PRAMIPEXOLE DIHYDROCHLORIDE tablet
Pramipexole Dihydrochloride by
Drug Labeling and Warnings
Pramipexole Dihydrochloride by is a Prescription medication manufactured, distributed, or labeled by Torrent Pharmaceuticals Limited, Torrent Pharma, Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use PRAMIPEXOLE DIHYDROCHLORIDE TABLETS safely and effectively. See full prescribing information for PRAMIPEXOLE DIHYDROCHLORIDE TABLETS.
PRAMIPEXOLE dihydrochloride tablets, for oral use
Initial U.S. Approval: 1997RECENT MAJOR CHANGES
Warnings and Precautions, Postural Deformity (5.6) 5/2018
Warnings and Precautions, Rhabdomyolysis (5.8) 5/2018
Warnings and Precautions,
Events Reported with Dopaminergic Therapy (5.10);
Melanoma Removed 5/2018
INDICATIONS AND USAGE
DOSAGE AND ADMINISTRATION
Parkinson's Disease-Normal Renal Function* (2.2)
Week
Dosage (mg)
Total Daily Dose (mg)
1
0.125 TID
0.375
2
0.25 TID
0.75
3
0.5 TID
1.5
4
0.75 TID
2.25
5
1 TID
3
6
1.25 TID
3.75
7
1.5 TID
4.5
* Doses should not be increased more frequently than every 5 to 7 days.
Titrate to effective dose. If used with levodopa, may need to reduce levodopa dose.
Parkinson's Disease-Impaired Renal Function (2.2)
Creatinine Clearance
Starting Dose (mg)
Maximum Dose (mg)
> 50 mL/min
0.125 TID
1.5 TID
30 to 50 mL/min
0.125 BID
0.75 TID
15 to 30 mL/min
0.125 QD
1.5 QD
< 15 mL/min and hemodialysis patients
Data not available
Restless Legs Syndrome* (2.3)
Titration Step
Dose (mg) 2 to 3 hours before bedtime
1
0.125
2 (if needed)
0.25
3 (if needed)
0.5
* Dosing interval is 4 to 7 days (14 days in patients with CrCl 20 to 60 mL/min)
DOSAGE FORMS AND STRENGTHS
Tablets: 0.125 mg, 0.25 mg, 0.5 mg, 0.75 mg, 1 mg, and 1.5 mg (3) (3)
CONTRAINDICATIONS
None (4)
WARNINGS AND PRECAUTIONS
- Falling asleep during activities of daily living: Sudden onset of sleep may occur without warning; advise patients to report symptoms (5.1)
- Symptomatic orthostatic hypotension: Monitor during dose escalation (5.2)
- Impulse control/Compulsive behaviors: Patients may experience compulsive behaviors and other intense urges (5.3)
- Hallucinations and Psychotic-like Behavior: May occur; risk increases with age (5.4)
- Dyskinesia: May be caused or exacerbated by pramipexole dihydrochloride tablets (5.5)
- Postural Deformity: Consider reducing the dose or discontinuing Pramipexole dihydrochloride tablets if postural deformity occurs (5.6)
ADVERSE REACTIONS
Most common adverse reactions (incidence >5% and greater than placebo): (6)
- Early PD without levodopa: nausea, dizziness, somnolence, insomnia, constipation, asthenia, and hallucinations (6.1)
- Advanced PD with levodopa: postural (orthostatic) hypotension, dyskinesia, extrapyramidal syndrome, insomnia, dizziness, hallucinations, accidental injury, dream abnormalities, confusion, constipation, asthenia, somnolence, dystonia, gait abnormality, hypertonia, dry mouth, amnesia, and urinary frequency (6.1)
- RLS: nausea, somnolence, fatigue, and headache (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Torrent Pharma Inc. at 1-800-912-95621 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch. (6)
DRUG INTERACTIONS
Dopamine antagonists: May diminish the effectiveness of pramipexole (7.1) (7)
USE IN SPECIFIC POPULATIONS
Pregnancy: Based on animal data, may cause fetal harm (8.1) (8)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 6/2018
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Parkinson’s Disease
1.2 Restless Legs Syndrome
2 DOSAGE AND ADMINISTRATION
2.1 General Dosing Considerations
2.2 Dosing for Parkinson's Disease
2.3 Dosing for Restless Legs Syndrome
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Falling Asleep During Activities of Daily Living and Somnolence
5.2 Symptomatic Orthostatic Hypotension
5.3 Impulse Control/Compulsive Behaviors
5.4 Hallucinations and Psychotic-like Behavior
5.5 Dyskinesia
5.6 Postural Deformity
5.7 Renal Impairment
5.8 Rhabdomyolysis
5.9 Retinal Pathology
5.10 Events Reported with Dopaminergic Therapy
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Post Marketing Experience
7 DRUG INTERACTIONS
7.1 Dopamine Antagonists
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
14.1 Parkinson’s Disease
14.2 Restless Legs Syndrome
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage and Handling
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 General Dosing Considerations
Pramipexole dihydrochloride tablets are taken orally, with or without food.
If a significant interruption in therapy with pramipexole dihydrochloride tablets has occurred, re-titration of therapy may be warranted.
2.2 Dosing for Parkinson's Disease
In all clinical studies, dosage was initiated at a subtherapeutic level to avoid intolerable adverse effects and orthostatic hypotension. Pramipexole dihydrochloride tablets should be titrated gradually in all patients. The dose should be increased to achieve a maximum therapeutic effect, balanced against the principal side effects of dyskinesia, hallucinations, somnolence, and dry mouth.
Dosing in Patients with Normal Renal Function
Initial Treatment
Doses should be increased gradually from a starting dose of 0.375 mg/day given in three divided doses and should not be increased more frequently than every 5 to 7 days. A suggested ascending dosage schedule that was used in clinical studies is shown in Table 1:
Table 1 Ascending Dosage Schedule of Pramipexole Dihydrochloride Tablets for Parkinson's Disease Week
Dosage (mg)
Total Daily Dose (mg)
1
0.125 three times a day
0.375
2
0.25 three times a day
0.75
3
0.5 three times a day
1.50
4
0.75 three times a day
2.25
5
1 three times a day
3.0
6
1.25 three times a day
3.75
7
1.5 three times a day
4.50
Pramipexole Dihydrochloride Tablets were effective and well tolerated over a dosage range of 1.5 to 4.5 mg/day administered in equally divided doses three times per day with or without concomitant levodopa (approximately 800 mg/day).
In a fixed-dose study in early Parkinson's disease patients, doses of 3 mg, 4.5 mg, and 6 mg per day of pramipexole dihydrochloride tablets were not shown to provide any significant benefit beyond that achieved at a daily dose of 1.5 mg/day. However, in the same fixed-dose study, the following adverse events were dose related: postural hypotension, nausea, constipation, somnolence, and amnesia. The frequency of these events was generally 2-fold greater than placebo for pramipexole doses greater than 3 mg/day. The incidence of somnolence reported with pramipexole at a dose of 1.5 mg/day was comparable to placebo.
When pramipexole dihydrochloride tablets are used in combination with levodopa, a reduction of the levodopa dosage should be considered. In a controlled study in advanced Parkinson's disease, the dosage of levodopa was reduced by an average of 27% from baseline.
Dosing in Patients with Renal Impairment
The recommended dosing of pramipexole dihydrochloride tablets in Parkinson's disease patients with renal impairment is provided in Table 2.
Table 2 Dosing of Pramipexole Dihydrochloride Tablets in Parkinson's Disease Patients with Renal Impairment Renal Status
Starting Dose (mg)
Maximum Dose (mg)
Normal to mild impairment
(creatinine Cl > 50 mL/min)
0.125 three times a day
1.5 three times a day
Moderate impairment
(creatinine Cl = 30 to 50 mL/min)
0.125 twice a day
0.75 three times a day
Severe impairment
(creatinine Cl = 15 to <30 mL/min)
0.125 once a day
1.5 once a day
Very severe impairment
(creatinine Cl < 15 mL/min and hemodialysis patients)
The use of pramipexole dihydrochloride tablets has not been adequately studied in this group of patients.
Pramipexole dihydrochloride tablets may be tapered off at a rate of 0.75 mg per day until the daily dose has been reduced to 0.75 mg. Thereafter, the dose may be reduced by 0.375 mg per day [ see Warnings and Precautions (5.10)].
2.3 Dosing for Restless Legs Syndrome
The recommended starting dose of pramipexole dihydrochloride tablets is 0.125 mg taken once daily 2 to 3 hours before bedtime. For patients requiring additional symptomatic relief, the dose may be increased every 4 to 7 days (Table 3). Although the dose of pramipexole dihydrochloride tablets was increased to 0.75 mg in some patients during long-term open-label treatment, there is no evidence that the 0.75 mg dose provides additional benefit beyond the 0.5 mg dose.
Table 3 Ascending Dosage Schedule of Pramipexole Dihydrochloride Tablets for RLS *If needed
Titration Step
Duration
Dose (mg) to be taken once daily, 2 to 3 hours before bedtime
1
4 to 7 days
0.125
2*
4 to 7 days
0.25
3*
4 to 7 days
0.5
Dosing in Patients with Renal Impairment
The duration between titration steps should be increased to 14 days in RLS patients with moderate and severe renal impairment (creatinine clearance 20 to 60 mL/min) [ see Clinical Pharmacology (12.3)].
Discontinuation of Treatment
In clinical trials of patients being treated for RLS with doses up to 0.75 mg once daily, pramipexole dihydrochloride tablets were discontinued without a taper. In a 26 week placebo-controlled clinical trial, patients reported a worsening of RLS symptom severity as compared to their untreated baseline when pramipexole dihydrochloride tablets treatment was suddenly withdrawn [ see Warnings and Precautions (5.10)].
-
3 DOSAGE FORMS AND STRENGTHS
- 0.125 mg: White to off white, round, flat, bevel edged, uncoated tablets, debossed with "91" on one side and plain on other side.
- 0.25 mg: Peach colored, round, flat, bevel edged, uncoated tablets with "9/2" debossed on one side and breakline on other side.
- 0.5 mg: Reddish brown colored, round, biconvex, uncoated tablets with "9/3" debossed on one side and breakline on other side.
- 0.75 mg: Yellow colored, round, flat, bevel edged, uncoated tablets with "84" debossed on one side and plain on other side.
- 1 mg: Light pink colored, round, flat, bevel edged, uncoated tablets with "9/4" debossed on one side and breakline on other side.
- 1.5 mg: White to off white, round, flat, bevel edged, uncoated tablets with "9/5" debossed on one side and breakline on other side.
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Falling Asleep During Activities of Daily Living and Somnolence
Patients treated with pramipexole have reported falling asleep while engaged in activities of daily living, including the operation of motor vehicles which sometimes resulted in accidents. Although many of these patients reported somnolence while on pramipexole dihydrochloride tablets, some perceived that they had no warning signs (sleep attack) such as excessive drowsiness, and believed that they were alert immediately prior to the event. Some of these events had been reported as late as one year after the initiation of treatment.
