KIMYRSA- oritavancin diphosphate injection, powder, lyophilized, for solution
Kimyrsa by
Drug Labeling and Warnings
Kimyrsa by is a Prescription medication manufactured, distributed, or labeled by Melinta Therapeutics, LLC. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use KIMYRSA safely and effectively. See full prescribing information for KIMYRSA.
KIMYRSA® (oritavancin) for injection, for intravenous use
Initial U.S. Approval: 2014INDICATIONS AND USAGE
KIMYRSA is a lipoglycopeptide antibacterial drug indicated for the treatment of adult patients with acute bacterial skin and skin structure infections caused or suspected to be caused by susceptible isolates of designated Gram-positive microorganisms. (1.1)
To reduce the development of drug-resistant bacteria and maintain the effectiveness of KIMYRSA and other antibacterial drugs, KIMYRSA should be used only to treat or prevent infections that are proven or strongly suspected to be caused by bacteria. (1.2)
DOSAGE AND ADMINISTRATION
- There are two oritavancin products (KIMYRSA and ORBACTIV®, another oritavancin product) that have differences in dose strength, duration of infusion and preparation instructions, including reconstitution and dilution instructions and compatible diluents (2.1, 2.2, 2.3, 2.4)
- Administer 1,200 mg of KIMYRSA as a single dose by intravenous infusion over 1 hour. (2.1, 5.3)
- Carefully follow the recommended dosage and dose preparation instructions for KIMYRSA in the full prescribing information. (2.1, 2.2, 2.3)
DOSAGE FORMS AND STRENGTHS
For injection: 1,200 mg of lyophilized powder in a single-dose vial for reconstitution. (3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- Coagulation test interference: Oritavancin has been shown to artificially prolong aPTT for up to 120 hours, and may prolong PT and INR for up to 12 hours and ACT for up to 24 hours. For patients who require aPTT monitoring within 120 hours of KIMYRSA dosing, consider a non-phospholipid dependent coagulation test such as a Factor Xa (chromogenic) assay or an alternative anticoagulant not requiring aPTT. (5.1, 7.2)
- Serious hypersensitivity reactions, including anaphylaxis, have been reported with the use of oritavancin products, including KIMYRSA. Discontinue infusion if signs of acute hypersensitivity occur. Carefully monitor patients with known hypersensitivity to glycopeptides. (5.2)
- Infusion Related Reactions: Infusion related reactions have been reported with the glycopeptide class of antimicrobial agents, including oritavancin products (e.g. KIMYRSA). Stopping or slowing the infusion may result in cessation of these reactions. (5.3)
- Clostridioides difficile-Associated Diarrhea: Evaluate patients if diarrhea occurs. (5.4)
- Concomitant warfarin use: Oritavancin has been shown to artificially prolong PT/INR for up to 12 hours (5.1). Patients should be monitored for bleeding if concomitantly receiving KIMYRSA and warfarin. (5.5)
- Osteomyelitis: Institute appropriate alternate antibacterial therapy in patients with confirmed or suspected osteomyelitis. (5.6)
ADVERSE REACTIONS
The most common adverse reactions (≥3%) in patients treated with oritavancin products were headache, nausea, vomiting, limb and subcutaneous abscesses, and diarrhea. The adverse reactions occurring in ≥2 patients receiving KIMYRSA were hypersensitivity, pruritus, chills and pyrexia. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Melinta Therapeutics at 1-844-633-6568 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 4/2025
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Acute Bacterial Skin and Skin Structure Infections
1.2 Usage
2 DOSAGE AND ADMINISTRATION
2.1 Dosage and Administration Overview
2.2 Recommended Dosage
2.3 Preparation of KIMYRSA for Intravenous Infusion
2.4 Compatibilities
2.5 Incompatibilities
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
4.1 Intravenous Unfractionated Heparin Sodium
4.2 Hypersensitivity
5 WARNINGS AND PRECAUTIONS
5.1 Coagulation Test Interference
5.2 Hypersensitivity
5.3 Infusion Related Reactions
5.4 Clostridioides difficile-Associated Diarrhea
5.5 Potential Risk of Bleeding with Concomitant Use of Warfarin
5.6 Osteomyelitis
5.7 Development of Drug Resistant Bacteria
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 Effect of KIMYRSA on CYP Substrates
7.2 Drug-Laboratory Test Interactions
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.4 Microbiology
12.6 Immunogenicity
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Acute Bacterial Skin and Skin Structure Infections (ABSSSI)
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied/Storage
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
1.1 Acute Bacterial Skin and Skin Structure Infections
KIMYRSA® is indicated for the treatment of adult patients with acute bacterial skin and skin structure infections (ABSSSI) caused by susceptible isolates of the following Gram-positive microorganisms:
Staphylococcus aureus (including methicillin-susceptible and methicillin-resistant isolates), Streptococcus pyogenes, Streptococcus agalactiae, Streptococcus dysgalactiae, Streptococcus anginosus group (includes S. anginosus, S. intermedius, and S. constellatus), and Enterococcus faecalis (vancomycin-susceptible isolates only).
1.2 Usage
To reduce the development of drug-resistant bacteria and maintain the effectiveness of KIMYRSA and other antibacterial drugs, KIMYRSA should be used only to treat or prevent infections that are proven or strongly suspected to be caused by susceptible bacteria. When culture and susceptibility information are available, they should be considered in selecting or modifying antibacterial therapy. In the absence of such data, local epidemiology and susceptibility patterns may contribute to the empiric selection of therapy.
-
2 DOSAGE AND ADMINISTRATION
2.1 Dosage and Administration Overview
There are two oritavancin products (KIMYRSA and ORBACTIV®, another oritavancin product) that:
- Are supplied in different dose strengths of oritavancin [see Dosage Forms and Strengths (3)].
- Have different recommended durations of infusion [see Dosage and Administration (2.2)].
- Have different preparation instructions, including differences in reconstitution, dilution, and compatible diluents [see Dosage and Administration (2.3, 2.4)].
Carefully follow the recommended dosage and dose preparation instructions for KIMYRSA in this prescribing information (PI) [see Dosage and Administration (2.1, 2.2, 2.3, 2.4)]. Refer to the ORBACTIV prescribing information for relevant information of the other oritavancin product.
2.2 Recommended Dosage
The recommended dosage of KIMYRSA is 1,200 mg administered as a single dose by intravenous infusion over 1 hour in patients 18 years and older [see Warnings and Precautions (5.3)].
