PAROXETINE tablet, film coated, extended release
Paroxetine by
Drug Labeling and Warnings
Paroxetine by is a Prescription medication manufactured, distributed, or labeled by Cadila Pharmaceuticals Limited. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use PAROXETINE EXTENDED-RELEASE TABLETS safely and effectively. See full prescribing information for PAROXETINE EXTENDED-RELEASE TABLETS.
PAROXETINE extended-release tablets, for oral use Initial U.S. Approval: 1992
WARNING: SUICIDAL THOUGHTS AND BEHAVIORS
See full prescribing information for complete boxed warning.
Increased risk of suicidal thoughts and behavior in pediatric and young adult patients taking antidepressants. Closely monitor all antidepressant- treated patients for clinical worsening and emergence of suicidal thoughts and behaviors. Paroxetine extended-release tablets are not approved for use in pediatric patients.
(5.1, 8.4)
INDICATIONS AND USAGE
Paroxetine extended-release tablets are selective serotonin reuptake inhibitor (SSRI) indicated in adults for the treatment of (1):
Major Depressive Disorder (MDD)
Panic Disorder (PD)
Social Anxiety Disorder (SAD)
Premenstrual Dysphoric Disorder (PMDD)
DOSAGE AND ADMINISTRATION
Swallow tablet whole; do not chew or crush. (2.1)
Recommended starting and maximum daily dosage: (2.2, 2.3)
Indication
Starting Dose
Maximum Dose
MDD
25 mg/day
62.5 mg/day
PD
12.5 mg/day
75 mg/day
SAD
12.5 mg/day
37.5 mg/dayPMDD
12.5 mg/day
25 mg/day
For PMDD, dose continuously or intermittently (luteal phase only). (2.3)
If inadequate response to starting dosage, titrate in 12.5 mg per day increments once weekly. (2.2, 2.3)
Elderly patients, patients with severe renal impairment or severe hepatic impairment: Starting dose is 12.5 mg per day. Do not exceed 50 mg per day for treatment of MDD and PD and 37.5 mg per day for treatment of SAD. (2.5) When discontinuing paroxetine extended-release tablets, reduce dose gradually. (2.7)
DOSAGE FORMS AND STRENGTHS
Extended-release tablets: 12.5 mg, 25 mg, and 37.5 mg tablets. (3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
Serotonin Syndrome: Increased risk when co-administered with other serotonergic agents, but also when taken alone. If occurs, discontinue paroxetine extended-release tablets and serotonergic agents and initiate supportive measures. (5.2)
Embryofetal Toxicity: May cause fetal harm. Meta-analysis of epidemiological studies have shown increased risk (less than 2-fold) of cardiovascular malformations with exposure during the first trimester. (5.4, 8.1)
Increased Risk of Bleeding: Concomitant use of aspirin, nonsteroidal anti- inflammatory drugs, other antiplatelet drugs, warfarin, and other anticoagulant drugs may increase risk. (5.5)
Activation of Mania/Hypomania: Screen patients for bipolar disorder. (5.6)
Seizures: Use with caution in patients with seizure disorders. (5.8)
Angle-Closure Glaucoma: Angle-closure glaucoma has occurred in patients with untreated anatomically narrow angles, treated with antidepressants (5.9)Sexual Dysfunction: Paroxetine extended-release tablets may cause symptoms of sexual dysfunction. (5.13)
ADVERSE REACTIONS
Most common adverse reactions (≥5% and at least twice placebo) in placebo- controlled MDD, PD, SAD, and PMDD clinical trials:
abnormal ejaculation, abnormal vision, asthenia, constipation, decreased appetite, diarrhea, dizziness, dry mouth, female genital disorder, impotence, insomnia, libido decreased, nausea, somnolence, sweating, tremor. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Cadila Pharmaceuticals Limited at 1-202-355-9785 (fax 1-202-355-9784) or www.cadilapharma.com or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
Drugs Highly Bound to Plasma Protein: Monitor for adverse reactions and reduce dosage of paroxetine extended-release tablets or other protein-bound drugs (e.g., warfarin) as warranted. (7)
Drugs Metabolized by CYP2D6: Reduce dosage of drugs metabolized by CYP2D6 as warranted. (7)
Concomitant use with Tamoxifen: Consider use of an alternative antidepressant with little or no CYP2D6 inhibition. (5.11, 7)USE IN SPECIFIC POPULATIONS
Pregnancy: SSRI use, particularly later in pregnancy, may increase the risk of persistent pulmonary hypertension and symptoms of poor adaptation (respiratory distress, temperature instability, feeding difficulty, hypotonia, irritability) in the neonate. (5.4, 8.1)
Nursing Mothers: Discontinue drug or nursing taking into consideration importance of drug to mother. (8.3)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 3/2024
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: SUICIDAL THOUGHTS AND BEHAVIORS
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Important Administration Instructions
2.2 Dosage in Patients with Major Depressive Disorder, Panic Disorder, and Social Anxiety Disorder
2.3 Dosage in Patients with Premenstrual Dysphoric Disorder
2.4 Screen for Bipolar Disorder Prior to Starting Paroxetine Extended-Release Tablets
2.5 Dosage Modifications for Elderly Patients, Patients with Severe Renal Impairment and Patients with Severe Hepatic Impairment
2.6 Switching Patients to or from a Monoamine Oxidase Inhibitor Antidepressant
2.7 Discontinuation of Treatment with Paroxetine Extended-Release Tablets
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Suicidal Thoughts and Behaviors in Adolescents and Young Adults
5.2 Serotonin Syndrome
5.3 Drug Interactions Leading to QT Prolongation
5.4 Embryofetal Toxicity
5.5 Increased Risk of Bleeding
5.6 Activation of Mania or Hypomania
5.7 Discontinuation Syndrome
5.8 Seizures
5.9 Angle-Closure Glaucoma
5.10 Hyponatremia
5.11 Reduction of Efficacy of Tamoxifen
5.12 Bone Fracture
5.13 Sexual Dysfunction
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Clinically Significant Drug Interactions with Paroxetine Extended-Release Tablets
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal and/or Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Major Depressive Disorder
14.2 Panic Disorder
14.3 Social Anxiety Disorder
14.4 Premenstrual Dysphoric Disorder
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: SUICIDAL THOUGHTS AND BEHAVIORS
Antidepressants increased the risk of suicidal thoughts and behaviors in pediatric and young adult patients in short-term studies. Closely monitor all antidepressant-treated patients for clinical worsening, and for emergence of suicidal thoughts and behaviors [see Warnings and Precautions (5.1)]. Paroxetine extended-release tablets are not approved for use in pediatric patients [see Use in Specific Populations (8.4)].
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Important Administration Instructions
Administer paroxetine extended-release tablets as a single daily dose in the morning, with or without food. Swallow tablets whole and do not chew or crush.
2.2 Dosage in Patients with Major Depressive Disorder, Panic Disorder, and Social Anxiety Disorder
The recommended initial dosage and maximum dosage of paroxetine extended-release tablets in patients with MDD, PD, and SAD are presented in Table 1.
In patients with an inadequate response, dosage may be increased in increments of 12.5 mg per day at intervals of at least 1 week, depending on tolerability.
Table 1: Recommended Daily Dosage of Paroxetine Extended-Release Tablets in Patients with MDD, PD, and SAD
Indication
Starting Dose
Maximum Dose
MDD
25 mg
62.5 mg
PD
12.5 mg
75 mg
SAD
12.5 mg
37.5 mg
2.3 Dosage in Patients with Premenstrual Dysphoric Disorder
The recommended starting dosage in women with PMDD is 12.5 mg per day. Paroxetine extended-release tablets may be administered either continuously (every day throughout the menstrual cycle) or intermittently (only during the luteal phase of the menstrual cycle, i.e., starting the daily dosage 14 days prior to the anticipated onset of menstruation and continuing through the onset of menses). Intermittent dosing is repeated with each new cycle.
In patients with an inadequate response, the dosage may be increased to the maximum recommended dosage of 25 mg per day, depending on tolerability. Institute dosage adjustments at intervals of at least 1 week.2.4 Screen for Bipolar Disorder Prior to Starting Paroxetine Extended-Release Tablets
Prior to initiating treatment with paroxetine extended-release tablets or another antidepressant, screen patients for a personal or family history of bipolar disorder, mania, or hypomania [see Warnings and Precautions (5.6)].
2.5 Dosage Modifications for Elderly Patients, Patients with Severe Renal Impairment and Patients with Severe Hepatic Impairment
The recommended initial dose of paroxetine extended-release tablets is 12.5 mg per day for elderly patients, patients with severe renal impairment, and patients with severe hepatic impairment. Reduce initial dose and increase up-titration intervals if necessary. Dosage should not exceed 50 mg per day for MDD or PD and should not exceed 37.5 mg per day for SAD [see Use in Specific Populations (8.5, 8.6)].
2.6 Switching Patients to or from a Monoamine Oxidase Inhibitor Antidepressant
At least 14 days must elapse between discontinuation of an monoamine oxidase inhibitor (MAOI) antidepressant and initiation of paroxetine extended-release tablets. In addition, at least 14 days must elapse after stopping paroxetine extended-release tablets before starting an MAOI antidepressant [see Contraindications (4), Warnings and Precautions (5.2)].
2.7 Discontinuation of Treatment with Paroxetine Extended-Release Tablets
Adverse reactions may occur upon discontinuation of paroxetine extended-release tablets [see Warnings and Precautions (5.7)]. Gradually reduce the dosage rather than stopping paroxetine extended-release tablets abruptly whenever possible.
-
3 DOSAGE FORMS AND STRENGTHS
Paroxetine extended-release tablets are available as:
12.5 mg yellow, biconvex, enteric film-coated, extended release, round tablets, debossed with “X1” on one side and plain on the other side. The tablets should be free from all physical defects.
25 mg pink, biconvex, enteric film-coated, extended release, round tablets debossed with "X2" on one side and plain on the other side. The tablets should be free from all physical defects.
37.5 mg blue, biconvex, enteric film-coated, extended release, round tablets debossed with "X3" on one side and plain on the other side. The tablets should be free from all physical defects. -
4 CONTRAINDICATIONS
Paroxetine extended-release tablets are contraindicated in patients:
Taking, or within 14 days of stopping, MAOIs (including the MAOIs linezolid and intravenous methylene blue) because of an increased risk of serotonin syndrome
[See Warnings and Precautions (5.2), Drug Interactions (7)].
Taking thioridazine because of risk of QT prolongation [see Warnings and Precautions (5.3), Drug Interactions (7)].
Taking pimozide because of risk of QT prolongation [see Warnings and Precautions (5.3), Drug Interactions (7)].
