MONOFERRIC- ferric derisomaltose solution
Monoferric by
Drug Labeling and Warnings
Monoferric by is a Prescription medication manufactured, distributed, or labeled by Pharmacosmos Therapeutics Inc., Millmount Healthcare Limited, d/b/a PCI Pharma Services. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use MONOFERRIC safely and effectively. See Full Prescribing Information for MONOFERRIC.
MONOFERRIC (ferric derisomaltose) injection, for intravenous use
Initial U.S. Approval: 2020INDICATIONS AND USAGE
DOSAGE AND ADMINISTRATION
- For patients weighing 50 kg or more: Administer 1,000 mg of Monoferric as an intravenous infusion.
- For patients weighing less than 50 kg: Administer Monoferric as 20 mg/kg actual body weight as an intravenous infusion.
- Repeat Monoferric treatment if iron deficiency anemia reoccurs. (2)
DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONS
Serious hypersensitivity to Monoferric or any of its components. (4)
WARNINGS AND PRECAUTIONS
- Hypersensitivity Reactions: Monitor patients for signs and symptoms of hypersensitivity during and after Monoferric administration for at least 30 minutes and until clinically stable following completion of the infusion. (5.1)
- Iron Overload: Do not administer Monoferric to patients with iron overload. (5.2)
ADVERSE REACTIONS
Most commonly reported adverse reactions (incidence ≥1%) are rash and nausea. (6)
To report SUSPECTED ADVERSE REACTIONS, contact Pharmacosmos at 1-888-828-0655 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 2/2020
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
2.2 Preparation and Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity Reactions
5.2 Iron Overload
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Post-marketing Experience
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage and Handling
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
For patients weighing 50 kg or more: Administer 1,000 mg of Monoferric by intravenous infusion over at least 20 minutes as a single dose. Repeat dose if iron deficiency anemia reoccurs.
For patients weighing less than 50 kg: Administer Monoferric as 20 mg/kg actual body weight by intravenous infusion over at least 20 minutes as a single dose. Repeat dose if iron deficiency anemia reoccurs.
The dosage of Monoferric is expressed in mg of elemental iron. Each mL of Monoferric contains 100 mg of elemental iron.
Only administer Monoferric when personnel and therapies are immediately available for the treatment of serious hypersensitivity reactions [Warnings and Precautions (5.1)].
2.2 Preparation and Administration
Inspect parenteral drug products visually for the absence of particulate matter and discoloration prior to administration. The product contains no preservatives.
Each vial of Monoferric is single-dose only. Discard unused portion.
- Withdraw the appropriate volume of Monoferric and dilute in 100 mL to 500 mL of 0.9% Sodium Chloride Injection, USP.
- Final diluted concentration should be more than 1 mg iron/mL.
- Compatibility of Monoferric with other drugs has not been established. Monoferric should not be mixed with or physically added to solutions containing other drugs.
- Administer the prepared solution via intravenous infusion over at least 20 minutes.
- Following dilution with 0.9% Sodium Chloride Injection, USP, Monoferric solution may be stored at room temperature for up to 8 hours.
- Extravasation of Monoferric may cause brown discoloration at the extravasation site which may be long lasting. Monitor for extravasation. If extravasation occurs, discontinue the Monoferric administration at that site.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
Monoferric is contraindicated in patients with a history of serious hypersensitivity to Monoferric or any of its components [see Warnings and Precautions (5.1), Description (11)]. Reactions have included shock, clinically significant hypotension, loss of consciousness, and/or collapse.
-
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity Reactions
Serious hypersensitivity reactions, including anaphylactic-type reactions, some of which have been life-threatening and fatal, have been reported in patients receiving Monoferric. Patients may present with shock, clinically significant hypotension, loss of consciousness, and/or collapse. Monitor patients for signs and symptoms of hypersensitivity during and after Monoferric administration for at least 30 minutes and until clinically stable following completion of the infusion. Only administer Monoferric when personnel and therapies are immediately available for the treatment of serious hypersensitivity reactions. Monoferric is contraindicated in patients with prior serious hypersensitivity reactions to Monoferric or any of its components [see Contraindications (4)]. In clinical trials in patients with IDA and CKD, serious or severe hypersensitivity were reported in 0.3% (6/2008) of the Monoferric treated subjects. These included 3 events of hypersensitivity in 3 patients; 2 events of infusion-related reactions in 2 patients and 1 event of asthma in one patient.
