NEMBUTAL SODIUM- pentobarbital sodium injection
Nembutal Sodium by
Drug Labeling and Warnings
Nembutal Sodium by is a Prescription medication manufactured, distributed, or labeled by Akron, Akorn Operating Company LLC. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
- SPL UNCLASSIFIED SECTION
-
DESCRIPTION
The barbiturates are nonselective central nervous system depressants which are primarily used as sedative hypnotics and also anticonvulsants in subhypnotic doses. The barbiturates and their sodium salts are subject to control under the Federal Controlled Substances Act (See “Drug Abuse and Dependence” section).
The sodium salts of amobarbital, pentobarbital, phenobarbital, and secobarbital are available as sterile parenteral solutions.
Barbiturates are substituted pyrimidine derivatives in which the basic structure common to these drugs is barbituric acid, a substance which has no central nervous system (CNS) activity. CNS activity is obtained by substituting alkyl, alkenyl, or aryl groups on the pyrimidine ring.
NEMBUTAL Sodium Solution (pentobarbital sodium injection) is a sterile solution for intravenous or intramuscular injection. Each mL contains pentobarbital sodium 50 mg, in a vehicle of propylene glycol, 40%, alcohol, 10% and water for injection, to volume. The pH is adjusted to approximately 9.5 with hydrochloric acid and/or sodium hydroxide.
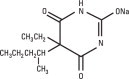
NEMBUTAL Sodium is a short-acting barbiturate, chemically designated as sodium 5-ethyl-5-(1-methylbutyl) barbiturate. The structural formula for pentobarbital sodium is:
The sodium salt occurs as a white, slightly bitter powder which is freely soluble in water and alcohol but practically insoluble in benzene and ether.
-
CLINICAL PHARMACOLOGY
Barbiturates are capable of producing all levels of CNS mood alteration from excitation to mild sedation, to hypnosis, and deep coma. Overdosage can produce death. In high enough therapeutic doses, barbiturates induce anesthesia.
Barbiturates depress the sensory cortex, decrease motor activity, alter cerebellar function, and produce drowsiness, sedation, and hypnosis.
Barbiturate-induced sleep differs from physiological sleep. Sleep laboratory studies have demonstrated that barbiturates reduce the amount of time spent in the rapid eye movement (REM) phase of sleep or dreaming stage. Also, Stages III and IV sleep are decreased. Following abrupt cessation of barbiturates used regularly, patients may experience markedly increased dreaming, nightmares, and/or insomnia. Therefore, withdrawal of a single therapeutic dose over 5 or 6 days has been recommended to lessen the REM rebound and disturbed sleep which contribute to drug withdrawal syndrome (for example, decrease the dose from 3 to 2 doses a day for 1 week).
In studies, secobarbital sodium and pentobarbital sodium have been found to lose most of their effectiveness for both inducing and maintaining sleep by the end of 2 weeks of continued drug administration at fixed doses. The short-, intermediate-, and, to a lesser degree, long-acting barbiturates have been widely prescribed for treating insomnia. Although the clinical literature abounds with claims that the short-acting barbiturates are superior for producing sleep while the intermediate-acting compounds are more effective in maintaining sleep, controlled studies have failed to demonstrate these differential effects. Therefore, as sleep medications, the barbiturates are of limited value beyond short-term use.
Barbiturates have little analgesic action at subanesthetic doses. Rather, in subanesthetic doses these drugs may increase the reaction to painful stimuli. All barbiturates exhibit anticonvulsant activity in anesthetic doses. However, of the drugs in this class, only phenobarbital, mephobarbital, and metharbital have been clinically demonstrated to be effective as oral anticonvulsants in subhypnotic doses.
Barbiturates are respiratory depressants. The degree of respiratory depression is dependent upon dose. With hypnotic doses, respiratory depression produced by barbiturates is similar to that which occurs during physiologic sleep with slight decrease in blood pressure and heart rate.
Studies in laboratory animals have shown that barbiturates cause reduction in the tone and contractility of the uterus, ureters, and urinary bladder. However, concentrations of the drugs required to produce this effect in humans are not reached with sedative-hypnotic doses.
Barbiturates do not impair normal hepatic function, but have been shown to induce liver microsomal enzymes, thus increasing and/or altering the metabolism of barbiturates and other drugs. (See “Precautions-Drug Interactions” section).
Pharmacokinetics:
Barbiturates are absorbed in varying degrees following oral, rectal, or parenteral administration. The salts are more rapidly absorbed than are the acids.
The onset of action for oral or rectal administration varies from 20 to 60 minutes. For IM administration, the onset of action is slightly faster. Following IV administration, the onset of action ranges from almost immediately for pentobarbital sodium to 5 minutes for phenobarbital sodium. Maximal CNS depression may not occur until 15 minutes or more after IV administration for phenobarbital sodium.
Duration of action, which is related to the rate at which the barbiturates are redistributed throughout the body, varies among persons and in the same person from time to time.
