PAMIDRONATE DISODIUM injection, powder, lyophilized, for solution
Pamidronate Disodium by
Drug Labeling and Warnings
Pamidronate Disodium by is a Prescription medication manufactured, distributed, or labeled by Areva Pharmaceuticals Inc., Cipla Limited. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
DESCRIPTION
Pamidronate disodium for Injection USP, is a bisphosphonate available in 30 mg, 60 mg or 90 mg vials for intravenous administration. Each 30-mg, 60-mg and 90-mg vial contains, respectively, 30 mg, 60 mg and 90 mg of sterile, lyophilized pamidronate disodium and 470 mg, 400 mg and 375 mg of mannitol, USP. The pH of a 1% solution of pamidronate disodium in distilled water is approximately 8.3. Pamidronate disodium, a member of the group of chemical compounds known as bisphosphonates, is an analog of pyrophosphate. Pamidronate disodium is designated chemically as phosphonic acid (3-amino-1-hydroxypropylidene) bis-, disodium salt, pentahydrate, and its structural formula is
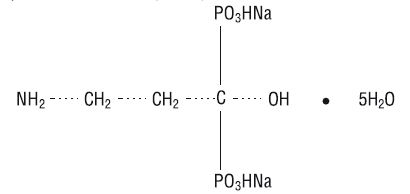
Pamidronate disodium is a white-to-practically-white powder. It is soluble in water and in 2N sodium hydroxide, sparingly soluble in 0.1N hydrochloric acid and in 0.1N acetic acid, and practically insoluble in organic solvents. Its molecular formula is C 3H 9NO 7P 2Na 25H 2O and its molecular weight is 369.1.
Inactive Ingredients: mannitol USP and phosphoric acid (for adjustment to pH 6.5 prior to lyophilization).
-
CLINICAL PHARMACOLOGY
The principal pharmacologic action of pamidronate disodium is inhibition of bone resorption. Although the mechanism of antiresorptive action is not completely understood, several factors are thought to contribute to this action. Pamidronate disodium adsorbs to calcium phosphate (hydroxyapatite) crystals in bone and may directly block dissolution of this mineral component of bone. In vitro studies also suggest that inhibition of osteoclast activity contributes to inhibition of bone resorption. In animal studies, at doses recommended for the treatment of hypercalcemia, pamidronate disodium inhibits bone resorption apparently without inhibiting bone formation and mineralization. Of relevance to the treatment of hypercalcemia of malignancy is the finding that pamidronate disodium inhibits the accelerated bone resorption that results from osteoclast hyperactivity induced by various tumors in animal studies.
Pharmacokinetics
Cancer patients (n=24) who had minimal or no bony involvement were given an intravenous infusion of 30, 60, or 90 mg of pamidronate disodium over 4 hours and 90 mg of pamidronate disodium over 24 hours ( Table 1).
Distribution
The mean ± SD body retention of pamidronate was calculated to be 54 ± 16% of the dose over 120 hours.
Excretion
After administration of 30, 60, and 90 mg of pamidronate disodium over 4 hours, and 90 mg of pamidronate disodium over 24 hours, an overall mean ± SD of 46 ± 16% of the drug was excreted unchanged in the urine within 120 hours. Cumulative urinary excretion was linearly related to dose. The mean ± SD elimination half-life is 28 ± 7 hours. Mean ± SD total and renal clearances of pamidronate were 107 ± 50 mL/min and 49 ± 28 mL/min, respectively. The rate of elimination from bone has not been determined.
Special Populations
There are no data available on the effects of age, gender, or race on the pharmacokinetics of pamidronate.
Renal Insufficiency
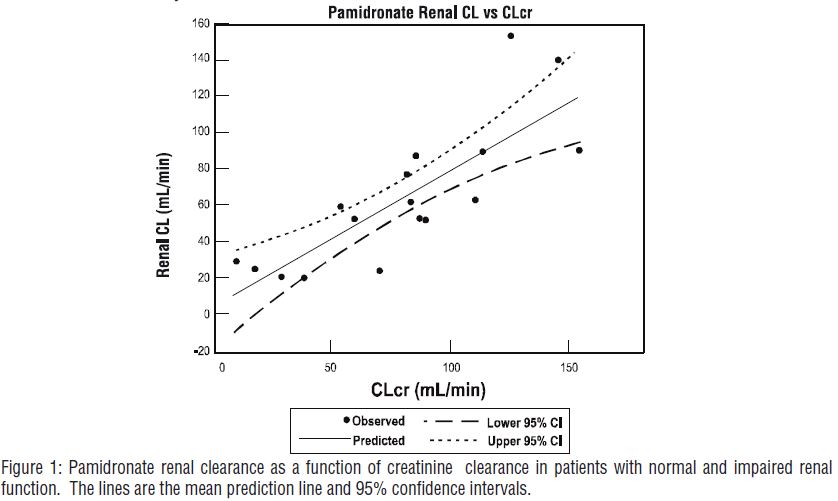
The pharmacokinetics of pamidronate were studied in cancer patients (n=19) with normal and varying degrees of renal impairment. Each patient received a single 90-mg dose of pamidronate disodium infused over 4 hours. The renal clearance of pamidronate in patients was found to closely correlate with creatinine clearance (see Figure 1). A trend toward a lower percentage of drug excreted unchanged in urine was observed in renally impaired patients. Adverse experiences noted were not found to be related to changes in renal clearance of pamidronate. Given the recommended dose, 90 mg infused over 4 hours, excessive accumulation of pamidronate in renally impaired patients is not anticipated if pamidronate disodium is administered on a monthly basis.
Hepatic Insufficiency
The pharmacokinetics of pamidronate were studied in male cancer patients at risk for bone metastases with normal hepatic function (n=6) and mild to moderate hepatic dysfunction (n=7). Each patient received a single 90-mg dose of pamidronate disodium infused over 4 hours. Although there was a statistically significant difference in the pharmacokinetics between patients with normal and impaired hepatic function, the difference was not considered clinically relevant. Patients with hepatic impairment exhibited higher mean AUC (53%) and C max (29%),and decreased plasma clearance (33%) values. Nevertheless, pamidronate was still rapidly cleared from the plasma. Drug levels were not detectable in patients by 12 to 36 hours after drug infusion. Because pamidronate disodium is administered on a monthly basis, drug accumulation is not expected. No changes in pamidronate disodium dosing regimen are recommended for patients with mild to moderate abnormal hepatic function. Pamidronate disodium has not been studied in patients with severe hepatic impairment.
Drug-Drug Interactions
There are no human pharmacokinetic data for drug interactions with pamidronate disodium.
Table 1 Mean (SD, CV%) Pamidronate Pharmacokinetic Parameters in Cancer Patients
(n=6 for each group)Dose
(infusion rate)Maximum
Concentration
(μg/mL)Percent of dose
excreted in urineTotal
Clearance
(mL/min)Renal
Clearance
(mL/min)30 mg
(4 hrs)0.73
(0.14, 19.1%)43.9
(14.0, 31.9%)136
(44, 32.4%)58
(27, 46.5%)60 mg
(4 hrs)1.44
(0.57, 39.6%)47.4
(47.4, 54.4%)88
(56, 63.6%)42
(28, 66.7%)90 mg
(4 hrs)2.61
(0.74, 28.3%)45.3
(25.8, 56.9%)103
(37, 35.9%)44
(16, 36.4%)90 mg
(24 hrs)1.38
(1.97, 142.7%)47.5
(10.2, 21.5%)101
(58, 57.4%)52
(42, 80.8%)After intravenous administration of radiolabeled pamidronate in rats, approximately 50%-60% of the compound was rapidly adsorbed by bone and slowly eliminated from the body by the kidneys. In rats given 10 mg/kg bolus injections of radiolabeled pamidronate disodium, approximately 30% of the compound was found in the liver shortly after administration and was then redistributed to bone or eliminated by the kidneys over 24-48 hours. Studies in rats injected with radiolabeled pamidronate disodium showed that the compound was rapidly cleared from the circulation and taken up mainly by bones, liver, spleen, teeth, and tracheal cartilage. Radioactivity was eliminated from most soft tissues within 1-4 days; was detectable in liver and spleen for 1 and 3 months, respectively; and remained high in bones, trachea, and teeth for 6 months after dosing. Bone uptake occurred preferentially in areas of high bone turnover. The terminal phase of elimination half-life in bone was estimated to be approximately 300 days.
Pharmacodynamics
Serum phosphate levels have been noted to decrease after administration of pamidronate disodium, presumably because of decreased release of phosphate from bone and increased renal excretion as parathyroid hormone levels, which are usually suppressed in hypercalcemia associated with malignancy, return toward normal. Phosphate therapy was administered in 30% of the patients in response to a decrease in serum phosphate levels. Phosphate levels usually returned toward normal within 7-10 days.
Urinary calcium/creatinine and urinary hydroxyproline/creatinine ratios decrease and usually return to within or below normal after treatment with Pamidronate disodium. These changes occur within the first week after treatment, as do decreases in serum calcium levels, and are consistent with an antiresorptive pharmacologic action.