Somnolence is a common occurrence in patients receiving pramipexole at doses above 1.5 mg/day (0.5 mg three times a day) for Parkinson's disease. In controlled clinical trials in RLS, patients treated with pramipexole dihydrochloride tablets at doses of 0.25 to 0.75 mg once a day, the incidence of somnolence was 6% compared to an incidence of 3% for placebo-treated patients [ see Adverse Reactions (6.1)].It has been reported that falling asleep while engaged in activities of daily living usually occurs in a setting of pre-existing somnolence, although patients may not give such a history. For this reason, prescribers should reassess patients for drowsiness or sleepiness, especially since some of the events occur well after the start of treatment. Prescribers should also be aware that patients may not acknowledge drowsiness or sleepiness until directly questioned about drowsiness or sleepiness during specific activities.
Before initiating treatment with pramipexole dihydrochloride tablets, advise patients of the potential to develop drowsiness and specifically ask about factors that may increase the risk for somnolence with pramipexole dihydrochloride tablets such as the use of concomitant sedating medications or alcohol, the presence of sleep disorders, and concomitant medications that increase pramipexole plasma levels (e.g., cimetidine) [ see Clinical Pharmacology (12.3)]. If a patient develops significant daytime sleepiness or episodes of falling asleep during activities that require active participation (e.g., conversations, eating, etc.), pramipexole dihydrochloride tablets should ordinarily be discontinued. If a decision is made to continue pramipexole dihydrochloride tablets, advise patients not to drive and to avoid other potentially dangerous activities that might result in harm if the patients become somnolent. While dose reduction reduces the degree of somnolence, there is insufficient information to establish that dose reduction will eliminate episodes of falling asleep while engaged in activities of daily living.
5.2 Symptomatic Orthostatic Hypotension
Dopamine agonists, in clinical studies and clinical experience, appear to impair the systemic regulation of blood pressure, with resulting orthostatic hypotension, especially during dose escalation. Parkinson's disease patients, in addition, appear to have an impaired capacity to respond to an orthostatic challenge. For these reasons, both Parkinson's disease patients and RLS patients being treated with dopaminergic agonists ordinarily require careful monitoring for signs and symptoms of orthostatic hypotension, especially during dose escalation, and should be informed of this risk.
In clinical trials of pramipexole, however, and despite clear orthostatic effects in normal volunteers, the reported incidence of clinically significant orthostatic hypotension was not greater among those assigned to pramipexole tablets than among those assigned to placebo. This result, especially with the higher doses used in Parkinson's disease, is clearly unexpected in light of the previous experience with the risks of dopamine agonist therapy.
While this finding could reflect a unique property of pramipexole, it might also be explained by the conditions of the study and the nature of the population enrolled in the clinical trials. Patients were very carefully titrated, and patients with active cardiovascular disease or significant orthostatic hypotension at baseline were excluded. Also, clinical trials in patients with RLS did not incorporate orthostatic challenges with intensive blood pressure monitoring done in close temporal proximity to dosing.
5.3 Impulse Control/Compulsive Behaviors
Case reports and the results of a cross-sectional study suggest that patients can experience intense urges to gamble, increased sexual urges, intense urges to spend money uncontrollably, binge eating, and/or other intense urges and the inability to control these urges while taking one or more of the medications, including pramipexole dihydrochloride tablets , that increase central dopaminergic tone and that are generally used for the treatment of Parkinson's disease. In some cases, although not all, these urges were reported to have stopped when the dose was reduced or the medication was discontinued. Because patients may not recognize these behaviors as abnormal it is important for prescribers to specifically ask patients or their caregivers about the development of new or increased gambling urges, sexual urges, uncontrolled spending or other urges while being treated with pramipexole dihydrochloride tablets. Physicians should consider dose reduction or stopping the medication if a patient develops such urges while taking pramipexole dihydrochloride tablets.
5.4 Hallucinations and Psychotic-like Behavior
In the three double-blind, placebo-controlled trials in early Parkinson's disease, hallucinations were observed in 9% (35 of 388) of patients receiving pramipexole dihydrochloride tablets, compared with 2.6% (6 of 235) of patients receiving placebo. In the four double-blind, placebo-controlled trials in advanced Parkinson's disease, where patients received pramipexole dihydrochloride tablets and concomitant levodopa, hallucinations were observed in 16.5% (43 of 260) of patients receiving pramipexole dihydrochloride tablets compared with 3.8% (10 of 264) of patients receiving placebo. Hallucinations were of sufficient severity to cause discontinuation of treatment in 3.1% of the early Parkinson's disease patients and 2.7% of the advanced Parkinson's disease patients compared with about 0.4% of placebo patients in both populations.
Age appears to increase the risk of hallucinations attributable to pramipexole. In the early Parkinson's disease patients, the risk of hallucinations was 1.9 times greater than placebo in patients younger than 65 years and 6.8 times greater than placebo in patients older than 65 years. In the advanced Parkinson's disease patients, the risk of hallucinations was 3.5 times greater than placebo in patients younger than 65 years and 5.2 times greater than placebo in patients older than 65 years.
Postmarketing reports with medication used to treat Parkinson's disease, including pramipexole dihydrochloride, indicate that patients may experience new or worsening mental status and behavioral changes, which may be severe, including psychotic-like behavior during treatment with pramipexole dihydrochloride or after starting or increasing the dose of pramipexole dihydrochloride. Other drugs prescribed to improve the symptoms of Parkinson's disease can have similar effects on thinking and behavior. This abnormal thinking and behavior can consist of one or more of a variety of manifestations including paranoid ideation, delusions, hallucinations, confusion, psychotic-like behavior, disorientation, aggressive behavior, agitation, and delirium.
Patients with a major psychotic disorder should ordinarily not be treated with dopamine agonists, including pramipexole dihydrochloride, because of the risk of exacerbating the psychosis. In addition, certain medications used to treat psychosis may exacerbate the symptoms of Parkinson's disease and may decrease the effectiveness of pramipexole dihydrochloride [ see Drug Interactions (7.1)].
In the RLS clinical trials, one pramipexole-treated patient (of 889) reported hallucinations; this patient discontinued treatment and the symptoms resolved.
5.6 Postural Deformity
Postural deformities, including antecollis, camptocormia (Bent Spine Syndrome), and pleurothotonus (Pisa Syndrome), have been reported in patients after starting or increasing the dose of pramipexole dihydrochloride. Postural deformity may occur several months after starting treatment or increasing the dose. Reducing the dose or discontinuing pramipexole dihydrochloride has been reported to improve postural deformity in some patients, and should be considered if postural deformity occurs.
5.7 Renal Impairment
Since pramipexole is eliminated through the kidneys, caution should be exercised when prescribing pramipexole dihydrochloride tablets to patients with renal impairment [ see Dosage and Administration (2.3), Use in Specific Populations (8.6), and Clinical Pharmacology (12.3)].
5.8 Rhabdomyolysis
A single case of rhabdomyolysis occurred in a 49-year-old male with advanced Parkinson's disease treated with pramipexole dihydrochloride tablets. The patient was hospitalized with an elevated CPK (10,631 IU/L). The symptoms resolved with discontinuation of the medication.
Advise patients to contact a physician if they experience any unexplained muscle pain, tenderness, or weakness, as these may be symptoms of rhabdomyolysis.
5.9 Retinal Pathology
A two-year open-label, randomized, parallel-group safety study of retinal deterioration and vision compared pramipexole dihydrochloride tablets and immediate-release ropinirole. Two hundred thirty four Parkinson's disease patients (115 on pramipexole, mean dose 3.0 mg/day and 119 on ropinirole, mean dose 9.5 mg/day) were evaluated using a panel of clinical ophthalmological assessments. Of 234 patients who were evaluable, 196 had been treated for two years and 29 were judged to have developed clinical abnormalities that were considered meaningful (19 patients in each treatment arm had received treatment for less than two years). There was no statistical difference in retinal deterioration between the treatment arms; however, the study was only capable of detecting a very large difference between treatments. In addition, because the study did not include an untreated comparison group (placebo treated), it is unknown whether the findings reported in patients treated with either drug are greater than the background rate in an aging population.
Animal Data
Pathologic changes (degeneration and loss of photoreceptor cells) were observed in the retina of albino rats in the 2-year carcinogenicity study. While retinal degeneration was not diagnosed in pigmented rats treated for 2 years, a thinning in the outer nuclear layer of the retina was slightly greater in rats given drug compared with controls. Evaluation of the retinas of albino mice, monkeys, and minipigs did not reveal similar changes. The potential significance of this effect in humans has not been established, but cannot be disregarded because disruption of a mechanism that is universally present in vertebrates (i.e., disk shedding) may be involved [see Nonclinical Toxicology (13.2)].
5.10 Events Reported with Dopaminergic Therapy
Although the events enumerated below may not have been reported in association with the use of pramipexole in its development program, they are associated with the use of other dopaminergic drugs. The expected incidence of these events, however, is so low that even if pramipexole caused these events at rates similar to those attributable to other dopaminergic therapies, it would be unlikely that even a single case would have occurred in a cohort of the size exposed to pramipexole in studies to date.
Hyperpyrexia and Confusion
Although not reported with pramipexole in the clinical development program, a symptom complex resembling the neuroleptic malignant syndrome (characterized by elevated temperature, muscular rigidity, altered consciousness, and autonomic instability), with no other obvious etiology, has been reported in association with rapid dose reduction, withdrawal of, or changes in dopaminergic therapy. If possible, avoid sudden discontinuation or rapid dose reduction in patients taking pramipexole dihydrochloride tablets. If the decision is made to discontinue pramipexole dihydrochloride tablets, the dose should be tapered to reduce the risk of hyperpyrexia and confusion [ see Dosage and Administration (2.2)].
Fibrotic Complications
Cases of retroperitoneal fibrosis, pulmonary infiltrates, pleural effusion, pleural thickening, pericarditis, and cardiac valvulopathy have been reported in patients treated with ergot-derived dopaminergic agents. While these complications may resolve when the drug is discontinued, complete resolution does not always occur.
Although these adverse events are believed to be related to the ergoline structure of these compounds, whether other, nonergot-derived dopamine agonists can cause them is unknown.
Cases of possible fibrotic complications, including peritoneal fibrosis, pleural fibrosis, and pulmonary fibrosis have been reported in the post marketing experience with pramipexole dihydrochloride tablets. While the evidence is not sufficient to establish a causal relationship between pramipexole dihydrochloride tablets and these fibrotic complications, a contribution of pramipexole dihydrochloride tablets cannot be completely ruled out.
Rebound and Augmentation in RLS
Reports in the literature indicate treatment of RLS with dopaminergic medications can result in rebound: a worsening of symptoms following treatment cessation with greater intensity than described before starting treatment. In a 26 week placebo controlled clinical trial in patients with RLS, a worsening of symptoms scores (IRLS) beyond their untreated baseline levels was reported more frequently by patients suddenly withdrawn from pramipexole dihydrochloride (up to 0.75 mg once daily) compared to the group assigned to placebo (10% vs. 2%, respectively). The worsening of RLS symptoms was considered generally mild.