No studies have demonstrated that the different routes of administration are equivalent with respect to bioavailability.
Barbiturates are weak acids that are absorbed and rapidly distributed to all tissues and fluids with high concentrations in the brain, liver, and kidneys. Lipid solubility of the barbiturates is the dominant factor in their distribution within the body. The more lipid soluble the barbiturate, the more rapidly it penetrates all tissues of the body. Barbiturates are bound to plasma and tissue proteins to a varying degree with the degree of binding increasing directly as a function of lipid solubility.
Phenobarbital has the lowest lipid solubility, lowest plasma binding, lowest brain protein binding, the longest delay in onset of activity, and the longest duration of action. At the opposite extreme is secobarbital which has the highest lipid solubility, plasma protein binding, brain protein binding, the shortest delay in onset of activity, and the shortest duration of action. Butabarbital is classified as an intermediate barbiturate.
The plasma half-life for pentobarbital in adults is 15 to 50 hours and appears to be dose dependent.
Barbiturates are metabolized primarily by the hepatic microsomal enzyme system, and the metabolic products are excreted in the urine, and less commonly, in the feces. Approximately 25 to 50 percent of a dose of aprobarbital or phenobarbital is eliminated unchanged in the urine, whereas the amount of other barbiturates excreted unchanged in the urine is negligible. The excretion of unmetabolized barbiturate is one feature that distinguishes the long-acting category from those belonging to other categories which are almost entirely metabolized. The inactive metabolites of the barbiturates are excreted as conjugates of glucuronic acid.
-
INDICATIONS AND USAGE
Parenteral:
- Sedatives.
- Hypnotics, for the short-term treatment of insomnia, since they appear to lose their effectiveness for sleep induction and sleep maintenance after 2 weeks (See “Clinical Pharmacology” section.)
- Preanesthetics.
- Anticonvulsant, in anesthetic doses, in the emergency control of certain acute convulsive episodes, e.g., those associated with status epilepticus, cholera, eclampsia, meningitis, tetanus, and toxic reactions to strychnine or local anesthetics.
- CONTRAINDICATIONS
-
WARNINGS
- Habit forming: Barbiturates may be habit forming. Tolerance, psychological and physical dependence may occur with continued use. (See “Drug Abuse and Dependence” and “Pharmacokinetics” sections.) Patients who have psychological dependence on barbiturates may increase the dosage or decrease the dosage interval without consulting a physician and may subsequently develop a physical dependence on barbiturates. To minimize the possibility of overdosage or the development of dependence, the prescribing and dispensing of sedative-hypnotic barbiturates should be limited to the amount required for the interval until the next appointment. Abrupt cessation after prolonged use in the dependent person may result in withdrawal symptoms, including delirium, convulsions, and possibly death. Barbiturates should be withdrawn gradually from any patient known to be taking excessive dosage over long periods of time. (See “Drug Abuse and Dependence” section.)
- IV administration: Too rapid administration may cause respiratory depression, apnea, laryngospasm, or vasodilation with fall in blood pressure.
- Acute or chronic pain: Caution should be exercised when barbiturates are administered to patients with acute or chronic pain, because paradoxical excitement could be induced or important symptoms could be masked. However, the use of barbiturates as sedatives in the postoperative surgical period and as adjuncts to cancer chemotherapy is well established.
-
Use in pregnancy: Barbiturates can cause fetal damage when administered to a pregnant woman. Retrospective, case-controlled studies have suggested a connection between the maternal consumption of barbiturates and a higher than expected incidence of fetal abnormalities. Following oral or parenteral administration, barbiturates readily cross the placental barrier and are distributed throughout fetal tissues with highest concentrations found in the placenta, fetal liver, and brain. Fetal blood levels approach maternal blood levels following parenteral administration.
Withdrawal symptoms occur in infants born to mothers who receive barbiturates throughout the last trimester of pregnancy. (See “Drug Abuse and Dependence” section.) If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to the fetus.
- Synergistic effects: The concomitant use of alcohol or other CNS depressants may produce additive CNS depressant effects.
-
Pediatric neurotoxicity: Published animal studies demonstrate that the administration of anesthetic and sedation drugs that block NMDA receptors and/or potentiate GABA activity increase neuronal apoptosis in the developing brain and result in long-term cognitive deficits when used for longer than 3 hours. The clinical significance of these findings is not clear. However, based on the available data, the window of vulnerability to these changes is believed to correlate with exposures in the third trimester of gestation through the first several months of life, but may extend out to approximately three years of age in humans (see “Precautions-Pregnancy and Pediatric Use” and “Animal Pharmacology and/or Toxicology”).
Some published studies in children suggest that similar deficits may occur after repeated or prolonged exposures to anesthetic agents early in life and may result in adverse cognitive or behavioral effects. These studies have substantial limitations, and it is not clear if the observed effects are due to the anesthetic/sedation drug administration or other factors such as the surgery or underlying illness.