5.2 Iron Overload
Excessive therapy with parenteral iron can lead to excess iron storage and possibly iatrogenic hemosiderosis or hemochromatosis. Monitor the hematologic response (hemoglobin and hematocrit) and iron parameters (serum ferritin and transferrin saturation) during parenteral iron therapy. Do not administer Monoferric to patients with iron overload [see Overdosage (10)].
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are discussed in greater detail in other sections of the labeling:
- Hypersensitivity Reactions [see Warnings and Precautions (5.1)].
- Iron Overload [see Warnings and Precautions (5.2)].
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, the adverse reaction rates observed cannot be directly compared to rates in other clinical trials and may not reflect the rates observed in clinical practice.
The safety of Monoferric was evaluated in 3008 patients with iron deficiency anemia enrolled in two randomized, actively-controlled trials. Trial 1 enrolled adult patients with iron deficiency anemia with intolerance to oral iron or had an unsatisfactory response to oral iron with a clinical need for repletion of iron stores. Eligible subjects were required to have a baseline hemoglobin of ≤11g/dl, transferrin saturation (TSAT) of less than 20% and serum ferritin level of <100 ng/mL. Trial 2 enrolled adult patients with non-dialysis dependent chronic kidney disease (CKD) with iron deficiency anemia [see Clinical Studies (14)]. Eligible subjects also had to have serum ferritin ≤200 μg/L or ≤300 ng/mL if TSAT ≤30%.
Trial 1 and Trial 2
In the two randomized, actively-controlled clinical trials, Trial 1 and Trial 2 [see Clinical Studies (14)], patients were randomized in a 2:1 ratio to intravenous Monoferric (n = 2008) or intravenous iron sucrose (n = 1000) respectively. Monoferric was administered as a single intravenous infusion of 1000 mg diluted in 100 mL 0.9 % sodium chloride and given over approximately 20 minutes (approximately 50 mg iron/min). Iron sucrose was administered as 200 mg undiluted intravenous injections over approximately 2-5 minutes and repeated according to standard practice or physician choice up to a maximum of five times (1000 mg) within the first two weeks starting at baseline.
The data described below reflect exposure to Monoferric in 2008 patients exposed to a 1000 mg single intravenous dose of Monoferric. The mean cumulative intravenous Iron exposure was 984 mg.
Trial 1 included 1483 patients with iron deficiency anemia in the safety analysis that had intolerance to oral iron or have had unsatisfactory response to oral iron or with a clinical need for rapid repletion of iron stores. Trial 2 included 1525 patients in the safety analysis who had non-dialysis dependent CKD. The mean (SD) age of the combined study population was 56.4 (18.3) years. The majority of patients were women (75.7%).
Adverse reactions were reported in 8.6% (172/2008) of patients treated with Monoferric.
Adverse reactions related to treatment and reported by ≥1% of the treated patients in the combined analysis of Trial 1 and 2 are listed in Table 1.
Table 1. Adverse Reactions (≥1%) in Patients Receiving Monoferric in Clinical Trials 1 and 2 Monoferric (N = 2008)
N (%)Iron sucrose (N = 1000)
N (%)Adverse Reaction Nausea 24 (1.2) 11 (1.1) Rash 21 (1) 1 (0.1) Adjudicated serious or severe hypersensitivity reactions were reported in 6/2008 (0.3%) patients in the Monoferric group.
Hypophosphatemia (serum phosphate <2.0 mg/dL) was reported in 3.5% of Monoferric-treated patients in Trials 1 & 2.
