TIBSOVO- ivosidenib tablet, film coated
TIBSOVO by
Drug Labeling and Warnings
TIBSOVO by is a Prescription medication manufactured, distributed, or labeled by Servier Pharmaceutical LLC. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use TIBSOVO safely and effectively. See full prescribing information for TIBSOVO.
TIBSOVO® (ivosidenib tablets), for oral use
Initial U.S. Approval: 2018WARNING: DIFFERENTIATION SYNDROME IN AML AND MDS
See full prescribing information for complete boxed warning.
Patients treated with TIBSOVO have experienced symptoms of differentiation syndrome, which can be fatal. If differentiation syndrome is suspected, initiate corticosteroid therapy and hemodynamic monitoring until symptom resolution (5.1, 6.1).
RECENT MAJOR CHANGES
INDICATIONS AND USAGE
TIBSOVO is an isocitrate dehydrogenase-1 (IDH1) inhibitor indicated for patients with a susceptible IDH1 mutation as detected by an FDA-approved test with:
Newly Diagnosed Acute Myeloid Leukemia (AML)
- In combination with azacitidine or as monotherapy for the treatment of newly diagnosed AML in adults 75 years or older, or who have comorbidities that preclude use of intensive induction chemotherapy (1.1).
Relapsed or refractory AML
- For the treatment of adult patients with relapsed or refractory AML (1.2).
Relapsed or refractory Myelodysplastic Syndromes (MDS)
- For the treatment of adult patients with relapsed or refractory myelodysplastic syndromes (1.3).
Locally Advanced or Metastatic Cholangiocarcinoma
- For the treatment of adult patients with locally advanced or metastatic cholangiocarcinoma who have been previously treated (1.4).
DOSAGE AND ADMINISTRATION
500 mg orally once daily with or without food until disease progression or unacceptable toxicity (2.2). Avoid a high-fat meal.
DOSAGE FORMS AND STRENGTHS
Tablets: 250 mg (3).
CONTRAINDICATIONS
None (4).
WARNINGS AND PRECAUTIONS
- QTc Interval Prolongation: Monitor electrocardiograms and electrolytes. If QTc interval prolongation occurs, dose reduce or withhold, then resume dose or permanently discontinue TIBSOVO (2.3, 5.2).
- Guillain-Barré Syndrome: Monitor patients for signs and symptoms of new motor and/or sensory findings. Permanently discontinue TIBSOVO in patients who are diagnosed with Guillain-Barré syndrome (2.3, 5.3).
ADVERSE REACTIONS
The most common adverse reactions including laboratory abnormalities (≥ 25%) in patients with AML are leukocytes decreased, diarrhea, hemoglobin decreased, platelets decreased, glucose increased, fatigue, alkaline phosphatase increased, edema, potassium decreased, nausea, vomiting, phosphate decreased, decreased appetite, sodium decreased, leukocytosis, magnesium decreased, aspartate aminotransferase increased, arthralgia, dyspnea, uric acid increased, abdominal pain, creatinine increased, mucositis, rash, electrocardiogram QT prolonged, differentiation syndrome, calcium decreased, neutrophils decreased, and myalgia (6.1).
The most common adverse reactions including laboratory abnormalities (≥25%) in patients with relapsed or refractory MDS are creatinine increased, hemoglobin decrease, arthralgia, albumin decreased, aspartate aminotransferase increased, fatigue, diarrhea, cough, sodium decreased, mucositis, decreased appetite, myalgia, phosphate decreased, pruritus, and rash (6.1).
The most common adverse reactions (≥15%) in patients with cholangiocarcinoma are fatigue, nausea, abdominal pain, diarrhea, cough, decreased appetite, ascites, vomiting, anemia, and rash (6.1).
The most common laboratory abnormalities (≥10%) in patients with cholangiocarcinoma are hemoglobin decreased, aspartate aminotransferase increased, and bilirubin increased (6.1).
To report SUSPECTED ADVERSE REACTIONS, contact Servier Pharmaceuticals at 1-800-807-6124 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- Strong or Moderate CYP3A4 Inhibitors: Reduce TIBSOVO dose with strong CYP3A4 inhibitors. Monitor patients for increased risk of QTc interval prolongation (2.4, 5.2, 7.1, 12.3).
- Strong CYP3A4 Inducers: Avoid concomitant use with TIBSOVO (7.1, 12.3).
- Sensitive CYP3A4 substrates: Avoid concomitant use with TIBSOVO (7.2, 12.3).
- QTc Prolonging Drugs: Avoid concomitant use with TIBSOVO. If co-administration is unavoidable, monitor patients for increased risk of QTc interval prolongation (5.2, 7.1).
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 10/2023
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: DIFFERENTIATION SYNDROME IN AML AND MDS
1 INDICATIONS AND USAGE
1.1 Newly Diagnosed Acute Myeloid Leukemia
1.2 Relapsed or Refractory Acute Myeloid Leukemia
1.3 Relapsed or Refractory Myelodysplastic Syndromes
1.4 Locally Advanced or Metastatic Cholangiocarcinoma
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection
2.2 Recommended Dosage
2.3 Monitoring and Dosage Modifications for Toxicities
2.4 Dosage Modification for Use with Strong CYP3A4 Inhibitors
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Differentiation Syndrome in AML and MDS
5.2 QTc Interval Prolongation
5.3 Guillain-Barré Syndrome
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on Ivosidenib
7.2 Effect of Ivosidenib on Other Drugs
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Newly Diagnosed AML
14.2 Relapsed or Refractory AML
14.3 Relapsed or Refractory MDS
14.4 Locally Advanced or Metastatic Cholangiocarcinoma
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: DIFFERENTIATION SYNDROME IN AML AND MDS
Patients treated with TIBSOVO have experienced symptoms of differentiation syndrome, which can be fatal. Symptoms may include fever, dyspnea, hypoxia, pulmonary infiltrates, pleural or pericardial effusions, rapid weight gain or peripheral edema, hypotension, and hepatic, renal, or multi-organ dysfunction. If differentiation syndrome is suspected, initiate corticosteroid therapy and hemodynamic monitoring until symptom resolution [see Warnings and Precautions (5.1) and Adverse Reactions (6.1)].
-
1 INDICATIONS AND USAGE
1.1 Newly Diagnosed Acute Myeloid Leukemia
TIBSOVO is indicated in combination with azacitidine or as monotherapy for the treatment of newly diagnosed acute myeloid leukemia (AML) with a susceptible isocitrate dehydrogenase-1 (IDH1) mutation as detected by an FDA-approved test in adults 75 years or older, or who have comorbidities that preclude use of intensive induction chemotherapy [see Dosage and Administration (2.1), Clinical Pharmacology (12.1) and Clinical Studies (14.1)].
1.2 Relapsed or Refractory Acute Myeloid Leukemia
TIBSOVO is indicated for the treatment of adult patients with relapsed or refractory acute myeloid leukemia (AML) with a susceptible isocitrate dehydrogenase-1 (IDH1) mutation as detected by an FDA-approved test [see Dosage and Administration (2.1), Clinical Pharmacology (12.1) and Clinical Studies (14.2)].
1.3 Relapsed or Refractory Myelodysplastic Syndromes
TIBSOVO is indicated for the treatment of adult patients with relapsed or refractory myelodysplastic syndromes (MDS) with a susceptible isocitrate dehydrogenase-1 (IDH1) mutation as detected by an FDA-approved test [see Dosage and Administration (2.1), Clinical Pharmacology (12.1) and Clinical Studies (14.3)].
1.4 Locally Advanced or Metastatic Cholangiocarcinoma
TIBSOVO is indicated for the treatment of adult patients with previously treated, locally advanced or metastatic cholangiocarcinoma with an isocitrate dehydrogenase-1 (IDH1) mutation as detected by an FDA-approved test [see Dosage and Administration (2.1), Clinical Pharmacology (12.1), and Clinical Studies (14.4)].
-
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection
Select patients for treatment with TIBSOVO based on the presence of IDH1 mutations [see Clinical Studies (14.1, 14.2, 14.3, 14.4)].
Information on FDA-approved tests for the detection of IDH1 mutations in AML, MDS, and cholangiocarcinoma is available at http://www.fda.gov/CompanionDiagnostics.
2.2 Recommended Dosage
The recommended dosage of TIBSOVO is 500 mg taken orally once daily until disease progression or unacceptable toxicity [see Clinical Studies (14.1, 14.2, 14.3, 14.4)].
For patients with AML or MDS without disease progression or unacceptable toxicity, continue TIBSOVO for a minimum of 6 months to allow time for clinical response.
- Administer TIBSOVO with or without food.
- Do not administer TIBSOVO with a high-fat meal [see Warnings and Precautions (5.2) and Clinical Pharmacology (12.3)].
- Do not split, crush, or chew TIBSOVO tablets.
- Administer TIBSOVO tablets orally about the same time each day.
- If a dose of TIBSOVO is vomited, do not administer a replacement dose; wait until the next scheduled dose is due.
- If a dose of TIBSOVO is missed or not taken at the usual time, administer the dose as soon as possible and at least 12 hours prior to the next scheduled dose. Return to the normal schedule the following day. Do not administer 2 doses within 12 hours.
Newly Diagnosed AML (Combination Regimen)
Start TIBSOVO administration on Cycle 1 Day 1 in combination with azacitidine 75 mg/m2 subcutaneously or intravenously once daily on Days 1-7 (or Days 1-5 and 8-9) of each 28-day cycle [see Clinical Studies (14.1)]. Refer to the Prescribing Information for azacitidine for additional dosing information.
2.3 Monitoring and Dosage Modifications for Toxicities
Obtain an electrocardiogram (ECG) prior to treatment initiation. Monitor ECGs at least once weekly for the first 3 weeks of therapy and then at least once monthly for the duration of therapy [see Warnings and Precautions (5.2)]. Manage any abnormalities promptly.
Interrupt dosing or reduce dose for toxicities. See Table 1 for dosage modification guidelines.
Table 1: Recommended Dosage Modifications for TIBSOVO Adverse Reactions Recommended Action - * Grade 1 is mild, Grade 2 is moderate, Grade 3 is severe, Grade 4 is life-threatening; grading based on Common Terminology Criteria for Adverse Events (CTCAE) version 4.03.
- Differentiation syndrome [see Warnings and Precautions (5.1)]
- If differentiation syndrome is suspected, administer systemic corticosteroids and initiate hemodynamic monitoring until symptom resolution and for a minimum of 3 days.
- Interrupt TIBSOVO if severe signs and/or symptoms persist for more than 48 hours after initiation of systemic corticosteroids.
- Resume TIBSOVO when signs and symptoms improve to Grade 2* or lower.
- Noninfectious leukocytosis (white blood cell [WBC] count greater than 25 × 109/L or an absolute increase in total WBC of greater than 15 × 109/L from baseline)
- Initiate treatment with hydroxyurea, as per standard institutional practices, and leukapheresis if clinically indicated.
- Taper hydroxyurea only after leukocytosis improves or resolves.
- Interrupt TIBSOVO if leukocytosis is not improved with hydroxyurea, and then resume TIBSOVO at 500 mg daily when leukocytosis has resolved.
- QTc interval greater than 480 msec to 500 msec [see Warnings and Precautions (5.2) and Drug Interactions (7.1)]
- Monitor and supplement electrolyte levels as clinically indicated.
- Review and adjust concomitant medications with known QTc interval-prolonging effects.
- Interrupt TIBSOVO.
- Restart TIBSOVO at 500 mg once daily after the QTc interval returns to less than or equal to 480 msec.
- Monitor ECGs at least weekly for 2 weeks following resolution of QTc prolongation.
- QTc interval greater than 500 msec [see Warnings and Precautions (5.2) and Drug Interactions (7.1)]
- Monitor and supplement electrolyte levels as clinically indicated.
- Review and adjust concomitant medications with known QTc interval-prolonging effects.
- Interrupt TIBSOVO.
- Resume TIBSOVO at a reduced dose of 250 mg once daily when QTc interval returns to within 30 msec of baseline or less than or equal to 480 msec.
- Monitor ECGs at least weekly for 2 weeks following resolution of QTc prolongation.