Anesthetic and sedation drugs are a necessary part of the care of children and pregnant women needing surgery, other procedures, or tests that cannot be delayed, and no specific medications have been shown to be safer than any other. Decisions regarding the timing of any elective procedures requiring anesthesia should take into consideration the benefits of the procedure weighed against the potential risks.
-
PRECAUTIONS
General:
Barbiturates may be habit forming. Tolerance and psychological and physical dependence may occur with continuing use. (See “Drug Abuse and Dependence” section.) Barbiturates should be administered with caution, if at all, to patients who are mentally depressed, have suicidal tendencies, or a history of drug abuse.
Elderly or debilitated patients may react to barbiturates with marked excitement, depression, and confusion. In some persons, barbiturates repeatedly produce excitement rather than depression.
In patients with hepatic damage, barbiturates should be administered with caution and initially in reduced doses. Barbiturates should not be administered to patients showing the premonitory signs of hepatic coma.
Parenteral solutions of barbiturates are highly alkaline. Therefore, extreme care should be taken to avoid perivascular extravasation or intra-arterial injection. Extravascular injection may cause local tissue damage with subsequent necrosis; consequences of intra-arterial injection may vary from transient pain to gangrene of the limb. Any complaint of pain in the limb warrants stopping the injection.
Information for the patient:
Practitioners should give the following information and instructions to patients receiving barbiturates.
- The use of barbiturates carries with it an associated risk of psychological and/or physical dependence. The patient should be warned against increasing the dose of the drug without consulting a physician.
- Barbiturates may impair mental and/or physical abilities required for the performance of potentially hazardous tasks (e.g., driving, operating machinery, etc.).
- Alcohol should not be consumed while taking barbiturates. Concurrent use of the barbiturates with other CNS depressants (e.g., alcohol, narcotics, tranquilizers, and antihistamines) may result in additional CNS depressant effects.
-
Effect of anesthetic and sedation drugs on early brain development
Studies conducted in young animals and children suggest repeated or prolonged use of general anesthetic or sedation drugs in children younger than 3 years may have negative effects on their developing brains. Discuss with parents and caregivers the benefits, risks, and timing and duration of surgery or procedures requiring anesthetic and sedation drugs. Because some animal data suggest that the window of vulnerability includes the 3rd trimester of pregnancy, discuss with pregnant women the benefits, risks, and timing and duration of surgery or procedures requiring anesthetic and sedation drugs. (See “Warnings-Pediatric Neurotoxicity”.)
Laboratory tests:
Prolonged therapy with barbiturates should be accompanied by periodic laboratory evaluation of organ systems, including hematopoietic, renal, and hepatic systems. (See “Precautions-General” and “Adverse Reactions” sections.)
Drug interactions:
Most reports of clinically significant drug interactions occurring with the barbiturates have involved phenobarbital. However, the application of these data to other barbiturates appears valid and warrants serial blood level determinations of the relevant drugs when there are multiple therapies.
- Anticoagulants: Phenobarbital lowers the plasma levels of dicumarol (name previously used: bishydroxycoumarin) and causes a decrease in anticoagulant activity as measured by the prothrombin time. Barbiturates can induce hepatic microsomal enzymes resulting in increased metabolism and decreased anticoagulant response of oral anticoagulants (e.g., warfarin, acenocoumarol, dicumarol, and phenprocoumon). Patients stabilized on anticoagulant therapy may require dosage adjustments if barbiturates are added to or withdrawn from their dosage regimen.
- Corticosteroids: Barbiturates appear to enhance the metabolism of exogenous corticosteroids probably through the induction of hepatic microsomal enzymes. Patients stabilized on corticosteroid therapy may require dosage adjustments if barbiturates are added to or withdrawn from their dosage regimen.
- Griseofulvin: Phenobarbital appears to interfere with the absorption of orally administered griseofulvin, thus decreasing its blood level. The effect of the resultant decreased blood levels of griseofulvin on therapeutic response has not been established. However, it would be preferable to avoid concomitant administration of these drugs.
-
Doxycycline: Phenobarbital has been shown to shorten the half-life of doxycycline for as long as 2 weeks after barbiturate therapy is discontinued.
This mechanism is probably through the induction of hepatic microsomal enzymes that metabolize the antibiotic. If phenobarbital and doxycycline are administered concurrently, the clinical response to doxycycline should be monitored closely. - Phenytoin, sodium valproate, valproic acid: The effect of barbiturates on the metabolism of phenytoin appears to be variable. Some investigators report an accelerating effect, while others report no effect. Because the effect of barbiturates on the metabolism of phenytoin is not predictable, phenytoin and barbiturate blood levels should be monitored more frequently if these drugs are given concurrently. Sodium valproate and valproic acid appear to decrease barbiturate metabolism; therefore, barbiturate blood levels should be monitored and appropriate dosage adjustments made as indicated.