With known hypersensitivity (e.g., anaphylaxis, angioedema, Stevens-Johnson syndrome) to paroxetine or to any of the inactive ingredients in paroxetine extended-release tablets [see Adverse Reactions (6.1, 6.2)]. -
5 WARNINGS AND PRECAUTIONS
5.1 Suicidal Thoughts and Behaviors in Adolescents and Young Adults
In pooled analyses of placebo-controlled trials of antidepressant drugs (SSRIs and other antidepressant classes) that included approximately 77,000 adult patients and 4,500 pediatric patients, the incidence of suicidal thoughts and behaviors in antidepressant-treated patients age 24 years and younger was greater than in placebo-treated patients. There was considerable variation in risk of suicidal thoughts and behaviors among drugs, but there was an increased risk identified in young patients for most drugs studied. There were differences in absolute risk of suicidal thoughts and behaviors across the different indications, with the highest incidence in patients with MDD. The drug-placebo differences in the number of cases of suicidal thoughts and behaviors per 1,000 patients treated are provided in Table 2.
Table 2: Risk Differences of the Number of Patients of Suicidal Thoughts and Behaviors in the Pooled Placebo-Controlled Trials of Antidepressants in Pediatric and Adult Patients
Age Range
Drug-Placebo Difference in Number of Patients of Suicidal Thoughts and Behaviors per 1,000 Patients Treated
Increases Compared to Placebo
<18 years old
14 additional patients
18 to 24 years old
5 additional patients
Decreases Compared to Placebo
25 to 64 years old
1 fewer patient
≥65 years old
6 fewer patients
It is unknown whether the risk of suicidal thoughts and behaviors in children, adolescents, and young adults extends to longer-term use, i.e., beyond four months. However, there is substantial evidence from placebo-controlled maintenance trials in adults with MDD that antidepressants delay the recurrence of depression and that depression itself is a risk factor for suicidal thoughts and behaviors.
Monitor all antidepressant-treated patients for any indication for clinical worsening and emergence of suicidal thoughts and behaviors, especially during the initial few months of drug therapy, and at times of dosage changes. Counsel family members or caregivers of patients to monitor for changes in behavior and to alert the healthcare provider.
Consider changing the therapeutic regimen, including possibly discontinuing paroxetine extended-release tablets, in patients whose depression is persistently worse, or who are experiencing emergent suicidal thoughts or behaviors.
5.2 Serotonin Syndrome
Serotonin-norepinephrine reuptake inhibitors (SNRIs) and SSRIs, including paroxetine extended-release tablets, can precipitate serotonin syndrome, a potentially life-threatening condition. The risk is increased with concomitant use of other serotonergic drugs (including triptans, tricyclic antidepressants, fentanyl, lithium, tramadol, meperidine, methadone, tryptophan, buspirone, amphetamines, and St. John's Wort) and with drugs that impair metabolism of serotonin, i.e., MAOIs [see Contraindications (4), Drug Interactions (7.1)]. Serotonin syndrome can also occur when these drugs are used alone.
Serotonin syndrome signs and symptoms may include mental status changes (e.g., agitation, hallucinations, delirium, and coma), autonomic instability (e.g., tachycardia, labile blood pressure, dizziness, diaphoresis, flushing, hyperthermia), neuromuscular symptoms (e.g., tremor, rigidity, myoclonus, hyperreflexia, incoordination), seizures, and gastrointestinal symptoms (e.g., nausea, vomiting, diarrhea).
The concomitant use of paroxetine extended-release tablets with MAOIs is contraindicated. In addition, do not initiate paroxetine extended-release tablets in a patient being treated with MAOIs such as linezolid or intravenous methylene blue. No reports involved the administration of methylene blue by other routes (such as oral tablets or local tissue injection). If it is necessary to initiate treatment with an MAOI such as linezolid or intravenous methylene blue in a patient taking paroxetine extended-release tablets, discontinue paroxetine extended-release tablets before initiating treatment with the MAOI [see Contraindications (4), Drug Interactions (7.1)].
Monitor all patients taking paroxetine extended-release tablets for the emergence of serotonin syndrome. Discontinue treatment with paroxetine extended-release tablets and any concomitant serotonergic agents immediately if the above symptoms occur, and initiate supportive symptomatic treatment. If concomitant use of paroxetine extended-release tablets with other serotonergic drugs is clinically warranted, inform patients of the increased risk for serotonin syndrome and monitor for symptoms.5.3 Drug Interactions Leading to QT Prolongation
The CYP2D6 inhibitory properties of paroxetine can elevate plasma levels of thioridazine and pimozide. Since thioridazine and pimozide given alone produce prolongation of the QTc interval and increase the risk of serious ventricular arrhythmias, the use of paroxetine extended-release tablets are contraindicated in combination with thioridazine and pimozide [see Contraindications (4), Drug Interactions (7), Clinical Pharmacology (12.3)].
5.4 Embryofetal Toxicity
Based on meta-analyses of epidemiological studies, exposure to paroxetine in the first trimester of pregnancy is associated with a less than 2-fold increase in the rate of cardiovascular malformations among infants. For women who intend to become pregnant or who are in their first trimester of pregnancy, Paroxetine extended-release tablets, should be initiated only after consideration of the other available treatment options [see Use in Specific Populations (8.1)].
5.5 Increased Risk of Bleeding
Drugs that interfere with serotonin reuptake inhibition, including paroxetine extended-release tablets, increase the risk of bleeding events. Concomitant use of aspirin, nonsteroidal anti-inflammatory drugs (NSAIDS), other antiplatelet drugs, warfarin, and other anticoagulants may add to this risk. Case reports and epidemiological studies (case-control and cohort design) have demonstrated an association between use of drugs that interfere with serotonin reuptake and the occurrence of gastrointestinal bleeding. Based on data from the published observational studies, exposure to SSRIs, particularly in the month before delivery, has been associated with a less than 2-fold increase in the risk of postpartum hemorrhage [see Use in Specific Populations (8.1)]. Bleeding events related to drugs that interfere with serotonin reuptake have ranged from ecchymoses, hematomas, epistaxis, and petechiae to life-threatening hemorrhages.
Inform patients about the increased risk of bleeding associated with the concomitant use of paroxetine extended-release tablets and antiplatelet agents or anticoagulants. For patients taking warfarin, carefully monitor the international normalized ratio.5.6 Activation of Mania or Hypomania
In patients with bipolar disorder, treating a depressive episode with paroxetine extended-release tablets or another antidepressant may precipitate a mixed/manic episode. During controlled clinical trials of immediate-release paroxetine hydrochloride, hypomania or mania occurred in approximately 1% of paroxetine-treated unipolar patients compared to 1.1% of active-control and 0.3% of placebo-treated unipolar patients. Prior to initiating treatment with paroxetine extended-release tablets, screen patients for any personal or family history of bipolar disorder, mania, or hypomania.
5.7 Discontinuation Syndrome
Adverse reactions after discontinuation of serotonergic antidepressants, particularly after abrupt discontinuation, include: nausea, sweating, dysphoric mood, irritability, agitation, dizziness, sensory disturbances (e.g., paresthesia, such as electric shock sensations), tremor, anxiety, confusion, headache, lethargy, emotional lability, insomnia, hypomania, tinnitus, and seizures. A gradual reduction in dosage rather than abrupt cessation is recommended whenever possible [See Dosage and Administration (2.7)].
Adverse reactions have been reported upon discontinuation of treatment with paroxetine in pediatric patients. The safety and effectiveness of paroxetine extended-release tablets in pediatric patients have not been established [see Boxed Warning, Warnings and Precautions (5.1), Use in Specific Populations (8.4)].5.8 Seizures
Paroxetine extended-release tablets has not been systematically evaluated in patients with seizure disorders. Patients with history of seizures were excluded from clinical studies. Paroxetine extended-release tablets should be prescribed with caution in patients with a seizure disorder and should be discontinued in any patient who develops seizures.
5.9 Angle-Closure Glaucoma
The pupillary dilation that occurs following use of many antidepressant drugs including paroxetine extended-release tablets may trigger an angle closure attack in a patient with anatomically narrow angles who does not have a patent iridectomy. Cases of angle-closure glaucoma associated with use of paroxetine hydrochloride tablets have been reported. Avoid use of antidepressants, including paroxetine extended-release tablets, in patients with untreated anatomically narrow angles.
5.10 Hyponatremia
Hyponatremia may occur as a result of treatment with SNRIs and SSRIs, including paroxetine extended-release tablets. Cases with serum sodium lower than 110 mmol/L have been reported. Signs and symptoms of hyponatremia include headache, difficulty concentrating, memory impairment, confusion, weakness, and unsteadiness, which may lead to falls. Signs and symptoms associated with more severe and/or acute cases have included hallucination, syncope, seizure, coma, respiratory arrest, and death. In many cases, this hyponatremia appears to be the result of the syndrome of inappropriate antidiuretic hormone secretion (SIADH).
In patients with symptomatic hyponatremia, discontinue paroxetine extended-release tablets and institute appropriate medical intervention. Elderly patients, patients taking diuretics, and those who are volume- depleted may be at greater risk of developing hyponatremia with SNRIs and SSRIs. [see Use in Specific Populations (8.5)].5.11 Reduction of Efficacy of Tamoxifen
Some studies have shown that the efficacy of tamoxifen, as measured by the risk of breast cancer relapse/mortality, may be reduced with concomitant use of paroxetine as a result of paroxetine’s irreversible inhibition of CYP2D6 and lower blood levels of tamoxifen [see Drug Interactions (7.1)]. One study suggests that the risk may increase with longer duration of coadministration. However, other studies have failed to demonstrate such a risk. When tamoxifen is used for the treatment or prevention of breast cancer, prescribers should consider using an alternative antidepressant with little or no CYP2D6 inhibition.
5.12 Bone Fracture
Epidemiological studies on bone fracture risk during exposure to some antidepressants, including SSRIs, have reported an association between antidepressant treatment and fractures. There are multiple possible causes for this observation, and it is unknown to what extent fracture risk is directly attributable to SSRI treatment.
5.13 Sexual Dysfunction
Use of SSRIs, including paroxetine extended-release tablets, may cause symptoms of sexual dysfunction [see Adverse Reactions (6.1)]. In male patients, SSRI use may result in ejaculatory delay or failure, decreased libido, and erectile dysfunction. In female patients, SSRI use may result in decreased libido and delayed or absent orgasm.
It is important for prescribers to inquire about sexual function prior to initiation of paroxetine extended-release tablets and to inquire specifically about changes in sexual function during treatment, because sexual function may not be spontaneously reported. When evaluating changes in sexual function, obtaining a detailed history (including timing of symptom onset) is important because sexual symptoms may have other causes, including the underlying psychiatric disorder. Discuss potential management strategies to support patients in making informed decisions about treatment.