- Consider re-escalating the dose of TIBSOVO to 500 mg daily if an alternative etiology for QTc prolongation can be identified.
- QTc interval prolongation with signs/symptoms of life-threatening arrhythmia [see Warnings and Precautions (5.2)]
- Discontinue TIBSOVO permanently.
- Guillain-Barré syndrome [see Warnings and Precautions (5.3)]
- Discontinue TIBSOVO permanently.
- Other Grade 3* adverse reactions
As monotherapy in AML and MDS: - Interrupt TIBSOVO until toxicity resolves to Grade 2* or lower.
- Resume TIBSOVO at 250 mg once daily; may increase to 500 mg once daily if toxicities resolve to Grade 1* or lower.
- If Grade 3* or higher toxicity recurs, discontinue TIBSOVO.
- Interrupt TIBSOVO until toxicity resolves to Grade 1* or lower, or baseline, then resume at 500 mg daily (Grade 3 toxicity) or 250 mg daily (Grade 4 toxicity).
- If Grade 3 toxicity recurs (a second time), reduce TIBSOVO dose to 250 mg daily until the toxicity resolves, then resume 500 mg daily.
- If Grade 3 toxicity recurs (a third time), or Grade 4 toxicity recurs, discontinue TIBSOVO.
Patients with AML or MDS
Assess blood counts and blood chemistries prior to the initiation of TIBSOVO, at least once weekly for the first month, once every other week for the second month, and once monthly for the duration of therapy.
Monitor blood creatine phosphokinase weekly for the first month of therapy.
2.4 Dosage Modification for Use with Strong CYP3A4 Inhibitors
If a strong CYP3A4 inhibitor must be coadministered, reduce the TIBSOVO dose to 250 mg once daily. If the strong inhibitor is discontinued, increase the TIBSOVO dose (after at least 5 half-lives of the strong CYP3A4 inhibitor) to the recommended dose of 500 mg once daily.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Differentiation Syndrome in AML and MDS
Differentiation syndrome is associated with rapid proliferation and differentiation of myeloid cells and may be life-threatening or fatal.
Symptoms of differentiation syndrome in patients treated with TIBSOVO included noninfectious leukocytosis, peripheral edema, pyrexia, dyspnea, pleural effusion, hypotension, hypoxia, pulmonary edema, pneumonitis, pericardial effusion, rash, fluid overload, tumor lysis syndrome and creatinine increased.
In the combination study AG120-C-009, 15% (11/71) patients with newly diagnosed AML treated with TIBSOVO plus azacitidine experienced differentiation syndrome [see Adverse Reactions (6.1)]. Of the 11 patients with newly diagnosed AML who experienced differentiation syndrome with TIBSOVO plus azacitidine 8 (73%) recovered. Differentiation syndrome occurred as early as 3 days after start of therapy and during the first month on treatment.
In the monotherapy clinical trial AG120-C-001, 25% (7/28) of patients with newly diagnosed AML and 19% (34/179) of patients with relapsed or refractory AML treated with TIBSOVO experienced differentiation syndrome [see Adverse Reactions (6.1)]. Of the 7 patients with newly diagnosed AML who experienced differentiation syndrome, 6 (86%) patients recovered. Of the 34 patients with relapsed or refractory AML who experienced differentiation syndrome, 27 (79%) patients recovered after treatment or after dose interruption of TIBSOVO. Differentiation syndrome occurred as early as 1 day and up to 3 months after TIBSOVO initiation and has been observed with or without concomitant leukocytosis.
In the monotherapy clinical trial AG120-C-001, 11% (2/19) of patients with relapsed or refractory MDS treated with TIBSOVO experienced differentiation syndrome [see Adverse Reactions (6.1)]. Of the 2 patients who experienced differentiation syndrome, both recovered after treatment or after dose interruption of TIBSOVO. Differentiation syndrome occurred as early as 1 day and up to 3 months after TIBSOVO initiation and has been observed with or without concomitant leukocytosis.
If differentiation syndrome is suspected, initiate dexamethasone 10 mg IV every 12 hours (or an equivalent dose of an alternative oral or IV corticosteroid) and hemodynamic monitoring until improvement [see Dosage and Administration (2.3)]. If concomitant noninfectious leukocytosis is observed, initiate treatment with hydroxyurea or leukapheresis, as clinically indicated. Taper corticosteroids and hydroxyurea after resolution of symptoms and administer corticosteroids for a minimum of 3 days. Symptoms of differentiation syndrome may recur with premature discontinuation of corticosteroid and/or hydroxyurea treatment. If severe signs and/or symptoms persist for more than 48 hours after initiation of corticosteroids, interrupt TIBSOVO until signs and symptoms are no longer severe [see Dosage and Administration (2.3)].
5.2 QTc Interval Prolongation
Patients treated with TIBSOVO can develop QT (QTc) prolongation and ventricular arrhythmias [see Clinical Pharmacology (12.2)].
Of the 71 patients with newly diagnosed AML treated with TIBSOVO in combination with azacitidine in the clinical trial (Study AG120-C-009), 10 (14%) were found to have a heart-rate corrected QT interval (using Fridericia's method) (QTcF) greater than 500 msec and 15 out of 69 (22%) had an increase from baseline QTcF greater than 60 msec [see Adverse Reactions (6.1)]. The clinical trial excluded patients with a QTcF ≥ 470 msec or other factors that increased the risk of QT prolongation or arrhythmic events (e.g. NYHA Class III or IV congestive heart failure, hypokalemia, family history of long QT interval syndrome).
Of the 265 patients with hematological malignancies, including patients with AML and MDS, treated with TIBSOVO monotherapy in the clinical trial (AG120-C-001), 9% were found to have a QTc interval greater than 500 msec and 14% of patients had an increase from baseline QTc greater than 60 msec [see Adverse Reactions (6.1)]. One patient developed ventricular fibrillation attributed to TIBSOVO. The clinical trial excluded patients with baseline QTc of ≥ 450 msec (unless the QTc ≥ 450 msec was due to a pre-existing bundle branch block) or with a history of long QT syndrome or uncontrolled or significant cardiovascular disease.
Of the 123 patients with cholangiocarcinoma treated with TIBSOVO in the clinical trial (Study AG120-C-005), 2% were found to have a QTc interval greater than 500 msec and 5% of patients had an increase from baseline QTc greater than 60 msec [see Adverse Reactions (6.1)]. The clinical trial excluded patients with a heart-rate corrected QT interval (using Fridericia's formula) (QTcF) ≥ 450 msec or other factors that increased the risk of QT prolongation or arrhythmic events (e.g., heart failure, hypokalemia, family history of long QT interval syndrome).
Concomitant use of TIBSOVO with drugs known to prolong the QTc interval (e.g., anti-arrhythmic medicines, fluoroquinolones, triazole anti-fungals, 5-HT3 receptor antagonists) and CYP3A4 inhibitors may increase the risk of QTc interval prolongation [see Drug Interactions (7.1), Clinical Pharmacology (12.2)]. Conduct monitoring of electrocardiograms (ECGs) and electrolytes [see Dosage and Administration (2.3)].
In patients with congenital long QTc syndrome, congestive heart failure, electrolyte abnormalities, or those who are taking medications known to prolong the QTc interval, more frequent monitoring may be necessary.
Interrupt TIBSOVO if QTc increases to greater than 480 msec and less than 500 msec. Interrupt and reduce TIBSOVO if QTc increases to greater than 500 msec. Permanently discontinue TIBSOVO in patients who develop QTc interval prolongation with signs or symptoms of life-threatening arrhythmia [See Dosage and Administration (2.3)].
5.3 Guillain-Barré Syndrome
Guillain-Barré syndrome can develop in patients treated with TIBSOVO. Guillain-Barré syndrome occurred in 0.8% (2/265) of patients treated with TIBSOVO in study AG120-C-001 [see Adverse Reactions (6.1)].
Monitor patients taking TIBSOVO for onset of new signs or symptoms of motor and/or sensory neuropathy such as unilateral or bilateral weakness, sensory alterations, paresthesias, or difficulty breathing. Permanently discontinue TIBSOVO in patients who are diagnosed with Guillain-Barré syndrome [see Dosage and Administration (2.3)].
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Differentiation Syndrome in AML and MDS [see Warnings and Precautions (5.1)]
- QTc Interval Prolongation [see Warnings and Precautions (5.2)]
- Guillain-Barré Syndrome [see Warnings and Precautions (5.3)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Acute Myeloid Leukemia
In AML, the safety population reflects exposure to TIBSOVO at 500 mg daily in combination with azacitidine or as monotherapy in patients in Studies AG120-C-009 (N=71) and AG120-C-001 (N=213), respectively [see Clinical Studies (14.1 and 14.2)]. In this safety population, the most common adverse reactions including laboratory abnormalities (≥ 25% in either trial) were leukocytes decreased, diarrhea, hemoglobin decreased, platelets decreased, glucose increased, fatigue, alkaline phosphatase increased, edema, potassium decreased, nausea, vomiting, phosphatase decreased, decreased appetite, sodium decreased, leukocytosis, magnesium decreased, aspartate aminotransferase increased, arthralgia, dyspnea, uric acid increased, abdominal pain, creatinine increased, mucositis, rash, electrocardiogram QT prolonged, differentiation syndrome, calcium decreased, neutrophils decreased, and myalgia.
Newly Diagnosed AML
TIBSOVO in Combination with Azacitidine
The safety of TIBSOVO was evaluated in AML patients treated in combination with azacitidine, in Study AG120-C-009 [see Clinical Studies (14.1)]. Patients received at least one dose of either TIBSOVO 500 mg daily (N=71) or placebo (N=73). Among patients who received TIBSOVO in combination with azacitidine, the median duration of exposure to TIBSOVO was 6 months (range 0 to 33 months). Thirty-four patients (48%) were exposed to TIBSOVO for at least 6 months and 22 patients (31%) were exposed for at least 1 year.
Common (≥ 5%) serious adverse reactions in patients who received TIBSOVO in combination with azacitidine included differentiation syndrome (8%).
Fatal adverse reactions occurred in 4% of patients who received TIBSOVO in combination with azacitidine, due to differentiation syndrome (3%) and one case of cerebral ischemia.
Adverse reactions leading to discontinuation of TIBSOVO in ≥2% of patients were differentiation syndrome (3%) and pulmonary embolism (3%).
The most common (>5%) adverse reactions leading to dose interruption of TIBSOVO were neutropenia (25%), electrocardiogram QT prolonged (7%), and thrombocytopenia (7%).
Adverse reactions leading to dose reduction of TIBSOVO included electrocardiogram QT prolonged (8%), neutropenia (8%), and thrombocytopenia (1%).
The most common adverse reactions and laboratory abnormalities observed in Study AG120-C-009 are shown in Tables 2 and 3.
Table 2: Adverse Reactions (≥10%) in Patients with AML Who Received TIBSOVO + azacitidine with a Difference Between Arms of ≥ 2% Compared with Placebo + azacitidine in AG120-C-009 TIBSOVO + Azacitidine
N=71Placebo + Azacitidine
N=73Body System
Adverse ReactionAll Grades
n (%)Grade ≥3
n (%)All Grades
n (%)Grade ≥3
n (%)- * Grouped term includes vomiting and retching.
- † Differentiation syndrome can be associated with other commonly reported events such as peripheral edema, leukocytosis, pyrexia, dyspnea, pleural effusion, hypotension, hypoxia, pulmonary edema, pneumonia, pericardial effusion, rash, fluid overload, tumor lysis syndrome, and creatinine increased.
- ‡ Grouped term includes leukocytosis, white blood cell count increased.
- § Grouped term includes hematoma, eye hematoma, catheter site hematoma, oral mucosa hematoma, spontaneous hematoma, application site hematoma, injection site hematoma, periorbital hematoma.
- ¶ Grouped term includes blood pressure increased, essential hypertension, and hypertension.
- # Grouped term includes pain in extremity, arthralgia, back pain, musculoskeletal stiffness, cancer pain, and neck pain.
- Þ Grouped term includes dyspnea, dyspnea exertional, hypoxia, respiration failure.