- Central nervous system depressants: The concomitant use of other central nervous system depressants, including other sedatives or hypnotics, antihistamines, tranquilizers, or alcohol, may produce additive depressant effects.
- Monoamine oxidase inhibitors (MAOI): MAOI prolong the effects of barbiturates probably because metabolism of the barbiturate is inhibited.
- Estradiol, estrone, progesterone and other steroidal hormones: Pretreatment with or concurrent administration of phenobarbital may decrease the effect of estradiol by increasing its metabolism. There have been reports of patients treated with antiepileptic drugs (e.g., phenobarbital) who became pregnant while taking oral contraceptives. An alternate contraceptive method might be suggested to women taking phenobarbital.
Carcinogenesis:
- Animal data. Phenobarbital sodium is carcinogenic in mice and rats after lifetime administration. In mice, it produced benign and malignant liver cell tumors. In rats, benign liver cell tumors were observed very late in life.
-
Human data. In a 29-year epidemiological study of 9,136 patients who were treated on an anticonvulsant protocol that included phenobarbital, results indicated a higher than normal incidence of hepatic carcinoma. Previously, some of these patients were treated with thorotrast, a drug that is known to produce hepatic carcinomas. Thus, this study did not provide sufficient evidence that phenobarbital sodium is carcinogenic in humans.
Data from one retrospective study of 235 children in which the types of barbiturates are not identified suggested an association between exposure to barbiturates prenatally and an increased incidence of brain tumor. (Gold, E., et al., “Increased Risk of Brain Tumors in Children Exposed to Barbiturates,” Journal of National Cancer Institute, 61:1031-1034, 1978).
Pregnancy:
- Teratogenic effects. Pregnancy Category D—See “Warnings-Use in Pregnancy” section.
- Nonteratogenic effects. Reports of infants suffering from long-term barbiturate exposure in utero included the acute withdrawal syndrome of seizures and hyperirritability from birth to a delayed onset of up to 14 days. (See “Drug Abuse and Dependence” section.)
- Published studies in pregnant primates demonstrate that the administration of anesthetic and sedation drugs that block NMDA receptors and/or potentiate GABA activity during the period of peak brain development increases neuronal apoptosis in the developing brain of the offspring when used for longer than 3 hours. There are no data on pregnancy exposures in primates corresponding to periods prior to the third trimester in humans.
In a published study, administration of an anesthetic dose of ketamine for 24 hours on Gestation Day 122 increased neuronal apoptosis in the developing brain of the fetus. In other published studies, administration of either isoflurane or propofol for 5 hours on Gestation Day 120 resulted in increased neuronal and oligodendrocyte apoptosis in the developing brain of the offspring. With respect to brain development, this time period corresponds to the third trimester of gestation in the human. The clinical significance of these findings is not clear; however, studies in juvenile animals suggest neuroapoptosis correlates with long-term cognitive deficits (see “Warnings-Pediatric Neurotoxicity”, “Precautions-Pediatric Use”, and “Animal Pharmacology and/or Toxicology”).
Labor and delivery:
Hypnotic doses of these barbiturates do not appear to significantly impair uterine activity during labor. Full anesthetic doses of barbiturates decrease the force and frequency of uterine contractions. Administration of sedative-hypnotic barbiturates to the mother during labor may result in respiratory depression in the newborn. Premature infants are particularly susceptible to the depressant effects of barbiturates. If barbiturates are used during labor and delivery, resuscitation equipment should be available.
Data are currently not available to evaluate the effect of these barbiturates when forceps delivery or other intervention is necessary. Also, data are not available to determine the effect of these barbiturates on the later growth, development, and functional maturation of the child.
Nursing mothers:
Caution should be exercised when a barbiturate is administered to a nursing woman since small amounts of barbiturates are excreted in the milk.
Pediatric use:
No adequate well-controlled studies have been conducted in pediatric patients; however, safety and effectiveness of pentobarbital in pediatric patients is supported by numerous studies and case reports cited in the literature.
Pediatric dosing information for Nembutal is described in the DOSAGE AND ADMINISTRATION section.
Published juvenile animal studies demonstrate that the administration of anesthetic and sedation drugs, such as Pentobarbital Sodium Injection USP, (Nembutal) that either block NMDA receptors or potentiate the activity of GABA during the period of rapid brain growth or synaptogenesis, results in widespread neuronal and oligodendrocyte cell loss in the developing brain and alterations in synaptic morphology and neurogenesis. Based on comparisons across species, the window of vulnerability to these changes is believed to correlate with exposures in the third trimester of gestation through the first several months of life, but may extend out to approximately 3 years of age in humans.
In primates, exposure to 3 hours of ketamine that produced a light surgical plane of anesthesia did not increase neuronal cell loss, however, treatment regimens of 5 hours or longer of isoflurane increased neuronal cell loss. Data from isoflurane-treated rodents and ketamine-treated primates suggest that the neuronal and oligodendrocyte cell losses are associated with prolonged cognitive deficits in learning and memory. The clinical significance of these nonclinical findings is not known, and healthcare providers should balance the benefits of appropriate anesthesia in pregnant women, neonates, and young children who require procedures with the potential risks suggested by the nonclinical data (see “Warnings-Pediatric Neurotoxicity”, “Precautions-Pregnancy”, and “Animal Pharmacology and/or Toxicology”.)