-
6 ADVERSE REACTIONS
The following adverse reactions are included in more detail in other sections of the prescribing information:
Hypersensitivity reactions to paroxetine [see Contraindications (4)]
Suicidal Thoughts and Behaviors [see Warnings and Precautions (5.1)]
Serotonin Syndrome [see Warnings and Precautions (5.2)]
Embryofetal and Neonatal Toxicity [see Warnings and Precautions (5.4)]
Increased Risk of Bleeding [see Warnings and Precautions (5.5)]
Activation of Mania/Hypomania [see Warnings and Precautions (5.6)]
Discontinuation Syndrome [see Warnings and Precautions (5.7)]
Seizures [see Warnings and Precautions (5.8)]
Angle-closure Glaucoma [see Warnings and Precautions (5.9)]
Hyponatremia [see Warnings and Precautions (5.10)]
Bone Fracture [see Warnings and Precautions (5.12)]Sexual Dysfunction [see Warnings and Precautions (5.13)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Safety data for paroxetine extended-release tablets is from 11 short-term, placebo-controlled clinical trials including 3 studies in patients with major depressive disorder (MDD) (Studies 1, 2, and 3), 3 studies in patients with panic disorder (PD) (Studies 4, 5, and 6), 1 study in patients with social anxiety disorder (SAD) (Study 7), and 4 studies in female patients with premenstrual dysphoric disorder (PMDD) (Studies 8, 9, 10, and 11) [see Clinical Studies (14)]. These 11 trials included 1,627 patients treated with paroxetine extended-release tablets.
Studies 1 and 2 were 12-week studies that enrolled patients 18 to 65 years old who received paroxetine extended-release tablets at doses ranging from 25 mg to 62.5 mg once daily. Study 3 was a 12-week study in patients 60 to 88 years old who received paroxetine extended-release tablets at doses ranging from 12.5 mg to 50 mg once daily.
Studies 4, 5, and 6 were 10-week studies in patients 19 to 72 years old who received paroxetine extended-release tablets at doses ranging from 12.5 mg to 75 mg once daily.
Study 7 was a 12-week study that enrolled adult patients who received paroxetine extended-release tablets at doses ranging from 12.5 mg to 37.5 mg once daily.
Studies 8, 9, and 10 were 12-week, placebo-controlled trials in female patients 18 to 46 years old who received paroxetine extended-release tablets at doses of 12.5 mg or 25 mg once daily. Study 11 was a 12-week placebo-controlled trial in patients 18 to 46 years old who received paroxetine extended-release tablets 2 weeks prior to the onset of menses (luteal phase dosing) at doses of 12.5 mg or 25 mg once daily.Adverse Reactions Leading to Discontinuation in Patients with MDD, PD, SAD, and PMDD In pooled studies in patients with MDD, PD and SAD, the most common adverse reactions leading to study withdrawal were: nausea (up to 4% of patients), asthenia, headache, depression, insomnia, and abnormal liver function tests (each occurring in up to 2% of patients), and dizziness, somnolence, and diarrhea (each occurring in up to 1% of patients).
In pooled studies for PMDD, the most common adverse reactions leading to study withdrawal were: nausea (occurring in up to 6% of patients), asthenia (occurring in up to 5% of patients), somnolence (occurring in up to 4% of patients), insomnia (occurring in approximately 2% of patients); and impaired concentration, dry mouth, dizziness, decreased appetite, sweating, tremor, yawn and diarrhea (occurring in less than or equal to 2% of patients).
Adverse Reactions in MDD, PD, and SAD
Table 3 presents the most common adverse reactions in paroxetine extended-release tablets-treated patients (incidence≥5% and greater than placebo within at least 1 of the indications) in controlled trials in patients with MDD, PD, and SAD.
Table 3. Adverse Reactions (≥5% of Patients Treated with Paroxetine Extended-Release Tablets and Greater than Placebo) in 10 to 12 Week Studies of MDD, PD, and SAD
MDD
18 to 65 year olds
MDD
≥60 years old
Panic Disorder
Social Anxiety Disorder
Body System/ Adverse Reaction
Paroxetine
Extended-
Release
Tablets (N=212)
%
Placebo (N=211)
%
Paroxetine
Extended-
Release
Tablets (N=104)
%
Placebo (N=109)
%
Paroxetine
Extended-
Release
Tablets (N=444)
%
Placebo (N=445)
%
Paroxetine
Extended-
Release
Tablets (N=186)
%
Placebo
(N=184)
%
Body as a Whole
Headache
27
20
17
13
NA
NA
23
17
Asthenia
14
9
15
14
15
10
18
7
Abdominal Pain
7
4
-
-
6
4
5
4
Back Pain
5
3
-
-
NA
NA
4
1
Digestive System
Nausea
22
10
-
-
23
17
22
6
Diarrhea
18
7
15
9
12
9
9
8
Dry Mouth
15
8
18
7
13
9
3
2
Constipation
10
4
13
5
9
6
5
2
Flatulence
6
4
-
-
NA
NA
NA
NA
Decreased Appetite
2
12
5
8
6
1
<1
Dyspepsia
NA
NA
13
10
NA
NA
2
<1
Musculoskel etal System
Myalgia
NA
NA
-
-
5
3
NA
NA
Nervous
System
Somnolence22
8
21
12
20
9
9
4
Insomnia17
9
10
8
20
11
9
4
Dizziness14
4
9
5
NA
NA
7
4
Libido
Decreased7
3
8
<1
9
4
1
NervousnessNA
NA
-
-
8
7
NA
NA
Tremor7
1
7
0
8
2
4
2
AnxietyNA
NA
-
-
5
4
2
1
Respiratory
System
SinusitisNA
NA
-
-
8
5
NA
NA
Yawn
0
-
-
3
0
2
0
Skin andAppendages
Sweating6
2
10
<1
7
2
14
3
Special
Senses
Abnormal Visiona 5
1
-
-
3
<1
2
0
Urogenital
System
Abnormal Ejaculationb,c
26
1
17
3
27
3
15
1
FemaleGenital Disorderb,d
10
<1
-
-
7
1
3
0
Impotenceb
5
3
9
3
10
1
9
0
Hyphen = the reaction listed occurred in <5% of patients treated with paroxetine extended-release tablets
NA = the adverse reaction listed did not occur in this group of patients
a Mostly blurred vision
b Based on the number of males or females
c Mostly anorgasmia or delayed ejaculation
d Mostly anorgasmia or delayed orgasm
Other Adverse Reactions Observed During the Premarketing Evaluation of Paroxetine Extended-Release Tablets
Adverse reactions from studies in MDD (not including Study 3 in elderly patients), PD, and SAD that occurred between 1% and 5% of patients treated with paroxetine extended-release tablets and at a rate greater than in placebo-treated patients include:, allergic reaction, tachycardia, vasodilatation, hypertension, migraine, vomiting, weight loss, weight gain, hypertonia, paresthesia, agitation, confusion, myoclonus, concentration impaired, depression, rhinitis, cough increased, bronchitis, photosensitivity, eczema, taste perversion, UTI, menstrual disorder, urinary frequency, urination impaired, and vaginitis.Adverse Reactions in Patients with PMDD
Table 4 displays adverse reactions that occurred (incidence of 5% or more and greater than placebo within at least 1 of the studies) in patients treated with paroxetine extended-release tablets in Studies 8, 9, 10, and 11.
Table 4 Adverse Reactions (≥5% of Patients Treated with Paroxetine Extended-Release Tablets and Greater than Placebo) in Pooled Studies PMDD (Studies 8, 9, 11), and in Study 10a,b,c
Body System/Adverse Reaction
% Reporting Adverse Reaction
Continuous Dosing
Studies 8, 9, and 10
Luteal Phase Dosing
Study 11
Paroxetine
Extended-
Release
Tablets
(n = 681)
%
Placebo
(n = 349) %
Paroxetine
Extended-
Release
Tablets
(n = 246)
%
Placebo
(n = 120)
%
Body as a Whole
Asthenia
17
6
15
4
Headache
15
12
NA
NA
Infection
6
4
NA
NA
Digestive System
Nausea
17
7
18
2
Diarrhea
6
2
6
0
Constipation
5
1
2
<1
Nervous System
Libido Decreased
12
5
9
6
Somnolence
9
2
3
<1
Insomnia
8
2
7
3
Dizziness
7
3
6
3
Tremor
4
<1
5
0
Skin and Appendages
Sweating
7
<1
6
<1
Urogenital System
Female Genital Disordersc
8
1
2
0
NA= the adverse reaction information is not available in this population.
a <1% means greater than zero and less than 1%.
b The luteal phase and continuous dosing PMDD trials were not designed for making direct comparisons between the 2 dosing regimens.
c Mostly anorgasmia or difficulty achieving orgasm.
Dose Dependent Adverse Reactions
Comparison of the incidence of adverse reactions (placebo vs. 12.5 mg paroxetine extended-release tablets vs. 25 mg paroxetine extended-release tablets) from studies 8, 9, 10 showed the following adverse reactions to be dose-related: Nausea, somnolence, sweating, dry mouth, dizziness, decreased appetite, tremor, impaired concentration, yawn, paresthesia, hyperkinesia, and vaginitis.Male and Female Sexual Dysfunction
Although changes in sexual desire, sexual performance, and sexual satisfaction often occur as manifestations of a psychiatric disorder, they may also be a consequence of SSRI treatment. However, reliable estimates of the incidence and severity of untoward experiences involving sexual desire, performance, and satisfaction are difficult to obtain, in part because patients and healthcare providers may be reluctant to discuss them. Accordingly, estimates of the incidence of untoward sexual experience and performance cited in labeling may underestimate their actual incidence.
The percentage of patients reporting symptoms of sexual dysfunction in the Studies 1 and 2 (nonelderly patients with MDD), 4, 5, 6, 7, 8, 9, 10, and 11 are presented in Table 5:
Table 5. Adverse Reactions Related To Sexual Dysfunction In Patients Treated With Paroxetine Extended-Release Tablets in Pooled 10 to 12 Week Studies of MDD, PD, SAD, and PMDD
Studies 1 and 2
%
Studies 4, 5, and 6
%
Study 7 %
Studies 8, 9, and 11 (Continuous Dosing)
%
Study 10 (Luteal Phase Dosing)
%
Paroxetine
Extended-
Release Tablets
Placebo
Paroxetine
Extended-
Release Tablets
Placebo
Paroxetine
Extended-
Release Tablets
Placebo
Paroxetine
Extended-
Release Tablets
Placebo
Paroxetine
Extended-
Release Tablets
Placebo
n (males)
78
78
162
194
88
97
NA
NA
NA
NA
Decreased Libido
10
5
9
6
13
1
NA
NA
NA
NA
Abnormal ejaculation
26
1
27
3
15
1
NA
NA
NA
NA
Impotence
5
3
10
1%
9
0
NA
NA
NA
NA
n (females)
134
133
282
251
98
87
681
349
246
120
Decreased Libido
4
2
8
2
4
1
12
5
9
6
Orgasmic Disturbance
10
<1
7
1
3
0
8
1
2
0
NA = the adverse reaction listed did not occur in this group of patients.
Paroxetine treatment has been associated with several cases of priapism. In those cases with a known outcome, patients recovered without sequelae.
Less Common Adverse Reactions
The following adverse reactions occurred during the clinical studies of paroxetine extended-release tablets and are not included elsewhere in the labeling.
