CARIMUNE NANOFILTERED- human immunoglobulin g injection, powder, lyophilized, for solution
Carimune by
Drug Labeling and Warnings
Carimune by is a Other medication manufactured, distributed, or labeled by CSL Behring AG. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
- SPL UNCLASSIFIED SECTION
-
BOXED WARNING
(What is this?)
WARNING: THROMBOSIS, RENAL DYSFUNCTION, or ACUTE RENAL FAILURE
- Thrombosis may occur with immune globulin products1-8, including Carimune NF. Risk factors may include: advanced age, prolonged immobilization, hypercoagulable conditions, history of venous or arterial thrombosis, use of estrogens, indwelling central vascular catheters, hyperviscosity, and cardiovascular risk factors. Thrombosis may occur in the absence of known risk factors (see PRECAUTIONS: Thrombosis and Information for Patients).
- Renal dysfunction, acute renal failure, osmotic nephrosis, and death may occur in predisposed patients with immune globulin intravenous (IGIV) products9–14, including Carimune NF. Patients predisposed to renal dysfunction include those with any degree of pre-existing renal insufficiency, diabetes mellitus, age greater than 65, volume depletion, sepsis, paraproteinemia, or patients receiving known nephrotoxic drugs. Renal dysfunction and acute renal failure occur more commonly in patients receiving IGIV products containing sucrose. Carimune NF contains sucrose.
- For patients at risk of thrombosis, renal dysfunction or acute renal failure, administer Carimune NF at the minimum dose and infusion rate practicable. Ensure adequate hydration in patients before administration. Monitor for signs and symptoms of thrombosis and assess blood viscosity in patients at risk for hyperviscosity (see DOSAGE AND ADMINISTRATION, and PRECAUTIONS: Thrombosis).
-
DESCRIPTION
Carimune® NF, Nanofiltered, Immune Globulin Intravenous (Human), is a sterile, highly purified polyvalent antibody product containing in concentrated form all the IgG antibodies which regularly occur in the donor population.15 This immunoglobulin preparation is produced by cold alcohol fractionation from the plasma of US donors. Part of the fractionation may be performed by another US-licensed manufacturer. Carimune® NF is made suitable for intravenous use by treatment at acid pH in the presence of trace amounts of pepsin.16,17 The manufacturing process by which Carimune® NF is prepared from plasma consists of fractionation and purification steps that comprise filtrations in the presence of filter aids. Four of these steps were validated for virus elimination of both enveloped and non-enveloped viruses. Additionally, the manufacturing process was investigated for its capacity to decrease the infectivity of an experimental agent of transmissible spongiform encephalopathy (TSE), considered as a model for the vCJD and CJD agents.18 To complement the existing virus elimination / inactivation mechanism in the Carimune® NF manufacturing process, nanofiltration (removing viruses via size-exclusion) was introduced as an additional virus removal step into the manufacturing process.19,20 Nanofiltration is performed prior to the viral inactivation step (pH 4 in presence of pepsin) in order to reduce the potential viral load before inactivation is performed. Treatment with pepsin at pH 4 rapidly inactivates enveloped viruses.21
The Carimune® NF manufacturing process provides a significant virus reduction capacity as shown in in vitro studies. The results, summarized in Table 1, demonstrate virus clearance during Carimune® NF manufacturing using model viruses for lipid enveloped and non-enveloped viruses.
Table 1. Virus Elimination and Inactivation Virus HIV BVDV PRV SFV SV BEV HIV: Human immunodeficiency virus, model for HIV 1 and HIV 2 BVDV: Bovine viral diarrhea virus, model for HCV (Hepatitis C virus) PRV: Pseudorabies virus, model for large, enveloped DNA viruses (e.g., herpes virus) SFV: Semliki Forest virus, model for HCV SV: Sindbis virus, model for HCV BEV: Bovine enterovirus, model for HAV (Hepatitis A virus) nt: not tested Genome RNA RNA DNA RNA RNA RNA Envelope Yes Yes Yes Yes Yes No Size (nm)
80–100 40–60 120–200 50–70 50–70 28–30 Fractionation & Depth filtration 15.5 nt 16.0 9.3 12.4 14.1 pH 4 / pepsin ≥ 6.1 ≥ 4.4 ≥ 5.3 ≥ 6.8 nt nt Nanofiltration ≥ 4.9 ≥ 4.5 ≥ 4.4 nt ≥ 7.5 ≥ 5.1 Overall reduction ≥ 26 ≥ 9 ≥ 25 ≥ 16 ≥ 19 ≥ 19 PRV and the two model viruses for HCV, BVDV and SFV, were inactivated within 1/10, and HIV within 1/2 of the incubation time (pH 4/pepsin treatment) used during production of Carimune® NF.
Several of the individual production steps in the Carimune® NF manufacturing process have been shown to decrease TSE infectivity of an experimental model agent. TSE reduction steps include precipitation (3.5 logs), depth filtrations (7.3 logs), and nanofiltration (4.4 logs). These studies provide reasonable assurance that low levels of CJD/vCJD agent infectivity, if present in the starting material, would be removed.
The preparation contains at least 96% of IgG and after reconstitution with a neutral unbuffered diluent has a pH of 6.6 ± 0.2. Most of the immunoglobulins are monomeric (7 S) IgG; the remainder consists of dimeric IgG and a small amount of polymeric IgG, traces of IgA and IgM and immunoglobulin fragments.22 The distribution of the IgG subclasses corresponds to that of normal serum.23-26 Final container lyophilized units are prepared so as to contain 3, 6, or 12 g protein with 1.67 g sucrose and less than 20 mg NaCl per gram of protein. The lyophilized preparation contains no preservative and may be reconstituted with sterile water, 5% dextrose or 0.9% saline to a solution with protein concentrations ranging from 3% to 12% (see Table 4). See Table 2 for calculated Carimune® NF osmolality (mOsm/kg) at each protein concentration. The patient's fluid, electrolyte, caloric requirements and renal function should be considered in selecting an appropriate diluent and concentration.
