TRUVADA- emtricitabine and tenofovir disoproxil fumarate tablet, film coated
Truvada by
Drug Labeling and Warnings
Truvada by is a Prescription medication manufactured, distributed, or labeled by Gilead Sciences, Inc. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use TRUVADA safely and effectively. See full prescribing information for TRUVADA.
TRUVADA® (emtricitabine and tenofovir disoproxil fumarate) tablets, for oral use
Initial U.S. Approval: 2004WARNING: POSTTREATMENT ACUTE EXACERBATION OF HEPATITIS B and RISK OF DRUG RESISTANCE WITH USE OF TRUVADA FOR HIV-1 PRE-EXPOSURE PROPHYLAXIS (PrEP) IN UNDIAGNOSED EARLY HIV-1 INFECTION
See full prescribing information for complete boxed warning.
- Severe acute exacerbations of hepatitis B virus (HBV) have been reported in HBV-infected patients who have discontinued TRUVADA. Hepatic function should be monitored closely in HBV-infected patients who discontinue TRUVADA. If appropriate, initiation of anti-hepatitis B therapy may be warranted. (5.1)
- TRUVADA used for HIV-1 PrEP must only be prescribed to individuals confirmed to be HIV-negative immediately prior to initial use and periodically during use. Drug-resistant HIV-1 variants have been identified with the use of TRUVADA for HIV-1 PrEP following undetected acute HIV-1 infection. Do not initiate TRUVADA for HIV-1 PrEP if signs or symptoms of acute HIV infection are present unless negative infection status is confirmed. (5.2)
RECENT MAJOR CHANGES
Indications and Usage Treatment of HIV-1 Infection (1.1) 05/2018 HIV-1 Pre-Exposure Prophylaxis (PrEP) (1.2) 05/2018 Dosage and Administration Testing Prior to Initiation of TRUVADA for Treatment of HIV-1 Infection or for HIV-1 PrEP (2.1) 05/2018 HIV-1 Screening for Individuals Receiving TRUVADA for HIV-1 PrEP (2.2) 05/2018 Recommended Dosage for HIV-1 PrEP (2.5) 05/2018 Warnings and Precautions Severe Acute Exacerbation of Hepatitis B in Patients with HBV Infection (5.1) 05/2018 Comprehensive Management to Reduce the Risk of Acquiring HIV-1 and Development of HIV-1 Resistance When TRUVADA Is Used for HIV-1 PrEP (5.2) 05/2018 New Onset or Worsening Renal Impairment (5.3) 05/2018 Risk of Adverse Reactions Due to Drug Interactions (5.7) 05/2018 Early Virologic Failure Removed 05/2018 INDICATIONS AND USAGE
TRUVADA is a two-drug combination of emtricitabine (FTC) and tenofovir disoproxil fumarate (TDF), both HIV-1 nucleoside analog reverse transcriptase inhibitors, and is indicated:
- in combination with other antiretroviral agents for the treatment of HIV-1 infection in adults and pediatric patients weighing at least 17 kg. (1.1)
- in combination with safer sex practices for HIV-1 pre-exposure prophylaxis (PrEP) to reduce the risk of sexually acquired HIV-1 in at-risk adults and adolescents weighing at least 35 kg. (1.2)
DOSAGE AND ADMINISTRATION
- Testing: Prior to or when initiating TRUVADA test for hepatitis B virus infection. Prior to initiation and during use of TRUVADA, on a clinically appropriate schedule, assess serum creatinine, estimated creatinine clearance, urine glucose, and urine protein in all patients. In patients with chronic kidney disease, also assess serum phosphorus. (2.1)
- HIV-1 Screening: Screen all patients for HIV-1 infection before initiating TRUVADA for HIV-1 PrEP and at least once every 3 months while taking TRUVADA. (2.2)
Treatment of HIV-1 Infection
- Recommended dosage in adults and pediatric patients weighing at least 35 kg: One TRUVADA tablet (containing 200 mg of FTC and 300 mg of TDF) once daily taken orally with or without food. (2.3)
- Recommended dosage in pediatric patients weighing at least 17 kg: One TRUVADA low-strength tablet (100 mg/150 mg, 133 mg/200 mg, or 167 mg/250 mg based on body weight) once daily taken orally with or without food. (2.4)
- Recommended dosage in renally impaired HIV-1 infected adult patients:
HIV-1 Pre-Exposure Prophylaxis (PrEP)
- Recommended dosage in HIV-1 uninfected adults and adolescents weighing at least 35 kg: One TRUVADA tablet (containing 200 mg of FTC and 300 mg of TDF) once daily taken orally with or without food. (2.5)
- Recommended dosage in renally impaired HIV-uninfected individuals: TRUVADA is not recommended in HIV-uninfected individuals if CrCl is below 60 mL/min. (2.6)
DOSAGE FORMS AND STRENGTHS
Tablets: 200 mg/300 mg, 167 mg/250 mg, 133 mg/200 mg, and 100 mg/150 mg of emtricitabine and tenofovir disoproxil fumarate, respectively. (3)
CONTRAINDICATIONS
TRUVADA for HIV-1 PrEP is contraindicated in individuals with unknown or positive HIV-1 status. (4)
WARNINGS AND PRECAUTIONS
- Comprehensive management to reduce the risk of acquiring HIV-1: Use as part of a comprehensive prevention strategy including other prevention measures; strictly adhere to dosing schedule. (5.2)
- Management to reduce the risk of acquiring HIV-1 drug resistance: refer to full prescribing information for additional detail. (5.2)
- New onset or worsening renal impairment: Can include acute renal failure and Fanconi syndrome. Avoid administering TRUVADA with concurrent or recent use of nephrotoxic drugs. (5.3)
- Immune reconstitution syndrome: May necessitate further evaluation and treatment. (5.4)
- Decreases in bone mineral density (BMD): Consider assessment of BMD in patients with a history of pathologic fracture or other risk factors for osteoporosis or bone loss. (5.5)
- Lactic acidosis/severe hepatomegaly with steatosis: Discontinue treatment in patients who develop symptoms or laboratory findings suggestive of lactic acidosis or pronounced hepatotoxicity. (5.6)
ADVERSE REACTIONS
- In HIV-1 infected patients, the most common adverse reactions (incidence greater than or equal to 10%) are diarrhea, nausea, fatigue, headache, dizziness, depression, insomnia, abnormal dreams, and rash. (6.1)
- In HIV-1 uninfected adults in PrEP trials, adverse reactions that were reported by more than 2% of TRUVADA subjects and more frequently than by placebo subjects were headache, abdominal pain, and weight decreased. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Gilead Sciences, Inc. at 1-800-445-3235 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch
DRUG INTERACTIONS
- Tenofovir disoproxil fumarate increases didanosine concentrations. Dose reduction and close monitoring for didanosine toxicity are warranted. (7.2)
- Coadministration decreases atazanavir concentrations. When coadministered with TRUVADA, use atazanavir given with ritonavir. (7.2)
- Coadministration of TRUVADA with certain HIV-1 protease inhibitors or certain drugs to treat HCV increases tenofovir concentrations. Monitor for evidence of tenofovir toxicity. (7.2)
- Consult Full Prescribing Information prior to and during treatment for important drug interactions. (7.2)
USE IN SPECIFIC POPULATIONS
Lactation: Women infected with HIV-1 or suspected of having acquired HIV-1 infection should be instructed not to breastfeed. (8.2)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 5/2018
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: POSTTREATMENT ACUTE EXACERBATION OF HEPATITIS B and RISK OF DRUG RESISTANCE WITH USE OF TRUVADA FOR HIV-1 PRE-EXPOSURE PROPHYLAXIS (PrEP) IN UNDIAGNOSED EARLY HIV-1 INFECTION
1 INDICATIONS AND USAGE
1.1 Treatment of HIV-1 Infection
1.2 HIV-1 Pre-Exposure Prophylaxis (PrEP)
2 DOSAGE AND ADMINISTRATION
2.1 Testing Prior to Initiation of TRUVADA for Treatment of HIV-1 Infection or for HIV-1 PrEP
2.2 HIV-1 Screening for Individuals Receiving TRUVADA for HIV-1 PrEP
2.3 Recommended Dosage for Treatment of HIV-1 Infection in Adults and Pediatric Patients Weighing at Least 35 kg
2.4 Recommended Dosage for Treatment of HIV-1 Infection in Pediatric Patients Weighing at Least 17 kg and Able to Swallow a Tablet
2.5 Recommended Dosage for HIV-1 PrEP
2.6 Dosage Adjustment in Patients with Renal Impairment
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Severe Acute Exacerbation of Hepatitis B in Patients with HBV Infection
5.2 Comprehensive Management to Reduce the Risk of Acquiring HIV-1 and Development of HIV-1 Resistance When TRUVADA Is Used for HIV-1 PrEP
5.3 New Onset or Worsening Renal Impairment
5.4 Immune Reconstitution Syndrome
5.5 Bone Loss and Mineralization Defects
5.6 Lactic Acidosis/Severe Hepatomegaly with Steatosis
5.7 Risk of Adverse Reactions Due to Drug Interactions
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Drugs Affecting Renal Function
7.2 Established and Significant Interactions
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.3 Pharmacokinetics
12.4 Microbiology
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
14.1 Overview of Clinical Trials
14.2 Clinical Trial Results for Treatment of HIV-1: Study 934
14.3 Clinical Trial Results for HIV-1 PrEP: iPrEx
14.4 Clinical Trial Results for HIV-1 PrEP: Partners PrEP
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: POSTTREATMENT ACUTE EXACERBATION OF HEPATITIS B and RISK OF DRUG RESISTANCE WITH USE OF TRUVADA FOR HIV-1 PRE-EXPOSURE PROPHYLAXIS (PrEP) IN UNDIAGNOSED EARLY HIV-1 INFECTION
Severe acute exacerbations of hepatitis B virus (HBV) have been reported in HBV-infected patients who have discontinued TRUVADA. Hepatic function should be monitored closely with both clinical and laboratory follow-up for at least several months in HBV-infected patients who discontinue TRUVADA. If appropriate, initiation of anti-hepatitis B therapy may be warranted [see Warnings and Precautions (5.1)].
TRUVADA used for HIV-1 PrEP must only be prescribed to individuals confirmed to be HIV-negative immediately prior to initiating and periodically (at least every 3 months) during use. Drug-resistant HIV-1 variants have been identified with use of TRUVADA for HIV-1 PrEP following undetected acute HIV-1 infection. Do not initiate TRUVADA for HIV-1 PrEP if signs or symptoms of acute HIV-1 infection are present unless negative infection status is confirmed [see Warnings and Precautions (5.2)].
-
1 INDICATIONS AND USAGE
1.1 Treatment of HIV-1 Infection
TRUVADA is indicated in combination with other antiretroviral agents for the treatment of HIV-1 infection in adults and pediatric patients weighing at least 17 kg [see Clinical Studies (14)].
1.2 HIV-1 Pre-Exposure Prophylaxis (PrEP)
TRUVADA is indicated in combination with safer sex practices for pre-exposure prophylaxis (PrEP) to reduce the risk of sexually acquired HIV-1 in at-risk adults and adolescents weighing at least 35 kg. Individuals must have a negative HIV-1 test immediately prior to initiating TRUVADA for HIV-1 PrEP [see Dosage and Administration (2.2)].
- If clinical symptoms consistent with acute viral infection are present and recent (<1 month) exposures are suspected, delay starting PrEP for at least one month and reconfirm HIV-1 status or use a test cleared by the FDA as an aid in the diagnosis of HIV-1 infection, including acute or primary HIV-1 infection [see Warnings and Precautions (5.2), Use in Specific Populations (8.4) and Clinical Studies (14.3, 14.4)].
When considering TRUVADA for HIV-1 PrEP, factors that help to identify individuals at risk may include:
- has partner(s) known to be HIV-1 infected, or
- engages in sexual activity within a high prevalence area or social network and has additional risk factors for HIV-1 acquisition, such as:
- inconsistent or no condom use
- diagnosis of sexually transmitted infections
- exchange of sex for commodities (such as money, food, shelter, or drugs)
- use of illicit drugs or alcohol dependence
- incarceration
- partner(s) of unknown HIV-1 status with any of the factors listed above
-
2 DOSAGE AND ADMINISTRATION
2.1 Testing Prior to Initiation of TRUVADA for Treatment of HIV-1 Infection or for HIV-1 PrEP
Prior to or when initiating TRUVADA, test patients for hepatitis B virus infection [see Warnings and Precautions (5.1)].
Prior to initiation and during use of TRUVADA, on a clinically appropriate schedule, assess serum creatinine, estimated creatinine clearance, urine glucose and urine protein in all patients. In patients with chronic kidney disease, also assess serum phosphorus [see Warnings and Precautions (5.3)].
2.2 HIV-1 Screening for Individuals Receiving TRUVADA for HIV-1 PrEP
Screen all patients for HIV-1 infection before initiating TRUVADA for HIV-1 PrEP and at least once every 3 months while taking TRUVADA [see Indications and Usage (1.2), Contraindications (4) and Warnings and Precautions (5.2)].
2.3 Recommended Dosage for Treatment of HIV-1 Infection in Adults and Pediatric Patients Weighing at Least 35 kg
TRUVADA is a two-drug fixed dose combination product containing emtricitabine (FTC) and tenofovir disoproxil fumarate (TDF). The recommended dosage of TRUVADA in adults and in pediatric patients weighing at least 35 kg is one tablet (containing 200 mg of FTC and 300 mg of TDF) once daily taken orally with or without food [see Clinical Pharmacology (12.3)].
2.4 Recommended Dosage for Treatment of HIV-1 Infection in Pediatric Patients Weighing at Least 17 kg and Able to Swallow a Tablet
The recommended oral dosage of TRUVADA for pediatric patients weighing at least 17 kg and who can swallow a tablet is presented in Table 1. Tablets should be taken once daily with or without food. Weight should be monitored periodically and the TRUVADA dose adjusted accordingly.
Table 1 Dosing for Treatment of HIV-1 Infection in Pediatric Patients Weighing 17 kg to less than 35 kg Body Weight (kg) Dosing of TRUVADA
(FTC/TDF)17 to less than 22 one 100 mg /150 mg tablet once daily 22 to less than 28 one 133 mg /200 mg tablet once daily 28 to less than 35 one 167 mg /250 mg tablet once daily 2.5 Recommended Dosage for HIV-1 PrEP
The dosage of TRUVADA in HIV-1 uninfected adults and adolescents weighing at least 35 kg is one tablet (containing 200 mg of FTC and 300 mg of TDF) once daily taken orally with or without food [see Clinical Pharmacology (12.3)].