Reactions are categorized by body system and listed in order of decreasing frequency according to the following definitions: Frequent adverse reactions are those occurring on 1 or more occasions in at least 1/100 patients; infrequent adverse reactions are those occurring in 1/100 to 1/1,000 patients; rare reactions are those occurring in fewer than 1/1,000 patients.
Cardiovascular System: Infrequent was postural hypotension.
Hemic and Lymphatic System: Rare was thrombocytopenia.
Metabolic and Nutritional Disorders: Infrequent were generalized edema and hypercholesteremia.
Nervous System: Infrequent were convulsion, akathisia, and manic reaction.
Psychiatric: Infrequent were hallucinations.
Skin and Appendages: Frequent was rash; infrequent was urticaria; rare was angioedema and erythema multiforme.
Urogenital System: Infrequent was urinary retention; rare was urinary incontinence.
6.2 Postmarketing Experience
The following reactions have been identified during post approval use of paroxetine. Because these reactions are reported voluntarily from a population of unknown size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Acute pancreatitis, elevated liver function tests (the most severe cases were deaths due to liver necrosis, and grossly elevated transaminases associated with severe liver dysfunction), Guillain-Barré syndrome, Stevens-Johnson syndrome, toxic epidermal necrolysis, drug reaction with eosinophilia and systemic symptoms (DRESS), priapism, syndrome of inappropriate ADH secretion (SIADH), prolactinemia and galactorrhea; extrapyramidal symptoms which have included akathisia, bradykinesia, cogwheel rigidity, dystonia, hypertonia, trismus; status epilepticus, acute renal failure, pulmonary hypertension, allergic alveolitis, anosmia, hyposmia, anaphylaxis, eclampsia, laryngismus, optic neuritis, porphyria, restless legs syndrome (RLS), ventricular fibrillation, ventricular tachycardia (including torsade de pointes), hemolytic anemia, events related to impaired hematopoiesis (including aplastic anemia, pancytopenia, bone marrow aplasia, and agranulocytosis), and vasculitic syndromes (such as Henoch-Schönlein purpura).
-
7 DRUG INTERACTIONS
7.1 Clinically Significant Drug Interactions with Paroxetine Extended-Release Tablets
Table 6 includes clinically significant drug interactions with Paroxetine Extended-Release Tablets.
Table 6: Clinically Significant Drug Interactions with Paroxetine Extended-Release Tablets
Monoamine Oxidase Inhibitors (MAOIs)
Clinical Impact
The concomitant use of SSRIs, including paroxetine extended-release tablets, and MAOIs increases the risk of serotonin syndrome.
Intervention
Paroxetine extended-release tablets are contraindicated in patients taking MAOIs, including MAOIs such as linezolid or intravenous methylene blue [see Dosage and Administration (2.6), Contraindications (4), Warnings and Precautions (5.2)].
Examples
selegiline, tranylcypromine, isocarboxazid, phenelzine, linezolid, methylene blue
Pimozide and Thioridazine
Clinical Impact
Increased plasma concentrations of pimozide and thioridazine, drugs with a narrow therapeutic index, may increase the risk of QTc prolongation and ventricular arrhythmias.
Intervention
Paroxetine extended-release tablets are contraindicated in patients taking pimozide or thioridazine [see Contraindications (4)].
Other Serotonergic Drugs
Clinical Impact
The concomitant use of serotonergic drugs with paroxetine extended-release tablets increases the risk of serotonin syndrome.
Intervention
Monitor patients for signs and symptoms of serotonin syndrome, particularly during treatment initiation and dosage increases. If serotonin syndrome occurs, consider discontinuation of paroxetine extended-release tablets and/or concomitant serotonergic drugs [see Warnings and Precautions (5.2)].
Examples
Other SSRIs, SNRIs, triptans, tricyclic antidepressants, opioids, lithium, tryptophan, buspirone, St. John’s Wort
Drugs that Interfere with Hemostasis (antiplatelet agents and anticoagulants)
Clinical Impact
The concurrent use of an antiplatelet agent or anticoagulant with
paroxetine extended-release tablets may potentiate the risk of bleeding.
Intervention
Inform patients of the increased risk of bleeding associated with the concomitant use of paroxetine extended-release tablets and antiplatelet agents and anticoagulants. For patients taking warfarin, carefully monitor the international normalized ratio [see Warnings and Precautions (5.5)].
Examples
aspirin, clopidogrel, heparin, warfarin
Drugs Highly Bound to Plasma Protein
Clinical Impact
Paroxetine extended-release tablets are highly bound to plasma protein. The concomitant use of paroxetine extended-release tablets with another drug that is highly bound to plasma protein may increase free concentrations of paroxetine extended-release tablets or other tightly-bound drugs in plasma.
Intervention
Monitor for adverse reactions and reduce dosage of paroxetine extended-release tablets or other protein-bound drugs as warranted.
Examples
warfarin
Drugs Metabolized by CYP2D6
Clinical Impact
Paroxetine extended-release tablets are a CYP2D6 inhibitor [see Clinical Pharmacology (12.3)]. The concomitant use of paroxetine extended-release tablets with a CYP2D6 substrate may increase the exposure of the CYP2D6 substrate.
Intervention
Decrease the dosage of a CYP2D6 substrate if needed with concomitant paroxetine extended-release tablets use. Conversely, an increase in dosage of a CYP2D6 substrate may be needed if paroxetine extended-release tablets are discontinued.
Examples
propafenone, flecainide, atomoxetine, desipramine, dextromethorphan, metoprolol, nebivolol, perphenazine, tolterodine, venlafaxine, risperidone.
Tamoxifen
Clinical Impact
Concomitant use of tamoxifen with paroxetine extended-release tablets may lead to reduced plasma concentrations of the active metabolite (endoxifen) and reduced efficacy of tamoxifen
Intervention
Consider use of an alternative antidepressant little or no CYP2D6 inhibition [see Warnings and Precautions (5.11)].
Fosamprenavir/Ritonavir
Clinical Impact
Co-administration of fosamprenavir/ritonavir with paroxetine significantly decreased plasma levels of paroxetine.
Intervention
Any dose adjustment should be guided by clinical effect (tolerability and efficacy).
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to antidepressants during pregnancy. Healthcare providers are encouraged to register patients by calling the National Pregnancy Registry for Antidepressants at 1-866-961-2388 or visiting online at https://womensmentalhealth.org/clinical-and-researchprograms/pregnancyregistry/antidepressants/.
Risk Summary
Based on data from published observational studies, exposure to SSRIs, particularly in the month before delivery, has been associated with a less than 2-fold increase in the risk of postpartum hemorrhage [see Warnings and Precautions (5.5) and Clinical Considerations].
Paroxetine extended-release tablets are associated with a less than 2-fold increase in cardiovascular malformations when administered to a pregnant woman during the first trimester. While individual epidemiological studies on the association between paroxetine use and cardiovascular malformations have reported inconsistent findings, some meta-analyses of epidemiological studies have identified an increased risk of cardiovascular malformations (see Data). There are risks of persistent pulmonary hypertension of the newborn (PPHN) (see Data) and/or poor neonatal adaptation with exposure to selective serotonin reuptake inhibitors (SSRIs), including Paroxetine extended-release tablets, during pregnancy. There also are risks associated with untreated depression in pregnancy (seeClinical Considerations). For women who intend to become pregnant or who are in their first trimester of pregnancy, paroxetine should be initiated only after consideration of the other available treatment options.
No evidence of treatment related malformations was observed in animal reproduction studies, when paroxetine was administered during the period of organogenesis at doses up to 50 mg/kg/day in rats and 6 mg/kg/day in rabbits. These doses are approximately 6 (rat) and less than 2 (rabbit) times the maximum recommended human dose (MRHD – 75 mg) on an mg/m2 basis. When paroxetine was administered to female rats during the last trimester of gestation and continued through lactation, there was an increase in the number of pup deaths during the first four days of lactation. This effect occurred at a dose of 1 mg/kg/day which is less than the MRHD on an mg/m2 basis (see Data).
The estimated background risks of major birth defects and miscarriage for the indicated populations are unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the US general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Clinical Considerations
Disease-associated maternal and/or embryo/fetal risk
Women who discontinue antidepressants during pregnancy are more likely to experience a relapse of major depression than women who continue antidepressants. This finding is from a prospective longitudinal study of 201 pregnant women with a history of major depressive disorder who were euthymic and taking antidepressants at the beginning of pregnancy. Consider the risks of untreated depression when discontinuing or changing treatment with antidepressant medication during pregnancy and postpartum.
Maternal Adverse Reactions
Use of Paroxetine extended-release tablets in the month before delivery may be associated with an increased risk of postpartum hemorrhage [see Warnings and Precautions (5.5)].
Fetal/Neonatal adverse reactions
Neonates exposed to Paroxetine extended-release tablets and other SSRIs late in the third trimester have developed complications requiring prolonged hospitalization, respiratory support, and tube feeding. Such complications can arise immediately upon delivery. Reported clinical findings have included respiratory distress, cyanosis, apnea, seizures, temperature instability, feeding difficulty, vomiting, hypoglycemia, hypotonia, hypertonia, hyperreflexia, tremors, jitteriness, irritability, and constant crying. These findings are consistent with either a direct toxic effect of SSRIs or possibly a drug discontinuation syndrome. It should be noted that, in some cases, the clinical picture is consistent with serotonin syndrome [see Warnings and Precautions (5.4)].
Data
Human Data
Published epidemiological studies on the association between first trimester paroxetine use and cardiovascular malformations have reported inconsistent results; however, meta-analyses of population-based cohort studies published between 1996-2017 indicate a less than 2-fold increased risk for overall cardiovascular malformations. Specific cardiac malformations identified in two meta-analyses include approximately 2 to 2.5-fold increased risk for right ventricular outflow tract defects. One meta-analysis also identified an increased risk (less than 2- fold) for bulbus cordis anomalies and anomalies of cardiac septal closure, and an increased risk for atrial septal defects (pooled OR 2.38, 95% CI 1.14-4.97). Important limitations of the studies included in these meta-analyses include potential confounding by indication, depression severity, and potential exposure misclassification.
Exposure to SSRIs, particularly later in pregnancy, may have an increased risk for PPHN. PPHN occurs in 1-2 per 1000 live births in the general population and is associated with substantial neonatal morbidity and mortality.
Animal Data
Reproduction studies were performed at doses up to 50 mg/kg/day in rats and 6 mg/kg/day in rabbits administered during organogenesis. These doses are approximately 6 (rat) and less than 2 (rabbit) times the maximum recommended human dose (MRHD – 75 mg) on an mg/m2 basis. These studies have revealed no evidence of malformations. However, in rats, there was an increase in pup deaths during the first 4 days of lactation when dosing occurred during the last trimester of gestation and continued throughout lactation. This effect occurred at a dose of 1 mg/kg/day which is less than the MRHD on an mg/m2 basis. The no-effect dose for rat pup mortality was not determined. The cause of these deaths is not known.
