CROMOLYN SODIUM solution, concentrate
CROMOLYN SODIUM by
Drug Labeling and Warnings
CROMOLYN SODIUM by is a Prescription medication manufactured, distributed, or labeled by Rising Pharma Holdings, Inc., Woodstock Sterile Solutions, Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
- SPL UNCLASSIFIED SECTION
-
DESCRIPTION:
Each 5 mL ampule of Cromolyn Sodium Oral Solution, Concentrate contains 100 mg cromolyn sodium, USP, in purified water. Cromolyn sodium is a hygroscopic, white powder having little odor. It may leave a slightly bitter aftertaste. Cromolyn Sodium Oral Solution, Concentrate is clear, colorless, and sterile. It is intended for oral use.
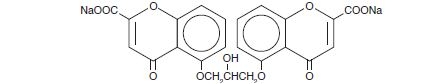
Chemically, cromolyn sodium is disodium 5,5’-[(2-hydroxy-trimethylene) dioxy]bis[4-oxo-4H-1-benzopyran-2-carboxylate] The empirical formula is C23H14Na2O11; the molecular weight is 512.34. Its chemical structure is:
Pharmacologic Category: Mast cell stabilizer
Therapeutic Category: Antiallergic
-
CLINICAL PHARMACOLOGY:
In vitro and in vivo animal studies have shown that cromolyn sodium inhibits the release of mediators from sensitized mast cells. Cromolyn sodium acts by inhibiting the release of histamine and leukotrienes (SRS-A) from the mast cell. Cromolyn sodium has no intrinsic vasoconstrictor, antihistamine, or glucocorticoid activity.
Cromolyn sodium is poorly absorbed from the gastrointestinal tract. No more than 1% of an administered dose is absorbed by humans after oral administration, the remainder being excreted in the feces. Very little absorption of cromolyn sodium was seen after oral administration of 500 mg by mouth to each of 12 volunteers. From 0.28 to 0.50% of the administered dose was recovered in the first 24 hours of urinary excretion in 3 subjects. The mean urinary excretion of an administered dose over 24 hours in the remaining 9 subjects was 0.45%.
-
CLINICAL STUDIES:
Four randomized, controlled clinical trials were conducted with Cromolyn Sodium Oral Solution, Concentrate in patients with either cutaneous or systemic mastocytosis; two of which utilized a placebo-controlled crossover design, one utilized an active controlled (chlorpheniramine plus cimetidine) crossover design, and one utilized a placebo-controlled parallel group design. Due to the rare nature of this disease, only 36 patients qualified for study entry, of whom 32 were considered evaluable. Consequently, formal statistical analyses were not performed. Clinically significant improvement in gastrointestinal symptoms (diarrhea, abdominal pain) were seen in the majority of patients with some improvement also seen for cutaneous manifestations (urticaria, pruritus, flushing) and cognitive function. The benefit seen with Cromolyn Sodium Oral Solution, Concentrate 200 mg QID was similar to chlorpheniramine (4 mg QID) plus cimetidine (300 mg QID) for both cutaneous and systemic symptoms of mastocytosis.
Clinical improvement occurred within 2-6 weeks of treatment initiation and persisted for 2-3 weeks after treatment withdrawal. Cromolyn Sodium Oral Solution, Concentrate did not affect urinary histamine levels or peripheral eosinophilia, although neither of these variables appeared to correlate with disease severity. Positive clinical benefits were also reported for 37 of 51 patients who received Cromolyn Sodium Oral Solution, Concentrate in United States and foreign humanitarian programs.
- INDICATIONS AND USAGE:
- CONTRAINDICATIONS:
- WARNINGS:
-
PRECAUTIONS:
Carcinogenesis, Mutagenesis, and Impairment of Fertility:
In carcinogenicity studies in mice, hamsters, and rats, cromolyn sodium had no neoplastic effects at intraperitoneal doses up to 150 mg/kg three days per week for 12 months in mice, at intraperitoneal doses up to 53 mg/kg three days per week for 15 weeks followed by 17.5 mg/kg three days per week for 37 weeks in hamsters, and at subcutaneous doses up to 75 mg/kg six days per week for 18 months in rats. These doses in mice, hamsters, and rats are less than the maximum recommended daily oral dose in adults and children on a mg/m2 basis.
Cromolyn sodium showed no mutagenic potential in Ames Salmonella/microsome plate assays, mitotic gene conversion in Saccharomyces cerevisiae and in an in vitro cytogenetic study in human peripheral lymphocytes.
In rats, cromolyn sodium showed no evidence of impaired fertility at subcutaneous doses up to 175 mg/kg in males (approximately equal to the maximum recommended daily oral dose in adults on a mg/m2 basis) and 100 mg/kg in females (less than the maximum recommended daily oral dose in adults on a mg/m2 basis).