Hypercalcemia of Malignancy
Osteoclastic hyperactivity resulting in excessive bone resorption is the underlying pathophysiologic derangement in metastatic bone disease and hypercalcemia of malignancy. Excessive release of calcium into the blood as bone is resorbed results in polyuria and gastrointestinal disturbances, with progressive dehydration and decreasing glomerular filtration rate. This, in turn, results in increased renal resorption of calcium, setting up a cycle of worsening systemic hypercalcemia. Correction of excessive bone resorption and adequate fluid administration to correct volume deficits are therefore essential to the management of hypercalcemia.
Most cases of hypercalcemia associated with malignancy occur in patients who have breast cancer; squamous-cell tumors of the lung or head and neck; renal-cell carcinoma; and certain hematologic malignancies, such as multiple myeloma and some types of lymphomas. A few less-common malignancies, including vasoactive intestinal-peptide-producing tumors and cholangiocarcinoma, have a high incidence of hypercalcemia as a metabolic complication. Patients who have hypercalcemia of malignancy can generally be divided into two groups, according to the pathophysiologic mechanism involved.
In humoral hypercalcemia, osteoclasts are activated and bone resorption is stimulated by factors such as parathyroid-hormone-related protein, which are elaborated by the tumor and circulate systemically. Humoral hypercalcemia usually occurs in squamous-cell malignancies of the lung or head and neck or in genitourinary tumors such as renal-cell carcinoma or ovarian cancer. Skeletal metastases may be absent or minimal in these patients.
Extensive invasion of bone by tumor cells can also result in hypercalcemia due to local tumor products that stimulate bone resorption by osteoclasts. Tumors commonly associated with locally mediated hypercalcemia include breast cancer and multiple myeloma.
Total serum calcium levels in patients who have hypercalcemia of malignancy may not reflect the severity of hypercalcemia, since concomitant hypoalbuminemia is commonly present. Ideally, ionized calcium levels should be used to diagnose and follow hypercalcemic conditions; however, these are not commonly or rapidly available in many clinical situations. Therefore, adjustment of the total serum calcium value for differences in albumin levels is often used in place of measurement of ionized calcium; several nomograms are in use for this type of calculation (see DOSAGE AND ADMINISTRATION).
Clinical Trials
In one double-blind clinical trial, 52 patients who had hypercalcemia of malignancy were enrolled to receive 30 mg, 60 mg, or 90 mg of pamidronate disodium as a single 24-hour intravenous infusion if their corrected serum calcium levels were ≥12.0 mg/dL after 48 hours of saline hydration.
The mean baseline-corrected serum calcium for the 30-mg, 60-mg, and 90-mg groups were 13.8 mg/dL,13.8 mg/dL, and 13.3 mg/dL, respectively.
The majority of patients (64%) had decreases in albumin-corrected serum calcium levels by 24 hours after initiation of treatment. Mean-corrected serum calcium levels at days 2-7 after initiation of treatment with pamidronate disodium were significantly reduced from baseline in all three dosage groups. As a result, by 7 days after initiation of treatment with pamidronate disodium, 40%, 61%, and 100% of the patients receiving 30 mg, 60 mg, and 90 mg of pamidronate disodium, respectively, had normal-corrected serum calcium levels. Many patients (33%-53%) in the 60-mg and 90-mg dosage groups continued to have normal-corrected serum calcium levels, or a partial response (≥15% decrease of corrected serum calcium from baseline), at Day 14.
In a second double-blind, controlled clinical trial, 65 cancer patients who had corrected serum calcium levels of ≥12.0 mg/ dL after at least 24 hours of saline hydration were randomized to receive either 60 mg of pamidronate disodium as a single 24-hour intravenous infusion or 7.5 mg/kg of etidronate disodium as a 2-hour intravenous infusion daily for 3 days. Thirty patients were randomized to receive pamidronate disodium and 35 to receive etidronate disodium.
The mean baseline-corrected serum calcium for the pamidronate disodium 60-mg and etidronate disodium groups were 14.6 mg/dL and 13.8 mg/dL, respectively.
By Day 7, 70% of the patients in the pamidronate disodium group and 41% of the patients in the etidronate disodium group had normal-corrected serum calcium levels (P<0.05). When partial responders (≥15% decrease of serum calcium from baseline) were also included, the response rates were 97% for the pamidronate disodium group and 65% for the etidronate disodium group (P<0.01). Mean-corrected serum calcium for the pamidronate disodium and etidronate disodium groups decreased from baseline values to 10.4 and 11.2 mg/dL, respectively, on Day 7. At Day 14, 43% of patients in the pamidronate disodium group and 18% of patients in the etidronate disodium group still had normal-corrected serum calcium levels, or maintenance of a partial response. For responders in the pamidronate disodium and etidronate disodium groups, the median duration of response was similar (7 and 5 days, respectively). The time course of effect on corrected serum calcium is summarized in the following table.
- * Comparison between treatment groups
Change in Corrected Serum Calcium by Time from Initiation of Treatment Time
(hr)Mean Change from Baseline in Corrected Serum Calcium (mg/dL) Pamidronate disodium Etidronate disodium P-Value * Baseline 14.6 13.8 24 -0.3 -0.5 48 -1.5 -1.1 72 -2.6 -2.0 96 -3.5 -2.0 <0.01 168 -4.1 -2.5 <0.01 In a third multicenter, randomized, parallel double-blind trial, a group of 69 cancer patients with hypercalcemia was enrolled to receive 60 mg of pamidronate disodium as a 4- or 24-hour infusion, which was compared to a saline-treatment group. Patients who had a corrected serum calcium level of ≥12.0 mg/dL after 24 hours of saline hydration were eligible for this trial.
The mean baseline-corrected serum calcium levels for pamidronate disodium 60-mg 4-hour infusion, pamidronate disodium 60-mg 24-hour infusion, and saline infusion were 14.2 mg/dL, 13.7 mg/dL, and 13.7 mg/dL, respectively.
By Day 7 after initiation of treatment, 78%, 61%, and 22% of the patients had normal-corrected serum calcium levels for the 60-mg 4-hour infusion, 60-mg 24-hour infusion, and saline infusion, respectively. At Day 14, 39% of the patients in the pamidronate disodium 60-mg 4-hour infusion group and 26% of the patients in the pamidronate disodium 60-mg 24-hour infusion group had normal-corrected serum calcium levels or maintenance of a partial response.
For responders, the median duration of complete responses was 4 days and 6.5 days for pamidronate disodium 60-mg 4-hour infusion and pamidronate disodium 60-mg 24-hour infusion, respectively.
In all three trials, patients treated with pamidronate disodium had similar response rates in the presence or absence of bone metastases. Concomitant administration of furosemide did not affect response rates.
Thirty-two patients who had recurrent or refractory hypercalcemia of malignancy were given a second course of 60 mg of pamidronate disodium over a 4- or 24-hour period. Of these, 41% showed a complete response and 16% showed a partial response to the retreatment, and these responders had about a 3-mg/dL fall in mean-corrected serum calcium levels 7 days after retreatment.
In a fourth multicenter, randomized, double-blind trial, 103 patients with cancer and hypercalcemia (corrected serum calcium ≥12.0 mg/dL) received 90 mg of pamidronate disodium as a 2-hour infusion. The mean baseline corrected serum calcium was 14.0 mg/dL. Patients were not required to receive IV hydration prior to drug administration, but all subjects did receive at least 500 mL of IV saline hydration concomitantly with the pamidronate infusion. By Day 10 after drug infusion, 70% of patients had normal corrected serum calcium levels (<10.8 mg/dL).
Paget's Disease
Paget’s disease of bone (osteitis deformans) is an idiopathic disease characterized by chronic, focal areas of bone destruction complicated by concurrent excessive bone repair, affecting one or more bones. These changes result in thickened but weakened bones that may fracture or bend under stress. Signs and symptoms may be bone pain, deformity, fractures, neurological disorders resulting from cranial and spinal nerve entrapment and from spinal cord and brain stem compression, increased cardiac output to the involved bone, increased serum alkaline phosphatase levels (reflecting increased bone formation) and/or urine hydroxyproline excretion (reflecting increased bone resorption).
Clinical Trials
In one double-blind clinical trial, 64 patients with moderate to severe Paget’s disease of bone were enrolled to receive 5 mg, 15 mg, or 30 mg of pamidronate disodium as a single 4-hour infusion on 3 consecutive days, for total doses of 15 mg, 45 mg, and 90 mg of pamidronate disodium.
The mean baseline serum alkaline phosphatase levels were 1,409 U/L, 983 U/L, and 1,085 U/L, and the mean baseline urine hydroxyproline/creatinine ratios were 0.25, 0.19, and 0.19 for the 15-mg, 45-mg, and 90-mg groups, respectively.