Table 2. Calculated Carimune® NF Osmolality (mOsm/kg) Concentration Diluent 3% 6% 9% 12% 0.9% NaCl 498 690 882 1074 5% Dextrose 444 636 828 1020 Sterile Water 192 384 576 768 -
CLINICAL PHARMACOLOGY
Carimune® NF contains a broad spectrum of antibody specificities against bacterial, viral, parasitic, and mycoplasma antigens, that are capable of both opsonization and neutralization of microbes and toxins. The 3 week half-life of Carimune® NF corresponds to that of Immune Globulin (Human) for intramuscular use, although individual variations in half-life have been observed.27,28
Appropriate doses of Carimune® NF restore abnormally low immunoglobulin G levels to the normal range. One hundred percent of the infused dose of IGIV-products is available in the recipient's circulation immediately after infusion. After approximately 6 days, equilibrium is reached between the intra- and extravascular compartments, with immunoglobulin G being distributed approximately 50% intravascular and 50% extravascular. In comparison, after the intramuscular injection of immune globulin, the IgG requires 2–5 days to reach its maximum concentration in the intravascular compartment. This concentration corresponds to about 40% of the injected dose.28
While Carimune® NF has been shown to be effective in some cases of Immune Thrombocytopenic Purpura (ITP) (see INDICATIONS AND USAGE), the mechanism of action in ITP has not been fully elucidated. Toxicity from overdose has not been observed on regimens of 0.4 g/kg body weight each day for 5 days.29-31 Sucrose is added to Carimune® NF for reasons of stability and solubility. Since sucrose is excreted unchanged in the urine when given intravenously, Carimune® NF may be given to diabetics without compensatory changes in insulin dosage regimen. Please see WARNINGS section.
-
INDICATIONS AND USAGE
Immunodeficiency
Carimune® NF is indicated for the maintenance treatment of patients with primary immunodeficiencies (PID), e.g., common variable immunodeficiency, X-linked agammaglobulinemia, severe combined immunodeficiency.30,32-34 Carimune® NF is preferable to intramuscular Immune Globulin (Human) preparations in treating patients who require an immediate and large increase in the intravascular immunoglobulin level28, in patients with limited muscle mass, and in patients with bleeding tendencies for whom intramuscular injections are contraindicated. The infusions must be repeated at regular intervals.
Please see DOSAGE AND ADMINISTRATION section.
Immune Thrombocytopenic Purpura (ITP)
Acute
A controlled study was performed in children in which Carimune® was compared with steroids for the treatment of acute (defined as less than 6 months duration) ITP. In this study sequential platelet levels of 30,000, 100,000, and 150,000/µL were all achieved faster with Carimune® than with steroids and without any of the side effects associated with steroids.29,35 However, it should be noted that many cases of acute ITP in childhood resolve spontaneously within weeks to months. Carimune® has been used with good results in the treatment of acute ITP in adult patients.36-38 In a study involving 10 adults with ITP of less than 16 weeks duration, Carimune® therapy raised the platelet count to the normal range after a 5 day course. This effect lasted a mean of over 173 days, ranging from 30 to 372 days.39
Chronic
Children and adults with chronic (defined as greater than 6 months duration) ITP have also shown an increase (sometimes temporary) in platelet counts upon administration of Carimune®.35,39-43 Therefore, in situations that require a rapid rise in platelet count, for example prior to surgery or to control excessive bleeding, use of Carimune® should be considered. In children with chronic ITP, Carimune® therapy resulted in a mean rise in platelet count of 312,000/µL with a duration of increase ranging from 2 to 6 months.40,43 Carimune® therapy may be considered as a means to defer or avoid splenectomy.42-44 In adults, Carimune® therapy has been shown to be effective in maintaining the platelet count in an acceptable range with or without periodic booster therapy. The mean rise in platelet count was 93,000/µL and the average duration of the increase was 20–24 days.39,40 However, it should be noted that not all patients will respond. Even in those patients who do respond, this treatment should not be considered to be curative.
-
CONTRAINDICATIONS
Carimune® NF is contraindicated in patients who have had an anaphylactic or severe systemic reaction to the administration of human immune globulin. Individuals with IgA deficiency, especially those who have known antibody against IgA, or hypersensitivity to immunoglobulins should only receive Carimune® NF with utmost caution due to the risk of severe immediate hypersensitivity reactions including anaphylaxis.
-
WARNINGS
Immune Globulin Intravenous (Human) (IGIV) products have been reported to be associated with renal dysfunction, acute renal failure, osmotic nephrosis, and death.9-14
Patients predisposed to acute renal failure include patients with:
- any degree of pre-existing renal insufficiency
- diabetes mellitus
- age greater than 65
- volume depletion
- sepsis
- paraproteinemia
- patients receiving known nephrotoxic drugs
In such patients, IGIV products should be administered at the minimum concentration available and the minimum rate of infusion practicable. While these reports of renal dysfunction and acute renal failure have been associated with the use of many of the licensed IGIV products, those containing sucrose as a stabilizer accounted for a disproportionate share of the total number. Carimune® NF contains sucrose. See PRECAUTIONS and DOSAGE AND ADMINISTRATION sections for important information intended to reduce the risk of acute renal failure.
IgA deficient patients, especially those with known antibodies against IgA, are at greater risk of developing severe hypersensitivity and anaphylactic reactions.
Because Carimune® NF is made from human blood, it may carry a risk of transmitting infectious agents, e.g., viruses, the variant Creutzfeldt-Jakob disease (vCJD) agent and, theoretically, the Creutzfeldt-Jakob disease (CJD) agent. The risk that such products will transmit an infectious agent has been reduced by screening plasma donors for prior exposure to certain viruses, by testing for the presence of certain current virus infections, and through the application of viral elimination/reduction steps such as alcohol fractionation in the presence of filter aids, nanofiltration and pH 4/pepsin treatment19-21 (see Table 1). All infections thought by a physician possibly to have been transmitted by Carimune® NF should be reported by lot number, by the physician, or other healthcare provider to CSL Behring Pharmacovigilance at 1-866-915-6958. The physician should discuss the risks and benefits of this product with the patient.
Patients with agamma- or extreme hypogammaglobulinemia who have never before received immunoglobulin substitution treatment or whose time from last treatment is greater than 8 weeks, may be at risk of developing inflammatory reactions on rapid infusion (greater than 2 mg/kg/min) of Carimune® NF. These reactions are manifested by a rise in temperature, chills, nausea, and vomiting. The patient's vital signs should be monitored continuously. The patient should be carefully observed throughout the infusion, since these reactions on rare occasions may lead to shock. Epinephrine and other appropriate resuscitative drugs and equipment should be available for treatment of an acute anaphylactic reaction.
-
PRECAUTIONS
Please see DOSAGE AND ADMINISTRATION below, for important information on Carimune® NF compatibility with other medications or fluids. Patients should not be volume depleted prior to the initiation of the infusion of IGIV. Periodic monitoring of renal function tests and urine output is particularly important in patients judged to have a potential increased risk for developing acute renal failure. Renal function, including measurement of blood urea nitrogen (BUN) and serum creatinine, should be assessed prior to the initial infusion of Carimune® NF and again at appropriate intervals thereafter. If renal function deteriorates, discontinuation of the product should be considered. For patients judged to be at risk for developing renal dysfunction, Carimune® NF should be infused at a rate less than 2 mg/kg/min.