Geriatric use:
Clinical studies of Nembutal have not included sufficient numbers of subjects aged 65 and over to determine whether elderly subjects respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal or cardiac function, and of concomitant disease or other drug therapy.
Elderly patients may react to barbiturates with marked excitement, depression, and confusion. In some persons, barbiturates repeatedly produce excitement rather than depression. Dosage should be reduced in the elderly because these patients may be more sensitive to barbiturates.
-
ADVERSE REACTIONS
The following adverse reactions and their incidence were compiled from surveillance of thousands of hospitalized patients. Because such patients may be less aware of certain of the milder adverse effects of barbiturates, the incidence of these reactions may be somewhat higher in fully ambulatory patients.
More than 1 in 100 patients. The most common adverse reaction estimated to occur at a rate of 1 to 3 patients per 100 is: Nervous System: Somnolence.
Less than 1 in 100 patients. Adverse reactions estimated to occur at a rate of less than 1 in 100 patients listed below, grouped by organ system, and by decreasing order of occurrence are:
Nervous system: Agitation, confusion, hyperkinesia, ataxia, CNS depression, nightmares, nervousness, psychiatric disturbance, hallucinations, insomnia, anxiety, dizziness, thinking abnormality.
Respiratory system: Hypoventilation, apnea.
Cardiovascular system: Bradycardia, hypotension, syncope.
Digestive system: Nausea, vomiting, constipation.
Other reported reactions: Headache, injection site reactions, hypersensitivity reactions (angioedema, skin rashes, exfoliative dermatitis), fever, liver damage, megaloblastic anemia following chronic phenobarbital use.
To report SUSPECTED ADVERSE REACTIONS, contact Oak Pharmaceuticals, Inc. at 1-800-932-5676 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
-
DRUG ABUSE AND DEPENDENCE
Pentobarbital sodium injection is subject to control by the Federal Controlled Substances Act under DEA schedule II.
Barbiturates may be habit forming. Tolerance, psychological dependence, and physical dependence may occur especially following prolonged use of high doses of barbiturates. Daily administration in excess of 400 milligrams (mg) of pentobarbital or secobarbital for approximately 90 days is likely to produce some degree of physical dependence. A dosage of from 600 to 800 mg taken for at least 35 days is sufficient to produce withdrawal seizures. The average daily dose for the barbiturate addict is usually about 1.5 grams. As tolerance to barbiturates develops, the amount needed to maintain the same level of intoxication increases; tolerance to a fatal dosage, however, does not increase more than two-fold. As this occurs, the margin between an intoxicating dosage and fatal dosage becomes smaller.
Symptoms of acute intoxication with barbiturates include unsteady gait, slurred speech, and sustained nystagmus. Mental signs of chronic intoxication include confusion, poor judgment, irritability, insomnia, and somatic complaints.
Symptoms of barbiturate dependence are similar to those of chronic alcoholism. If an individual appears to be intoxicated with alcohol to a degree that is radically disproportionate to the amount of alcohol in his or her blood the use of barbiturates should be suspected. The lethal dose of a barbiturate is far less if alcohol is also ingested.
The symptoms of barbiturate withdrawal can be severe and may cause death. Minor withdrawal symptoms may appear 8 to 12 hours after the last dose of a barbiturate. These symptoms usually appear in the following order: anxiety, muscle twitching, tremor of hands and fingers, progressive weakness, dizziness, distortion in visual perception, nausea, vomiting, insomnia, and orthostatic hypotension. Major withdrawal symptoms (convulsions and delirium) may occur within 16 hours and last up to 5 days after abrupt cessation of these drugs. Intensity of withdrawal symptoms gradually declines over a period of approximately 15 days. Individuals susceptible to barbiturate abuse and dependence include alcoholics and opiate abusers, as well as other sedative-hypnotic and amphetamine abusers.
Drug dependence to barbiturates arises from repeated administration of a barbiturate or agent with barbiturate-like effect on a continuous basis, generally in amounts exceeding therapeutic dose levels. The characteristics of drug dependence to barbiturates include: (a) a strong desire or need to continue taking the drug; (b) a tendency to increase the dose; (c) a psychic dependence on the effects of the drug related to subjective and individual appreciation of those effects; and (d) a physical dependence on the effects of the drug requiring its presence for maintenance of homeostasis and resulting in a definite, characteristic, and self-limited abstinence syndrome when the drug is withdrawn.
