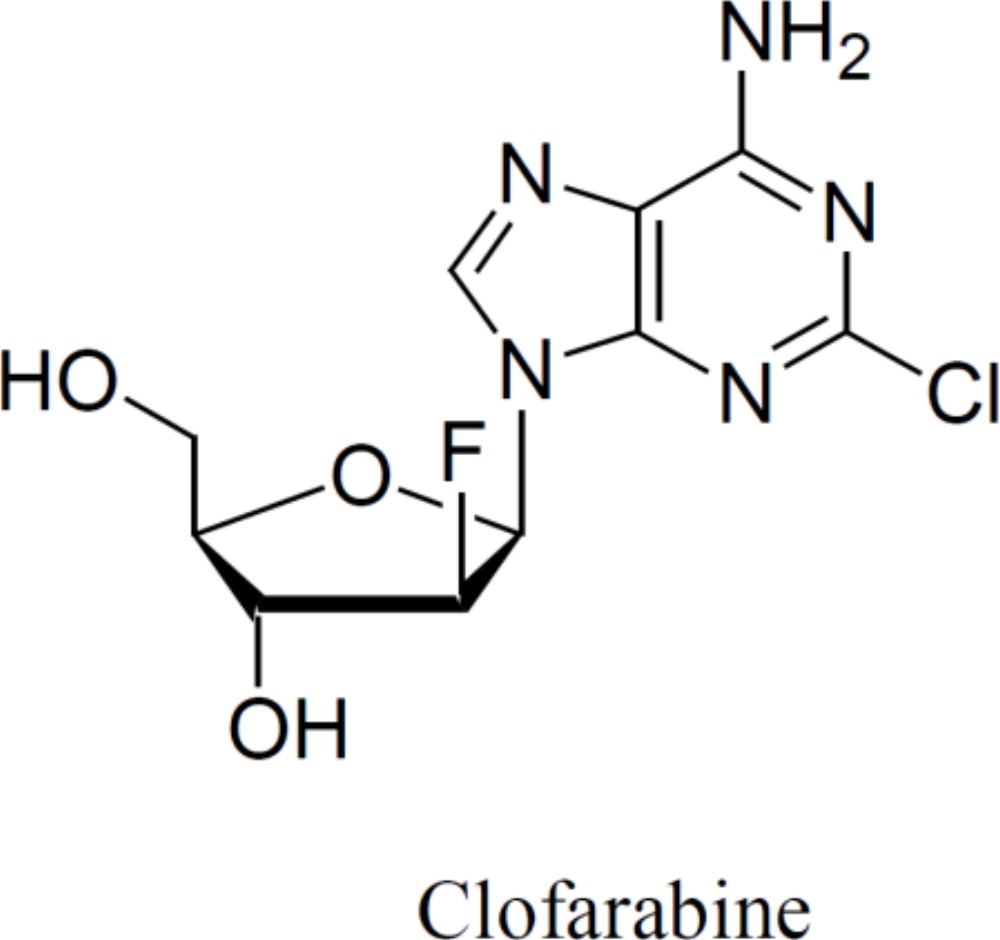
Clofarabine Injection (1 mg per mL) is supplied in a 20 mL single-dose vial. The 20 mL vial contains 20 mg clofarabine formulated in 20 mL unbuffered normal saline (comprised of Water for Injection, USP and Sodium Chloride, USP). The pH range of the solution is 4.5 to 7.5. The solution is sterile, clear and practically colorless, and is preservative-free.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Clofarabine is sequentially metabolized intracellularly to the 5′-monophosphate metabolite by deoxycytidine kinase and mono- and di-phospho-kinases to the active 5′-triphosphate metabolite. Clofarabine has affinity for the activating phosphorylating enzyme, deoxycytidine kinase, equal to or greater than that of the natural substrate, deoxycytidine. Clofarabine inhibits DNA synthesis by decreasing cellular deoxynucleotide triphosphate pools through an inhibitory action on ribonucleotide reductase, and by terminating DNA chain elongation and inhibiting repair through incorporation into the DNA chain by competitive inhibition of DNA polymerases. The affinity of clofarabine triphosphate for these enzymes is similar to or greater than that of deoxyadenosine triphosphate. In preclinical models, clofarabine has demonstrated the ability to inhibit DNA repair by incorporation into the DNA chain during the repair process. Clofarabine 5′-triphosphate also disrupts the integrity of mitochondrial membrane, leading to the release of the pro-apoptotic mitochondrial proteins, cytochrome C and apoptosis-inducing factor, leading to programmed cell death.