Information for Patients
- Instruct patients to immediately report symptoms of decreased urine output, sudden weight gain, fluid retention/edema, and/or shortness of breath (which may suggest kidney damage) to their physicians.
- Instruct patients to immediately report symptoms of thrombosis. These symptoms may include: pain and/or swelling of an arm or leg with warmth over the affected area, discoloration of an arm or leg, unexplained shortness of breath, chest pain or discomfort that worsens on deep breathing, unexplained rapid pulse, numbness or weakness on one side of the body.
Laboratory Tests
IGIV recipients should be monitored for clinical signs and symptoms of hemolysis. IGIV recipients should be monitored for pulmonary adverse reactions. If Transfusion-Related Acute Lung Injury (TRALI) is suspected, appropriate tests should be performed for the presence of anti-neutrophil antibodies in both the product and patient serum. Baseline assessment of blood viscosity should be considered in patients at risk for hyperviscosity, including those with cryoglobulins, fasting chylomicronemia/markedly high triacylglycerols (triglycerides), or monoclonal gammopathies.
Pregnancy
Animal reproduction studies have not been conducted with Carimune® NF. It is also not known whether Carimune® NF can cause fetal harm when administered to a pregnant woman or can affect reproduction capacity. Carimune® NF should be given to a pregnant woman only if clearly needed.38 Intact immune globulins such as those contained in Carimune® NF cross the placenta from maternal circulation increasingly after 30 weeks gestation.45,46 In cases of maternal ITP where Carimune® was administered to the mother prior to delivery, the platelet response and clinical effect were similar in the mother and neonate.38,46-55
Pediatric Use
High dose administration of Carimune® in pediatric patients with acute or chronic Immune Thrombocytopenic Purpura did not reveal any pediatric-specific hazard.29 Antibodies in Immune Globulin Intravenous (Human) may impair the efficacy of live attenuated viral vaccines such as measles, rubella, and mumps.56-58 Immunizing physicians should be informed of recent therapy with Immune Globulin Intravenous (Human) so that appropriate precautions may be taken.
Geriatric Use
Carimune® NF should be used with caution in patients over 65 years of age and judged to be at increased risk of developing renal insufficiency (see DOSAGE AND ADMINISTRATION). In the absence of prospective data, recommended doses should not be exceeded and the concentration and infusion rate selected should be the minimum practicable. The product should be infused at a rate less than 2 mg/kg/min.
Aseptic Meningitis Syndrome
An aseptic meningitis syndrome (AMS) has been reported to occur infrequently in association with Immune Globulin Intravenous (Human) (IGIV) treatment. The syndrome usually begins within several hours to two days following IGIV treatment. It is characterized by symptoms and signs including severe headache, nuchal rigidity, drowsiness, fever, photophobia, painful eye movements, and nausea and vomiting. Cerebrospinal fluid (CSF) studies are frequently positive with pleocytosis. Patients exhibiting such symptoms and signs should receive a thorough neurological examination, including CSF studies, to rule out other causes of meningitis. AMS may occur more frequently in association with high dose (2 g/kg) IGIV treatment. Discontinuation of IGIV treatment has resulted in remission of AMS within several days without sequelae.
Hemolysis
Immune Globulin Intravenous (Human) (IGIV) products can contain blood group antibodies which may act as hemolysins and induce in vivo coating of red blood cells with immunoglobulin, causing a positive direct antiglobulin reaction and, rarely, hemolysis.59-61 Hemolytic anemia can develop subsequent to IGIV therapy due to enhanced RBC sequestration62 (see ADVERSE REACTIONS). IGIV recipients should be monitored for clinical signs and symptoms of hemolysis (see PRECAUTIONS: Laboratory Tests).
Transfusion-Related Acute Lung Injury (TRALI)
There have been reports of noncardiogenic pulmonary edema Transfusion-Related Acute Lung Injury (TRALI) in patients administered IGIV.63 TRALI is characterized by severe respiratory distress, pulmonary edema, hypoxemia, normal left ventricular function, and fever and typically occurs within 1–6 hours after transfusion. Patients with TRALI may be managed by using oxygen therapy with adequate ventilatory support.
IVIG recipients should be monitored for pulmonary adverse reactions. If TRALI is suspected, appropriate tests should be performed for the presence of anti-neutrophil antibodies in both the product and patient serum (see PRECAUTIONS: Laboratory Tests).
Thrombosis
Thrombosis may occur following treatment with immune globulin products1-8, including Carimune NF. Risk factors may include: advanced age, prolonged immobilization, hypercoagulable conditions, history of venous or arterial thrombosis, use of estrogens, indwelling central vascular catheters, hyperviscosity, and cardiovascular risk factors. Thrombosis may occur in the absence of known risk factors.
Consider baseline assessment of blood viscosity in patients at risk for hyperviscosity, including those with cryoglobulins, fasting chylomicronemia/markedly high triacylglycerols (triglycerides), or monoclonal gammopathies. For patients at risk of thrombosis, administer Carimune NF at the minimum dose and infusion rate practicable. Ensure adequate hydration in patients before administration. Monitor for signs and symptoms of thrombosis and assess blood viscosity in patients at risk for hyperviscosity (see BOXED WARNING, DOSAGE AND ADMINISTRATION, PRECAUTIONS: Information for Patients).
-
ADVERSE REACTIONS
Increases in creatinine and blood urea nitrogen (BUN) have been observed as soon as one to two days following infusion. Progression to oliguria or anuria, requiring dialysis has been observed. Types of severe renal adverse events that have been seen following IGIV therapy include: acute renal failure, acute tubular necrosis, proximal tubular nephropathy and osmotic nephrosis.9-14,64,71–73
Inflammatory adverse reactions have been described in agammaglobulinemic and hypogammaglobulinemic patients who have never received immunoglobulin substitution therapy before or in patients whose time from last treatment is greater than 8 weeks and whose initial infusion rate exceeds 2 mg/kg/min.
This occurs in approximately 10% of such cases. Such reactions may also be observed in some patients during chronic substitution therapy.
Reactions, which may become apparent only 30 minutes to 1 hour after the beginning of the infusion, are as follows: flushing of the face, feelings of tightness in the chest, chills, fever, dizziness, nausea, diaphoresis, and hypotension or hypertension. In such cases, the infusion should be slowed or temporarily stopped until the symptoms subside. The infusion may then be resumed at a lower rate that is comfortable for the patient. If anaphylaxis or other severe reactions occur, the infusion should be stopped immediately.
Arthralgia, myalgia, and transient skin reactions (such as rash, erythema, pruritus, urticaria, eczema or dermatitis) have also been reported.