The effects of pamidronate disodium on serum alkaline phosphatase (SAP) and urine hydroxyproline/creatinine ratios (UOHP/C) are summarized in the following table.
Percent of Patients With Significant % Decreases in SAP and UOHP/C SAP UOHP/C % Decrease 15 mg 45 mg 90 mg 15 mg 45 mg 90 mg ≥50 26 33 60 15 47 72 ≥30 40 65 83 35 57 85 The median maximum percent decreases from baseline in serum alkaline phosphatase and urine hydroxyproline/creatinine ratios were 25%, 41%, and 57%, and 25%, 47%, and 61% for the 15-mg, 45-mg, and 90-mg groups, respectively. The median time to response (≥50% decrease) for serum alkaline phosphatase was approximately 1 month for the 90-mg group, and the response duration ranged from 1 to 372 days.
No statistically significant differences between treatment groups, or statistically significant changes from baseline were observed for the bone pain response, mobility, and global evaluation in the 45-mg and 90-mg groups. Improvement in radiologic lesions occurred in some patients in the 90-mg group.
Twenty-five patients who had Paget’s disease were retreated with 90 mg of pamidronate disodium. Of these, 44% had a ≥50% decrease in serum alkaline phosphatase from baseline after treatment, and 39% had a ≥50% decrease in urine hydroxyproline/creatinine ratio from baseline after treatment.
Osteolytic Bone Metastases of Breast Cancer and Osteolytic Lesions of Multiple Myeloma
Osteolytic bone metastases commonly occur in patients with multiple myeloma or breast cancer. These cancers demonstrate a phenomenon known as osteotropism, meaning they possess an extraordinary affinity for bone. The distribution of osteolytic bone metastases in these cancers is predominantly in the axial skeleton, particularly the spine, pelvis, and ribs, rather than the appendicular skeleton, although lesions in the proximal femur and humerus are not uncommon. This distribution is similar to the red bone marrow in which slow blood flow possibly assists attachment of metastatic cells. The surface-to-volume ratio of trabecular bone is much higher than cortical bone, and therefore disease processes tend to occur more floridly in trabecular bone than at sites of cortical tissue.
These bone changes can result in patients having evidence of osteolytic skeletal destruction leading to severe bone pain that requires either radiation therapy or narcotic analgesics (or both) for symptomatic relief. These changes also cause pathologic fractures of bone in both the axial and appendicular skeleton. Axial skeletal fractures of the vertebral bodies may lead to spinal cord compression or vertebral body collapse with significant neurologic complications. Also, patients may experience episode(s) of hypercalcemia.
Clinical Trials
In a double-blind, randomized, placebo-controlled trial, 392 patients with advanced multiple myeloma were enrolled to receive pamidronate disodium or placebo in addition to their underlying antimyeloma therapy to determine the effect of pamidronate disodium on the occurrence of skeletal-related events (SREs). SREs were defined as episodes of pathologic fractures, radiation therapy to bone, surgery to bone, and spinal cord compression. Patients received either 90 mg of pamidronate disodium or placebo as a monthly 4-hour intravenous infusion for 9 months. Of the 392 patients, 377 were evaluable for efficacy (196 pamidronate disodium, 181 placebo). The proportion of patients developing any SRE was significantly smaller in the pamidronate disodium group (24% vs 41%, P<0.001), and the mean skeletal morbidity rate (#SRE/year) was significantly smaller for pamidronate disodium patients than for placebo patients (mean: 1.1 vs 2.1, P<.02). The times to the first SRE occurrence, pathologic fracture, and radiation to bone were significantly longer in the pamidronate disodium group (P=.001, .006, and .046, respectively). Moreover, fewer pamidronate disodium patients suffered any pathologic fracture (17% vs 30%, P=.004) or needed radiation to bone (14% vs 22%, P=.049).
In addition, decreases in pain scores from baseline occurred at the last measurement for those pamidronate disodium patients with pain at baseline (P=.026) but not in the placebo group. At the last measurement, a worsening from baseline was observed in the placebo group for the Spitzer quality of life variable (P<.001) and ECOG performance status (P<.011) while there was no significant deterioration from baseline in these parameters observed in pamidronate disodium-treated patients.*
After 21 months, the proportion of patients experiencing any skeletal event remained significantly smaller in the pamidronate disodium group than the placebo group (P=.015). In addition, the mean skeletal morbidity rate (#SRE/year) was 1.3 vs 2.2 for pamidronate disodium patients vs placebo patients (P=.008), and time to first SRE was significantly longer in the pamidronate disodium group compared to placebo (P=.016). Fewer pamidronate disodium patients suffered vertebral pathologic fractures (16% vs 27%, P=.005). Survival of all patients was not different between treatment groups.
Two double-blind, randomized, placebo-controlled trials compared the safety and efficacy of 90 mg of pamidronate disodium infused over 2 hours every 3 to 4 weeks for 24 months to that of placebo in preventing SREs in breast cancer patients with osteolytic bone metastases who had one or more predominantly lytic metastases of at least 1 cm in diameter: one in patients being treated with antineoplastic chemotherapy and the second in patients being treated with hormonal antineoplastic therapy at trial entry.
382 patients receiving chemotherapy were randomized, 185 to pamidronate disodium and 197 to placebo. 372 patients receiving hormonal therapy were randomized, 182 to pamidronate disodium and 190 to placebo. All but three patients were evaluable for efficacy. Patients were followed for 24 months of therapy or until they went off study. Median duration of follow-up was 13 months in patients receiving chemotherapy and 17 months in patients receiving hormone therapy. Twenty-five percent of the patients in the chemotherapy study and 37% of the patients in the hormone therapy study received pamidronate disodium for 24 months. The efficacy results are shown in the table below:
PD = Pamidronate Disodium - * Fractures and radiation to bone were two of several secondary endpoints. The statistical significance of these analyses may be overestimated since numerous analyses were performed.
- † NR = Not Reached
Breast Cancer Patients
Receiving ChemotherapyBreast Cancer Patients
Receiving Hormonal TherapyAny SRE Radiation Fractures Any SRE Radiation Fractures PD P PD P PD P PD P PD P PD P N 185 195 185 195 185 195 182 189 182 189 182 189 Skeletal Morbidity
Rate
(#SRE/year)
Mean2.5 3.7 0.8 1.3 1.6 2.2 2.4 3.6 0.6 1.2 1.6 2.2 P-Value <.001 <.001 * .018 * .021 .013 * .040 * Proportion of
patients having
an SRE46% 65% 28% 45% 36% 49% 55% 63% 31% 40% 45% 55% P-Value <.001 <.001 * .014 * .094 .058 * .054 * Median Time to
SRE (months)13.9 7.0 NR † 14.2 25.8 13.3 10.9 7.4 NR † 23.4 20.6 12.8 P-Value <.001 <.001 * .009 * .118 .016 * .113 * Bone lesion response was radiographically assessed at baseline and at 3, 6, and 12 months. The complete + partial response rate was 33% in pamidronate disodium patients and 18% in placebo patients treated with chemotherapy (P=.001). No difference was seen between pamidronate disodium and placebo in hormonally-treated patients.
Pain and analgesic scores, ECOG performance status and Spitzer quality of life index were measured at baseline and periodically during the trials. The changes from baseline to the last measurement carried forward are shown in the following table:
- * The statistical significance of analyses of these secondary endpoints of pain, quality of life, and performance status in all three trials may be overestimated since numerous analyses were performed.
Mean Change (Δ) from Baseline at Last Measurement Breast Cancer Patients
Receiving ChemotherapyBreast Cancer Patients
Receiving Hormonal TherapyPD P PD vs P PD P PD vs P N Mean Δ N Mean Δ P-Value * N Mean Δ N Mean Δ P-Value * Pain Score 175 +0.93 183 +1.69 .050 173 +0.50 179 +1.60 .007 Analgesic Score 175 +0.74 183 +1.55 .009 173 +0.90 179 +2.28 <.001 ECOG PS 178 +0.81 186 +1.19 .002 175 +0.95 182 +0.90 .773 Spitzer QOL 177 -1.76 185 -2.21 .103 173 -1.86 181 -2.05 .409 Decreases in pain, analgesic scores and ECOG PS, and increases in Spitzer QOL indicate an improvement from baseline. -
INDICATIONS AND USAGE
Hypercalcemia of Malignancy
Pamidronate disodium for Injection USP, in conjunction with adequate hydration, is indicated for the treatment of moderate or severe hypercalcemia associated with malignancy, with or without bone metastases. Patients who have either epidermoid or non-epidermoid tumors respond to treatment with Pamidronate disodium for Injection USP. Vigorous saline hydration, an integral part of hypercalcemia therapy, should be initiated promptly and an attempt should be made to restore the urine output to about 2 L/day throughout treatment. Mild or asymptomatic hypercalcemia may be treated with conservative measures (i.e., saline hydration, with or without loop diuretics). Patients should be hydrated adequately throughout the treatment, but overhydration, especially in those patients who have cardiac failure, must be avoided. Diuretic therapy should not be employed prior to correction of hypovolemia. The safety and efficacy of Pamidronate disodium for Injection USP in the treatment of hypercalcemia associated with hyperparathyroidism or with other non-tumor-related conditions has not been established.