2.3 Preparation of KIMYRSA for Intravenous Infusion
There are two oritavancin products (KIMYRSA and ORBACTIV, another oritavancin product) that have differences in dose strengths, duration of infusion, reconstitution and dilution instructions, and compatible diluents. Carefully follow the reconstitution, and dilution instructions with the appropriate compatible diluent for KIMYRSA specified in this prescribing information. Refer to the ORBACTIV prescribing information for relevant information of the other oritavancin product.
KIMYRSA is intended for intravenous infusion, only after reconstitution and dilution.
One KIMYRSA 1,200 mg single-dose vial needs to be reconstituted and diluted to prepare a single 1,200 mg intravenous dose.
Reconstitution: Aseptic technique should be used to reconstitute one KIMYRSA 1,200 mg vial.
- Add 40 mL of sterile water for injection (SWFI) to reconstitute the vial to provide a 30 mg/mL solution.
- Gently swirl the contents to avoid foaming and ensure that all KIMYRSA powder is completely dissolved to form a reconstituted solution.
- Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. The reconstituted vial should appear to be a clear, colorless to pink solution, free of visible particles.
Dilution: Use 0.9% sodium chloride injection or 5% dextrose in sterile water (D5W) for dilution to prepare the final intravenous solution for infusion. Since no preservative or bacteriostatic agent is present in KIMYRSA, aseptic technique must be used in preparing the final intravenous solution as follows:
- Withdraw and discard 40 mL from a 250 mL intravenous bag of 0.9% sodium chloride injection or D5W.
- Withdraw 40 mL of the reconstituted vial of KIMYRSA and add to the intravenous bag of 0.9% sodium chloride injection or D5W to bring the bag volume to 250 mL. This yields a concentration of 4.8 mg/mL.
Discard any unused portion of the reconstituted solution remaining in the vial.
Storage and Use of Intravenous Solution: Diluted intravenous solution in an infusion bag should be used within 4 hours when stored at room temperature, or used within 12 hours when refrigerated at 2 to 8°C (36 to 46°F). The combined storage time (reconstituted solution in the vial and diluted solution in the bag) and 1 hour infusion time should not exceed 4 hours at room temperature or 12 hours if refrigerated.
2.4 Compatibilities
KIMYRSA solution for administration by 1-hour infusion is compatible with:
- 0.9% sodium chloride injection
- 5% dextrose in sterile water (D5W)
2.5 Incompatibilities
Drugs formulated at a basic or neutral pH may be incompatible with KIMYRSA. KIMYRSA should not be administered simultaneously with commonly used intravenous drugs through a common intravenous port. If the same intravenous line is used for sequential infusion of additional medications, the line should be flushed before and after infusion of KIMYRSA with 0.9% sodium chloride injection or D5W.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
4.1 Intravenous Unfractionated Heparin Sodium
Use of intravenous unfractionated heparin sodium is contraindicated for 120 hours (5 days) after KIMYRSA administration because the activated partial thromboplastin time (aPTT) test results may remain falsely elevated for up to 120 hours (5 days) after KIMYRSA administration [see Warnings and Precautions (5.1) and Drug Interactions (7.2)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Coagulation Test Interference
Oritavancin has been shown to artificially prolong aPTT for up to 120 hours, PT and INR for up to 12 hours, and activated clotting time (ACT) for up to 24 hours following administration of a single 1,200 mg dose by binding to and preventing action of the phospholipid reagents commonly used in laboratory coagulation tests. Oritavancin has also been shown to elevate D-dimer concentrations up to 72 hours after oritavancin administration.
For patients who require aPTT monitoring within 120 hours of KIMYRSA dosing, a non-phospholipid dependent coagulation test such as a Factor Xa (chromogenic) assay or an alternative anticoagulant not requiring aPTT monitoring may be considered [see Contraindications (4.1) and Drug Interactions (7.2)].
Oritavancin has no effect on the coagulation system in vivo.
5.2 Hypersensitivity
Serious hypersensitivity reactions, including anaphylaxis, have been reported with the use of oritavancin products, including KIMYRSA. If an acute hypersensitivity reaction occurs during KIMYRSA infusion, discontinue KIMYRSA immediately and institute appropriate supportive care. Before using KIMYRSA, inquire carefully about previous hypersensitivity reactions to glycopeptides. Due to the possibility of cross-sensitivity, carefully monitor for signs of hypersensitivity during KIMYRSA infusion in patients with a history of glycopeptide allergy. In the Phase 3 ABSSSI clinical trials, the median onset of hypersensitivity reactions in oritavancin-treated patients was 1.2 days and the median duration of these reactions was 2.4 days [see Adverse Reactions (6.1)].
5.3 Infusion Related Reactions
Infusion related reactions have been reported with the glycopeptide class of antimicrobial agents, including oritavancin products (e.g. KIMYRSA), including flushing of the upper body, urticaria, pruritus and/or rash [see Adverse Reactions (6.1)]. Infusion reactions characterized by chest pain, back pain, chills and tremor have been observed with the use of oritavancin, including after the administration of more than one dose of oritavancin during a single course of therapy.
Stopping or slowing the infusion may result in cessation of these reactions. The safety and effectiveness of more than one dose of KIMYRSA during a single course of therapy have not been established [see Dosage and Administration (2.2)].
5.4 Clostridioides difficile-Associated Diarrhea
Clostridioides difficile-associated diarrhea (CDAD) has been reported for nearly all systemic antibacterial drugs, including oritavancin products (e.g. KIMYRSA), and may range in severity from mild diarrhea to fatal colitis. Treatment with antibacterial agents alters the normal flora of the colon and may permit overgrowth of C. difficile.
C. difficile produces toxins A and B which contribute to the development of CDAD. Hypertoxin-producing strains of C. difficile cause increased morbidity and mortality, as these infections can be refractory to antibacterial therapy and may require colectomy. CDAD must be considered in all patients who present with diarrhea following antibacterial use. Careful medical history is necessary because CDAD has been reported to occur more than 2 months after the administration of antibacterial agents.
If CDAD is suspected or confirmed, antibacterial use not directed against C. difficile may need to be discontinued. Appropriate fluid and electrolyte management, protein supplementation, antibacterial treatment of C. difficile, and surgical evaluation should be instituted as clinically indicated.
5.5 Potential Risk of Bleeding with Concomitant Use of Warfarin
Oritavancin has been shown to artificially prolong prothrombin time (PT) and international normalized ratio (INR) for up to 12 hours, making the monitoring of the anticoagulation effect of warfarin unreliable up to 12 hours after an oritavancin dose [see Warnings and Precautions (5.1)].
Patients should be monitored for bleeding if concomitantly receiving KIMYRSA and warfarin [see Drug Interactions (7.1)].