Pregnancy:
Pregnancy Category B. In reproductive studies in pregnant mice, rats, and rabbits, cromolyn sodium produced no evidence of fetal malformations at subcutaneous doses up to 540 mg/kg in mice (approximately equal to the maximum recommended daily oral dose in adults on a mg/m2 basis) and 164 mg/kg in rats (less than the maximum recommended daily oral dose in adults on a mg/m2 basis) or at intravenous doses up to 485 mg/kg in rabbits (approximately 4 times the maximum recommended daily oral dose in adults on a mg/m2 basis). There are, however, no adequate and well controlled studies in pregnant women.
Because animal reproduction studies are not always predictive of human response, this drug should be used during pregnancy only if clearly needed.
Drug Interaction During Pregnancy: In pregnant mice, cromolyn sodium alone did not cause significant increases in resorptions or major malformations at subcutaneous doses up to 540 mg/kg (approximately equal to the maximum recommended daily oral dose in adults on a mg/m2 basis). Isoproterenol alone increased both resorptions and major malformations (primarily cleft palate) at a subcutaneous dose of 2.7 mg/kg (approximately 7 times the maximum recommended daily inhalation dose in adults on a mg/m2 basis). The incidence of major malformations increased further when cromolyn sodium at a subcutaneous dose of 540 mg/kg was added to isoproterenol at a subcutaneous dose of 2.7 mg/kg. No such interaction was observed in rats or rabbits.
Nursing Mothers:
It is not known whether this drug is excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when Cromolyn Sodium Oral Solution, Concentrate is administered to a nursing woman.
Pediatric Use:
In adult rats no adverse effects of cromolyn sodium were observed at oral doses up to 6144 mg/kg (approximately 25 times the maximum recommended daily oral dose in adults on a mg/m2 basis). In neonatal rats, cromolyn sodium increased mortality at oral doses of 1000 mg/kg or greater (approximately 9 times the maximum recommended daily oral dose in infants on a mg/m2 basis) but not at doses of 300 mg/kg or less (approximately 3 times the maximum recommended daily oral dose in infants on a mg/m2 basis). Plasma and kidney concentrations of cromolyn after oral administration to neonatal rats were up to 20 times greater than those in older rats. In term infants up to six months of age, available clinical data suggest that the dose should not exceed 20 mg/kg/day. The use of this product in pediatric patients less than two years of age should be reserved for patients with severe disease in which the potential benefits clearly outweigh the risks.
Geriatric Use:
Clinical studies of Cromolyn Sodium Oral Solution, Concentrate did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
-
ADVERSE REACTIONS:
Most of the adverse events reported in mastocytosis patients have been transient and could represent symptoms of the disease. The most frequently reported adverse events in mastocytosis patients who have received Cromolyn Sodium Oral Solution, Concentrate during clinical studies were headache and diarrhea, each of which occurred in 4 of the 87 patients. Pruritus, nausea, and myalgia were each reported in 3 patients and abdominal pain, rash, and irritability in 2 patients each. One report of malaise was also recorded.
To report SUSPECTED ADVERSE REACTIONS, contact Rising Pharmaceuticals, Inc. at 1-866-562-4597 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Other Adverse Events: Additional adverse events have been reported during studies in other clinical conditions and from worldwide postmarketing experience. In most cases the available information is incomplete and attribution to the drug cannot be determined. The majority of these reports involve the gastrointestinal system and include: diarrhea, nausea, abdominal pain, constipation, dyspepsia, flatulence, glossitis, stomatitis, vomiting, dysphagia, esophagospasm.
Other less commonly reported events (the majority representing only a single report) include the following:
Skin:
pruritus, rash, urticaria/angioedema, erythema/ burning, photosensitivity
Musculoskeletal:
arthralgia, myalgia, stiffness/weakness of legs
Neurologic:
headache, dizziness, hypoesthesia, paresthesia, migraine, convulsions, flushing
Psychiatric:
psychosis, anxiety, depression, hallucinations, behavior change, insomnia, nervousness
Heart Rate:
tachycardia, premature ventricular contractions (PVCs), palpitations
Respiratory:
pharyngitis, dyspnea
Miscellaneous:
fatigue, edema, unpleasant taste, chest pain, postprandial lightheadedness and lethargy, dysuria, urinary frequency, purpura, hepatic function test abnormal, polycythemia, neutropenia, pancytopenia, tinnitus, lupus erythematosus (LE) syndrome
-
DOSAGE AND ADMINISTRATION:
NOT FOR INHALATION OR INJECTION. SEE DIRECTIONS FOR USE.
The usual starting dose is as follows:
Adults and Adolescents (13 Years and Older): Two ampules four times daily, taken one-half hour before meals and at bedtime.
Children 2-12 Years: One ampule four times daily, taken one-half hour before meals and at bedtime.
Pediatric Patients Under 2 Years: Not recommended.
If satisfactory control of symptoms is not achieved within two to three weeks, the dosage may be increased but should not exceed 40 mg/kg/day. Patients should be advised that the effect of Cromolyn Sodium Oral Solution, Concentrate therapy is dependent upon its administration at regular intervals, as directed.
