FUZEON by Genentech, Inc. / F. Hoffmann-La Roche Ltd / Roche Diagnostics GmbH FUZEON- enfuvirtide kit
FUZEON by
Drug Labeling and Warnings
FUZEON by is a Prescription medication manufactured, distributed, or labeled by Genentech, Inc., F. Hoffmann-La Roche Ltd, Roche Diagnostics GmbH. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use FUZEON safely and effectively. See full prescribing information for FUZEON.
FUZEON® (enfuvirtide) for Injection
Initial U.S. Approval: 2003INDICATIONS AND USAGE
FUZEON is an HIV-1 gp41 fusion inhibitor indicated for use in combination with other antiretroviral agents for the treatment of HIV-1 infection in treatment-experienced patients with HIV-1 replication despite ongoing antiretroviral therapy. (1)
DOSAGE AND ADMINISTRATION
- Adults: Recommended dosage of FUZEON is 90 mg (1 mL) twice daily injected subcutaneously into the upper arm, anterior thigh, or abdomen. (2.2)
- Pediatric Patients (weighing at least 11kg): Recommended dose of 2 mg/kg twice daily up to a maximum dose of 90 mg twice daily injected subcutaneously. Weight should be monitored periodically and the FUZEON dose should be adjusted accordingly. (2.3)
- FUZEON must only be reconstituted with 1 mL of Sterile Water for Injection provided in the Convenience Kit. (2.4)
- Reconstituted FUZEON must be injected immediately or kept refrigerated in the original vial. It must be used within 24 hours. (2.4)
DOSAGE FORMS AND STRENGTHS
- Lyophilized powder: 108 mg of enfuvirtide per single-dose vial. (3)
CONTRAINDICATIONS
- Hypersensitivity to FUZEON or any of its components. (4)
WARNINGS AND PRECAUTIONS
- Injection Site Reaction: 98% of subjects experienced at least one injection site reaction during FUZEON treatment in randomized, controlled, open-label, multicenter trials. Manifestations included pain and discomfort, erythema, nodules and cysts, and ecchymosis. (5.1)
- Biojector® 2000: Administration of FUZEON with Biojector 2000 may result in neuralgia and/or paresthesia, bruising and hematomas. (5.2)
- Post-Injection Bleeding: Patients receiving anticoagulants or persons with hemophilia, or other coagulation disorders, may have a higher risk of post-injection bleeding. (5.3)
- Hypersensitivity: FUZEON should be discontinued immediately upon signs and symptoms of systemic hypersensitivity reactions. (5.4)
- Pneumonia: Monitor for signs and symptoms of pneumonia in HIV-infected patients, especially those predisposed to pneumonia (e.g., low initial CD4 cell count). (5.5)
- Immune Reconstitution: Patients treated with combination antiretroviral therapy, including FUZEON, may experience immune reconstitution syndrome requiring further evaluation and treatment. (5.7)
ADVERSE REACTIONS
Most common adverse reactions are local injection site reactions, diarrhea, nausea, and fatigue. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Genentech at 1-888-835-2555 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
USE IN SPECIFIC POPULATIONS
- Lactation: Breastfeeding is not recommended due to risk of HIV-1 transmission. (8.2)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 12/2019
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 General Dosing Information
2.2 Recommended Dosage for Adults
2.3 Recommended Dosage for Pediatric Patients
2.4 Preparation
2.5 Assessment Prior to Administration
2.6 Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Local Injection Site Reactions (ISRs)
5.2 Administration with Biojector® 2000
5.3 Post-Injection Bleeding
5.4 Hypersensitivity Reactions
5.5 Pneumonia
5.6 Non-HIV Infected Individuals
5.7 Immune Reconstitution Syndrome
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
8.7 Renal Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.3 Pharmacokinetics
12.4 Microbiology
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Antiretroviral-experienced Adult Subjects
14.2 Treatment-experienced Pediatric Subjects
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 General Dosing Information
FUZEON is available in a single-dose lyophilized powder for injection containing 108 mg enfuvirtide per vial.
FUZEON is administered subcutaneously into the upper arm, anterior thigh or abdomen after reconstituting the lyophilized powder containing 108 mg enfuvirtide with 1 mL of Sterile Water for Injection [see Dosage and Administration (2.5)]. Patients should contact their healthcare provider for any questions regarding the administration of FUZEON by calling the toll-free number 1-877-4-FUZEON (1-877-438-9366) or visiting the FUZEON website, www.FUZEON.com.
2.2 Recommended Dosage for Adults
The recommended dosage of FUZEON is 90 mg (1 mL) twice daily injected subcutaneously into the upper arm, anterior thigh or abdomen [see Dosage and Administration (2.5 and 2.6)].
2.3 Recommended Dosage for Pediatric Patients
The recommended dosage of FUZEON in pediatric patients weighing at least 11 kg is 2 mg per kg twice daily up to a maximum dose of 90 mg twice daily injected subcutaneously into the upper arm, anterior thigh or abdomen [see Dosage and Administration (2.5 and 2.6) and Use in Specific Populations (8.4)]. Table 1 contains dosing recommendations for FUZEON based on body weight. Weight should be monitored periodically and the FUZEON dose adjusted accordingly.
Table 1 Pediatric Dosing Recommendations Weighing at Least 11 Kg Weight Recommended Daily Dosage (mg) Injection Volume
(mL)Kilograms (kg) 11.0 to 15.5 27 mg twice daily 0.3 mL twice daily 15.6 to 20.0 36 mg twice daily 0.4 mL twice daily 20.1 to 24.5 45 mg twice daily 0.5 mL twice daily 24.6 to 29.0 54 mg twice daily 0.6 mL twice daily 29.1 to 33.5 63 mg twice daily 0.7 mL twice daily 33.6 to 38.0 72 mg twice daily 0.8 mL twice daily 38.1 to 42.5 81 mg twice daily 0.9 mL twice daily ≥42.6 90 mg twice daily 1.0 mL twice daily 2.4 Preparation
FUZEON for injection can be administered by patients after training by medical professional using aseptic technique. Refer patients to FUZEON Injection Instructions for step by step instructions during self-administration.
A vial is suitable for single-dose only; unused portions must be discarded.
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration. Patients should return product to pharmacy if there is evidence of particulate matter after mixing FUZEON with sterile water as described below.
- Remove the flip-off cap from the single-dose Sterile Water for Injection vile and from the FUZEON vial.
- Wipe each vial with a new sterile alcohol swab and let the tops air-dry.
- Using the 3 mL (large) syringe with the plunger pulled back to the 1 mL mark, slowly inject the air into the sterile water vial.
- Insert sterile syringe needle into the vial through the center of the stopper.
- Turn the vial upside down and draw 1 mL of the sterile water into the syringe then remove the needle and syringe from the vial.
- Insert the syringe with sterile water into the FUZEON vial at an angle.
- Inject the sterile water slowly, so that it drips down the side of the vial into the FUZEON powder.
- Never shake the vial but gently tap the FUZEON vial with fingertip for 10 seconds to start dissolving the powder.
- Then gently roll the FUZEON vial between hands to reduce the mixing time, making sure no FUZEON is stuck to the vial wall.
- Once the powder starts to dissolve, just set it aside and it will completely dissolve; it could take up to 45 minutes for the powder to completely dissolve and become a solution.
- When completely mixed, the FUZEON solution should be clear, colorless and without bubbles or particulate matter. If the FUZEON is foamy or jelled, allow more time for it to dissolve.
FUZEON contains no preservatives. Once reconstituted, FUZEON should be injected immediately or kept refrigerated in the original vial until use. Reconstituted FUZEON must be used within 24 hours. Refrigerated reconstituted solution should be brought to room temperature before injection and the vial should be inspected visually again to ensure that the contents are fully dissolved in solution and that the solution is clear, colorless, and without bubbles or particulate matter.
The subsequent dose of FUZEON can be reconstituted in advance but must be stored in the refrigerator in the original vial and used within 24 hours.
2.5 Assessment Prior to Administration
Each injection should be given at a site different from the preceding injection site, and only where there is no current injection site reaction from an earlier dose.
Do not inject FUZEON:
- Near anatomical areas where large nerves course close to the skin, such as near the elbow, knee, groin or the inferior or medial section of the buttocks.
- Directly over or near skin abnormalities such as moles, scar tissue, bruises, surgical scars, tattoos or burn sites.
- Directly over a blood vessel.
- Near the naval.
2.6 Administration
- Clean the injection site with a new sterile alcohol pad.
- Clean the FUZEON vial top again, using a new sterile alcohol pad.
- Using the 1 mL (small) syringe with plunger pulled back to 1 mL, insert the syringe with needle into the vial FUZEON solution.
- Before turning the vial upside down, slowly inject the air into the FUZEON.
- Gently turn the vial upside down and slowly pull the plunger to get 1 mL of FUZEON solution and remove the needle and syringe from the vial.
- Pinch and hold a fold of skin around the injection site and pierce the skin. The needle should be inserted most of the way in. Slowly push the plunger all the way to inject FUZEON.
- Remove the needle from the injection site.
- Instruct patients how to safely discard the syringe and needle.
- Cover the injection site with a small bandage if needed.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
FUZEON is contraindicated in patients with known hypersensitivity to FUZEON or any of its components [see Warnings and Precautions (5.4)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Local Injection Site Reactions (ISRs)
The majority of subjects (98%) receiving FUZEON in randomized, controlled, open-label, multicenter clinical trials had at least one local injection site reaction; ISRs occurred throughout treatment with FUZEON. Manifestations may include pain and discomfort, induration, erythema, nodules and cysts, pruritus, and ecchymosis [see Adverse Reactions (6)]. Reactions are often present at more than one injection site. Patients must be familiar with the FUZEON Injection Instructions in order to know how to inject FUZEON appropriately and how to monitor carefully for signs or symptoms of cellulitis or local infection.