Gastrointestinal disorders Nausea 30 (42) 2 (3) 28 (38) 3 (4) Vomiting* 29 (41) 0 20 (27) 1 (1) Investigations Electrocardiogram QT prolonged 14 (20) 7 (10) 5 (7) 2 (3) Psychiatric Disorders Insomnia 13 (18) 1 (1) 9 (12) 0 Blood system and lymphatic system disorders Differentiation Syndrome† 11 (15) 7 (10) 6 (8) 6 (8) Leukocytosis‡ 9 (13) 0 1 (1) 0 Vascular disorders Hematoma§ 11 (15) 0 3 (4) 0 Hypertension¶ 9 (13) 3 (4) 6 (8) 4 (5) Musculoskeletal and connective tissue disorders Arthralgia# 21 (30) 3 (4) 6 (8) 1 (1) Respiratory, thoracic and mediastinal disorders DyspneaÞ 14 (20) 2 (3) 11 (15) 4 (5) Nervous system disorders Headache 8 (11) 0 2 (3) 0 Table 3: Select Laboratory Abnormalities*, † (≥10%) That Worsened from Baseline in Patients with AML Who Received TIBSOVO + azacitidine in AG120-C-009 TIBSOVO + Azacitidine
N=71Placebo + Azacitidine
N=73Parameter All Grades
n (%)Grade ≥ 3
n (%)All Grades
n (%)Grade ≥ 3
n (%)- * Laboratory abnormality is defined as new or worsened by at least one grade from baseline, or if baseline is unknown.
- † The denominator used to calculate percentages is the number of treated subjects who can be evaluated for CTCAE criteria for each parameter in each arm.
Hematology Parameters Leukocytes decreased 46 (65) 39 (55) 47 (64) 42 (58) Platelets decreased 41 (58) 30 (42) 52 (71) 42 (58) Hemoglobin decreased 40 (56) 33 (46) 48 (66) 42 (58) Neutrophils decreased 18 (25) 16 (23) 25 (35) 23 (32) Lymphocytes increased 17 (24) 1 (1) 7 (10) 1 (1) Chemistry Parameters Glucose increased 40 (56) 9 (13) 34 (47) 8 (11) Phosphate decreased 29 (41) 7 (10) 25 (34) 9 (12) Aspartate Aminotransferase increased 26 (37) 0 17 (23) 0 Magnesium decreased 25 (35) 0 19 (26) 0 Alkaline Phosphatase increased 23 (32) 0 21 (29) 0 Potassium increased 17 (24) 2 (3) 9 (12) 1 (1) TIBSOVO Monotherapy
The safety profile of single-agent TIBSOVO was studied in 28 adults with newly diagnosed AML treated with 500 mg daily [see Clinical Studies (14.1)]. The median duration of exposure to TIBSOVO was 4.3 months (range 0.3 to 40.9 months). Ten patients (36%) were exposed to TIBSOVO for at least 6 months and 6 patients (21%) were exposed for at least 1 year.
Common (≥ 5%) serious adverse reactions included differentiation syndrome (18%), electrocardiogram QT prolonged (7%), and fatigue (7%). There was one case of posterior reversible encephalopathy syndrome (PRES).
Common (≥ 10%) adverse reactions leading to dose interruption included electrocardiogram QT prolonged (14%) and differentiation syndrome (11%). Two (7%) patients required a dose reduction due to electrocardiogram QT prolonged. One patient each required permanent discontinuation due to diarrhea and PRES.
The most common adverse reactions reported in the trial are shown in Table 4.
Table 4: Adverse Reactions Reported in ≥ 10% (Any Grade) or ≥ 5% (Grade ≥ 3) of Patients with Newly Diagnosed AML in AG120-C-001 TIBSOVO (500 mg daily)
N=28Body System
Adverse ReactionAll Grades
n (%)Grade ≥ 3
n (%)- * Grouped term includes abdominal pain, upper abdominal pain, abdominal discomfort, and abdominal tenderness.
- † Grouped term includes aphthous ulcer, esophageal pain, esophagitis, gingival pain, gingivitis, mouth ulceration, mucosal inflammation, oral pain, oropharyngeal pain, proctalgia, and stomatitis.
- ‡ Grouped term includes asthenia and fatigue.
- § Grouped term includes edema, face edema, fluid overload, fluid retention, hypervolemia, peripheral edema, and swelling face.
- ¶ Grouped term includes leukocytosis, hyperleukocytosis, and increased white blood cell count.
- # Differentiation syndrome can be associated with other commonly reported events such as peripheral edema, leukocytosis, pyrexia, dyspnea, pleural effusion, hypotension, hypoxia, pulmonary edema, pneumonia, pericardial effusion, rash, fluid overload, tumor lysis syndrome, and creatinine increased.
- Þ Grouped term includes arthralgia, back pain, musculoskeletal stiffness, neck pain, and pain in extremity.
- ß Grouped term includes myalgia, muscular weakness, musculoskeletal pain, musculoskeletal chest pain, musculoskeletal discomfort, and myalgia intercostal.
- à Grouped term includes dyspnea, dyspnea exertional, hypoxia, and respiratory failure.
- è Grouped term includes cough, productive cough, and upper airway cough syndrome.
- ð Grouped term includes burning sensation, lumbosacral plexopathy, neuropathy peripheral, paresthesia, and peripheral motor neuropathy.
- ø Grouped term includes dermatitis acneiform, dermatitis, rash, rash maculo-papular, urticaria, rash erythematous, rash macular, rash pruritic, rash generalized, rash papular, skin exfoliation, and skin ulcer.
Gastrointestinal disorders Diarrhea 17 (61) 2 (7) Nausea 10 (36) 2 (7) Abdominal pain* 8 (29) 1 (4) Constipation 6 (21) 1 (4) Vomiting 6 (21) 1 (4) Mucositis† 6 (21) 0 Dyspepsia 3 (11) 0 General disorders and administration site conditions Fatigue‡ 14 (50) 4 (14) Edema§ 12 (43) 0 Metabolism and nutrition disorders Decreased appetite 11 (39) 1 (4) Blood system and lymphatic system disorders Leukocytosis¶ 10 (36) 2 (7) Differentiation Syndrome# 7 (25) 3 (11) Musculoskeletal and connective tissue disorders ArthralgiaÞ 9 (32) 1 (4) Myalgiaß 7 (25) 1 (4) Respiratory, thoracic, and mediastinal disorders Dyspneaà 8 (29) 1 (4) Coughè 4 (14) 0 Investigations Electrocardiogram QT prolonged 6 (21) 3 (11) Weight decreased 3 (11) 0 Nervous system disorders Dizziness 6 (21) 0 Neuropathyð 4 (14) 0 Headache 3 (11) 0 Skin and subcutaneous tissue disorders Pruritus 4 (14) 1 (4) Rashø 4 (14) 1 (4) Changes in selected post-baseline laboratory values that were observed in patients with newly diagnosed AML are shown in Table 5.
Table 5: Most Common (≥ 10%) or ≥ 5% (Grade ≥ 3) New or Worsening Laboratory Abnormalities Reported in Patients with Newly Diagnosed AML* in AG120-C-001 TIBSOVO (500 mg daily)
N=28Parameter All Grades
n (%)Grade ≥ 3
n (%)- * Laboratory abnormality is defined as new or worsened by at least one grade from baseline, or if baseline is unknown.
Hemoglobin decreased 15 (54) 12 (43) Alkaline phosphatase increased 13 (46) 0 Potassium decreased 12 (43) 3 (11) Sodium decreased 11 (39) 1 (4) Uric acid increased 8 (29) 1 (4) Aspartate aminotransferase increased 8 (29) 1 (4) Creatinine increased 8 (29) 0 Magnesium decreased 7 (25) 0 Calcium decreased 7 (25) 1 (4) Phosphate decreased 6 (21) 2 (7) Alanine aminotransferase increased 4 (14) 1 (4) Relapsed or Refractory AML
The safety profile of single-agent TIBSOVO was studied in 179 adults with relapsed or refractory AML treated with 500 mg daily [see Clinical Studies (14.2)].
The median duration of exposure to TIBSOVO was 3.9 months (range 0.1 to 39.5 months). Sixty-five patients (36%) were exposed to TIBSOVO for at least 6 months and 16 patients (9%) were exposed for at least 1 year.
Serious adverse reactions (≥ 5%) were differentiation syndrome (10%), leukocytosis (10%), and electrocardiogram QT prolonged (7%). There was one case of progressive multifocal leukoencephalopathy (PML).
The most common adverse reactions leading to dose interruption were electrocardiogram QT prolonged (7%), differentiation syndrome (3%), leukocytosis (3%) and dyspnea (3%). Five out of 179 patients (3%) required a dose reduction due to an adverse reaction.
Adverse reactions leading to a dose reduction included electrocardiogram QT prolonged (1%), diarrhea (1%), nausea (1%), decreased hemoglobin (1%), and increased transaminases (1%).
Adverse reactions leading to permanent discontinuation included Guillain-Barré syndrome (1%), rash (1%), stomatitis (1%), and creatinine increased (1%).
The most common adverse reactions reported in the trial are shown in Table 6.
Table 6: Adverse Reactions Reported in ≥ 10% (Any Grade) or ≥ 5% (Grade ≥ 3) of Patients with Relapsed or Refractory AML TIBSOVO (500 mg daily)
N=179Body System
Adverse ReactionAll Grades
n (%)Grade ≥ 3
n (%)- * Grouped term includes asthenia and fatigue.
- † Grouped term includes peripheral edema, edema, fluid overload, fluid retention, and face edema.
- ‡ Grouped term includes angina pectoris, chest pain, chest discomfort, and non-cardiac chest pain
- § Grouped term includes leukocytosis, hyperleukocytosis, and increased white blood cell count.
- ¶ Differentiation syndrome can be associated with other commonly reported events such as peripheral edema, leukocytosis, pyrexia, dyspnea, pleural effusion, hypotension, hypoxia, pulmonary edema, pneumonia, pericardial effusion, rash, fluid overload, tumor lysis syndrome, and creatinine increased.
- # Grouped term includes arthralgia, back pain, musculoskeletal stiffness, neck pain, and pain in extremity.
- Þ Grouped term includes myalgia, muscular weakness, musculoskeletal pain, musculoskeletal chest pain, musculoskeletal discomfort, and myalgia intercostal.
- ß Grouped term includes aphthous ulcer, esophageal pain, esophagitis, gingival pain, gingivitis, mouth ulceration, mucosal inflammation, oral pain, oropharyngeal pain, proctalgia, and stomatitis.
- à Grouped term includes vomiting and retching.
- è Grouped term includes abdominal pain, upper abdominal pain, abdominal discomfort, and abdominal tenderness.
- ð Grouped term includes dyspnea, respiratory failure, hypoxia, and dyspnea exertional.
- ø Grouped term includes cough, productive cough, and upper airway cough syndrome.
- ý Grouped term includes dermatitis acneiform, dermatitis, rash, rash maculo-papular, urticaria, rash erythematous, rash macular, rash pruritic, rash generalized, rash papular, skin exfoliation, and skin ulcer.
- £ Grouped term includes ataxia, burning sensation, gait disturbance, Guillain-Barré syndrome, neuropathy peripheral, paresthesia, peripheral sensory neuropathy, peripheral motor neuropathy, and sensory disturbance.
- ¥ Grouped term includes hypotension and orthostatic hypotension.
General disorders and administration site conditions Fatigue* 69 (39) 6 (3) Edema† 57 (32) 2 (1) Pyrexia 41 (23) 2 (1) Chest pain‡ 29 (16) 5 (3) Blood system and lymphatic system disorders Leukocytosis§ 68 (38) 15 (8) Differentiation Syndrome¶ 34 (19) 23 (13) Musculoskeletal and connective tissue disorders Arthralgia# 64 (36) 8 (4) MyalgiaÞ 33 (18) 1 (1) Gastrointestinal disorders Diarrhea 60 (34) 4 (2) Nausea 56 (31) 1 (1) Mucositisß 51 (28) 6 (3) Constipation 35 (20) 1 (1) Vomitingà 32 (18) 2 (1) Abdominal painè 29 (16) 2 (1) Respiratory, thoracic, and mediastinal disorders Dyspneað 59 (33) 16 (9) Coughø 40 (22) 1 (<1) Pleural effusion 23 (13) 5 (3) Investigations Electrocardiogram QT prolonged 46 (26) 18 (10) Skin and subcutaneous tissue disorders Rashý 46 (26) 4 (2) Metabolism and nutrition disorders Decreased appetite 33 (18) 3 (2) Tumor lysis syndrome 14 (8) 11 (6) Nervous system disorders Headache 28 (16) 0 Neuropathy£ 21 (12) 2 (1) Vascular disorders Hypotension¥ 22 (12) 7 (4) Changes in selected post-baseline laboratory values that were observed in patients with relapsed or refractory AML are shown in Table 7.