Maintenance Dose:
Once a therapeutic response has been achieved, the dose may be reduced to the minimum required to maintain the patient with a lower degree of symptomatology. To prevent relapses, the dosage should be maintained.
Administration:
Cromolyn Sodium Oral Solution, Concentrate should be administered as a solution at least 1/2 hour before meals and at bedtime after preparation according to the following directions:
- Break open ampule(s) and squeeze liquid contents of ampule(s) into a glass of water.
- Stir solution.
- Drink all of the liquid.
-
HOW SUPPLIED:
Cromolyn Sodium Oral Solution, Concentrate is an unpreserved, colorless solution supplied in a low density polyethylene plastic unit dose ampule with 8 ampules per foil pouch.
Each 5 mL ampule contains 100 mg cromolyn sodium, USP, in purified water.
NDC: 16571-600-96 96 ampules x 5 mL
(12 pouches x 8 ampules)Cromolyn Sodium Oral Solution, Concentrate should be stored between 20° – 25°C (68° – 77°F) [see USP Controlled Room Temperature] and protected from light. Do not use if it contains a precipitate or becomes discolored. Keep out of the reach of children.
Store ampules in foil pouch until ready for use. - SPL UNCLASSIFIED SECTION
-
PATIENT INSTRUCTIONS
Cromolyn Sodium
Oral Solution, ConcentrateFor Oral Use Only – Not for Inhalation or Injection.
MUST BE DILUTED.
How to Use Cromolyn Sodium Oral Solution, Concentrate:
As with all prescription drugs, follow the directions for dosage that your physician recommends.
The effect of Cromolyn Sodium Oral Solution, Concentrate therapy is dependent upon its administration at REGULAR intervals, for as long as recommended by your physician.
Usual Starting Dose:
Adults and Adolescents (13 Years and Older):
Two ampules four times daily, taken one-half hour before meals and at bedtime.Children 2-12 Years:
One ampule four times daily, taken one-half hour before meals and at bedtime.Note:
Your physician may decide to increase OR decrease your dosage to achieve optimum results with Cromolyn Sodium Oral Solution, Concentrate. However, do not change your dose or stop taking Cromolyn Sodium Oral Solution, Concentrate without first consulting your physician.Care & Storage:
Cromolyn Sodium Oral Solution, Concentrate should be stored between 15°-30°C (59°-86°F) and protected from light. Do not use if it contains a precipitate (particles or cloudiness) or becomes discolored. Keep out of the reach of children.
Store ampules in foil pouch until ready for use.Recycling Information: Cromolyn Sodium Oral Solution, Concentrate ampules are made with a low density polyethylene plastic (recycling material code:
 LDPE).
LDPE).Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
Directions for Use: 
1. Open foil
pouch by tearing
at serrated edge as
shown.
2. Remove
ampule(s) from
the strip.
3. Open the ampule
by twisting off the
tabbed top section.
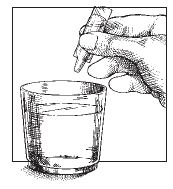
4. Squeeze liquid
contents into a glass
of water. Stir
solution. Drink all
of the liquid.
Discard the empty ampule.
Manufactured by:
Catalent Pharma Solutions, LLC
Woodstock, IL 60098
Distributed by:
Rising Pharmaceuticals, Inc.
Saddle Brook, NJ 07663Rev. 08/18
STW-RIS60101 -
Cromolyn Sodium Oral Concentrate Carton
Contains: 96 (12 pouches x
8 - five mL unit dose ampules)
SterileRising® NDC: 16571-600-96
Cromolyn Sodium
Oral Solution, Concentrate
100 mg/5 mLFOR ORAL SOLUTION ONLY –
NOT FOR INHALATION OR INJECTIONRx Only
Must Be DilutedStore ampules in foil pouch until ready to use
DESCRIPTION: Each 5 mL ampule contains 100 mg cromolyn sodium, USP, in purified water.
NOTE: See package circular for full prescribing information including contraindications, warnings and precautions.
Cromolyn Sodium Oral Solution, Concentrate should be stored between 20° - 25°C (68° - 77°F) [see USP Controlled Room Temperature] and protect from light. Do not use if it contains a precipitate or becomes discolored. Keep out of the reach of children.

-
INGREDIENTS AND APPEARANCE
CROMOLYN SODIUM
cromolyn sodium solution, concentrateProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 16571-600 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength CROMOLYN SODIUM (UNII: Q2WXR1I0PK) (CROMOLYN - UNII:Y0TK0FS77W) CROMOLYN SODIUM 100 mg in 5 mL Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 16571-600-96 12 in 1 CARTON 12/05/2011 1 8 in 1 POUCH 1 5 mL in 1 AMPULE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA202583 12/05/2011 Labeler - Rising Pharmaceuticals, Inc. (835513529) Registrant - Rising Pharmaceuticals, Inc. (835513529) Establishment Name Address ID/FEI Business Operations Catalent Pharma Solutions, LLC 043911403 ANALYSIS(16571-600) , MANUFACTURE(16571-600) , PACK(16571-600)
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.