5.2 Administration with Biojector® 2000
Nerve pain (neuralgia and/or paresthesia) lasting up to 6 months associated with administration at anatomical sites where large nerves course close to the skin, bruising and hematomas have occurred with use of the Biojector 2000 needle-free device for administration of FUZEON.
5.3 Post-Injection Bleeding
Patients receiving anticoagulants or persons with hemophilia, or other coagulation disorders, may have a higher risk of post-injection bleeding.
5.4 Hypersensitivity Reactions
Systemic hypersensitivity reactions have been associated with FUZEON therapy and may recur on re-challenge. Hypersensitivity reactions have occurred in <1% of subjects studied and have included combinations of: rash, fever, nausea and vomiting, chills, rigors, hypotension, and/or elevated serum liver transaminases. Other adverse events that may be immune mediated and have been reported in subjects receiving FUZEON include primary immune complex reaction, respiratory distress, glomerulonephritis, and Guillain-Barre syndrome. Patients developing signs and symptoms suggestive of a systemic hypersensitivity reaction should discontinue FUZEON and should seek medical evaluation immediately. Therapy with FUZEON should not be restarted following systemic signs and symptoms consistent with a hypersensitivity reaction. Risk factors that may predict the occurrence or severity of hypersensitivity to FUZEON have not been identified.
5.5 Pneumonia
An increased rate of bacterial pneumonia was observed in subjects treated with FUZEON in the Phase 3 clinical trials compared to the control arm. The incidence of pneumonia was 2.7% or 3.2 events/100 patient-years in subjects receiving FUZEON+background regimen. On analysis of all diagnoses of pneumonia (pneumonia, bacterial pneumonia, bronchopneumonia, and related terms) in T20-301 and T20-302, an increased rate of bacterial pneumonia was observed in subjects treated with FUZEON compared to the control arm (6.9%, 6.7 pneumonia events per 100 patient-years versus 0.6 events per 100 patient-years, respectively). Approximately half of the study subjects with pneumonia required hospitalization. Three subject deaths in the FUZEON arm were attributed to pneumonia; all three had serious concomitant AIDS-related illnesses that contributed to their deaths. Risk factors for pneumonia included low initial CD4 lymphocyte count, high initial viral load, intravenous drug use, smoking, and a prior history of lung disease.
Because it was unclear whether the higher incidence rate of pneumonia was related to FUZEON use, an observational study in 1850 HIV-infected patients (740 FUZEON treated patients and 1110 non-FUZEON treated patients) was conducted to evaluate the risk of pneumonia in patients treated with FUZEON. A total of 123 patients had a confirmed or probable pneumonia event in this study (62 in the FUZEON treatment arm with 1962 patient-years of observation and 61 in the non-FUZEON treatment arm with 3378 patient-years of observation). The incidence of pneumonia was 3.2 events/100 patient-years in the FUZEON treatment arm and 1.8 events/100 patient-years in the non-FUZEON treatment arm. The hazard ratio, adjusting for other baseline risk factors, was 1.34 (95% C.I. = 0.90 – 2.00). Based on this observational study, it is not possible to exclude an increased risk of pneumonia in patients treated with FUZEON compared to non-FUZEON treated patients.
It is unclear if the increased incidence of pneumonia is related to FUZEON use. However, because of these findings, patients with HIV-1 infection should be carefully monitored for signs and symptoms of pneumonia, especially if they have underlying conditions which may predispose them to pneumonia. Risk factors for pneumonia included low initial CD4 cell count, high initial viral load, intravenous drug use, smoking, and a prior history of lung disease.
5.6 Non-HIV Infected Individuals
There is a theoretical risk that FUZEON use may lead to the production of anti-enfuvirtide antibodies which cross react with HIV gp41. This could result in a false positive HIV test with an ELISA assay; a confirmatory western blot test would be expected to be negative. FUZEON has not been studied in non-HIV infected individuals.
5.7 Immune Reconstitution Syndrome
Immune reconstitution syndrome has been reported in patients treated with combination antiretroviral therapy, including FUZEON. During the initial phase of combination antiretroviral treatment, patients whose immune system responds may develop an inflammatory response to indolent or residual opportunistic infections (such as Mycobacterium avium infection, cytomegalovirus, Pneumocystis jirovecii pneumonia [PCP] or tuberculosis), which may necessitate further evaluation and treatment.
Autoimmune disorders (such as Graves' disease, polymyositis, and Guillain-Barré syndrome) have also been reported to occur in the setting of immune reconstitution, however, the time to onset is more variable, and can occur many months after initiation of treatment.
-
6 ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections:
- Administration with Biojector® 2000 [see Warnings and Precautions (5.2)]
- Hypersensitivity Reactions [see Warnings and Precautions (5.4)]
- Pneumonia [see Warnings and Precautions (5.5)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The overall safety profile of FUZEON is based on 2131 subjects who received at least 1 dose of FUZEON during various clinical trials. This includes 2051 adults, 658 of whom received the recommended dose for greater than 48 weeks, and 63 pediatric subjects.
Assessment of treatment-emergent adverse events is based on the pooled data from the two randomized, controlled, open-label, multicenter trials in treatment-experienced subjects, T20-301 (TORO 1) and T20-302 (TORO 2).
Local Injection Site Reactions
Local injection site reactions were the most frequent adverse events associated with the use of FUZEON. In T20-301 and T20-302, 98% of subjects had at least one local injection site reaction (ISR). A total of 7% of subjects discontinued treatment with FUZEON because of ISRs (4%) or difficulties with injecting FUZEON (3%) such as injection fatigue and inconvenience. Eighty-five percent of subjects experienced their first ISR during the initial week of treatment; ISRs continued to occur throughout treatment with FUZEON. For most subjects the severity of signs and symptoms associated with ISRs did not change during the 48 weeks of treatment. The majority of ISRs were associated with erythema, induration, the presence of nodules or cysts, and mild to moderate pain at the injection site (Table 2). In addition, the average duration of individual ISRs was between three and seven days in 41% of subjects and more than seven days in 24% of subjects. Also, the numbers of ISRs per subject at any one time was between six to 14 ISRs in 26% of subjects and more than 14 ISRs in 1.3% of subjects. Infection at the injection site (including abscess and cellulitis) was reported in 1.7% of adult subjects.
Table 2 Summary of Individual Signs/Symptoms Characterizing Local Injection Site Reactions to Enfuvirtide in Studies T20-301 and T20-302 Combined (% of Subjects) Through 48 Weeks N=663 Event Category Any Severity Grade % of Subjects with Grade 3 Reactions % of Subjects with Grade 4 Reactions - * Grade 3 = severe pain requiring prescription non-topical analgesics or limiting usual activities.
Grade 4 = severe pain requiring hospitalization or prolongation of hospitalization, resulting in death, or persistent or significant disability/incapacity, or life-threatening, or medically significant.- † Grade 3 = refractory to topical treatment or requiring oral or parenteral treatment.
Grade 4 = not applicable.Pain/Discomfort * 96% 11% 0% Induration 90% 39%
>25 but <50 mm18%
≥50 mmErythema 91% 22%
>50 but <85 mm10%
≥85 mmNodules and Cysts 80% 23%
>3 cm average diameter0.2%
DrainingPruritus † 65% 3% NA Ecchymosis 52% 5%
>3 but ≤5 cm2%
>5 cmOther Adverse Events
In T20-301 and T20-302, after study week 8, subjects on background alone who met protocol defined criteria for virological failure were permitted to revise their background regimens and add FUZEON. Exposure on FUZEON+background was 557 patient-years, and to background alone 162 patient-years. Due to this difference in exposure, safety results are expressed as the number of patients with an adverse event per 100 patient-years of exposure. For FUZEON+background, adverse events are also displayed by percent of subjects.
The events most frequently reported in subjects receiving FUZEON+background regimen, excluding ISRs, were diarrhea (38 per 100 patient-years or 31.7%), nausea (27 per 100 patient-years or 22.8%), and fatigue (24 per 100 patient-years or 20.2%). These events were also commonly observed in subjects that received background regimen alone: diarrhea (73 per 100 patient-years), nausea (50 per 100 patient-years), and fatigue (38 per 100 patient-years).
Treatment-emergent adverse events, regardless of causality and excluding ISRs, from Phase 3 studies are summarized for adult subjects, in Table 3. Any Grade 2 or above events occurring at ≥2 percent of subjects and at a higher rate in subjects treated with FUZEON are summarized in Table 3; events that occurred at a higher rate in the control arms are not displayed.
Rates of adverse events for subjects who switched to FUZEON after virological failure were similar.
Table 3 Rates of Treatment-Emergent Adverse Events* (≥Grade 2) Reported in ≥2% of Subjects Treated with FUZEON† (Pooled Studies T20-301/T20-302 at 48 Weeks) Adverse Event
(by System Organ Class)FUZEON+Background Regimen
(N=663)FUZEON+Background Regimen
(N=663)Background Regimen
(N=334)663 subjects total 557 total patient-years 162 total patient-years % frequency rate/100 patient-years rate/100 patient-years - * Excludes Injection Site Reactions
- † Events listed occurred more frequently in subjects treated with FUZEON (based on rates/100 patient-years).