8.2 Lactation
Risk Summary
Data from the published literature report the presence of paroxetine in human milk (see Data). There are reports of agitation, irritability, poor feeding and poor weight gain in infants exposed to paroxetine through breast milk (see Clinical Considerations). There are no data on the effects of paroxetine on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for Paroxetine extended-release tablets and any potential adverse effects on the breastfed infant from Paroxetine extended-release tablets or the underlying maternal condition.
Clinical Considerations
Infants exposed to Paroxetine extended-release tablets should be monitored for agitation, irritability, poor feeding and poor weight gain.
Data
Published literature suggests the presence of paroxetine in human milk with relative infant doses ranging between 0.4% to 2.2%, and a milk: plasma ratio of <1. No significant amounts were detected in the plasma of infants after breastfeeding.
8.3 Females and Males of Reproductive Potential
Infertility
Male
Based on findings from clinical studies, paroxetine may affect sperm quality which may impair fertility; it is not known if this effect is reversible [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of paroxetine extended-release tablets in pediatric patients have not been established [see Boxed Warning, Warnings and Precautions (5.1)].
Three placebo-controlled trials in 752 pediatric patients with MDD have been conducted with immediate-release paroxetine, and effectiveness was not established in pediatric patients.
Decreased appetite and weight loss have been observed in association with the use of SSRIs.
In placebo-controlled clinical trials conducted with pediatric patients, the following adverse reactions were reported in at least 2% of pediatric patients treated with immediate-release paroxetine hydrochloride and at a rate at least twice that for pediatric patients receiving placebo: emotional lability (including self-harm, suicidal thoughts, attempted suicide, crying, and mood fluctuations), hostility, decreased appetite, tremor, sweating, hyperkinesia, and agitation.
Adverse reactions upon discontinuation of treatment with immediate-release paroxetine hydrochloride in the pediatric clinical trials that included a taper phase regimen, which occurred in at least 2% of patients and at a rate at least twice that of placebo, were: emotional lability (including suicidal ideation, suicide attempt, mood changes, and tearfulness), nervousness, dizziness, nausea, and abdominal pain.
8.5 Geriatric Use
SSRIs and SNRIs, including paroxetine extended-release tablets, have been associated with cases of clinically significant hyponatremia in elderly patients, who may be at greater risk for this adverse reaction [see Warnings and Precautions (5.9)].
In premarketing clinical trials with immediate-release paroxetine hydrochloride, 17% of paroxetine treated patients (approximately 700) were 65 years or older. Pharmacokinetic studies revealed a decreased clearance in the elderly, and a lower starting dose is recommended; however, no overall differences in safety or effectiveness were observed between these subjects and younger subjects [see Dosage and Administration (2.5), Clinical Pharmacology (12.3)].
8.6 Renal and/or Hepatic Impairment
Increased plasma concentrations of paroxetine occur in patients with renal and hepatic impairment. The initial dosage of Paroxetine extended-release tablets should be reduced in patients with severe renal impairment and patients with severe hepatic impairment [see Dosage and Administration (2.5) and Clinical Pharmacology (12.3)].
-
10 OVERDOSAGE
The following have been reported with paroxetine tablet overdosage:
Seizures, which may be delayed, and altered mental status including coma.
Cardiovascular toxicity, which may be delayed, including QRS and QTc interval prolongation. Hypertension most commonly seen, but rarely can see hypotension alone or with co-ingestants including alcohol.
Serotonin syndrome (patients with a multiple drug overdosage with other proserotonergic drugs may have a higher risk).
Gastrointestinal decontamination with activated charcoal should be considered in patients who present early after a paroxetine overdose.
Consider contacting a Poison Center (1-800-222-1222) or a medical toxicologist for additional overdosage management recommendations.
-
11 DESCRIPTION
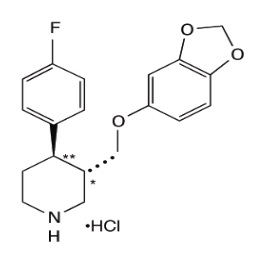
Paroxetine extended-release tablets, contains paroxetine hydrochloride hemihydrate, USP an SSRI. It is the hydrochloride salt of a phenylpiperidine compound identified chemically as (3S,4R)-3-[(1,3 benzodioxol-5-yloxy)methyl]-4-(4-fluorophenyl) piperidine hydrochloride hemihydrate or Piperidine, 3-[(1,3-benzodioxol-5-yloxy)methyl]- 4-( 4-fluorophenyl)-, hydrochloride, (3S-trans)- and has the empirical formula of C19H20FNO3HCl1/2 H2O. The molecular weight is 374.8 g/mol (329.4 g/mol as free base). The structural formula of paroxetine hydrochloride is:
Paroxetine hydrochloride hemihydrate, USP is a white to off white solid (or) powder, having a melting point range of 120°C to 142°C and soluble in methanol and in alcohol; slightly soluble in water.
Paroxetine extended-release tablets are intended for oral administration. Each film-coated, extended-release tablet contains paroxetine hydrochloride USP equivalent to paroxetine 12.5 mg, 25 mg and 37.5 mg. One layer of the tablet consists of a degradable barrier layer and the other contains the active material in a hydrophilic matrix.
Inactive ingredients consist of ferric oxide yellow, glyceryl behenate, hypromellose, lactose monohydrate, magnesium stearate, methacrylic acid – ethyl acrylate copolymer (1:1) type A, polyethylene glycols, polysorbate 80, polyvinylpyrrolidone, silicon dioxide, sodium lauryl sulfate, talc, titanium dioxide, triethyl citrate, film-coating material contains - FD&C Yellow #6/sunset yellow FCF aluminum lake, FD&C blue #2/Indigo carmine AL 3% to 5% and D&C Yellow #10 aluminum lake for 12.5 mg; D&C Red #30/Helendon pink aluminum lake for 25 mg and FD&C blue #2/Indigo carmine AL 3% to 5% and FD&C blue #2 Indigo carmine aluminum lake for 37.5 mg.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
The mechanism of action of paroxetine in the treatment of major depressive disorder (MDD), panic disorder (PD), social anxiety disorder (SAD), and premenstrual dysphoric disorder (PMDD) is unknown, but is presumed to be linked to potentiation of serotonergic activity in the central nervous system resulting from inhibition of neuronal reuptake of serotonin (5-HT).
12.2 Pharmacodynamics
Studies at clinically relevant doses in humans have demonstrated that paroxetine blocks the uptake of serotonin into human platelets. In vitro studies in animals also suggest that paroxetine is a potent and highly selective inhibitor of neuronal serotonin reuptake (SSRI) and has only very weak effects on norepinephrine and dopamine neuronal reuptake.
12.3 Pharmacokinetics
Absorption
Tablets of paroxetine extended-release tablets contain a degradable polymeric matrix designed to control the dissolution rate of paroxetine over a period of approximately 4 to 5 hours. In addition to controlling the rate of drug release in vivo, an enteric coat delays the start of drug release until tablets of paroxetine extended-release tablets have left the stomach.
Paroxetine extended-release tablets are completely absorbed after oral dosing of a solution of the hydrochloride salt. In a study in which normal male and female subjects (n = 23) received single oral doses of paroxetine extended-release tablets at 4 dosage strengths (12.5 mg, 25 mg, 37.5 mg, and 50 mg), paroxetine Cmax and AUC0-inf increased disproportionately with dose (as seen also with immediate-release formulations). Mean Cmax and AUC0-inf values at these doses were 2.0, 5.5, 9.0, and 12.5 ng/mL, and 121, 261, 338, and 540 nghr. /mL, respectively. Tmax was observed typically between 6 and 10 hours post-dose, reflecting a reduction in absorption rate compared with immediate-release formulations. The bioavailability of 25 mg paroxetine extended-release tablets are not affected by food.
Distribution
Paroxetine distributes throughout the body, including the CNS, with only 1% remaining in the plasma.
Approximately 95% and 93% of paroxetine is bound to plasma protein at 100 ng/mL and 400 ng/mL, respectively. Under clinical conditions, paroxetine concentrations would normally be less than
400 ng/mL. Paroxetine does not alter the in vitro protein binding of phenytoin or warfarin.Elimination
Metabolism
The mean elimination half-life of paroxetine was 15 to 20 hours throughout a range of single doses of paroxetine extended-release tablets (12.5 mg, 25 mg, 37.5 mg, and 50 mg). During repeated administration of paroxetine extended-release tablets (25 mg once daily), steady state was reached within 2 weeks (i.e., comparable to immediate-release formulations). In a repeat-dose study in which normal male and female subjects (n = 23) received paroxetine extended-release tablets (25 mg daily), mean steady state Cmax, Cmin, and AUC0-24 values were 30 ng/mL, 20 ng/mL, and 550 nghr./mL, respectively.
Based on studies using immediate-release formulations, steady-state drug exposure based on AUC0-24 was several-fold greater than would have been predicted from single-dose data. The excess accumulation is a consequence of the fact that 1 of the enzymes that metabolizes paroxetine is readily saturable.
In steady-state dose proportionality studies involving elderly and nonelderly patients, at doses of the immediate-release formulation of 20 mg to 40 mg daily for the elderly and 20 mg to 50 mg daily for the nonelderly, some nonlinearity was observed in both populations, again reflecting a saturable metabolic pathway (Figure 3).
Paroxetine is extensively metabolized after oral administration. The principal metabolites are polar and conjugated products of oxidation and methylation, which are readily cleared. Conjugates with glucuronic acid and sulfate predominate, and major metabolites have been isolated and identified. Data indicate that the metabolites have no more than 1/50 the potency of the parent compound at inhibiting serotonin uptake. The metabolism of paroxetine is accomplished in part by CYP2D6. Saturation of this enzyme at clinical doses appears to account for the nonlinearity of paroxetine kinetics with increasing dose and increasing duration of treatment. The role of this enzyme in paroxetine metabolism also suggests potential drug-drug interactions [see Drug Interactions (7.1)].
Excretion
Approximately 64% of a 30-mg oral solution dose of paroxetine was excreted in the urine with 2% as the parent compound and 62% as metabolites over a 10-day post-dosing period. About 36% was excreted in the feces (probably via the bile), mostly as metabolites and less than 1% as the parent compound over the 10-day post-dosing period.The elimination half-life is approximately 15 to 20 hours after a single dose of paroxetine extended-release tablets. Paroxetine metabolism is mediated in part by CYP2D6, and the metabolites are primarily excreted in the urine and to some extent in the feces. Pharmacokinetic behavior of paroxetine has not been evaluated in subjects who are deficient in CYP2D6 (poor metabolizers).
Drug Interaction Studies
There are clinically significant, known drug interactions between paroxetine and other drugs [see Drug Interactions (7)].