Treatment of barbiturate dependence consists of cautious and gradual withdrawal of the drug. Barbiturate-dependent patients can be withdrawn by using a number of different withdrawal regimens. In all cases withdrawal takes an extended period of time. One method involves substituting a 30 mg dose of phenobarbital for each 100 to 200 mg dose of barbiturate that the patient has been taking. The total daily amount of phenobarbital is then administered in 3 to 4 divided doses, not to exceed 600 mg daily. Should signs of withdrawal occur on the first day of treatment, a loading dose of 100 to 200 mg of phenobarbital may be administered IM in addition to the oral dose. After stabilization on phenobarbital, the total daily dose is decreased by 30 mg a day as long as withdrawal is proceeding smoothly. A modification of this regimen involves initiating treatment at the patient's regular dosage level and decreasing the daily dosage by 10 percent if tolerated by the patient.
Infants physically dependent on barbiturates may be given phenobarbital 3 to 10 mg/kg/day. After withdrawal symptoms (hyperactivity, disturbed sleep, tremors, hyperreflexia) are relieved, the dosage of phenobarbital should be gradually decreased and completely withdrawn over a 2-week period.
-
OVERDOSAGE
The toxic dose of barbiturates varies considerably. In general, an oral dose of 1 gram of most barbiturates produces serious poisoning in an adult. Death commonly occurs after 2 to 10 grams of ingested barbiturate. Barbiturate intoxication may be confused with alcoholism, bromide intoxication, and with various neurological disorders.
Acute overdosage with barbiturates is manifested by CNS and respiratory depression which may progress to Cheyne-Stokes respiration, areflexia, constriction of the pupils to a slight degree (though in severe poisoning they may show paralytic dilation), oliguria, tachycardia, hypotension, lowered body temperature, and coma. Typical shock syndrome (apnea, circulatory collapse, respiratory arrest, and death) may occur.
In extreme overdose, all electrical activity in the brain may cease, in which case a “flat” EEG normally equated with clinical death cannot be accepted. This effect is fully reversible unless hypoxic damage occurs. Consideration should be given to the possibility of barbiturate intoxication even in situations that appear to involve trauma.
Complications such as pneumonia, pulmonary edema, cardiac arrhythmias, congestive heart failure, and renal failure may occur. Uremia may increase CNS sensitivity to barbiturates. Differential diagnosis should include hypoglycemia, head trauma, cerebrovascular accidents, convulsive states, and diabetic coma. Blood levels from acute overdosage for some barbiturates are listed in Table 1.
Table 1. Concentration of Barbiturate in the Blood Versus Degree of CNS Depression * Categories of degree of depression in nontolerant persons:
1. Under the influence and appreciably impaired for purposes of driving a motor vehicle or performing tasks requiring alertness and unimpaired judgment and reaction time.
2. Sedated, therapeutic range, calm, relaxed, and easily aroused.
3. Comatose, difficult to arouse, significant depression of respiration.
4. Compatible with death in aged or ill persons or in presence of obstructed airway, other toxic agents, or exposure to cold.
5. Usual lethal level, the upper end of the range includes those who received some supportive treatment.
Blood barbiturate level in ppm (μg/mL) Barbiturate Onset/duration Degree of depression in nontolerant persons* 1 2 3 4 5 Pentobarbital Fast/short ≤2 0.5 to 3 10 to 15 12 to 25 15 to 40 Secobarbital Fast/short ≤2 0.5 to 5 10 to 15 15 to 25 15 to 40 Amobarbital Intermediate/
intermediate≤3 2 to 10 30 to 40 30 to 60 40 to 80 Butabarbital Intermediate/
intermediate≤5 3 to 25 40 to 60 50 to 80 60 to 100 Phenobarbital Slow/long ≤10 5 to 40 50 to 80 70 to 120 100 to 200 Treatment of overdosage is mainly supportive and consists of the following:
- Maintenance of an adequate airway, with assisted respiration and oxygen administration as necessary.
- Monitoring of vital signs and fluid balance.
- Fluid therapy and other standard treatment for shock, if needed.
- If renal function is normal, forced diuresis may aid in the elimination of the barbiturate. Alkalinization of the urine increases renal excretion of some barbiturates, especially phenobarbital, also aprobarbital and mephobarbital (which is metabolized to phenobarbital).
- Although not recommended as a routine procedure, hemodialysis may be used in severe barbiturate intoxications or if the patient is anuric or in shock.
- Patient should be rolled from side to side every 30 minutes.
- Antibiotics should be given if pneumonia is suspected.
- Appropriate nursing care to prevent hypostatic pneumonia, decubiti, aspiration, and other complications of patients with altered states of consciousness.
-
DOSAGE AND ADMINISTRATION
Dosages of barbiturates must be individualized with full knowledge of their particular characteristics and recommended rate of administration. Factors of consideration are the patient's age, weight, and condition. Parenteral routes should be used only when oral administration is impossible or impractical.