Augmentation has also been described during therapy for RLS. Augmentation refers to the earlier onset of symptoms in the evening (or even the afternoon), increase in symptoms, and spread of symptoms to involve other extremities. In a 26 week placebo controlled clinical trial in patients with RLS, augmentation was reported with greater frequency by patients treated with Pramipexole dihydrochloride tablets (up to 0.75 mg once daily) compared to patients who received placebo (12% vs. 9%, respectively). The incidence of augmentation increased with increasing duration of exposure to pramipexole dihydrochloride and to placebo.
The frequency and severity of augmentation and/or rebound after longer-term use of pramipexole dihydrochloride tablets and the appropriate management of these events have not been adequately evaluated in controlled clinical trials.
-
6 ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections of the labeling:
- Falling Asleep During Activities of Daily Living and Somnolence [ see Warnings and Precautions (5.1) ] .
- Symptomatic Orthostatic Hypotension [ see Warnings and Precautions (5.2) ] .
- Impulse Control/Compulsive Behaviors [ see Warnings and Precautions (5.3) ] .
- Hallucinations and Psychotic-like Behavior [ see Warnings and Precautions (5.4) ] .
- Dyskinesia [ see Warnings and Precautions (5.5) ].
- Postural Deformity [ see Warnings and Precautions (5.6) ].
- Rhabdomyolysis [ see Warnings and Precautions (5.8) ] .
- Retinal Pathology [ see Warnings and Precautions (5.9) ] .
- Events Reported with Dopaminergic Therapy [ see Warnings and Precautions (5.10) ] .
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
Parkinson's Disease
During the premarketing development of pramipexole, patients with either early or advanced Parkinson's disease were enrolled in clinical trials. Apart from the severity and duration of their disease, the two populations differed in their use of concomitant levodopa therapy. Patients with early disease did not receive concomitant levodopa therapy during treatment with pramipexole; those with advanced Parkinson's disease all received concomitant levodopa treatment. Because these two populations may have differential risks for various adverse reactions, this section will, in general, present adverse-reaction data for these two populations separately.
Because the controlled trials performed during premarketing development all used a titration design, with a resultant confounding of time and dose, it was impossible to adequately evaluate the effects of dose on the incidence of adverse reactions.
Early Parkinson's Disease
In the three double-blind , placebo-controlled trials of patients with early Parkinson's disease, the most common adverse reactions (>5%) that were numerically more frequent in the group treated with pramipexole dihydrochloride tablets were nausea, dizziness, somnolence, insomnia, constipation, asthenia, and hallucinations.
Approximately 12% of 388 patients with early Parkinson's disease and treated with pramipexole dihydrochloride tablets who participated in the double-blind, placebo-controlled trials discontinued treatment due to adverse reactions compared with 11% of 235 patients who received placebo. The adverse reactions most commonly causing discontinuation of treatment were related to the nervous system (hallucinations [3.1% on pramipexole dihydrochloride tablets vs 0.4% on placebo]; dizziness [2.1% on pramipexole dihydrochloride tablets vs 1% on placebo]; somnolence [1.6% on pramipexole dihydrochloride tablets vs 0% on placebo]; headache and confusion [1.3% and 1.0%, respectively, on pramipexole dihydrochloride tablets vs 0% on placebo]) and gastrointestinal system (nausea [2.1% on pramipexole dihydrochloride tablets vs 0.4% on placebo]).
Adverse-reaction Incidence in Controlled Clinical Studies in Early Parkinson's Disease: Table 4 lists adverse reactions that occurred in the double-blind, placebo-controlled studies in early Parkinson's disease that were reported by ≥1% of patients treated with pramipexole dihydrochloride tablets and were numerically more frequent than in the placebo group. In these studies, patients did not receive concomitant levodopa.
Table 4 Adverse-Reactions in Pooled Double-Blind, Placebo-Controlled Trials with Pramipexole Dihydrochloride Tablets in Early Parkinson's Disease Body System/
Adverse Reaction
Pramipexole Dihydrochloride Tablets
(N=388)
%
Placebo
(N=235)
%
Nervous System
Dizziness
25
24
Somnolence
22
9
Insomnia
17
12
Hallucinations
9
3
Confusion
4
1
Amnesia
4
2
Hypesthesia
3
1
Dystonia
2
1
Akathisia
2
0
Thinking abnormalities
2
0
Decreased libido
1
0
Myoclonus
1
0
Digestive System
Nausea
28
18
Constipation
14
6
Anorexia
4
2
Dysphagia
2
0
Body as a Whole
Asthenia
14
12
General edema
5
3
Malaise
2
1
Reaction unevaluable
2
1
Fever
1
0
Metabolic & Nutritional System
Peripheral edema
5
4
Decreased weight
2
0
Special Senses
Vision abnormalities
3
0
Urogenital System
Impotence
2
1
In a fixed-dose study in early Parkinson's disease, occurrence of the following reactions increased in frequency as the dose increased over the range from 1.5 mg/day to 6 mg/day: postural hypotension, nausea, constipation, somnolence, and amnesia. The frequency of these reactions was generally 2-fold greater than placebo for pramipexole doses greater than 3 mg/day. The incidence of somnolence with pramipexole at a dose of 1.5 mg/day was comparable to that reported for placebo.
Advanced Parkinson's Disease
In the four double-blind, placebo-controlled trials of patients with advanced Parkinson's disease, the most common adverse reactions (>5%) that were numerically more frequent in the group treated with pramipexole dihydrochloride tablets and concomitant levodopa were postural (orthostatic) hypotension, dyskinesia, extrapyramidal syndrome, insomnia, dizziness, hallucinations, accidental injury, dream abnormalities, confusion, constipation, asthenia, somnolence, dystonia, gait abnormality, hypertonia, dry mouth, amnesia, and urinary frequency.
Approximately 12% of 260 patients with advanced Parkinson's disease who received pramipexole dihydrochloride tablets and concomitant levodopa in the double-blind, placebo-controlled trials discontinued treatment due to adverse reactions compared with 16% of 264 patients who received placebo and concomitant levodopa. The reactions most commonly causing discontinuation of treatment were related to the nervous system (hallucinations [2.7% on pramipexole dihydrochloride tablets vs 0.4% on placebo]; dyskinesia [1.9% on pramipexole dihydrochloride tablets vs 0.8% on placebo]) and cardiovascular system (postural [orthostatic] hypotension [2.3% on pramipexole dihydrochloride tablets vs 1.1% on placebo]).
Adverse-reaction Incidence in Controlled Clinical Studies in Advanced Parkinson's Disease: Table 5 lists adverse reactions that occurred in the double-blind, placebo-controlled studies in advanced Parkinson's disease that were reported by ≥1% of patients treated with pramipexole dihydrochloride tablets and were numerically more frequent than in the placebo group. In these studies, pramipexole dihydrochloride tablets or placebo was administered to patients who were also receiving concomitant levodopa.
Table 5 Adverse-Reactions in Pooled Double-Blind, Placebo-Controlled Trials with Pramipexole Dihydrochloride Tablets in Advanced Parkinson's Disease Body System/
Adverse Event
Pramipexole Dihydrochloride Tablets
(N=260)
%
Placebo
(N=264)
%
Nervous System
Dyskinesia
47
31
Extrapyramidal syndrome
28
26
Insomnia
27
22
Dizziness
26
25
Hallucinations
17
4
Dream abnormalities
11
10
Confusion
10
7
Somnolence
9
6
Dystonia
8
7
Gait abnormalities
7
5
Hypertonia
7
6
Amnesia
6
4
Akathisia
3
2
Thinking abnormalities
3
2
Paranoid reaction
2
0
Delusions
1
0
Sleep disorders
1
0
Cardiovascular System
Postural hypotension
53
48
Body as a Whole
Accidental injury
17
15
Asthenia
10
8
General edema
4
3
Chest pain
3
2
Malaise
3
2
Digestive System
Constipation
10
9
Dry mouth
7
3
Urogenital System
Urinary frequency
6
3
Urinary tract infection
4
3
Urinary incontinence
2
1
Respiratory System
Dyspnea
4
3
Rhinitis
3
1
Pneumonia
2
0
Special Senses
Accommodation abnormalities
4
2
Vision abnormalities
3
1
Diplopia
1
0
Musculoskeletal System
Arthritis
3
1
Twitching
2
0
Bursitis
2
0
Myasthenia
1
0
Metabolic & Nutritional System
Peripheral edema
2
1
Increased creatine PK
1
0
Skin & Appendages
Skin disorders
2
1
Pramipexole dihydrochloride tablets for treatment of RLS have been evaluated for safety in 889 patients, including 427 treated for over six months and 75 for over one year.
The overall safety assessment focuses on the results of three double-blind, placebo-controlled trials, in which 575 patients with RLS were treated with pramipexole dihydrochloride tablets for up to 12 weeks. The most common adverse reactions with pramipexole dihydrochloride tablets in the treatment of RLS (observed in >5% of pramipexole-treated patients and at a rate at least twice that observed in placebo-treated patients) were nausea and somnolence . Occurrences of nausea and somnolence in clinical trials were generally mild and transient.
Approximately 7% of 575 patients treated with pramipexole dihydrochloride tablets during the double-blind periods of three placebo-controlled trials discontinued treatment due to adverse reactions compared to 5% of 223 patients who received placebo. The adverse reaction most commonly causing discontinuation of treatment was nausea (1%).
Table 6 lists reactions that occurred in three double-blind, placebo-controlled studies in RLS patients that were reported by ≥2% of patients treated with pramipexole dihydrochloride tablets and were numerically more frequent than in the placebo group.
Table 6 Adverse-Reactions in Pooled Double-Blind, Placebo-Controlled Trials with Pramipexole Dihydrochloride Tablets in Restless Legs Syndrome Body System/Adverse Reaction
Pramipexole Dihydrochloride Tablets
0.125 to 0.75 mg/day (N=575)
%
Placebo
(N=223)
%
Gastrointestinal disorders
Nausea
16
5
Constipation
4
1
Diarrhea
3
1
Dry mouth
3
1
Nervous system disorders
Headache
16
15
Somnolence
6
3
General disorders and administration site conditions
Fatigue
9
7
Infections and infestations
Influenza
3
1
Table 7 summarizes data for adverse reactions that appeared to be dose related in the 12-week fixed dose study.