Immediate anaphylactoid and hypersensitivity reactions due to previous sensitization of the recipient to certain antigens, most commonly IgA, may be observed in exceptional cases, described under CONTRAINDICATIONS.30,31,65 In patients with ITP, who receive higher doses (0.4 g/kg/day or greater), 2.9% of infusions may result in adverse reactions.21 Headache, generally mild, is the most common symptom noted, occurring during or following 2% of infusions. A few cases of usually mild hemolysis have been reported after infusion of intravenous immunoglobulin products.59-61 These were attributed to transferal of blood group (e.g., anti-D) antibodies.
Postmarketing
The following adverse reactions have been identified and reported during the post-approval use of IGIV products:
Respiratory
Apnea, Acute Respiratory Distress Syndrome (ARDS), Transfusion-Related Acute Lung Injury (TRALI), cyanosis, hypoxemia, pulmonary edema, dyspnea, bronchospasm
Cardiovascular
Cardiac arrest, thromboembolism, vascular collapse, hypotension
Neurological
Coma, loss of consciousness, seizures, tremor
Integumentary
Stevens-Johnson syndrome, epidermolysis, erythema multiforme, bullous dermatitis
Hematologic
Pancytopenia, leukopenia, hemolysis, positive direct antiglobulin (Coombs) test
General/Body as a Whole
Pyrexia, rigors
Musculoskeletal
Back pain
Gastrointestinal
Hepatic dysfunction, abdominal painBecause postmarketing reporting of these reactions is voluntary and the at-risk populations are of uncertain size, it is not always possible to reliably estimate the frequency of the reaction or establish a causal relationship to exposure to the product. Such is also the case with literature reports authored independently.66
-
DOSAGE AND ADMINISTRATION
It is generally advisable not to dilute plasma derivatives with other infusable drugs. Carimune® NF should be given by a separate infusion line. No other medications or fluids should be mixed with Carimune® NF preparation.
Carimune® NF should be used with caution in patients with pre-existing renal insufficiency and in patients judged to be at increased risk of developing renal insufficiency (including, but not limited to those with diabetes mellitus, age greater than 65, volume depletion, paraproteinemia, sepsis, and patients receiving known nephrotoxic drugs). In these cases especially it is important to assure that patients are not volume depleted prior to Carimune® NF infusion. No prospective data are presently available to identify a maximum safe dose, concentration, and rate of infusion in patients determined to be at increased risk of acute renal failure. In the absence of prospective data, recommended doses should not be exceeded and the concentration and infusion rate selected should be the minimum practicable. For patients judged to be at risk for developing renal dysfunction, Carimune® NF should be infused at a rate less than 2 mg/kg/min.
For patients judged to be at an increased risk for thrombosis, a maximum infusion rate of less than 2 mg/kg/min for patients is recommended (see PRECAUTIONS: Thrombosis).
If side effects occur, the infusion should be stopped or slowed until the symptoms subside.
Adult and Child Substitution Therapy
The recommended dose of Carimune® NF in primary immunodeficiency is 0.4 to 0.8 g/kg of body weight administered once every three to four weeks by intravenous infusion.
The first infusion of Carimune® NF in previously untreated agammaglobulinemic or hypogammaglobulinemic patients must be given as a 3% immunoglobulin solution (see Reconstitution). Subsequent infusions may be administered at a higher concentration if the patient shows good tolerance.
An initial infusion rate of 0.5 mg/kg/min is recommended. If tolerated, after 30 minutes, the rate may be increased to 1 mg/kg/min for the next 30 minutes. Thereafter, the rate may be gradually increased in a stepwise manner up to a maximum of 3 mg/kg/min as tolerated. Refer to Table 3 for the corresponding infusion rates in mg/kg/min or mL/kg/min for all product concentrations.
The first infusion of Carimune® NF in previously untreated agammaglobulinemic and hypogammaglobulinemic patients may lead to systemic side effects. The nature of these effects has not been fully elucidated. Some of them may be due to the release of proinflammatory cytokines by activated macrophages in immunodeficient recipients.67,68 Subsequent administration of Carimune® NF to immunodeficient patients as well as to normal individuals usually does not cause further untoward side effects.
Therapy of Idiopathic Thrombocytopenic Purpura (ITP)
Induction
The recommended dose of Carimune® NF for the treatment of ITP is 0.4 g/kg of body weight on 2–5 consecutive days. An immunoglobulin solution of 6% (see Reconstitution) is recommended for use in ITP.
The recommended initial infusion rate for the treatment of ITP is 0.5 mg/kg/min. If tolerated, after 30 minutes, the rate may be increased to 1 mg/kg/min for the next 30 minutes. Thereafter, the rate may be gradually increased in a stepwise manner up to a maximum of 3 mg/kg/min as tolerated. Refer to Table 3 for the corresponding infusion rates in mg/kg/min or mL/kg/min for all product concentrations.
Acute ITP – Childhood
In acute ITP of childhood, if an initial platelet count response to the first two doses is adequate (30–50,000/µL), therapy may be discontinued after the second day of the 5 day course.35
Maintenance – Chronic ITP
In adults and children, if after induction therapy the platelet count falls to less than 30,000/µL and/or the patient manifests clinically significant bleeding, 0.4 g/kg of body weight may be given as a single infusion. If an adequate response does not result, the dose can be increased to 0.8–1 g/kg of body weight given as a single infusion.36,69,70
Table 3. Infusion Rates for Carimune® NF Concentrations Concentration
(%)Initial Infusion Rate:
0.5 mg/kg/min1 mg/kg/min 2 mg/kg/min* Maximum Infusion Rate†:
3 mg/kg/min- * Maximum infusion rate for patients at risk of renal dysfunction or thromboembolic events.
- † For patients not at risk of renal dysfunction of thromboembolic events.