5.6 Osteomyelitis
In Phase 3 ABSSSI clinical trials, more cases of osteomyelitis were reported in the oritavancin treated arm than in the vancomycin-treated arm. Monitor patients treated with KIMYRSA for signs and symptoms of osteomyelitis. If osteomyelitis is suspected or diagnosed, institute appropriate alternate antibacterial therapy [see Adverse Reactions (6.1)].
5.7 Development of Drug Resistant Bacteria
Prescribing KIMYRSA in the absence of a proven or strongly suspected bacterial infection or a prophylactic indication is unlikely to provide benefit to the patient and increases the risk of the development of drug-resistant bacteria [see Patient Counseling Information (17)].
-
6 ADVERSE REACTIONS
The following adverse reactions are also discussed in the Warnings and Precautions section of labeling:
Hypersensitivity Reactions [see Warnings and Precautions (5.2)]
Infusion Related Reactions [see Warnings and Precautions (5.3)]
Clostridioides difficile-Associated Diarrhea [see Warnings and Precautions (5.4)]
Osteomyelitis [see Warnings and Precautions (5.6)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of oritavancin products cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of KIMYRSA has been established from adequate and well-controlled trials of another oritavancin product, ORBACTIV (hereinafter referred to as oritavancin), in patients with ABSSSI, and a study of KIMYRSA in patients with ABSSSI.
Oritavancin has been evaluated in two, double-blind, controlled ABSSSI clinical trials, which included 976 adult patients treated with a single 1,200 mg intravenous dose of oritavancin and 983 patients treated with intravenous vancomycin for 7 to 10 days. The median age of patients treated with oritavancin was 45.6 years, ranging between 18 and 89 years of age with 8.8% ≥65 years of age. Patients treated with oritavancin were predominantly male (65.4%), 64.4% were Caucasian, 5.8% were African American, and 28.1% were Asian. Safety was evaluated for up to 60 days after dosing.
In the pooled ABSSSI clinical trials, serious adverse reactions were reported in 57/976 (5.8%) patients treated with oritavancin and 58/983 (5.9%) treated with vancomycin. The most commonly reported serious adverse reaction was cellulitis in both treatment groups: 11/976 (1.1%) in oritavancin and 12/983 (1.2%) in the vancomycin arms, respectively.
The most commonly reported adverse reactions (≥3%) in patients receiving a single 1,200 mg dose of oritavancin in the pooled ABSSSI clinical trials were: headache, nausea, vomiting, limb and subcutaneous abscesses, and diarrhea.
In the pooled ABSSSI clinical trials, oritavancin was discontinued due to adverse reactions in 36/976 (3.7%) of patients; the most common reported reactions leading to discontinuation were cellulitis (4/976, 0.4%) and osteomyelitis (3/976, 0.3%).
Table 1 provides selected adverse reactions occurring in ≥1.5% of patients receiving oritavancin in the pooled ABSSSI clinical trials. There were 540 (55.3%) patients in the oritavancin arm and 559 (56.9%) patients in the vancomycin arm, who reported ≥1 adverse reaction.
Table 1: Incidence of Selected Adverse Reactions Occurring in ≥1.5% of Patients Receiving Oritavancin in the Pooled ABSSSI Clinical Trials Adverse Reactions Oritavancin
N=976 (%)Vancomycin
N=983 (%)Gastrointestinal disorders Diarrhea 36 (3.7) 32 (3.4) Nausea 97 (9.9) 103 (10.5) Vomiting 45 (4.6) 46 (4.7) Nervous system disorders Dizziness 26 (2.7) 26 (2.6) Headache 69 (7.1) 66 (6.7) General disorders and administration Infusion site phlebitis 24 (2.5) 15 (1.5) Infusion site reaction 19 (1.9) 34 (3.5) Infections and infestations Abscess (limb and subcutaneous) 37 (3.8) 23 (2.3) Investigations Alanine aminotransferase increased 27 (2.8) 15 (1.5) Aspartate aminotransferase increased 18 (1.8) 15 (1.5) Cardiac disorders Tachycardia 24 (2.5) 11 (1.1) The following selected adverse reactions were reported in oritavancin-treated patients at a rate of less than 1.5%:
Blood and lymphatic system disorders: anemia, eosinophilia
General disorders and administration site conditions: infusion site erythema, extravasation, induration, pruritus, rash, edema peripheral
Immune system disorders: hypersensitivity
Infections and infestations: osteomyelitis
Investigations: total bilirubin increased, hyperuricemia
Metabolism and nutrition disorders: hypoglycemia
Musculoskeletal and connective tissue disorders: tenosynovitis, myalgia
Respiratory, thoracic and mediastinal disorders: bronchospasm, wheezing
Skin and subcutaneous tissue disorders: urticaria, angioedema, erythema multiforme, pruritus, leucocytoclastic vasculitis, rash.
KIMYRSA has been evaluated in a randomized, open-label, multi-center ABSSSI study which included 50 adult patients treated with a single 1,200 mg intravenous dose of KIMYRSA administered by intravenous infusion over 1 hour, and 52 patients treated with a single 1,200 mg intravenous dose of oritavancin administered by intravenous infusion over 3 hours.
Selected adverse reactions occurring in ≥2 patients receiving either KIMYRSA or oritavancin in the open-label, multi-center ABSSSI study were diarrhea, nausea, vomiting, hypersensitivity, pruritus, chills, headache and pyrexia.
-
7 DRUG INTERACTIONS
7.1 Effect of KIMYRSA on CYP Substrates
A screening drug-drug interaction study indicated that oritavancin is a nonspecific, weak inhibitor (CYP2C9 and CYP2C19) or inducer (CYP3A4 and CYP2D6) of several CYP isoforms [see Clinical Pharmacology (12.3)]. A drug-drug interaction study that assessed the interaction potential of a single 1,200 mg dose of oritavancin on the pharmacokinetics of S-warfarin (CYP2C9 probe substrate) showed no effect of oritavancin on S-warfarin Cmax or AUC.
Avoid administering KIMYRSA concomitantly with drugs that are predominantly metabolized by one of the affected CYP450 enzymes, as co-administration may increase or decrease concentrations of those drugs. Patients should be closely monitored for signs of toxicity or lack of efficacy if they have been given KIMYRSA while on a potentially affected compound (e.g., patients should be monitored for bleeding if concomitantly receiving KIMYRSA and warfarin).