Table 7 Dose-Related Adverse Reactions in a 12-Week Double-Blind, Placebo-Controlled Fixed Dose Study in Restless Legs Syndrome (Occurring in >5% of all Patients in the Treatment Phase) Body System/Adverse Reaction
Pramipexole Dihydrochloride Tablets
0.25 mg
(N=88)
%
Pramipexole Dihydrochloride Tablets
0.5 mg
(N=80)
%
Pramipexole Dihydrochloride Tablets
0.75 mg
(N=90)
%
Placebo
(N=86)
%
Gastrointestinal disorders
Nausea
11
19
27
5
Diarrhea
3
1
7
0
Dyspepsia
3
1
4
7
Psychiatric disorders
Insomnia
9
9
13
9
Abnormal dreams
2
1
8
2
General disorders and administration site conditions
Fatigue
3
5
7
5
Musculoskeletal and connective tissue disorders
Pain in extremity
3
3
7
1
Infections and infestations
Influenza
1
4
7
1
Respiratory, thoracic and mediastinal disorders
Nasal congestion
0
3
6
1
Adverse Reactions: Relationship to Age, Gender, and Race
Among the adverse reactions in patients treated with pramipexole dihydrochloride tablets, hallucination appeared to exhibit a positive relationship to age in patients with Parkinson's disease. Although no gender-related differences were observed in Parkinson's disease patients, nausea and fatigue, both generally transient, were more frequently reported by female than male RLS patients. Less than 4% of patients enrolled were non-Caucasian: therefore, an evaluation of adverse reactions related to race is not possible.
Laboratory Tests
During the development of pramipexole dihydrochloride tablets, no systematic abnormalities on routine laboratory testing were noted.
6.2 Post Marketing Experience
In addition to the adverse events reported during clinical trials, the following adverse reactions have been identified during post-approval use of pramipexole dihydrochloride tablets, primarily in Parkinson's disease patients. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure. Decisions to include these reactions in labeling are typically based on one or more of the following factors: (1) seriousness of the reaction, (2) frequency of reporting, or (3) strength of causal connection to pramipexole tablets.
Cardiac Disorders: cardiac failure
Gastrointestinal Disorders: vomiting
Metabolism and Nutrition Disorders: syndrome of inappropriate antidiuretic hormone secretion (SIADH), weight increase
Musculoskeletal and Connective Tissue Disorders: postural deformity [ see Warnings and Precautions (5.6)]
Nervous System Disorders: syncope
Skin and Subcutaneous Tissue Disorders: skin reactions (including erythema, rash, pruritus, urticaria)
- 7 DRUG INTERACTIONS
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
There are no adequate data on the developmental risk associated with the use of Pramipexole dihydrochloride tablets in pregnant women. No adverse developmental effects were observed in animal studies in which pramipexole was administered to rabbits during pregnancy. Effects on embryofetal development could not be adequately assessed in pregnant rats; however, postnatal growth was inhibited at clinically relevant exposures [see Data].
In the U.S. general population, the estimated background risk of major birth defects and of miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively. The background risk of major birth defects and miscarriage for the indicated population is unknown.
Data
Animal Data
Oral administration of pramipexole (0.1, 0.5, or 1.5 mg/kg/day) to pregnant rats during the period of organogenesis resulted in a high incidence of total resorption of embryos at the highest dose tested. This increase in embryolethality is thought to result from the prolactin-lowering effect of pramipexole; prolactin is necessary for implantation and maintenance of early pregnancy in rats but not rabbits or humans. Because of pregnancy disruption and early embryonic loss in this study, the teratogenic potential of pramipexole could not be adequately assessed in rats. The highest no-effect dose for embryolethality in rats was associated with maternal plasma drug exposures (AUC) approximately equal to those in humans receiving the maximum recommended human dose (MRHD) of 4.5 mg/day. There were no adverse effects on embryo-fetal development following oral administration of pramipexole (0.1, 1, and 10 mg/kg/day) to pregnant rabbits during organogenesis (plasma AUC up to approximately 70 times that in humans at the MRHD). Postnatal growth was inhibited in the offspring of rats treated with pramipexole (0.1, 0.5, or 1.5 mg/kg/day) during the latter part of pregnancy and throughout lactation. The no-effect dose for adverse effects on offspring growth (0.1 mg/kg/day) was associated with maternal plasma drug exposures lower than that in humans at the MRHD.
8.2 Lactation
There are no data on the presence of pramipexole in human milk, the effects of pramipexole on the breastfed infant, or the effects of pramipexole on milk production. However, inhibition of lactation is expected because pramipexole inhibits secretion of prolactin in humans. Pramipexole or metabolites, or both, are present in rat milk [see Data].
The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for pramipexole dihydrochloride and any potential adverse effects on the breastfed infant from pramipexole dihydrochloride or from the underlying maternal condition.
Data
In a study of radio-labeled pramipexole, pramipexole or metabolites, or both, were present in rat milk at concentrations three to six times higher than those in maternal plasma.
8.4 Pediatric Use
Safety and effectiveness of pramipexole dihydrochloride tablets in pediatric patients has not been established.
8.5 Geriatric Use
Pramipexole total oral clearance is approximately 30% lower in subjects older than 65 years compared with younger subjects, because of a decline in pramipexole renal clearance due to an age-related reduction in renal function. This resulted in an increase in elimination half-life from approximately 8.5 hours to 12 hours.
In clinical studies with Parkinson's disease patients, 38.7% of patients were older than 65 years. There were no apparent differences in efficacy or safety between older and younger patients, except that the relative risk of hallucination associated with the use of pramipexole dihydrochloride tablets were increased in the elderly.
In clinical studies with RLS patients, 22% of patients were at least 65 years old. There were no apparent differences in efficacy or safety between older and younger patients.
8.6 Renal Impairment
The elimination of pramipexole is dependent on renal function. Pramipexole clearance is extremely low in dialysis patients, as a negligible amount of pramipexole is removed by dialysis. Caution should be exercised when administering pramipexole dihydrochloride tablets to patients with renal disease [ see Dosage and Administration (2.2), Warnings and Precautions (5.7), and Clinical Pharmacology (12.3)].
-
10 OVERDOSAGE
There is no clinical experience with significant overdosage. One patient took 11 mg/day of pramipexole for 2 days in a clinical trial for an investigational use. Blood pressure remained stable although pulse rate increased to between 100 and 120 beats/minute. No other adverse reactions were reported related to the increased dose.
There is no known antidote for overdosage of a dopamine agonist. If signs of central nervous system stimulation are present, a phenothiazine or other butyrophenone neuroleptic agent may be indicated; the efficacy of such drugs in reversing the effects of overdosage has not been assessed. Management of overdose may require general supportive measures along with gastric lavage, intravenous fluids, and electrocardiogram monitoring.
-
11 DESCRIPTION
Pramipexole dihydrochloride tablets contain pramipexole, a nonergot dopamine agonist. The chemical name of pramipexole dihydrochloride is ( S)-2-amino-4,5,6,7-tetrahydro-6-(propylamino)benzothiazole dihydrochloride monohydrate. Its empirical formula is C 10 H 17 N 3 S · 2HCl · H 2O, and its molecular weight is 302.26.
The structural formula is:
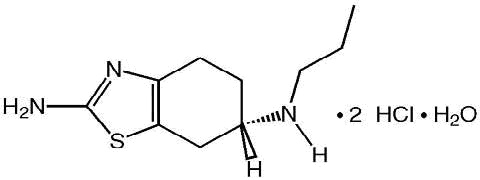
Pramipexole dihydrochloride monohydrate, USP is a white to off-white powder substance. Melting occurs in the range of 296°C to 301°C, with decomposition. Pramipexole dihydrochloride monohydrate, USP is more than 20% soluble in water, about 8% in methanol, about 0.5% in ethanol, and practically insoluble in dichloromethane.
Pramipexole dihydrochloride tablets, for oral administration, contain 0.125 mg, 0.25 mg, 0.5 mg, 0.75 mg, 1 mg, or 1.5 mg of pramipexole dihydrochloride monohydrate. Inactive ingredients consist of colloidal silicon dioxide, corn starch, ferric oxide red (0.25, 0.5 and 1 mg tablets), ferric oxide yellow (0.25 and 0.75 mg tablets), magnesium stearate, mannitol, povidone K-30 and pregelatinized maize starch.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Pramipexole is a non-ergot dopamine agonist with high relative in vitro specificity and full intrinsic activity at the D 2 subfamily of dopamine receptors, binding with higher affinity to D 3 than to D 2 or D 4 receptor subtypes.
Parkinson's Disease
The precise mechanism of action of pramipexole as a treatment for Parkinson's disease is unknown, although it is believed to be related to its ability to stimulate dopamine receptors in the striatum. This conclusion is supported by electrophysiologic studies in animals that have demonstrated that pramipexole influences striatal neuronal firing rates via activation of dopamine receptors in the striatum and the substantia nigra, the site of neurons that send projections to the striatum. The relevance of D 3 receptor binding in Parkinson's disease is unknown.
Restless Legs Syndrome (RLS)
The precise mechanism of action of pramipexole dihydrochloride tablets as a treatment for RLS is unknown. Although the pathophysiology of RLS is largely unknown, neuropharmacological evidence suggests primary dopaminergic system involvement. Positron Emission Tomographic (PET) studies suggest that a mild striatal presynaptic dopaminergic dysfunction may be involved in the pathogenesis of RLS.
12.2 Pharmacodynamics
The effect of pramipexole on the QT interval of the ECG was investigated in a clinical study in 60 healthy male and female volunteers. All subjects initiated treatment with 0.375 mg extended release pramipexole tablets administered once daily, and were up-titrated every 3 days to 2.25 mg and 4.5 mg daily, a faster rate of titration than recommended in the label. No dose- or exposure-related effect on mean QT intervals was observed; however, the study did not have a valid assessment of assay sensitivity. The effect of pramipexole on QTc intervals at higher exposures achieved either due to drug interactions (e.g., with cimetidine), renal impairment, or at higher doses has not been systematically evaluated.
Although mean values remained within normal reference ranges throughout the study, supine systolic blood pressure (SBP), diastolic blood pressure (DBP), and pulse rate for subjects treated with pramipexole generally increased during the rapid up-titration phase, by 10 mmHg, 7 mmHg, and 10 bpm higher than placebo, respectively. Higher SBP, DBP, and pulse rates compared to placebo were maintained until the pramipexole doses were tapered; values on the last day of tapering were generally similar to baseline values. Such effects have not been observed in clinical studies with Parkinson's disease patients, who were titrated according to labeled recommendations.
12.3 Pharmacokinetics
Pramipexole displays linear pharmacokinetics over the clinical dosage range. Its terminal half-life is about 8 hours in young healthy volunteers and about 12 hours in elderly volunteers. Steady-state concentrations are achieved within 2 days of dosing.
Absorption
Pramipexole is rapidly absorbed, reaching peak concentrations in approximately 2 hours. The absolute bioavailability of pramipexole is greater than 90%, indicating that it is well absorbed and undergoes little presystemic metabolism. Food does not affect the extent of pramipexole absorption, although the time of maximum plasma concentration (T max) is increased by about 1 hour when the drug is taken with a meal.
Distribution
Pramipexole is extensively distributed, having a volume of distribution of about 500 L (coefficient of variation [CV]=20%). It is about 15% bound to plasma proteins. Pramipexole distributes into red blood cells as indicated by an erythrocyte-to-plasma ratio of approximately 2.
Metabolism
Pramipexole is metabolized only to a negligible extent (<10%). No specific active metabolite has been identified in human plasma or urine.