Figure 1. Impact of Paroxetine on the Pharmacokinetics of Co-Administered Drugs (log scale)
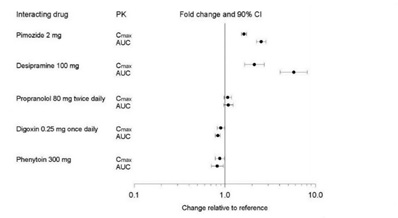
Figure 2. Impact of Co-Administered Drugs on the Pharmacokinetics of Paroxetine
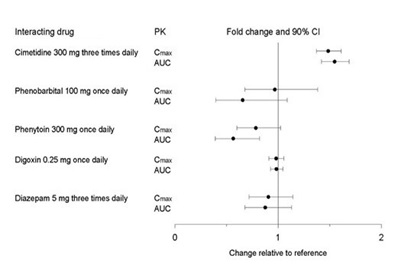
Theophylline: Reports of elevated theophylline levels associated with immediate-release paroxetine treatment have been reported. While this interaction has not been formally studied, it is recommended that theophylline levels be monitored when these drugs are concurrently administered.
Drugs Metabolized by Cytochrome CYP3A4
An in vivo interaction study involving the coadministration under steady-state conditions of paroxetine and terfenadine, a substrate for CYP3A4, revealed no effect of paroxetine on terfenadine pharmacokinetics. In addition, in vitro studies have shown ketoconazole, a potent inhibitor of CYP3A4 activity, to be at least 100 times more potent than paroxetine as an inhibitor of the metabolism of several substrates for this enzyme, including terfenadine, astemizole, cisapride, triazolam, and cyclosporine. Paroxetine’s extent of inhibition of CYP3A4 activity is not expected to be of clinical significance.Specific Populations
The impact of specific populations on the pharmacokinetics of paroxetine are shown in Figure 3.
Figure 3. Impact of Specific Population on the Pharmacokinetics of Paroxetine (log scale)
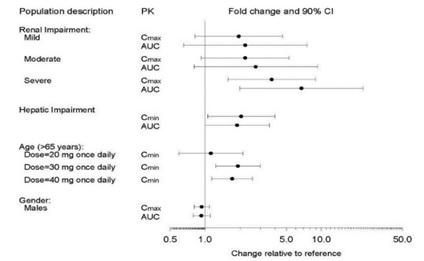
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Two-year carcinogenicity studies were conducted in rodents given paroxetine in the diet at 1, 5, and 25 mg/kg/day (mice) and 1, 5, and 20 mg/kg/day (rats). These doses are up to approximately 1.6 (mouse) and 2.5 (rat) times the MRHD on an mg/m2 basis. There was a significantly greater number of male rats in the high-dose group with reticulum cell sarcomas (1/100, 0/50, 0/50, and 4/50 for control, low-, middle-, and high-dose groups, respectively) and a significantly increased linear trend across dose groups for the occurrence of lymphoreticular tumors in male rats. Female rats were not affected. Although there was a dose-related increase in the number of tumors in mice, there was no drug-related increase in the number of mice with tumors. The relevance of these findings to humans is unknown.
Mutagenesis
Paroxetine produced no genotoxic effects in a battery of 5 in vitro and 2 in vivo assays that included the following: Bacterial mutation assay, mouse lymphoma mutation assay, unscheduled DNA synthesis assay, and tests for cytogenetic aberrations in vivo in mouse bone marrow and in vitro in human lymphocytes and in a dominant lethal test in rats.
Impairment of Fertility
A reduced pregnancy rate was found in reproduction studies in rats at a dose of paroxetine of 15 mg/kg/day, which is approximately twice the MRHD on an mg/m2 basis. Irreversible lesions occurred in the reproductive tract of male rats after dosing in toxicity studies for 2 to 52 weeks. These lesions consisted of vacuolation of epididymal tubular epithelium at 50 mg/kg/day and atrophic changes in the seminiferous tubules of the testes with arrested spermatogenesis at 25 mg/kg/day (approximately 6 and 3 times the MRHD on an mg/m2 basis).
-
14 CLINICAL STUDIES
14.1 Major Depressive Disorder
The efficacy of paroxetine extended-release tablets as a treatment for major depressive disorder (MDD) was established in two 12-week, multicenter, randomized, double-blind, placebo-controlled, flexible dose studies with paroxetine extended-release tablets (Study 1 and Study 2) in adult patients who met Diagnostic and Statistical Manual of Mental Disorders (DSM)-IV criteria for MDD. Study 1 and 2 included patients 18 to 65 years old who received paroxetine extended-release tablets doses of 25 to 62.5 mg/day (N= 212) or placebo (N= 211) once daily compared to immediate-release paroxetine 20 to 50 mg (N=217). A third 12-week, multicenter, randomized, double-blind, placebo-controlled, flexible dose study with paroxetine extended-release tablets (Study 3) included elderly patients, ranging in age from 60 to 88 years old and used paroxetine extended-release tablets doses of 12.5 to 50 mg/day (N=104) or placebo (N=109) once daily compared to immediate- release paroxetine 10 to 40 mg (N=106). In all three studies, paroxetine extended-release tablets were statistically superior to placebo in improving depressive symptoms as measured by the following: the mean change from baseline in the Hamilton Depression Rating Scale (HDRS) total score at Week 12, the mean change from baseline in the Hamilton Depressed Mood item score at Week 12, and the mean change from baseline in the Clinical Global Impression (CGI)–Severity of Illness score.
Long-term efficacy of paroxetine for treatment of MDD in outpatients was established with one randomized withdrawal study with immediate-release paroxetine. Patients who responded to immediate-release paroxetine (HDRS total score <8) during an initial 8-week open-label treatment phase were then randomized to continue immediate-release paroxetine or placebo, for up to 1 year. Patients treated with immediate-release paroxetine demonstrated a statistically significant lower relapse rate during the withdrawal phase (15%) compared to those on placebo (39%). Effectiveness was similar for male and female patients.14.2 Panic Disorder
The effectiveness of paroxetine extended-release tablets in the treatment of panic disorder (PD) was evaluated in three 10-week, multicenter, flexible-dose studies (Studies 4, 5, and 6) comparing paroxetine extended-release tablets (12.5 to 75 mg daily) to placebo in adult outpatients 19 to 72 years of age who met panic disorder (with or without agoraphobia) criteria according to DSM-IV. These trials were assessed on the basis of their outcomes on 3 variables: (1) the proportions of patients free of full panic attacks at Week 10; (2) change from baseline to Week 10 in the median number of full panic attacks; and (3) change from baseline to Week 10 in the median Clinical Global Impression Severity score. For Studies 4 and 5, paroxetine extended-release tablets was superior to placebo on 2 of these 3 variables. Study 6 failed to consistently demonstrate a statistically significant difference between paroxetine extended-release tablets and placebo on any of these variables.
For all 3 studies, the mean dose of paroxetine extended-release tablets for completers at Week 10 was approximately 50 mg/day. Subgroup analyses did not indicate that there were any differences in treatment outcomes as a function of age or gender.
Long-term maintenance effects of paroxetine in patients with PD were demonstrated in a randomized-withdrawal study using immediate-release paroxetine. Patients who were responders during a 10-week, double-blind trial (followed by a 3-month double-blind maintenance phase) of immediate-release paroxetine were re-randomized to continue immediate-release paroxetine or placebo in a 3-month, double-blind withdrawal phase. Patients randomized to immediate-release paroxetine were statistically significantly less likely to relapse than placebo-treated patients.14.3 Social Anxiety Disorder
The efficacy of paroxetine extended-release tablets as a treatment for social anxiety disorder (SAD) was established, in part, on the basis of extrapolation from the established effectiveness of immediate- release paroxetine in the treatment of SAD. In addition, the effectiveness of paroxetine extended-release tablets in the treatment of SAD was demonstrated in one 12-week, multicenter, double-blind, flexible-dose, placebo-controlled study of adult outpatients with a primary diagnosis of SAD by DSM-IV criteria (Study 7). In Study 7, the effectiveness of paroxetine extended-release tablets (12.5 to 37.5 mg daily) compared to placebo was evaluated on the basis of (1) change from baseline in the Liebowitz Social Anxiety Scale (LSAS) total score at Week 12 and (2) the proportion of responders who scored 1 or 2 (very much improved or much improved) on the CGI Global Improvement score at Week 12.
In Study 7, paroxetine extended-release tablets demonstrated statistically significant superiority over placebo on both the change on LSAS total score at Week 12 and the CGI Improvement responder criterion at Week 12. For patients who completed the trial, 64% of patients treated with paroxetine extended-release tablets compared to 35% of patients treated with placebo were CGI Improvement responders at Week 12.
Subgroup analyses did not indicate that there were any differences in treatment outcomes as a function of gender. Subgroup analyses of studies utilizing the immediate-release formulation of paroxetine generally did not indicate differences in treatment outcomes as a function of age, race, or gender.14.4 Premenstrual Dysphoric Disorder
The effectiveness of paroxetine extended-release tablets for the treatment of Premenstrual Dysphoric Disorder (PMDD) utilizing a continuous dosing regimen has been established in 2 placebo-controlled trials in female patients ages 18 to 46 (Studies 8 and 9 [N=672]). Patients in these trials met DSM-IV criteria for PMDD. Of 1,030 patients including Study 10, who were treated with daily doses of paroxetine extended-release tablets 12.5 or 25 mg/day, or placebo continuously throughout the menstrual cycle for a period of 3 menstrual cycles, the mean duration of the PMDD symptoms was approximately 11 ± 7 years. Patients on systemic hormonal contraceptives were excluded from these trials. Therefore, the efficacy of paroxetine extended-release tablets in combination with systemic (including oral) hormonal contraceptives for the continuous daily treatment of PMDD is unknown.
The VAS score is a patient-rated instrument that mirrors the diagnostic criteria of PMDD as identified in the DSM-IV, and includes assessments for mood, physical symptoms, and other symptoms associated with PMDD. In Studies 8 and 9, 12.5 mg/day and 25 mg/day of paroxetine extended-release tablets were statistically significantly more effective than placebo as measured by change from baseline to Month 3 on the luteal phase VAS score.
In an additional study employing luteal phase dosing (Study 11), patients (N = 366) were treated for the 2 weeks prior to the onset of menses with 12.5 or 25 mg/day of paroxetine extended-release tablets or placebo for a period of 3 months. In this trial, 12.5 mg/day and 25 mg/day of paroxetine extended-release tablets, as luteal phase dosing, was statistically significantly more effective than placebo as measured by change from baseline to luteal phase VAS score at Month 3.
There is insufficient information to determine the effect of race or age on outcome in Studies 8, 9, 10, and 11. -
16 HOW SUPPLIED/STORAGE AND HANDLING
Paroxetine extended-release tablets are supplied as follows:
Paroxetine extended-release tablets 12.5 mg are yellow, biconvex, enteric film-coated, extended- release, round tablets, debossed with “X1” on one side and plain on the other side. The tablets should be free from all physical defects.