2.6 Dosage Adjustment in Patients with Renal Impairment
Treatment of HIV-1 Infection
Table 2 provides dosage interval adjustment for patients with renal impairment. No dosage adjustment is necessary for HIV-1 infected patients with mild renal impairment (creatinine clearance 50–80 mL/min). The safety and effectiveness of the dosing interval adjustment recommendations in patients with moderate renal impairment (creatinine clearance 30–49 mL/min) have not been clinically evaluated; therefore, clinical response to treatment and renal function should be closely monitored in these patients [see Warnings and Precautions (5.3)].
No data are available to make dosage recommendations in pediatric patients with renal impairment.
Table 2 Dosage Interval Adjustment for HIV-1 Infected Adult Patients with Altered Creatinine Clearance Creatinine Clearance (mL/min)* ≥50 30–49 <30
(Including Patients Requiring Hemodialysis)- * Calculated using ideal (lean) body weight
Recommended Dosing Interval Every 24 hours Every 48 hours TRUVADA is not recommended. HIV-1 PrEP
TRUVADA for HIV-1 PrEP is not recommended in HIV-1 uninfected individuals with estimated creatinine clearance below 60 mL/min [see Warnings and Precautions (5.3)].
If a decrease in estimated creatinine clearance is observed in uninfected individuals while using TRUVADA for HIV-1 PrEP, evaluate potential causes and re-assess potential risks and benefits of continued use [see Warnings and Precautions (5.3)].
-
3 DOSAGE FORMS AND STRENGTHS
TRUVADA tablets are available in four dose strengths.
- 100 mg/150 mg Tablets: 100 mg of emtricitabine (FTC) and 150 mg of tenofovir disoproxil fumarate (TDF) (equivalent to 123 mg of tenofovir disoproxil): blue, oval shaped, film coated, debossed with "GSI" on one side and with "703" on the other side.
- 133 mg/200 mg Tablets: 133 mg of FTC and 200 mg of TDF (equivalent to 163 mg of tenofovir disoproxil): blue, rectangular shaped, film coated, debossed with "GSI" on one side and with "704" on the other side.
- 167 mg/250 mg Tablets: 167 mg of FTC and 250 mg of TDF (equivalent to 204 mg of tenofovir disoproxil): blue, modified capsule shaped, film coated, debossed with "GSI" on one side and with "705" on the other side.
- 200 mg/300 mg Tablets: 200 mg of FTC and 300 mg of TDF (equivalent to 245 mg of tenofovir disoproxil): blue, capsule shaped, film coated, debossed with "GILEAD" on one side and with "701" on the other side.
-
4 CONTRAINDICATIONS
TRUVADA for HIV-1 PrEP is contraindicated in individuals with unknown or positive HIV-1 status [see Warnings and Precautions (5.2)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Severe Acute Exacerbation of Hepatitis B in Patients with HBV Infection
All patients should be tested for the presence of chronic hepatitis B virus (HBV) before or when initiating TRUVADA [see Dosage and Administration (2.1)].
Severe acute exacerbations of hepatitis B (e.g., liver decompensation and liver failure) have been reported in HBV-infected patients who have discontinued TRUVADA. Patients infected with HBV who discontinue TRUVADA should be closely monitored with both clinical and laboratory follow-up for at least several months after stopping treatment. If appropriate, initiation of anti-hepatitis B therapy may be warranted, especially in patients with advanced liver disease or cirrhosis, since posttreatment exacerbation of hepatitis may lead to hepatic decompensation and liver failure. HBV-uninfected individuals should be offered vaccination.
5.2 Comprehensive Management to Reduce the Risk of Acquiring HIV-1 and Development of HIV-1 Resistance When TRUVADA Is Used for HIV-1 PrEP
Use TRUVADA for HIV-1 PrEP only as part of a comprehensive prevention strategy that includes other prevention measures, such as safer sex practices, because TRUVADA is not always effective in preventing acquisition of HIV-1 [see Clinical Studies (14.3 and 14.4)].
- Counsel uninfected individuals about safer sex practices that include consistent and correct use of condoms, knowledge of their HIV-1 status and that of their partner(s), the importance of virologic suppression in their partner(s) with HIV-1, and regular testing for other sexually transmitted infections that can facilitate HIV-1 transmission (such as syphilis, chlamydia, and gonorrhea).
- Inform uninfected individuals about and support their efforts in reducing sexual risk behavior.
Use TRUVADA to reduce the risk of acquiring HIV-1 only in individuals confirmed to be HIV-negative. HIV-1 resistance substitutions may emerge in individuals with undetected HIV-1 infection who are taking only TRUVADA, because TRUVADA alone does not constitute a complete regimen for HIV-1 treatment [see Microbiology (12.4)]; therefore, care should be taken to minimize drug exposure in HIV-infected individuals.
- Many HIV-1 tests, such as rapid tests, detect anti-HIV antibodies and may not identify HIV-1 during the acute stage of infection. Prior to initiating TRUVADA for HIV-1 PrEP, evaluate seronegative individuals for current or recent signs or symptoms consistent with acute viral infections (e.g., fever, fatigue, myalgia, skin rash) and ask about potential exposure events (e.g., unprotected, or condom broke during, sex with an HIV-1 infected partner) that may have occurred within the last month.
- If clinical symptoms consistent with acute viral infection are present and recent (<1 month) exposures are suspected, delay starting HIV-1 PrEP for at least one month and reconfirm HIV-1 status or use a test approved or cleared by the FDA as an aid in the diagnosis of HIV-1 infection, including acute or primary HIV-1 infection.
While using TRUVADA for HIV-1 PrEP, HIV-1 screening tests should be repeated at least every 3 months, and upon diagnosis of any sexually transmitted infections. Some individuals, such as adolescents, may benefit from more frequent visits and counseling [see Use in Specific Populations (8.4)].
- If a screening test indicates possible HIV-1 infection, or if symptoms consistent with acute HIV-1 infection develop following a potential exposure event, convert the HIV-1 PrEP regimen to an HIV treatment regimen until negative infection status is confirmed using a test approved or cleared by the FDA as an aid in the diagnosis of HIV-1, including acute or primary HIV-1 infection.
Counsel uninfected individuals to strictly adhere to the recommended TRUVADA dosing schedule. The effectiveness of TRUVADA in reducing the risk of acquiring HIV-1 is strongly correlated with adherence, as demonstrated by measurable drug levels in clinical trials [see Use in Specific Populations (8.4), Microbiology (12.4), and Clinical Studies (14.3 and 14.4)].
5.3 New Onset or Worsening Renal Impairment
Emtricitabine and tenofovir are principally eliminated by the kidney. Renal impairment, including cases of acute renal failure and Fanconi syndrome (renal tubular injury with severe hypophosphatemia), has been reported with the use of TDF, a component of TRUVADA [see Adverse Reactions (6.2)].
Prior to initiation and during use of TRUVADA, on a clinically appropriate schedule, assess serum creatinine, estimated creatinine clearance, urine glucose, and urine protein in all patients. In patients with chronic kidney disease, also assess serum phosphorus.
TRUVADA should be avoided with concurrent or recent use of a nephrotoxic agent (e.g., high-dose or multiple non-steroidal anti-inflammatory drugs [NSAIDs]) [see Drug Interactions (7.1)]. Cases of acute renal failure after initiation of high-dose or multiple NSAIDs have been reported in HIV-infected patients with risk factors for renal dysfunction who appeared stable on TDF. Some patients required hospitalization and renal replacement therapy. Alternatives to NSAIDs should be considered, if needed, in patients at risk for renal dysfunction.
Persistent or worsening bone pain, pain in extremities, fractures, and/or muscular pain or weakness may be manifestations of proximal renal tubulopathy and should prompt an evaluation of renal function in patients at risk of renal dysfunction.
Treatment of HIV-1 Infection
Dosing interval adjustment of TRUVADA and close monitoring of renal function are recommended in all patients with estimated creatinine clearance 30–49 mL/min [see Dosage and Administration (2.6)]. No safety or efficacy data are available in patients with renal impairment who received TRUVADA using these dosing guidelines, so the potential benefit of TRUVADA therapy should be assessed against the potential risk of renal toxicity. TRUVADA is not recommended in patients with estimated creatinine clearance below 30 mL/min or patients requiring hemodialysis.
HIV-1 PrEP
TRUVADA for HIV-1 PrEP is not recommended in uninfected individuals with estimated creatinine clearance less than 60 mL/min. If a decrease in estimated creatinine clearance is observed while using TRUVADA for HIV-1 PrEP, evaluate potential causes and re-assess potential risks and benefits of continued use [see Dosage and Administration (2.6)].
5.4 Immune Reconstitution Syndrome
Immune reconstitution syndrome has been reported in HIV-1 infected patients treated with combination antiretroviral therapy, including TRUVADA. During the initial phase of combination antiretroviral treatment, HIV-1 infected patients whose immune system responds may develop an inflammatory response to indolent or residual opportunistic infections (such as Mycobacterium avium infection, cytomegalovirus, Pneumocystis jirovecii pneumonia [PCP], or tuberculosis), which may necessitate further evaluation and treatment.
Autoimmune disorders (such as Graves' disease, polymyositis, and Guillain-Barré syndrome) have also been reported to occur in the setting of immune reconstitution; however, the time to onset is more variable and can occur many months after initiation of treatment.
5.5 Bone Loss and Mineralization Defects
Bone Mineral Density
In clinical trials in HIV-1 infected adults and in a clinical trial of HIV-1 uninfected individuals, TDF (a component of TRUVADA) was associated with slightly greater decreases in bone mineral density (BMD) and increases in biochemical markers of bone metabolism, suggesting increased bone turnover relative to comparators [see Adverse Reactions (6.1)]. Serum parathyroid hormone levels and 1,25 Vitamin D levels were also higher in subjects receiving TDF.
Clinical trials evaluating TDF in pediatric and adolescent subjects were conducted. Under normal circumstances, BMD increases rapidly in pediatric patients. In HIV-1 infected subjects aged 2 years to less than 18 years, bone effects were similar to those observed in adult subjects and suggest increased bone turnover. Total body BMD gain was less in the TDF-treated HIV-1 infected pediatric subjects as compared to the control groups. Similar trends were observed in adolescent subjects aged 12 years to less than 18 years treated for chronic hepatitis B. In all pediatric trials, skeletal growth (height) appeared to be unaffected.
The effects of TDF-associated changes in BMD and biochemical markers on long-term bone health and future fracture risk are unknown. Assessment of BMD should be considered for adult and pediatric patients who have a history of pathologic bone fracture or other risk factors for osteoporosis or bone loss. Although the effect of supplementation with calcium and vitamin D was not studied, such supplementation may be beneficial. If bone abnormalities are suspected, appropriate consultation should be obtained.
Mineralization Defects
Cases of osteomalacia associated with proximal renal tubulopathy, manifested as bone pain or pain in extremities and which may contribute to fractures, have been reported in association with TDF use [see Adverse Reactions (6.1)]. Arthralgia and muscle pain or weakness have also been reported in cases of proximal renal tubulopathy. Hypophosphatemia and osteomalacia secondary to proximal renal tubulopathy should be considered in patients at risk of renal dysfunction who present with persistent or worsening bone or muscle symptoms while receiving TDF-containing products [see Warnings and Precautions (5.3)].
5.6 Lactic Acidosis/Severe Hepatomegaly with Steatosis
Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported with the use of nucleoside analogs, including FTC and TDF, components of TRUVADA, alone or in combination with other antiretrovirals. Treatment with TRUVADA should be suspended in any patient or uninfected individual who develops clinical or laboratory findings suggestive of lactic acidosis or pronounced hepatotoxicity (which may include hepatomegaly and steatosis even in the absence of marked transaminase elevations).
5.7 Risk of Adverse Reactions Due to Drug Interactions
The concomitant use of TRUVADA and other drugs may result in known or potentially significant drug interactions, some of which may lead to possible clinically significant adverse reactions from greater exposures of concomitant drugs [see Drug Interactions (7.2)].
See Table 7 for steps to prevent or manage these possible and known significant drug interactions, including dosing recommendations. Consider the potential for drug interactions prior to and during therapy with TRUVADA; review concomitant medications during therapy with TRUVADA; and monitor for adverse reactions associated with the concomitant drugs.
-
6 ADVERSE REACTIONS
The following adverse reactions are discussed in other sections of the labeling:
- Severe Acute Exacerbations of Hepatitis B in Patients with HBV Infection [see Warnings and Precautions (5.1)].
- New Onset or Worsening Renal Impairment [see Warnings and Precautions (5.3)].
- Immune Reconstitution Syndrome [see Warnings and Precautions (5.4)].
- Bone Loss and Mineralization Defects [see Warnings and Precautions (5.5)].
- Lactic Acidosis/Severe Hepatomegaly with Steatosis [see Warnings and Precautions (5.6)].
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Adverse Reactions from Clinical Trials Experience in HIV-1 Infected Subjects
Clinical Trials in Adult Subjects
In Study 934, 511 antiretroviral-naïve subjects received efavirenz (EFV) administered in combination with either FTC+TDF (N=257) or zidovudine (AZT)/lamivudine (3TC) (N=254) for 144 weeks. The most common adverse reactions (incidence greater than or equal to 10%, all grades) included diarrhea, nausea, fatigue, headache, dizziness, depression, insomnia, abnormal dreams, and rash. Table 3 provides the treatment-emergent adverse reactions (Grades 2–4) occurring in greater than or equal to 5% of subjects treated in any treatment group.
Skin discoloration, manifested by hyperpigmentation, occurred in 3% of subjects taking FTC+TDF, and was generally mild and asymptomatic. The mechanism and clinical significance are unknown.
Table 3 Selected Adverse Reactions* (Grades 2–4) Reported in ≥5% in Any Treatment Group in Study 934 (0–144 Weeks) FTC+TDF+EFV† AZT/3TC+EFV N=257 N=254 - * Frequencies of adverse reactions are based on all treatment-emergent adverse events, regardless of relationship to study drug.
- † From Weeks 96 to 144 of the trial, subjects received TRUVADA with efavirenz in place of FTC+TDF with efavirenz.
- ‡ Rash event includes rash, exfoliative rash, rash generalized, rash macular, rash maculo-papular, rash pruritic, and rash vesicular.
Fatigue 9% 8% Depression 9% 7% Nausea 9% 7% Diarrhea 9% 5% Dizziness 8% 7% Upper respiratory tract infections 8% 5% Sinusitis 8% 4% Rash event‡ 7% 9% Headache 6% 5% Insomnia 5% 7% Nasopharyngitis 5% 3% Vomiting 2% 5% Laboratory Abnormalities: Laboratory abnormalities observed in this trial were generally consistent with those seen in other trials of TDF and/or FTC (Table 4).