3% 0.0167 mL/kg/min 0.033 mL/kg/min 0.067 mL/kg/min 0.10 mL/kg/min 6% 0.008 mL/kg/min 0.0167 mL/kg/min 0.033 mL/kg/min 0.050 mL/kg/min 9% 0.006 mL/kg/min 0.011 mL/kg/min 0.022 mL/kg/min 0.033 mL/kg/min 12% 0.004 mL/kg/min 0.008 mL/kg/min 0.016 mL/kg/min 0.025 mL/kg/min Reconstitution
(see also pictures next page)
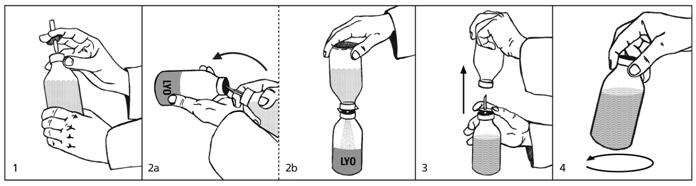
1. Remove the protective plastic caps from the lyophilisate (LYO) and diluent bottles and disinfect both rubber stoppers with alcohol. Remove the protective cover from one end of the transfer set and insert the exposed needle through the rubber stopper into the bottle containing the diluent (picture 1). 2a. and 2b. Remove the second protective cover from the other end of the transfer set. Grasp both bottles as shown in picture 2a, quickly plunge the diluent bottle onto the lyophilisate bottle and bring the bottles into an upright position. Only if this is done quickly and the bottles are immediately brought into an upright position can the vacuum in the lyophilisate bottle be maintained, thus speeding up reconstitution and facilitating the transfer. Allow the diluent to flow into the lyophilisate bottle (picture 2b). 3. Once the appropriate amount of diluent is transferred (see Table 4), lift the diluent bottle off the spike to release the vacuum (picture 3). This will reduce foaming and facilitate dissolution. Remove the spike. 4. Swirl vigorously but do not shake, otherwise foam will form which is very slow to subside (picture 4). The lyophilisate dissolves within a few minutes. To reconstitute Carimune® NF from the individual vial package, or when using other diluents or higher concentrations, Table 4 indicates the volume of sterile diluent required. Observing aseptic technique, this volume should be drawn into a sterile hypodermic syringe and needle. The diluent is then injected into the corresponding Carimune® NF vial size.
Table 4. Required Diluent Volume* Target Concentration 6 g Vial 12 g Vial - * In patients judged to be at increased risk of developing renal insufficiency and thromboembolic events, the concentration and infusion rate of Carimune® NF should be the minimum practicable.
- † Container not large enough to permit this concentration.
3% 200 mL † 6% 100 mL 200 mL 9% 66 mL 132 mL 12% 50 mL 100 mL If large doses of Carimune® NF are to be administered, several reconstituted vials of identical concentration and diluent may be pooled in an empty sterile glass or plastic i.v. infusion container using aseptic technique.
Carimune® NF normally dissolves within a few minutes, though in exceptional cases it may take up to 20 minutes.
DO NOT SHAKE! Excessive shaking will cause foaming.
Any undissolved particles should respond to careful rotation of the bottle. Avoid foaming. Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. Filtering of Carimune® NF is acceptable but not required. Pore sizes of 15 microns or larger will be less likely to slow infusion, especially with higher Carimune® NF concentrations. Antibacterial filters (0.2 microns) may be used. When reconstitution of Carimune® NF occurs outside of sterile laminar air flow conditions, administration must begin promptly with partially used vials discarded. When reconstitution is carried out in a sterile laminar flow hood using aseptic technique, administration may begin within 24 hours provided the solution has been refrigerated during that time. Do not freeze Carimune® NF solution.
PROCEED WITH INFUSION ONLY IF SOLUTION IS CLEAR AND AT APPROXIMATELY ROOM TEMPERATURE.
-
HOW SUPPLIED
Carimune® NF is available as a white lyophilized powder in 6 and 12 g size vials. The only diluents which may be used to reconstitute the product are sterile (0.9%) Sodium Chloride Injection USP, 5% Dextrose, or Sterile Water.
Each product presentation includes a package insert and the following components:
Presentation Carton
NDC NumberComponents 6 g 44206-417-06 - Carimune NF in a single-use vial [NDC: 44206-417-91]
- One double-ended transfer spike for reconstitution
12 g 44206-418-12 - Carimune NF in a single-use vial [NDC: 44206-418-92]
- One double-ended transfer spike for reconstitution
-
REFERENCES
- Dalakas MC: High-dose intravenous immunoglobulin and serum viscosity: risk of precipitating thromboembolic events. Neurology 1994; 44:223–226.
- Caress JB, Cartwright MS, Donofrio PD, Peacock JE: The clinical features of 16 cases of stroke associated with administration of IVIg. Neurology 2003; 60:1822–1824.
- Woodruff RK, Grigg AP, Firkin FC, Smith IL: Fatal thrombotic events during treatment of autoimmune thrombocytopenia with intravenous immunoglobulin in elderly patients. Lancet 1986; 2:217–218.
- Jordan S, Cunningham-Rundles C, McEwan R: Utility of intravenous immune globulin in kidney transplantation: efficacy, safety, and cost implications. Am J Transplant 2003; 3:653–664.
- Wolberg AS, Kon RH, Monroe DM, Hoffman M: Coagulation factor XI is a contaminant in intravenous immunoglobulin preparations. Am J Hematol 2000; 65:30–34.
- Zaidan R, Al Moallem M, Wani BA, Shameena AR, Al Tahan AR, Daif AK, Al Rajeh S: Thrombosis complicating high dose intravenous immunoglobulin: report of three cases and review of the literature. Eur J Neurology 2003; 10:367–372.
- Okuda D, Flaster M, Frey J, Sivakumar, K: Arterial thrombosis induced by IVIg and its treatment with tPA. Neurology 2003; 60:1825–1826.
- Dalakas MC, Clark WM: Strokes, thromboembolic events, and IVIg. Rare incidents blemish an excellent safety record. Neurology 2003; 60:1736–1737.
- Winward DB, Brophy MT: Acute renal failure after administration of intravenous immunoglobulin: Review of the literature and case report. Pharmacotherapy 1995; 15:765–772.
- Cantú TG, Hoehn-Saric EW, Burgess KM, Racusen L, Scheel P: Acute renal failure associated with immunoglobulin therapy. Am J Kidney Dis 1995; 25:228–234.
- Cayco AV, Perazella MA, Hayslett JP: Renal insufficiency after intravenous immune globulin therapy: a report of two cases and an analysis of the literature. J Amer Soc Nephrology 1997; 8:1788–1793.
- Rault R, Piraino B, Johnston JR, Oral A: Pulmonary and renal toxicity of intravenous immunoglobulin. Clin Nephrol 1991, 36:83–86.
- Michail S, Nakopoulou L, Stravrianopoulos I, Stamatiadis D, Avdikou K, Vaiopoulos G, Stathakis C: Acute renal failure associated with immunoglobulin administration. Nephrol Dial Transplant 1997; 12:1497–99.
- Ashan N, Wiegand LA, Abendroth CS, Manning EC: Acute renal failure following immunoglobulin therapy. Am J Nephrol 1996; 16:532–6.
- Gardi A: Quality control in the production of an immunoglobulin for intravenous use. Blut 1984; 48:337–344.
- Römer J, Morgenthaler JJ, Scherz R, et al: Characterization of various immunoglobulin-preparations for intravenous application. I. Protein composition and antibody content. Vox Sang 1982; 42:62–73.
- Römer J, Späth PJ, Skvaril F, et al: Characterization of various immunoglobulin preparations for intravenous application. II. Complement activation and binding to Staphylococcus protein A. Vox Sang 1982; 42:74–80.