Weight Decreased 6.6% 7.9 6.2 Sinusitis 6.0% 7.2 4.9 Abdominal Pain 3.9% 4.7 3.7 Cough 3.9% 4.7 2.5 Herpes Simplex 3.5% 4.1 3.7 Appetite Decreased 3.2% 3.8 2.5 Pancreatitis 3.0% 3.6 2.5 Pain in Limb 2.9% 3.4 3.1 Pneumonia (see text below) 2.7% 3.2 0.6 Myalgia 2.7% 3.2 1.2 Influenza-Like Illness 2.4% 2.9 1.9 Folliculitis 2.4% 2.9 2.5 Anorexia 2.3% 2.7 1.9 Dry Mouth 2.1% 2.5 1.9 Conjunctivitis 2.0% 2.3 1.9 Less Common Events
The following adverse events have been reported in 1 or more subjects; however, a causal relationship to FUZEON has not been established.
Immune System Disorders: worsening abacavir hypersensitivity reaction
Renal and Urinary Disorders: glomerulonephritis; tubular necrosis; renal insufficiency; renal failure (including fatal cases)
Blood and Lymphatic Disorders: thrombocytopenia; neutropenia; fever; lymphadenopathy
Endocrine and Metabolic: hyperglycemia
Infections: sepsis; herpes simplex
Nervous System Disorders: taste disturbance; Guillain-Barre syndrome (fatal); sixth nerve palsy; peripheral neuropathy
Cardiac Disorders: unstable angina pectoris
Gastrointestinal Disorders: constipation; abdominal pain upper
General: asthenia
Hepatobiliary Disorders: toxic hepatitis; hepatic steatosis
Investigations: increased amylase; increased lipase; increased AST; increased GGT; increased triglycerides
Psychiatric Disorders: insomnia; depression; anxiety; suicide attempt
Respiratory, Thoracic, and Mediastinal Disorders: pneumopathy; respiratory distress; cough
Skin and Subcutaneous Tissue Disorders: pruritus
Laboratory Abnormalities
Table 4 shows the treatment-emergent laboratory abnormalities that occurred in at least 2 subjects per 100 patient-years and more frequently in those receiving FUZEON+background regimen than background regimen alone from T20-301 and T20-302.
Table 4 Treatment-Emergent Laboratory Abnormalities in ≥2% of Subjects Receiving FUZEON* (Pooled Studies T20-301 and T20-302 at 48 Weeks) Laboratory Parameters Grading FUZEON+Background Regimen
(N=663)FUZEON+Background Regimen
(N=663)Background Regimen
(N=334)663 subjects total 557 total patient-years 162 total patient-years % frequency rate/100 patient-years rate/100 patient-years - * Events listed occurred more frequently in subjects treated with FUZEON (based on rates/100 patient-years).
Eosinophilia 1-2 × ULN (0.7 × 109/L) 0.7-1.4 × 109/L 9.1% 10.8 3.7 >2 × ULN (0.7 × 109/L) >1.4 × 109/L 1.8% 2.2 1.8 ALT Grade 3 >5-10 × ULN 4.1% 4.8 4.3 Grade 4 >10 × ULN 1.2% 1.4 1.2 Creatine Phosphokinase (U/L) Grade 3 >5-10 × ULN 6.9% 8.3 8.0 Grade 4 >10 × ULN 2.6% 3.1 8.6 Adverse Events in Pediatric Patients
FUZEON has been studied in 63 pediatric subjects 5 through 16 years of age with duration of FUZEON exposure ranging from 1 dose to 134 weeks. Adverse experiences seen during clinical trials were similar to those observed in adult subjects, although infections at site of injection (cellulitis or abscess) were more frequent in adolescents than in adults, with 4 events occurring in 3 of 28 (11%) subjects.
6.2 Postmarketing Experience
The following adverse reaction has been identified during post approval use of FUZEON. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in individuals exposed to antiretroviral medicines during pregnancy. Healthcare providers are encouraged to register patients by calling the Antiretroviral Pregnancy Registry (APR) at 1-800-258-4263.
Risk Summary
Prospective pregnancy data from the APR are not sufficient to adequately assess the risk of birth defects or fetal outcomes. Limited number of reports on the use of enfuvirtide during pregnancy has been submitted to the APR and the number of exposures to enfuvirtide is insufficient to make a risk assessment compared to a reference population. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. The estimated background rate for major birth defects is 2.7% in the U.S. reference population of the Metropolitan Atlanta Congenital Defects Program (MACDP). The estimated rate of miscarriage is not reported in the APR. The estimated background rate of miscarriage in the U.S. general population is 15–20%.
In animal reproduction studies, no adverse developmental effects were observed with subcutaneous administration of enfuvirtide at exposures greater than or equal to approximately 2 times higher than human exposure at the recommended human dose (RHD) based on surface area (see Data).
Data
Animal Data
In the embryofetal development studies, enfuvirtide was administered by subcutaneous injection to pregnant rats at doses up to 500 mg/kg/day from gestation days 6 to 17, and to pregnant rabbits at doses up to 30 mg/kg/day from gestation days 6 to 18. No embryofetal toxicities were observed at doses up to the highest doses tested (27 times and 3.2 times higher than human exposure at the RHD in rats and rabbits, respectively, based on surface area).
In the pre/postnatal development study, enfuvirtide was administered by subcutaneous injection to pregnant rats at doses up to 30 mg/kg/day from gestation day 6 to postnatal day 20. No toxicities were observed at doses up to 30 mg/kg/day (1.6 times higher than human exposure at the RHD based on surface area).
8.2 Lactation
Risk Summary
The Centers for Disease Control and Prevention recommends that HIV-1 infected mothers not breastfeed their infants to avoid the risk of postnatal transmission of HIV-1.
There are no human data available regarding the presence of enfuvirtide or its metabolites (amino acids and peptide fragments) in human milk, the effects on the breastfed infant, or the effects on milk production. When enfuvirtide was administered to lactating rats, enfuvirtide was likely present in the milk (see Data).
Because of both the potential for (1) HIV-1 transmission (in HIV-negative infants), (2) developing viral resistance (in HIV-positive infants) and (3) adverse reactions in breastfed infants similar to those seen in adults, instruct mothers not to breast-feed if they are receiving FUZEON.
8.4 Pediatric Use
Use of FUZEON in pediatric patients weighing at least 11kg is supported by evidence from adequate and well-controlled studies of FUZEON in adult and by two pediatric studies evaluating the safety, pharmacokinetics and efficacy of FUZEON in subjects 6 years of age and older:
- T20-204 was an open-label, multicenter trial that evaluated the safety and antiviral activity of FUZEON in 11, treatment-experienced pediatric subjects 6 to 12 years (median age of 9 years) [see Clinical Pharmacology (12.3) and Clinical Studies (14.2)].
- T20-310 was an open-label, multicenter trial that evaluated the pharmacokinetics, safety, and antiviral activity of FUZEON in 52, treatment-experienced pediatric subjects 5 years and older (median age of 12 years) [see Clinical Pharmacology (12.3) and Clinical Studies (14.2)].
Overall, the adverse experiences, including ISRs in the 63 pediatric subjects were similar to those observed in adult subjects, although infections at site of injection (cellulitis or abscess) were more frequent in adolescents than in adults, with 4 events occurring in 3 of 28 (11%) subjects [see Adverse Reactions (6.1)].
8.5 Geriatric Use
Clinical studies of FUZEON did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. In general, appropriate caution should be exercised in the administration and monitoring of FUZEON in elderly patients reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
8.6 Hepatic Impairment
No dosage adjustments of FUZEON are needed in patients with hepatic impairment [see Clinical Pharmacology (12.3)].
8.7 Renal Impairment
No dosage adjustments of FUZEON are needed in patients with renal impairment [see Clinical Pharmacology (12.3)].
-
10 OVERDOSAGE
There are no reports of human experience of acute overdose with FUZEON. The highest dose administered to 12 subjects in a clinical trial was 180 mg as a single-dose subcutaneously. There is no specific antidote for overdose with FUZEON. Treatment of overdose should consist of general supportive measures.
-
11 DESCRIPTION
FUZEON (enfuvirtide) is an inhibitor of the fusion of HIV-1 with CD4 cells. Enfuvirtide is a linear 36-amino acid synthetic peptide with the N-terminus acetylated and the C-terminus is a carboxamide. It is composed of naturally occurring L-amino acid residues.
Enfuvirtide is a white to off-white amorphous solid. It has negligible solubility in pure water and the solubility increases in aqueous buffers (pH 7.5) to 85-142 g/100 mL. The empirical formula of enfuvirtide is C204H301N51O64, and the molecular weight is 4492. It has the following primary amino acid sequence:
CH3CO-Tyr-Thr-Ser-Leu-Ile-His-Ser-Leu-Ile-Glu-Glu-Ser-Gln-Asn-Gln-Gln-Glu-Lys-Asn-Glu-Gln-Glu-Leu-Leu-Glu-Leu-Asp-Lys-Trp-Ala-Ser-Leu-Trp-Asn-Trp-Phe-NH2 and the following structural formula:
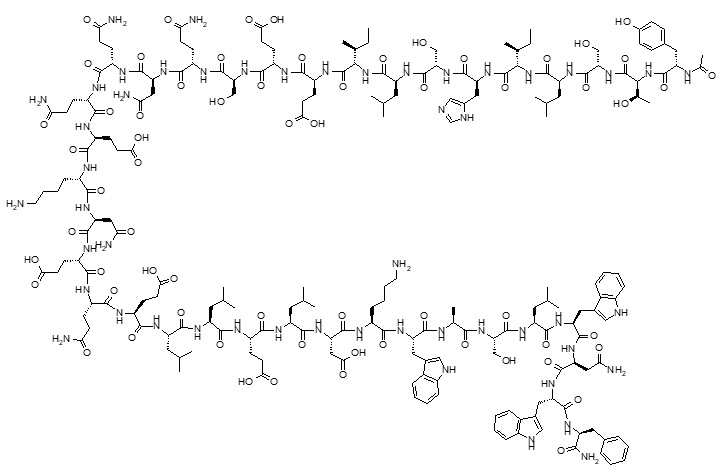
The drug product, FUZEON (enfuvirtide) for Injection, is a white to off-white, sterile, lyophilized powder. Each single-dose vial contains 108 mg of enfuvirtide for the delivery of 90 mg. Prior to subcutaneous administration, the contents of the vial are reconstituted with 1 mL of Sterile Water for Injection to provide the delivery of 1 mL of the solution. Each 1 mL of the reconstituted solution contains approximately 90 mg of enfuvirtide with approximate amounts of the following excipients: 22.55 mg of mannitol, 2.39 mg of sodium carbonate (anhydrous), and sodium hydroxide and hydrochloric acid for pH adjustment as needed. The reconstituted solution has an approximate pH of 9.0.