Intramuscular Administration: IM injection of the sodium salts of barbiturates should be made deeply into a large muscle, and a volume of 5 mL should not be exceeded at any one site because of possible tissue irritation. After IM injection of a hypnotic dose, the patient's vital signs should be monitored. The usual adult dosage of NEMBUTAL Sodium Solution is 150 to 200 mg as a single IM injection; the recommended pediatric dosage ranges from 2 to 6 mg/kg as a single IM injection not to exceed 100 mg.
Intravenous Administration: NEMBUTAL Sodium Solution should not be admixed with any other medication or solution. IV injection is restricted to conditions in which other routes are not feasible, either because the patient is unconscious (as in cerebral hemorrhage, eclampsia, or status epilepticus), or because the patient resists (as in delirium), or because prompt action is imperative. Slow IV injection is essential, and patients should be carefully observed during administration. This requires that blood pressure, respiration, and cardiac function be maintained, vital signs be recorded, and equipment for resuscitation and artificial ventilation be available. The rate of IV injection should not exceed 50 mg/min for pentobarbital sodium.
There is no average intravenous dose of NEMBUTAL Sodium Solution (pentobarbital sodium injection) that can be relied on to produce similar effects in different patients. The possibility of overdose and respiratory depression is remote when the drug is injected slowly in fractional doses.
A commonly used initial dose for the 70 kg adult is 100 mg. Proportional reduction in dosage should be made for pediatric or debilitated patients. At least one minute is necessary to determine the full effect of intravenous pentobarbital. If necessary, additional small increments of the drug may be given up to a total of from 200 to 500 mg for normal adults.
Anticonvulsant use: In convulsive states, dosage of NEMBUTAL Sodium Solution should be kept to a minimum to avoid compounding the depression which may follow convulsions. The injection must be made slowly with due regard to the time required for the drug to penetrate the blood-brain barrier.
-
HOW SUPPLIED
NEMBUTAL Sodium Solution (pentobarbital sodium injection, USP) is available in the following sizes:
20-mL multiple-dose vial, 1 g per vial (NDC: 76478-501-20); and 50-mL multiple-dose vial, 2.5 g per vial (NDC: 76478-501-50).
Each mL contains:
Pentobarbital Sodium, derivative of barbituric acid ……………….. 50 mg
Propylene glycol ………………………………………………… 40% v/v
Alcohol ………………………………………………………….. 10%
Water for Injection ………………………………………………. qs(pH adjusted to approximately 9.5 with hydrochloric acid and/or sodium hydroxide.)
Vial stoppers are latex free.
Exposure of pharmaceutical products to heat should be minimized. Avoid excessive heat. Protect from freezing. It is recommended that the product be stored at 20° to 25°C (68° to 77°F), however, brief excursions are permitted between 15° to 30°C (59° to 86°F). See USP controlled room temperature.
-
ANIMAL PHARMACOLOGY AND/OR TOXICOLOGY
Published studies in animals demonstrate that the use of anesthetic agents during the period of rapid brain growth or synaptogenesis results in widespread neuronal and oligodendrocyte cell loss in the developing brain and alterations in synaptic morphology and neurogenesis. Based on comparisons across species, the window of vulnerability to these changes is believed to correlate with exposures in the third trimester through the first several months of life, but may extend out to approximately 3 years of age in humans.
In primates, exposure to 3 hours of exposure to an anesthetic regimen that produced a light surgical plane of anesthesia did not increase neuronal cell loss, however, treatment regimens of 5 hours or longer increased neuronal cell loss. Data in rodents and in primates suggest that the neuronal and oligodendrocyte cell losses are associated with subtle but prolonged cognitive deficits in learning and memory. The clinical significance of these nonclinical findings is not known, and healthcare providers should balance the benefits of appropriate anesthesia in neonates and young children who require procedures against the potential risks suggested by the nonclinical data (see “Warnings-Pediatric Neurotoxicity” and “Precautions-Pregnancy and Pediatric Use”).
AKORN
Distributed by: Akorn, Inc.
Lake Forest, IL 60045OAK
Manufactured for: Oak Pharmaceuticals, Inc.