Clofarabine is cytotoxic to rapidly proliferating and quiescent cancer cell types in vitro.
12.3 Pharmacokinetics
The population pharmacokinetics of clofarabine were studied in 40 pediatric patients aged 2 to 19 years (21 males/19 females) with relapsed or refractory acute lymphoblastic leukemia (ALL) or acute myelogenous leukemia (AML). At the given 52 mg/m2 dose, similar concentrations were obtained over a wide range of body surface areas (BSAs). Clofarabine was 47% bound to plasma proteins, predominantly to albumin. Based on non-compartmental analysis, systemic clearance and volume of distribution at steady-state were 28.8 L/h/m2 and 172 L/m2, respectively. The terminal half-life was 5.2 hours. No apparent difference in pharmacokinetics was observed between patients with ALL and AML or between males and females.
No relationship between clofarabine or clofarabine triphosphate exposure and toxicity or response was found in this population.
Based on 24-hour urine collections in the pediatric studies, 49% to 60% of the dose is excreted in the urine unchanged. In vitro studies using isolated human hepatocytes indicate very limited metabolism (0.2%). The pathways of non-hepatic elimination remain unknown.
Clofarabine has not been studied in patients with hepatic impairment.
Drug-Drug Interactions
In vitro studies suggested that clofarabine undergoes limited metabolism and does not inhibit or induce major CYP enzymes. CYP inhibitors and inducers are unlikely to affect the metabolism of clofarabine. Clofarabine is unlikely to affect the metabolism of CYP substrates. However, no in vivo drug interaction studies have been conducted.
An in vitro transporter study suggested that clofarabine is a substrate of human transporters OAT1, OAT3, and OCT1. A preclinical study using perfused rat kidney demonstrated that the renal excretion of clofarabine was decreased by cimetidine, an inhibitor of the hOCT2. Although the clinical implications of this finding have not been determined, signs of clofarabine toxicity should be monitored when administered with other hOAT1, hOAT3, hOCT1 and hOCT2 substrates or inhibitors.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Clofarabine has not been tested for carcinogenic potential.
Clofarabine was clastogenic in the in vitro mammalian cell chromosome aberration assay (CHO cells) and in the in vivo rat micronucleus assay. Clofarabine was not mutagenic in the bacterial mutation assay (Ames test).
Studies in mice, rats, and dogs have demonstrated dose-related adverse effects on male reproductive organs. Seminiferous tubule and testicular degeneration and atrophy were reported in male mice receiving intraperitoneal doses of 3 mg/kg/day (approximately 0.2-times the recommended human dose based on body surface area [BSA]). Rats receiving 25 mg/kg/day (approximately 3-times the recommended human dose based on BSA) in a 6-month intravenous study had testicular findings of bilateral degeneration of the seminiferous epithelium with retained spermatids and atrophy of interstitial cells. In a 6-month intravenous dog study, cell degeneration of the epididymis and degeneration of the seminiferous epithelium in the testes were observed at 0.375 mg/kg/day (approximately 0.1-times the recommended human dose on a BSA basis). Ovarian atrophy or degeneration and uterine mucosal apoptosis were observed in female mice at 75 mg/kg/day (approximately 4-times the recommended human dose on a mg/m2 basis), the only dose administered to female mice.
-
14 CLINICAL STUDIES
Seventy-eight (78) pediatric patients with ALL were exposed to clofarabine. Seventy (70) of the patients received the recommended pediatric dose of clofarabine 52 mg/m2 daily for 5 days as an intravenous infusion.