Elimination
Urinary excretion is the major route of pramipexole elimination, with 90% of a pramipexole dose recovered in urine, almost all as unchanged drug. The renal clearance of pramipexole is approximately 400 mL/min (CV=25%), approximately three times higher than the glomerular filtration rate. Thus, pramipexole is secreted by the renal tubules, probably by the organic cation transport system.
Pharmacokinetics in Specific Populations
Because therapy with pramipexole dihydrochloride tablets are initiated at a low dose and gradually titrated upward according to clinical tolerability to obtain the optimum therapeutic effect, adjustment of the initial dose based on gender, weight, race, or age is not necessary. However, renal insufficiency, which can cause a large decrease in the ability to eliminate pramipexole, may necessitate dosage adjustment [ see Dosage and Administration (2.2)].
Gender
Pramipexole clearance is about 30% lower in women than in men, but this difference can be accounted for by differences in body weight. There is no difference in half-life between males and females.
Age
Pramipexole clearance decreases with age as the half-life and clearance are about 40% longer and 30% lower, respectively, in elderly (aged 65 years or older) compared with young healthy volunteers (aged less than 40 years). This difference is most likely due to the reduction in renal function with age, since pramipexole clearance is correlated with renal function, as measured by creatinine clearance.
Race
No racial differences in metabolism and elimination have been identified.
Parkinson's Disease Patients
A cross-study comparison of data suggests that the clearance of pramipexole may be reduced by about 30% in Parkinson's disease patients compared with healthy elderly volunteers. The reason for this difference appears to be reduced renal function in Parkinson's disease patients, which may be related to their poorer general health. The pharmacokinetics of pramipexole were comparable between early and advanced Parkinson's disease patients.
Restless Legs Syndrome Patients
A cross-study comparison of data suggests that the pharmacokinetic profile of pramipexole administered once daily in RLS patients is similar to the pharmacokinetic profile of pramipexole in healthy volunteers.
Hepatic Impairment
The influence of hepatic insufficiency on pramipexole pharmacokinetics has not been evaluated. Because approximately 90% of the recovered dose is excreted in the urine as unchanged drug, hepatic impairment would not be expected to have a significant effect on pramipexole elimination.
Renal Impairment
Clearance of pramipexole was about 75% lower in patients with severe renal impairment (creatinine clearance approximately 20 mL/min) and about 60% lower in patients with moderate impairment (creatinine clearance approximately 40 mL/min) compared with healthy volunteers [ see Warnings and Precautions (5.7) and Dosage and Administration (2.2)]. In patients with varying degrees of renal impairment, pramipexole clearance correlates well with creatinine clearance. Therefore, creatinine clearance can be used as a predictor of the extent of decrease in pramipexole clearance.
Drug Interactions
Carbidopa/levodopa: Carbidopa/levodopa did not influence the pharmacokinetics of pramipexole in healthy volunteers (N=10). Pramipexole did not alter the extent of absorption (AUC) or the elimination of carbidopa/levodopa, although it caused an increase in levodopa C max by about 40% and a decrease in T max from 2.5 to 0.5 hours.
Selegiline: In healthy volunteers (N=11), selegiline did not influence the pharmacokinetics of pramipexole.
Amantadine: Population pharmacokinetic analyses suggest that amantadine may slightly decrease the oral clearance of pramipexole.
Cimetidine: Cimetidine, a known inhibitor of renal tubular secretion of organic bases via the cationic transport system, caused a 50% increase in pramipexole AUC and a 40% increase in half-life (N=12).
Probenecid: Probenecid, a known inhibitor of renal tubular secretion of organic acids via the anionic transporter, did not noticeably influence pramipexole pharmacokinetics (N=12).
Other drugs eliminated via renal secretion: Population pharmacokinetic analysis suggests that coadministration of drugs that are secreted by the cationic transport system (e.g., cimetidine, ranitidine, diltiazem, triamterene, verapamil, quinidine, and quinine) decreases the oral clearance of pramipexole by about 20%, while those secreted by the anionic transport system (e.g., cephalosporins, penicillins, indomethacin, hydrochlorothiazide, and chlorpropamide) are likely to have little effect on the oral clearance of pramipexole. Other known organic cation transport substrates and/or inhibitors (e.g., cisplatin and procainamide) may also decrease the clearance of pramipexole.
CYP interactions: Inhibitors of cytochrome P450 enzymes would not be expected to affect pramipexole elimination because pramipexole is not appreciably metabolized by these enzymes in vivo or in vitro. Pramipexole does not inhibit CYP enzymes CYP1A2, CYP2C9, CYP2C19, CYP2E1, and CYP3A4. Inhibition of CYP2D6 was observed with an apparent Ki of 30 μM, indicating that pramipexole will not inhibit CYP enzymes at plasma concentrations observed following the clinical dose of 4.5 mg/day (1.5 mg TID).
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Two-year carcinogenicity studies with pramipexole have been conducted in mice and rats. Pramipexole was administered in the diet to mice at doses up to 10 mg/kg/day (or approximately 10 times the maximum recommended human dose (MRHD) for Parkinson's disease of 4.5 mg/day on a mg/m 2 basis). Pramipexole was administered in the diet to rats at doses up to 8 mg/kg/day. These doses were associated with plasma AUCs up to approximately 12 times that in humans at the MRHD. No significant increases in tumors occurred in either species.
Pramipexole was not mutagenic or clastogenic in a battery of in vitro (bacterial reverse mutation, V79/HGPRT gene mutation, chromosomal aberration in CHO cells) and in vivo (mouse micronucleus) assays.
In rat fertility studies, pramipexole at a dose of 2.5 mg/kg/day (5 times the MRHD on a mg/m 2basis), prolonged estrus cycles and inhibited implantation. These effects were associated with reductions in serum levels of prolactin, a hormone necessary for implantation and maintenance of early pregnancy in rats.
13.2 Animal Toxicology and/or Pharmacology
Pathologic changes (degeneration and loss of photoreceptor cells) were observed in the retina of albino rats in the 2-year carcinogenicity study with pramipexole. These findings were first observed during week 76 and were dose-dependent in animals receiving 2 or 8 mg/kg/day (plasma AUCs equal to 2.5 and 12.5 times that in humans at the MRHD). In a similar study of pigmented rats with 2 years exposure to pramipexole at 2 or 8 mg/kg/day, retinal degeneration was not observed. Animals given drug had thinning in the outer nuclear layer of the retina that was only slightly greater (by morphometric analysis) than that seen in control rats.
Investigative studies demonstrated that pramipexole reduced the rate of disk shedding from the photoreceptor rod cells of the retina in albino rats, which was associated with enhanced sensitivity to the damaging effects of light. In a comparative study, degeneration and loss of photoreceptor cells occurred in albino rats after 13 weeks of treatment with 25 mg/kg/day of pramipexole (54 times the MRHD on a mg/m 2basis) and constant light (100 lux) but not in pigmented rats exposed to the same dose and higher light intensities (500 lux). Thus, the retina of albino rats is considered to be uniquely sensitive to the damaging effects of pramipexole and light. Similar changes in the retina did not occur in a 2-year carcinogenicity study in albino mice treated with 0.3, 2, or 10 mg/kg/day (0.3, 2.2 and 11 times the MRHD on a mg/m 2basis). Evaluation of the retinas of monkeys given 0.1, 0.5, or 2.0 mg/kg/day of pramipexole (0.4, 2.2, and 8.6 times the MRHD on a mg/m 2basis) for 12 months and minipigs given 0.3, 1, or 5 mg/kg/day of pramipexole for 13 weeks also detected no changes.
The potential significance of this effect in humans has not been established, but cannot be disregarded because disruption of a mechanism that is universally present in vertebrates (i.e., disk shedding) may be involved.
Fibro-osseous Proliferative Lesions in Mice
An increased incidence of fibro-osseous proliferative lesions occurred in the femurs of female mice treated for 2 years with 0.3, 2.0, or 10 mg/kg/day (0.3, 2.2, and 11 times the MRHD on a mg/m 2basis). Similar lesions were not observed in male mice or rats and monkeys of either sex that were treated chronically with pramipexole. The significance of this lesion to humans is not known.
-
14 CLINICAL STUDIES
14.1 Parkinson’s Disease
The effectiveness of pramipexole dihydrochloride tablets in the treatment of Parkinson's disease was evaluated in a multinational drug development program consisting of seven randomized, controlled trials. Three were conducted in patients with early Parkinson's disease who were not receiving concomitant levodopa, and four were conducted in patients with advanced Parkinson's disease who were receiving concomitant levodopa. Among these seven studies, three studies provide the most persuasive evidence of pramipexole's effectiveness in the management of patients with Parkinson's disease who were and were not receiving concomitant levodopa. Two of these three trials enrolled patients with early Parkinson's disease (not receiving levodopa), and one enrolled patients with advanced Parkinson's disease who were receiving maximally tolerated doses of levodopa.
In all studies, the Unified Parkinson's Disease Rating Scale (UPDRS), or one or more of its subparts, served as the primary outcome assessment measure. The UPDRS is a four-part multi-item rating scale intended to evaluate mentation (part I), Activities of Daily Living (ADL) (part II), motor performance (part III), and complications of therapy (part IV).
Part II of the UPDRS contains 13 questions relating to ADL, which are scored from 0 (normal) to 4 (maximal severity) for a maximum (worst) score of 52. Part III of the UPDRS contains 27 questions (for 14 items) and is scored as described for part II. It is designed to assess the severity of the cardinal motor findings in patients with Parkinson's disease (e.g., tremor, rigidity, bradykinesia, postural instability, etc.), scored for different body regions, and has a maximum (worst) score of 108.
Studies in Patients with Early Parkinson's Disease
Patients (N=599) in the two studies of early Parkinson's disease had a mean disease duration of 2 years, limited or no prior exposure to levodopa (generally none in the preceding 6 months), and were not experiencing the "on-off" phenomenon and dyskinesia characteristic of later stages of the disease.
One of the two early Parkinson's disease studies (N=335) was a double-blind, placebo-controlled, parallel trial consisting of a 7-week dose-escalation period and a 6-month maintenance period. Patients could be on selegiline, anticholinergics, or both, but could not be on levodopa products or amantadine. Patients were randomized to pramipexole dihydrochloride tablets or placebo. Patients treated with pramipexole dihydrochloride tablets had a starting daily dose of 0.375 mg and were titrated to a maximally tolerated dose, but no higher than 4.5 mg/day in three divided doses. At the end of the 6-month maintenance period, the mean improvement from baseline on the UPDRS part II (ADL) total score was 1.9 in the group receiving pramipexole dihydrochloride tablets and -0.4 in the placebo group, a difference that was statistically significant. The mean improvement from baseline on the UPDRS part III total score was 5.0 in the group receiving pramipexole dihydrochloride tablets and -0.8 in the placebo group, a difference that was also statistically significant. A statistically significant difference between groups in favor of pramipexole dihydrochloride tablets were seen beginning at week 2 of the UPDRS part II (maximum dose 0.75 mg/day) and at week 3 of the UPDRS part III (maximum dose 1.5 mg/day).