® Trademark of Oak Pharmaceuticals, Inc.OPNM00N Rev. 04/19
-
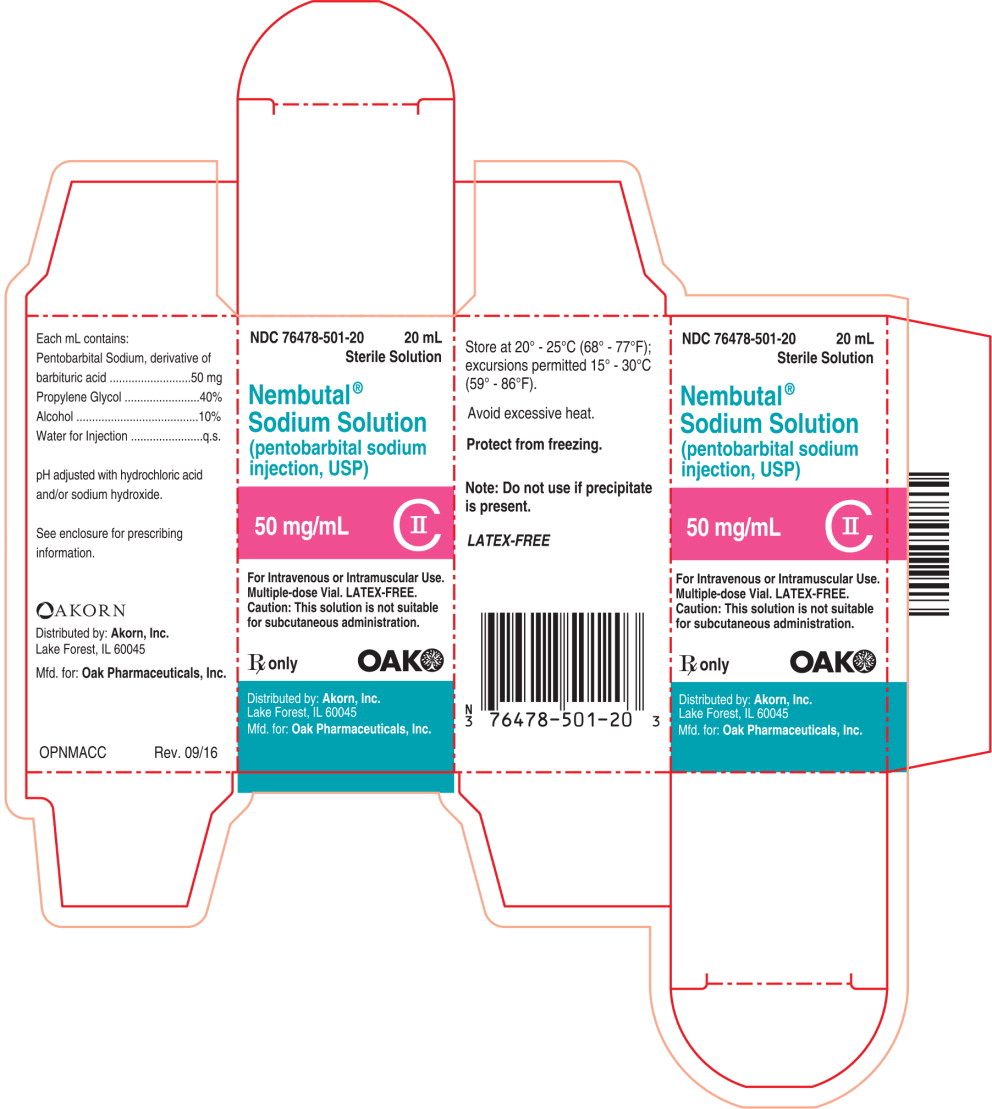
PRINCIPAL DISPLAY PANEL
Principal Display Panel Text for Container Label:
NDC: 76478-501-20 20 mL Sterile Solution
Nembutal®
Sodium Solution
(pentobarbital sodium injection, USP)
50 mg/mL CII
For Intravenous or Intramuscular Use.
Multiple-dose Vial. LATEX-FREE
Rx only Oak Logo
-
PRINCIPAL DISPLAY PANEL
Principal Display Panel Text for Carton Label:
NDC: 76478-501-20 20 mL
Sterile Solution
Nembutal®
Sodium Solution
(pentobarbital sodium
injection, USP)
50 mg/mL CII
For Intravenous or Intramuscular Use.
Multiple-dose Vial. LATEX-FREE.
Caution: This solution is not suitable
for subcutaneous administration.
Rx only Oak Logo
-
INGREDIENTS AND APPEARANCE
NEMBUTAL SODIUM
pentobarbital sodium injectionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 76478-501 Route of Administration INTRAVENOUS, INTRAMUSCULAR DEA Schedule CII Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength pentobarbital sodium (UNII: NJJ0475N0S) (pentobarbital - UNII:I4744080IR) pentobarbital sodium 50 mg in 1 mL Inactive Ingredients Ingredient Name Strength Propylene Glycol (UNII: 6DC9Q167V3) Alcohol (UNII: 3K9958V90M) Water (UNII: 059QF0KO0R) Hydrochloric Acid (UNII: QTT17582CB) Sodium Hydroxide (UNII: 55X04QC32I) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 76478-501-20 1 in 1 CARTON 09/19/1973 1 20 mL in 1 VIAL; Type 0: Not a Combination Product 2 NDC: 76478-501-50 1 in 1 CARTON 09/19/1973 2 50 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA083246 09/19/1973 Labeler - Oak Pharmaceuticals, Inc. (Subsidiary of Akorn, Inc.) (968937719) Establishment Name Address ID/FEI Business Operations Akorn, Inc. 063434679 PACK(76478-501) , LABEL(76478-501) Establishment Name Address ID/FEI Business Operations Akorn, Inc. 155135783 MANUFACTURE(76478-501) , ANALYSIS(76478-501) , STERILIZE(76478-501)

© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.