Table 4 Significant Laboratory Abnormalities Reported in ≥1% of Subjects in Any Treatment Group in Study 934 (0–144 Weeks) FTC+TDF+EFV* AZT/3TC+EFV N=257 N=254 - * From Weeks 96 to 144 of the trial, subjects received TRUVADA with efavirenz in place of FTC+TDF with efavirenz.
Any ≥ Grade 3 Laboratory Abnormality 30% 26% Fasting Cholesterol (>240 mg/dL) 22% 24% Creatine Kinase
(M: >990 U/L)
(F: >845 U/L)9% 7% Serum Amylase (>175 U/L) 8% 4% Alkaline Phosphatase (>550 U/L) 1% 0% AST
(M: >180 U/L)
(F: >170 U/L)3% 3% ALT
(M: >215 U/L)
(F: >170 U/L)2% 3% Hemoglobin (<8.0 mg/dL) 0% 4% Hyperglycemia (>250 mg/dL) 2% 1% Hematuria (>75 RBC/HPF) 3% 2% Glycosuria (≥3+) <1% 1% Neutrophils (<750/mm3) 3% 5% Fasting Triglycerides (>750 mg/dL) 4% 2% Clinical Trials in Pediatric Subjects
Emtricitabine: In addition to the adverse reactions reported in adults, anemia and hyperpigmentation were observed in 7% and 32%, respectively, of pediatric subjects (3 months to less than 18 years of age) who received treatment with FTC in the larger of two open-label, uncontrolled pediatric trials (N=116).
Tenofovir Disoproxil Fumarate: In pediatric clinical trials (Studies 352 and 321) conducted in 184 HIV-1 infected subjects 2 to less than 18 years of age, the adverse reactions observed in pediatric subjects who received treatment with TDF were consistent with those observed in clinical trials of TDF in adults.
In Study 352 (2 to less than 12 years of age), 89 pediatric subjects received TDF for a median exposure of 104 weeks. Of these, 4 subjects discontinued from the trial due to adverse reactions consistent with proximal renal tubulopathy. Three of these 4 subjects presented with hypophosphatemia and had decreases in total body or spine BMD Z-score [see Warnings and Precautions (5.5)]. Total body BMD gain at Week 48 was less in the TDF group compared to the stavudine (d4T) or zidovudine (AZT) treatment groups. The mean rate of BMD gain in lumbar spine was similar between treatment groups. One TDF-treated subject and none of the d4T- or AZT-treated subjects experienced significant (greater than 4%) lumbar spine BMD loss at Week 48. Changes from baseline in BMD Z-scores were −0.012 for lumbar spine and −0.338 for total body in the 64 subjects who were treated with TDF for 96 weeks.
In Study 321 (12 to less than 18 years of age), the mean rate of BMD gain at Week 48 was less in the TDF compared to the placebo treatment group. Six TDF-treated subjects and one placebo-treated subject had significant (greater than 4%) lumbar spine BMD loss at Week 48. Changes from baseline BMD Z-scores were −0.341 for lumbar spine and −0.458 for total body in the 28 subjects who were treated with TDF for 96 weeks.
In both trials, skeletal growth (height) appeared to be unaffected.
Adverse Reactions from Clinical Trial Experience in Uninfected Subjects Taking TRUVADA for HIV-1 PrEP
Clinical Trials in Adult Subjects
The safety profile of TRUVADA for HIV-1 PrEP was comparable to that observed in clinical trials of HIV-infected subjects based on two randomized placebo-controlled clinical trials (iPrEx, Partners PrEP) in which 2,830 HIV-1 uninfected adults received TRUVADA once daily for HIV-1 PrEP. Subjects were followed for a median of 71 weeks and 87 weeks, respectively. Table 5 provides a list of selected adverse events that occurred in 2% or more of subjects in any treatment group in the iPrEx trial, with an incidence greater than placebo.
Table 5 Selected Adverse Events (All Grades) Reported in ≥2% in Any Treatment Group in the iPrEx Trial and Greater than Placebo FTC/TDF
(N=1251)Placebo
(N=1248)Headache 7% 6% Abdominal pain 4% 2% Weight decreased 3% 2% In the Partners PrEP trial, the frequency of adverse events in the TRUVADA treatment group was generally either less than or the same as in the placebo group.
Laboratory Abnormalities: Table 6 provides a list of Grade 2–4 laboratory abnormalities observed in the iPrEx and Partners PrEP trials. Six subjects in the TDF-containing arms of the Partners PrEP trial discontinued from the trial due to an increase in serum creatinine compared with no discontinuations in the placebo group. One subject in the TRUVADA arm of the iPrEx trial discontinued from the trial due to an increase in serum creatinine and another subject discontinued due to low serum phosphorus. Grades 2−3 proteinuria (2–4+) and/or glycosuria (3+) occurred in less than 1% of subjects treated with TRUVADA in the iPrEx trial and Partners PrEP trial.
Table 6 Laboratory Abnormalities (Highest Toxicity Grade Reported for Each Subject) in the iPrEx Trial and Partners PrEP Trial iPrEx Trial Partners PrEP Trial Grade 2–4* FTC/TDF
(N=1251)Placebo
(N=1248)FTC/TDF
(N=1579)Placebo
(N=1584)- * Grading is per DAIDS criteria.
Creatinine (>1.4 × ULN) <1% <1% <1% <1% Phosphorus (<2.0 mg/dL) 10% 8% 9% 9% AST (>2.6 × ULN) 5% 5% <1% <1% ALT (>2.6 × ULN) 7% 7% <1% <1% Hemoglobin (<9.4 mg/dL) 1% 2% 2% 2% Neutrophils (<750/mm3) <1% <1% 5% 3% Changes in Bone Mineral Density: In clinical trials of HIV-1 uninfected individuals, decreases in BMD were observed. In the iPrEx trial, a substudy of 503 subjects found mean changes from baseline in BMD ranging from –0.4% to –1.0% across total hip, spine, femoral neck, and trochanter in the TRUVADA group compared with the placebo group, which returned toward baseline after discontinuation of treatment. Thirteen percent of TRUVADA-treated subjects versus 6% of placebo-treated subjects lost at least 5% of BMD at the spine during treatment. Bone fractures were reported in 1.7% of the TRUVADA group compared with 1.4% in the placebo group. No correlation between BMD and fractures was noted [see Clinical Studies (14.3)]. The Partners PrEP trial found similar fracture rates between the treatment and placebo groups (0.8% and 0.6%, respectively); no BMD evaluations were performed in this trial [see Clinical Studies (14.4)].
Clinical Trials in Adolescent Subjects
In a single-arm, open-label clinical trial (ATN113), in which 67 HIV-1 uninfected adolescent (15 to 18 years of age) men who have sex with men received TRUVADA once daily for HIV-1 PrEP, the safety profile of TRUVADA was similar to that observed in adults. Median duration to exposure of TRUVADA was 47 weeks [see Use in Specific Populations (8.4)].
In the ATN113 trial, median BMD increased from baseline to Week 48, +2.58% for lumbar spine and +0.72% for total body. One subject had significant (greater than or equal to 4%) total body BMD loss at Week 24. Median changes from baseline BMD Z-scores were 0.0 for lumbar spine and −0.2 for total body at Week 48. Three subjects showed a worsening (change from > −2 to ≤ −2) from baseline in their lumbar spine or total body BMD Z-scores at Week 24 or 48. Interpretation of these data, however, may be limited by the low rate of adherence to TRUVADA by Week 48.
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of TDF. No additional adverse reactions have been identified during postapproval use of FTC. Because postmarketing reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Immune System Disorders
allergic reaction, including angioedemaMetabolism and Nutrition Disorders
lactic acidosis, hypokalemia, hypophosphatemiaRespiratory, Thoracic, and Mediastinal Disorders
dyspneaGastrointestinal Disorders
pancreatitis, increased amylase, abdominal painHepatobiliary Disorders
hepatic steatosis, hepatitis, increased liver enzymes (most commonly AST, ALT gamma GT)Skin and Subcutaneous Tissue Disorders
rashMusculoskeletal and Connective Tissue Disorders
rhabdomyolysis, osteomalacia (manifested as bone pain and which may contribute to fractures), muscular weakness, myopathyRenal and Urinary Disorders
acute renal failure, renal failure, acute tubular necrosis, Fanconi syndrome, proximal renal tubulopathy, interstitial nephritis (including acute cases), nephrogenic diabetes insipidus, renal insufficiency, increased creatinine, proteinuria, polyuriaGeneral Disorders and Administration Site Conditions
astheniaThe following adverse reactions, listed under the body system headings above, may occur as a consequence of proximal renal tubulopathy: rhabdomyolysis, osteomalacia, hypokalemia, muscular weakness, myopathy, hypophosphatemia.
-
7 DRUG INTERACTIONS
7.1 Drugs Affecting Renal Function
FTC and tenofovir are primarily excreted by the kidneys by a combination of glomerular filtration and active tubular secretion [see Clinical Pharmacology (12.3)]. No drug-drug interactions due to competition for renal excretion have been observed; however, coadministration of TRUVADA with drugs that are eliminated by active tubular secretion may increase concentrations of FTC, tenofovir, and/or the coadministered drug. Some examples include, but are not limited to, acyclovir, adefovir dipivoxil, cidofovir, ganciclovir, valacyclovir, valganciclovir, aminoglycosides (e.g., gentamicin), and high-dose or multiple NSAIDs [see Warnings and Precautions (5.3)]. Drugs that decrease renal function may increase concentrations of FTC and/or tenofovir.
7.2 Established and Significant Interactions
Table 7 provides a listing of established or clinically significant drug interactions. The drug interactions described are based on studies conducted with either TRUVADA, the components of TRUVADA (FTC and TDF) as individual agents and/or in combination, or are predicted drug interactions that may occur with TRUVADA [see Clinical Pharmacology (12.3)].
Table 7 Established and Significant* Drug Interactions: Alteration in Dose or Regimen May Be Recommended Based on Drug Interaction Trials Concomitant Drug Class: Drug Name Effect on Concentration Clinical Comment b. ↑=Increase, ↓=Decrease - * This table is not all inclusive.
- † Indicates that a drug-drug interaction trial was conducted.
NRTI:
didanosine†↑ didanosine Patients receiving TRUVADA and didanosine should be monitored closely for didanosine-associated adverse reactions. Discontinue didanosine in patients who develop didanosine-associated adverse reactions. Higher didanosine concentrations could potentiate didanosine-associated adverse reactions, including pancreatitis, and neuropathy. Suppression of CD4+ cell counts has been observed in patients receiving TDF with didanosine 400 mg daily.
In patients weighing greater than 60 kg, reduce the didanosine dose to 250 mg when it is coadministered with TRUVADA. Data are not available to recommend a dose adjustment of didanosine for adult or pediatric patients weighing less than 60 kg. When coadministered, TRUVADA and Videx EC may be taken under fasted conditions or with a light meal (less than 400 kcal, 20% fat).HIV-1 Protease Inhibitors:
atazanavir†↓ atazanavir When coadministered with TRUVADA, atazanavir 300 mg should be given with ritonavir 100 mg. lopinavir/ritonavir†
atazanavir/ritonavir†
darunavir/ritonavir†↑ tenofovir Monitor patients receiving TRUVADA concomitantly with lopinavir/ritonavir, ritonavir-boosted atazanavir, or ritonavir-boosted darunavir for TDF-associated adverse reactions. Discontinue TRUVADA in patients who develop TDF-associated adverse reactions. Hepatitis C Antiviral Agents:
sofosbuvir/velpatasvir†
sofosbuvir/velpatasvir/voxilaprevir†↑ tenofovir Monitor patients receiving TRUVADA concomitantly with EPCLUSA® (sofosbuvir/velpatasvir) or VOSEVI® (sofosbuvir/velpatasvir/voxilaprevir) for adverse reactions associated with TDF. ledipasvir/sofosbuvir† Monitor patients receiving TRUVADA concomitantly with HARVONI® (ledipasvir/sofosbuvir) without an HIV-1 protease inhibitor/ritonavir or an HIV-1 protease inhibitor/cobicistat combination for adverse reactions associated with TDF. In patients receiving TRUVADA concomitantly with HARVONI and an HIV-1 protease inhibitor/ritonavir or an HIV-1 protease inhibitor/cobicistat combination, consider an alternative HCV or antiretroviral therapy, as the safety of increased tenofovir concentrations in this setting has not been established. If coadministration is necessary, monitor for adverse reactions associated with TDF. -
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to TRUVADA during pregnancy. Healthcare providers are encouraged to register patients by calling the Antiretroviral Pregnancy Registry (APR) at 1-800-258-4263.
Risk Summary
Data on the use of TRUVADA during pregnancy from observational studies have shown no increased risk of major birth defects. Available data from the APR show no increase in the overall risk of major birth defects with first trimester exposure for emtricitabine (FTC) (2.3%) or tenofovir disoproxil fumarate (TDF) (2.1%) compared with the background rate for major birth defects of 2.7% in a U.S. reference population of the Metropolitan Atlanta Congenital Defects Program (MACDP) (see Data). The rate of miscarriage for individual drugs is not reported in the APR. In the U.S. general population, the estimated background risk of miscarriage in clinically recognized pregnancies is 15–20%.
In animal reproduction studies, no adverse developmental effects were observed when the components of TRUVADA were administered separately at doses/exposures ≥60 (FTC), ≥14 (TDF) and 2.7 (tenofovir) times those of the recommended daily dose of TRUVADA (see Data).
Clinical Considerations
Disease-associated maternal and/or embryo/fetal risk
HIV-1 PrEP: Published studies indicate an increased risk of HIV-1 infection during pregnancy and an increased risk of mother to child transmission during acute HIV-1 infection. In women at risk of acquiring HIV-1, consideration should be given to methods to prevent acquisition of HIV, including continuing or initiating TRUVADA for HIV-1 PrEP, during pregnancy.
Data
Human Data
TRUVADA for HIV-1 PrEP: In an observational study based on prospective reports to the APR, 78 HIV-seronegative women exposed to TRUVADA during pregnancy delivered live-born infants with no major malformations. All except for one were first trimester exposures, and the median duration of exposure was 10.5 weeks. There were no new safety findings in the women receiving TRUVADA for HIV-1 PrEP compared with HIV-1 infected women treated with other antiretroviral medications.
Emtricitabine: Based on prospective reports to the APR of 3,749 exposures to FTC-containing regimens during pregnancy resulting in live births (including 2,614 exposed in the first trimester and 1,135 exposed in the second/third trimester), there was no increase in overall major birth defects with FTC compared with the background birth defect rate of 2.7% in a U.S. reference population of the MACDP. The prevalence of major birth defects in live births was 2.3% (95% CI: 1.8% to 2.9%) with first trimester exposure to FTC-containing regimens and 2.1% (95% CI: 1.4% to 3.1%) with the second/third trimester exposure to FTC-containing regimens.