6.2 Post-marketing Experience
Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure. The following adverse reactions have been most commonly reported from the post-marketing spontaneous reports with Monoferric:
Cardiac disorders: Tachycardia
Gastrointestinal disorders: Abdominal pain, nausea and vomiting, constipation, diarrhea
General disorders and administration site conditions: Fatigue, pyrexia, chest pain, chills, Fishbane reaction, extravasation, influenza like symptoms, injection site reactions
Immune System disorders: Anaphylactic/anaphylactoid reaction, hypersensitivity
Investigations: Hepatic enzymes increased
Musculoskeletal and connective tissue disorders: Back pain, muscle spasms, arthralgia, myalgia
Nervous system disorders: Dizziness, headache, paresthesia, dysgeusia, seizure, loss of consciousness, syncope
Psychiatric disorders: Anxiety
Respiratory, thoracic and mediastinal disorders: Dyspnea, cough
Skin and subcutaneous tissue disorders: Erythema, urticaria, discoloration skin, rash, pruritus, sweating
Vascular disorders: Hypertension, hypotension, flushing, phlebitis
Extravasation of Monoferric at the injection site that may lead to irritation of the skin and potentially long lasting brown discoloration at the site of injection has also been reported.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no available data on Monoferric use in pregnant women to evaluate for a drug-associated risk of major birth defects, miscarriage or adverse maternal or fetal outcomes. Published studies on the use of intravenous iron products in pregnant women have not reported an association with adverse developmental outcomes. However, these studies cannot establish or exclude the absence of any drug-related risk during pregnancy because the studies were not designed to assess for the risk of major birth defects [see Data]. There are risks to the mother and fetus associated with untreated iron deficiency anemia (IDA) in pregnancy [see Clinical Considerations]. Iron complexes have been reported to be teratogenic and embryocidal in non-iron depleted pregnant animals. The findings in animals may be due to iron overload and may not be applicable to patients with iron deficiency. Animal reproduction studies of ferric derisomaltose administered to rats and rabbits during the period of organogenesis caused adverse developmental outcomes including structural abnormalities and embryo-fetal mortality at doses approximately 0.09 and 0.4 times the maximum recommended human dose (MRHD) of 1000 mg, respectively, based on body surface area [see Data].
The estimated background risk of major birth defects and miscarriage for the indicated populations is unknown. Adverse outcomes in pregnancy occur regardless of the health of the mother or the use of medications. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Data
Animal Data
Iron complexes have been reported to be teratogenic and embryocidal in non-anemic pregnant animals at single doses above 125 mg iron/kg body weight. The highest recommended dose in human clinical use is 20 mg iron/kg body weight.
In a combined fertility and embryo-fetal development study in rats, ferric derisomaltose was administered intravenously to female rats 14 days prior to cohabitation and through gestation day (GD) 17 at doses of 3, 11, and 32 mg Fe/kg/day. The doses of 11 and 32 mg Fe/kg/day (approximately 0.1 and 0.3 times the MRHD of 1000 mg, based on body surface area (BSA)) resulted in an increase in the incidence of skeletal developmental delays.
Ferric derisomaltose was administered intravenously to pregnant rabbits during organogenesis, from GD7 to GD20, at doses of 11, 25 and 43 mg Fe/kg/day. The dose of 43 mg Fe/kg/day (approximately 0.8 times the MRHD of 1000 mg, based on BSA) resulted in increased maternal mortality, abortion, and premature delivery, and increased postimplantation loss. Adverse developmental findings at this dose included fetal mortality, reduced fetal weights, and fetal developmental variations and malformations (including domed head, cleft palate, macroglossia, hydrocephaly, small brain). Fetal malformations and reduced fetal weights were also noted in the 25 mg Fe/kg/day group (approximately 0.5 times the MRHD based on BSA).
8.2 Lactation
Risk Summary
The available data on the use of Monoferric in lactating women demonstrate that iron is present in breast milk. However, the data do not inform the potential exposure of iron for the breastfed child or the effects on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for Monoferric in addition to any potential adverse effects on the breastfed child from the drug or from the underlying maternal condition.
8.5 Geriatric Use
Of the 3934 patients in clinical studies of Monoferric, 29% were 65 years and over, while 13% were 75 years and over. No overall differences in safety or effectiveness were observed between these patients and younger subjects, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
-
10 OVERDOSAGE
Excessive dosages of Monoferric may lead to accumulation of iron in storage sites potentially leading to hemosiderosis and hemochromatosis. Avoid use of Monoferric in patients with iron overload [see Warnings and Precautions (5.2)].
-
11 DESCRIPTION
Monoferric is an iron replacement product containing ferric derisomaltose for intravenous infusion. Ferric derisomaltose is an iron carbohydrate complex with a matrix structure composed of interchanging layers of ferric hydroxide and the carbohydrate derisomaltose. Derisomaltose consists of linear, hydrogenated isomaltooligosaccharides with an average molecular weight of 1000 Da and a narrow molecular weight distribution that is almost devoid of mono- and disaccharides.