Paget's Disease
Pamidronate disodium for Injection USP is indicated for the treatment of patients with moderate to severe Paget’s disease of bone. The effectiveness of Pamidronate disodium for Injection USP was demonstrated primarily in patients with serum alkaline phosphatase ≥3 times the upper limit of normal. Pamidronate disodium for Injection USP therapy in patients with Paget’s disease has been effective in reducing serum alkaline phosphatase and urinary hydroxyproline levels by ≥50% in at least 50% of patients, and by ≥30% in at least 80% of patients. Pamidronate disodium for Injection USP therapy has also been effective in reducing these biochemical markers in patients with Paget’s disease who failed to respond, or no longer responded to other treatments.
Osteolytic Bone Metastases of Breast Cancer and Osteolytic Lesions of Multiple Myeloma
Pamidronate disodium for Injection USP is indicated, in conjunction with standard antineoplastic therapy, for the treatment of osteolytic bone metastases of breast cancer and osteolytic lesions of multiple myeloma. The Pamidronate disodium for Injection USP treatment effect appeared to be smaller in the study of breast cancer patients receiving hormonal therapy than in the study of those receiving chemotherapy, however, overall evidence of clinical benefit has been demonstrated (see CLINICAL PHARMACOLOGY, Osteolytic Bone Metastases of Breast Cancer and Osteolytic Lesions of Multiple Myeloma, Clinical Trials section).
- CONTRAINDICATIONS
-
WARNINGS
Deterioration in Renal Function
Bisphosphonates, including pamidronate disodium, have been associated with renal toxicity manifested as deterioration of renal function and potential renal failure.
DUE TO THE RISK OF CLINICALLY SIGNIFICANT DETERIORATION IN RENAL FUNCTION, WHICH MAY PROGRESS TO RENAL FAILURE, SINGLE DOSES OF PAMIDRONATE DISODIUM SHOULD NOT EXCEED 90 MG (see DOSAGE AND ADMINISTRATION for appropriate infusion durations). Renal deterioration, progression to renal failure, and dialysis have been reported in patients after the initial or a single dose of pamidronate disodium.
Focal segmental glomerulosclerosis (including the collapsing variant) with or without nephrotic syndrome, which may lead to renal failure, has been reported in pamidronate disodium-treated patients, particularly in the setting of multiple myeloma and breast cancer. Some of these patients had gradual improvement in renal status after pamidronate disodium was discontinued.
Patients who receive pamidronate disodium should have serum creatinine assessed prior to each treatment. Patients treated with pamidronate disodium for bone metastases should have the dose withheld if renal function has deteriorated. (See DOSAGE AND ADMINISTRATION.)
PREGNANCY
Bisphosphonates, such as pamidronate disodium, are incorporated into the bone matrix, from where they are gradually released over periods of weeks to years. Pamidronate disodium may cause fetal harm when administered to a pregnant woman.
In reproductive studies in rats and rabbits, pamidronate doses equivalent to 0.6 to 8.3 times the highest human recommended dose resulted in maternal toxicity and embryo/fetal effects. There are no adequate and well-controlled studies of pamidronate disodium in pregnant women. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, apprise the patient of the potential hazard to the fetus (See PRECAUTIONS, Pregnancy Category D)
-
PRECAUTIONS
General
Standard hypercalcemia-related metabolic parameters, such as serum levels of calcium, phosphate, magnesium, and potassium, should be carefully monitored following initiation of therapy with pamidronate disodium. Cases of asymptomatic hypophosphatemia (12%), hypokalemia (7%), hypomagnesemia (11%), and hypocalcemia (5%-12%), were reported in pamidronate disodium-treated patients. Rare cases of symptomatic hypocalcemia (including tetany) have been reported in association with pamidronate disodium therapy. If hypocalcemia occurs, short-term calcium therapy may be necessary. In Paget’s disease of bone, 17% of patients treated with 90 mg of pamidronate disodium showed serum calcium levels below 8 mg/dL.
Patients with a history of thyroid surgery may have relative hypoparathyroidism that may predispose to hypocalcemia with pamidronate disodium.
Renal Insufficiency
Pamidronate disodium is excreted intact primarily via the kidney, and the risk of renal adverse reactions may be greater in patients with impaired renal function. Patients who receive pamidronate disodium should have serum creatinine assessed prior to each treatment. In patients receiving pamidronate disodium for bone metastases, who show evidence of deterioration in renal function, pamidronate disodium treatment should be withheld until renal function returns to baseline (see WARNINGS and DOSAGE AND ADMINISTRATION).
In clinical trials, patients with renal impairment (serum creatinine >3.0 mg/dL) have not been studied. Limited pharmacokinetic data exist in patients with creatinine clearance <30 ml/min (See Clinical Pharmacology, Pharmacokinetics.) For the treatment of bone metastases, the use of pamidronate disodium in patients with severe renal impairment is not recommended. In other indications, clinical judgment should determine whether the potential benefit outweighs the potential risk in such patients.
Osteonecrosis of the Jaw
Osteonecrosis of the jaw (ONJ) has been reported predominantly in cancer patients treated with intravenous bisphosphonates, including pamidronate disodium. Many of these patients were also receiving chemotherapy and corticosteroids which may be risk factors for ONJ. Postmarketing experience and the literature suggest a greater frequency of reports of ONJ based on tumor type (advanced breast cancer, multiple myeloma), and dental status (dental extraction, periodontal disease, local trauma including poorly fitting dentures). Many reports of ONJ involved patients with signs of local infection including osteomyelitis.
Cancer patients should maintain good oral hygiene and should have a dental examination with preventive dentistry prior to treatment with bisphosphonates.
While on treatment, these patients should avoid invasive dental procedures if possible. For patients who develop ONJ while on bisphosphonate therapy, dental surgery may exacerbate the condition. For patients requiring dental procedures, there are no data available to suggest whether discontinuation of bisphosphonate treatment reduces the risk of ONJ. Clinical judgment of the treating physician should guide the management plan of each patient based on individual benefit/risk assessment (See Adverse Reactions).
Musculoskeletal Pain
In post-marketing experience, severe and occasionally incapacitating bone, joint, and/or muscle pain has been reported in patients taking bisphosphonates. This category of drugs includes Pamidronate disodium for Injection USP. The time to onset of symptoms varied from one day to several months after starting the drug. Most patients had relief of symptoms after stopping. A subset had recurrence of symptoms when rechallenged with the same drug or another bisphosphonate.
Atypical Fractures of the Femur
Atypical subtrochanteric and diaphyseal femoral fractures have been reported in patients receiving bisphosphonate therapy, including pamidronate disodium. These fractures can occur anywhere in the femoral shaft from just below the lesser trochanter to just above the supracondylar flare and are transverse or short oblique in orientation without evidence of comminution. These fractures occur after minimal or no trauma. Patients may experience thigh or groin pain weeks to months before presenting with a completed femoral fracture. Fractures are often bilateral; therefore the contralateral femur should be examined in bisphosphonate- treated patients who have sustained a femoral shaft fracture. Poor healing of these fractures has also been reported. A number of case reports noted that patients were also receiving treatment with glucocorticoids (such as prednisone or dexamethasone) at the time of fracture. Causality with bisphosphonate therapy has not been established.
Any patient with a history of bisphosphonate exposure who presents with thigh or groin pain in the absence of trauma should be suspected of having an atypical fracture and should be evaluated. Discontinuation of Pamidronate disodium USP therapy in patients suspected to have an atypical femur fracture should be considered pending evaluation of the patient, based on an individual benefit risk assessment. It is unknown whether the risk of atypical femur fracture continues after stopping therapy.
Laboratory Tests
Patients who receive Pamidronate disodium USP should have serum creatinine assessed prior to each treatment. Serum calcium, electrolytes, phosphate, magnesium, and CBC, differential, and hematocrit/hemoglobin must be closely monitored in patients treated with Pamidronate disodium USP. Patients who have preexisting anemia, leukopenia, or thrombocytopenia should be monitored carefully in the first 2 weeks following treatment. Patients receiving Pamidronate disodium USP may be at risk for anemia, leukopenia or thrombocytopenia and should have regular hematology assessments.
Drug Interactions
Concomitant administration of a loop diuretic had no effect on the calcium-lowering action of pamidronate disodium.
Caution is indicated when Pamidronate disodium USP is used with other potentially nephrotoxic drugs.
In multiple myeloma patients, the risk of renal function deterioration may be increased when Pamidronate disodium USP is used in combination with thalidomide.