Tenofovir Disoproxil Fumarate: Based on prospective reports from the APR of 4,817 exposures to TDF-containing regimens during pregnancy resulting in live births (including 3,342 exposed in the first trimester and 1,475 exposed in the second/third trimester), there was no increase in overall major birth defects with TDF compared with the background birth defect rate of 2.7% in a U.S. reference population of the MACDP. The prevalence of major birth defects in live births was 2.3% (95% CI: 1.8% to 2.8%) with first trimester exposure to TDF-containing regimens, and 2.1% (95% CI: 1.4% to 3.0%) with the second/third trimester exposure to TDF-containing regimens.
Methodologic limitations of the APR include the use of MACDP as the external comparator group. The MACDP population is not disease-specific, evaluates women and infants from a limited geographic area, and does not include outcomes for births that occurred at <20 weeks gestation.
Additionally, published observational studies on emtricitabine and tenofovir exposure in pregnancy have not shown an increased risk for major malformations.
Animal Data
Emtricitabine: FTC was administered orally to pregnant mice (at 0, 250, 500, or 1,000 mg/kg/day), and rabbits (at 0, 100, 300, or 1,000 mg/kg/day) through organogenesis (on gestation days 6 through 15, and 7 through 19, respectively). No significant toxicological effects were observed in embryo-fetal toxicity studies performed with FTC in mice at exposures (AUC) approximately 60 times higher and in rabbits at approximately 120 times higher than human exposures at the recommended daily dose. In a pre/postnatal development study in mice, FTC was administered orally at doses up to 1,000 mg/kg/day; no significant adverse effects directly related to drug were observed in the offspring exposed daily from before birth (in utero) through sexual maturity at daily exposures (AUC) of approximately 60 times higher than human exposures at the recommended daily dose.
Tenofovir Disoproxil Fumarate: TDF was administered orally to pregnant rats (at 0, 50, 150, or 450 mg/kg/day) and rabbits (at 0, 30, 100, or 300 mg/kg/day) through organogenesis (on gestation days 7 through 17, and 6 through 18, respectively). No significant toxicological effects were observed in embryo-fetal toxicity studies performed with TDF in rats at doses up to 14 times the human dose based on body surface area comparisons and in rabbits at doses up to 19 times the human dose based on body surface area comparisons. In a pre/postnatal development study in rats, TDF was administered orally through lactation at doses up to 600 mg/kg/day; no adverse effects were observed in the offspring at tenofovir exposures of approximately 2.7 times higher than human exposures at the recommended daily dose of TRUVADA.
8.2 Lactation
Risk Summary
Based on published data, FTC and tenofovir have been shown to be present in human breast milk (see Data). It is not known if the components of TRUVADA affect milk production or have effects on the breastfed child.
Treatment of HIV-1 Infection:
The Centers for Disease Control and Prevention recommend that HIV-1 infected mothers not breastfeed their infants to avoid risking postnatal transmission of HIV-1.
Because of the potential for: (1) HIV transmission (in HIV-negative infants); (2) developing viral resistance (in HIV-positive infants); and (3) adverse reactions in a breastfed infant similar to those seen in adults, instruct mothers not to breastfeed if they are taking TRUVADA for the treatment of HIV-1.
HIV-1 PrEP:
In HIV-uninfected women, the developmental and health benefits of breastfeeding and the mother's clinical need for TRUVADA for HIV-1 PrEP should be considered along with any potential adverse effects on the breastfed child from TRUVADA and the risk of HIV-1 acquisition due to nonadherence and subsequent mother to child transmission.
Women should not breastfeed if acute HIV-1 infection is suspected because of the risk of HIV-1 transmission to the infant.
Data
HIV-1 PrEP: In a study of 50 breastfeeding women who received TRUVADA for HIV-1 PrEP between 1 and 24 weeks postpartum (median 13 weeks), after 7 days of treatment, tenofovir was undetectable but FTC was detectable in the plasma of most infants. In these infants, the average FTC plasma concentration was less than 1% of the FTC Cmax observed in HIV-infected infants (up to 3 months of age) receiving the therapeutic dose of FTC (3 mg/kg/day). There were no serious adverse events. Two infants (4%) had an adverse event of mild diarrhea which resolved.
8.4 Pediatric Use
Treatment of HIV-1 Infection
No pediatric clinical trial was conducted to evaluate the safety and efficacy of TRUVADA in patients with HIV-1 infection. Data from previously conducted trials with the individual drug products, FTC and TDF, were relied upon to support dosage recommendations for TRUVADA. For additional information, consult the prescribing information for EMTRIVA and VIREAD.
TRUVADA should only be administered to HIV-1 infected pediatric patients with body weight greater than or equal to 17 kg and who are able to swallow a tablet. Because it is a fixed-dose combination tablet, TRUVADA cannot be adjusted for patients of lower weight [see Warnings and Precautions (5.5), Adverse Reactions (6.1) and Clinical Pharmacology (12.3)]. TRUVADA is not approved for use in pediatric patients weighing less than 17 kg.
HIV-1 PrEP
The safety and effectiveness of TRUVADA for HIV-1 PrEP in at-risk adolescents weighing at least 35 kg is supported by data from adequate and well-controlled studies of TRUVADA for HIV-1 PrEP in adults with additional data from safety and pharmacokinetic studies in previously conducted trials with the individual drug products, FTC and TDF, in HIV-1 infected adults and pediatric subjects [see Dosage and Administration (2.5), Adverse Reactions (6.1), Clinical Pharmacology (12.3 and 12.4), and Clinical Studies (14.3 and 14.4)].
Safety, adherence, and resistance were evaluated in a single-arm, open-label clinical trial (ATN113) in which 67 HIV-1 uninfected at-risk adolescent men who have sex with men received TRUVADA once daily for HIV-1 PrEP. The mean age of subjects was 17 years (range 15 to 18 years); 46% were Hispanic, 52% Black, and 37% White. The safety profile of TRUVADA in ATN113 was similar to that observed in the adult HIV-1 PrEP trials [see Adverse Reactions (6.1)].
In the ATN113 trial, HIV-1 seroconversion occurred in 3 subjects. Tenofovir diphosphate levels in dried blood spot assays indicate that these subjects had poor adherence. No tenofovir- or FTC-associated HIV-1 resistance substitutions were detected in virus isolated from the 3 subjects who seroconverted [see Microbiology (12.4)].
Adherence to study drug, as demonstrated by tenofovir diphosphate levels in dried blood spot assays, declined markedly after Week 12 once subjects switched from monthly to quarterly visits, suggesting that adolescents may benefit from more frequent visits and counseling.
8.5 Geriatric Use
Clinical trials of FTC, TDF, or TRUVADA did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects.
8.6 Renal Impairment
Treatment of HIV-1 Infection
The dosing interval for TRUVADA should be modified in HIV-infected adult patients with estimated creatinine clearance of 30–49 mL/min. TRUVADA is not recommended in patients with estimated creatinine clearance below 30 mL/min and in patients with end-stage renal disease requiring dialysis [see Dosage and Administration (2.6)].
HIV-1 PrEP
TRUVADA for HIV-1 PrEP is not recommended in HIV-1 uninfected individuals with estimated creatinine clearance below 60 mL/min. If a decrease in estimated creatinine clearance is observed in uninfected individuals while using TRUVADA for HIV-1 PrEP, evaluate potential causes and re-assess potential risks and benefits of continued use [see Dosage and Administration (2.6)].
-
10 OVERDOSAGE
If overdose occurs, the patient must be monitored for evidence of toxicity, and standard supportive treatment applied as necessary.
-
11 DESCRIPTION
TRUVADA tablets are fixed-dose combination tablets containing emtricitabine (FTC) and tenofovir disoproxil fumarate (TDF). FTC is a synthetic nucleoside analog of cytidine. TDF is converted in vivo to tenofovir, an acyclic nucleoside phosphonate (nucleotide) analog of adenosine 5′-monophosphate. Both FTC and tenofovir exhibit inhibitory activity against HIV-1 reverse transcriptase.
Emtricitabine: The chemical name of FTC is 5-fluoro-1-(2R,5S)-[2-(hydroxymethyl)-1,3-oxathiolan-5-yl]cytosine. FTC is the (-) enantiomer of a thio analog of cytidine, which differs from other cytidine analogs in that it has a fluorine in the 5-position.
It has a molecular formula of C8H10FN3O3S and a molecular weight of 247.24. It has the following structural formula:
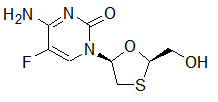
FTC is a white to off-white crystalline powder with a solubility of approximately 112 mg/mL in water at 25 °C. The partition coefficient (log p) for emtricitabine is −0.43 and the pKa is 2.65.
Tenofovir Disoproxil Fumarate: TDF is a fumaric acid salt of the bis-isopropoxycarbonyloxymethyl ester derivative of tenofovir. The chemical name of tenofovir DF is 9-[(R)-2 [[bis[[(isopropoxycarbonyl)oxy]- methoxy]phosphinyl]methoxy]propyl]adenine fumarate (1:1). It has a molecular formula of C19H30N5O10P ∙ C4H4O4 and a molecular weight of 635.52. It has the following structural formula:
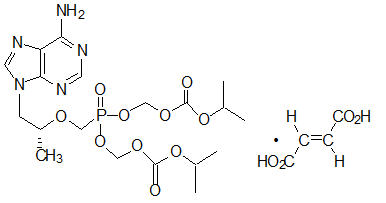
Tenofovir disoproxil fumarate is a white to off-white crystalline powder with a solubility of 13.4 mg/mL in water at 25 °C. The partition coefficient (log p) for tenofovir disoproxil is 1.25 and the pKa is 3.75. All dosages are expressed in terms of TDF except where otherwise noted.
TRUVADA tablets are for oral administration, and are available in the following strengths:
- Film-coated tablet containing 200 mg of FTC and 300 mg of TDF (which is equivalent to 245 mg of tenofovir disoproxil) as active ingredients
- Film-coated tablet containing 167 mg of FTC and 250 mg of TDF (which is equivalent to 204 mg of tenofovir disoproxil) as active ingredients
- Film-coated tablet containing 133 mg of FTC and 200 mg of TDF (which is equivalent to 163 mg of tenofovir disoproxil) as active ingredients
- Film-coated tablet containing 100 mg of FTC and 150 mg of TDF (which is equivalent to 123 mg of tenofovir disoproxil) as active ingredients
All strengths of TRUVADA tablets also include the following inactive ingredients: croscarmellose sodium, lactose monohydrate, magnesium stearate, microcrystalline cellulose, and pregelatinized starch (gluten free). The 200 mg/300 mg strength tablets are coated with Opadry II Blue Y-30-10701, which contains FD&C Blue #2 aluminum lake, hypromellose 2910, lactose monohydrate, titanium dioxide, and triacetin. The 167 mg/250 mg, 133 mg/200 mg, and 100 mg/150 mg strength tablets are coated with Opadry II Blue, which contains FD&C Blue #2 aluminum lake, hypromellose 2910, lactose monohydrate, titanium dioxide, and triacetin.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
TRUVADA is a fixed-dose combination of antiviral drugs FTC and TDF [see Microbiology (12.4)].
12.3 Pharmacokinetics
TRUVADA: One TRUVADA tablet was comparable to one FTC capsule (200 mg) plus one TDF tablet (300 mg) following single-dose administration to fasting healthy subjects (N=39).
Emtricitabine: The pharmacokinetic properties of FTC are summarized in Table 8. Following oral administration of FTC, FTC is rapidly absorbed with peak plasma concentrations occurring at 1–2 hours postdose. Less than 4% of FTC binds to human plasma proteins in vitro, and the binding is independent of concentration over the range of 0.02–200 μg/mL. Following administration of radiolabelled FTC, approximately 86% is recovered in the urine and 13% is recovered as metabolites. The metabolites of FTC include 3′-sulfoxide diastereomers and their glucuronic acid conjugate. Emtricitabine is eliminated by a combination of glomerular filtration and active tubular secretion. Following a single oral dose of FTC, the plasma FTC half-life is approximately 10 hours.
Tenofovir Disoproxil Fumarate: The pharmacokinetic properties of TDF are summarized in Table 8. Following oral administration of TDF, maximum tenofovir serum concentrations are achieved in 1.0 ± 0.4 hour. Less than 0.7% of tenofovir binds to human plasma proteins in vitro, and the binding is independent of concentration over the range of 0.01–25 µg/mL. Approximately 70–80% of the intravenous dose of tenofovir is recovered as unchanged drug in the urine. Tenofovir is eliminated by a combination of glomerular filtration and active tubular secretion. Following a single oral dose of TDF, the terminal elimination half-life of tenofovir is approximately 17 hours.
Table 8 Single Dose Pharmacokinetic Parameters for FTC and Tenofovir in Adults* FTC Tenofovir - * NC=Not calculated
- † Median (range)
- ‡ Mean (± SD)
- § Data presented as steady state values
Fasted Oral Bioavailability† (%) 92 (83.1–106.4) 25 (NC–45.0) Plasma Terminal Elimination Half-Life† (hr) 10 (7.4–18.0) 17 (12.0–25.7) Cmax‡ (μg/mL) 1.8±0.72§ 0.30±0.09 AUC‡ (μg∙hr/mL) 10.0±3.12§ 2.29±0.69 CL/F‡ (mL/min) 302±94 1043±115 CLrenal‡ (mL/min) 213±89 243±33 Effects of Food on Oral Absorption
TRUVADA may be administered with or without food. Administration of TRUVADA following a high fat meal (784 kcal; 49 grams of fat) or a light meal (373 kcal; 8 grams of fat) delayed the time of tenofovir Cmax by approximately 0.75 hour. The mean increases in tenofovir AUC and Cmax were approximately 35% and 15%, respectively, when administered with a high fat or light meal, compared to administration in the fasted state. In previous safety and efficacy trials, TDF (tenofovir) was taken under fed conditions. FTC systemic exposures (AUC and Cmax) were unaffected when TRUVADA was administered with either a high fat or a light meal.
Specific Populations
Race
Pediatric Patients
Treatment of HIV-1 Infection: The pharmacokinetic data for tenofovir and FTC following administration of TRUVADA in pediatric subjects weighing 17 kg and above are not available. The dosage recommendations of TRUVADA in this population are based on the dosage recommendations of FTC and TDF in this population. Refer to the EMTRIVA and VIREAD prescribing information for pharmacokinetic information on the individual products in pediatric patients.