Table 7: Most Common (≥ 10%) or ≥ 5% (Grade ≥ 3) New or Worsening Laboratory Abnormalities Reported in Patients with Relapsed or Refractory AML* TIBSOVO (500 mg daily)
N=179Parameter All Grades
n (%)Grade ≥ 3
n (%)- * Laboratory abnormality is defined as new or worsened by at least one grade from baseline, or if baseline is unknown.
Hemoglobin decreased 108 (60) 83 (46) Sodium decreased 69 (39) 8 (4) Magnesium decreased 68 (38) 0 Uric acid increased 57 (32) 11 (6) Potassium decreased 55 (31) 11 (6) Alkaline phosphatase increased 49 (27) 1 (1) Aspartate aminotransferase increased 49 (27) 1 (1) Phosphate decreased 45 (25) 15 (8) Creatinine increased 42 (23) 2 (1) Alanine aminotransferase increased 26 (15) 2 (1) Bilirubin increased 28 (16) 1 (1) Relapsed or Refractory Myelodysplastic Syndromes
The safety of TIBSOVO was evaluated in 19 adults with relapsed or refractory MDS treated with 500 mg daily in AG120-C-001 [see Clinical Studies (14.3)]. The median duration of exposure to TIBSOVO was 9.3 months (range 3.3 to 78.8 months). Fourteen patients (74%) were exposed to TIBSOVO for at least 6 months and 8 patients (42%) were exposed for at least 1 year.
Serious adverse reactions in ≥ 5% included differentiation syndrome (11%), fatigue (5%), and rash (5%).
Permanent discontinuation of TIBSOVO due to an adverse reaction occurred in 5% of patients. The adverse reaction which resulted in permanent discontinuation of TIBSOVO was fatigue.
Adverse reactions leading to dosage interruption of TIBSOVO occurred in 16% of patients. Adverse reactions which required dosage interruption in ≥ 5% were differentiation syndrome, leukocytosis, and rash.
Dose reductions of TIBSOVO due to an adverse reaction occurred in 16% of patients. Adverse reactions which required a dose reduction in ≥ 5% included differentiation syndrome, fatigue, and rash.
The most common (≥ 25%) adverse reactions, including laboratory abnormalities, were creatinine increased, hemoglobin decrease, arthralgia, albumin decreased, aspartate aminotransferase increased, fatigue, diarrhea, cough, sodium decreased, mucositis, decreased appetite, myalgia, phosphate decreased, pruritus, and rash.
Table 8 summarizes the adverse reactions in AG120-C-001.
Table 8: Adverse Reactions ≥ 10% in Patients with Relapsed or Refractory MDS in AG120-C-001 TIBSOVO (500 mg daily)
N=19Body System
Adverse ReactionAll Grades
%Grade 3 or 4
%- * Grouped term includes arthralgia, back pain, pain in extremity, flank pain, joint swelling, and neck pain.
- † Grouped term includes myalgia, muscle spasms, muscle discomfort, and musculoskeletal chest pain.
- ‡ Grouped term includes fatigue and asthenia.
- § Grouped term includes dyspnea and dyspnea exertional.
- ¶ Grouped term includes oropharyngeal pain, gingivitis, mouth ulceration, stomatitis.
- # Grouped term includes rash, catheter site erythema, and urticaria.
- Þ Grouped term includes leukocytosis, hyperleukocytosis, and white blood cell count increased.
Musculoskeletal and connective tissue disorders Arthralgia* 42 16 Myalgia† 26 0 General disorders and administration site conditions Fatigue‡ 37 11 Respiratory, thoracic, and mediastinal disorders Cough 32 0 Dyspnea§ 21 0 Gastrointestinal disorders Diarrhea 32 0 Mucositis¶ 26 5 Constipation 16 0 Nausea 16 0 Skin and subcutaneous tissue disorders Pruritus 26 0 Rash# 26 0 Metabolism and nutrition disorders Decreased appetite 26 0 Blood system and lymphatic system disorders LeukocytosisÞ 16 5 Differentiation Syndrome 11 0 Nervous system disorders Headache 16 0 Vascular disorders Hypertension 16 16 Investigations Electrocardiogram QT prolonged 11 0 Table 9 summarizes laboratory abnormalities in AG120-C-001.
Table 9: Select Laboratory Abnormalities (≥ 15%) That Worsened from Baseline in Patients with Relapsed or Refractory MDS in AG120-C-001 TIBSOVO*
N=19Laboratory Abnormality All Grades
%Grade 3 or 4
%- * Laboratory abnormality is defined as new or worsened by at least one grade from baseline, or if baseline is unknown.
Creatinine increased 95 5 Hemoglobin decreased 42 32 Albumin decreased 37 0 Aspartate Aminotransferase increased 37 5 Sodium decreased 32 5 Phosphate decreased 26 5 Alanine Aminotransferase increased 21 5 Bilirubin increased 21 0 Magnesium decreased 21 0 Alkaline Phosphatase increased 16 0 Potassium increased 16 0 Locally Advanced or Metastatic Cholangiocarcinoma
The safety of TIBSOVO was studied in patients with previously treated, locally advanced or metastatic cholangiocarcinoma in Study AG120-C-005 [see Clinical Studies (14.4)]. Patients received at least one dose of either TIBSOVO 500 mg daily (N=123) or placebo (N=59). The median duration of treatment was 2.8 months (range 0.1 to 34.4 months) with TIBSOVO.
Serious adverse reactions occurred in 34% of patients receiving TIBSOVO. Serious adverse reactions in ≥2% of patients in the TIBSOVO arm were pneumonia, ascites, hyperbilirubinemia, and jaundice cholestatic.
Fatal adverse reactions occurred in 4.9% of patients receiving TIBSOVO, including sepsis (1.6%) and pneumonia, intestinal obstruction, pulmonary embolism, and hepatic encephalopathy (each 0.8%).
TIBSOVO was permanently discontinued in 7% of patients. The most common adverse reactions leading to permanent discontinuation was acute kidney injury (1.6%).
Dose interruptions due to adverse reactions occurred in 29% of patients treated with TIBSOVO. The most common (>2%) adverse reactions leading to dose interruption were hyperbilirubinemia, alanine aminotransferase increased, aspartate aminotransferase increased, ascites, and fatigue.
Dose reductions of TIBSOVO due to an adverse reaction occurred in 4.1% of patients. Adverse reactions leading to dose reduction were electrocardiogram QT prolonged (3.3%) and neuropathy peripheral (0.8%).
The most common adverse reactions (≥15%) were fatigue, nausea, abdominal pain, diarrhea, cough, decreased appetite, ascites, vomiting, anemia, and rash.
Adverse reactions and laboratory abnormalities observed in Study AG120-C-005 are shown in Tables 10 and 11.
Table 10: Adverse Reactions Occurring in ≥ 10% of Patients Receiving TIBSOVO in Study AG120-C-005 TIBSOVO (500 mg daily)
N=123Placebo
N=59Body System
Adverse ReactionAll Grades
n (%)Grade ≥ 3
n (%)All Grades
n (%)Grade ≥ 3
n (%)- * Grouped term includes asthenia and fatigue.
- † Grouped term includes abdominal pain, abdominal pain upper, abdominal discomfort, abdominal pain lower, epigastric discomfort, abdominal tenderness, and gastrointestinal pain.
- ‡ Grouped term includes vomiting and retching.
- § Grouped term includes cough and productive cough.
- ¶ Grouped term includes rash, rash maculo-papular, erythema, rash macular, dermatitis exfoliative generalized, drug eruption, and drug hypersensitivity.
- # Grouped term includes neuropathy peripheral, peripheral sensory neuropathy, and paresthesia.
General disorders and administration site conditions Fatigue* 53 (43) 4 (3) 18 (31) 3 (5) Gastrointestinal disorders Nausea 51 (41) 3 (2) 17 (29) 1 (2) Diarrhea 43 (35) 0 10 (17) 0 Abdominal pain† 43 (35) 3 (2) 13 (22) 2 (3) Ascites 28 (23) 11 (9) 9 (15) 4 (7) Vomiting‡ 28 (23) 3 (2) 12 (20) 0 Respiratory, thoracic, and mediastinal disorders Cough§ 33 (27) 0 5 (9) 0 Metabolism and nutrition disorders Decreased appetite 30 (24) 2 (2) 11 (19) 0 Blood and lymphatic system disorders Anemia 22 (18) 8 (7) 3 (5) 0 Skin and subcutaneous tissue disorders Rash¶ 19 (15) 1 (1) 4 (7) 0 Nervous system disorders Headache 16 (13) 0 4 (7) 0 Neuropathy peripheral# 13 (11) 0 0 0 Investigations Electrocardiogram QT prolonged 12 (10) 2 (2) 2 (3) 0 Table 11: Selected Laboratory Abnormalities Occurring in ≥ 10% of Patients Receiving TIBSOVO in Study AG120-C-005* TIBSOVO
(500 mg daily)
N=123Placebo
N=59Parameter All Grades
n (%)Grade ≥ 3
n (%)All Grades
n (%)Grade ≥ 3
n (%)- * Laboratory abnormality is defined as new or worsened by at least one grade from baseline, or baseline is unknown.
AST increased 41 (34) 5 (4) 14 (24) 1 (2) Bilirubin increased 36 (30) 15 (13) 11 (19) 2 (3) Hemoglobin decreased 48 (40) 8 (7) 14 (25) 0 -
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on Ivosidenib
Strong or Moderate CYP3A4 Inhibitors Clinical Impact - Co-administration of TIBSOVO with strong or moderate CYP3A4 inhibitors increased ivosidenib plasma concentrations [see Clinical Pharmacology (12.3)].
- Increased ivosidenib plasma concentrations may increase the risk of QTc interval prolongation [see Warnings and Precautions (5.2)].
Prevention or Management - Consider alternative therapies that are not strong or moderate CYP3A4 inhibitors during treatment with TIBSOVO.
- If co-administration of a strong CYP3A4 inhibitor is unavoidable, reduce TIBSOVO to 250 mg once daily [see Dosage and Administration (2.3)].
- Monitor patients for increased risk of QTc interval prolongation [see Warnings and Precautions (5.2)].
Strong CYP3A4 Inducers Clinical Impact - Co-administration of TIBSOVO with strong CYP3A4 inducers decreased ivosidenib plasma concentrations [see Clinical Pharmacology (12.3)].
Prevention or Management - Avoid co-administration of strong CYP3A4 inducers with TIBSOVO.
QTc Prolonging Drugs Clinical Impact - Co-administration of TIBSOVO with QTc prolonging drugs may increase the risk of QTc interval prolongation [see Warnings and Precautions (5.2)].
Prevention or Management - Avoid co-administration of QTc prolonging drugs with TIBSOVO or replace with alternative therapies.
- If co-administration of a QTc prolonging drug is unavoidable, monitor patients for increased risk of QTc interval prolongation [see Warnings and Precautions (5.2)].
7.2 Effect of Ivosidenib on Other Drugs
Ivosidenib induces CYP3A4 and may induce CYP2C9. Co-administration will decrease concentrations of drugs that are sensitive CYP3A4 substrates and may decrease concentrations of drugs that are sensitive CYP2C9 substrates [see Clinical Pharmacology (12.3)]. Use alternative therapies that are not sensitive substrates of CYP3A4 and CYP2C9 during TIBSOVO treatment. If co-administration of TIBSOVO with sensitive CYP3A4 substrates or CYP2C9 substrates is unavoidable, monitor patients for loss of therapeutic effect of these drugs.