7.2 Drug-Laboratory Test Interactions
Prolongation of Certain Laboratory Coagulation Tests
KIMYRSA may artificially prolong certain laboratory coagulation tests (see Table 2) by binding to and preventing the action of the phospholipid reagents which activate coagulation in commonly used laboratory coagulation tests [see Contraindications (4.1) and Warnings and Precautions (5.1, 5.5)]. For patients who require monitoring of anticoagulation effect within the indicated time after KIMYRSA dosing, a non-phospholipid dependent coagulation test such as a Factor Xa (chromogenic) assay or an alternative anticoagulant not requiring aPTT monitoring may be considered.
Oritavancin does not interfere with coagulation in vivo. In addition, oritavancin does not affect tests that are used for diagnosis of Heparin Induced Thrombocytopenia (HIT).
Table 2: Coagulation Tests Affected and Unaffected by Oritavancin Elevated by Oritavancin Unaffected by Oritavancin Prothrombin time (PT) up to 12 hours Chromogenic Factor Xa Assay International normalized ratio (INR) up to 12 hours Thrombin Time (TT) Activated partial thromboplastin time (aPTT) up to 120 hours Activated clotting time (ACT) up to 24 hours Silica clot time (SCT) up to 18 hours Dilute Russell's viper venom time (DRVVT) up to 72 hours D-dimer up to 72 hours Positive Indirect and Direct Antiglobulin Tests (IAT/DAT)
Positive IAT/DAT were noted with administration of oritavancin products, including KIMYRSA, in studies with healthy volunteers and patients with ABSSSI. Positive IAT may interfere with cross-matching before blood transfusion [see Clinical Pharmacology (12.6)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no available data on KIMYRSA use in pregnant women to evaluate for a drug- associated risk of major birth defects, miscarriage or adverse maternal or fetal outcomes. In animal reproduction studies, no effects on embryo-fetal development or survival were observed in pregnant rats or rabbits treated at the highest doses throughout organogenesis with intravenous oritavancin, at doses equivalent to 25% of the single clinical dose of 1,200 mg (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
Reproduction studies performed in rats and rabbits have revealed no evidence of harm to the fetus due to oritavancin at the highest doses administered throughout organogenesis, 30 mg/kg/day (gestation days 6-17) and 15 mg/kg/day (gestation days 7-19), respectively. Those doses would be equivalent to a human dose of 300 mg, or 25% of the single clinical dose of 1,200 mg. Higher doses were not evaluated in nonclinical developmental and reproductive toxicology studies.
8.2 Lactation
Risk Summary
There are no data on the presence of oritavancin in human milk, the effects on the breastfed- child, or the effects on milk production. Oritavancin is present in the breast milk of rats (see Data). When a drug is present in animal milk, it is likely that the drug will be present in human milk.
The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for KIMYRSA and any potential adverse effects on the breast-fed child from KIMYRSA or from the underlying maternal condition.
8.4 Pediatric Use
Safety and effectiveness of KIMYRSA in pediatric patients (younger than 18 years of age) have not been established.
8.5 Geriatric Use
The pooled Phase 3 ABSSSI clinical trials of oritavancin did not include sufficient numbers of subjects aged 65 and older to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
8.6 Renal Impairment
No dosage adjustment of KIMYRSA is needed in patients with mild or moderate renal impairment [see Clinical Pharmacology (12.3)]. The pharmacokinetics of KIMYRSA in severe renal impairment have not been evaluated. Oritavancin is not removed from blood by hemodialysis.
8.7 Hepatic Impairment
No dosage adjustment of KIMYRSA is needed in patients with mild or moderate hepatic impairment. The pharmacokinetics of KIMYRSA in patients with severe hepatic insufficiency has not been studied [see Dosage and Administration (2.2), Clinical Pharmacology (12.3)].
- 10 OVERDOSAGE
-
11 DESCRIPTION
KIMYRSA (oritavancin) for injection contains oritavancin diphosphate, a semisynthetic lipoglycopeptide antibacterial drug for intravenous infusion.
The chemical name for oritavancin is [4"R]-22-O-(3-amino-2,3,6-trideoxy-3-C-methyl-α-L-arabino-hexopyranosyl)-N3''-[(4'-chloro[1,1'-biphenyl]-4-yl)methyl] vancomycin phosphate [1:2] [salt]. The empirical formula of oritavancin diphosphate is C86H97N10O26Cl32H3PO4 and the molecular weight is 1989.09. The chemical structure is represented below:
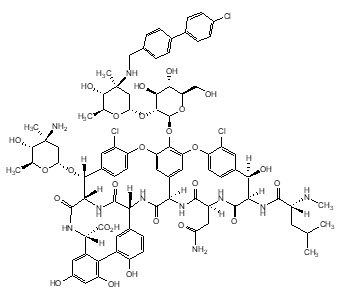
∙2H3PO4
KIMYRSA for injection is supplied as a sterile white to off-white or pink lyophilized powder in a single-dose clear glass vial that contains 1,200 mg of oritavancin (equivalent to 1331.16 mg oritavancin diphosphate) and the following inactive ingredients: hydroxypropyl-β-cyclodextrin (HPβCD) (2400 mg), mannitol (800 mg) and phosphoric acid or sodium hydroxide (to adjust pH 4.0 to 6.0).
The vial is reconstituted with sterile water for injection and further diluted with 0.9% sodium chloride injection or 5% dextrose in sterile water (D5W) for intravenous infusion. Both the reconstituted solution and the diluted solution for infusion should be a clear, colorless to pink solution, free of visible particles [see Dosage and Administration (2.3)].
-
12 CLINICAL PHARMACOLOGY
12.2 Pharmacodynamics
The antimicrobial activity of oritavancin appears to correlate with the ratio of area under the concentration-time curve to minimal inhibitory concentration (AUC/MIC) based on animal models of infection. The AUC from time zero to 72 hours correlates with antimicrobial activity in both preclinical and clinical studies.
Exposure-response analyses from both preclinical and clinical studies support the treatment of clinically relevant gram-positive microorganisms (e.g. S. aureus and S. pyogenes) causative of ABSSSI with a single 1,200 mg dose of oritavancin.
12.3 Pharmacokinetics
The mean (±SD) pharmacokinetic parameters of oritavancin products (KIMYRSA and oritavancin) in patients with ABSSSI are presented in Table 3.
Table 3: Mean (±SD) Pharmacokinetic Parameters following a single 1,200 mg dose of KIMYRSA by intravenous infusion over 1 hour (N= 50) and Oritavancin by intravenous infusion over 3 hours (N=50) in Patients with ABSSSI Pharmacokinetic Parameter KIMYRSA (1 hour)
Mean (± SD)Oritavancin (3 hour)
Mean (± SD)Cmax, Maximum plasma concentration; AUC0-72, Area under the plasma concentration-time curve from time zero to 72 hours; SD, Standard deviation. Cmax (µg/mL) 148 (±43.0) 112 (±34.5) AUC0-72 (h∙µg /mL) 1460 (±511) 1470 (±582) Oritavancin exhibits linear pharmacokinetics at a dose up to 1,200 mg. The mean, population-predicted oritavancin concentration-time profile displays a multi-exponential decline with a long terminal plasma half-life.