Ferric derisomaltose has an average molecular weight of 155,000 Da and has the following empirical formula:
{FeO(1-3X) (OH)(1+3X) (C6H5O73-)X}, (H20)T, -
(C6H10O6)R(-C6H10O5-)Z(C6H13O5)R, (NaCl)Y
X = 0.0311; T = 0.25; R = 0.14; Z = 0.49; Y = 0.14
Iron atoms placed in the electronegative cavities of the 3-D structure between and within the derisomaltose molecules. A schematic representation is presented below
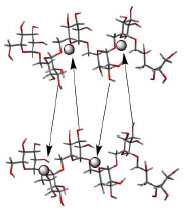
Monoferric is a sterile, dark brown, non-transparent aqueous solution with pH 5.0-7.0, containing ferric derisomaltose dissolved in water for injections and filled into Type I glass vials.
Each 1 mL of solution contains 100 mg of elemental iron as ferric derisomaltose in water for injection.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Ferric derisomaltose is a complex of iron (III) hydroxide and derisomaltose, an iron carbohydrate oligosaccharide that releases iron. Iron binds to transferrin for transport to erythroid precursor cells to be incorporated into hemoglobin.
12.2 Pharmacodynamics
Serum ferritin peaks approximately 7 days after an intravenous dose of Monoferric and slowly returns to stable levels after about 4 weeks.
12.3 Pharmacokinetics
The pharmacokinetics of total iron (derisomaltose-bound plus transferrin-bound iron) were evaluated in adult patients with IDA.
After a single dose of Monoferric, maximum concentration (Cmax) and area under the concentration time curve (AUC) of serum total iron increased approximately proportionally over the 100 to 1000 mg dose range. After a 1000 mg single dose, the Cmax and AUCinf of total iron (geometric mean and CV%) of serum total iron were 408 (10.5) μg/mL and 17730 (22.1) μg.h /mL.
Distribution
Circulating iron is removed from the plasma by cells of the reticuloendothelial system. The iron is bound to the available protein moieties to form hemosiderin or ferritin, the physiological storage forms of iron, or to a lesser extent, to the transport molecule transferrin.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been conducted.
Iron oligosaccharide, an earlier formulation of ferric derisomaltose, was not genotoxic in an in vitro bacterial reverse mutation assay, an in vitro chromosomal aberrations test and an in vivo mouse micronucleus assay.
In a combined fertility and embryo-fetal development study in rats, ferric derisomaltose was administered intravenously to male rats 28 days prior to mating and through cohabitation and to female rats 14 days prior to cohabitation and through GD 17. Doses administered were 2, 6, or 19 mg Fe/kg/day in males and 3, 11, or 32 mg Fe/kg/day in females. There was no effect on male or female fertility in rats at up to 19 mg Fe/kg/day (approximately 0.2 times the MRHD of 1000 mg, based on BSA) in males and up to 32 mg Fe/kg/day (approximately 0.3 times the MRHD of 1000 mg based on BSA) in females.
-
14 CLINICAL STUDIES
The safety and efficacy of Monoferric for treatment of iron deficiency anemia (IDA) were evaluated in two randomized, open-label, actively-controlled clinical trials performed in a total of 3050 patients with IDA of different etiology. Trial 1 included patients with IDA who had intolerance to oral iron or who had had unsatisfactory response to oral iron or for whom there was a clinical need for rapid repletion of iron stores. Trial 2 included patients with IDA who had non-dialysis dependent chronic kidney disease (NDD-CKD). In these two 8-Week trials, patients were randomized 2:1 to treatment with Monoferric or iron sucrose. Monoferric was intravenously administered as a single dose of 1000 mg.
Iron Deficiency Anemia in Patients Who Had Intolerance to Oral Iron or Who Had Unsatisfactory Response to Oral Iron
In Trial 1 (NCT02940886) 1512 adult patients with IDA caused by different etiologies, who had documented intolerance or lack of response to oral iron or screening hemoglobin (Hb) measurement sufficiently low to require repletion of iron stores were randomized in a 2:1 ratio to treatment with Monoferric or iron sucrose. Adult patients aged ≥18 years with baseline Hb ≤11 g/dL, TSAT <20 %, and s-ferritin <100 ng/mL were eligible for enrollment. The median age of patients was 44 years (range 18-91) and 89 % were women.