-
12 CLINICAL PHARMACOLOGY
12.3 Pharmacokinetics
The pharmacokinetic properties of enfuvirtide were evaluated in HIV-1 infected adult and pediatric subjects.
Absorption
Following a 90-mg single subcutaneous injection of FUZEON into the abdomen in 12 HIV-1 infected subjects, the mean (±SD) Cmax was 4.59 ± 1.5 mg/L, AUC was 55.8 ± 12.1 mg∙h/L and the median Tmax was 8 hours (ranged from 3 to 12 h). The absolute bioavailability (using a 90-mg intravenous dose as a reference) was 84.3% ± 15.5%. Following 90-mg twice daily dosing of FUZEON subcutaneously in combination with other antiretroviral agents in 11 HIV-1 infected subjects, the mean (±SD) steady-state Cmax was 5.0 ± 1.7 mg/L, Ctrough was 3.3 ± 1.6 mg/L, AUC0-12h was 48.7 ± 19.1 mg∙h/L, and the median Tmax was 4 hours (ranged from 4 to 8 h).
Absorption of the 90-mg dose was comparable when injected into the subcutaneous tissue of the abdomen, thigh or arm.
Distribution
The mean (±SD) steady-state volume of distribution after intravenous administration of a 90-mg dose of FUZEON (N=12) was 5.5 ± 1.1 L.
Enfuvirtide is approximately 92% bound to plasma proteins in HIV-infected plasma over a concentration range of 2 to 10 mg/L. It is bound predominantly to albumin and to a lower extent to α-1 acid glycoprotein.
The CSF levels of enfuvirtide (measured from 2 hours to 18 hours after administration of enfuvirtide) in 4 HIV-infected subjects were below the limit of quantification (0.025 mg/L).
Metabolism/Elimination
As a peptide, enfuvirtide is expected to undergo catabolism to its constituent amino acids, with subsequent recycling of the amino acids in the body pool.
Mass balance studies to determine elimination pathway(s) of enfuvirtide have not been performed in humans.
In vitro studies with human microsomes and hepatocytes indicate that enfuvirtide undergoes hydrolysis to form a deamidated metabolite at the C-terminal phenylalanine residue, M3. The hydrolysis reaction is not NADPH dependent. The M3 metabolite is detected in human plasma following administration of enfuvirtide, with an AUC ranging from 2.4% to 15% of the enfuvirtide AUC.
Following a 90-mg single subcutaneous dose of enfuvirtide (N=12) the mean ±SD elimination half-life of enfuvirtide is 3.8 ± 0.6 h and the mean ±SD apparent clearance was 0.0248 ± 0.0041 L/h/kg. Following 90-mg twice daily dosing of FUZEON subcutaneously in combination with other antiretroviral agents in 11 HIV-1 infected subjects, the mean ±SD apparent clearance was 0.0306 ± 0.0106 L/h/kg.
Specific Populations
No clinically significant changes in pharmacokinetics have been observed based on renal impairment (any degree of renal impairment, including hemodialysis), gender, adult weight, or race.
Pharmacokinetic studies of enfuvirtide have not been conducted in subjects with hepatic impairment or subjects over 65 years of age.
Pediatric Patients
The pharmacokinetics of enfuvirtide has been studied in 25 pediatric subjects aged 5 through 16 years. Enfuvirtide pharmacokinetics were determined in the presence of concomitant medications including antiretroviral agents. A dose of 2 mg/kg twice daily (maximum 90 mg twice daily) provided enfuvirtide plasma concentrations similar to those obtained in adult subjects receiving 90 mg twice daily.
In the 25 pediatric subjects receiving the 2 mg/kg twice daily dose, the mean ± SD steady-state AUC0-12 was 54.3 ± 23.5 mg∙h/L, Cmax was 6.14 ± 2.48 mg/L, and Ctrough was 2.93 ± 1.55 mg/L [see Use in Specific Populations (8.4)].
Drug Interaction Studies
Based on the results from an in vitro human microsomal study, enfuvirtide is not an inhibitor of CYP450 enzymes. In an in vivo human metabolism study (N=12), FUZEON at the recommended dose of 90 mg twice daily did not alter the metabolism of CYP3A4, CYP2D6, CYP1A2, CYP2C19 or CYP2E1 substrates.
Based on the available data, co-administration of FUZEON and other drugs which are inducers or inhibitors of CYP450 is not expected to alter the pharmacokinetics of enfuvirtide. No dose adjustments are needed when FUZEON is co-administered with other antiretroviral and non-antiretroviral drugs.
Table 5 shows the results of the drug-drug interaction studies conducted between FUZEON and the following drugs: ritonavir, saquinavir/ritonavir, and rifampin.
Table 5 Effect of Ritonavir, Saquinavir/Ritonavir, and Rifampin on the Steady-State Pharmacokinetics of Enfuvirtide (90 mg bid)* Coadministered Drug Dose of Coadministered Drug N % Change of Enfuvirtide Pharmacokinetic Parameters†‡ (90% CI) Cmax AUC Ctrough - * All studies were performed in HIV-1+ subjects using a sequential crossover design.
- † ↑= Increase; ↓ = Decrease; ⇔ = No Effect (↑ or ↓ <10%)
- ‡ No interactions were clinically significant.
Ritonavir 200 mg, q12h, 4 days 12 ↑24
(↑9 to ↑41)↑22
(↑8 to ↑37)↑14
(↑2 to ↑28)Saquinavir/Ritonavir 1000/100 mg, q12h, 4 days 12 ⇔ ↑14
(↑5 to ↑24)↑26
(↑17 to↑35)Rifampin 600 mg, qd, 10 days 12 ⇔ ⇔ ↓15
(↓22 to ↓7)12.4 Microbiology
Mechanism of Action
Enfuvirtide interferes with the entry of HIV-1 into cells by inhibiting fusion of viral and cellular membranes. Enfuvirtide binds to the first heptad-repeat (HR1) in the gp41 subunit of the viral envelope glycoprotein and prevents the conformational changes required for the fusion of viral and cellular membranes.
Antiviral Activity in Cell Culture
The antiviral activity of enfuvirtide was assessed by infecting different CD4+ cell types with laboratory and clinical isolates of HIV-1. The median EC50 value for baseline clinical isolates was 4.10 nM (ranged from 0.089 to 107 nM; 0.4 to 480 ng/mL) by the cMAGI assay (n=130) and was 55.9 nM (1.56 to 1675 nM; 7 to 7526 ng/mL) by a recombinant phenotypic entry assay (n=627). Enfuvirtide was similarly active in cell culture against clades A, AE, C, D, F, and G (median EC50 value was 7.01 nM; range 3.78 to 27.9 nM; 17-126 ng/mL), and R5, X4, and dual tropic viruses. Enfuvirtide has no activity against HIV-2.
Enfuvirtide did not exhibit antagonism in cell culture assays when combined with individual members of various antiretroviral classes, including non-nucleoside reverse transcriptase inhibitors (NNRTIs: efavirenz), nucleos(t)ide reverse transcriptase inhibitors (NRTIs: lamivudine, zidovudine), and protease inhibitors (PIs: indinavir, nelfinavir).
Drug Resistance
HIV-1 isolates with reduced susceptibility to enfuvirtide have been selected in cell culture. Genotypic analysis of these resistant isolates showed amino acid substitutions at the enfuvirtide binding HR1 domain positions 36 to 38 of the HIV-1 envelope glycoprotein gp41. Phenotypic analysis of site-directed mutants in positions 36 to 38 in an HIV-1 molecular clone showed a 5-fold to 684-fold decrease in susceptibility to enfuvirtide.
In clinical trials, HIV-1 isolates with reduced susceptibility to enfuvirtide have been recovered from subjects failing a FUZEON containing regimen. Post treatment HIV-1 virus from 277 subjects experiencing protocol defined virological failure at 48 weeks exhibited a median decrease in susceptibility to enfuvirtide of 33.4-fold (range 0.4-6318-fold) relative to their respective baseline virus. Of these, 249 had decreases in susceptibility to enfuvirtide of greater than 4-fold and all but 3 of those 249 encoded substitutions at gp41 HR1 domain amino acids 36 to 45. HR1 substitutions associated with resistance to enfuvirtide include G36D/E/S/V, I37V, V38A/E/G/M, Q39R, Q40H, N42D/Q/T, N43D/H/K/Q/S, L44M, and L45M. Combinations of HR1 resistance-associated substitutions can lead to greater reductions in susceptibility. Substitutions or polymorphisms in other regions of gp41 have been associated with resistance (e.g., HR2 domain N126K, E137K, and S138A substitutions) and these may affect susceptibility to enfuvirtide.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis and Mutagenesis
Carcinogenicity studies have not been conducted with enfuvirtide.
Enfuvirtide was not genotoxic in in vivo and in vitro assays including a bacterial reverse mutation assay, a mammalian cell forward gene mutation assay in AS52 Chinese Hamster ovary cells, and an in vivo mouse micronucleus assay.