Distribution
Oritavancin is approximately 85% bound to human plasma proteins.
Based on population pharmacokinetic (PK) analysis, the population mean total volume of distribution is estimated to be approximately 87.6 L, indicating that oritavancin is extensively distributed into the tissues.
Exposures of oritavancin in skin blister fluid were approximately 20% of those in plasma (AUC0-24) after single 800 mg dose in healthy subjects.
Metabolism/Excretion
Non-clinical studies, including in vitro human liver microsome studies, indicated that oritavancin is not metabolized. No mass balance study has been conducted in humans. In humans, oritavancin is slowly excreted unchanged in feces and urine, with less than 1% and 5% of the dose recovered in feces and urine, respectively, after 2 weeks of collection.
Oritavancin has a terminal half-life of approximately 245 hours and a clearance of 0.445 L/h, based on population PK analyses.
Specific Populations
No dosage adjustments of KIMYRSA are required for patients with mild-to-moderate renal or mild-to-moderate hepatic impairment or other subpopulations, including age, gender, race, and weight.
Renal Impairment
The PK of oritavancin was examined in the Phase 3 ABSSSI trials in patients with normal renal function, CrCL ≥80 mL/min (n=238), mild renal impairment, CrCL 50-79 mL/min (n=48), and moderate renal impairment, CrCL 30-49 mL/min (n=11). Population PK analysis indicated that mild-to-moderate renal impairment had no clinically relevant effect on the exposure of oritavancin. No dedicated studies in dialysis patients have been conducted.
The solubilizer HPβCD is excreted in urine. Clearance of HPβCD may be reduced in patients with renal impairment. The clinical significance of this finding is unknown.
Dosage adjustment of KIMYRSA is not needed in patients with mild or moderate renal impairment. The PK of oritavancin in patients with severe renal impairment have not been evaluated.
Hepatic Impairment
The PK of oritavancin was evaluated in study of subjects with moderate hepatic impairment (Child-Pugh Class B) (n=20) and compared with healthy subjects (n=20) matched for gender, age, and weight. There were no relevant changes in PK of oritavancin in subjects with moderate hepatic impairment.
Dosage adjustment of KIMYRSA is not needed in patients with mild or moderate hepatic impairment. The PK of oritavancin in patients with severe hepatic insufficiency has not been studied.
Drug Interactions
In vitro studies with human liver microsomes showed that oritavancin inhibited the activities of cytochrome P450 (CYP) enzymes 1A2, 2B6, 2D6, 2C9, 2C19, and 3A4. The observed inhibition of multiple CYP isoforms by oritavancin in vitro is likely to be reversible and noncompetitive. In vitro studies indicate that oritavancin is neither a substrate nor an inhibitor of the efflux transporter P-glycoprotein (P-gp).
Drugs that Inhibit or Induce CYP450 Enzymes
A screening drug-drug interaction study was conducted in healthy volunteers (n=16) evaluating the concomitant administration of a single 1,200 mg dose of oritavancin with probe substrates for several CYP450 enzymes. The results showed that oritavancin is a weak inducer of CYP3A4 (a decrease of 18% in the mean AUC of midazolam) and CYP2D6 (decrease of 31% in the ratio of dextromethorphan to dextrorphan concentrations in the urine after administration of dextromethorphan). Oritavancin was also a weak inhibitor of CYP2C19 (increase of 15% in the ratio of omeprazole to 5-OH-omeprazole concentrations in the plasma after administration of omeprazole) and CYP2C9 (with an increase of 31% in the mean AUC of warfarin) [see Warnings and Precautions (5.5), and Drug Interactions (7.1)].
In the screening drug-drug interaction study, co-administration of oritavancin resulted in an increase of 18% in the ratio of 1-methylxanthine + 1 methylurate + 5-acetylamino-6-formylamino-3-methyluracil (1X + 1U + AFMU) to 1,7-dimethylurate (17U) concentrations in the urine after administration of caffeine (CYP1A2 probe substrate), and an increase of 16% in the ratio of AFMU to (1X +1U) concentrations in the urine after administration of caffeine (N-Acetyltransferase- 2 probe substrate). Co-administration of oritavancin did not change the mean systemic exposure of caffeine metabolite (Xanthine oxidase probe substrate).
A study to assess the drug-drug interaction potential of a single 1,200 mg dose of oritavancin on the pharmacokinetics of S-warfarin following a single dose was conducted in 36 healthy subjects. S-warfarin PK was evaluated following a single dose of warfarin 25 mg given alone, or administered at the start, 24, or 72 hours after a single 1,200 mg oritavancin dose. The results showed no effect of oritavancin on S-warfarin Cmax or AUC.
12.4 Microbiology
KIMYRSA is a semi-synthetic, lipoglycopeptide antibacterial drug. KIMYRSA exerts a concentration-dependent bactericidal activity in vitro against S. aureus, S. pyogenes, and E. faecalis.
Mechanism of Action
Oritavancin has three mechanisms of action: (i) inhibition of the transglycosylation (polymerization) step of cell wall biosynthesis by binding to the stem peptide of peptidoglycan precursors; (ii) inhibition of the transpeptidation (crosslinking) step of cell wall biosynthesis by binding to the peptide bridging segments of the cell wall; and (iii) disruption of bacterial membrane integrity, leading to depolarization, permeabilization, and cell death. These multiple mechanisms contribute to the concentration-dependent bactericidal activity of oritavancin.
Resistance
In serial passage studies, resistance to oritavancin was observed in isolates of S. aureus and E. faecalis. Resistance to oritavancin was not observed in clinical studies.
Interaction with Other Antimicrobial Agents
In in vitro studies, oritavancin exhibits synergistic bactericidal activity in combination with gentamicin, moxifloxacin or rifampicin against isolates of methicillin-susceptible S. aureus (MSSA), with gentamicin or linezolid against isolates of heterogeneous vancomycin-intermediate S. aureus (hVISA), VISA, and vancomycin-resistant S. aureus (VRSA), and with rifampin against isolates of VRSA. In vitro studies demonstrated no antagonism between oritavancin and gentamicin, moxifloxacin, linezolid or rifampin.