Carcinogenesis, Mutagenesis, Impairment of Fertility
In a 104-week carcinogenicity study with daily oral administration of pamidronate in rats, there was a positive dose response relationship for benign adrenal pheochromocytoma in males (P<0.00001). Although this condition was also observed in females, the incidence was not statistically significant. When the dose calculations were adjusted to account for the limited oral bioavailability of pamidronate in rats, systemic exposure with the lowest daily dose associated with adrenal pheochromocytoma resulted in systemic exposures that were similar to the systemic exposure achieved at the intended clinical dose. Adrenal pheochromocytoma was also observed in low numbers in the control animals and is considered a relatively common spontaneous neoplasm in the rat. Pamidronate given daily by oral administration was not carcinogenic in an 80-week study in mice.
Pamidronate was nonmutagenic in six mutagenicity assays, including: the Ames bacterial mutagenicity assay (with and without metabolic activation), nucleus-anomaly test, sister-chromatid-exchange study, point-mutation test, and micronucleus test in the rat.
In rats, decreased fertility occurred in first-generation offspring of parents who had received 150 mg/kg of pamidronate orally; however, this occurred only when animals were mated with members of the same dose group. Pamidronate has not been administered intravenously in such a study.
Animal Toxicology
In both rats and dogs, nephropathy has been associated with intravenous (bolus and infusion) administration of pamidronate.
Two 7-day intravenous infusion studies were conducted in the dog wherein pamidronate was given for 1, 4, or 24 hours at doses of 1-20 mg/kg for up to 7 days. In the first study, the compound was well tolerated at 3 mg/kg (1.7 x highest recommended human dose [HRHD] for a single intravenous infusion) when administered for 4 or 24 hours, but renal findings such as elevated BUN and creatinine levels and renal tubular necrosis occurred when 3 mg/kg was infused for 1 hour and at doses of ≥10 mg/kg. In the second study, slight renal tubular necrosis was observed in 1 male at 1 mg/kg when infused for 4 hours. Additional findings included elevated BUN levels in several treated animals and renal tubular dilation and/or inflammation at ≥1 mg/kg after each infusion time.
Pamidronate was given to rats at doses of 2, 6, and 20 mg/kg and to dogs at doses of 2, 4, 6, and 20 mg/kg as a 1-hour infusion, once a week, for 3 months followed by a 1-month recovery period. In rats, nephrotoxicity was observed at ≥6 mg/kg and included increased BUN and creatinine levels and tubular degeneration and necrosis. These findings were still present at 20 mg/kg at the end of the recovery period. In dogs, moribundity/death and renal toxicity occurred at 20 mg/kg as did kidney findings of elevated BUN and creatinine levels at ≥6 mg/kg and renal tubular degeneration at ≥4 mg/kg. The kidney changes were partially reversible at 6 mg/kg. In both studies, the dose level that produced no adverse renal effects was considered to be 2 mg/kg (1.1 x HRHD for a single intravenous infusion).
Pregnancy Category D
(See WARNINGS)
There are no adequate and well-controlled studies in pregnant women.
Pamidronate disodium USP may cause fetal harm when administered to a pregnant woman. Bisphosphonates, such as pamidronate disodium, are incorporated into the bone matrix, from where they are gradually released over periods of weeks to years. The extent of bisphosphonate incorporation into adult bone, and hence, the amount available for release back into the systemic circulation, is directly related to the total dose and duration of bisphosphonate use. Although there are no data on fetal risk in humans, bisphosphonates do cause fetal harm in animals, and animal data suggest that uptake of bisphosphonates into fetal bone is greater than into maternal bone. Therefore, there is a theoretical risk of fetal harm (e.g., skeletal and other abnormalities) if a woman becomes pregnant after completing a course of bisphosphonate therapy. The impact of variables such as time between cessation of bisphosphonate therapy to conception, the particular bisphosphonate used, and the route of administration (intravenous versus oral) on this risk has not been established. If Pamidronate disodium USP is used during pregnancy or if the patient becomes pregnant while taking or after taking this drug, the patient should be apprised of the potential hazard to the fetus.
Intravenous bolus dosing of pregnant rats and rabbits with pamidronate resulted in maternal toxicity and embryo/fetal effects when given during organogenesis at doses of 0.6 to 8.3 times the highest recommended human dose for a single intravenous infusion. Pamidronate can cross the placenta in rats, and has produced marked maternal and nonteratogenic embryo/fetal effects in both rats and rabbits.
Nursing Mothers
It is not known whether pamidronate is excreted in human milk. Because many drugs are excreted in human milk, and because of the potential for serious adverse reactions in nursing infants from Pamidronate disodium USP, a decision should be made to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
Pediatric Use
Safety and effectiveness of pamidronate disodium in pediatric patients have not been established.
Geriatric Use
Of the total number of subjects in clinical studies of pamidronate disodium, approximately 20% were 65 and over, while approximately 15% were 75 and over. No overall differences in safety or effectiveness were observed between these subjects and younger subjects, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
-
ADVERSE REACTIONS
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may reflect the rates observed in practice.
Clinical Studies
Hypercalcemia of Malignancy
Transient mild elevation of temperature by at least 1°C was noted 24 to 48 hours after administration of pamidronate disodium in 34% of patients in clinical trials. In the saline trial, 18% of patients had a temperature elevation of at least 1°C 24 to 48 hours after treatment.
Drug-related local soft-tissue symptoms (redness, swelling or induration and pain on palpation) at the site of catheter insertion were most common in patients treated with 90 mg of pamidronate disodium. Symptomatic treatment resulted in rapid resolution in all patients.
Rare cases of uveitis, iritis, scleritis, and episcleritis have been reported, including one case of scleritis, and one case of uveitis upon separate rechallenges.
Five of 231 patients (2%) who received pamidronate disodium during the four U.S. controlled hypercalcemia clinical studies were reported to have had seizures, 2 of whom had preexisting seizure disorders. None of the seizures were considered to be drug-related by the investigators. However, a possible relationship between the drug and the occurrence of seizures cannot be ruled out. It should be noted that in the saline arm 1 patient (4%) had a seizure.
There are no controlled clinical trials comparing the efficacy and safety of 90-mg pamidronate disodium over 24 hours to 2 hours in patients with hypercalcemia of malignancy. However, a comparison of data from separate clinical trials suggests that the overall safety profile in patients who received 90-mg pamidronate disodium over 24 hours is similar to those who received 90-mg pamidronate disodium over 2 hours. The only notable differences observed were an increase in the proportion of patients in the pamidronate disodium 24-hour group who experienced fluid overload and electrolyte/ mineral abnormalities.
At least 15% of patients treated with pamidronate disodium for hypercalcemia of malignancy also experienced the following adverse events during a clinical trial:
General: Fluid overload, generalized pain
Cardiovascular: Hypertension
Gastrointestinal: Abdominal pain, anorexia, constipation, nausea, vomiting
Genitourinary: Urinary tract infection
Musculoskeletal: Bone pain
Laboratory Abnormality: Anemia, hypokalemia, hypomagnesemia, hypophosphatemiaMany of these adverse experiences may have been related to the underlying disease state. The following table lists the adverse experiences considered to be treatment-related during comparative, controlled U.S. trials.
Treatment-Related Adverse Experiences Reported in Three U.S. Controlled Clinical Trials Percent of Patients
Pamidronate Disodium InjectionEtidronate Disodium Saline 60 mg
over 4 hr60 mg
over 24 hr90 mg
over 24 hr7.5 mg/kg
x 3 daysn = 23 n = 73 n = 17 n = 35 n = 23 General Edema 0 1 0 0 0 Fatigue 0 0 12 0 0 Fever 26 19 18 9 0 Fluid overload 0 0 0 6 0 Infusion-site reaction 0 4 18 0 0 Moniliasis 0 0 6 0 0 Rigors 0 0 0 0 4 Gastrointestinal Abdominal pain 0 1 0 0 0 Anorexia 4 1 12 0 0 Constipation 4 0 6 3 0 Diarrhea 0 1 0 0 0 Dyspepsia 4 0 0 0 0 Gastrointestinal hemorrhage 0 0 6 0 0 Nausea 4 0 18 6 0 Stomatitis 0 1 0 3 0 Vomiting 4 0 0 0 0 Respiratory Dyspnea 0 0 0 3 0 Rales 0 0 6 0 0 Rhinitis 0 0 6 0 0 Upper respiratory infection 0 3 0 0 0 CNS Anxiety 0 0 0 0 4 Convulsions 0 0 0 3 0 Insomnia 0 1 0 0 0 Nervousness 0 0 0 0 4 Psychosis 4 0 0 0 0 Somnolence 0 1 6 0 0 Taste perversion 0 0 0 3 0 Cardiovascular Atrial fibrillation 0 0 6 0 4 Atrial flutter 0 1 0 0 0 Cardiac failure 0 1 0 0 0 Hypertension 0 0 6 0 4 Syncope 0 0 6 0 0 Tachycardia 0 0 6 0 4 Endocrine Hypothyroidism 0 0 6 0 0 Hemic and Lymphatic Anemia 0 0 6 0 0 Leukopenia 4 0 0 0 0 Neutropenia 0 1 0 0 0 Thrombocytopenia 0 1 0 0 0 Musculoskeletal Myalgia 0 1 0 0 0 Urogenital Uremia 4 0 0 0 0 Laboratory Abnormalities Hypocalcemia 0 1 12 0 0 Hypokalemia 4 4 18 0 0 Hypomagnesemia 4 10 12 3 4 Hypophosphatemia 0 9 18 3 0 Abnormal liver function 0 0 0 3 0 Paget's Disease
Transient mild elevation of temperature >1°C above pretreatment baseline was noted within 48 hours after completion of treatment in 21% of the patients treated with 90 mg of pamidronate disodium in clinical trials.