HIV-1 PrEP: The pharmacokinetic data for tenofovir and FTC following administration of TRUVADA in HIV-1 uninfected adolescents weighing 35 kg and above are not available. The dosage recommendations of TRUVADA for HIV-1 PrEP in this population are based on safety and adherence data from the ATN113 trial [see Use in Specific Populations (8.4)] and known pharmacokinetic information in HIV-infected adolescents taking TDF and FTC for treatment.
Geriatric Patients
Pharmacokinetics of FTC and tenofovir have not been fully evaluated in the elderly (65 years of age and older).
Patients with Renal Impairment
The pharmacokinetics of FTC and tenofovir are altered in subjects with renal impairment [see Warnings and Precautions (5.3)]. In adult subjects with creatinine clearance below 50 mL/min, Cmax and AUC0–∞ of FTC and tenofovir were increased. No data are available to make dosage recommendations in pediatric patients with renal impairment.
Patients with Hepatic Impairment
The pharmacokinetics of tenofovir following a 300 mg dose of TDF have been studied in non-HIV infected subjects with moderate to severe hepatic impairment. There were no substantial alterations in tenofovir pharmacokinetics in subjects with hepatic impairment compared with unimpaired subjects. The pharmacokinetics of TRUVADA or FTC have not been studied in subjects with hepatic impairment; however, FTC is not significantly metabolized by liver enzymes, so the impact of liver impairment should be limited.
Assessment of Drug Interactions
The steady state pharmacokinetics of FTC and tenofovir were unaffected when FTC and TDF were administered together versus each agent dosed alone.
In vitro studies and clinical pharmacokinetic drug-drug interaction trials have shown that the potential for CYP mediated interactions involving FTC and tenofovir with other medicinal products is low.
TDF is a substrate of P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP) transporters. When TDF is coadministered with an inhibitor of these transporters, an increase in absorption may be observed.
No clinically significant drug interactions have been observed between FTC and famciclovir, indinavir, stavudine, TDF, and zidovudine (Tables 9 and 10). Similarly, no clinically significant drug interactions have been observed between TDF and efavirenz, methadone, nelfinavir, oral contraceptives, ribavirin, or sofosbuvir in trials conducted in healthy volunteers (Tables 11 and 12).
Table 9 Drug Interactions: Changes in Pharmacokinetic Parameters for FTC in the Presence of the Coadministered Drug* Coadministered Drug Dose of Coadministered Drug (mg) FTC Dose (mg) N % Change of FTC Pharmacokinetic Parameters† (90% CI) Cmax AUC Cmin - * All interaction trials conducted in healthy volunteers
- † ↑ = Increase; ⇔ = No Effect; NA = Not Applicable
TDF 300 once daily × 7 days 200 once daily × 7 days 17 ⇔ ⇔ ↑ 20
(↑ 12 to ↑ 29)Zidovudine 300 twice daily × 7 days 200 once daily × 7 days 27 ⇔ ⇔ ⇔ Indinavir 800 × 1 200 × 1 12 ⇔ ⇔ NA Famciclovir 500 × 1 200 × 1 12 ⇔ ⇔ NA Stavudine 40 × 1 200 × 1 6 ⇔ ⇔ NA Table 10 Drug Interactions: Changes in Pharmacokinetic Parameters for Coadministered Drug in the Presence of FTC* Coadministered Drug Dose of Coadministered Drug (mg) FTC Dose (mg) N % Change of Coadministered Drug Pharmacokinetic Parameters† (90% CI) Cmax AUC Cmin - * All interaction trials conducted in healthy volunteers
- † ↑ = Increase; ⇔ = No Effect; NA = Not Applicable
TDF 300 once daily × 7 days 200 once daily × 7 days 17 ⇔ ⇔ ⇔ Zidovudine 300 twice daily × 7 days 200 once daily × 7 days 27 ↑ 17
(↑ 0 to ↑ 38)↑ 13
(↑ 5 to ↑ 20)⇔ Indinavir 800 × 1 200 × 1 12 ⇔ ⇔ NA Famciclovir 500 × 1 200 × 1 12 ⇔ ⇔ NA Stavudine 40 × 1 200 × 1 6 ⇔ ⇔ NA Table 11 Drug Interactions: Changes in Pharmacokinetic Parameters for Tenofovir* in the Presence of the Coadministered Drug Coadministered Drug Dose of Coadministered Drug (mg) N % Change of Tenofovir Pharmacokinetic Parameters†
(90% CI)Cmax AUC Cmin - * Subjects received VIREAD 300 mg once daily.
- † Increase = ↑; Decrease = ↓; No Effect = ⇔
- ‡ Reyataz Prescribing Information.
- § Prezista Prescribing Information.
- ¶ Data generated from simultaneous dosing with HARVONI (ledipasvir/sofosbuvir). Staggered administration (12 hours apart) provided similar results.
- # Comparison based on exposures when administered as atazanavir/ritonavir + FTC/TDF.
- Þ Comparison based on exposures when administered as darunavir/ritonavir + FTC/TDF.
- ß Study conducted with ATRIPLA (efavirenz/FTC/TDF) coadministered with HARVONI.
- à Study conducted with COMPLERA (FTC/rilpivirine/TDF) coadministered with HARVONI.
- è Study conducted with TRUVADA (FTC/TDF) + dolutegravir coadministered with HARVONI.
- ð Study conducted with ATRIPLA coadministered with SOVALDI® (sofosbuvir).
- ø Study conducted with COMPLERA coadministered with EPCLUSA; coadministration with EPCLUSA also results in comparable increases in tenofovir exposures when TDF is administered as ATRIPLA, STRIBILD, TRUVADA + atazanavir/ritonavir, or TRUVADA + darunavir/ritonavir.
- ý Administered as raltegravir + FTC/TDF.
- £ Comparison based on exposures when administered as darunavir + ritonavir + FTC/TDF.
- ¥ Study conducted with additional voxilaprevir 100 mg to achieve voxilaprevir exposures expected in HCV-infected patients
- ΠAptivus Prescribing Information.
Atazanavir‡ 400 once daily × 14 days 33 ↑ 14
(↑ 8 to ↑ 20)↑ 24
(↑ 21 to ↑ 28)↑ 22
(↑ 15 to ↑ 30)Atazanavir/ Ritonavir‡ 300/100 once daily 12 ↑ 34
(↑ 20 to ↑ 51)↑ 37
(↑ 30 to ↑ 45)↑ 29
(↑ 21 to ↑ 36)Darunavir/ Ritonavir§ 300/100 twice daily 12 ↑ 24
(↑ 8 to ↑ 42)↑ 22
(↑ 10 to ↑ 35)↑ 37
(↑ 19 to ↑ 57)Indinavir 800 three times daily × 7 days 13 ↑ 14
(↓ 3 to ↑ 33)⇔ ⇔ Ledipasvir/ Sofosbuvir¶, # 90/400 once daily × 10 days 24 ↑ 47
(↑ 37 to ↑ 58)↑ 35
(↑ 29 to ↑42 )↑ 47
(↑ 38 to ↑ 57)Ledipasvir/ Sofosbuvir¶, Þ 23 ↑ 64
(↑ 54 to ↑ 74)↑ 50
(↑ 42 to ↑ 59)↑ 59
(↑ 49 to ↑ 70)Ledipasvir/ Sofosbuvirß 90/400 once daily × 14 days 15 ↑ 79
(↑ 56 to ↑ 104)↑ 98
(↑ 77 to ↑ 123)↑ 163
(↑ 132 to ↑ 197)Ledipasvir/ Sofosbuvirà 90/400 once daily × 10 days 14 ↑ 32
(↑ 25 to ↑ 39 )↑ 40
(↑ 31 to ↑ 50 )↑ 91
(↑ 74 to ↑ 110)Ledipasvir/ Sofosbuvirè 90/400 once daily × 10 days 29 ↑ 61
(↑ 51 to ↑ 72)↑ 65
(↑ 59 to ↑ 71)↑ 115
(↑ 105 to ↑ 126)Lopinavir/ Ritonavir 400/100 twice daily × 14 days 24 ⇔ ↑ 32
(↑ 25 to ↑ 38)↑ 51
(↑ 37 to ↑ 66)Saquinavir/ Ritonavir 1000/100 twice daily × 14 days 35 ⇔ ⇔ ↑ 23
(↑ 16 to ↑ 30)Sofosbuvirð 400 single dose 16 ↑ 25
(↑ 8 to ↑ 45)⇔ ⇔ Sofosbuvir/ Velpatasvirø 400/100 once daily 24 ↑ 44
(↑ 33 to ↑ 55)↑ 40
(↑ 34 to ↑ 46)↑ 84
(↑ 76 to ↑ 92)Sofosbuvir/ Velpatasvirý 400/100 once daily 30 ↑ 46
(↑ 39 to ↑ 54)↑ 40
(↑ 34 to ↑ 45)↑ 70
(↑ 61 to ↑ 79)Sofosbuvir/ Velpatasvir/Voxilaprevir£ 400/100/100 + Voxilaprevir¥ 100 once daily 29 ↑ 48
(↑ 36 to ↑ 61)↑ 39
(↑ 32 to ↑ 46)↑ 47
(↑ 38 to ↑ 56)Tacrolimus 0.05 mg/kg twice daily × 7 days 21 ↑ 13
(↑ 1 to ↑ 27)⇔ ⇔ Tipranavir/ RitonavirŒ 500/100 twice daily 22 ↓ 23
(↓ 32 to ↓ 13)↓ 2
(↓ 9 to ↑ 5)↑ 7
(↓ 2 to ↑ 17)750/200 twice daily (23 doses) 20 ↓ 38
(↓ 46 to ↓ 29)↑ 2
(↓ 6 to ↑ 10)↑ 14
(↑ 1 to ↑ 27)No effect on the pharmacokinetic parameters of the following coadministered drugs was observed with TRUVADA: abacavir, didanosine (buffered tablets), FTC, entecavir, and lamivudine.
Table 12 Drug Interactions: Changes in Pharmacokinetic Parameters for Coadministered Drug in the Presence of Tenofovir Coadministered Drug Dose of Coadministered Drug (mg) N % Change of Coadministered Drug Pharmacokinetic Parameters*
(90% CI)Cmax AUC Cmin - * Increase = ↑; Decrease = ↓; No Effect = ⇔; NA = Not Applicable
- † Reyataz Prescribing Information.
- ‡ In HIV-infected subjects, addition of TDF to atazanavir 300 mg plus ritonavir 100 mg resulted in AUC and Cmin values of atazanavir that were 2.3- and 4-fold higher than the respective values observed for atazanavir 400 mg when given alone.
- § Prezista Prescribing Information.
- ¶ Videx EC Prescribing Information. Subjects received didanosine enteric-coated capsules. When didanosine 250 mg enteric-coated capsules were administered with TDF, systemic exposures of didanosine were similar to those seen with the 400 mg enteric-coated capsules alone under fasted conditions.
- # 373 kcal, 8.2 g fat
- Þ Compared with didanosine (enteric-coated) 400 mg administered alone under fasting conditions.
- ß Increases in AUC and Cmin are not expected to be clinically relevant; hence, no dose adjustments are required when TDF and ritonavir-boosted saquinavir are coadministered.
- à Aptivus Prescribing Information.
Abacavir 300 once 8 ↑ 12
(↓ 1 to ↑ 26)⇔ NA Atazanavir† 400 once daily × 14 days 34 ↓ 21
(↓ 27 to ↓ 14)↓ 25
(↓ 30 to ↓ 19)↓ 40
(↓ 48 to ↓ 32)Atazanavir† Atazanavir/Ritonavir 300/100 once daily × 42 days 10 ↓ 28
(↓ 50 to ↑ 5)↓ 25‡
(↓ 42 to ↓ 3)↓ 23‡
(↓ 46 to ↑ 10)Darunavir§ Darunavir/Ritonavir 300/100 once daily 12 ↑ 16
(↓ 6 to ↑ 42)↑ 21
(↓ 5 to ↑ 54)↑ 24
(↓ 10 to ↑ 69)Didanosine¶ 250 once, simultaneously with TDF and a light meal# 33 ↓ 20Þ
(↓ 32 to ↓ 7)⇔Þ NA Emtricitabine 200 once daily × 7 days 17 ⇔ ⇔ ↑ 20
(↑ 12 to ↑ 29)Indinavir 800 three times daily × 7 days 12 ↓ 11
(↓ 30 to ↑ 12)⇔ ⇔ Entecavir 1 once daily × 10 days 28 ⇔ ↑ 13
(↑ 11 to ↑ 15)⇔ Lamivudine 150 twice daily × 7 days 15 ↓ 24
(↓ 34 to ↓ 12)⇔ ⇔ Lopinavir Lopinavir/Ritonavir 400/100 twice daily × 14 days 24 ⇔ ⇔ ⇔ Ritonavir ⇔ ⇔ ⇔ Saquinavir Saquinavir/Ritonavir 1000/100 twice daily × 14 days 32 ↑ 22
(↑ 6 to ↑41)↑ 29ß
(↑ 12 to ↑ 48)↑ 47ß
(↑ 23 to ↑ 76)Ritonavir ⇔ ⇔ ↑ 23
(↑ 3 to ↑ 46)Tacrolimus 0.05 mg/kg twice daily × 7 days 21 ⇔ ⇔ ⇔ Tipranavirà Tipranavir/Ritonavir 500/100 twice daily 22 ↓ 17
(↓ 26 to ↓ 6)↓ 18
(↓ 25 to ↓ 9)↓ 21
(↓ 30 to ↓ 10)Tipranavir/Ritonavir 750/200 twice daily (23 doses) 20 ↓ 11
(↓ 16 to ↓ 4)↓ 9
(↓ 15 to ↓ 3)↓ 12
(↓ 22 to 0)12.4 Microbiology
Mechanism of Action
Emtricitabine: FTC, a synthetic nucleoside analog of cytidine, is phosphorylated by cellular enzymes to form emtricitabine 5'-triphosphate (FTC-TP), which inhibits the activity of the HIV-1 reverse transcriptase (RT) by competing with the natural substrate deoxycytidine 5'-triphosphate and by being incorporated into nascent viral DNA which results in chain termination. FTC-TP is a weak inhibitor of mammalian DNA polymerases α, β, ε and mitochondrial DNA polymerase γ.
Tenofovir Disoproxil Fumarate: TDF is an acyclic nucleoside phosphonate diester analog of adenosine monophosphate. TDF requires initial diester hydrolysis for conversion to tenofovir and subsequent phosphorylations by cellular enzymes to form tenofovir diphosphate (TFV-DP), which inhibits the activity of HIV-1 RT by competing with the natural substrate deoxyadenosine 5′-triphosphate and, after incorporation into DNA, by DNA chain termination. TFV-DP is a weak inhibitor of mammalian DNA polymerases α, β, and mitochondrial DNA polymerase γ.