- Gregori L, Maring JA, MacAuley C et al: Partitioning of TSE infectivity during ethanol fractionation of human plasma. Biologicals 2004; 32:1–10.
- Omar A, and Kempf C: Removal of neutralized model Parvoviruses and Enteroviruses in human IgG solutions by nanofiltration. Transfusion 2002; 42:1005–1010.
- Späth P, Kempf C, and Gold R: Herstellung, Verträglichkeit und Virussicherheit von intravenösem Immunglobulin. In "Immunglobuline in der Neurobiologie" (P. Berlit, ed.), Steinkopff Verlag, Darmstadt, BRD 2001, pp 1–42.
- Kempf C, Morgenthaler JJ, Rentsch M, and Omar A: Viral safety and manufacturing of an intravenous immunoglobulin. In "Intravenous Immunoglobulin Research and Therapy" Kazatchkine and Morell, eds. Parthenon Publishing Group. 1996, pp 11–18.
- Römer J, Späth PJ: Molecular composition of immunoglobulin preparations and its relation to complement activation, in Nydegger UE (ed): Immunohemotherapy: A Guide to Immunoglobulin Prophylaxis and Therapy. London, Academic Press 1981, pp 123–130.
- Skvaril F, Roth-Wicky B, and Barandun S: IgG subclasses in human-g-globulin preparations for intravenous use and their reactivity with Staphylococcus protein A. Vox Sang 1980; 38:147.
- Skvaril F: Qualitative and quantitative aspects of IgG subclasses in i.v. immunoglobulin preparations, in Nydegger UE (ed): Immunohemotherapy: A Guide to Immunoglobulin Prophylaxis and Therapy. London, Academic Press, 1981, pp 113–122.
- Skvaril F, and Barandun S: In vitro characterization of immunoglobulins for intravenous use, in Alving BM, Finlayson JS (eds): Immunoglobulins: Characteristics and Uses of Intravenous Preparations, DHHS Publication No. (FDA)-80-9005. US Government Printing Office, 1980, pp 201–206.
- Burckhardt JJ, Gardi A, Oxelius V, et al: Immunoglobulin G subclass distribution in three human intravenous immunoglobulin preparations. Vox Sang 1989; 57:10–14.
- Morell A, and Skvaril F: Struktur und biologische Eigenschaften von Immunglobulinen und g-Globulin-Präparaten. II. Eigenschaften von g-Globulin-Präparaten. Schweiz Med Wochenschr 1980; 110:80.
- Morell A, Schürch B, Ryser D, et al: In vivo behaviour of gamma globulin preparations. Vox Sang 1980; 38:272.
- Imbach P, Barandun S, d'Apuzzo V, et al: High-dose intravenous gamma globulin for idiopathic thrombocytopenic purpura in childhood. Lancet 1981; 1:1228.
- Barandun S, Morell A, Skvaril F: Clinical experiences with immunoglobulin for intravenous use, in Alving BM, Finlayson JS (eds): Immunoglobulins: Characteristics and Uses of Intravenous Preparations. DHHS Publication No. (FDA)-80-9005. US Government Printing Office, 1980, pp 31–35.
- Schiff R, Sedlak D, Buckley R: Rapid infusion of Sandoglobulin™ in patients with primary humoral immunodeficiency. J Allergy Clin Immunol 88:61, 1991.
- Joller PW, Barandun S, Hitzig WH: Neue Möglichkeiten der Immunglobulin-Ersatztherapie bei Antikörpermangel-Syndrom. Schweiz Med Wochenschr 1980; 110:1451.
- Barandun S, Imbach P, Morell A, et al: Clinical indications for immunoglobulin infusion, in Nydegger UE (ed): Immunohemotherapy: A Guide to Immunoglobulin Prophylaxis and Therapy. London, Academic Press, 1981, pp 275–282.
- Cunningham-Rundles C, Smithwick EM, Siegal FP, et al: Treatment of primary humoral immunodeficiency disease with intravenous (pH 4.0 treated) gamma globulin, in Nydegger UE (ed): Guide to Immunoglobulin Prophylaxis and Therapy. London, Academic Press, 1981, pp 283–290.
- Imbach P, Wagner HP, Berchtold W, et al: Intravenous immunoglobulin versus oral corticosteroids in acute immune thrombocytopenic purpura in childhood. Lancet 1985; 2:464.
- Fehr J, Hofmann V, Kappeler U: Transient reversal of thrombocytopenia in idiopathic thrombocytopenic purpura by high-dose intravenous gamma globulin. N Engl J Med 1982; 306:1254.
- Müller-Eckhardt C, Küenzlen E, Thilo-Körner D, et al: High-dose intravenous immunoglobulin for posttransfusion purpura. N Engl J Med 1983; 308:287.
- Wenske G, Gaedicke G, Küenzlen E, et al: Treatment of idiopathic thrombocytopenic purpura in pregnancy by high-dose intravenous immunoglobulin. Blut 1983; 46:347–353.
- Newland AC, Treleaven JG, Minchinton B, et al: High-dose intravenous IgG in adults with autoimmune thrombocytopenia. Lancet 1983; 1:84–87.
- Bussel JB, Kimberly RP, Inman RD, et al: Intravenous gammaglobulin for chronic idiopathic thrombocytopenic purpura. Blood 1983; 62:480–486.
- Abe T, Matsuda J, Kawasugi K, et al: Clinical effect of intravenous immunoglobulin in chronic idiopathic thrombocytopenic purpura. Blut 1983; 47:69–75.
- Bussel JB, Schulman I, Hilgartner MW, et al: Intravenous use of gamma globulin in the treatment of chronic immune thrombocytopenic purpura as a means to defer splenectomy. J Pediatr 1983; 103:651–654.
- Imholz B, et al: Intravenous immunoglobulin (i.v. IgG) for previously treated acute or for chronic idiopathic thrombocytopenic purpura (ITP) in childhood: A prospective multicenter study. Blut 1988; 56:63–68.
- Lusher JM, and Warrier I: Use of intravenous gamma globulin in children with idiopathic thrombocytopenic purpura and other immune thrombocytopenias. Am J Med 1987; 83 (suppl 4A):10–16.
- Hammarstrom L, and Smith CI: Placental transfer of intravenous immunoglobulin. Lancet 1986; 1:681.
- Sidiropoulos D, et al: Transplacental passage of intravenous immunoglobulin in the last trimester of pregnancy. J Pediatr 1986; 109:505–508.
- Wenske G, et al: Idiopathic thrombocytopenic purpura in pregnancy and neonatal period. Blut 1984; 48:377–382.
- Fabris P, et al: Successful treatment of a steroid-resistant form of idiopathic thrombocytopenic purpura in pregnancy with high doses of intravenous immunoglobulins. Acta Haemat 1987; 77:107–110.