NDC: 71209-094-01 Bottle of 30’s
NDC: 71209-094-10 Bottle of 500’s
Paroxetine extended-release tablets 25 mg are pink, biconvex, enteric film-coated, extended- release, round tablets debossed with "X2" on one side and plain on the other side. The tablets should be free from all physical defects.
NDC: 71209-095-01 Bottle of 30’s
NDC: 71209-095-10 Bottle of 500’s
Paroxetine extended-release tablets 37.5 mg are blue, biconvex, enteric film-coated, extended- release, round tablets debossed with "X3" on one side and plain on the other side. The tablets should be free from all physical defects.
NDC: 71209-096-01 Bottle of 30’s
NDC: 71209-096-10 Bottle of 500’s
Store at or below 20°C to 25°C (68°F to 77°F) excursions permitted between 15° to 30°C (59° to 86°F) [see USP Controlled Room Temperature].
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Suicidal Thoughts and Behaviors
Advise patients and caregivers to look for the emergence of suicidality, especially early during treatment and when the dosage is adjusted up or down, and instruct them to report such symptoms to the healthcare provider [see Boxed Warning and Warnings and Precautions (5.1)].Important Administration Instructions
Instruct patients to swallow paroxetine extended-release tablets whole and to not chew or crush the tablets [see Dosage and Administration (2.1)].Serotonin Syndrome
Caution patients about the risk of serotonin syndrome, particularly with the concomitant use of paroxetine extended-release tablets with other serotonergic drugs including triptans, tricyclic antidepressants, opioids, lithium, tryptophan, buspirone, amphetamines, St. John’s Wort, and with drugs that impair metabolism of serotonin (in particular, MAOIs, both those intended to treat psychiatric disorders and also others, such as linezolid). Instruct patients to contact their health care provider or report to the emergency room if they experience signs or symptoms of serotonin syndrome [see Warnings and Precautions (5.2), Drug Interactions (7.1)].
Concomitant Medications
Advise patients to inform their physician if they are taking, or plan to take, any prescription or over-the-counter drugs, since there is a potential for drug-drug interactions [see Warning and Precautions (5.3), Drug Interactions (7)].
Increased Risk of Bleeding
Inform patients about the concomitant use of paroxetine extended-release tablets with aspirin, NSAIDs, other antiplatelet drugs, warfarin, or other anticoagulants because the combined use has been associated with an increased risk of bleeding. Advise patients to inform their health care providers if they are taking or planning to take any prescription or over-the counter medications that increase the risk of bleeding [see Warnings and Precautions (5.5)].
Activation of Mania/Hypomania
Advise patients and their caregivers to observe for signs of activation of mania/hypomania and instruct them to report such symptoms to the healthcare provider [see Warnings and Precautions (5.6)].Sexual Dysfunction
Advise patients that use of paroxetine extended-release tablets may cause symptoms of sexual dysfunction in both male and female patients. Inform patients that they should discuss any changes in sexual function and potential management strategies with their healthcare provider [see Warnings and Precautions (5.13)].Discontinuation Syndrome
Advise patients not to abruptly discontinue paroxetine extended-release tablets and to discuss any tapering regimen with their healthcare provider. Inform patients that adverse reactions can occur when paroxetine extended-release tablets are discontinued [See Warnings and Precautions (5.7)].
Embryo-Fetal Toxicity
Advise women to notify their healthcare provider if they become pregnant or intend to become pregnant during treatment with Paroxetine extended-release tablets. Advise women of risks associated with first trimester use of Paroxetine extended-release tablets and that use later in pregnancy may lead to an increased risk for neonatal complications requiring prolonged hospitalization, respiratory support, tube feeding, and/or persistent pulmonary hypertension of the newborn (PPHN) [see Warnings and Precautions (5.4), Use in Specific Populations (8.1)]. Advise women that there is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to Paroxetine extended-release tablets during pregnancy [see Warnings and Precautions (5.4), Use in Specific Populations (8.1)].
Lactation
Advise breastfeeding women using Paroxetine extended-release tablets to monitor infants for agitation, irritability, poor feeding and poor weight gain and to seek medical care if they notice these signs [see Use in Specific Populations (8.2)].
Females and Males of Reproductive Potential
Advise men that Paroxetine extended-release tablets may affect sperm quality, which may impair fertility; it is unknown if this effect is reversible [see Use in Specific Populations (8.3)]
Allergic Reactions
Advise patients to notify their healthcare provider if they develop an allergic reaction such as rash, hives, swelling, or difficulty breathing [see Adverse Reactions (6.1, 6.2)].Manufactured by:
Cadila Pharmaceuticals Limited
1389, Trasad Road, Dholka-382225,
District - Ahmedabad,
Gujarat State, INDIA.
All registered trademarks in this document are the property of their respective owners.
Revised: March 2024
Medication Guide
Paroxetine Extended-Release Tablets
(pa rox' e teen)What is the most important information I should know about paroxetine extended-release tablets?
Paroxetine extended-release tablets can cause serious side effects, including:Increased risk of suicidal thoughts or actions. Antidepressant medicines may increase suicidal thoughts and actions in some children and young adults within the first few months of treatment or when the dose is changed. Paroxetine extended-release tablets are not for use in people younger than 18 years of age.
How can I watch for and try to prevent suicidal thoughts and actions?
o Depression or other serious mental illnesses are the most important causes of suicidal thoughts and actions.
o Pay close attention to any changes, especially sudden changes in mood, behavior, thoughts or feelings or if you develop suicidal thoughts or actions. This is very important when an antidepressant medicine is started or when the dose is changed.
o Call your healthcare provider right away to report new or sudden changes in mood, behavior, thoughts or feelings or if you develop suicidal thoughts or actions.
o Keep all follow-up visits with your healthcare provider as scheduled. Call your healthcare provider between visits as needed, especially if you have concerns about symptoms.
Call your healthcare provider or get emergency medical help right away if you have any of the following symptoms, especially if they are new, worse, or worry you:o attempts to commit suicide
o acting on dangerous impulses
o acting aggressive or violent o thoughts about suicide or dying
o new or worse depression
o new or worse anxiety or panic attacks
o feeling agitated, restless, angry, or irritable
o trouble sleeping
o an increase in activity and talking more than what is normal for you o other unusual changes in behavior or mood
What is paroxetine extended-release tablets?
Paroxetine extended-release tablets are a prescription medicine used in adults to treat:
A certain type of depression called Major Depressive Disorder (MDD)
Panic Disorder
Social Anxiety Disorder (SAD)
Premenstrual Dysphoric Disorder (PMDD)
Do not take paroxetine extended-release tablets if you:
take a monoamine oxidase inhibitor (MAOI)
have stopped taking an MAOI in the last 14 days
are being treated with the antibiotic linezolid or intravenous methylene blue
are taking thioridazine
are taking pimozide
are allergic to paroxetine or any of the ingredients in paroxetine extended-release tablets. See the end of this Medication Guide for a complete list of ingredients in paroxetine extended-release tablets.
Ask your healthcare provider or pharmacist if you are not sure if you take an MAOI or one of these medicines, including intravenous methylene blue.
Do not start taking an MAOI for at least 14 days after you stop treatment with paroxetine extended-release tablets.
Before taking paroxetine extended-release tablets, tell your healthcare provider about all your medical conditions, including if you:
have heart problems
have or had bleeding problems
have, or have a family history of bipolar disorder, mania or hypomaniahave or had seizures or convulsions
have glaucoma (high pressure in the eye)
have low sodium levels in your blood
have bone problems
have kidney or liver problems
are pregnant or plan to become pregnant. Paroxetine extended-release tablets may harm your unborn baby.o Taking Paroxetine extended-release tablets during your first trimester of pregnancy may cause your baby to be at an increased risk of having a heart problem (cardiac malformations) at birth.
o Taking Paroxetine extended-release tablets during your third trimester of pregnancy may cause your baby to have breathing, temperature, and feeding problems, low muscle tone (floppy baby syndrome), and irritability after birth and may cause your baby to be at an increased risk of a serious lung problem at birth. Talk to your healthcare provider about the risk to your unborn baby if you take Paroxetine extended-release tablets during pregnancy.
o Tell your healthcare provider right away if you become pregnant or think you are pregnant during treatment with paroxetine extended-release tablets.
o There is a pregnancy registry for females who are exposed to Paroxetine extended-release tablets during pregnancy. The purpose of the registry is to collect information about the health of females exposed to Paroxetine extended-release tablets and their baby. If you become pregnant during treatment with Paroxetine extended-release tablets talk to your healthcare provider about registering with the National Pregnancy Registry for Antidepressants at 1-866-961-2388 or visit online at https://womensmentalhealth.org/clinical-and-research-programs/pregnancyregistry/antidepressants/.
are breastfeeding or plan to breastfeed. Paroxetine passes into your breast milk. Talk to your healthcare provider about the best way to feed your baby during treatment with paroxetine extended-release tablets.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
Paroxetine extended-release tablets and some other medicines may affect each other causing possible serious side effects. Paroxetine extended-release tablets may affect the way other medicines work and other medicines may affect the way paroxetine extended-release tablets works.
Especially tell your healthcare provider if you take:
medicines used to treat migraine headaches called triptans
tricyclic antidepressants
lithium
tramadol, fentanyl, meperidine, methadone, or other opioids
tryptophan
buspirone
amphetamines
St. John’s Wort
medicines that can affect blood clotting such as aspirin, nonsteroidal anti-inflammatory drugs (NSAIDs), or warfarin
diuretics
tamoxifen
Ask your healthcare provider if you are not sure if you are taking any of these medicines. Your healthcare provider can tell you if it is safe to take paroxetine extended-release tablets with your other medicines.
Do not start or stop any other medicines during treatment with paroxetine extended-release tablets without talking to your healthcare provider first. Stopping paroxetine extended-release tablets suddenly may cause you to have serious side effects. See, “What are the possible side effects of paroxetine extended-release tablets?”
Know the medicines you take. Keep a list of them to show to your healthcare provider and pharmacist when you get a new medicine.
How should I take paroxetine extended-release tablets?
Take paroxetine extended-release tablets exactly as your healthcare provider tell you to. Your healthcare provider may need to change the dose of paroxetine extended-release tablets until it is the right dose for you.
Take paroxetine extended-release tablets 1 time each day in the morning.
Take Paroxetine extended-release tablets with or without food.
Swallow paroxetine extended-release tablets whole. Do not chew or crush paroxetine extended-release tablets.
If you take too much paroxetine extended-release tablets, call your poison control center at 1-800-222-1222 or got to the nearest hospital emergency room right away.
What are possible side effects of paroxetine extended-release tablets?
Paroxetine extended-release tablets can cause serious side effects, including:
See, “What is the most important information I should know about paroxetine extended release tablets?”