The second early Parkinson's disease study (N=264) was a double-blind, placebo-controlled, parallel trial consisting of a 6-week dose-escalation period and a 4-week maintenance period. Patients could be on selegiline, anticholinergics, amantadine, or any combination of these, but could not be on levodopa products. Patients were randomized to 1 of 4 fixed doses of pramipexole dihydrochloride tablets (1.5 mg, 3.0 mg, 4.5 mg, or 6.0 mg per day) or placebo. At the end of the 4-week maintenance period, the mean improvement from baseline on the UPDRS part II total score was 1.8 in the patients treated with pramipexole dihydrochloride tablets, regardless of assigned dose group, and 0.3 in placebo-treated patients. The mean improvement from baseline on the UPDRS part III total score was 4.2 in patients treated with pramipexole dihydrochloride tablets and 0.6 in placebo-treated patients. No dose-response relationship was demonstrated. The between-treatment differences on both parts of the UPDRS were statistically significant in favor of pramipexole dihydrochloride tablets for all doses.
No differences in effectiveness based on age or gender were detected. There were too few non-Caucasian patients to evaluate the effect of race. Patients receiving selegiline or anticholinergics had responses similar to patients not receiving these drugs.
Studies in Patients with Advanced Parkinson's Disease
In the advanced Parkinson's disease study, the primary assessments were the UPDRS and daily diaries that quantified amounts of "on" and "off" time.
Patients in the advanced Parkinson's disease study (N=360) had a mean disease duration of 9 years, had been exposed to levodopa for long periods of time (mean 8 years), used concomitant levodopa during the trial, and had "on-off" periods.
The advanced Parkinson's disease study was a double-blind, placebo-controlled, parallel trial consisting of a 7-week dose-escalation period and a 6-month maintenance period. Patients were all treated with concomitant levodopa products and could additionally be on concomitant selegiline, anticholinergics, amantadine, or any combination. Patients treated with pramipexole dihydrochloride tablets had a starting dose of 0.375 mg/day and were titrated to a maximally tolerated dose, but no higher than 4.5 mg/day in three divided doses. At selected times during the 6-month maintenance period, patients were asked to record the amount of "off," "on," or "on with dyskinesia" time per day for several sequential days. At the end of the 6-month maintenance period, the mean improvement from baseline on the UPDRS part II total score was 2.7 in the group treated with pramipexole dihydrochloride tablets and 0.5 in the placebo group, a difference that was statistically significant. The mean improvement from baseline on the UPDRS part III total score was 5.6 in the group treated with pramipexole dihydrochloride tablets and 2.8 in the placebo group, a difference that was statistically significant. A statistically significant difference between groups in favor of pramipexole dihydrochloride tablets were seen at week 3 of the UPDRS part II (maximum dose 1.5 mg/day) and at week 2 of the UPDRS part III (maximum dose 0.75 mg/day). Dosage reduction of levodopa was allowed during this study if dyskinesia (or hallucinations) developed; levodopa dosage reduction occurred in 76% of patients treated with pramipexole dihydrochloride tablets versus 54% of placebo patients. On average, the levodopa dose was reduced 27%.
The mean number of "off" hours per day during baseline was 6 hours for both treatment groups. Throughout the trial, patients treated with pramipexole dihydrochloride tablets had a mean of 4 "off" hours per day, while placebo-treated patients continued to experience 6 "off" hours per day.
No differences in effectiveness based on age or gender were detected. There were too few non-Caucasian patients to evaluate the effect of race.
14.2 Restless Legs Syndrome
The efficacy of pramipexole dihydrochloride tablets in the treatment of RLS was evaluated in a multinational drug development program consisting of 4 randomized, double-blind, placebo-controlled trials. This program included approximately 1000 patients with moderate to severe RLS; patients with RLS secondary to other conditions (e.g., pregnancy, renal failure, and anemia) were excluded. All patients were administered pramipexole dihydrochloride tablets (0.125 mg, 0.25 mg, 0.5 mg, or 0.75 mg) or placebo once daily 2 to 3 hours before going to bed. Across the 4 studies, the mean duration of RLS was 4.6 years (range of 0 to 56 years), mean age was approximately 55 years (range of 18 to 81 years), and approximately 66.6% were women.
Key diagnostic criteria for RLS are: an urge to move the legs usually accompanied or caused by uncomfortable and unpleasant leg sensations; symptoms begin or worsen during periods of rest or inactivity such as lying or sitting; symptoms are partially or totally relieved by movement such as walking or stretching at least as long as the activity continues; and symptoms are worse or occur only in the evening or night. Difficulty falling asleep may frequently be associated with symptoms of RLS.
The two outcome measures used to assess the effect of treatment were the International RLS Rating Scale (IRLS Scale) and a Clinical Global Impression - Improvement (CGI-I) assessment. The IRLS Scale contains 10 items designed to assess the severity of sensory and motor symptoms, sleep disturbance, daytime somnolence, and impact on activities of daily living and mood associated with RLS. The range of scores is 0 to 40, with 0 being absence of RLS symptoms and 40 the most severe symptoms. The CGI-I is designed to assess clinical progress (global improvement) on a 7-point scale.
In Study 1, fixed doses of pramipexole dihydrochloride tablets were compared to placebo in a study of 12 weeks duration. A total of 344 patients were randomized equally to the 4 treatment groups. Patients treated with pramipexole dihydrochloride tablets (n=254) had a starting dose of 0.125 mg/day and were titrated to one of the three randomized doses (0.25, 0.5, 0.75 mg/day) in the first three weeks of the study. The mean improvement from baseline on the IRLS Scale total score and the percentage of CGI-I responders for each of the pramipexole dihydrochloride tablets treatment groups compared to placebo are summarized in Table 8. All treatment groups reached statistically significant superiority compared to placebo for both endpoints. There was no clear evidence of a dose-response across the 3 randomized dose groups.
Table 8 Mean Changes from Baseline to Week 12 in IRLS Score and CGI-I (Study 1) *CGI-I responders = "much improved" and "very much improved"
Pramipexole Dihydrochloride Tablets
0.25 mg
Pramipexole Dihydrochloride Tablets
0.5 mg
Pramipexole Dihydrochloride Tablets
0.75 mg
Pramipexole Dihydrochloride Tablets
Total
Placebo
No. Patients
88
79
87
254
85
IRLS score
-13.1
-13.4
-14.4
-13.6
-9.4
CGI-I responders*
74.7%
67.9%
72.9%
72.0%
51.2%
Study 2 was a randomized-withdrawal study, designed to demonstrate the sustained efficacy of pramipexole for treatment of RLS after a period of six months. RLS patients who responded to pramipexole dihydrochloride tablets treatment in a preceding 6-month open-label treatment phase (defined as having a CGI-I rating of "very much improved" or "much improved" compared to baseline and an IRLS score of 15 or below) were randomized to receive either continued active treatment (n=78) or placebo (n=69) for 12 weeks. The primary endpoint of this study was time to treatment failure, defined as any worsening on the CGI-I score along with an IRLS Scale total score above 15.
In patients who had responded to 6-month open label treatment with pramipexole dihydrochloride tablets, the administration of placebo led to a rapid decline in their overall conditions and return of their RLS symptoms. At the end of the 12-week observation period, 85% of patients treated with placebo had failed treatment, compared to 21% treated with blinded pramipexole, a difference that was highly statistically significant. The majority of treatment failures occurred within 10 days of randomization. For the patients randomized, the distribution of doses was: 7 on 0.125 mg, 44 on 0.25 mg, 47 on 0.5 mg, and 49 on 0.75 mg.
Study 3 was a 6-week study, comparing a flexible dose of pramipexole dihydrochloride tablets to placebo. In this study, 345 patients were randomized in a 2:1 ratio to pramipexole dihydrochloride tablets or placebo. The mean improvement from baseline on the IRLS Scale total score was -12 for pramipexole dihydrochloride tablet-treated patients and -6 for placebo-treated patients. The percentage of CGI-I responders was 63% for pramipexole dihydrochloride tablet-treated patients and 32% for placebo-treated patients. The between-group differences were statistically significant for both outcome measures. For the patients randomized to Pramipexole dihydrochloride tablets, the distribution of achieved doses was: 35 on 0.125 mg, 51 on 0.25 mg, 65 on 0.5 mg, and 69 on 0.75 mg.
Study 4 was a 3-week study, comparing 4 fixed doses of pramipexole dihydrochloride tablets, 0.125 mg, 0.25 mg, 0.5 mg, and 0.75 mg, to placebo. Approximately 20 patients were randomized to each of the 5 dose groups. The mean improvement from baseline on the IRLS Scale total score and the percentage of CGI-I responders for each of the pramipexole dihydrochloride tablets treatment groups compared to placebo are summarized in Table 9. In this study, the 0.125 mg dose group was not significantly different from placebo. On average, the 0.5 mg dose group performed better than the 0.25 mg dose group, but there was no difference between the 0.5 mg and 0.75 mg dose groups.
Table 9 Mean Changes from Baseline to Week 3 in IRLS Score and CGI-I (Study 4) *CGI-I responders = "much improved" and "very much improved"
Pramipexole Dihydrochloride Tablets
0.125 mg
Pramipexole Dihydrochloride Tablets
0.25 mg
Pramipexole Dihydrochloride Tablets
0.5 mg
Pramipexole Dihydrochloride Tablets
0.75 mg
Pramipexole Dihydrochloride Tablets
Total
Placebo
No. Patients
21
22
22
21
86
21
IRLS score
-11.7
-15.3
-17.6
-15.2
-15.0
-6.2
CGI-I responders*
61.9%
68.2%
86.4%
85.7%
75.6%
42.9%
No differences in effectiveness based on age or gender were detected. There were too few non-Caucasian patients to evaluate the effect of race.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
Pramipexole Dihydrochloride Tablets are available as follows:
Pramipexole Dihydrochloride Tablets 0.125 mg are white to off white, round, flat, bevel edged, uncoated tablets with "91" debossed on one side and plain on other side.
Bottles of 30 NDC: 13668-091-30
Bottles of 90 NDC: 13668-091-90
Bottles of 500 NDC: 13668-091-05
Bottles of 6000 NDC: 13668-091-42
100 Unit Dose Tablets NDC: 13668-091-74
Pramipexole Dihydrochloride Tablets 0.25 mg are peach colored, round, flat, bevel edged, uncoated tablets with "9/2" debossed on one side and breakline on other side.
Bottles of 30 NDC: 13668-092-30
Bottles of 90 NDC: 13668-092-90
Bottles of 500 NDC: 13668-092-05
Bottles of 5500 NDC: 13668-092-41
100 Unit Dose Tablets NDC: 13668-092-74
Pramipexole Dihydrochloride Tablets 0.5 mg are reddish brown colored, round, biconvex, uncoated tablets with "9/3" debossed on one side and breakline on other side.