- Coller BS, et al: Management of severe ITP during pregnancy with intravenous immunoglobulin (IVIgG). Clin Res 1985; 33:545A.
- Tchernia G, et al: Management of immune thrombocytopenia in pregnancy: Response to infusions of immunoglobulins. Am J Obstet Gynecol 1984; 148:225–226.
- Newland AC, et al: Intravenous IgG for autoimmune thrombocytopenia in pregnancy. N Engl J Med 1984; 310:261–262.
- Morgenstern GR, et al: Autoimmune thrombocytopenia in pregnancy: New approach to management. Br Med J 1983; 287:584.
- Ciccimarra F, et al: Treatment of neonatal passive immune thrombocytopenia. J Pediat 1984; 105:677–678.
- Rose VL, and Gordon LI: Idiopathic thrombocytopenic purpura in pregnancy. Successful management with immunoglobulin infusion. JAMA 1985; 254:2626–2628.
- Gounder MP, et al: Intravenous gammaglobulin therapy in the management of a patient with idiopathic thrombocytopenic purpura and a warm autoimmune erythrocyte panagglutinin during pregnancy. Obstet Gynecol 1986; 67:741–746.
- Siber GR, Werner BG, Halsey NA, et al: Interference of immune globulin with measles and rubella immunisation. J Pediatr 1993; 122:204–211.
- American Academy of Pediatrics, Committee on Infectious Diseases: Recommended timing of routine measles immunization for children who have recently received immune globulin preparations. Pediatrics 1994; 93:682–685.
- Centers of Disease Control and Prevention. Measles, mumps, and rubella-vaccine use and strategies for elimination of measles, rubella, and congenital rubella syndrome and control of mumps: recommendations of the advisory committee on immunization practices (ACIP). MMWR, Morbidity and Mortality Weekly Report. May 22, 1998; vol 47/No. RR-8, 1–57.
- Copelan EA, Strohn PL, Kennedy MS, Tutschka PJ: Hemolysis following intravenous immune globulin therapy. Transfusion 1986; 26:410–412.
- Thomas MJ, Misbah SA, Chapel HM, Jones M, Elrington G, Newsom-Davis J: Hemolysis after high-dose intravenous Ig. Blood 1993; 15:3789.
- Wilson JR, Bhoopalam N, Fisher M. Hemolytic anemia associated with intravenous immunoglobulin. Muscle & Nerve 1997; 20:1142–1145.
- Kessary-Shoham H, Levy Y, Shoenfeld Y, Lorber M, Gershon H: In vivo administration of intravenous immunoglobulin (IVIg) can lead to enhanced erythrocyte sequestration. J Autoimmun 1999; 13:129–135.
- Rizk A, Gorson KC, Kenney L, Weinstein R: Transfusion-related acute lung injury after the infusion of IVIG. Transfusion 2001; 41:264–268.
- Phillips AO: Renal failure and intravenous immunoglobulin [letter; comment]. Clin Nephrol 1992; 37:217.
- Cunningham-Rundles C, Day NK, Wahn V, et al: Reactions to intravenous gamma globulin infusions and immune complex formation, in Nydegger UE (ed): Immunohemotherapy: A Guide to Immunoglobulin Prophylaxis and Therapy. London, Academic Press, 1981, pp 447–449.
- Pierce LR, Jain N: Risks associated with the use of intravenous immunoglobulin. Trans Med Rev 2003; 17:241–251.
- Aukrust P, Froland SS, Liabakk N-B, Müller F., et al: Release of cytokines, soluble cytokine receptors, and interleukin-1 receptor antagonist after intravenous immunoglobulin administration in vivo. Blood 1994; 84:2136–2143.
- Bagdasarian A, Tonetta S, Harel W, Mamidi R., Uemura Y: IVIG adverse reactions: potential role of cytokines and vasoactive substances. Vox Sang 1998; 74:74–82.
- Bussel JB, Pham LC, Hilgartner MW, et al: Long-term maintenance of adults with ITP using intravenous gamma globulin. Abstract, American Society of Hematology. New Orleans, December, 1985.
- Imbach PA, Kühne T, Holländer G: Immunologic aspects in the pathogenesis and treatment of immune thrombocytopenic purpura in children. Current opinion in Pediatrics 1997; 9:35–40.
- Anderson W, Bethea W: Renal lesions following administration of hypertonic solutions of sucrose. JAMA 1940; 114:1983–1987.
- Lindberg H, Wald A: Renal lesions following the administration of hypertonic solutions: Arch Intern Med 1939; 63:907–918.
- Rigdon RH, Cardwell ES: Renal lesions following the intravenous injection of hypertonic solution of sucrose: A clinical and experimental study. Arch Intern Med 1942; 69:670–690.