-
14 CLINICAL STUDIES
14.1 Antiretroviral-experienced Adult Subjects
T20-301 and T20-302 were randomized, controlled, open-label, multicenter trials in HIV-1 infected subjects. Subjects were required to have either (1) viremia despite 3 to 6 months prior therapy with a nucleoside reverse transcriptase inhibitor (NRTI), non-nucleoside reverse transcriptase inhibitor (NNRTI), and protease inhibitor (PI) or (2) viremia and documented resistance or intolerance to at least one member in each of the NRTI, NNRTI, and PI classes.
All subjects received an individualized background regimen consisting of 3 to 5 antiretroviral agents selected on the basis of the subject's prior treatment history and baseline genotypic and phenotypic viral resistance measurements. Subjects were then randomized at a 2:1 ratio to FUZEON 90 mg twice daily with background regimen or background regimen alone.
After week 8, subjects on either treatment arm who met protocol defined criteria for virological failure were permitted to revise their background regimens; those on background regimen alone were also permitted to add FUZEON.
Demographic characteristics for studies T20-301 and T20-302 are shown in Table 6. Subjects had prior exposure to a median of 12 antiretrovirals for a median of 7 years.
Table 6 T20-301 and T20-302 Pooled Subject Demographics FUZEON+Background Regimen
N=663Background Regimen
N=334Sex Male 90% 90% Female 10% 10% Race White 89% 89% Black 8% 7% Mean Age (yr)
(range)42
(16-67)43
(24-82)Median Baseline HIV-1 RNA (log10 copies/mL)
(range)5.2
(3.5-6.7)5.1
(3.7-7.1)Median Baseline CD4 Cell Count (cells/mm3)
(range)89
(1-994)97
(1-847)The disposition and efficacy outcomes of T20-301 and T20-302 are shown in Table 7.
Table 7 Outcomes at Week 48 (Pooled Studies T20-301 and T20-302) - * Includes never responded, rebound, and missing RNA data.
- † Includes study discontinuation for virological failure and insufficient response as per the judgment of the investigator.
- ‡ Includes difficulties with injection, such as injection fatigue and inconvenience.
- § Includes lost to follow-up, treatment refusal, and non-compliance.
Outcomes FUZEON+Background Regimen
90 mg bid
N=663Background Regimen
N=334Virological Responder
(at least 1 log10 below baseline)304 (46%) 61 (18%) Virological Non-responder: - Switch
0 220 (66%) - Completed 48 weeks randomized regimen*
191 (29%) 12 (4%) Continued Background Regimen
(N=112)Switched to FUZEON
(N=220)Discontinued due to insufficient treatment response† 37 (5%) 13 (12%) 22 (10%) Discontinued due to adverse reactions/intercurrent illness/labs 46 (7%) 9 (8%) 13 (6%) Deaths 15 (2%) 5 (4%) 2 (1%) Discontinued due to injection: - Injection site reactions
27 (4%) NA 10 (5%) - Difficulty with injecting FUZEON‡
18 (3%) NA 2 (1%) Discontinued due to other reasons§ 25 (4%) 14 (13%) 6 (3%) At 48 weeks, 154 (23%) of subjects in the FUZEON+background regimen and 27 (8%) in the background regimen alone had HIV-1 RNA levels <50 copies/mL, and 225 (34%) of subjects receiving FUZEON+background regimen had HIV-1 RNA levels <400 copies/mL compared to 44 (13%) in the background regimen alone. Subjects achieving HIV-1 RNA levels <50 copies/mL were included in the <400 copies/mL category and both categories were incorporated in the overall virologic responder category of achieving HIV-1 RNA at least 1 log10 below baseline.
The mean log change in HIV-1 RNA from baseline was -1.4 log10 copies/mL in subjects receiving FUZEON+background and -0.5 in those receiving background alone. The mean change in CD4 cell count from baseline to week 48 was +91 cells/mm3 in the FUZEON+background arm and +45 cells/mm3 in the background alone arm.
Subjects in the FUZEON+background arm achieved a better virologic and immunologic outcome than subjects in the background alone arm across all subgroups based on baseline CD4 cell count, baseline HIV-1 RNA, number of prior ARVs or number of active ARVs in the background regimen.
14.2 Treatment-experienced Pediatric Subjects
Sixty-three HIV-1 infected pediatric subjects ages 5 through 16 years have received FUZEON in two open-label, single-arm clinical trials.
T20-204 was an open-label, multicenter trial that evaluated the safety and antiviral activity of FUZEON in treatment-experienced pediatric subjects. Eleven subjects from 6 to 12 years were enrolled (median age of 9 years). Median baseline CD4 cell count was 495 cells/µL and the median baseline HIV-1 RNA was 4.6 log10 copies/mL.
Ten of the 11 study subjects completed 48 weeks of chronic therapy. At week 48, 6/11 (55%) subjects had ≥1 log10 decline in HIV-1 RNA and 4/11 (36%) subjects were below 400 copies/mL of HIV-1 RNA. The median changes from baseline (for the As Treated population) in HIV-1 RNA and CD4 cell count were -1.48 log10 copies/mL and +122 cells/µL, respectively.
T20-310 was an open-label, multicenter trial that evaluated the pharmacokinetics, safety, and antiviral activity of FUZEON in treatment-experienced pediatric subjects. Fifty-two subjects from 5 through 16 years were enrolled (median age of 12 years). Median baseline CD4 cell count was 117 cells/µL and the median baseline HIV-1 RNA was 5.0 log10 copies/mL.
Thirty-two of the 52 study subjects completed 48 weeks of chronic therapy. At week 48, 17/52 (33%) of subjects had ≥1 log10 decline in HIV-1 RNA, 11/52 (21%) of subjects were below 400 copies/mL of HIV-1 RNA and 5/52 (10%) were below 50 copies/mL. The median changes from baseline (for the As Treated population) in HIV-1 RNA and CD4 cell count were -1.17 log10 copies/mL and +106 cells/µL, respectively.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
FUZEON (enfuvirtide) for Injection is a white to off-white, sterile, lyophilized powder and it is packaged in a single-dose clear glass vial containing 108 mg of enfuvirtide for the delivery of approximately 90 mg/1 mL when reconstituted with 1 mL of Sterile Water for Injection.
FUZEON is available in a Convenience Kit containing 60 single-dose vials of FUZEON (90 mg strength), 60 vials (2 cartons of 30 each) of Sterile Water for Injection (1 mL per vial), 60 reconstitution syringes (3 cc), 60 administration syringes (1 cc), Package Insert, Patient Package Insert, and Injection Instructions (NDC: 0004-0381-40).
-
17 PATIENT COUNSELING INFORMATION
See FDA-Approved Patient Labeling (Patient Information, Instructions for Use)
To assure safe and effective use of FUZEON, the following information and instructions should be given to patients:
Injection Site Reactions
Inform patients that injection site reactions occur in almost all patients taking FUZEON. Patients must be familiar with the FUZEON Injection Instructions for instructions on how to appropriately inject FUZEON and how to carefully monitor for signs or symptoms of cellulitis or local infection. Instruct patients when to contact their healthcare provider about these reactions [see Warnings and Precautions (5.1)].
Administration with Biojector® 2000
Inform patients that nerve pain (neuralgia and/or paresthesia) associated with administration at anatomical sites where large nerves course close to the skin, bruising and hematomas have occurred with use of the Biojector 2000 needle-free device for administration of FUZEON [see Warnings and Precautions (5.2)].
Post-injection Bleeding
Advise patients about the risk of post-injection bleeding if they are receiving anticoagulants or have coagulation disorders such as hemophilia [see Warnings and Precautions (5.3)].
Systemic Hypersensitivity
Advise patients of the possibility of a systemic hypersensitivity reaction to FUZEON. Advise patients to discontinue therapy and immediately seek medical evaluation if they develop signs/symptoms of systemic hypersensitivity such as combinations of rash, fever, nausea and vomiting, chills, rigors, and/or hypotension [see Warnings and Precautions (5.4)].
Pneumonia
Advise patients that an increased rate of bacterial pneumonia was observed in subjects treated with FUZEON in clinical trials. Advise patients to seek medical evaluation immediately if they develop signs or symptoms suggestive of pneumonia (cough with fever, rapid breathing, shortness of breath) [see Warnings and Precautions (5.5)].
Pregnancy Exposure Registry
Advise patients that there is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to antiretroviral medicines during pregnancy [see Use in Specific Populations (8.1)].
Lactation
Instruct mothers with HIV-1 infection not to breastfeed because HIV-1 can be passed to the baby in breast milk [see Use in Specific Populations (8.2)].
Important Dosing and Administration Instructions
- Inform patients that FUZEON must be taken as part of a combination antiretroviral regimen and that use of FUZEON alone may lead to rapid development of virus resistant to FUZEON and possibly other agents of the same class.
- Instruct patients and caregivers in the use of aseptic technique when administering FUZEON in order to avoid injection site infections. Appropriate training for FUZEON reconstitution and self-injection must be given by a healthcare provider, including a careful review of the FUZEON Patient Package Insert and FUZEON Injection Instructions. The first injection should be performed under the supervision of an appropriately qualified healthcare provider. It is recommended that the patient and/or caregiver's understanding and use of aseptic injection techniques and procedures be periodically re-evaluated.
- Instruct patients and caregivers on the preferred anatomical sites for administration (upper arm, abdomen, anterior thigh). FUZEON should not be injected near any anatomical areas where large nerves course close to the skin, such as near the elbow, knee, groin or the inferior or medial sections of the buttocks, skin abnormalities, including directly over a blood vessel, into moles, scar tissue, bruises, or near the navel, surgical scars, tattoos or burn sites.