Do not administer TIBSOVO with anti-fungal agents that are substrates of CYP3A4 due to expected loss of antifungal efficacy.
Co-administration of TIBSOVO may decrease the concentrations of hormonal contraceptives, consider alternative methods of contraception in patients receiving TIBSOVO.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on animal embryo-fetal toxicity studies, TIBSOVO may cause fetal harm when administered to a pregnant woman. There are no available data on TIBSOVO use in pregnant women to inform a drug-associated risk of major birth defects and miscarriage. In animal embryo-fetal toxicity studies, oral administration of ivosidenib to pregnant rats and rabbits during organogenesis was associated with embryo-fetal mortality and alterations to growth starting at 2 times the steady state clinical exposure based on the AUC at the recommended human dose (see Data). If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, advise the patient of the potential risk to a fetus.
The background risk of major birth defects and miscarriage for the indicated population is unknown. Adverse outcomes in pregnancy occur regardless of the health of the mother or the use of medications. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2%-4% and 15%-20%, respectively.
Data
Animal Data
Ivosidenib administered to pregnant rats at a dose of 500 mg/kg/day during organogenesis (gestation days 6-17) was associated with adverse embryo-fetal effects including lower fetal weights, and skeletal variations. These effects occurred in rats at approximately 2 times the human exposure at the recommended dose of 500 mg daily.
In pregnant rabbits treated during organogenesis (gestation days 7-20), ivosidenib was maternally toxic at doses of 180 mg/kg/day (exposure approximately 3.9 times the human exposure at the recommended dose of 500 mg daily) and caused spontaneous abortions as well as decreased fetal weights, skeletal variations, and visceral variations.
8.2 Lactation
Risk Summary
There are no data on the presence of ivosidenib or its metabolites in human milk, the effects on the breastfed child, or the effects on milk production. Because many drugs are excreted in human milk and because of the potential for adverse reactions in breastfed children, advise women not to breastfeed during treatment with TIBSOVO and for 1 month after the last dose.
8.4 Pediatric Use
The safety and effectiveness of TIBSOVO in pediatric patients have not been established.
8.5 Geriatric Use
Of the 304 patients who received TIBSOVO in the clinical studies for AML and MDS, 75% were 65 years of age or older and 35% were 75 years or older.
Of the 124 patients with cholangiocarcinoma treated with TIBSOVO in Study AG120-C-005, 37% were 65 years of age or older and 11% were 75 years or older.
No overall differences in effectiveness or safety were observed between patients who were 65 years and older compared to younger patients.
8.6 Renal Impairment
No modification of the starting dose is recommended for patients with mild or moderate renal impairment (eGFR ≥ 30 mL/min/1.73m2, MDRD). The pharmacokinetics and safety of ivosidenib in patients with severe renal impairment (eGFR < 30 mL/min/1.73m2, MDRD) or renal impairment requiring dialysis are unknown [see Clinical Pharmacology (12.3)]. For patients with pre-existing severe renal impairment or who are requiring dialysis, consider the risks and potential benefits before initiating treatment with TIBSOVO.
8.7 Hepatic Impairment
No modification of the starting dose is recommended for patients with mild or moderate (Child-Pugh A or B) hepatic impairment [see Clinical Pharmacology (12.3)]. The pharmacokinetics and safety of ivosidenib in patients with severe hepatic impairment (Child-Pugh C) are unknown. For patients with pre-existing severe hepatic impairment, consider the risks and potential benefits before initiating treatment with TIBSOVO.
-
11 DESCRIPTION
TIBSOVO (ivosidenib) is an inhibitor of isocitrate dehydrogenase 1 (IDH1) enzyme. The chemical name is (2S)-N-{(1S)-1-(2-chlorophenyl)-2-[(3,3-difluorocyclobutyl)-amino]-2-oxoethyl}-1-(4-cyanopyridin-2-yl)-N-(5-fluoropyridin-3-yl)-5-oxopyrrolidine-2-carboxamide. The chemical structure is:
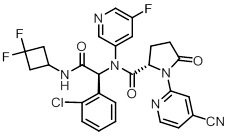
The molecular formula is C28H22ClF3N6O3 and the molecular weight is 583.0 g/mol. Ivosidenib is practically insoluble in aqueous solutions between pH 1.2 and 7.4.
TIBSOVO (ivosidenib) is available as a film-coated 250 mg tablet for oral administration. Each tablet contains the following inactive ingredients: colloidal silicon dioxide, croscarmellose sodium, hypromellose acetate succinate, magnesium stearate, microcrystalline cellulose, and sodium lauryl sulfate. The tablet coating includes FD&C blue #2, hypromellose, lactose monohydrate, titanium dioxide, and triacetin.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Ivosidenib is a small molecule inhibitor that targets the mutant isocitrate dehydrogenase 1 (IDH1) enzyme. In patients with AML, susceptible IDH1 mutations are defined as those leading to increased levels of 2-hydroxyglutarate (2-HG) in the leukemia cells and where efficacy is predicted by 1) clinically meaningful remissions with the recommended dose of ivosidenib and/or 2) inhibition of mutant IDH1 enzymatic activity at concentrations of ivosidenib sustainable at the recommended dosage according to validated methods. The most common of such mutations in patients with AML are R132H and R132C substitutions.
Ivosidenib was shown to inhibit selected IDH1 R132 mutants at much lower concentrations than wild-type IDH1 in vitro. Inhibition of the mutant IDH1 enzyme by ivosidenib led to decreased 2-HG levels and induced myeloid differentiation in vitro and in vivo in mouse xenograft models of IDH1-mutated AML. In blood samples from patients with AML with mutated IDH1, ivosidenib decreased 2-HG levels ex-vivo, reduced blast counts, and increased percentages of mature myeloid cells.
In a patient-derived xenograft intra-hepatic cholangiocarcinoma mouse model with IDH1 R132C, ivosidenib reduced 2-HG levels.
12.2 Pharmacodynamics
Multiple doses of ivosidenib 500 mg daily were observed to decrease plasma 2-HG concentrations in patients with hematological malignancies and cholangiocarcinoma to levels similar to those observed at baseline in healthy subjects. In bone marrow of patients with hematological malignancies and in tumor biopsy of patients with cholangiocarcinoma, the mean [% coefficient of variation (%CV)] reduction in 2-HG concentrations were 93.1% (11.1%) and 82.2% (32.4%), respectively.
Cardiac Electrophysiology
The mean increase in QTc was 17 msec (UCI: 20 msec) following administration of TIBSOVO 500 mg in patients with newly diagnosed AML and patients with relapsed or refractory AML. The increase in QTc interval was concentration-dependent [see Warnings and Precautions (5.2)]. A similar mean increase following administration of TIBSOVO 500 mg daily was observed in patients with relapsed or refractory MDS and in patients with solid tumors, including patients with cholangiocarcinoma. Co-administration with moderate or strong CYP3A inhibitors is expected to further increase QTc interval prolongation from baseline.
12.3 Pharmacokinetics
The AUC and Cmax of ivosidenib increase in a less than dose-proportional manner from 200 mg to 1,200 mg daily (0.4 to 2.4 times the approved recommended dosage). The following ivosidenib pharmacokinetic parameters (Table 12) were observed following administration of ivosidenib 500 mg as a single dose or daily dose (for steady state), unless otherwise specified. The steady-state pharmacokinetics of ivosidenib 500 mg were comparable between patients with newly diagnosed AML, relapsed or refractory AML, and relapsed or refractory MDS, and were lower in patients with cholangiocarcinoma.
Table 12: Pharmacokinetics of ivosidenib Newly diagnosed AML treated with a combination of TIBSOVO and azacitidine Relapsed or refractory AML treated with TIBSOVO Relapsed or refractory MDS treated with TIBSOVO Cholangiocarcinoma treated with TIBSOVO - * PK parameters expressed as mean (%CV).
- † Following administration of a single dose in healthy subjects, a high-fat meal (approximately 900 to 1,000 calories, 500 to 600 fat calories, 250 carbohydrate calories and 150 protein calories).
- ‡ Data from a single radiolabeled ivosidenib dose in healthy subjects.
PK parameters Single dose Cmax (ng/mL)* 4,820 (39%) 4,503 (38%) 4,020 (31%) 4,060 (45%) Steady state Cmax (ng/mL)* 6,145 (34%) 6,551 (44%) 5,820 (37%) 4,799 (33%) Steady state AUC (ng∙hr/mL)* 106,326 (41%) 117,348 (50%) 103,770 (40%) 86,382 (34%) Steady state PK Within 14 days Accumulation Cmax 1.2 1.5 1.4 1.2 AUC 1.6 1.9 2 1.5 Absorption Median Tmax (hr) 2 3 3 2 Effect of Food† Cmax 1.98-fold (90% CI: 1.79, 2.19) AUC 1.24-fold (90% CI: 1.16, 1.33) Distribution In vitro protein binding 92 to 96% Apparent volume of distribution at steady state (L)* 504 (22%) 403 (35%) 552 (26%) 706 (45%) Elimination Apparent clearance at steady state (L/hr)* 4.6 (35%) 5.6 (35%) 5.1 (35%) 6.1 (31%) Terminal half-life at steady state (hr)* 98 (42%) 58 (42%) 96 (43%) 129 (102%) Metabolism Plasma‡ >92% of total radioactivity as ivosidenib Metabolic pathways Major CYP3A4 Minor N-dealkylation and hydrolytic pathways Excretion‡ Urine 17% (10% as unchanged ivosidenib) Feces 77% (67% as unchanged ivosidenib) Specific Populations
No clinically significant effects on the pharmacokinetics of ivosidenib were observed based on age (18 years to 89 years), sex, race (White, Asian, Black or African American), body weight (38 to 150 kg), ECOG performance status, mild or moderate hepatic impairment (Child-Pugh A or B) or mild or moderate renal impairment (eGFR ≥30 mL/min/1.73m2, MDRD). The pharmacokinetics of ivosidenib in patients with severe renal impairment (eGFR <30 mL/min/1.73m2, MDRD) or renal impairment requiring dialysis or severe hepatic impairment (Child-Pugh C) is unknown.
Drug Interaction Studies
Clinical Studies and Model-Based Approaches
Effect of Strong or Moderate CYP3A4 Inhibitors on Ivosidenib
Co-administration of 250 mg ivosidenib with a strong CYP3A4 inhibitor (200 mg itraconazole once daily for 18 days) increased ivosidenib single-dose AUC by 269% (90% CI: 245%, 295%) with no change in Cmax in healthy subjects. Co-administration of 500 mg ivosidenib with the moderate CYP3A4 inhibitor fluconazole (dosed to steady-state) increases ivosidenib single-dose AUC by 173% with no change in Cmax. Co-administration of fluconazole following multiple daily ivosidenib doses is predicted to increase ivosidenib steady-state Cmax by 152% and AUC by 190%.
Effect of Strong CYP3A4 Inducers on Ivosidenib
Co-administration of ivosidenib with a strong CYP3A4 inducer (600 mg rifampin once daily for 15 days) is predicted to decrease ivosidenib steady-state AUC by 33%.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been conducted with ivosidenib. Ivosidenib was not mutagenic in an in vitro bacterial reverse mutation (Ames) assay. Ivosidenib was not clastogenic in an in vitro human lymphocyte micronucleus assay, or in an in vivo rat bone marrow micronucleus assay. Fertility studies in animals have not been conducted with ivosidenib. In repeat-dose toxicity studies up to 90 days in duration with twice daily oral administration of ivosidenib in rats, uterine atrophy was reported in females at non-tolerated dose levels.