The efficacy of Monoferric was established based upon the change in Hb from baseline to week 8. Non-inferiority was demonstrated for change in Hb from baseline to Week 8 (Table 2).
Table 2. Change in Hemoglobin Endpoints in Trial 1 Trial 1 Monoferric
N = 1009Iron sucrose
N = 503Difference - * Least square mean
- † The estimate is from a mixed model for repeated measures with treatment, week, treatment-by-week and stratum as fixed effects and baseline Hb and baseline-by-week as covariates.
Mean change in Hb from Baseline to Week 8
Mean* (95% CI), g/dL
(Primary endpoint)2.49
(2.41;2.56)2.49
(2.38;2.59)Estimate†: 0.00 (95% CI -0.13;0.13)
Non-inferiority confirmedIron Deficiency Anemia in Patients with Non-Hemodialysis Dependent Chronic Kidney Disease (NDD-CKD)
Trial 2 (NCT02940860) was a randomized controlled trial in 1538 patients with NDD-CKD who were randomized in a 2:1 ratio to treatment with Monoferric or iron sucrose respectively. Adult patients aged ≥18 years with Hb ≤11 g/dL, s-ferritin ≤100 ng/mL (or ≤300 ng/mL if TSAT ≤30%), chronic renal impairment with eGFR between 15-59 mL/min, and either no ESAs or ESAs at a stable dose (+/-20 %) for 4 weeks before randomization were eligible for enrollment. The median age of patients was 69 years (range 25-97), 63% were female.
The efficacy of Monoferric was established based upon the demonstration of non-inferiority for change in hemoglobin from baseline to Week 8 (Table 3).
Table 3. Change in Hemoglobin Endpoints in Trial 2 Trial 2 Monoferric
N = 1027Iron sucrose
N = 511Difference - * Least square mean
- † The estimate is from a mixed model for repeated measures with treatment, week, treatment-by-week and stratum as fixed effects and baseline Hb and baseline-by-week as covariates.
Mean change in Hb from Baseline to Week 8
Mean* (95% CI), g/dL
(Primary endpoint)1.22
(1.14;1.31)1.14
(1.03;1.26)Estimate†: 0.08 (95% CI -0.06;0.23)
Non-inferiority confirmed -
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
Monoferric injection is a sterile, dark brown, non-transparent aqueous solution supplied in cartons as single-dose vials (10 mL, 5 mL or 1 mL) in the following configurations:
Vial size Number of vials per carton NDC 1,000 mg/10 mL 1 73594-9310-1 500 mg/5 mL 1 73594-9305-1 100 mg/mL 5 73594-9301-2 16.2 Storage and Handling
Store at 20° to 25°C (68° to 77°F); excursions permitted to 15° to 30°C (59° to 86°F). See the USP controlled room temperature.
Do not freeze.
When added to an infusion bag containing 0.9% Sodium Chloride Injection, USP, Monoferric solution may be stored for up to 8 hours at room temperature.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Prior History of Allergies to Parenteral Iron Products
Question patients regarding any prior history of reactions to parenteral iron products [see Warnings and Precautions (5.1)].
Hypersensitivity Reactions
Advise patients to report any signs and symptoms of hypersensitivity that may develop during and following Monoferric administration, such as rash, itching, dizziness, lightheadedness, swelling and breathing problems [see Warnings and Precautions (5.1)].
- SPL UNCLASSIFIED SECTION
-
PATIENT PACKAGE INSERT
This Patient Information has been approved by the U.S. Food and Drug Administration. Issued: 02/2020 Patient Information
MONOFERRIC (mon-oh-fer-ik)
(ferric derisomaltose)
InjectionWhat is MONOFERRIC?
MONOFERRIC is a prescription iron replacement medicine used to treat iron deficiency anemia in adults who have:- intolerance to oral iron or who have not responded well to treatment with oral iron
- non-dialysis dependent chronic kidney disease
Who should not receive MONOFERRIC?
Do not receive MONOFERRIC if you are allergic to ferric derisomaltose or any of the ingredients in MONOFERRIC.
See the end of this leaflet for a complete list of ingredients in MONOFERRIC.Before receiving MONOFERRIC, tell your healthcare provider about all of your medical conditions, including if you: - have had an allergic reaction to IV iron
- are pregnant or plan to become pregnant. It is not known if MONOFERRIC will harm your unborn baby.