- Instruct patients and caregivers in the proper techniques for preparation, injection and disposal of needles and syringes (including not recapping needles) in order to avoid needle stick injuries. Advise patients not to reuse needles or syringes, and of safe disposal procedures, including the use of a puncture-resistant container, for disposal of used needles and syringes. Instruct patients on the safe disposal of full containers as per local requirements. Caregivers who experience an accidental needle stick after patient injection should contact a healthcare provider immediately.
- Inform patients to contact their healthcare provider for any questions regarding the administration of FUZEON.
- Inform patients not to change the dose or dosing schedule of FUZEON or any antiretroviral medication without consulting their healthcare provider.
- Inform patients to contact their healthcare provider immediately if they stop taking FUZEON or any other drug in their antiretroviral regimen.
- Inform patients that they can obtain more information on the self-administration of FUZEON at www.FUZEON.com or by calling 1-877-4-FUZEON (1-877-438-9366).
-
SPL UNCLASSIFIED SECTION
FUZEON is a trademark of Hoffmann-La Roche Inc.
FUZEON has been jointly developed by Alexion Pharmaceuticals, Inc. and Hoffmann-La Roche Inc. FUZEON is manufactured by Hoffmann-La Roche Inc.
Distributed by:
Genentech USA, Inc.
A Member of the Roche Group
1 DNA Way
South San Francisco, CA 94080® 2019 Genentech, Inc. and Alexion Pharmaceuticals, Inc. All rights reserved.
-
PATIENT PACKAGE INSERT
Patient Information
FUZEON® (few'-zee-on)
(enfuvirtide) InjectionThis Patient Information has been approved by the U.S. Food and Drug Administration. Revised: 12/2019 What is FUZEON?
FUZEON is a prescription medicine used in combination with other antiretroviral medicines to treat Human Immunodeficiency Virus-1 (HIV-1) infection in people who have taken other antiretroviral medicines and whose HIV-1 levels have continued to increase while on treatment.
HIV-1 is the virus that causes Acquired Immune Deficiency Syndrome (AIDS).
It is not known if FUZEON is safe and effective for use in children under 6 years of age.
Do not use FUZEON if you are allergic to enfuvirtide or any of the ingredients in FUZEON. See the end of this leaflet for a complete list of ingredients in FUZEON.
Before using FUZEON, tell your healthcare provider about all your medical conditions, including if you:
- have bleeding problems
- have or had lung problems
- have a low CD4 count
- smoke or use intravenous (IV) street drugs
- are pregnant or plan to become pregnant. It is not known if FUZEON can harm your unborn baby. Tell your healthcare provider if you become pregnant during treatment with FUZEON.
- Pregnancy Registry: There is a pregnancy registry for women who use antiretroviral medicines during pregnancy. The purpose of this registry is to collect information about the health of you and your baby. Talk to your healthcare provider about how you can take part in this registry.
- are breastfeeding or plan to breastfeed. Do not breastfeed if you use FUZEON.
- You should not breastfeed if you have HIV-1 because of the risk of passing HIV-1 to your baby.
- It is not known if FUZEON passes into your breast milk.
- Talk to your healthcare provider about the best way to feed your baby during treatment with FUZEON.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
Especially tell your healthcare provider if you take medicines that affect blood clotting.
Some medicines interact with FUZEON. Keep a list of your medicines to show to your healthcare provider and pharmacist when you get a new medicine.
You can ask your healthcare provider or pharmacist for a list of medicines that interact with FUZEON.
Do not start a new medicine without telling your healthcare provider. Your healthcare provider can tell you if it is safe to use FUZEON with other medicines.
How should I use FUZEON?
- Read the FUZEON "Injection Instructions" for information about how to prepare and inject FUZEON.
- Use FUZEON exactly as your healthcare provider tells you to.
- Do not stop using FUZEON or change your dose without first talking with your healthcare provider.
- Your healthcare provider should show you how to prepare and inject FUZEON before you inject it for the first time. Do not use FUZEON until you have been shown how to inject FUZEON the right way.
- FUZEON is injected under the skin (subcutaneously) of your stomach (abdomen), outer thigh, or upper arm.
- Change (rotate) your injection site with each injection. Do not use the same site for each injection.
- Do not inject FUZEON into areas with scars, moles, bruises, tattoos, burns, blood vessels, or in the area within 2 inches of your belly button (naval).
- FUZEON must be used in combination with other antiretroviral medicines. Do not use FUZEON as your only antiretroviral medicine.
- See your healthcare provider regularly while using FUZEON.
- If you take too much FUZEON, call your healthcare provider right away.
What should I avoid while using FUZEON?
- Do not drive, operate heavy machinery, or do other dangerous activities until you know how FUZEON affects you.
What are the possible side effects of FUZEON?
FUZEON may cause serious side effects, including:
- Injection site reactions. Injection site reactions including pain and discomfort, redness, rash, itching and bruising have happened in people who use FUZEON. Call your healthcare provider right away if you have pain, redness, or swelling around the injection site that does not go away within a few days or gets worse.
- Nerve pain (neuralgia) or numbness, burning, or prickling feeling of your skin (paresthesia) have happened in people who use the Biojector 2000 needle-free device to give their FUZEON dose. These symptoms can last up to 6 months.
- Bleeding after your injection. People who take medicines that affect blood clotting (anticoagulants) or people with hemophilia or other blood clotting problems may have a higher risk.
- Allergic reactions. Stop using FUZEON and call your healthcare provider or go to the nearest hospital emergency room right away if you develop any of these signs and symptoms of an allergic reaction:
- rash
- fever
- chills
- trouble breathing
- hives
- nausea and vomiting
- swelling of your face, eyes, lips or mouth
- low blood pressure
- Pneumonia. Pneumonia which can be serious and cause hospitalization and death has happened in people who use FUZEON. Call your healthcare provider or go to the nearest hospital emergency room right away if you develop any of these signs or symptoms of pneumonia:
- cough with fever
- fast breathing
- shortness of breath
- Changes in your immune system (Immune Reconstitution Syndrome) can happen when an HIV-1 infected person starts taking antiretroviral medicines including FUZEON. Your immune system may get stronger and begin to fight infections that have been hidden in your body for a long time. Tell your healthcare provider right away if you start having any new symptoms after starting FUZEON.
The most common side effects of FUZEON include:
- local injection site reactions
- diarrhea
- nausea
- tiredness
- weight loss
- sinus problems
- stomach pain
- cough
- herpes simplex
- decreased appetite
- pancreas problems
- pain in arms and legs
- pneumonia
- pain and numbness in feet or legs
- flu-like symptoms
- infected hair follicle
- dry mouth
- eye infection
These are not all the possible side effects of FUZEON.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store FUZEON?
- Store FUZEON vials that have not been mixed with sterile water at room temperature between 68°F to 77°F (20°C to 25°C).
- Store FUZEON that has been mixed with sterile water in the original vial and in the refrigerator between 36°F to 48°F (2°C to 8°C) for up to 24 hours. Throw away (discard) any unused FUZEON left in the vial after 24 hours.
Keep FUZEON and all medicines out of the reach of children.
General information about the safe and effective use of FUZEON
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use FUZEON for a condition for which it was not prescribed. Do not give FUZEON to other people, even if they have the same symptoms you have. It may harm them. You can ask your healthcare provider or pharmacist for information about FUZEON that is written for health professionals.
What are the ingredients in FUZEON?
Active ingredient: enfuvirtide
Inactive ingredients: mannitol, sodium carbonate, sodium hydroxide, and hydrochloric acid
Distributed by:
Genentech USA, Inc.
1 DNA Way, South San Francisco, CA 94080-4990© 2019 Genentech, Inc. and Alexion Pharmaceuticals, Inc. All rights reserved. FUZEON is a trademark of Hoffmann-La Roche Inc.
FUZEON has been jointly developed by Alexion Pharmaceuticals, Inc. and Hoffmann-La Roche Inc. FUZEON is manufactured by Hoffmann-La Roche Inc.
More information go to www.FUZEON.com or call 1-877-438-9366.
-
INSTRUCTIONS FOR USE
Instructions for Use
FUZEON™ (few'-zee-on)
(enfuvirtide)
Injection, for subcutaneous use
108 mg vial1 Before You Begin
Important Information:
- Your healthcare provider should show you how to prepare and inject FUZEON before you inject it for the first time. Do not inject yourself or someone else until you have been shown how to inject FUZEON the right way.
- Call your healthcare provider if you have any questions about how to inject FUZEON the right way. You may also call 1-877-438-9366 or visit www.FUZEON.com.
- Do not reuse or share syringes or needles with other people. You may give other people a serious infection or get a serious infection from them.
- The instructions below are for mixing a single dose. If you want to mix 2 doses at the same time you will need 2 vials of FUZEON, 2 vials of sterile water, new sterile alcohol pads and syringes.
How should I store FUZEON?
- Store FUZEON vials that have not been mixed with sterile water at room temperature between 68°F to 77°F (20°C to 25°C).
- Store FUZEON that has been mixed with sterile water in the original vial and in the refrigerator between 36°F to 46°F (2°C to 8°C) for up to 24 hours. Throw away (discard) any unused FUZEON left in the vial after 24 hours.
- Keep FUZEON and all medicines out of the reach of children.
2 Getting Started
Gather Supplies: Supplies you will need to give your FUZEON injection:
- A clean flat surface like a table
- One vial of FUZEON—at room temperature
- One 1 mL vial of sterile water
- One 3 mL (large) syringe with a 1-inch needle
- One 1 mL (small) syringe with a 1/2-inch needle
- Sterile alcohol pads (not supplied in this kit; may be obtained from your pharmacy)
- One sharps container for throwing away used syringes and needles. See "Disposing of your used syringes and needles" at the end of these instructions.