- SPL UNCLASSIFIED SECTION
-
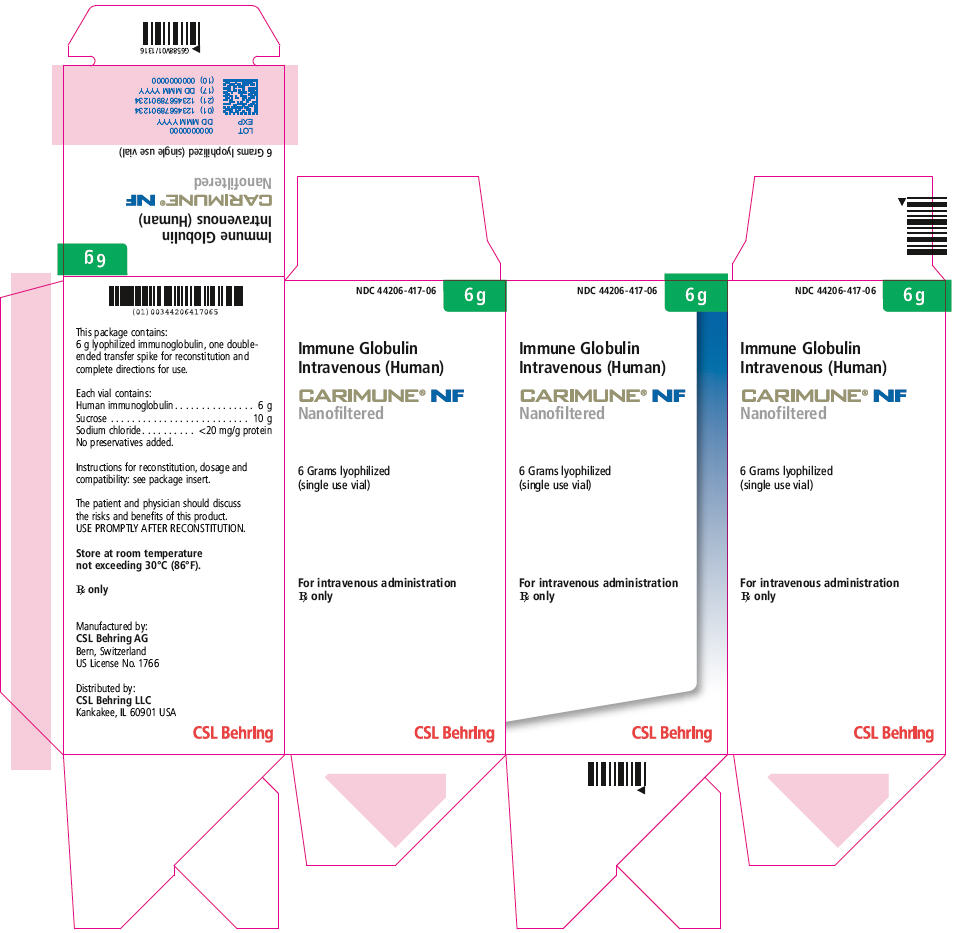
PRINCIPAL DISPLAY PANEL - 6 g Vial Carton
NDC: 44206-417-06
6 g
Immune Globulin
Intravenous (Human)CARIMUNE® NF
Nanofiltered6 Grams lyophilized
(single use vial)For intravenous administration
Rx onlyCSL Behring
-
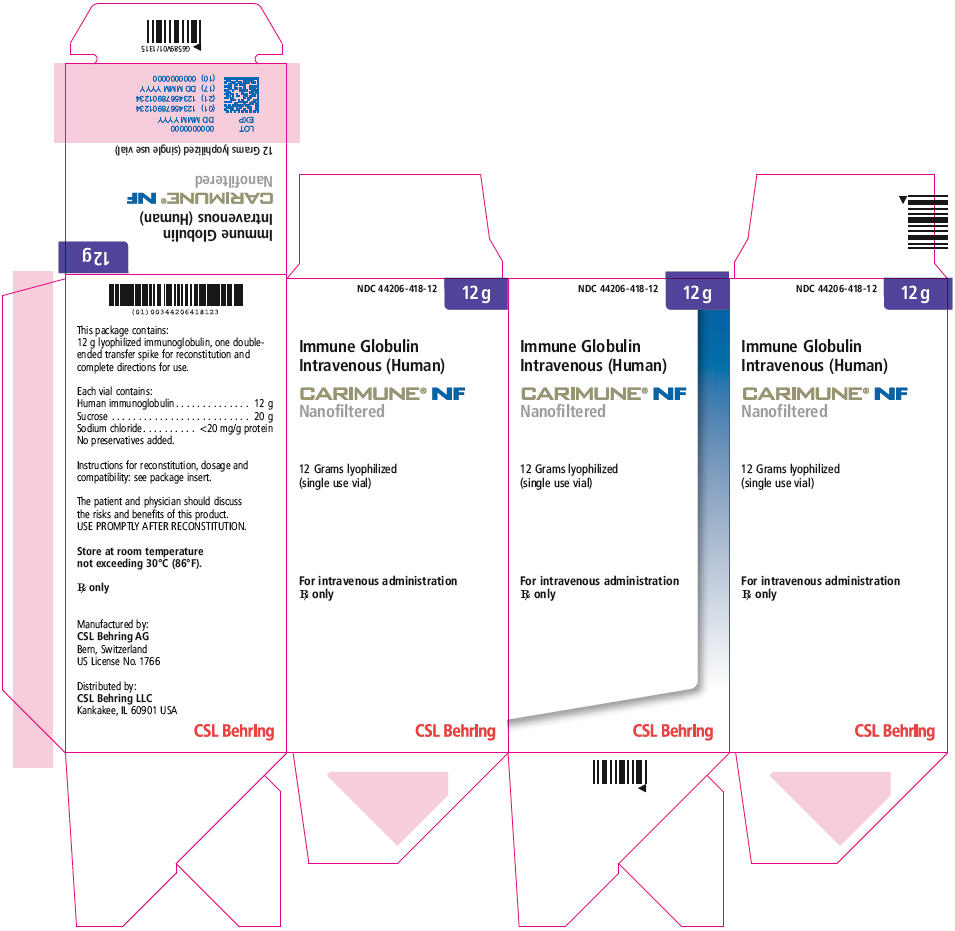
PRINCIPAL DISPLAY PANEL - 12 g Vial Carton
NDC: 44206-418-12
12 g
Immune Globulin
Intravenous (Human)CARIMUNE® NF
Nanofiltered12 Grams lyophilized
(single use vial)For intravenous administration
Rx onlyCSL Behring
-
INGREDIENTS AND APPEARANCE
CARIMUNE NANOFILTERED
human immunoglobulin g injection, powder, lyophilized, for solutionProduct Information Product Type PLASMA DERIVATIVE Item Code (Source) NDC: 44206-417 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength HUMAN IMMUNOGLOBULIN G (UNII: 66Y330CJHS) (HUMAN IMMUNOGLOBULIN G - UNII:66Y330CJHS) HUMAN IMMUNOGLOBULIN G 6 g Inactive Ingredients Ingredient Name Strength Sucrose (UNII: C151H8M554) 10 g Sodium Chloride (UNII: 451W47IQ8X) 20 mg Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 44206-417-06 1 in 1 CARTON 1 NDC: 44206-417-91 1 in 1 VIAL, SINGLE-USE; Type 9: Other Type of Part 3 Combination Product (e.g., Drug/Device/Biological Product) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA102367 02/10/2009 05/31/2021 CARIMUNE NANOFILTERED
human immunoglobulin g injection, powder, lyophilized, for solutionProduct Information Product Type PLASMA DERIVATIVE Item Code (Source) NDC: 44206-418 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength HUMAN IMMUNOGLOBULIN G (UNII: 66Y330CJHS) (HUMAN IMMUNOGLOBULIN G - UNII:66Y330CJHS) HUMAN IMMUNOGLOBULIN G 12 g Inactive Ingredients Ingredient Name Strength Sucrose (UNII: C151H8M554) 20 g Sodium Chloride (UNII: 451W47IQ8X) 20 mg Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 44206-418-12 1 in 1 CARTON 1 NDC: 44206-418-92 1 in 1 VIAL, SINGLE-USE; Type 9: Other Type of Part 3 Combination Product (e.g., Drug/Device/Biological Product) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA102367 02/10/2009 05/31/2021 Labeler - CSL Behring AG (481152762) Establishment Name Address ID/FEI Business Operations CSL Behring AG 481152762 MANUFACTURE
Trademark Results [Carimune]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 CARIMUNE 76345581 2782715 Live/Registered |
CSL BEHRING AG 2001-12-06 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.