-
14 CLINICAL STUDIES
14.1 Newly Diagnosed AML
Newly Diagnosed AML in Combination with Azacitidine
The efficacy of TIBSOVO was evaluated in a randomized (1:1), multicenter, double-blind, placebo-controlled clinical trial (Study AG120-C-009, NCT03173248) of 146 adult patients with newly-diagnosed AML with an IDH1 mutation who were 75 years or older, or had comorbidities that precluded the use of intensive induction chemotherapy based on at least one of the following criteria: baseline Eastern Cooperative Oncology Group (ECOG) performance status of 2, severe cardiac or pulmonary disease, hepatic impairment with bilirubin > 1.5 times the upper limit of normal, creatinine clearance < 45 mL/min, or other comorbidity. IDH1 mutations were confirmed centrally using the Abbott RealTime™ IDH1 Assay. Local diagnostic tests were permitted for screening and randomization provided a bone marrow or peripheral blood sample was sent for central confirmation. Gene mutation analysis to document IDH1 mutated disease from a bone marrow or peripheral blood sample was conducted for all patients. Patients were randomized to receive either TIBSOVO 500 mg or matched placebo orally once daily on Days 1-28 in combination with azacitidine 75 mg/m2/day either subcutaneously or intravenously on Days 1-7 or Days 1-5 and 8-9 of each 28-day cycle beginning on Cycle 1 Day 1. Patients were treated for a minimum of 6 cycles unless they experienced disease progression, unacceptable toxicity or undergoing hematopoietic stem cell transplantation. Baseline demographic and disease characteristics are shown in Table 13.
Table 13: Baseline Demographic and Disease Characteristics in Patients with Newly Diagnosed AML (Study AG120-C-009) Demographic and Disease Characteristics TIBSOVO + azacitidine
(500 mg daily)
N=72Placebo + azacitidine
N=74ECOG PS: Eastern Cooperative Oncology Group Performance Status; MPN = Myeloproliferative Neoplasm; MDS = Myelodysplastic syndrome - * Using confirmatory Abbott RealTime IDH1 assay testing results.
- † Cytogenetic risk status: National Comprehensive Cancer Network (NCCN) guidelines.
- ‡ Patients were defined as transfusion dependent at baseline if they received any red blood cell or platelet transfusion within 56 days prior to the first dose of TIBSOVO.
Demographics Age (Years) Median (Min, Max) 76 (58, 84) 76 (45, 94) Age Categories, n (%) <65 years 4 (6) 4 (5) ≥65 years to <75 years 29 (40) 27 (36) ≥75 years 39 (54) 43 (58) Sex, n (%) Male 42 (58) 38 (51) Female 30 (42) 36 (49) Race, n (%) Asian 15 (21) 19 (26) White 12 (17) 12 (16) Black or African American 0 2 (3) Other 1 (1) 1 (1) Not provided 44 (61) 40 (54) Disease Characteristics ECOG PS, n (%) 0 14 (19) 10 (14) 1 32 (44) 40 (54) 2 26 (36) 24 (32) IDH1 Mutation, n (%)* R132C 45 (63) 51 (69) R132H 14 (19) 12 (16) R132G 6 (8) 4 (5) R132L 3 (4) 0 R132S 2 (3) 6 (8) Wild type 1 (1) 0 Missing 1 (1) 1 (1) Cytogenetic risk status† n (%) Favorable 3 (4) 7 (9) Intermediate 48 (67) 44 (59) Poor 16 (22) 20 (27) Other 3 (4) 1 (1) Missing 2 (3) 2 (3) Transfusion Dependent at Baseline‡, n (%) 39 (54) 40 (54) Type of AML, n (%) De novo AML 54 (75) 53 (72) Secondary AML 18 (25) 21 (28) Therapy-related AML 2 (3) 1 (1) MDS related 10 (14) 12 (16) MPN related 4 (6) 8 (11) Efficacy was established on the basis of event-free survival (EFS), overall survival (OS), and rate and duration of complete remission (CR). EFS was defined as the time from randomization until treatment failure, relapse from remission, or death from any cause, whichever occurred first. Treatment failure was defined as failure to achieve CR by 24 weeks. The efficacy results are shown in Table 14 and Figure 1.
Table 14: Efficacy Results in Patients with Newly Diagnosed AML (Study AG120-C-009) Endpoint TIBSOVO
(500 mg daily) + azacitidine
N=72Placebo + azacitidine
N=74Abbreviations: EFS = Event free survival; CI: confidence interval; OS = Overall survival; CR = Complete remission; CRh = Complete remission with partial hematologic recovery; NE = Not estimable. The 2-sided p-value boundaries for EFS, OS, CR, and CR+CRh are 0.0095, 0.0034, 0.0174, and 0.0174, respectively. - * Hazard ratio is estimated using a Cox's proportional hazards model stratified by the randomization stratification factors (AML status and geographic region) with Placebo+ azacitidine as the denominator.
- † Two-sided p-value is calculated from the log-rank test stratified by the randomization stratification factors (AML status and geographic region).
- ‡ CI of percentage is calculated with the Clopper and Pearson (exact Binomial) method.
- § Mantel-Haenszel estimate of risk difference in percentage between TIBSOVO + azacitidine and Placebo+ azacitidine is calculated.
- ¶ Two-sided p-value is calculated from the Cochran-Mantel-Haenszel test stratified by the randomization stratification factors (AML status and geographic region).
EFS, events (%) 47 (65) 62 (84) Treatment Failure 43 (60) 59 (80) Relapse 3 (4) 2 (3) Death 1 (1) 1 (1) Hazard ratio* (95% CI) 0.35 (0.17, 0.72) p-value† 0.0038 OS events (%) 28 (39) 46 (62) Median OS (95% CI) months 24.0 (11.3, 34.1) 7.9 (4.1, 11.3) Hazard ratio* (95% CI) 0.44 (0.27, 0.73) p-value† 0.0010 CR, n (%) 34 (47) 11 (15) 95% CI‡ (35, 59) (8, 25) Risk difference§ (95% CI), (%) 31 (17, 46) p-value¶ <0.0001 Median duration of CR (95% CI), months NE (13.0, NE) 11.2 (3.2, NE) CR +CRh, n (%) 37 (51) 13 (18) 95% CI‡ (39, 63) (10, 28) Risk difference§ (95% CI), (%) 33 (18, 47) p-value¶ <0.0001 Median duration of CR + CRh (95% CI), months NE (13.0, NE) 9.2 (5.8, NE) Figure 1: Kaplan-Meier Curve for Overall Survival in AG120-C-009
The median time to first CR for TIBSOVO with azacitidine was 4 months (range, 1.7 to 11.9 months).
The median time to first CR + CRh for TIBSOVO with azacitidine was 4 months (range, 1.7 to 11.9 months).
Monotherapy in Newly Diagnosed AML
The efficacy of TIBSOVO was evaluated in an open-label, single-arm, multicenter clinical trial (Study AG120-C-001, NCT02074839) that included 28 adult patients with newly diagnosed AML with an IDH1 mutation. IDH1 mutations were identified by a local or central diagnostic test and confirmed retrospectively using the Abbott RealTime™ IDH1 Assay. The cohort included patients who were age 75 years or older or who had comorbidities that precluded the use of intensive induction chemotherapy based on at least one of the following criteria: baseline ECOG performance status of ≥ 2, severe cardiac or pulmonary disease, hepatic impairment with bilirubin > 1.5 times the upper limit of normal, or creatinine clearance < 45 mL/min. TIBSOVO was given orally at a starting dose of 500 mg daily until disease progression, development of unacceptable toxicity, or undergoing hematopoietic stem cell transplantation. Two (7%) of the 28 patients went on to stem cell transplantation following TIBSOVO treatment.
The baseline demographic and disease characteristics are shown in Table 15.
Table 15: Baseline Demographic and Disease Characteristics in Patients with Newly Diagnosed AML (Study AG120-C-001) Demographic and Disease Characteristics TIBSOVO (500 mg daily)
N=28ECOG PS: Eastern Cooperative Oncology Group Performance Status. ELN: European Leukemia Net - * Using confirmatory Abbott RealTime IDH1 assay testing results.
- † Patients were defined as transfusion dependent at baseline if they received any transfusion occurring within 56 days prior to the first dose of TIBSOVO.
- ‡ AML with myelodysplasia-related changes.
Demographics Age (Years) Median (Min, Max) 77 (64, 87) Age Categories, n (%) <65 years 1 (4) ≥65 years to <75 years 8 (29) ≥75 years 19 (68) Sex, n (%) Male 15 (54) Female 13 (46) Race, n (%) White 24 (86) Black or African American 2 (7) Asian 0 Native Hawaiian/Other Pacific Islander 0 Other/Not provided 2 (7) Disease Characteristics ECOG PS, n (%) 0 6 (21) 1 16 (57) 2 5 (18) 3 1 (4) IDH1 Mutation, n (%)* R132C 24 (86) R132H 2 (7) R132G 1 (4) R132L 1 (4) R132S 0 ELN Risk Category, n (%) Favorable 0 Intermediate 9 (32) Adverse 19 (68) Transfusion Dependent at Baseline†, n (%) 17 (61) Type of AML, n (%) De novo AML 6 (21) AML-MRC‡ 19 (68) Therapy-related AML 3 (11) Prior Hypomethylating Agent for Antecedent Hematologic Disorder 13 (46) Efficacy was established on the basis of the rate of complete remission (CR) or complete remission with partial hematologic recovery (CRh), the duration of CR+CRh, and the rate of conversion from transfusion dependence to transfusion independence. The efficacy results are shown in Table 16. The median follow-up was 8.1 months (range, 0.6 to 40.9 months) and median treatment duration was 4.3 months (range, 0.3 to 40.9 months).
Table 16: Efficacy Results in Patients with Newly Diagnosed AML (Study AG120-C-001) Endpoint TIBSOVO (500 mg daily)
N=28CI: confidence interval, NE: not estimable - * CR (complete remission) was defined as <5% blasts in the bone marrow, no evidence of disease, and full recovery of peripheral blood counts (platelets >100,000/microliter and absolute neutrophil counts [ANC] >1,000/microliter).
- † DOCR (duration of CR), DOCRh (duration of CRh), and DOCR+CRh (duration of CR+CRh) was defined as time since first response of CR, CRh or CR/CRh, respectively, to relapse or death, whichever is earlier. + indicates censored observation.
- ‡ The median durations of CR and CR+CRh were not estimable, with 5 patients (41.7%) who achieved CR or CRh remaining on TIBSOVO treatment (treatment duration range: 20.3 to 40.9 months).
- § CRh (complete remission with partial hematological recovery) was defined as <5% blasts in the bone marrow, no evidence of disease, and partial recovery of peripheral blood counts (platelets >50,000/microliter and ANC >500/microliter).
CR* n (%) 8 (28.6) 95% CI (13.2, 48.7) Median DOCR† (months) NE‡ 95% CI (4.2, NE) CRh§ n (%) 4 (14.3) 95% CI (4.0, 32.7) Observed DOCRh† (months) 2.8, 4.6, 8.3, 15.7+ CR+CRh n (%) 12 (42.9) 95% CI (24.5, 62.8) Median DOCR+CRh† (months) NE‡ 95% CI (4.2, NE) For patients who achieved a CR or CRh, the median time to CR or CRh was 2.8 months (range, 1.9 to 12.9 months). Of the 12 patients who achieved a best response of CR or CRh, 11 (92%) achieved a first response of CR or CRh within 6 months of initiating TIBSOVO.
Among the 17 patients who were dependent on red blood cell (RBC) and/or platelet transfusions at baseline, 7 (41.2%) became independent of RBC and platelet transfusions during any 56-day post-baseline period. Of the 11 patients who were independent of both RBC and platelet transfusions at baseline, 6 (54.5%) remained transfusion independent during any 56-day post-baseline period.
14.2 Relapsed or Refractory AML
The efficacy of TIBSOVO was evaluated in an open-label, single-arm, multicenter clinical trial (Study AG120-C-001, NCT02074839) of 174 adult patients with relapsed or refractory AML with an IDH1 mutation. IDH1 mutations were identified by a local or central diagnostic test and confirmed retrospectively using the Abbott RealTime™ IDH1 Assay. TIBSOVO was given orally at a starting dose of 500 mg daily until disease progression, development of unacceptable toxicity, or undergoing hematopoietic stem cell transplantation. Twenty-one (12%) of the 174 patients went on to stem cell transplantation following TIBSOVO treatment.
The baseline demographic and disease characteristics are shown in Table 17.