3 Prepare Your FUZEON Dose
- Check the expiration date printed on the FUZEON vial label.
- Check to make sure that none of the items in your kit have been opened.
- Check that the FUZEON vial is not cracked or damaged.
-
Do not use the FUZEON vial, properly throw it away and get a new one if:
- the expiration date printed on the FUZEON vial label has passed
- the items in your FUZEON kit have been opened
- the FUZEON vial is cracked or damaged
Step 1: - Wash your hands well using soap and warm water and dry them with a clean towel.
- Once your hands are clean, do not touch anything other than the medicine, supplies and the area around the injection site.
Step 2: -
Gather your syringes. You will need 2 syringes:
- One 3 mL (large) syringe for mixing FUZEON
- One 1 mL (small) syringe for giving your FUZEON injection
The safety syringes have a green colored piece of plastic that is attached to the needle. This piece of plastic is a safety feature that covers the needle after use.
Your healthcare provider may recommend other types of syringes for use with FUZEON.
Step 3:- Open the syringe packages, remove the syringes, and throw away the syringe packages.
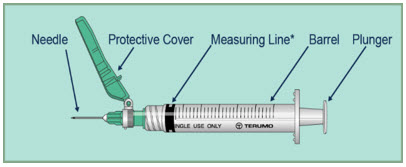
*The measuring line of the syringe is the edge line of the plunger closest to the needle.
Step 4: - Take the caps off the vials and throw away the vial caps.
- Wipe each vial top with a new sterile alcohol pad and let the tops air-dry.
- Do not touch the tops of the vials once they have been cleaned with an alcohol pad.
- If you accidentally touch the rubber tops after cleaning them, clean them again with a new sterile alcohol pad.
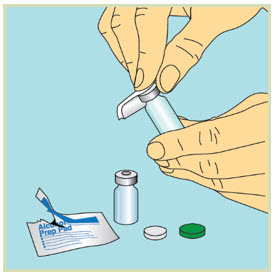
4 Mixing FUZEON
- Do not mix FUZEON with tap water. Use only the sterile water provided to mix FUZEON.
- Do not mix anything or any other medicine in the same syringe as FUZEON.
- Do not touch the needle when holding the syringe. If you touch the needle, you will need to start over with a new syringe. If you run out of syringes, call your pharmacy.
- To save time, you can mix both of your daily doses of FUZEON at the same time, but you will need to keep the second vial of mixed FUZEON in the refrigerator. Write the date and time on the vial when mixed if you are mixing the dose to be used later.
- Before using the dose of refrigerated FUZEON, be sure it is clear and allow it to warm to room temperature. You may want to hold it in your hand to help bring it to room temperature before you inject it. Do not microwave the vial or put it in hot water.
- Do not store mixed FUZEON in the syringe.
Step 5: -
Draw up the sterile water.
- Pick up the 3 mL (large) syringe.
- Hold the clear plastic cap and tighten the needle with a gentle clockwise twist. Do not use too much force as the needle may loosen.
- Using your index finger, pull the green protection cover away from the capped needle.
- Pull the clear plastic cap straight off.
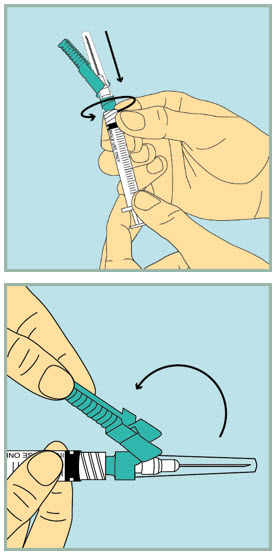
Step 6: - Slowly pull the plunger back until the tip of the plunger reaches the 1 mL line.
Step 7: - Push the needle into the rubber stopper of the sterile water vial.
- Turn the vial upside down.
- Slowly push the plunger all the way in to inject the air into the vial. Do not remove the needle from the vial.
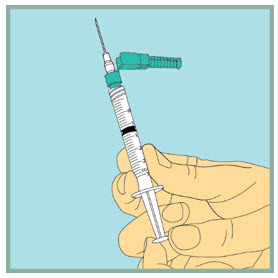
Step 8: - Make sure the tip of the needle is always below the surface of the sterile water to help keep air bubbles from entering the syringe.
- Slowly pull the plunger back until the tip of the plunger reaches the 1 mL line on the syringe.
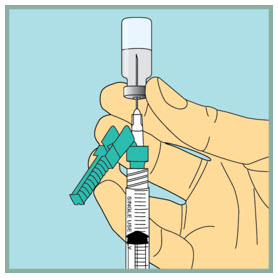
Step 9: - Gently tap or flick the barrel and push and pull the plunger to remove extra air and bubbles. To be sure you end up with 1 mL of sterile water in the syringe, you may need to pull the plunger past the 1 mL line.
- Check to make sure you have 1 mL of sterile water in the syringe. If you have more than 1 mL in the syringe, adjust the amount of sterile water in the syringe by pushing the plunger in until it is 1 mL.
- Carefully remove the needle from the vial.
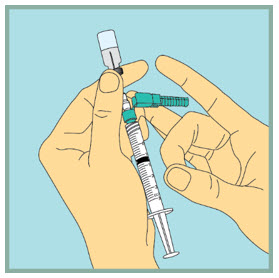
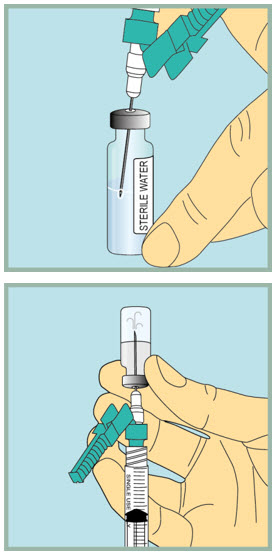
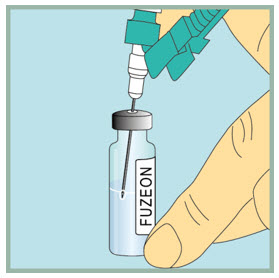
Step 10: -
Inject sterile water into the FUZEON vial.
- Gently tap the FUZEON vial to loosen the powder.
- Insert the needle of the syringe filled with sterile water into the FUZEON vial at an angle.
- Point the needle toward the side of the FUZEON vial and slowly push the plunger all the way down to inject the sterile water.
- When injecting the sterile water into the FUZEON vial, the sterile water should drip down the side of the vial into the FUZEON powder.
- Remove the needle from the vial.
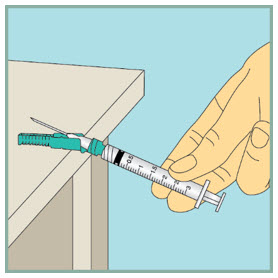
Step 11: - Using 1 hand, gently press the green protective cover against a flat surface until you hear a click and the needle is re-covered. Do not use your hand to re-cover the needle.
- Throw away (dispose of) the used syringe in the sharps container.
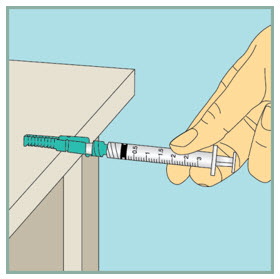
Step 12: -
Gently mix FUZEON.
- Gently tap the FUZEON vial with your fingertip for 10 seconds to start dissolving the powder.
- Next, gently roll the FUZEON vial between your hands to reduce the mixing time.
- Once the powder starts to dissolve, just set it aside and it will completely dissolve. After tapping and gently rolling the FUZEON vial, it could take up to 45 minutes for the FUZEON powder to fully dissolve.
- Do not shake the FUZEON vial. Shaking will make the medicine foam and it will take much longer to dissolve.
- Make sure no FUZEON is stuck to the vial wall.
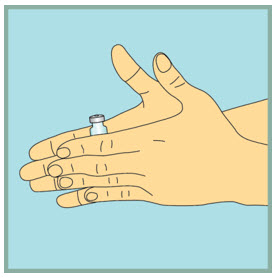
Step 13: -
Inspect the FUZEON liquid.
- Important: Completely dissolved and mixed FUZEON should be clear, colorless and without bubbles or particles. If the FUZEON is foamy or jelled, allow more time for it to dissolve.
- If the FUZEON liquid is cloudy, discolored, or you see any particles in it once it is completely mixed and you have waited 45 minutes, do not use that vial. Properly throw it away and get a new one.
- If you see bubbles, gently tap the vial until they disappear.

5 Choose and prepare your injection site
- Inject FUZEON exactly as your healthcare provider has shown you.
- Inject FUZEON just under the skin (subcutaneous). FUZEON should never be given directly into your veins (intravenous) or directly into your muscle (intramuscular).
- You should change (rotate) your injection site with each injection. Do not use the same injection site 2 times in a row.
- Talk with your healthcare provider if you have any questions about where on your body to inject FUZEON.
Step 14: -
Choose your injection site.
The sites you may choose for your injection are:- your stomach area (abdomen)
- your upper thigh
- the back of your arm. To inject in the back of your arm, you will need someone to help you.
Do not inject:- into areas where the skin is tender, bruised, burned or areas with moles, scars or tattoos
- over a blood vessel
- within 2 inches of the belly-button (naval)
- near the elbow, knee, groin, or the lower or inner buttocks
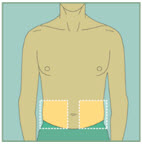 Abdomen
Abdomen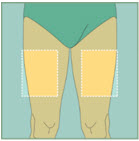 Upper Thighs
Upper Thighs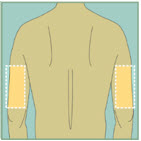 Upper Arms
Upper ArmsStep 15: -
Prepare your skin.