Serotonin syndrome. A potentially life-threatening problem called serotonin syndrome can happen when you take paroxetine extended-release tablets with certain other medicines. See, “Who should not take paroxetine extended-release tablets?” Call your healthcare provider or go to the nearest hospital emergency room right away if you have any of the following signs and symptoms of serotonin syndrome:
o agitation o sweating o seeing or hearing things that are not real (hallucinations) o flushing o confusion o high body temperature (hyperthermia) o coma o shaking (tremors), stiff muscles, or muscle twitching o fast heart beat o loss of coordination o changes in blood pressure o seizures o dizziness o nausea, vomiting, diarrhea
Medicine interactions. Taking paroxetine extended-release tablets with certain other medicines including thioridazine and pimozide may increase the risk of developing a serious heart problem
called QT prolongation.
Abnormal bleeding. Taking paroxetine extended-release tablets with aspirin, NSAIDs, or blood thinners may add to this risk. Tell your healthcare provider about any unusual bleeding or bruising.
Manic episodes. Manic episodes may happen in people with bipolar disorder who take paroxetine extended-release tablets. Symptoms may include:
o greatly increased energy o severe problems sleeping o racing thoughts o reckless behavior o unusually grand ideas o excessive happiness or irritability o talking more or faster than usual
Discontinuation syndrome. Suddenly stopping paroxetine extended-release tablets may cause you to have serious side effects. Your healthcare provider may want to decrease your dose slowly. Symptoms may include:
o Nausea o electric shock feeling (paresthesia) o tiredness o sweating o tremor o problems sleeping o changes in your mood o anxiety o ringing in your ears (tinnitus) o irritability and agitation o confusion o seizures o dizziness o headache
Seizures (convulsions).
Eye problems (angle-closure glaucoma). Paroxetine extended-release tablets may cause a type of eye problem called angle-closure glaucoma in people with certain other eye conditions. You may want to undergo an eye examination to see if you are at risk and receive preventative treatment if you are.
Low sodium levels in your blood (hyponatremia). Low sodium levels in your blood that may be serious and may cause death, can happen during treatment with paroxetine extended-release tablets. Elderly people and people who take certain medicines may be at a greater risk for developing low sodium levels in your blood. Signs and symptoms may include:
o headache
o difficulty concentrating
o memory changes
o confusion
o weakness and unsteadiness on your feet which can lead to falls
In more severe or more sudden cases, signs and symptoms include:
o seeing or hearing things that are not real (hallucinations)
o fainting
o seizures
o coma
o stopping breathing (respiratory arrest)
Bone fractures.Sexual problems (dysfunction). Taking selective serotonin reuptake inhibitors (SSRIs), including paroxetine extended-release tablets, may cause sexual problems.
Symptoms in males may include:
o Delayed ejaculation or inability to have an ejaculation
o Decreased sex drive
o Problems getting or keeping an erection
Symptoms in females may include:
o Decreased sex drive
o Delayed orgasm or inability to have an orgasm
Talk to your healthcare provider if you develop any changes in your sexual function or if you have any questions or concerns about sexual problems during treatment with paroxetine extended-release tablets. There may be treatments your healthcare provider can suggest.
The most common side effects paroxetine extended-release tablets include:
male and female sexual function problems dry mouth blurred vision problems sleeping weakness (asthenia) nausea constipation sleepiness decreased appetite sweating diarrhea tremor
These are not all the possible side effects of paroxetine extended-release tablets. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store paroxetine extended-release tablets?
Store paroxetine extended-release tablets at room temperature between 68°F to 77°F (20°C to 25°C).
Keep paroxetine extended-release tablets and all medicines out of the reach of children.
General information about the safe and effective use of paroxetine extended-release tablets.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not take paroxetine extended-release tablets for a condition for which it was not prescribed. Do not give paroxetine extended-release tablets to other people, even if they have the same symptoms that you have. It may harm them. You may ask your healthcare provider or pharmacist for information about paroxetine extended-release tablets that is written for healthcare professionals.
What are the ingredients in paroxetine extended-release tablets?
Active ingredient: Paroxetine hydrochloride hemihydrate, USP
Inactive ingredients: ferric oxide yellow, glyceryl behenate, hypromellose, lactose monohydrate, magnesium stearate, methacrylic acid – ethyl acrylate copolymer (1:1) type A, polyethylene glycols, polysorbate 80, polyvinylpyrrolidone, silicon dioxide, sodium lauryl sulfate, talc, titanium dioxide, triethyl citrate, film-coating material contains - FD&C Yellow #6/sunset yellow FCF aluminum lake, FD&C blue #2/Indigo carmine AL 3% to 5% and D&C Yellow #10 aluminum lake for 12.5 mg; D&C Red #30/Helendon pink aluminum lake for 25 mg and FD&C blue #2/Indigo carmine AL 3% to 5% and FD&C blue #2 Indigo carmine aluminum lake for 37.5 mg.
All registered trademarks in this document are the property of their respective owners.
This Medication Guide has been approved by the U.S. Food and Drug Administration.
Manufactured by:
Cadila Pharmaceuticals Limited
1389, Trasad Road, Dholka-382225,
District - Ahmedabad,
Gujarat State, INDIA.Revised: March 2024
-
PACKAGE LABEL.PRINCIPAL DISPLAY PANEL
Paroxetine Extended-Release Tablets - Container Label 30 Tablets
NDC 71209-094-01
Paroxetine Extended-Release Tablets
12.5 mg
Pharmacist: Dispense the accompanying Medication Guide to each patient.
RX only 30 Tablets
Paroxetine Extended-Release Tablets - Container Label 500 Tablets
NDC 71209- 094-10
Paroxetine Extended-Release Tablets
12.5 mg
Pharmacist: Dispense the accompanying Medication Guide to each patient.
RX only 500 Tablets

Paroxetine Extended-Release Tablets - Container Label 30 Tablets
NDC 71209- 095-01
Paroxetine Extended-Release Tablets
25 mg
Pharmacist: Dispense the accompanying Medication Guide to each patient.
RX only 30 Tablets

Paroxetine Extended-Release Tablets - Container Label 500 Tablets
NDC 71209- 095-10
Paroxetine Extended-Release Tablets
25 mg
Pharmacist: Dispense the accompanying Medication Guide to each patient.
RX only 500 Tablets

Paroxetine Extended-Release Tablets - Container Label 30 Tablets
NDC 71209- 096-01
Paroxetine Extended-Release Tablets
37.5 mg
Pharmacist: Dispense the accompanying Medication Guide to each patient.
RX only 30 Tablets
Paroxetine Extended-Release Tablets - Container Label 500 Tablets
NDC 71209-096-10
Paroxetine Extended-Release Tablets
37.5 mg
Pharmacist: Dispense the accompanying Medication Guide to each patient.
RX only 500 Tablets

-
INGREDIENTS AND APPEARANCE
PAROXETINE
paroxetine tablet, film coated, extended releaseProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 71209-094 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength PAROXETINE HYDROCHLORIDE HEMIHYDRATE (UNII: X2ELS050D8) (PAROXETINE - UNII:41VRH5220H) PAROXETINE 12.5 mg Inactive Ingredients Ingredient Name Strength FERRIC OXIDE YELLOW (UNII: EX438O2MRT) GLYCERYL DIBEHENATE (UNII: R8WTH25YS2) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) POLYSORBATE 80 (UNII: 6OZP39ZG8H) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) SODIUM LAURYL SULFATE (UNII: 368GB5141J) TALC (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) TRIETHYL CITRATE (UNII: 8Z96QXD6UM) METHACRYLIC ACID - ETHYL ACRYLATE COPOLYMER (1:1) TYPE A (UNII: NX76LV5T8J) POVIDONE K30 (UNII: U725QWY32X) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) HYPROMELLOSES (UNII: 3NXW29V3WO) Product Characteristics Color YELLOW Score no score Shape ROUND Size 7mm Flavor Imprint Code X1 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 71209-094-01 30 in 1 BOTTLE; Type 0: Not a Combination Product 02/14/2020 2 NDC: 71209-094-10 500 in 1 BOTTLE; Type 0: Not a Combination Product 02/14/2020 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA212645 02/14/2020 PAROXETINE
paroxetine tablet, film coated, extended releaseProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 71209-095 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength PAROXETINE HYDROCHLORIDE HEMIHYDRATE (UNII: X2ELS050D8) (PAROXETINE - UNII:41VRH5220H) PAROXETINE 25 mg Inactive Ingredients Ingredient Name Strength FERRIC OXIDE YELLOW (UNII: EX438O2MRT) GLYCERYL DIBEHENATE (UNII: R8WTH25YS2) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) POLYSORBATE 80 (UNII: 6OZP39ZG8H) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) SODIUM LAURYL SULFATE (UNII: 368GB5141J) TALC (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) TRIETHYL CITRATE (UNII: 8Z96QXD6UM) METHACRYLIC ACID - ETHYL ACRYLATE COPOLYMER (1:1) TYPE A (UNII: NX76LV5T8J) POVIDONE K30 (UNII: U725QWY32X) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) HYPROMELLOSES (UNII: 3NXW29V3WO) Product Characteristics Color PINK Score no score Shape ROUND Size 7mm Flavor Imprint Code X2 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 71209-095-01 30 in 1 BOTTLE; Type 0: Not a Combination Product 02/14/2020 2 NDC: 71209-095-10 500 in 1 BOTTLE; Type 0: Not a Combination Product 02/14/2020 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA212645 02/14/2020 PAROXETINE
paroxetine tablet, film coated, extended releaseProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 71209-096 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength PAROXETINE HYDROCHLORIDE HEMIHYDRATE (UNII: X2ELS050D8) (PAROXETINE - UNII:41VRH5220H) PAROXETINE 37.5 mg Inactive Ingredients Ingredient Name Strength FERRIC OXIDE YELLOW (UNII: EX438O2MRT) GLYCERYL DIBEHENATE (UNII: R8WTH25YS2) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) POLYSORBATE 80 (UNII: 6OZP39ZG8H) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) SODIUM LAURYL SULFATE (UNII: 368GB5141J) TALC (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) TRIETHYL CITRATE (UNII: 8Z96QXD6UM) METHACRYLIC ACID - ETHYL ACRYLATE COPOLYMER (1:1) TYPE A (UNII: NX76LV5T8J) POVIDONE K30 (UNII: U725QWY32X) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) HYPROMELLOSES (UNII: 3NXW29V3WO) Product Characteristics Color BLUE Score no score Shape ROUND Size 8mm Flavor Imprint Code X3 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 71209-096-01 30 in 1 BOTTLE; Type 0: Not a Combination Product 02/14/2020 2 NDC: 71209-096-10 500 in 1 BOTTLE; Type 0: Not a Combination Product 02/14/2020 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA212645 02/14/2020 Labeler - Cadila Pharmaceuticals Limited (862257719) Establishment Name Address ID/FEI Business Operations Cadila Pharmaceuticals Limited 918451696 ANALYSIS(71209-094, 71209-095, 71209-096) , LABEL(71209-094, 71209-095, 71209-096) , MANUFACTURE(71209-094, 71209-095, 71209-096) , PACK(71209-094, 71209-095, 71209-096)
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.