Table 17: Baseline Demographic and Disease Characteristics in Patients with Relapsed or Refractory AML (Study AG120-C-001) Demographic and Disease Characteristics TIBSOVO (500 mg daily)
N=174ECOG PS: Eastern Cooperative Oncology Group Performance Status. - * Using confirmatory Abbott RealTime IDH1 assay testing results.
- † Patients were defined as transfusion dependent at baseline if they received any transfusion occurring within 56 days prior to the first dose of TIBSOVO.
Demographics Age (Years) Median (Min, Max) 67 (18, 87) Age Categories, n (%) <65 years 63 (36) ≥65 years to <75 years 71 (41) ≥75 years 40 (23) Sex, n (%) Male 88 (51) Female 86 (49) Race, n (%) White 108 (62) Black or African American 10 (6) Asian 6 (3) Native Hawaiian/Other Pacific Islander 1 (1) Other/Not provided 49 (28) Disease Characteristics ECOG PS, n (%) 0 36 (21) 1 97 (56) 2 39 (22) 3 2 (1) IDH1 Mutation, n (%)* R132C 102 (59) R132H 43 (25) R132G 12 (7) R132S 10 (6) R132L 7 (4) Cytogenetic Risk Status, n (%) Intermediate 104 (60) Poor 47 (27) Missing/Unknown 23 (13) Relapse Type Primary refractory 64 (37) Refractory relapse 45 (26) Untreated relapse 65 (37) Relapse Number 0 64 (37) 1 83 (48) 2 21 (12) ≥3 6 (3) Prior Stem Cell Transplantation for AML, n (%) 40 (23) Transfusion Dependent at Baseline†, n (%) 110 (63) Median Number of Prior Therapies (Min, Max) 2 (1, 6) Type of AML, n (%) De novo AML 116 (67) Secondary AML 58 (33) Efficacy was established on the basis of the rate of complete remission (CR) plus complete remission with partial hematologic recovery (CRh), the duration of CR+CRh, and the rate of conversion from transfusion dependence to transfusion independence. The efficacy results are shown in Table 18. The median follow-up was 8.3 months (range, 0.2 to 39.5 months) and median treatment duration was 4.1 months (range, 0.1 to 39.5 months).
Table 18: Efficacy Results in Patients with Relapsed or Refractory AML (Study AG120-C-001) Endpoint TIBSOVO (500 mg daily)
N=174CI: confidence interval - * CR (complete remission) was defined as <5% blasts in the bone marrow, no evidence of disease, and full recovery of peripheral blood counts (platelets >100,000/microliter and absolute neutrophil counts [ANC] >1,000/microliter).
- † DOCR (duration of CR), DOCRh (duration of CRh), and DOCR+CRh (duration of CR+CRh) was defined as time since first response of CR, CRh or CR/CRh, respectively, to relapse or death, whichever is earlier.
- ‡ CRh (complete remission with partial hematological recovery) was defined as <5% blasts in the bone marrow, no evidence of disease, and partial recovery of peripheral blood counts (platelets >50,000/microliter and ANC >500/microliter).
- § CR+CRh rate appeared to be consistent across all baseline demographic and baseline disease characteristics with the exception of number of prior regimens.
CR* n (%) 43 (24.7) 95% CI (18.5, 31.8) Median DOCR† (months) 10.1 95% CI (6.5, 22.2) CRh‡ n (%) 14 (8.0) 95% CI (4.5, 13.1) Median DOCRh† (months) 3.6 95% CI (1, 5.5) CR+CRh§ n (%) 57 (32.8) 95% CI (25.8, 40.3) Median DOCR+CRh† (months) 8.2 95% CI (5.6, 12) For patients who achieved a CR or CRh, the median time to CR or CRh was 2 months (range, 0.9 to 5.6 months). Of the 57 patients who achieved a best response of CR or CRh, all achieved a first response of CR or CRh within 6 months of initiating TIBSOVO.
Among the 110 patients who were dependent on red blood cell (RBC) and/or platelet transfusions at baseline, 41 (37.3%) became independent of RBC and platelet transfusions during any 56-day post-baseline period. Of the 64 patients who were independent of both RBC and platelet transfusions at baseline, 38 (59.4%) remained transfusion independent during any 56-day post-baseline period.
14.3 Relapsed or Refractory MDS
The efficacy of TIBSOVO was evaluated in an open-label, single-arm, multicenter study (study AG120-C-001, NCT02074839) of 18 adult patients with relapsed or refractory MDS with an IDH1 mutation. IDH1 mutations were detected in peripheral blood or bone marrow by a local or central diagnostic test and confirmed retrospectively using the Abbott RealTime™ IDH1 Assay. TIBSOVO was given orally at a starting dose of 500 mg daily continuous for 28-day cycles until disease progression, development of unacceptable toxicity, or undergoing hematopoietic stem cell transplantation. One (6%) of the 18 patients went on to stem cell transplantation following TIBSOVO treatment.
The baseline demographic and disease characteristics are shown in Table 19.
Table 19: Baseline Demographic and Disease Characteristics in Patients with Relapsed or Refractory MDS (Study AG120-C-001) Demographic and Disease Characteristics TIBSOVO (500 mg daily)
N=18ECOG PS: Eastern Cooperative Oncology Group Performance Status. - * Using confirmatory Abbott RealTime IDH1 assay testing results.
Demographics Age (Years) Median (Min, Max) 74 (61, 82) Age Categories, n (%) <65 years 3 (17) ≥65 years to <75 years 7 (39) ≥75 years 8 (44) Sex, n (%) Male 14 (78) Female 4 (22) Race, n (%) White 14 (78) Black or African American 1 (6) Not Reported 3 (17) Disease Characteristics ECOG PS, n (%) 0 5 (28) 1 10 (56) 2 3 (17) IDH1 Mutation, n (%)* R132C 9 (50) R132H 5 (28) R132G 2 (11) R132L 1 (6) R132S 1 (6) Cytogenetic Risk Status, n (%) Good 4 (22) Intermediate 8 (44) Poor 5 (28) Missing 1 (6) Baseline Bone Marrow Blasts, n (%) < 5% 7 (39) ≥ 5% 11 (61) Prior Therapies Intensive chemotherapy 3 (17) Non-intensive chemotherapy 15 (83) 1 line of HMA-based therapy 14 (78) 2 lines of HMA-based therapy 1 (6) Efficacy was established on the basis of the rate of complete remission (CR) or partial remission (PR) as per the 2006 International Working Group response criteria for MDS, the duration of CR+PR, and the rate of conversion from transfusion dependence to transfusion independence. All observed responses were CRs. The efficacy results are shown in Table 20. The median follow-up was 27.1 months (range 3.7 to 88.7 months) and median duration of exposure to TIBSOVO was 8.3 months (range 3.3 to 78.8 months).
Table 20: Efficacy Results in Patients with Relapsed or Refractory MDS (Study AG120-C-001) Endpoint TIBSOVO (500 mg daily)
N=18CI: confidence interval, CR: complete remission, NE: not estimable, derived based on Kaplan-Meier method. - * CR responders with baseline bone marrow blast < 5% was 43% (3/7).
- † Duration of CR (DOCR) = date of first documented CR (lasted at least 4 weeks) to date of first documented confirmed relapse or death, whichever is earlier.
- ‡ + indicates censored observation.
CR* n (%) 7 (38.9) 95% CI (17.3, 64.3) DOCR† (months) median (range) NE (1.9, 80.8+‡) For patients who achieved a CR, the median time to CR was 1.9 months (range, 1.0 to 5.6 months).
Among the 9 patients who were dependent on red blood cell (RBC) and/or platelet transfusions at baseline, 6 (67%) became independent of RBC and platelet transfusions during any 56-day post-baseline period. Of the 9 patients who were independent of both RBC and platelet transfusions at baseline, 7 (78%) remained transfusion independent during any 56-day post-baseline period.
14.4 Locally Advanced or Metastatic Cholangiocarcinoma
The efficacy of TIBSOVO was evaluated in a randomized (2:1), multicenter, double-blind, placebo-controlled clinical trial (Study AG120-C-005, NCT02989857) of 185 adult patients with locally advanced or metastatic cholangiocarcinoma with an IDH1 mutation whose disease had progressed following at least 1 but not more than 2 prior regimens, including at least one gemcitabine- or 5-FU-containing regimen. Patients were randomized to receive either TIBSOVO 500 mg orally once daily or matched placebo until disease progression or unacceptable toxicity. Randomization was stratified by number of prior therapies (1 or 2). Eligible patients who were randomized to placebo were allowed to cross over to receive TIBSOVO after documented radiographic disease progression. Patients with IDH1 mutations were selected using a central diagnostic next generation sequencing assay. Tumor imaging assessments were performed every 6 weeks for the first 8 assessments and every 8 weeks thereafter.
The major efficacy outcome measure was Progression Free Survival (PFS) as determined by independent review committee (IRC) according to Response Evaluation Criteria in Solid Tumors (RECIST) v1.1.
The median age was 62 years (range: 33 to 83); 63% were female; 57% were White, 12% Asian, 1.1% Black, 0.5% Native Hawaiian/Other Pacific Islander, 0.5% American Indian or Alaska Native, 28% race missing/not reported; and 37% had an ECOG performance status of 0 (37%) or 1 (62%). All patients received at least 1 prior line of systemic therapy and 47% received two prior lines. Most patients had intrahepatic cholangiocarcinoma (91%) at diagnosis and 92% had metastatic disease. Across both arms, 70% patients had an R132C mutation, 15% had an R132L mutation, 12% had an R132G mutation, 1.1% had an R132H mutation, and 1.6% had an R132S mutation.
The efficacy results are shown in Table 21 and Figure 2. The study demonstrated a statistically significant improvement in PFS.
Table 21: Efficacy Results in Patients with Locally Advanced or Metastatic Cholangiocarcinoma in AG120-C-005 Endpoint TIBSOVO
(500 mg daily)Placebo IRC: Independent Review Committee; CI: Confidence Interval - * Hazard ratio is calculated from stratified Cox regression model. Stratified by number of prior lines of therapy.
- † P-value is calculated from the one-sided stratified log-rank test. Stratified by number of prior lines of therapy.
- ‡ OS results are based on the final analysis of OS (based on 150 deaths) which occurred 16 months after the final analysis of PFS. The median OS (95% CI) for TIBSOVO was 10.3 (7.8, 12.4) months; and placebo was 7.5 (4.8, 11.1) months without adjusting for crossover. In the analysis of OS, 70% of the patients randomized to placebo had crossed over to receive TIBSOVO after radiographic disease progression.
Progression-Free Survival by IRC Assessment N=124 N=61 Events, n (%) 76 (61) 50 (82) Progressive Disease 64 (52) 44 (72) Death 12 (10) 6 (10) Hazard ratio (95% CI)* 0.37 (0.25, 0.54) p-value† <0.0001 Objective Response Rate, n (%) 3 (2.4) 0 Overall Survival‡ N=126 N=61 Deaths, n (%) 100 (79) 50 (82) Hazard ratio (95% CI)* 0.79 (0.56, 1.12) p-value† 0.093 Figure 2: Kaplan-Meier Plot of Progression-Free Survival per Independent Review Committee - Before Crossover (ITT)
-
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
250 mg tablet: Blue oval-shaped film-coated tablet debossed "IVO" on one side and "250" on the other side.
- 60-count bottles of 250 mg tablets with a desiccant canister (NDC: 72694-617-60)
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Differentiation Syndrome in AML and MDS
Advise patients being treated with TIBSOVO of the risks of developing differentiation syndrome as early as 1 day after start of therapy and during the first 3 months on treatment. Ask patients to immediately report any symptoms suggestive of differentiation syndrome, such as fever, cough or difficulty breathing, rash, decreased urinary output, low blood pressure, rapid weight gain, or swelling of their arms or legs, to their healthcare provider for further evaluation [see Boxed Warning and Warnings and Precautions (5.1)].
QTc Interval Prolongation
Inform patients of symptoms that may be indicative of significant QTc interval prolongation including dizziness, lightheadedness, and fainting. Advise patients to report these symptoms and the use of all medications to their healthcare provider [see Warnings and Precautions (5.2)].