Drug-related musculoskeletal pain and nervous system symptoms (dizziness, headache, paresthesia, increased sweating) were more common in patients with Paget’s disease treated with 90 mg of pamidronate disodium than in patients with hypercalcemia of malignancy treated with the same dose.
Adverse experiences considered to be related to trial drug, which occurred in at least 5% of patients with Paget’s disease treated with 90 mg of pamidronate disodium in two U.S. clinical trials, were fever, nausea, back pain, and bone pain.
At least 10% of all pamidronate disodium-treated patients with Paget’s disease also experienced the following adverse experiences during clinical trials:
Osteolytic Bone Metastases of Breast Cancer and Osteolytic Lesions of Multiple Myeloma
The most commonly reported (>15%) adverse experiences occurred with similar frequencies in the pamidronate disodium-and placebo-treatment groups, and most of these adverse experiences may have been related to the underlying disease state or cancer therapy.
Commonly Reported Adverse Experiences in Three U.S. Controlled Clinical Trials Pamidronate
disodium
90 mg
over 4 hoursPlacebo Pamidronate
disodium
90 mg
over 2 hoursPlacebo All
Pamidronate
disodium
90 mgPlacebo N = 205 N = 187 N = 367 N = 386 N = 572 N = 573 % % % % % % General Asthenia 16.1 17.1 25.6 19.2 22.2 18.5 Fatigue 31.7 28.3 40.3 28.8 37.2 29 Fever 38.5 38 38.1 32.1 38.5 34 Metastases 1.0 3.0 31.3 24.4 20.5 17.5 Pain 13.2 11.8 15.0 18.1 14.3 16.1 Digestive System Anorexia 17.1 17.1 31.1 24.9 26.0 22.3 Constipation 28.3 31.7 36.0 38.6 33.2 35.1 Diarrhea 26.8 26.8 29.4 30.6 28.5 29.7 Dyspepsia 17.6 13.4 18.3 15.0 22.6 17.5 Nausea 35.6 37.4 63.5 59.1 53.5 51.8 Pain Abdominal 19.5 16.0 24.3 18.1 22.6 17.5 Vomiting 16.6 19.8 46.3 39.1 35.7 32.8 Hemic and Lymphatic Anemia 47.8 41.7 39.5 36.8 42.5 38.4 Granulocytopenia 20.5 15.5 19.3 20.5 19.8 18.8 Thrombocytopenia 16.6 17.1 12.5 14.0 14.0 15.0 Musculoskeletal System Arthralgias 10.7 7.0 15.3 12.7 13.6 10.8 Myalgia 25.4 15.0 26.4 22.5 26.0 20.1 Skeletal Pain 61 71.7 70 75.4 66.8 74 CNS Anxiety 7.8 9.1 18.0 16.8 14.3 14.3 Headache 24.4 19.8 27.2 23.6 26.2 22.3 Insomnia 17.1 17.2 25.1 19.4 22.2 19.0 Respiratory System Coughing 26.3 22.5 25.3 19.7 25.7 20.6 Dyspnea 22.0 21.4 35.1 24.4 30.4 23.4 Pleural Effusion 2.9 4.3 15.0 9.1 10.7 7.5 Sinusitis 14.6 16.6 16.1 10.4 15.6 12.0 Upper Respiratory Tract Infection 32.2 28.3 19.6 20.2 24.1 22.9 Urogenital System Urinary Tract Infection 15.6 9.1 20.2 17.6 18.5 15.6 Of the toxicities commonly associated with chemotherapy, the frequency of vomiting, anorexia, and anemia were slightly more common in the pamidronate disodium patients whereas stomatitis and alopecia occurred at a frequency similar to that in placebo patients. In the breast cancer trials, mild elevations of serum creatinine occurred in 18.5% of pamidronate disodium patients and 12.3% of placebo patients. Mineral and electrolyte disturbances, including hypocalcemia, were reported rarely and in similar percentages of pamidronate disodium-treated patients compared with those in the placebo group. The reported frequencies of hypocalcemia, hypokalemia, hypophosphatemia, and hypomagnesemia for pamidronate disodium-treated patients were 3.3%, 10.5%, 1.7%, and 4.4%, respectively, and for placebo-treated patients were 1.2%, 12%, 1.7%, and 4.5%, respectively. In previous hypercalcemia of malignancy trials, patients treated with pamidronate disodium (60 or 90 mg over 24 hours) developed electrolyte abnormalities more frequently (see ADVERSE REACTIONS, Hypercalcemia of Malignancy).
Arthralgias and myalgias were reported slightly more frequently in the pamidronate disodium group than in the placebo group (13.6% and 26% vs 10.8% and 20.1%, respectively).
In multiple myeloma patients, there were five pamidronate disodium-related serious and unexpected adverse experiences. Four of these were reported during the 12-month extension of the multiple myeloma trial. Three of the reports were of worsening renal function developing in patients with progressive multiple myeloma or multiple myeloma-associated amyloidosis. The fourth report was the adult respiratory distress syndrome developing in a patient recovering from pneumonia and acute gangrenous cholecystitis. One pamidronate disodium-treated patient experienced an allergic reaction characterized by swollen and itchy eyes, runny nose, and scratchy throat within 24 hours after the sixth infusion.
In the breast cancer trials, there were four pamidronate disodium-related adverse experiences, all moderate in severity, that caused a patient to discontinue participation in the trial. One was due to interstitial pneumonitis, another to malaise and dyspnea. One pamidronate disodium patient discontinued the trial due to a symptomatic hypocalcemia. Another pamidronate disodium patient discontinued therapy due to severe bone pain after each infusion, which the investigator felt was trial-drug-related.
Renal Toxicity
In a study of the safety and efficacy of pamidronate disodium 90 mg (2-hour infusion) vs Zometa 4 mg (15-minute infusion) in bone metastases patients with multiple myeloma or breast cancer, renal deterioration was defined as an increase in serum creatinine of 0.5 mg/dL for patients with normal baseline creatinine (<1.4 mg/dL) or an increase of 1.0 mg/dL for patients with an abnormal baseline creatinine (≥1.4 mg/dL). The following are data on the incidence of renal deterioration in patients in this trial. See table below.
- * Patients were randomized following the 15 minute infusion amendment for the Zometa arm.
Incidence of Renal Function Deterioration in Multiple Myeloma and Breast Cancer Patients with Normal and Abnormal Serum Creatinine at Baseline * Patient Population/Baseline
CreatininePamidronate Disodium 90 mg/2 hours Zometa 4 mg/15 minutes n/N (%) n/N (%) Normal 20/246 (8.1%) 23/246 (9.3%) Abnormal 2/22 (9.1%) 1/26 (3.8%) Total 22/268 (8.2%) 24/272 (8.8%) Post-Marketing Experience
The following adverse reactions have been reported during post-approval use of pamidronate disodium. Because these reports are from a population of uncertain size and are subject to confounding factors, it is not possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
The following adverse reactions have been reported in post-marketing use: General: reactivation of Herpes simplex and Herpes zoster, influenza-like symptoms; CNS: confusion and visual hallucinations, sometimes in the presence of electrolyte imbalance; Skin: rash, pruritus; Special senses: conjunctivitis, orbital inflammation; Renal and urinary disorders: focal segmental glomerulosclerosis including the collapsing variant, nephrotic syndrome, renal tubular disorders (RTD), tubulointerstitial nephritis and glomerulonephropathies; Laboratory abnormalities: hyperkalemia, hypernatremia, hematuria. Rare instances of allergic manifestations have been reported, including hypotension, dyspnea, or angioedema, and, very rarely, anaphylactic shock. Pamidronate disodium for Injection USP is contraindicated in patients with clinically significant hypersensitivity to pamidronate disodium or other bisphosphonates (see CONTRAINDICATIONS). Respiratory, thoracic and mediastinal disorders: adult respiratory distress syndrome (ARDS), interstitial lung disease (ILD). Musculoskeletal and connective tissue disorders: severe and occasionally incapacitating bone, joint, and/or muscle pain.
Cases of osteonecrosis (primarily involving the jaw) have been reported predominantly in cancer patients treated with intravenous bisphosphonates, including pamidronate disodium. Many of these patients were also receiving chemotherapy and corticosteroids which may be risk factors for ONJ. Data suggest a greater frequency of reports of ONJ in certain cancers, such as advanced breast cancer and multiple myeloma. The majority of the reported cases are in cancer patients following invasive dental procedures, such as tooth extraction. It is therefore prudent to avoid invasive dental procedures as recovery may be prolonged. (See PRECAUTIONS.)