Antibacterial Activity
Oritavancin has been shown to be active against most isolates of the following microorganisms, both in vitro and in clinical infections [see Indications and Usage (1.1)].
Gram-positive Bacteria
Staphylococcus aureus (including methicillin-resistant isolates)
Streptococcus agalactiae
Streptococcus anginosus group (includes S. anginosus, S. intermedius, and S. constellatus)
Streptococcus dysgalactiae
Streptococcus pyogenes
Enterococcus faecalis (vancomycin-susceptible isolates only)
The following in vitro data are available, but their clinical significance is unknown. At least 90 percent of the following bacteria exhibit an in vitro minimum inhibitory concentration (MIC) less than or equal to the susceptible breakpoint for oritavancin against isolates of a similar organism group. However, the efficacy of oritavancin in treating clinical infections due to these bacteria has not been established in adequate and well-controlled clinical trials.
12.6 Immunogenicity
Positive indirect and direct antiglobulin tests (IAT/DAT) have been noted with the administration of KIMYRSA and oritavancin in studies with healthy subjects and patients with ABSSSI. The observed incidence of anti-drug antibodies is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of anti-drug antibodies in the studies described below with the incidence of anti-drug antibodies in other studies, including those of KIMYRSA or other oritavancin products.
In a randomized, open-label, multi-center ABSSSI study, positive IAT/DAT were reported in 9.6% (5/52) of subjects who received ORBACTIV, another oritavancin product, and 2% (1/50) of subjects who received KIMYRSA. Oritavancin-dependent red blood cell (RBC) antibodies were detected when tested in the presence of drug for three subjects in the oritavancin group 14 days after dosing.
In a multiple-dose study with oritavancin in healthy volunteers, 90% (9/10) of subjects had a positive IAT 14 days after the second infusion.
In a healthy volunteer study, 66% (22/32) of subjects receiving KIMYRSA had a positive IAT 15 days after receiving dosing and one subject had a positive DAT at 8 days after dosing.
There were no reports of hemolysis in subjects who had positive IAT/DAT in any of the above referenced studies.
An in vitro study further evaluated 403 stored plasma samples from 12 healthy subjects and from 101 patients with ABSSSI from previously conducted clinical studies at multiple timepoints in healthy subjects and in patients with ABSSSI. In samples collected during the 15- and 8-day windows following single-dose treatment in healthy volunteers (Study MDCO-ORI-15-01) and in patients with ABSSSI (Study ML-ORI-201), respectively, low level (<8) direct agglutinating (IgM) anti-oritavancin antibodies were detected in 19 of 113 (17%) subjects. There was no identified clinically significant effect of anti-drug antibodies on pharmacokinetics, pharmacodynamics, safety, or effectiveness of single-dose oritavancin. No hematologic adverse events were reported for subjects with a positive titer.
There were no identified clinically significant effects of anti-drug antibodies on pharmacokinetics, pharmacodynamics, safety, or effectiveness of KIMYRSA over the treatment duration of 14 days.
Positive IAT may interfere with cross-matching before blood transfusion [see Drug Interactions (7.2)].
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Long term studies in animals have not been conducted to determine the carcinogenic potential of oritavancin.
No mutagenic or clastogenic potential of oritavancin was found in a battery of tests, including an Ames assay, in vitro chromosome aberration assay in Chinese hamster ovary cells, in vitro forward mutation assay in mouse lymphoma cells and an in vivo mouse micronucleus assay.
Oritavancin did not affect the fertility or reproductive performance of male rats (exposed to daily doses up to 30 mg/kg for at least 4 weeks) and female rats (exposed to daily doses up to 30 mg/kg for at least 2 weeks prior to mating). Those daily doses would be equivalent to a human dose of 300 mg, or 25% of clinical dose. Higher doses were not evaluated in nonclinical fertility studies.
-
14 CLINICAL STUDIES
14.1 Acute Bacterial Skin and Skin Structure Infections (ABSSSI)
A total of 1987 adults with clinically documented ABSSSI suspected or proven to be due to Gram-positive pathogens were randomized into two identically designed, randomized, double-blind, multi-center, multinational, non-inferiority trials (Trial 1 and Trial 2) comparing a single 1,200 mg intravenous dose of oritavancin to intravenous vancomycin (1 g or 15 mg/kg every 12 hours) for 7 to 10 days. The primary analysis population (modified intent to treat, mITT) included all randomized patients who received any study drug. Patients could receive concomitant aztreonam or metronidazole for suspected Gram-negative and anaerobic infection, respectively. Patient demographic and baseline characteristics were balanced between treatment groups. Approximately 64% of patients were Caucasian and 65% were males. The mean age was 45 years and the mean body mass index was 27 kg/m2. Across both trials, approximately 60% of patients were enrolled from the United States and 27% of patients from Asia. A history of diabetes was present in 14% of patients. The types of ABSSSI across both trials included cellulitis/erysipelas (40%), wound infection (29%), and major cutaneous abscesses (31%). Median infection area at baseline across both trials was 266.6 cm2. The primary endpoint in both trials was early clinical response (responder), defined as cessation of spread or reduction in size of baseline lesion, absence of fever, and no rescue antibacterial drug at 48 to 72 hours after initiation of therapy.
Table 4 provides the efficacy results for the primary endpoint in Trial 1 and Trial 2 in the primary analysis population.
Table 4: Clinical Response Rates in ABSSSI Trials using Responders*, † at 48-72 Hours after Initiation of Therapy Oritavancin
n /N (%)Vancomycin
n /N (%)Difference (95% CI)‡ - * Cessation of spread or reduction in size of baseline lesion, absence of fever (<37.7°C) and no rescue antibacterial drug at 48 to 72 hours.
- † Patients who died at 48 to 72 hours, after initiation of therapy or who had increase in lesion size at 48 to 72 hours, after initiation of therapy or who used non-study antibacterial therapy during first 72 hours or who had an additional, unplanned, surgical procedure or who had missing measurements during the first 72 hours from initiation of study drug were classified as non-responders.
- ‡ 95% CI based on the Normal approximation to Binomial distribution.
Trial 1 391/475 (82.3) 378/479 (78.9) 3.4 (-1.6, 8.4) Trial 2 403/503 (80.1) 416/502 (82.9) -2.7 (-7.5, 2.0) A key secondary endpoint in these two ABSSSI trials evaluated the percentage of patients achieving a 20% or greater reduction in lesion area from baseline at 48-72 hours after initiation of therapy. Table 5 summarizes the findings for this endpoint in the two ABSSSI trials.