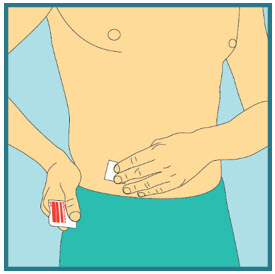
- Clean the injection site with a new sterile alcohol pad. Start in the center, apply pressure and clean in a circular motion, working outward. Allow the site to air-dry. Do not touch the cleaned area again before injecting.
6 Inject FUZEON
Step 16: -
Draw up your FUZEON dose.
- Clean the FUZEON vial top again, using a new sterile alcohol pad. Allow it to air-dry.
- Pick up the 1 mL (small) syringe. Be sure the capped needle is tight by pushing it down slightly while twisting it clockwise.
- Using your index finger, pull the green protective cover away from the capped needle.
- Pull the clear plastic cap straight off.
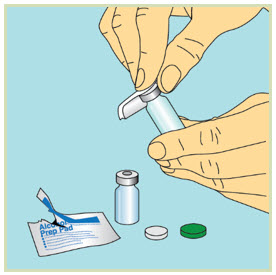
- Slowly pull back the plunger until the tip of the plunger reaches the 1 mL line.
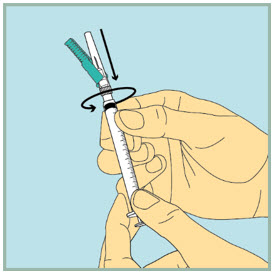
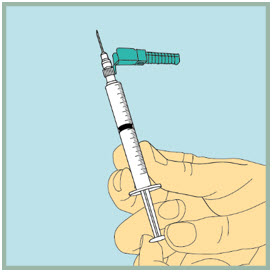
Step 17: - Insert the needle of the syringe into the vial of mixed FUZEON.
- Gently turn the vial upside down.
- Slowly push the plunger all the way down to inject the air into the FUZEON vial. Do not remove the needle from the vial.
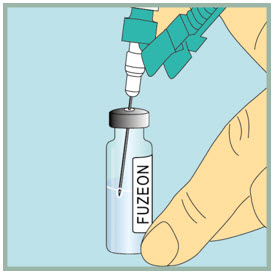
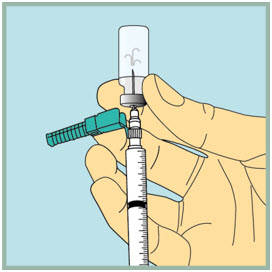
Step 18: - Make sure the tip of the needle is always below the surface of the FUZEON liquid to help keep air bubbles from entering the syringe.
- Slowly pull the plunger down until the tip of the plunger reaches the 1 mL line.
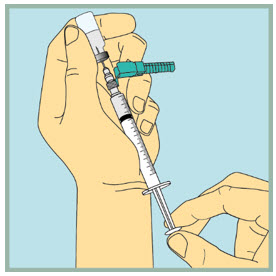
Step 19: - Gently tap or flick the barrel, and push and pull the plunger to remove extra air and bubbles. To be sure you end up with 1 mL of FUZEON in the syringe, you may need to pull the plunger past the 1mL mark.
- Carefully remove the needle and syringe from the vial.
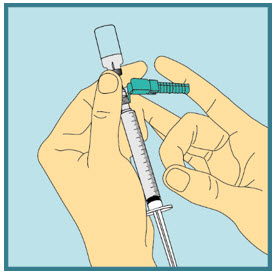
Step 20: -
Inject your FUZEON dose.
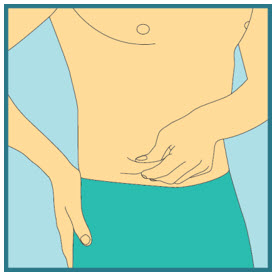
- With 1 hand gently pinch and hold a fold of skin around the cleaned injection site.
- With the other hand, insert the needle into your skin at a 45-degree angle. The needle should be inserted most of the way in. Gently let go of the skin. Make sure the needle stays in place.
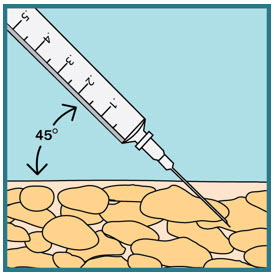
Step 21: - Slowly push the plunger all the way down until all the FUZEON medicine is injected.
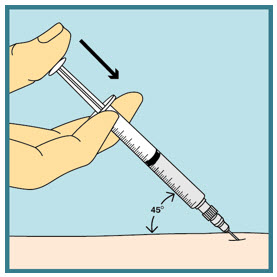
Step 22: - Remove the needle from your skin.
- Press a dry cotton ball or gauze over the injection site for a few seconds. Do not rub the injection site. You may cover the injection site with a small adhesive bandage if needed.
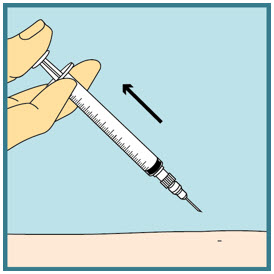
Step 23: -
After your injection.
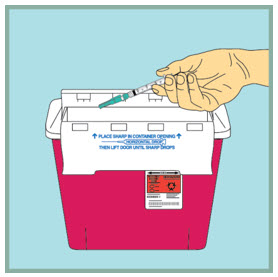
- Using 1 hand, gently press the colored protective cover of needle against a flat surface until you hear a click and the needle is re-covered. Do not use your hand to re-cover the needle.
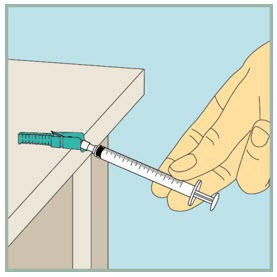
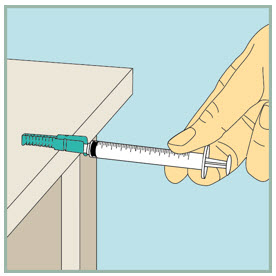
Dispose of (throw away) your used FUZEON needle and syringe:
- Put your used FUZEON needle and syringe in an FDA-cleared sharps disposal container right away after use. Do not throw away (dispose of) your FUZEON needle and syringe in your household trash.
- If you do not have an FDA-cleared sharps disposal container, you may use a household container that is:
- made of a heavy-duty plastic,
- can be closed with a tight-fitting, puncture-resistant lid, without sharps being able to come out,
- upright and stable during use,
- leak-resistant,
- properly labeled to warn of hazardous waste inside the container.
- When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used needles and syringes. For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA's website at: www.fda.gov/safesharpsdisposal
- Do not recycle your used sharps disposal container.
Rx only
FUZEON has been jointly developed by Alexion Pharmaceuticals, Inc. and Hoffmann-La Roche Inc.
FUZEON is manufactured by Hoffmann-La Roche Inc.Distributed by:
Genentech USA, Inc.
A Member of the Roche Group
1 DNA Way
South San Francisco, CA 94080© 2019 Genentech, Inc. and Alexion Pharmaceuticals, Inc. All rights reserved.
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
Revised: 12/2019 - SPL UNCLASSIFIED SECTION
-
PRINCIPAL DISPLAY PANEL - Kit Carton
NDC: 0004-0381-40
Fuzeon®
(enfuvirtide) for Injection90 mg
For Subcutaneous use after reconstitution.
Each vial contains 108 mg enfuvirtide to provide
delivery of 90 mg.
60 Single Use VialsGTIN 00300040381405
Rx only
Package Contains:
Monthly Convenience Kit
60 Vials, FUZEON® 90 mg vial,
NDC: 0004-0380-01
60 Vials, Sterile Water for Injection
1 mL per vial
60 1cc Syringes (for administration)
60 3cc Syringes (for reconstitution)Genentech

-
INGREDIENTS AND APPEARANCE
FUZEON
enfuvirtide kitProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0004-0381 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0004-0381-40 1 in 1 CARTON 04/10/2012 Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 60 VIAL, SINGLE-USE 60 mL Part 2 60 VIAL, SINGLE-DOSE 60 mL Part 1 of 2 FUZEON
enfuvirtide injection, powder, lyophilized, for solutionProduct Information Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength Enfuvirtide (UNII: 19OWO1T3ZE) (Enfuvirtide - UNII:19OWO1T3ZE) Enfuvirtide 90 mg in 1 mL Inactive Ingredients Ingredient Name Strength Mannitol (UNII: 3OWL53L36A) sodium carbonate (UNII: 45P3261C7T) Sodium hydroxide (UNII: 55X04QC32I) Hydrochloric acid (UNII: QTT17582CB) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 1 mL in 1 VIAL, SINGLE-USE ; Type 1: Convenience Kit of Co-Package Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA021481 04/10/2012 Part 2 of 2 STERILE WATER
water injectionProduct Information Route of Administration SUBCUTANEOUS Inactive Ingredients Ingredient Name Strength Water (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 1 mL in 1 VIAL, SINGLE-DOSE; Type 1: Convenience Kit of Co-Package Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA021481 04/10/2012 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA021481 04/10/2012 Labeler - Genentech, Inc. (080129000) Establishment Name Address ID/FEI Business Operations F. Hoffmann-La Roche Ltd 485244961 PACK(0004-0381) , LABEL(0004-0381) , ANALYSIS(0004-0381) Establishment Name Address ID/FEI Business Operations Roche Diagnostics GmbH 315028860 MANUFACTURE(0004-0381) , ANALYSIS(0004-0381) Establishment Name Address ID/FEI Business Operations Sharp Corporation 143696495 PACK(0004-0381) , LABEL(0004-0381)
Trademark Results [FUZEON]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 FUZEON 76119460 2742964 Live/Registered |
Hoffmann-La Roche Inc. 2000-08-30 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.