Antiviral Activity
Emtricitabine and Tenofovir Disoproxil Fumarate: No antagonism was observed in combination studies evaluating the cell culture antiviral activity of FTC and tenofovir together.
Emtricitabine: The antiviral activity of FTC against laboratory and clinical isolates of HIV-1 was assessed in lymphoblastoid cell lines, the MAGI-CCR5 cell line, and peripheral blood mononuclear cells. The 50% effective concentration (EC50) values for FTC were in the range of 0.0013–0.64 µM (0.0003–0.158 µg/mL). In drug combination studies of FTC with nucleoside RT inhibitors (abacavir, lamivudine, stavudine, zidovudine), non-nucleoside RT inhibitors (delavirdine, efavirenz, nevirapine), and protease inhibitors (amprenavir, nelfinavir, ritonavir, saquinavir), no antagonism was observed. Emtricitabine displayed antiviral activity in cell culture against HIV-1 clades A, B, C, D, E, F, and G (EC50 values ranged from 0.007–0.075 µM) and showed strain-specific activity against HIV-2 (EC50 values ranged from 0.007–1.5 µM).
Tenofovir Disoproxil Fumarate: The antiviral activity of tenofovir against laboratory and clinical isolates of HIV-1 was assessed in lymphoblastoid cell lines, primary monocyte/macrophage cells, and peripheral blood lymphocytes. The EC50 values for tenofovir were in the range of 0.04–8.5 µM. In drug combination studies of tenofovir with nucleoside RT inhibitors (abacavir, didanosine, lamivudine, stavudine, zidovudine), non-nucleoside RT inhibitors (delavirdine, efavirenz, nevirapine), and protease inhibitors (amprenavir, indinavir, nelfinavir, ritonavir, saquinavir), no antagonism was observed. Tenofovir displayed antiviral activity in cell culture against HIV-1 clades A, B, C, D, E, F, G, and O (EC50 values ranged from 0.5–2.2 µM) and showed strain-specific activity against HIV-2 (EC50 values ranged from 1.6 µM to 5.5 µM).
Prophylactic Activity in a Nonhuman Primate Model of HIV-1 Transmission
Emtricitabine and Tenofovir Disoproxil Fumarate: The prophylactic activity of the combination of daily oral FTC and TDF was evaluated in a controlled study of macaques inoculated once weekly for 14 weeks with SIV/HIV-1 chimeric virus (SHIV) applied to the rectal surface. Of the 18 control animals, 17 became infected after a median of 2 weeks. In contrast, 4 of the 6 animals treated daily with oral FTC and TDF remained uninfected and the two infections that did occur were significantly delayed until 9 and 12 weeks and exhibited reduced viremia. An M184I-expressing FTC-resistant variant emerged in 1 of the 2 macaques after 3 weeks of continued drug exposure.
Resistance
Emtricitabine and Tenofovir Disoproxil Fumarate: HIV-1 isolates with reduced susceptibility to the combination of FTC and tenofovir have been selected in cell culture. Genotypic analysis of these isolates identified the M184V/I and/or K65R amino acid substitutions in the viral RT. In addition, a K70E substitution in the HIV-1 RT has been selected by tenofovir and results in reduced susceptibility to tenofovir.
In Study 934, a clinical trial of treatment-naïve subjects [see Clinical Studies (14.2)], resistance analysis was performed on HIV-1 isolates from all confirmed virologic failure subjects with greater than 400 copies/mL of HIV-1 RNA at Week 144 or early discontinuation. Development of efavirenz resistance-associated substitutions occurred most frequently and was similar between the treatment arms. The M184V amino acid substitution, associated with resistance to FTC and lamivudine, was observed in 2/19 analyzed subject isolates in the FTC+TDF group and in 10/29 analyzed subject isolates in the zidovudine/lamivudine group. Through 144 weeks of Study 934, no subjects have developed a detectable K65R or K70E substitution in their HIV-1 as analyzed through standard genotypic analysis.
Emtricitabine: FTC-resistant isolates of HIV-1 have been selected in cell culture and in vivo. Genotypic analysis of these isolates showed that the reduced susceptibility to FTC was associated with a substitution in the HIV-1 RT gene at codon 184 which resulted in an amino acid substitution of methionine by valine or isoleucine (M184V/I).
Tenofovir Disoproxil Fumarate: HIV-1 isolates with reduced susceptibility to tenofovir have been selected in cell culture. These viruses expressed a K65R substitution in RT and showed a 2- to 4-fold reduction in susceptibility to tenofovir.
In treatment-naïve subjects, isolates from 8/47 (17%) analyzed subjects developed the K65R substitution in the TDF arm through 144 weeks; 7 occurred in the first 48 weeks of treatment and 1 at Week 96. In treatment-experienced subjects, 14/304 (5%) isolates from subjects failing TDF through Week 96 showed greater than 1.4-fold (median 2.7) reduced susceptibility to tenofovir. Genotypic analysis of the resistant isolates showed a K65R amino acid substitution in the HIV-1 RT.
iPrEx Trial: In the iPrEx trial, a clinical trial of HIV-1 seronegative adult subjects [see Clinical Studies (14.3)], no amino acid substitutions associated with resistance to FTC or TDF were detected at the time of seroconversion among 48 subjects in the TRUVADA group and 83 subjects in the placebo group who became infected with HIV-1 during the trial. Ten subjects were observed to be HIV-1 infected at time of enrollment. The M184V/I substitutions associated with resistance to FTC were observed in 3 of the 10 subjects (2 of 2 in the TRUVADA group and 1 of 8 in the placebo group). One of the two subjects in the TRUVADA group harbored wild type virus at enrollment and developed the M184V substitution 4 weeks after enrollment. The other subject had indeterminate resistance at enrollment but was found to have the M184I substitution 4 weeks after enrollment.
Partners PrEP Trial: In the Partners PrEP trial, a clinical trial of HIV-1 seronegative adult subjects [see Clinical Studies (14.4)], no variants expressing amino acid substitutions associated with resistance to FTC or TDF were detected at the time of seroconversion among 12 subjects in the TRUVADA group, 15 subjects in the TDF group, and 51 subjects in the placebo group. Fourteen subjects were observed to be HIV-1 infected at the time of enrollment (3 in the TRUVADA group, 5 in the TDF group, and 6 in the placebo group). One of the three subjects in the TRUVADA group who was infected with wild type virus at enrollment selected an M184V expressing virus by Week 12. Two of the five subjects in the TDF group had tenofovir-resistant viruses at the time of seroconversion; one subject infected with wild type virus at enrollment developed a K65R substitution by Week 16, while the second subject had virus expressing the combination of D67N and K70R substitutions upon seroconversion at Week 60, although baseline virus was not genotyped and it is unclear if the resistance emerged or was transmitted. Following enrollment, 4 subjects (2 in the TDF group, 1 in the TRUVADA group, and 1 in the placebo group) had virus expressing K103N or V106A substitutions, which confer high-level resistance to NNRTIs but have not been associated with FTC or TDF and may have been present in the infecting virus.
ATN113 Trial: In ATN113, a clinical trial of HIV-1 seronegative adolescent subjects [see Use in Specific Populations (8.4)], no amino acid substitutions associated with resistance to FTC or TDF were detected at the time of seroconversion from any of the 3 subjects who became infected with HIV-1 during the trial. All 3 subjects who seroconverted were nonadherent to the recommended TRUVADA dosage.
Cross Resistance
Emtricitabine and Tenofovir Disoproxil Fumarate: Cross-resistance among certain NRTIs has been recognized. The M184V/I and/or K65R substitutions selected in cell culture by the combination of FTC and tenofovir are also observed in some HIV-1 isolates from subjects failing treatment with tenofovir in combination with either FTC or lamivudine, and either abacavir or didanosine. Therefore, cross-resistance among these drugs may occur in patients whose virus harbors either or both of these amino acid substitutions.
Emtricitabine: FTC-resistant isolates (M184V/I) were cross-resistant to lamivudine but retained susceptibility in cell culture to the NRTIs didanosine, stavudine, tenofovir, and zidovudine, and to NNRTIs (delavirdine, efavirenz, and nevirapine). HIV-1 isolates containing the K65R substitution, selected in vivo by abacavir, didanosine, and tenofovir, demonstrated reduced susceptibility to inhibition by FTC. Viruses harboring substitutions conferring reduced susceptibility to stavudine and zidovudine (M41L, D67N, K70R, L210W, T215Y/F, K219Q/E), or didanosine (L74V) remained sensitive to FTC. HIV-1 containing the K103N substitution associated with resistance to NNRTIs was susceptible to FTC.
Tenofovir Disoproxil Fumarate: The K65R and K70E substitutions selected by tenofovir are also selected in some HIV-1 infected patients treated with abacavir or didanosine. HIV-1 isolates with the K65R and K70E substitutions also showed reduced susceptibility to FTC and lamivudine. Therefore, cross-resistance among these NRTIs may occur in patients whose virus harbors the K65R or K70E substitutions. HIV-1 isolates from subjects (N=20) whose HIV-1 expressed a mean of 3 zidovudine-associated RT amino acid substitutions (M41L, D67N, K70R, L210W, T215Y/F, or K219Q/E/N) showed a 3.1-fold decrease in the susceptibility to tenofovir. Subjects whose virus expressed an L74V substitution without zidovudine resistance-associated substitutions (N=8) had reduced response to TDF. Limited data are available for patients whose virus expressed a Y115F substitution (N=3), Q151M substitution (N=2), or T69 insertion (N=4), all of whom had a reduced response.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Emtricitabine: In long-term oral carcinogenicity studies of FTC, no drug-related increases in tumor incidence were found in mice at doses up to 750 mg/kg/day (26 times the human systemic exposure at the therapeutic dose of 200 mg/day) or in rats at doses up to 600 mg/kg/day (31 times the human systemic exposure at the therapeutic dose).
FTC was not genotoxic in the reverse mutation bacterial test (Ames test), or the mouse lymphoma or mouse micronucleus assays.
FTC did not affect fertility in male rats at approximately 140-fold or in male and female mice at approximately 60-fold higher exposures (AUC) than in humans given the recommended 200 mg daily dose. Fertility was normal in the offspring of mice exposed daily from before birth (in utero) through sexual maturity at daily exposures (AUC) of approximately 60-fold higher than human exposures at the recommended 200 mg daily dose.
Tenofovir Disoproxil Fumarate: Long-term oral carcinogenicity studies of TDF in mice and rats were carried out at exposures up to approximately 16 times (mice) and 5 times (rats) those observed in humans at the therapeutic dose for HIV-1 infection. At the high dose in female mice, liver adenomas were increased at exposures 16 times that in humans. In rats, the study was negative for carcinogenic findings at exposures up to 5 times that observed in humans at the therapeutic dose.
TDF was mutagenic in the in vitro mouse lymphoma assay and negative in an in vitro bacterial mutagenicity test (Ames test). In an in vivo mouse micronucleus assay, TDF was negative when administered to male mice.
There were no effects on fertility, mating performance, or early embryonic development when TDF was administered to male rats at a dose equivalent to 10 times the human dose based on body surface area comparisons for 28 days prior to mating and to female rats for 15 days prior to mating through day 7 of gestation. There was, however, an alteration of the estrous cycle in female rats.
13.2 Animal Toxicology and/or Pharmacology
Tenofovir and TDF administered in toxicology studies to rats, dogs, and monkeys at exposures (based on AUCs) greater than or equal to 6-fold those observed in humans caused bone toxicity. In monkeys the bone toxicity was diagnosed as osteomalacia. Osteomalacia observed in monkeys appeared to be reversible upon dose reduction or discontinuation of tenofovir. In rats and dogs, the bone toxicity manifested as reduced bone mineral density. The mechanism(s) underlying bone toxicity is unknown.
Evidence of renal toxicity was noted in four animal species. Increases in serum creatinine, BUN, glycosuria, proteinuria, phosphaturia, and/or calciuria and decreases in serum phosphate were observed to varying degrees in these animals. These toxicities were noted at exposures (based on AUCs) 2–20 times higher than those observed in humans. The relationship of the renal abnormalities, particularly the phosphaturia, to the bone toxicity is not known.
-
14 CLINICAL STUDIES
14.1 Overview of Clinical Trials
The efficacy and safety of TRUVADA have been evaluated in the studies summarized in Table 13.
Table 13 Trials Conducted with TRUVADA for HIV-1 Treatment and HIV-1 PrEP Trial Population Study Arms (N)* Timepoint - * Randomized and dosed.
- † Randomized, open label, active-controlled trial.
- ‡ Randomized, double-blind, placebo-controlled trial.
Study 934† (NCT00112047) HIV-infected, treatment-naïve adults FTC+TDF + efavirenz (257) zidovudine/lamivudine + efavirenz (254) 48 Weeks iPrEx‡
(NCT00458393)HIV-seronegative men or transgender women who have sex with men TRUVADA (1,251)
Placebo (1,248)4,237 person-years Partners PrEP‡
(NCT00557245)HIV serodiscordant heterosexual couples TRUVADA (1,583)
Placebo (1,586)7,827 person-years 14.2 Clinical Trial Results for Treatment of HIV-1: Study 934
Data through 144 weeks are reported for Study 934, a randomized, open-label, active-controlled multicenter trial comparing FTC+TDF administered in combination with efavirenz (EFV) versus zidovudine (AZT)/lamivudine (3TC) fixed-dose combination administered in combination with EFV in 511 antiretroviral-naïve adult subjects. From Weeks 96 to 144 of the trial, subjects received TRUVADA with EFV in place of FTC+TDF with EFV. Subjects had a mean age of 38 years (range 18–80); 86% were male, 59% were Caucasian, and 23% were Black. The mean baseline CD4+ cell count was 245 cells/mm3 (range 2–1,191) and median baseline plasma HIV-1 RNA was 5.01 log10 copies/mL (range 3.56–6.54). Subjects were stratified by baseline CD4+ cell count (< or ≥200 cells/mm3); 41% had CD4+ cell counts <200 cells/mm3 and 51% of subjects had baseline viral loads >100,000 copies/mL. Treatment outcomes through 48 and 144 weeks for those subjects who did not have EFV resistance at baseline are presented in Table 14.
Table 14 Virologic Outcomes of Randomized Treatment at Weeks 48 and 144 (Study 934) Outcomes At Week 48 At Week 144 FTC+TDF +EFV
(N=244)AZT/3TC +EFV
(N=243)FTC+TDF +EFV
(N=227)*AZT/3TC +EFV
(N=229)*- * Subjects who were responders at Week 48 or Week 96 (HIV-1 RNA <400 copies/mL) but did not consent to continue trial after Week 48 or Week 96 were excluded from analysis.