Bottles of 30 NDC: 13668-093-30
Bottles of 90 NDC: 13668-093-90
Bottles of 500 NDC: 13668-093-05
Bottles of 4000 NDC: 13668-093-40
100 Unit Dose Tablets NDC: 13668-093-74
Pramipexole Dihydrochloride Tablets 0.75 mg are yellow colored, round, flat, bevel edged, uncoated tablets with "84" debossed on one side and plain on other side.
Bottles of 30 NDC: 13668-184-30
Bottles of 90 NDC: 13668-184-90
Bottles of 500 NDC: 13668-184-05
Bottles of 2500 NDC: 13668-184-31
100 Unit Dose Tablets NDC: 13668-184-74
Pramipexole Dihydrochloride Tablets 1 mg are light pink colored, round, flat, bevel edged, uncoated tablets with "9/4" debossed on one side and breakline on other side.
Bottles of 30 NDC: 13668-094-30
Bottles of 90 NDC: 13668-094-90
Bottles of 500 NDC: 13668-094-05
Bottles of 2500 NDC: 13668-094-31
100 Unit Dose Tablets NDC: 13668-094-74
Pramipexole Dihydrochloride Tablets 1.5 mg are white to off white, round, flat, bevel edged, uncoated tablets with "9/5" debossed on one side and breakline on other side.
Bottles of 30 NDC: 13668-095-30
Bottles of 90 NDC: 13668-095-90
Bottles of 500 NDC: 13668-095-05
Bottles of 1500 NDC: 13668-095-15
100 Unit Dose Tablets NDC: 13668-095-74
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Dosing Instructions
Instruct patients to take pramipexole dihydrochloride tablets only as prescribed. If a dose is missed, advise patients not to double their next dose.
Pramipexole dihydrochloride tablets can be taken with or without food. If patients develop nausea, advised that taking pramipexole dihydrochloride tablets with food may reduce the occurrence of nausea.
Pramipexole is the active ingredient that is in both pramipexole dihydrochloride tablets and extended-release pramipexole tablets. Ensure that patients do not take both extended-release pramipexole and pramipexole dihydrochloride tablets.
Sedating Effects
Alert patients to the potential sedating effects associated with pramipexole dihydrochloride tablets, including somnolence and the possibility of falling asleep while engaged in activities of daily living. Since somnolence is a frequent adverse reaction with potentially serious consequences, patients should neither drive a car nor engage in other potentially dangerous activities until they have gained sufficient experience with pramipexole dihydrochloride tablets to gauge whether or not it affects their mental and/or motor performance adversely. Advise patients that if increased somnolence or new episodes of falling asleep during activities of daily living (e.g., conversations or eating) are experienced at any time during treatment, they should not drive or participate in potentially dangerous activities until they have contacted their physician. Because of possible additive effects, advise caution when patients are taking other sedating medications or alcohol in combination with pramipexole dihydrochloride tablets and when taking concomitant medications that increase plasma levels of pramipexole (e.g., cimetidine) [ see Warnings and Precautions (5.1)].
Impulse Control Symptoms Including Compulsive Behaviors
Alert patients and their caregivers to the possibility that they may experience intense urges to spend money uncontrollably, intense urges to gamble, increased sexual urges, binge eating and/or other intense urges and the inability to control these urges while taking pramipexole dihydrochloride tablets. [ see Warnings and Precautions (5.3)].
Hallucinations and Psychotic-like Behavior
Inform patients that hallucinations and other psychotic-like behavior can occur and that the elderly are at a higher risk than younger patients with Parkinson's disease [ see Warnings and Precautions (5.4)].
Postural (Orthostatic) Hypotension
Advise patients that they may develop postural (orthostatic) hypotension, with or without symptoms such as dizziness, nausea, fainting or blackouts, and sometimes, sweating. Hypotension may occur more frequently during initial therapy. Accordingly, caution patients against rising rapidly after sitting or lying down, especially if they have been doing so for prolonged periods and especially at the initiation of treatment with pramipexole dihydrochloride tablets [ see Warnings and Precautions (5.2)].
Pregnancy
Because the teratogenic potential of pramipexole has not been completely established in laboratory animals, and because experience in humans is limited, advise women to notify their physicians if they become pregnant or intend to become pregnant during therapy [ see Use in Specific Populations (8.1)].
Lactation
Because of the possibility that pramipexole may be excreted in breast milk, advise women to notify their physicians if they intend to breast-feed or are breast-feeding an infant [ see Use in Specific Populations (8.2)].

TORRENT PHARMACEUTICALS LTD., Indrad-382 721, Dist. Mehsana, INDIA.
Or
TORRENT PHARMACEUTICALS LTD., Bharuch-392130, INDIA.
For:
TORRENT PHARMA INC., 150 Allen Road, Suite 102, Basking Ridge, NJ 07920.
8062604 Revised May 2018
Pramipexole Dihydrochloride (PRAM-i-PEX-ole dye-HYE-droe-KLOR-ide) Tablets
Read this Patient Information before you start taking pramipexole dihydrochloride tablets and each time you get a refill. There may be new information. This information does not take the place of talking with your doctor about your medical condition or your treatment.
What are pramipexole dihydrochloride tablets?
Pramipexole dihydrochloride tablets are prescription medicine used to treat:
- signs and symptoms of Parkinson's disease (PD)
- moderate to severe primary Restless Legs Syndrome (RLS)
It is not known if pramipexole dihydrochloride tablets are safe and effective in children.
What should I tell my doctor before taking pramipexole dihydrochloride tablets?
- Before taking pramipexole dihydrochloride tablets, tell your doctor if you:
feel sleepy during the day from a sleep problem other than Restless Legs Syndrome.
have low blood pressure, or if you feel dizzy or faint, especially when getting up from sitting or lying down
have trouble controlling your muscles (dyskinesia)
have kidney problems
drink alcohol. Alcohol can increase the chance that pramipexole dihydrochloride tablets will make you feel sleepy or fall asleep when you should be awake.
have any other medical conditions
are pregnant or plan to become pregnant. It is not known if pramipexole dihydrochloride tablets will harm your unborn baby.
are breastfeeding or plan to breastfeed. It is not known if pramipexole dihydrochloride tablets passes into your breast milk. You and your doctor should decide if you will take pramipexole dihydrochloride tablets or breastfeed. You should not do both.
Tell your doctor about all the medicines you take, including prescription and nonprescription medicines, vitamins, and herbal supplements.
The combination of pramipexole dihydrochloride tablets and other medicines may affect each other and may cause side effects. Pramipexole dihydrochloride tablets may affect the way other medicines work, and other medicines may affect how pramipexole dihydrochloride tablets work.
Especially tell your doctor if you take:
- medicines called neuroleptics (phenothiazines, butyrophenones, thioxanthenes) or metoclopramide. Pramipexole dihydrochloride tablets may not work as well if you take these medicines.
- extended-release pramipexole (MIRAPEX ER). Pramipexole is the active ingredient in both pramipexole dihydrochloride tablets and extended-release pramipexole tablets (MIRAPEX ER). If you are taking extended-release pramipexole tablets (MIRAPEX ER), you should not take pramipexole dihydrochloride tablets.
- any other medicines that make you sleepy or may increase the effects of pramipexole dihydrochloride tablets, such as cimetidine (Tagamet).
Ask your doctor for a list of these medicines if you are not sure.
Know the medicines you take. Keep a list of them and show it to your doctor and pharmacist when you get a new medicine.
How should I take pramipexole dihydrochloride tablets?
- Take pramipexole dihydrochloride tablets exactly as your doctor tells you to take it.
- Your doctor will tell you how much pramipexole dihydrochloride tablets to take and when to take it. Do not take more or less pramipexole dihydrochloride tablets than your doctor tells you to.
- Your doctor may change your dose if needed.
- Pramipexole dihydrochloride tablets can be taken with or without food. Taking pramipexole dihydrochloride tablets with food may lower your chances of getting nausea.
- If you take more pramipexole dihydrochloride tablets than your doctor recommends, call your doctor or go to the nearest hospital emergency room right away.
- If you miss a dose, do not double your next dose . Skip the dose you missed and take your next regular dose.
- If you have Parkinson's disease and your doctor tells you to stop taking pramipexole dihydrochloride tablets, you should stop pramipexole dihydrochloride tablets slowly as directed by your doctor. If you stop pramipexole dihydrochloride tablets too quickly you may have withdrawal symptoms such as:
confusion
severe muscle stiffness
Do not stop taking pramipexole dihydrochloride tablets without talking to your doctor.
What should I avoid while taking pramipexole dihydrochloride tablets?
- Do not drink alcohol while taking pramipexole dihydrochloride tablets. It can increase your chance of having serious side effects. See "What are the possible side effects of pramipexole dihydrochloride tablets?"
- Do not drive a car, operate a machine, or do other dangerous activities until you know how pramipexole dihydrochloride tablets affects you. Sleepiness caused by pramipexole dihydrochloride tablets can happen as late as 1 year after you start your treatment.
What are the possible side effects of pramipexole dihydrochloride tablets?
Pramipexole dihydrochloride tablets may cause serious side effects, including:
falling asleep during normal daily activities. Pramipexole dihydrochloride tablets may cause you to fall asleep while you are doing daily activities such as driving, talking with other people, or eating.
Some people taking the medicine in pramipexole dihydrochloride tablets have had car accidents because they fell asleep while driving.
Some patients did not feel sleepy before they fell asleep while driving. You could fall asleep without any warning.
Tell your doctor right away if you fall asleep while you are doing activities such as talking, eating, driving, or if you feel sleepier than normal for you.
low blood pressure when you sit or stand up quickly. You may have:
dizziness
nausea
fainting
sweating
Sit and stand up slowly after you have been sitting or lying down.
unusual urges. Some people who take certain medicines to treat Parkinson's disease, including pramipexole dihydrochloride tablets, have reported problems, such as gambling, compulsive eating, compulsive buying, and increased sex drive.
If you or your family members notice that you are developing unusual urges or behaviors, talk to your doctor.
hallucinations and other psychotic-like behavior (seeing visions, hearing sounds or feeling sensations that are not real, confusion, excessive suspicion, aggressive behavior, agitation, delusional beliefs and disorganized thinking). Your chance of having hallucinations and other psychotic-like behavior is higher if you are elderly (age 65 or older).
If you have hallucinations or other psychotic-like changes, talk with your doctor right away.
uncontrolled sudden movements (dyskinesia) .
If you have new dyskinesia or your existing dyskinesia gets worse tell your doctor.
posture changes. Talk with your doctor if you have posture changes you cannot control. These may include your neck bending forward, bending forward at the waist, or tilting sideways when you sit, stand, or walk.
The most common side effects in people taking pramipexole dihydrochloride tablet for Restless Legs Syndrome are nausea and headache.
The most common side effects in people taking pramipexole dihydrochloride tablets for Parkinson's disease are:
- nausea
- dizziness
- insomnia
- constipation
- muscle weakness
- abnormal dreams
- confusion
- memory problems(amnesia)
- urinating more often than normal
These are not all the possible side effects of pramipexole dihydrochloride tablets. Tell your doctor if you have any side effect that bothers you.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store pramipexole dihydrochloride tablets?