Table 5: Clinical Response Rates* in ABSSSI Trials using Reduction in Lesion Area of 20% or Greater at 48-72 Hours after Initiation of Therapy Oritavancin
n /N (%)Vancomycin
n /N (%)Difference (95% CI)† - * Patients who died at 48 to 72 hours, after initiation of therapy or who had increase in lesion size at 48 to 72 hours, after initiation of therapy or who used non-study antibacterial therapy during first 72 hours or who had an additional, unplanned, surgical procedure or who had missing measurements during the first 72 hours from initiation of study drug were classified as non-responders.
- † 95% CI based on the Normal approximation to Binomial distribution.
Trial 1 413/475 (86.9) 397/479 (82.9) 4.1 (-0.5, 8.6) Trial 2 432/503 (85.9) 428/502 (85.3) 0.6 (-3.7, 5.0) Another secondary efficacy endpoint in the two trials was investigator-assessed clinical success at post therapy evaluation at Day 14 to 24 (7 to 14 days from end of blinded therapy). A patient was categorized as a clinical success if the patient experienced a complete or nearly complete resolution of baseline signs and symptoms related to primary ABSSSI site (erythema, induration/edema, purulent drainage, fluctuance, pain, tenderness, local increase in heat/warmth) such that no further treatment with antibacterial drugs was needed.
Table 6 summarizes the findings for this endpoint in the mITT and clinically evaluable population in these two ABSSSI trials. Note that there are insufficient historical data to establish the magnitude of drug effect for antibacterial drugs compared with placebo at the post therapy visits. Therefore, comparisons of oritavancin to vancomycin based on clinical success rates at these visits cannot be utilized to establish non-inferiority conclusions.
Table 6: Clinical Success Rates* in ABSSSI Trials at the Follow-Up Visit (7-14 days after end of therapy) Oritavancin
n /N (%)Vancomycin
n /N (%)Difference (95% CI)† - * Clinical success was defined if the patient experienced a complete or nearly complete resolution of baseline signs and symptoms as described above.
- † 95% CI based on the Normal approximation to Binomial distribution.
- ‡ mITT population consisted of all randomized patients who received study drug; CE population consisted of all mITT patients who did not have violations of inclusion and exclusion criteria, completed treatment and had investigator assessment at the Follow-Up Visit.
Trial 1 mITT‡ 378/475 (79.6) 383/479 (80.0) -0.4 (-5.5, 4.7) CE‡ 362/394 (91.9) 370/397 (93.2) -1.3 (-5.0,2.3) Trial 2 mITT‡ 416/503 (82.7) 404/502 (80.5) 2.2 (-2.6, 7.0) CE‡ 398/427 (93.2) 387/408 (94.9) -1.6 (-4.9,1.6) Outcomes by Baseline Pathogen: Table 7 shows outcomes in patients with an identified baseline pathogen in the microbiological Intent-to-Treat (microITT) population in a pooled analysis of Trial 1 and Trial 2. The outcomes shown in the table are clinical response rates at 48 to 72 hours and clinical success rates at follow-up study day 14 to 24.
Table 7: Outcomes by Baseline Pathogen (microITT) At 48-72 hours Study day 14 to 24 Early Clinical Responder* ≥ 20% reduction in lesion size† Clinical Success‡ Pathogen§ Oritavancin
n/N (%)Vancomycin
n/N (%)Oritavancin
n/N (%)Vancomycin
n/N (%)Oritavancin
n/N (%)Vancomycin
n/N (%)- * Early clinical response defined as a composite of the cessation of spread or reduction in size of baseline lesion, absence of fever and no rescue antibacterial drug at 48-72 hours.
- † Patients achieving a 20% or greater reduction in lesion area from baseline at 48-72 hours after initiation of therapy.
- ‡ Clinical success was defined if the patient experienced a complete or nearly complete resolution of baseline signs and symptoms as described above.
- § Baseline bacteremia in the oritavancin arm with relevant microorganisms causing ABSSSI included four subjects with MSSA and seven subjects with MRSA. Eight of these eleven subjects were responders at 48 to 72 hours after initiation of therapy.
Staphylococcus aureus 388/472 (82.2) 395/473 (83.5) 421/472 (89.2) 407/473 (86.0) 390/472 (82.6) 398/473 (84.1) Methicillin-susceptible 222/268 (82.8) 233/272 (85.7) 231/268 (86.2) 232/272 (85.3) 220/268 (82.1) 229/272 (84.2) Methicillin-resistant 166/204 (81.4) 162/201 (80.6) 190/204 (93.1) 175/201 (87.1) 170/204 (83.3) 169/201 (84.1) Streptococcus pyogenes 21/31 (67.7) 23/32 (71.9) 24/31 (77.4) 24/32 (75.0) 25/31 (80.6) 23/32 (71.9) Streptococcus agalactiae 7/8 (87.5) 12/12 (100.0) 8/8 (100.0) 12/12 (100.0) 7/8 (87.5) 11/12 (91.7) Streptococcus dysgalactiae 7/9 (77.8) 6/6 (100.0) 6/9 (66.7) 5/6 (83.3) 7/9 (77.8) 3/6 (50.0) Streptococcus anginosus group 28/33 (84.8) 40/45 (88.9) 29/33 (87.9) 42/45 (93.3) 25/33 (75.8) 38/45 (84.4) Enterococcus faecalis 11/13 (84.6) 10/12 (83.3) 10/13 (76.9) 8/12 (66.7) 8/13 (61.5) 9/12 (75.0) -
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied/Storage
KIMYRSA is supplied as a sterile white to off-white or pink lyophilized powder in single-dose clear glass vials containing 1,200 mg of oritavancin. One vial is packaged in a carton to supply a single 1,200 mg dose treatment (NDC: 70842-225-01).
KIMYRSA vials should be stored at 20ºC to 25ºC (68ºF to 77ºF); excursions permitted to 15ºC to 30ºC (59ºF to 86ºF) [see USP, Controlled Room Temperature (CRT)].
-
17 PATIENT COUNSELING INFORMATION
Allergic Reactions
Patients should be advised that allergic reactions, including serious allergic reactions, could occur and that serious reactions require immediate treatment. They should inform their healthcare provider about any previous hypersensitivity reactions to oritavancin products, other glycopeptides (vancomycin, telavancin, or dalbavancin) or other allergens.
Diarrhea
Patients should be advised that diarrhea is a common problem caused by antibacterial drugs including KIMYRSA, which usually resolves when the drug is discontinued. Sometimes, frequent watery or bloody diarrhea may occur and may be a sign of a more serious intestinal infection. If severe watery or bloody diarrhea develops, patients should contact their healthcare provider.