Guillain-Barré Syndrome
Inform patients of symptoms that may be indicative of Guillain-Barré syndrome, including new signs or symptoms of motor and/or sensory neuropathy, such as weakness or tingling sensation in the legs, arms, or upper body, numbness and pain on one side or both sides of the body, changes to any sensory function, or burning or prickling sensation, or difficulty breathing. Advise patients to report these symptoms to their healthcare provider [see Warnings and Precautions (5.3)].
Drug Interactions
Advise patients to inform their healthcare providers of all concomitant medications, including over-the-counter medications, vitamins, and herbal products [see Drug Interactions (7)].
Tumor Lysis Syndrome
Advise patients on the risks of developing tumor lysis syndrome. Advise patients on the importance of maintaining high fluid intake, and the need for frequent monitoring of blood chemistry values [see Adverse Reactions (6.1)].
Gastrointestinal Adverse Reactions
Advise patients on the risks of experiencing gastrointestinal reactions such as diarrhea, nausea, mucositis, constipation, vomiting, decreased appetite, ascites and abdominal pain. Ask patients to report these events to their healthcare provider and advise patients how to manage them [see Adverse Reactions (6.1)].
Lactation
Advise women not to breastfeed during treatment with TIBSOVO and for 1 month after the last dose [see Use in Specific Populations (8.2)].
Dosing and Storage Instructions
- Advise patients to swallow tablets whole and to not split, crush, or chew TIBSOVO tablets.
- Advise patients to avoid taking TIBSOVO with a high-fat meal.
- Instruct patients that if a dose of TIBSOVO is vomited, not to take an additional dose, and wait until the next scheduled dose is due. If a dose of TIBSOVO is missed or not taken at the usual time, instruct patients to take the dose as soon as possible unless the next dose is due within 12 hours. Patients can return to the normal schedule the following day.
- Store TIBSOVO at room temperature from 20°C to 25°C (68°F to 77°F).
- Advise patients not to remove the desiccant canister from the bottle.
-
SPL UNCLASSIFIED SECTION
Manufactured for Servier Pharmaceuticals LLC, Boston, MA 02210
Servier and the Servier logo are registered trademarks of Les Laboratoires Servier.
TIBSOVO is a registered trademark of Servier Pharmaceuticals LLC, a wholly owned, indirect subsidiary of Les Laboratoires Servier.
© 2023 Servier Pharmaceuticals LLC
AG-PI-006 -
MEDICATION GUIDE
This Medication Guide has been approved by the U.S. Food and Drug Administration. Revised: OCT 2023 MEDICATION GUIDE
TIBSOVO® (tib-SOH-voh)
(ivosidenib) tabletsWhat is the most important information I should know about TIBSOVO?
TIBSOVO may cause serious side effects, including: - Differentiation Syndrome. Differentiation syndrome is a common condition that affects your blood cells and may be life-threatening or lead to death. Differentiation syndrome in adults with acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS) has happened as early as 1 day and up to 3 months after starting TIBSOVO. Call your healthcare provider or go to the nearest hospital emergency room right away if you develop any of the following symptoms of differentiation syndrome during treatment with TIBSOVO:
- fever
- cough
- trouble breathing
- rash
- decreased urination
- dizziness or lightheadedness
- rapid weight gain
- swelling of your arms or legs
If you develop signs and symptoms of differentiation syndrome, your healthcare provider may treat you with a corticosteroid medicine or a medicine called hydroxyurea and may monitor you in the hospital. See "What are the possible side effects of TIBSOVO?" for more information about side effects. What is TIBSOVO? TIBSOVO is a prescription medicine used to treat adults with an isocitrate dehydrogenase-1 (IDH1) mutation with: - acute myeloid leukemia (AML):
- with newly diagnosed AML treated in combination with azacitidine, or alone, who are 75 years or older or who have health problems that prevent the use of certain chemotherapy treatments.
- when the disease has come back or has not improved after previous treatment(s).
- myelodysplastic syndrome (MDS):
- when the disease has come back or has not improved after previous treatment(s).
- bile duct cancer (cholangiocarcinoma):
- with bile duct cancer that has spread and
- who have already received previous treatment(s).
Your healthcare provider will perform a test to make sure that TIBSOVO is right for you. It is not known if TIBSOVO is safe and effective in children. Before taking TIBSOVO, tell your healthcare provider about all of your medical conditions, including if you: - have any heart problems, including a condition called long QT syndrome.
- have problems with abnormal electrolytes, such as sodium, potassium, calcium or magnesium levels.
- have nervous system problems.
- have problems with your kidneys or are on dialysis.
- have any liver disorders, including cirrhosis.
- are pregnant or plan to become pregnant. TIBSOVO may cause harm to your unborn baby. You should avoid becoming pregnant during treatment with TIBSOVO. Tell your healthcare provider right away if you become pregnant or think you might be pregnant during treatment with TIBSOVO.
- are breastfeeding or plan to breastfeed. It is not known if TIBSOVO passes into your breast milk. Do not breastfeed during your treatment with TIBSOVO and for 1 month after your last dose of TIBSOVO.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. Especially tell your healthcare provider if you take hormonal contraceptives. TIBSOVO may affect how hormonal contraceptives work and may cause them to not work as well. How should I take TIBSOVO? - Take TIBSOVO exactly as your healthcare provider tells you to.
- Do not change your dose or stop taking TIBSOVO without talking to your healthcare provider.
- Take TIBSOVO 1 time a day about the same time each day.
- Swallow TIBSOVO tablets whole. Do not split, crush, or chew the tablet.
- TIBSOVO can be taken with or without food.
- Do not take TIBSOVO with a high-fat meal. An example of a high-fat meal includes 2 eggs fried in butter, 2 strips of bacon, 2 slices of white bread with butter, 1 croissant with 1 slice of cheese, and 8 ounces of whole milk (approximately 1,000 calories and 58 grams of fat).
- If you vomit after taking a dose of TIBSOVO, do not take an additional dose. Take your next dose at your usual time.
- If you miss a dose of TIBSOVO or did not take it at the usual time, take your dose as soon as possible and at least 12 hours before your next dose. Return to your normal schedule the following day. Do not take 2 doses of TIBSOVO within 12 hours.
What are the possible side effects of TIBSOVO? TIBSOVO may cause serious side effects, including: - See "What is the most important information I should know about TIBSOVO?"
- Changes in the electrical activity of your heart called QTc prolongation. QTc prolongation can cause irregular heartbeats that can be life-threatening. Your healthcare provider will check the electrical activity of your heart with a test called an electrocardiogram (ECG) before and during treatment with TIBSOVO. Tell your healthcare provider right away if you feel dizzy, lightheaded, or faint.
- Guillain-Barré syndrome has happened in people treated with TIBSOVO. Your healthcare provider will monitor you for nervous system problems and will permanently stop your treatment with TIBSOVO if you develop Guillain-Barré syndrome. Tell your healthcare provider right away if you develop any signs or symptoms of Guillain-Barré syndrome, including:
- weakness or tingling feeling in your legs, arms, or upper body
- numbness and pain on one side or both sides of your body
- any changes in your ability to see, touch, hear, or taste
- burning or prickling sensation
- difficulty breathing
The most common side effects of TIBSOVO when used in combination with azacitidine or alone in adults with AML include: - changes in certain blood cell counts
- diarrhea
- increased blood sugar
- fatigue
- changes in certain liver function tests
- swelling of arms or legs
- decreased levels of electrolytes in the blood
- nausea
- vomiting
- decreased appetite
- joint pain
- shortness of breath
- uric acid increased
- stomach (abdominal) pain
- changes in certain kidney function tests
- pain or sores in your mouth or throat
- rash
- irregular heart rhythm or heartbeat (QTc prolongation)
- differentiation syndrome
- muscle pain
The most common side effects of TIBSOVO when used in adults with Myelodysplastic Syndromes (MDS) include: - changes in certain kidney function tests
- changes in certain blood cell counts
- joint pain, back pain, or neck pain
- decreased levels of albumin in the blood
- changes in liver function tests
- fatigue
- diarrhea
- cough
- decreased levels of sodium in the blood
- pain or sores in your mouth or throat
- decreased appetite
- muscle pain
- decreased levels of phosphorus in the blood
- itchy skin
- rash
The most common side effects of TIBSOVO in adults with Cholangiocarcinoma include:
- fatigue
- nausea
- abdominal pain
- diarrhea
- cough
- decreased appetite
- fluid and swelling in your stomach area
- vomiting
- hemoglobin decreased (anemia)
- rash
- changes in liver function tests
Your healthcare provider will do blood tests before you start and during treatment with TIBSOVO. Your healthcare provider may decrease, temporarily hold, or permanently stop your treatment with TIBSOVO if you develop certain side effects. TIBSOVO may cause fertility problems in females and males, which may affect your ability to have children. Talk to your healthcare provider if you have concerns about fertility. These are not all of the possible side effects of TIBSOVO. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. How should I store TIBSOVO? - Store TIBSOVO at room temperature between 68°F to 77°F (20°C to 25°C).
- Do not remove the desiccant canister from the bottle.
Keep TIBSOVO and all medicines out of the reach of children. General information about the safe and effective use of TIBSOVO Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not take TIBSOVO for conditions for which it was not prescribed. Do not give TIBSOVO to other people, even if they have the same symptoms you have. It may harm them. You can ask your pharmacist or healthcare provider for information about TIBSOVO that is written for healthcare professionals. What are the ingredients in TIBSOVO? Active ingredient: ivosidenib Inactive ingredients: colloidal silicon dioxide, croscarmellose sodium, hypromellose acetate succinate, magnesium stearate, microcrystalline cellulose, and sodium lauryl sulfate. The tablet coating includes FD&C blue #2, hypromellose, lactose monohydrate, titanium dioxide, and triacetin. Manufactured for Servier Pharmaceuticals LLC, Boston, MA 02210 Servier and the Servier logo are registered trademarks of Les Laboratoires Servier. TIBSOVO is a registered trademark of Servier Pharmaceuticals LLC, a wholly owned, indirect subsidiary of Les Laboratoires Servier. © 2023 Servier Pharmaceuticals LLC For more information go to www.TIBSOVO.com or call 1-800-807-6124. AG-MG-006 -
PRINCIPAL DISPLAY PANEL - 250 mg Tablet Bottle Carton
NDC: 72694-617-60
TIBSOVO®
(ivosidenib tablets)250 mg
Dispense the enclosed
Medication Guide to each patientSwallow tablets whole. Do not split,
crush, or chew the tablet.1 Bottle containing 60 tablets
Rx only
-
INGREDIENTS AND APPEARANCE
TIBSOVO
ivosidenib tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 72694-617 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ivosidenib (UNII: Q2PCN8MAM6) (ivosidenib - UNII:Q2PCN8MAM6) ivosidenib 250 mg Inactive Ingredients Ingredient Name Strength Silicon dioxide (UNII: ETJ7Z6XBU4) Croscarmellose sodium (UNII: M28OL1HH48) HYPROMELLOSE ACETATE SUCCINATE 06081224 (3 MPA.S) (UNII: 6N003M473W) Magnesium stearate (UNII: 70097M6I30) Microcrystalline cellulose (UNII: OP1R32D61U) sodium lauryl sulfate (UNII: 368GB5141J) FD&C BLUE NO. 2 (UNII: L06K8R7DQK) Hypromellose, unspecified (UNII: 3NXW29V3WO) Lactose Monohydrate (UNII: EWQ57Q8I5X) Titanium Dioxide (UNII: 15FIX9V2JP) Triacetin (UNII: XHX3C3X673) Product Characteristics Color BLUE Score no score Shape OVAL Size 18mm Flavor Imprint Code IVO;250 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 72694-617-60 1 in 1 CARTON 10/19/2021 1 60 in 1 BOTTLE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA211192 10/19/2021 Labeler - Servier Pharmaceutical LLC (116608503)
Trademark Results [TIBSOVO]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 TIBSOVO 87556035 5587521 Live/Registered |
Agios Pharmaceuticals, Inc. 2017-08-04 |
 TIBSOVO 87556017 5587520 Live/Registered |
Agios Pharmaceuticals, Inc. 2017-08-04 |
 TIBSOVO 87122179 5444093 Live/Registered |
Agios Pharmaceuticals, Inc. 2016-07-31 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.