- are breastfeeding or plan to breastfeed. MONOFERRIC passes into your breast milk and may harm your baby. Talk to your healthcare provider about the best way to feed your baby during treatment with MONOFERRIC.
Know the medicines you take. Keep a list of them to show to your healthcare provider and pharmacist when you get a new medicine.How will I receive MONOFERRIC?
MONOFERRIC is given into your vein (intravenously) by your healthcare provider over at least 20 minutes.What are the possible side effects of MONOFERRIC?
MONOFERRIC may cause serious side effects, including:- Allergic (hypersensitivity) reactions. Serious life-threatening allergic reactions have happened in people who receive MONOFERRIC. Symptoms of an allergic reaction including rash, itching, hives, dizziness, lightheadedness, breathing problems and low blood pressure have also happened during treatment with MONOFERRIC. Tell your healthcare provider right away if you develop any of the above symptoms of a serious allergic reaction or if you have ever had any unusual or allergic reaction to any IV iron in the past.
- Too much iron stored in your body (iron overload). Your healthcare provider should check the iron level in your blood before you start and during treatment with MONOFERRIC.
These are not all the possible side effects of MONOFERRIC.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.General information about MONOFERRIC
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. You can ask your pharmacist or healthcare provider for information about MONOFERRIC that is written for health professionals.What are the ingredients in MONOFERRIC?
Active ingredient: ferric derisomaltose
Inactive ingredients: water for injection
Manufactured under license from Pharmacosmos A/S, Denmark
Distributed by: Pharmacosmos Therapeutics Inc., Morristown, NJ 07960
For more information go to www.pharmacosmos.com or call 1-888-828-0655. - PRINCIPAL DISPLAY PANEL - 100 mg/mL Vial Box
- PRINCIPAL DISPLAY PANEL - 500 mg/5 mL Vial Box
- PRINCIPAL DISPLAY PANEL - 1000 mg/10 mL Vial Box
-
INGREDIENTS AND APPEARANCE
MONOFERRIC
ferric derisomaltose solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 73594-9301 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength Ferric derisomaltose (UNII: AHU547PI9H) (Ferric derisomaltose - UNII:AHU547PI9H) Ferric derisomaltose 100 mg in 1 mL Inactive Ingredients Ingredient Name Strength Hydrochloric acid (UNII: QTT17582CB) Sodium hydroxide (UNII: 55X04QC32I) Water (UNII: 059QF0KO0R) Nitrogen (UNII: N762921K75) Product Characteristics Color BROWN (Dark brown, non-transparent (opaque) solution) Score Shape Size Flavor Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 73594-9301-2 5 in 1 BOX 01/16/2020 1 NDC: 73594-9301-3 1 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA208171 01/16/2020 MONOFERRIC
ferric derisomaltose solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 73594-9305 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength Ferric derisomaltose (UNII: AHU547PI9H) (Ferric derisomaltose - UNII:AHU547PI9H) Ferric derisomaltose 500 mg in 5 mL Inactive Ingredients Ingredient Name Strength Hydrochloric acid (UNII: QTT17582CB) Sodium hydroxide (UNII: 55X04QC32I) Water (UNII: 059QF0KO0R) Nitrogen (UNII: N762921K75) Product Characteristics Color BROWN (Dark brown, non-transparent (opaque) solution) Score Shape Size Flavor Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 73594-9305-1 1 in 1 BOX 01/16/2020 1 5 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA208171 01/16/2020 MONOFERRIC
ferric derisomaltose solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 73594-9310 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength Ferric derisomaltose (UNII: AHU547PI9H) (Ferric derisomaltose - UNII:AHU547PI9H) Ferric derisomaltose 1000 mg in 10 mL Inactive Ingredients Ingredient Name Strength Hydrochloric acid (UNII: QTT17582CB) Sodium hydroxide (UNII: 55X04QC32I) Water (UNII: 059QF0KO0R) Nitrogen (UNII: N762921K75) Product Characteristics Color BROWN (Dark brown, non-transparent (opaque) solution) Score Shape Size Flavor Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 73594-9310-1 1 in 1 BOX 01/16/2020 1 10 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA208171 01/16/2020 Labeler - Pharmacosmos Therapeutics Inc. (117344166)



Trademark Results [Monoferric]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 MONOFERRIC 87839247 5673033 Live/Registered |
Pharmacosmos A/S 2018-03-19 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.