- † Subjects achieved and maintained confirmed HIV-1 RNA <400 copies/mL through Weeks 48 and 144.
- ‡ Includes confirmed viral rebound and failure to achieve confirmed <400 copies/mL through Weeks 48 and 144.
- § Includes lost to follow-up, subject withdrawal, noncompliance, protocol violation, and other reasons.
Responder† 84% 73% 71% 58% Virologic failure‡ 2% 4% 3% 6% Rebound 1% 3% 2% 5% Never suppressed 0% 0% 0% 0% Change in antiretroviral regimen 1% 1% 1% 1% Death <1% 1% 1% 1% Discontinued due to adverse event 4% 9% 5% 12% Discontinued for other reasons§ 10% 14% 20% 22% Through Week 48, 84% and 73% of subjects in the FTC+TDF group and the AZT/3TC group, respectively, achieved and maintained HIV-1 RNA <400 copies/mL (71% and 58% through Week 144). The difference in the proportion of subjects who achieved and maintained HIV-1 RNA <400 copies/mL through 48 weeks is largely due to the higher number of discontinuations due to adverse events and other reasons in the AZT/3TC group in this open-label trial. In addition, 80% and 70% of subjects in the FTC+TDF group and the AZT/3TC group, respectively, achieved and maintained HIV-1 RNA <50 copies/mL through Week 48 (64% and 56% through Week 144). The mean increase from baseline in CD4+ cell count was 190 cells/mm3 in the FTC+TDF group and 158 cells/mm3 in the AZT/3TC group at Week 48 (312 and 271 cells/mm3 at Week 144).
Through 48 weeks, 7 subjects in the FTC+TDF group and 5 subjects in the AZT/3TC group experienced a new CDC Class C event (10 and 6 subjects through 144 weeks).
14.3 Clinical Trial Results for HIV-1 PrEP: iPrEx
The iPrEx trial was a randomized, double-blind, placebo-controlled multinational study evaluating TRUVADA in 2,499 HIV-seronegative men or transgender women who have sex with men and with evidence of high-risk behavior for HIV-1 infection. Evidence of high-risk behavior included any one of the following reported to have occurred up to six months prior to study screening: no condom use during anal intercourse with an HIV-1 positive partner or a partner of unknown HIV status; anal intercourse with more than 3 sex partners; exchange of money, gifts, shelter, or drugs for anal sex; sex with male partner and diagnosis of sexually transmitted infection; no consistent use of condoms with sex partner known to be HIV-1 positive.
All subjects received monthly HIV-1 testing, risk-reduction counseling, condoms, and management of sexually transmitted infections. Of the 2,499 enrolled subjects, 1,251 received TRUVADA and 1,248 received placebo. The mean age of subjects was 27 years; 5% were Asian, 9% Black, 18% White, and 72% Hispanic/Latino.
Subjects were followed for 4,237 person-years. The primary outcome measure was the incidence of documented HIV seroconversion. At the end of treatment, emergent HIV-1 seroconversion was observed in 131 subjects, of which 48 occurred in the TRUVADA group and 83 occurred in the placebo group, indicating a 42% (95% CI: 18–60%) reduction in risk. Risk reduction was found to be higher (53%; 95% CI: 34–72%) among subjects who reported previous unprotected anal intercourse (URAI) at screening (732 and 753 subjects reported URAI within the last 12 weeks at screening in the TRUVADA and placebo groups, respectively). In a post-hoc case control study of plasma and intracellular drug levels in about 10% of study subjects, risk reduction appeared to be greatest in subjects with detectable intracellular tenofovir diphosphate concentrations. Efficacy was therefore strongly correlated with adherence.
14.4 Clinical Trial Results for HIV-1 PrEP: Partners PrEP
The Partners PrEP trial was a randomized, double-blind, placebo-controlled 3-arm trial conducted in 4,758 HIV-1 serodiscordant heterosexual couples in Kenya and Uganda to evaluate the efficacy and safety of TDF (N=1,589) and FTC/TDF (N=1,583) versus (parallel comparison) placebo (N=1,586) in preventing HIV-1 acquisition by the uninfected partner.
All uninfected partner subjects received monthly HIV-1 testing, evaluation of adherence, assessment of sexual behavior, and safety evaluations. Women were also tested monthly for pregnancy. Women who became pregnant during the trial had study drug interrupted for the duration of the pregnancy and while breastfeeding. The uninfected partner subjects were predominantly male (61–64% across study drug groups) and had a mean age of 33–34 years.
Following 7,827 person-years of follow-up, 82 emergent HIV-1 seroconversions were reported, with an overall observed seroincidence rate of 1.05 per 100 person-years. Of the 82 seroconversions, 13 and 52 occurred in partner subjects randomized to TRUVADA and placebo, respectively. Two of the 13 seroconversions in the TRUVADA arm and 3 of the 52 seroconversions in the placebo arm occurred in women during treatment interruptions for pregnancy. The risk reduction for TRUVADA relative to placebo was 75% (95% CI: 55–87%). In a post-hoc case control study of plasma drug levels in about 10% of study subjects, risk reduction appeared to be greatest in subjects with detectable plasma tenofovir concentrations. Efficacy was therefore strongly correlated with adherence.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
TRUVADA tablets are available in bottles containing 30 tablets with child-resistant closure as follows:
- 100 mg of FTC and 150 mg of TDF (equivalent to 123 mg of tenofovir disoproxil) tablets are blue, oval shaped, film coated, debossed with "GSI" on one side and with "703" on the other side (NDC: 61958-0703-1).
- 133 mg of FTC and 200 mg of TDF (equivalent to 163 mg of tenofovir disoproxil) tablets are blue, rectangular shaped, film coated, debossed with "GSI" on one side and with "704" on the other side (NDC: 61958-0704-1).
- 167 mg of FTC and 250 mg of TDF (equivalent to 204 mg of tenofovir disoproxil) tablets are blue, modified capsule shaped, film coated, debossed with "GSI" on one side and with "705" on the other side (NDC: 61958-0705-1).
- 200 mg of FTC and 300 mg of TDF (equivalent to 245 mg of tenofovir disoproxil) tablets are blue, capsule shaped, film coated, debossed with "GILEAD" on one side and with "701" on the other side (NDC: 61958-0701-1).
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Important Information for Uninfected Individuals Taking TRUVADA for HIV-1 PrEP
Encourage use of the Agreement Form for Initiating TRUVADA for PrEP of Sexually Acquired HIV-1 Infection.
In addition, advise HIV-uninfected individuals about the following [see Warnings and Precautions (5.2)]:
- The need to confirm that they are HIV-negative before starting to take TRUVADA to reduce the risk of acquiring HIV-1.
- That HIV-1 resistance substitutions may emerge in individuals with undetected HIV-1 infection who are taking TRUVADA, because TRUVADA alone does not constitute a complete regimen for HIV-1 treatment.
- The importance of taking TRUVADA on a regular dosing schedule and to strictly adhere to the recommended dosing schedule to reduce the risk of acquiring HIV-1. Uninfected individuals who miss doses are at greater risk of acquiring HIV-1 than those who do not miss doses.
- That TRUVADA should only be used as part of a complete prevention strategy including other prevention measures.
- To use condoms consistently and correctly to lower the chances of sexual contact with any body fluids such as semen, vaginal secretions, or blood.
- The importance of knowing their HIV status and the status of their partner(s).
- The importance of virologic suppression in their partner(s) with HIV-1.
- The need to get tested regularly for HIV-1 (at least every 3 months, or more frequently for some individuals such as adolescents) and to ask their partner(s) to get tested as well.
- To report any symptoms of acute HIV-1 infection (flu-like symptoms) to their healthcare provider immediately.
- That the signs and symptoms of acute infection include fever, headache, fatigue, arthralgia, vomiting, myalgia, diarrhea, pharyngitis, rash, night sweats, and adenopathy (cervical and inguinal).
- To get tested for other sexually transmitted infections, such as syphilis, chlamydia, and gonorrhea, that may facilitate HIV-1 transmission.
- To assess their sexual risk behavior and get support to help reduce sexual risk behavior.
Severe Acute Exacerbation of Hepatitis B in Patients Infected with HBV
Inform patients that severe acute exacerbations of hepatitis B have been reported in patients infected with hepatitis B virus (HBV) who have discontinued TRUVADA. Advise patients not to discontinue TRUVADA without first informing their healthcare provider. All patients should be tested for HBV infection before or when starting TRUVADA and those who are infected with HBV need close medical follow-up for several months after stopping TRUVADA to monitor for exacerbations of hepatitis [see Warnings and Precautions (5.1)].
New Onset or Worsening Renal Impairment
Inform patients that renal impairment, including cases of acute renal failure and Fanconi syndrome, has been reported in association with the use of TDF, a component of TRUVADA. Advise patients to avoid TRUVADA with concurrent or recent use of a nephrotoxic agent (e.g., high-dose or multiple NSAIDs) [see Warnings and Precautions (5.3)]. The dosing interval of TRUVADA may need adjustment in HIV-1 infected patients with renal impairment. TRUVADA for HIV-1 PrEP should not be used in HIV-1 uninfected individuals if estimated creatinine clearance is less than 60 mL/min. If a decrease in estimated creatinine clearance is observed in uninfected individuals while using TRUVADA for HIV-1 PrEP, evaluate potential causes and re-assess potential risks and benefits of continued use [see Dosage and Administration (2.6)].
Immune Reconstitution Syndrome
Inform patients that in some patients with advanced HIV infection (AIDS), signs and symptoms of inflammation from previous infections may occur soon after anti-HIV treatment is started. It is believed that these symptoms are due to an improvement in the body's immune response, enabling the body to fight infections that may have been present with no obvious symptoms. Advise patients to inform their healthcare provider immediately of any symptoms of infection [see Warnings and Precautions (5.4)].
Bone Loss and Mineralization Defects
Inform patients that decreases in bone mineral density have been observed with the use of TDF or TRUVADA. Consider bone monitoring in patients and uninfected individuals who have a history of pathologic bone fracture or at risk for osteopenia [see Warnings and Precautions (5.5)].
Lactic Acidosis and Severe Hepatomegaly
Inform patients and uninfected individuals that lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported. Treatment with TRUVADA should be suspended in any person who develops clinical symptoms suggestive of lactic acidosis or pronounced hepatotoxicity [see Warnings and Precautions (5.6)].
Drug Interactions
Advise patients that TRUVADA may interact with many drugs; therefore, advise patients to report to their healthcare provider the use of any other medication, including other HIV drugs and drugs for treatment of hepatitis C virus [see Warnings and Precautions (5.7) and Drug Interactions (7)].
Dosage Recommendations for Treatment of HIV-1 Infection
Inform patients that it is important to take TRUVADA with other antiretroviral drugs for the treatment of HIV-1 on a regular dosing schedule with or without food and to avoid missing doses as it can result in development of resistance.
Pregnancy Registry
Inform patients using TRUVADA for HIV-1 treatment or HIV-1 PrEP that there is an antiretroviral pregnancy registry to monitor fetal outcomes of pregnant women exposed to TRUVADA [see Use in Specific Populations (8.1)].
Lactation
Instruct mothers not to breastfeed if they are taking TRUVADA for the treatment of HIV-1 infection or if acute HIV-1 infection is suspected in a mother taking TRUVADA for HIV-1 PrEP because of the risk of passing the HIV-1 virus to the baby. In HIV-uninfected women, the benefits and risks of TRUVADA while breastfeeding should be evaluated, including the risk of HIV-1 acquisition due to medication nonadherence and subsequent mother to child transmission [see Use in Specific Populations (8.2)].
-
SPL UNCLASSIFIED SECTION
TRUVADA, ATRIPLA, COMPLERA, EMTRIVA, EPCLUSA, HARVONI, SOVALDI, STRIBILD, and VIREAD are trademarks of Gilead Sciences, Inc., or its related companies. All other trademarks referenced herein are the property of their respective owners.
© 2018 Gilead Sciences, Inc. All rights reserved.
Manufactured for and distributed by:
Gilead Sciences, Inc.
Foster City, CA 9440421752-GS-032
-
MEDICATION GUIDE
This Medication Guide has been approved by the U.S. Food and Drug Administration. Revised: May 2018 Medication Guide
TRUVADA® (tru-VAH-dah)
(emtricitabine and tenofovir disoproxil fumarate)
tabletsRead this Medication Guide before you start taking TRUVADA and each time you get a refill. There may be new information. This information does not take the place of talking to your healthcare provider about your medical condition or your treatment.
This Medication Guide provides information about two different ways that TRUVADA may be used. See the section "What is TRUVADA?" for detailed information about how TRUVADA may be used.What is the most important information I should know about TRUVADA?
TRUVADA can cause serious side effects, including:-
Worsening of Hepatitis B virus infection (HBV). Your healthcare provider will test you for HBV before starting treatment with TRUVADA. If you have HBV infection and take TRUVADA, your HBV may get worse (flare-up) if you stop taking TRUVADA. A "flare-up" is when your HBV infection suddenly returns in a worse way than before.
- Do not stop taking TRUVADA without first talking to your healthcare provider.
- Do not run out of TRUVADA. Refill your prescription or talk to your healthcare provider before your TRUVADA is all gone.
- If you stop taking TRUVADA, your healthcare provider will need to check your health often and do blood tests regularly for several months to check your HBV infection, or give you a medication to treat hepatitis B. Tell your healthcare provider about any new or unusual symptoms you may have after you stop taking TRUVADA.
Other important information for people who take TRUVADA to help reduce their risk of getting HIV-1 infection:
Before taking TRUVADA to reduce your risk of getting HIV-1 infection:- You must be HIV-negative to start TRUVADA. You must get tested to make sure that you do not already have HIV-1 infection.
- Do not take TRUVADA to reduce the risk of getting HIV-1 unless you are confirmed to be HIV-negative.
- Many HIV-1 tests can miss HIV-1 infection in a person who has recently become infected. If you have flu-like symptoms, you could have recently become infected with HIV-1. Tell your healthcare provider if you had a flu-like illness within the last month before starting TRUVADA or at any time while taking TRUVADA. Symptoms of new HIV-1 infection include:
- tiredness
- fever
- joint or muscle aches
- headache
- sore throat
- vomiting or diarrhea
- rash
- night sweats
- enlarged lymph nodes in the neck or groin
While you are taking TRUVADA to reduce your risk of getting HIV-1: - Just taking TRUVADA may not keep you from getting HIV-1.