Atypical subtrochanteric and diaphyseal femoral fractures have been reported with bisphosphonate therapy, including pamidronate disodium. (See PRECAUTIONS.)
-
OVERDOSAGE
There have been several cases of drug maladministration of intravenous pamidronate disodium in hypercalcemia patients with total doses of 225 mg to 300 mg given over 2 ½ to 4 days. All of these patients survived, but they experienced hypocalcemia that required intravenous and/or oral administration of calcium. Single doses of Pamidronate disodium USP should not exceed 90 mg and the duration of the intravenous infusion should be no less than 2 hours. (See WARNINGS.)
In addition, one obese woman (95 kg) who was treated with 285 mg of pamidronate disodium/day for 3 days experienced high fever (39.5°C), hypotension (from 170/90 mmHg to 90/60 mmHg), and transient taste perversion, noted about 6 hours after the first infusion. The fever and hypotension were rapidly corrected with steroids.
If overdosage occurs, symptomatic hypocalcemia could also result; such patients should be treated with short-term intravenous calcium.
-
DOSAGE AND ADMINISTRATION
Hypercalcemia of Malignancy
Consideration should be given to the severity of as well as the symptoms of hypercalcemia. Vigorous saline hydration alone may be sufficient for treating mild, asymptomatic hypercalcemia. Overhydration should be avoided in patients who have potential for cardiac failure. In hypercalcemia associated with hematologic malignancies, the use of glucocorticoid therapy may be helpful.
Moderate Hypercalcemia
The recommended dose of Pamidronate disodium for Injection USP in moderate hypercalcemia (corrected serum calcium* of approximately 12-13.5 mg/dL) is 60 to 90 mg given as a SINGLE-DOSE, intravenous infusion over 2 to 24 hours. Longer infusions (i.e., >2 hours) may reduce the risk for renal toxicity, particularly in patients with preexisting renal insufficiency.
Severe Hypercalcemia
The recommended dose of Pamidronate disodium for Injection USP in severe hypercalcemia (corrected serum calcium*>13.5 mg/dL) is 90 mg given as a SINGLE-DOSE, intravenous infusion over 2 to 24 hours. Longer infusions (i.e., >2 hours) may reduce the risk for renal toxicity, particularly in patients with preexisting renal insufficiency.
_____________________________________________________________________________
*Albumin-corrected serum calcium (CCa,mg/dL) = serum calcium, mg/dL + 0.8 (4.0-serum albumin, g/dL).
Retreatment
A limited number of patients have received more than one treatment with pamidronate disodium for hypercalcemia. Retreatment with Pamidronate disodium for Injection USP, in patients who show complete or partial response initially, may be carried out if serum calcium does not return to normal or remain normal after initial treatment. It is recommended that a minimum of 7 days elapse before retreatment, to allow for full response to the initial dose. The dose and manner of retreatment is identical to that of the initial therapy.
Paget's Disease
The recommended dose of Pamidronate disodium for Injection USP in patients with moderate to severe Paget’s disease of bone is 30 mg daily, administered as a 4-hour infusion on 3 consecutive days for a total dose of 90 mg.
Osteolytic Bone Lesions of Multiple Myeloma
The recommended dose of Pamidronate disodium for Injection USP in patients with osteolytic bone lesions of multiple myeloma is 90 mg administered as a 4-hour infusion given on a monthly basis.
Patients with marked Bence-Jones proteinuria and dehydration should receive adequate hydration prior to Pamidronate disodium for Injection USP infusion.
Limited information is available on the use of Pamidronate disodium for Injection USP in multiple myeloma patients with a serum creatinine ≥3.0 mg/dL.
Patients who receive Pamidronate disodium for Injection USP should have serum creatinine assessed prior to each treatment. Treatment should be withheld for renal deterioration. In a clinical study, renal deterioration was defined as follows:
- For patients with normal baseline creatinine, increase of 0.5 mg/dL.
- For patients with abnormal baseline creatinine, increase of 1 mg/dL.
In this clinical study, pamidronate disodium treatment was resumed only when the creatinine returned to within 10% of the baseline value.
The optimal duration of therapy is not yet known, however, in a study of patients with myeloma, final analysis after 21 months demonstrated overall benefits (see CLINICAL TRIALS section).
Osteolytic Bone Metastases of Breast Cancer
The recommended dose of Pamidronate disodium for Injection USP in patients with osteolytic bone metastases is 90 mg administered over a 2-hour infusion given every 3-4 weeks.
Pamidronate disodium has been frequently used with doxorubicin, fluorouracil, cyclophosphamide, methotrexate, mitoxantrone, vinblastine, dexamethasone, prednisone, melphalan, vincristine, megesterol, and tamoxifen. It has been given less frequently with etoposide, cisplatin, cytarabine, paclitaxel, and aminoglutethimide.
Patients who receive Pamidronate disodium for Injection USP should have serum creatinine assessed prior to each treatment. Treatment should be withheld for renal deterioration. In a clinical study, renal deterioration was defined as follows:
- For patients with normal baseline creatinine, increase of 0.5 mg/dL.
- For patients with abnormal baseline creatinine, increase of 1 mg/dL.
In this clinical study, pamidronate disodium treatment was resumed only when the creatinine returned to within 10% of the baseline value.
The optimal duration of therapy is not known, however, in two breast cancer studies, final analyses performed after 24 months of therapy demonstrated overall benefits (see CLINICAL TRIALS section).
Calcium and Vitamin D Supplementation
In the absence of hypercalcemia, patients with predominantly lytic bone metastases or multiple myeloma, who are at risk of calcium or vitamin D deficiency, and patients with Paget’s disease of the bone, should be given oral calcium and vitamin D supplementation in order to minimize the risk of hypocalcemia.
Reconstitution
Pamidronate disodium for Injection USP is reconstituted by adding 10 mL of Sterile Water for Injection, USP, to each vial, resulting in a solution of 30 mg/10 mL, 60mg/10mL or 90 mg/10 mL. The pH of the reconstituted solution is 6.0-7.4. The drug should be completely dissolved before the solution is withdrawn.
Method of Administration
DUE TO THE RISK OF CLINICALLY SIGNIFICANT DETERIORATION IN RENAL FUNCTION, WHICH MAY PROGRESS TO RENAL FAILURE, SINGLE DOSES OF PAMIDRONATE DISODIUM FOR INJECTION USP SHOULD NOT EXCEED 90 MG. (SEE WARNINGS.)
There must be strict adherence to the intravenous administration recommendations for Pamidronate disodium for Injection USP in order to decrease the risk of deterioration in renal function.
Hypercalcemia of Malignancy
The daily dose must be administered as an intravenous infusion over at least 2 to 24 hours for the 60-mg and 90-mg doses. The recommended dose should be diluted in 1000 mL of sterile 0.45% or 0.9% Sodium Chloride, USP, or 5% Dextrose Injection, USP. This infusion solution is stable for up to 24 hours at room temperature.
Paget’s Disease
The recommended daily dose of 30 mg should be diluted in 500 mL of sterile 0.45% or 0.9% Sodium Chloride, USP, or 5% Dextrose Injection, USP, and administered over a 4-hour period for 3 consecutive days.
Osteolytic Bone Metastases of Breast Cancer
The recommended dose of 90 mg should be diluted in 250 mL of sterile 0.45% or 0.9% Sodium Chloride, USP, or 5% Dextrose Injection, USP, and administered over a 2-hour period every 3-4 weeks.
Osteolytic Bone Lesions of Multiple Myeloma
The recommended dose of 90 mg should be diluted in 500 mL of sterile 0.45% or 0.9% Sodium Chloride, USP, or 5% Dextrose Injection, USP, and administered over a 4-hour period on a monthly basis.
Pamidronate disodium for Injection USP must not be mixed with calcium-containing infusion solutions, such as Ringer’s solution, and should be given in a single intravenous solution and line separate from all other drugs.
Note: Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit.
Pamidronate disodium for Injection USP reconstituted with Sterile Water for Injection may be stored under refrigeration at 2°C-8°C (36°F-46°F) for up to 24 hours.
-
HOW SUPPLIED
Pamidronate disodium for Injection USP is available as a white, sterile, lyophilized cake contained in a vial with an externally lacquered, light blue, tear-off, flip-off seal, containing 30 mg pamidronate disodium USP and 470 mg mannitol USP, packaged one vial per carton (NDC No. 59923-601-10).
Pamidronate disodium for Injection USP is available as a white, sterile, lyophilized cake contained in a vial with an externally lacquered, white, tear-off, flip-off seal, containing 60 mg pamidronate disodium USP and 400 mg mannitol USP, packaged one vial per carton (NDC No. 59923-602-10).