- Store pramipexole dihydrochloride tablets at 20°-25°C (68°-77°F); excursions permitted to 15°-30°C (59°-86°F) [see USP Controlled Room Temperature].
- Keep pramipexole dihydrochloride tablets out of the light.
- Keep pramipexole dihydrochloride tablets and all medicines out of the reach of children.
General Information about the safe and effective use of pramipexole dihydrochloride tablets.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use pramipexole dihydrochloride tablets for a condition for which it was not prescribed. Do not give pramipexole dihydrochloride tablets to other people, even if they have the same symptoms that you have. It may harm them.
This Patient Information leaflet summarizes the most important information about pramipexole dihydrochloride tablets. If you would like more information, talk with your doctor. You can ask your pharmacist or doctor for information about pramipexole dihydrochloride tablets that is written for healthcare professionals.
For additional information, you may also call at 1-800-912-9561.
What are the ingredients in pramipexole dihydrochloride tablets?
Active Ingredient: pramipexole dihydrochloride monohydrate, USP
Inactive Ingredients: colloidal silicon dioxide, corn starch, ferric oxide red (0.25, 0.5 and 1 mg tablets), ferric oxide yellow (0.25 and 0.75 mg tablets), magnesium stearate, mannitol, povidone K-30 and pregelatinized maize starch.
This Patient Information has been approved by the U.S. Food and Drug Administration.
Trademarks are the property of their respective owners.

TORRENT PHARMACEUTICALS LTD., Indrad-382 721, Dist. Mehsana, INDIA.
Or
TORRENT PHARMACEUTICALS LTD., Bharuch-392130, INDIA.
For:
TORRENT PHARMA INC., 150 Allen Road, Suite 102, Basking Ridge, NJ 07920.
8062604 Revised May 2018
- PACKAGE LABEL.PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
PRAMIPEXOLE DIHYDROCHLORIDE
pramipexole dihydrochloride tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 13668-091 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength PRAMIPEXOLE DIHYDROCHLORIDE (UNII: 3D867NP06J) (PRAMIPEXOLE - UNII:83619PEU5T) PRAMIPEXOLE DIHYDROCHLORIDE 0.125 mg Inactive Ingredients Ingredient Name Strength MAGNESIUM STEARATE (UNII: 70097M6I30) MANNITOL (UNII: 3OWL53L36A) POVIDONE K30 (UNII: U725QWY32X) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) STARCH, PREGELATINIZED CORN (UNII: O8232NY3SJ) Product Characteristics Color white (white to off white) Score no score Shape ROUND Size 5mm Flavor Imprint Code 91 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 13668-091-42 6000 in 1 BOTTLE; Type 0: Not a Combination Product 10/08/2010 2 NDC: 13668-091-05 500 in 1 BOTTLE; Type 0: Not a Combination Product 10/08/2010 3 NDC: 13668-091-90 90 in 1 BOTTLE; Type 0: Not a Combination Product 10/08/2010 4 NDC: 13668-091-30 30 in 1 BOTTLE; Type 0: Not a Combination Product 10/08/2010 5 NDC: 13668-091-74 100 in 1 CARTON; Type 0: Not a Combination Product 10/08/2010 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA090865 10/08/2010 PRAMIPEXOLE DIHYDROCHLORIDE
pramipexole dihydrochloride tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 13668-092 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength PRAMIPEXOLE DIHYDROCHLORIDE (UNII: 3D867NP06J) (PRAMIPEXOLE - UNII:83619PEU5T) PRAMIPEXOLE DIHYDROCHLORIDE 0.25 mg Inactive Ingredients Ingredient Name Strength FERRIC OXIDE RED (UNII: 1K09F3G675) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) MAGNESIUM STEARATE (UNII: 70097M6I30) MANNITOL (UNII: 3OWL53L36A) POVIDONE K30 (UNII: U725QWY32X) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) STARCH, PREGELATINIZED CORN (UNII: O8232NY3SJ) Product Characteristics Color yellow (peach) Score 2 pieces Shape ROUND Size 6mm Flavor Imprint Code 9;2 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 13668-092-41 5500 in 1 BOTTLE; Type 0: Not a Combination Product 10/08/2010 2 NDC: 13668-092-05 500 in 1 BOTTLE; Type 0: Not a Combination Product 10/08/2010 3 NDC: 13668-092-90 90 in 1 BOTTLE; Type 0: Not a Combination Product 10/08/2010 4 NDC: 13668-092-30 30 in 1 BOTTLE; Type 0: Not a Combination Product 10/08/2010 5 NDC: 13668-092-74 100 in 1 CARTON; Type 0: Not a Combination Product 10/08/2010 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA090865 10/08/2010 PRAMIPEXOLE DIHYDROCHLORIDE
pramipexole dihydrochloride tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 13668-093 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength PRAMIPEXOLE DIHYDROCHLORIDE (UNII: 3D867NP06J) (PRAMIPEXOLE - UNII:83619PEU5T) PRAMIPEXOLE DIHYDROCHLORIDE 0.5 mg Inactive Ingredients Ingredient Name Strength FERRIC OXIDE RED (UNII: 1K09F3G675) MAGNESIUM STEARATE (UNII: 70097M6I30) MANNITOL (UNII: 3OWL53L36A) POVIDONE K30 (UNII: U725QWY32X) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) STARCH, PREGELATINIZED CORN (UNII: O8232NY3SJ) Product Characteristics Color brown (reddish brown) Score 2 pieces Shape ROUND Size 7mm Flavor Imprint Code 9;3 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 13668-093-30 30 in 1 BOTTLE; Type 0: Not a Combination Product 10/08/2010 2 NDC: 13668-093-74 100 in 1 CARTON; Type 0: Not a Combination Product 10/08/2010 3 NDC: 13668-093-40 4000 in 1 BOTTLE; Type 0: Not a Combination Product 10/08/2010 4 NDC: 13668-093-05 500 in 1 BOTTLE; Type 0: Not a Combination Product 10/08/2010 5 NDC: 13668-093-90 90 in 1 BOTTLE; Type 0: Not a Combination Product 10/08/2010 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA090865 10/08/2010 PRAMIPEXOLE DIHYDROCHLORIDE
pramipexole dihydrochloride tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 13668-184 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength PRAMIPEXOLE DIHYDROCHLORIDE (UNII: 3D867NP06J) (PRAMIPEXOLE - UNII:83619PEU5T) PRAMIPEXOLE DIHYDROCHLORIDE 0.75 mg Inactive Ingredients Ingredient Name Strength FERRIC OXIDE YELLOW (UNII: EX438O2MRT) MAGNESIUM STEARATE (UNII: 70097M6I30) MANNITOL (UNII: 3OWL53L36A) POVIDONE K30 (UNII: U725QWY32X) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) STARCH, PREGELATINIZED CORN (UNII: O8232NY3SJ) Product Characteristics Color yellow Score no score Shape ROUND Size 7mm Flavor Imprint Code 84 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 13668-184-31 2500 in 1 BOTTLE; Type 0: Not a Combination Product 10/08/2010 2 NDC: 13668-184-05 500 in 1 BOTTLE; Type 0: Not a Combination Product 10/08/2010 3 NDC: 13668-184-90 90 in 1 BOTTLE; Type 0: Not a Combination Product 10/08/2010 4 NDC: 13668-184-30 30 in 1 BOTTLE; Type 0: Not a Combination Product 10/08/2010 5 NDC: 13668-184-74 100 in 1 CARTON; Type 0: Not a Combination Product 10/08/2010 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA090865 10/08/2010 PRAMIPEXOLE DIHYDROCHLORIDE
pramipexole dihydrochloride tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 13668-094 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength PRAMIPEXOLE DIHYDROCHLORIDE (UNII: 3D867NP06J) (PRAMIPEXOLE - UNII:83619PEU5T) PRAMIPEXOLE DIHYDROCHLORIDE 1 mg Inactive Ingredients Ingredient Name Strength FERRIC OXIDE RED (UNII: 1K09F3G675) MAGNESIUM STEARATE (UNII: 70097M6I30) MANNITOL (UNII: 3OWL53L36A) POVIDONE K30 (UNII: U725QWY32X) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) STARCH, PREGELATINIZED CORN (UNII: O8232NY3SJ) Product Characteristics Color pink (light pink) Score 2 pieces Shape ROUND Size 8mm Flavor Imprint Code 9;4 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 13668-094-74 100 in 1 CARTON; Type 0: Not a Combination Product 10/08/2010 2 NDC: 13668-094-31 2500 in 1 BOTTLE; Type 0: Not a Combination Product 10/08/2010 3 NDC: 13668-094-05 500 in 1 BOTTLE; Type 0: Not a Combination Product 10/08/2010 4 NDC: 13668-094-90 90 in 1 BOTTLE; Type 0: Not a Combination Product 10/08/2010 5 NDC: 13668-094-30 30 in 1 BOTTLE; Type 0: Not a Combination Product 10/08/2010 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA090865 10/08/2010 PRAMIPEXOLE DIHYDROCHLORIDE
pramipexole dihydrochloride tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 13668-095 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength PRAMIPEXOLE DIHYDROCHLORIDE (UNII: 3D867NP06J) (PRAMIPEXOLE - UNII:83619PEU5T) PRAMIPEXOLE DIHYDROCHLORIDE 1.5 mg Inactive Ingredients Ingredient Name Strength MAGNESIUM STEARATE (UNII: 70097M6I30) MANNITOL (UNII: 3OWL53L36A) POVIDONE K30 (UNII: U725QWY32X) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) STARCH, PREGELATINIZED CORN (UNII: O8232NY3SJ) Product Characteristics Color white (white to off white) Score 2 pieces Shape ROUND Size 10mm Flavor Imprint Code 9;5 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 13668-095-15 1500 in 1 BOTTLE; Type 0: Not a Combination Product 10/08/2010 2 NDC: 13668-095-05 500 in 1 BOTTLE; Type 0: Not a Combination Product 10/08/2010 3 NDC: 13668-095-90 90 in 1 BOTTLE; Type 0: Not a Combination Product 10/08/2010 4 NDC: 13668-095-30 30 in 1 BOTTLE; Type 0: Not a Combination Product 10/08/2010 5 NDC: 13668-095-74 100 in 1 CARTON; Type 0: Not a Combination Product 10/08/2010 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA090865 10/08/2010 Labeler - Torrent Pharmaceuticals Limited (916488547) Registrant - Torrent Pharma, Inc. (790033935) Establishment Name Address ID/FEI Business Operations Torrent Pharmaceuticals Limited 864147745 analysis(13668-091, 13668-092, 13668-093, 13668-184, 13668-094, 13668-095) , manufacture(13668-091, 13668-092, 13668-093, 13668-184, 13668-094, 13668-095) , pack(13668-091, 13668-092, 13668-093, 13668-184, 13668-094, 13668-095) Establishment Name Address ID/FEI Business Operations Torrent Pharmaceuticals Limited 916488547 manufacture(13668-091, 13668-092, 13668-093, 13668-184, 13668-094, 13668-095)





© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.