Dose Escalation Study in Pediatric Patients with Hematologic Malignancies
The safety and efficacy of clofarabine were evaluated in pediatric patients with refractory or relapsed hematologic malignancies in an open-label, dose-escalation, noncomparative study (NCT00042341, A Phase II, Open Label Study of Clofarabine in Pediatric Patients With Refractory or Relapsed Acute Lymphoblastic Leukemia). The starting dose of clofarabine was 11.25 mg/m2/day intravenous infusion daily × 5 and escalated to 70 mg/m2/day intravenous infusion daily × 5. This dosing schedule was repeated every 2 to 6 weeks depending on toxicity and response. Nine of 17 ALL patients were treated with clofarabine 52 mg/m2 daily for 5 days. In the 17 ALL patients there were 2 complete remissions (12%) and 2 partial remissions (12%) at varying doses. Dose-limiting toxicities in this study were reversible hyperbilirubinemia and elevated transaminase levels and skin rash, experienced at 70 mg/m2. As a result of this study, the recommended dose for subsequent study in pediatric patients was determined to be 52 mg/m2/day for 5 days.
Single-Arm Study in Pediatric ALL
Clofarabine was evaluated in an open-label, single-arm study (NCT00042341) of 61 pediatric patients with relapsed/refractory ALL. Patients received a dose of 52 mg/m2 intravenous infusion over 2 hours for 5 consecutive days repeated every 2 to 6 weeks for up to 12 cycles. There was no dose escalation in this study.
All patients had disease that had relapsed after and/or was refractory to two or more prior therapies. Most patients, 38/61 (62%), had received >2 prior regimens and 18/61 (30%) of the patients had undergone at least 1 prior transplant. The median age of the treated patients was 12 years, 61% were male, 39% were female, 44% were White, 38% were Hispanic or Latino, 12% were Black or African-American, 2% were Asian and 5% were Other race.
The overall remission (OR) rate (Complete Remission [CR] + CR in the absence of total platelet recovery [CRp]) was evaluated. CR was defined as no evidence of circulating blasts or extramedullary disease, an M1 bone marrow (≤5% blasts), and recovery of peripheral counts [platelets ≥100 × 109/L and absolute neutrophil count (ANC) ≥1 × 109/L]. CRp was defined as meeting all criteria for CR except for recovery of platelet counts to ≥100 × 109/L. Partial Response (PR) was also determined, defined as complete disappearance of circulating blasts, an M2 bone marrow (≥5% and ≤25% blasts), and appearance of normal progenitor cells or an M1 marrow that did not qualify for CR or CRp. Duration of remission was also evaluated. Transplantation rate was not a study endpoint.
Response rates for these studies were determined by an unblinded Independent Response Review Panel (IRRP).
Table 3 summarizes results for the pediatric ALL study. Responses were seen in both pre-B and T-cell immunophenotypes of ALL. The median cumulative dose was 530 mg (range 29-2,815 mg) in 1 (41%), 2 (44%) or 3 or more (15%) cycles. The median number of cycles was 2 (range 1-12). The median time between cycles was 28 days with a range of 12 to 55 days.
Table 3: Results in Single-Arm Pediatric ALL CR = Complete response
CRp = Complete response without platelet recovery
1 Does not include 4 patients who were transplanted (duration of response, including response after transplant, in these 4 patients was 28.6 to 107.7 weeks).
N=61 CR % [95% CI] 11.5 (4.7, 22.2) CRp % [95% CI] 8.2 (2.7, 18.1) Median Duration of CR plus CRp (range in weeks)1 10.7 (4.3 to 58.6) The median duration of CR, including patients who received transplantation, was 47.9 weeks (range 4.3 to 107.7+), where + indicates the patient was still in CR by the study end.
Of 35 patients who were refractory to their immediately preceding induction regimen, 6 (17%) achieved a CR or CRp. Of 18 patients who had at least 1 prior hematopoietic stem cell transplant (HSCT), 5 (28%) achieved a CR or CRp.
Among the 12 patients who achieved at least a CRp, 6 patients achieved the best response after 1 cycle of clofarabine, 5 patients required 2 courses and 1 patient achieved a CR after 3 cycles of therapy.