Pamidronate disodium for Injection USP is available as a white, sterile, lyophilized cake contained in a vial with an externally lacquered, dark green, tear-off, flip-off seal, containing 90 mg pamidronate disodium USP and 375 mg mannitol USP, packaged one vial per carton (NDC No. 59923-603-10).
Store at 20° - 25° C (68°-77° F) [See USP Controlled Room Temperature].
Areva
Manufactured for
AREVA Pharmaceuticals, Inc.,
7112 Areva Dr. N.E.
Georgetown, IN 47122Mfd. by Cipla Ltd. Made in India
© 2011 AREVA Pharmaceuticals, Inc.
Toll-free number: 1-855-853-4760
Revised: July 2015
-
PRINCIPAL DISPLAY PANEL
Rx only
NDC: 59923-601-10
Pamidronate Disodium for Injection USP
30 mg/vial
For Intravenous Infusion
Further dilution required.
Important: dilution and administration differs for each indication.
Sterile, Lyophilized
1 vial
Areva
Each Vial Contains:
Pamidronate disodium USP 30 mg; mannitol USP, 470 mg in lyophilized form; and phosphoric acid to adjust pH.
The pH of the reconstituted solution is 6.0 to 7.0.
Do not mix with calcium-containing infusion solutions.
Important: Reconstitution differs for each indication.
See package insert for Dosage and Administration and Preparation of Solution.
Pamidronate Disodium reconstituted with Sterile Water for Injection USP, may be stored under refrigeration at 2°C - 8°C (36°F - 46°F) for up to 24 hours.
Store at 20° - 25°C (68° - 77°F) [see USP Controlled Room Temperature].
Rev. 09/16
Code No.: GO/DRUGS/704
Manufactured for
AREVA Pharmaceuticals, Inc.,
Georgetown, IN 47122
Mfd. by Cipla Ltd.
Made in India
© 2011 AREVA Pharmaceuticals, Inc.
21057194
Rx only
NDC: 59923-601-10
Pamidronate Disodium for Injection USP
30 mg/vial
For Intravenous Infusion
Further dilution required.
Important: dilution and administration differs for each indication.
Sterile, Lyophilized
1 vial
Areva
Each Vial Contains:
Pamidronate disodium USP 30 mg; mannitol USP 470 mg in lyophilized form; and phosphoric acid to adjust pH.
The pH of the reconstituted solution is 6.0 – 7.0.
Do not mix with calcium–containing infusion solutions.
Store at 20° - 25°C (68° - 77°F) [see USP Controlled Room Temperature].
Code No.: GO/DRUGS/704
Manufactured for
AREVA Pharmaceuticals, Inc.,
Georgetown, IN 47122
Mfd. by Cipla Ltd. Made in India
© 2011 AREVA Pharmaceuticals
Rev. 07/15
207735
Rx only
NDC: 59923-602-10
Pamidronate Disodium for Injection USP
60 mg/vial
For Intravenous Infusion
Lyophilized Further dilution required.
Important: dilution and administration differs for each indication.
Sterile, Lyophilized
1 vial
Areva
Each Vial Contains:
Pamidronate disodium 60 mg USP; mannitol USP, 400 mg in lyophilized form; and phosphoric acid to adjust pH.
The pH of the reconstituted solution is 6.0 to 7.0.
Do not mix with calcium-containing infusion solutions.
Important: Reconstitution differs for each indication.
See package insert for Dosage and Administration and Preparation of Solution.
Pamidronate Disodium reconstituted with Sterile Water for Injection USP, may be stored under refrigeration at 2°C - 8°C (36°F - 46°F) for up to 24 hours.
Store at 20° - 25°C (68° - 77°F)
[see USP Controlled Room Temperature].
Rev. 09/16
Code No.: GO/DRUGS/704
Manufactured for
AREVA Pharmaceuticals,
Elizabethtown, KY 42701
Mfd. by Cipla Ltd.
Made in India
© 2011 AREVA Pharmaceuticals
21043497

Rx only
NDC: 59923-602-10
Pamidronate Disodium for Injection USP
60 mg/vial
For Intravenous Infusion
Further dilution required.
Important: dilution and administration differs for each indication.
Sterile, Lyophilized
1 vial
Areva
Each Vial Contains:
Pamidronate disodium USP 60 mg; mannitol USP 400 mg in lyophilized form; and phosphoric acid to adjust pH.
The pH of the reconstituted solution is 6.0 – 7.0.
Do not mix with calcium–containing infusion solutions.
Store at 20° - 25°C (68° - 77°F) [see USP Controlled Room Temperature].
Code No.: GO/DRUGS/704
Manufactured for
AREVA Pharmaceuticals,
Georgetown, IN 47122
Mfd. by Cipla Ltd.
Made in India
© 2011 AREVA Pharmaceuticals Inc.
Rev.07/15
207736
Rx only
NDC: 59923-603-10
Pamidronate Disodium for Injection USP
90 mg/vial
For Intravenous Infusion Sterile,
Lyophilized Further dilution required.
Important: dilution and administration differs for each indication.
Sterile, Lyophilized
1 vial
Areva
Each Vial Contains:
Pamidronate disodium USP 90 mg; mannitol USP, 375 mg in lyophilized form; and phosphoric acid to adjust pH.
The pH of the reconstituted solution is 6.0 to 7.0.
Do not mix with calcium-containing infusion solutions.
Important: Reconstitution differs for each indication.
See package insert for Dosage and Administration and Preparation of Solution.
Pamidronate Disodium reconstituted with Sterile Water for Injection USP, may be stored under refrigeration at 2°C - 8°C (36°F - 46°F) for up to 24 hours.
Store at 20° - 25°C (68° - 77°F) [see USP Controlled Room Temperature].
Rev.09/16
Code No.: GO/DRUGS/704
Manufactured for
AREVA Pharmaceuticals, Inc.,
Georgetown, IN 47122
Mfd. by Cipla Ltd.
Made in India
© 2011 AREVA Pharmaceuticals
21057195
Rx only
NDC: 59923-603-10
Pamidronate Disodium for Injection USP
90 mg/vial
For Intravenous Infusion
Further dilution required.
Important: dilution and administration differs for each indication.
Sterile, Lyophilized
1 vial
Areva
Each Vial Contains:
Pamidronate disodium USP 90 mg; mannitol USP 375 mg in lyophilized form; and phosphoric acid to adjust pH.
The pH of the reconstituted solution is 6.0 – 7.0.
Do not mix with calcium–containing infusion solutions.
Store at 20° - 25°C (68° - 77°F) [see USP Controlled Room Temperature].
Code No.: GO/DRUGS/704
Manufactured for
AREVA Pharmaceuticals, Inc.,
Georgetown, IN 47122
Mfd. by Cipla Ltd. Made in India
© 2011 AREVA Pharmaceuticals, Inc.
Rev. 07/15
207737

-
INGREDIENTS AND APPEARANCE
PAMIDRONATE DISODIUM
pamidronate disodium injection, powder, lyophilized, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 59923-601 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength PAMIDRONATE DISODIUM (UNII: 8742T8ZQZA) (PAMIDRONIC ACID - UNII:OYY3447OMC) PAMIDRONATE DISODIUM 3 mg in 1 mL Inactive Ingredients Ingredient Name Strength MANNITOL (UNII: 3OWL53L36A) WATER (UNII: 059QF0KO0R) PHOSPHORIC ACID (UNII: E4GA8884NN) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 59923-601-10 1 in 1 CARTON 08/06/2013 1 10 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA077433 08/06/2013 PAMIDRONATE DISODIUM
pamidronate disodium injection, powder, lyophilized, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 59923-602 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength PAMIDRONATE DISODIUM (UNII: 8742T8ZQZA) (PAMIDRONIC ACID - UNII:OYY3447OMC) PAMIDRONATE DISODIUM 6 mg in 1 mL Inactive Ingredients Ingredient Name Strength MANNITOL (UNII: 3OWL53L36A) WATER (UNII: 059QF0KO0R) PHOSPHORIC ACID (UNII: E4GA8884NN) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 59923-602-10 1 in 1 CARTON 08/06/2013 1 10 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA077433 08/06/2013 PAMIDRONATE DISODIUM
pamidronate disodium injection, powder, lyophilized, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 59923-603 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength PAMIDRONATE DISODIUM (UNII: 8742T8ZQZA) (PAMIDRONIC ACID - UNII:OYY3447OMC) PAMIDRONATE DISODIUM 9 mg in 1 mL Inactive Ingredients Ingredient Name Strength MANNITOL (UNII: 3OWL53L36A) WATER (UNII: 059QF0KO0R) PHOSPHORIC ACID (UNII: E4GA8884NN) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 59923-603-10 1 in 1 CARTON 08/06/2013 1 10 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA077433 08/06/2013 Labeler - Areva Pharmaceuticals Inc. (833189835) Establishment Name Address ID/FEI Business Operations Cipla Limited 650072015 manufacture(59923-601, 59923-602, 59923-603)
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.