Development of Antibacterial Resistance
Patients should be counseled that antibacterial drugs including KIMYRSA should only be used to treat bacterial infections. They do not treat viral infections (e.g. the common cold). When KIMYRSA is prescribed to treat a bacterial infection, patients should be told that although it is common to feel better early in the course of therapy, the medication should be taken exactly as directed. Skipping doses or not completing the full course of therapy may (1) decrease the effectiveness of the immediate treatment and (2) increase the likelihood that bacteria will develop resistance and will not be treatable by KIMYRSA or other antibacterial drugs in the future.
- SPL UNCLASSIFIED SECTION
-
PRINCIPAL DISPLAY PANEL - 1,200 mg Vial Label
Rx only
NDC: 70842-225-01Kimyrsa®
(oritavancin) for injection1,200 mg per vial
Must be reconstituted and
further diluted to a final
volume of 250 mLFor Intravenous Infusion Only
Single-dose Vial
Discard Unused Portion
-
PRINCIPAL DISPLAY PANEL - 1,200 mg Vial Carton
Rx only
NDC: 70842-225-01Kimyrsa®
(oritavancin)
for injection1,200 mg per vial
Must be reconstituted
and further diluted
to a final volume
of 250 mLFor Intravenous Infusion Only
Single-dose Vial
Discard Unused Portion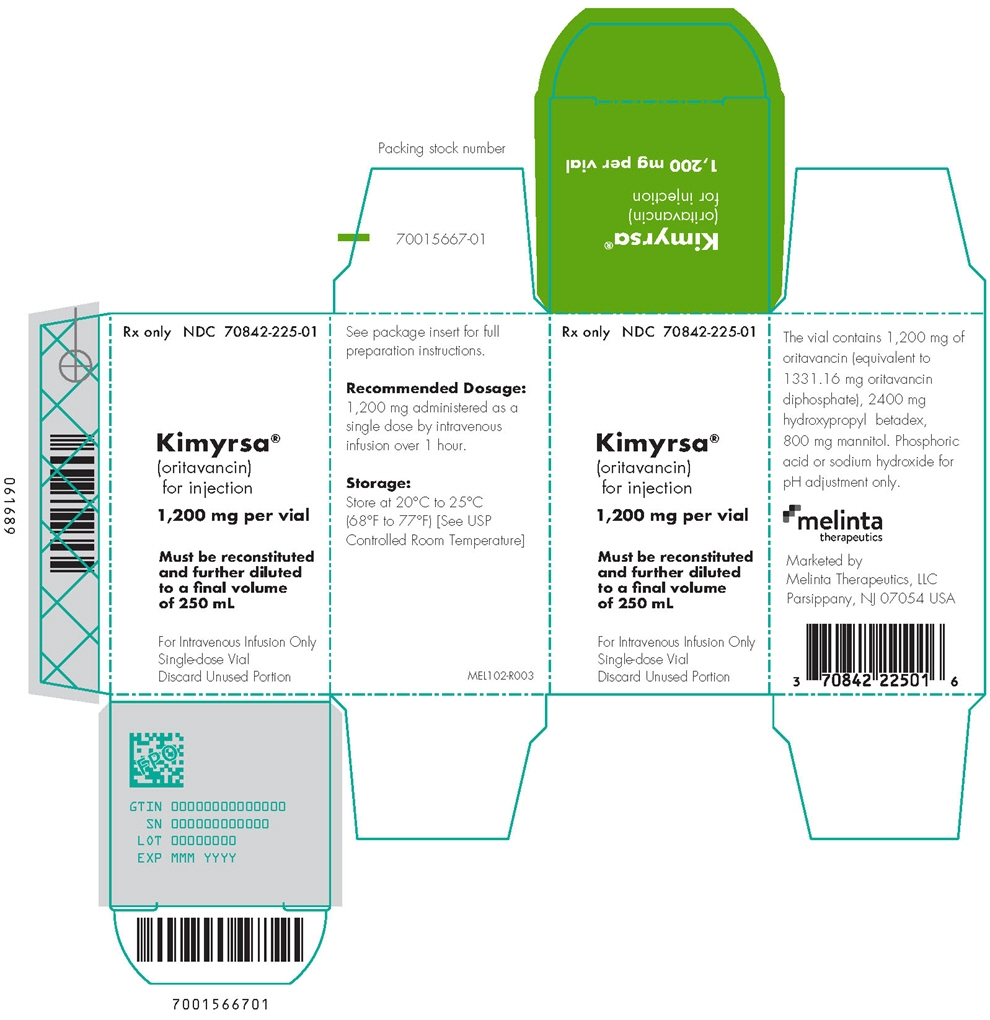
-
INGREDIENTS AND APPEARANCE
KIMYRSA
oritavancin diphosphate injection, powder, lyophilized, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 70842-225 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ORITAVANCIN DIPHOSPHATE (UNII: VL1P93MKZN) (ORITAVANCIN - UNII:PUG62FRZ2E) ORITAVANCIN 1200 mg in 40 mL Inactive Ingredients Ingredient Name Strength HYDROXYPROPYL BETADEX (UNII: 1I96OHX6EK) MANNITOL (UNII: 3OWL53L36A) PHOSPHORIC ACID (UNII: E4GA8884NN) SODIUM HYDROXIDE (UNII: 55X04QC32I) Product Characteristics Color WHITE (white to off-white or pink) Score Shape Size Flavor Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 70842-225-01 1 in 1 CARTON 03/23/2021 1 40 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA214155 03/23/2021 Labeler - Melinta Therapeutics, LLC (034132850) Establishment Name Address ID/FEI Business Operations Patheon Italia SpA 338336589 ANALYSIS(70842-225) , MANUFACTURE(70842-225) , PACK(70842-225) Establishment Name Address ID/FEI Business Operations Pharma Packaging Solutions, LLC dba Tjoapack, LLC 928861723 PACK(70842-225) , LABEL(70842-225) Establishment Name Address ID/FEI Business Operations AbbVie Inc. 078521674 ANALYSIS(70842-225) , API MANUFACTURE(70842-225) Establishment Name Address ID/FEI Business Operations BioConvergence LLC DBA Singota Solutions 620582002 MANUFACTURE(70842-225) Establishment Name Address ID/FEI Business Operations Frontage Laboratories, Inc. 832653625 ANALYSIS(70842-225) Establishment Name Address ID/FEI Business Operations PPD Development, L.P. 838082055 ANALYSIS(70842-225)
Trademark Results [Kimyrsa]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 KIMYRSA 90039970 not registered Live/Pending |
Melinta Therapeutics, Inc. 2020-07-07 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.