- 15 REFERENCES
-
16 HOW SUPPLIED/STORAGE AND HANDLING
Clofarabine Injection is a sterile, clear and practically colorless solution, is preservative-free and is free from foreign matter. It is supplied in single-dose flint vials containing 20 mg of clofarabine in 20 mL of solution as follows:
NDC Clofarabine Injection (1 mg per mL) Package Factor 71288-128-20 20 mg per 20 mL Single-Dose Vial 1 vial per carton The pH range of the solution is 4.5 to 7.5.
Storage Conditions
Vials containing undiluted Clofarabine Injection should be stored at 25°C (77°F); excursions permitted between 15° to 30°C (59° to 86°F). Do not freeze. Retain in carton until contents are used.
Clofarabine Injection is a hazardous drug. Follow applicable special handling and disposal procedures.1
Discard unused portion.
Sterile, Nonpyrogenic, Preservative-free.
The container closure is not made with natural rubber latex. -
17 PATIENT COUNSELING INFORMATION
Hematologic Toxicity
Advise patients to return for regular blood counts and to report any symptoms associated with hematologic toxicity (such as weakness, fatigue, pallor, shortness of breath, easy bruising, petechiae, purpura, fever) to their physician [see Warnings and Precautions (5.1)].
Infection
Advise patients of the signs or symptoms of infection (e.g., fever) and report to the physician immediately if any occur [see Warnings and Precautions (5.3)].
Hepatic and Renal Toxicity
Advise patients to avoid medications including over the counter and herbal medications, which may be hepatotoxic or nephrotoxic, during the 5 days of clofarabine administration. Also, advise patients of the possibility of developing liver function abnormalities and to immediately report signs or symptoms of jaundice. Advise patients of the signs or symptoms of renal failure/acute renal failure [see Warnings and Precautions (5.7, 5.8)].
Systemic Inflammatory Response Syndrome (SIRS)/Capillary Leak Syndrome
Advise patients of the signs or symptoms of SIRS, such as fever, tachycardia, tachypnea, dyspnea and symptoms suggestive of hypotension [see Warnings and Precautions (5.5)].
Pregnancy
Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.11), Use in Specific Populations (8.3)]. Advise female patients of reproductive potential to use effective contraception during treatment with clofarabine and for 6 months after the last dose [see Use in Specific Populations (8.3)]. Advise males with female partners of reproductive potential to use effective contraception during treatment with clofarabine and for 3 months after the last dose [see Use in Specific Populations (8.3), Nonclinical Toxicology (13.1)].
Lactation
Advise females not to breastfeed during treatment with clofarabine and for 2 weeks after the last dose [see Use in Specific Populations (8.2)].
Gastrointestinal Disorders
Advise patients that they may experience nausea, vomiting, and/or diarrhea with clofarabine. If these symptoms are significant, they should seek medical attention [see Warnings and Precautions (5.9)].
Rash
Advise patients that they may experience skin rash with clofarabine. If this symptom is significant, they should seek medical attention.
meitheal®
Mfd. for Meitheal Pharmaceuticals
Chicago, IL 60631 (USA)
©2022 Meitheal Pharmaceuticals Inc.Mfd. by Kindos Pharmaceuticals Co., Ltd.
Chengdu, China 611731Revised: September 2022
LB-279-V2
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
CLOFARABINE
clofarabine injectionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 71288-128 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength clofarabine (UNII: 762RDY0Y2H) (clofarabine - UNII:762RDY0Y2H) clofarabine 1 mg in 1 mL Inactive Ingredients Ingredient Name Strength sodium chloride (UNII: 451W47IQ8X) 9 mg in 1 mL water (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 71288-128-20 1 in 1 CARTON 10/23/2020 1 20 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA213461 10/23/2020 Labeler - Meitheal Pharmaceuticals Inc. (080548348) Establishment Name Address ID/FEI Business Operations Kindos Pharmaceuticals Co., Ltd. 529111185 MANUFACTURE(71288-128)


© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.