- You must continue using safer sex practices while you are taking TRUVADA to reduce your risk of getting HIV-1.
-
You must stay HIV-negative to keep taking TRUVADA to reduce your risk of infection.
- Know your HIV-1 status and the HIV-1 status of your partners.
- Practice safer sex by using a latex or polyurethane condom to lower the chance of sexual contact with semen, vaginal fluids, or blood.
- Ask your partners with HIV-1 if they are taking anti-HIV-1 medicine and have an undetectable viral load. An undetectable viral load is when the amount of virus in the blood is too low to be measured in a lab test. To maintain an undetectable viral load, your partners must keep taking anti-HIV-1 medicine every day. Your risk of getting HIV-1 is lower if your partners with HIV-1 are taking effective treatment.
- Get tested for HIV-1 at least every 3 months or when your healthcare provider tells you.
- Get tested for other sexually transmitted infections such as syphilis, chlamydia, and gonorrhea. These infections make it easier for HIV-1 to infect you.
- If you think you were exposed to HIV-1, tell your healthcare provider right away. They may want to do more tests to be sure you are still HIV-negative.
- Get information and support to help reduce risky sexual behavior.
- Have fewer sex partners.
- Do not miss any doses of TRUVADA. Missing doses may increase your risk of getting HIV-1 infection.
- If you do become HIV-positive, you need more medicine than TRUVADA alone to treat HIV-1. TRUVADA by itself is not a complete treatment for HIV-1.
- If you have HIV-1 and take only TRUVADA, over time your HIV-1 may become harder to treat.
What is TRUVADA?
TRUVADA is a prescription medicine that is used to:- treat HIV-1 infection when used with other anti-HIV-1 medicines in adults and children who weigh at least 37 pounds (at least 17 kg).
- help reduce the risk of getting HIV-1 infection when used with safer sex practices in adults and adolescents who weigh at least 77 pounds (at least 35 kg).
HIV is the virus that causes AIDS (Acquired Immune Deficiency Syndrome).
TRUVADA contains the medicines emtricitabine and tenofovir disoproxil fumarate.
It is not known if TRUVADA is safe and effective in children with HIV-1 infection who weigh less than 37 pounds (less than 17 kg).For people taking TRUVADA to reduce the risk of getting HIV-1 infection:
Do not take TRUVADA to help reduce your risk of getting HIV-1 if:- you already have HIV-1 infection. If you are HIV-positive, you need to take other medicines with TRUVADA to treat HIV-1. TRUVADA by itself is not a complete treatment for HIV-1.
- you do not know your HIV-1 infection status. You may already be HIV-positive. You need to take other HIV-1 medicines with TRUVADA to treat HIV-1.
What should I tell my healthcare provider before taking TRUVADA?
Before taking TRUVADA, tell your healthcare provider about all of your medical conditions, including if you:- have liver problems, including HBV infection
- have kidney problems or receive kidney dialysis treatment
- have bone problems
- are pregnant or plan to become pregnant. It is not known if TRUVADA can harm your unborn baby. Tell your healthcare provider if you become pregnant during treatment with TRUVADA.
Pregnancy Registry: There is a pregnancy registry for women who take TRUVADA during pregnancy. The purpose of this registry is to collect information about the health of you and your baby. Talk to your healthcare provider about how you can take part in this registry. - are breastfeeding or plan to breastfeed. TRUVADA can pass to your baby in your breast milk.
- Do not breastfeed if you have HIV-1 or if you think you have recently become infected with HIV-1 because of the risk of passing HIV-1 to your baby.
- If you take TRUVADA to reduce the risk of getting HIV-1, talk with your healthcare provider about the best way to feed your baby.
Some medicines may interact with TRUVADA. Keep a list of your medicines and show it to your healthcare provider and pharmacist when you get a new medicine.- You can ask your healthcare provider or pharmacist for a list of medicines that interact with TRUVADA.
- Do not start a new medicine without telling your healthcare provider. Your healthcare provider can tell you if it is safe to take TRUVADA with other medicines.
How should I take TRUVADA? - Take TRUVADA exactly as your healthcare provider tells you to take it.
- If you take TRUVADA to treat HIV-1 infection, you need to take other HIV-1 medicines. Your healthcare provider will tell you what medicines to take and how to take them.
- Take TRUVADA 1 time each day with or without food.
- Children who take TRUVADA are prescribed a lower strength tablet than adults. Children should swallow the TRUVADA tablet. Tell your healthcare provider if your child cannot swallow the tablet, because they may need a different HIV-1 medicine.
- Your healthcare provider will change the dose of TRUVADA as needed based on your child's weight.
- Take TRUVADA at the same time each day to help keep TRUVADA blood levels constant.
- Do not miss a dose of TRUVADA. Missing a dose lowers the amount of medicine in your blood. Refill your TRUVADA prescription before you run out of medicine.
- Do not change your dose or stop taking TRUVADA without first talking with your healthcare provider. Stay under a healthcare provider's care when taking TRUVADA.
- If you take too much TRUVADA, call your healthcare provider or go to the nearest hospital emergency room right away.
What are the possible side effects of TRUVADA?
TRUVADA may cause serious side effects, including:- See "What is the most important information I should know about TRUVADA?"
- New or worse kidney problems, including kidney failure. Your healthcare provider should do blood and urine tests to check your kidneys before you start and during treatment with TRUVADA. Your healthcare provider may tell you to take TRUVADA less often, or to stop taking TRUVADA if you get new or worse kidney problems.
- Changes in your immune system (Immune Reconstitution Syndrome) can happen when an HIV-1 infected person starts taking HIV-1 medicines. Your immune system may get stronger and begin to fight infections that have been hidden in your body for a long time. Tell your healthcare provider right away if you start having any new symptoms after starting TRUVADA for treatment of HIV-1 infection.
- Bone problems can happen in some people who take TRUVADA. Bone problems include bone pain, or softening or thinning of bones, which may lead to fractures. Your healthcare provider may need to do tests to check your bones.
- Too much lactic acid in your blood (lactic acidosis). Too much lactic acid is a serious but rare medical emergency that can lead to death. Tell your healthcare provider right away if you get these symptoms: weakness or being more tired than usual, unusual muscle pain, being short of breath or fast breathing, stomach pain with nausea and vomiting, cold or blue hands and feet, feel dizzy or lightheaded, or a fast or abnormal heartbeat.
- Severe liver problems. In rare cases, severe liver problems can happen that can lead to death. Tell your healthcare provider right away if you get these symptoms: skin or the white part of your eyes turns yellow, dark "tea-colored" urine, light-colored stools, loss of appetite for several days or longer, nausea, or stomach-area pain.
- diarrhea
- nausea
- tiredness
- headache
- dizziness
- depression
- problems sleeping
- abnormal dreams
- rash
Common side effects in people who take TRUVADA to reduce the risk of getting HIV-1 infection include: - headache
- stomach-area (abdomen) pain
- decreased weight
These are not all the possible side effects of TRUVADA.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.How should I store TRUVADA? - Store TRUVADA at room temperature between 68°F to 77°F (20°C to 25°C).
- Keep TRUVADA in its original container.
- Keep the container tightly closed.
- Do not use TRUVADA if seal over bottle opening is broken or missing.
General information about TRUVADA.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use TRUVADA for a condition for which it was not prescribed. Do not give TRUVADA to other people, even if they have the same symptoms you have. It may harm them. You can ask your healthcare provider or pharmacist for information about TRUVADA that is written for health professionals.What are the ingredients in TRUVADA?
Active ingredients: emtricitabine and tenofovir disoproxil fumarate.
Inactive ingredients: croscarmellose sodium, lactose monohydrate, magnesium stearate, microcrystalline cellulose, and pregelatinized starch (gluten free). The 200 mg/300 mg strength tablets are coated with Opadry II Blue Y-30-10701, which contains FD&C Blue #2 aluminum lake, hypromellose 2910, lactose monohydrate, titanium dioxide, and triacetin. The 167 mg/250 mg, 133 mg/200 mg, and 100 mg/150 mg strength tablets are coated with Opadry II Blue, which contains FD&C Blue #2 aluminum lake, hypromellose 2910, lactose monohydrate, titanium dioxide, and triacetin.
Manufactured for and distributed by:
Gilead Sciences, Inc.
Foster City, CA 94404
TRUVADA is a trademark of Gilead Sciences, Inc., or its related companies.
For more information, call 1-800-445-3235 or go to www.TRUVADA.com.
© 2018 Gilead Sciences, Inc. All rights reserved.
21752-GS-032 -
Worsening of Hepatitis B virus infection (HBV). Your healthcare provider will test you for HBV before starting treatment with TRUVADA. If you have HBV infection and take TRUVADA, your HBV may get worse (flare-up) if you stop taking TRUVADA. A "flare-up" is when your HBV infection suddenly returns in a worse way than before.
-
PRINCIPAL DISPLAY PANEL - 30 Tablet Bottle Label
NDC: 61958-0701-1
Truvada®
(emtricitabine and tenofovir
disoproxil fumarate)
Tablets30 tablets
Rx only
DISPENSER: Each time Truvada® is dispensed
give the patient the attached Medication Guide.
- PRINCIPAL DISPLAY PANEL - 100 mg/150 mg Tablet Bottle Label
- PRINCIPAL DISPLAY PANEL - 133 mg/200 mg Tablet Bottle Label
- PRINCIPAL DISPLAY PANEL - 167 mg/250 mg Tablet Bottle Label
-
INGREDIENTS AND APPEARANCE
TRUVADA
emtricitabine and tenofovir disoproxil fumarate tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 61958-0701 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength emtricitabine (UNII: G70B4ETF4S) (emtricitabine - UNII:G70B4ETF4S) emtricitabine 200 mg tenofovir disoproxil fumarate (UNII: OTT9J7900I) (tenofovir anhydrous - UNII:W4HFE001U5) tenofovir disoproxil fumarate 300 mg Inactive Ingredients Ingredient Name Strength croscarmellose sodium (UNII: M28OL1HH48) lactose monohydrate (UNII: EWQ57Q8I5X) magnesium stearate (UNII: 70097M6I30) microcrystalline cellulose (UNII: OP1R32D61U) starch, corn (UNII: O8232NY3SJ) water (UNII: 059QF0KO0R) FD&C Blue No. 2 (UNII: L06K8R7DQK) aluminum oxide (UNII: LMI26O6933) hypromellose, unspecified (UNII: 3NXW29V3WO) titanium dioxide (UNII: 15FIX9V2JP) triacetin (UNII: XHX3C3X673) Product Characteristics Color BLUE Score no score Shape OVAL (Capsule-shaped) Size 19mm Flavor Imprint Code GILEAD;701 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 61958-0701-1 30 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 08/02/2004 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA021752 08/02/2004 TRUVADA
emtricitabine and tenofovir disoproxil fumarate tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 61958-0703 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength emtricitabine (UNII: G70B4ETF4S) (emtricitabine - UNII:G70B4ETF4S) emtricitabine 100 mg tenofovir disoproxil fumarate (UNII: OTT9J7900I) (tenofovir anhydrous - UNII:W4HFE001U5) tenofovir disoproxil fumarate 150 mg Inactive Ingredients Ingredient Name Strength croscarmellose sodium (UNII: M28OL1HH48) lactose monohydrate (UNII: EWQ57Q8I5X) magnesium stearate (UNII: 70097M6I30) microcrystalline cellulose (UNII: OP1R32D61U) starch, corn (UNII: O8232NY3SJ) water (UNII: 059QF0KO0R) FD&C Blue No. 2 (UNII: L06K8R7DQK) aluminum oxide (UNII: LMI26O6933) hypromellose, unspecified (UNII: 3NXW29V3WO) titanium dioxide (UNII: 15FIX9V2JP) triacetin (UNII: XHX3C3X673) Product Characteristics Color BLUE Score no score Shape OVAL Size 14mm Flavor Imprint Code GSI;703 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 61958-0703-1 30 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 03/10/2016 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA021752 03/10/2016 TRUVADA
emtricitabine and tenofovir disoproxil fumarate tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 61958-0704 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength emtricitabine (UNII: G70B4ETF4S) (emtricitabine - UNII:G70B4ETF4S) emtricitabine 133 mg tenofovir disoproxil fumarate (UNII: OTT9J7900I) (tenofovir anhydrous - UNII:W4HFE001U5) tenofovir disoproxil fumarate 200 mg Inactive Ingredients Ingredient Name Strength croscarmellose sodium (UNII: M28OL1HH48) lactose monohydrate (UNII: EWQ57Q8I5X) magnesium stearate (UNII: 70097M6I30) microcrystalline cellulose (UNII: OP1R32D61U) starch, corn (UNII: O8232NY3SJ) water (UNII: 059QF0KO0R) FD&C Blue No. 2 (UNII: L06K8R7DQK) aluminum oxide (UNII: LMI26O6933) hypromellose, unspecified (UNII: 3NXW29V3WO) titanium dioxide (UNII: 15FIX9V2JP) triacetin (UNII: XHX3C3X673) Product Characteristics Color BLUE Score no score Shape OVAL (Rectangular-shaped) Size 16mm Flavor Imprint Code GSI;704 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 61958-0704-1 30 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 03/10/2016 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA021752 03/10/2016 TRUVADA
emtricitabine and tenofovir disoproxil fumarate tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 61958-0705 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength emtricitabine (UNII: G70B4ETF4S) (emtricitabine - UNII:G70B4ETF4S) emtricitabine 167 mg tenofovir disoproxil fumarate (UNII: OTT9J7900I) (tenofovir anhydrous - UNII:W4HFE001U5) tenofovir disoproxil fumarate 250 mg Inactive Ingredients Ingredient Name Strength croscarmellose sodium (UNII: M28OL1HH48) lactose monohydrate (UNII: EWQ57Q8I5X) magnesium stearate (UNII: 70097M6I30) microcrystalline cellulose (UNII: OP1R32D61U) starch, corn (UNII: O8232NY3SJ) water (UNII: 059QF0KO0R) FD&C Blue No. 2 (UNII: L06K8R7DQK) aluminum oxide (UNII: LMI26O6933) hypromellose, unspecified (UNII: 3NXW29V3WO) titanium dioxide (UNII: 15FIX9V2JP) triacetin (UNII: XHX3C3X673) Product Characteristics Color BLUE Score no score Shape OVAL (Capsule-shaped) Size 18mm Flavor Imprint Code GSI;705 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 61958-0705-1 30 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 03/10/2016 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA021752 03/10/2016 Labeler - Gilead Sciences, Inc (185049848)

Trademark Results [Truvada]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 TRUVADA 78239720 2915213 Live/Registered |
Gilead Sciences, Inc. 2003-04-18 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.