TEZRULY- terazosin solution
TEZRULY by
Drug Labeling and Warnings
TEZRULY by is a Prescription medication manufactured, distributed, or labeled by ANI Pharmaceuticals, Inc., Novitium Pharma LLC. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use TEZRULY safely and effectively. See full prescribing information for TEZRULY.
TEZRULY (terazosin) oral solution
Initial U.S. Approval: 1993
INDICATIONS AND USAGE
TEZRULYTM (terazosin) is an alpha-1 adrenoceptor antagonist indicated for: (1)
- The treatment of signs and symptoms of benign prostatic hyperplasia (BPH) (1.1)
- The treatment of hypertension alone or with other antihypertensive agents, to lower blood pressure. Lowering blood pressure reduces the risk of fatal and nonfatal cardiovascular events, primarily strokes and myocardial infarction. (1.2)
DOSAGE AND ADMINISTRATION
- For the treatment of BPH: Initiate therapy at 1 mg orally once daily at bedtime. Titrate the dose upwards step-wise from 2 mg to 10 mg once daily. Doses of 10 mg once daily are generally required for a clinical response. Data is insufficient to support doses greater than 20 mg once daily. (2.1)
- For the treatment hypertension: Initiate therapy at 1 mg orally once daily at bedtime. Usual recommended dose range is 1 mg to 5 mg once daily. If response is substantially diminished at 24 hours, increase the dose or use twice daily. Dose may be titrated as needed up to 20 mg once daily. (2.2)
- If terazosin is discontinued for more than a few days, restart using the initial dosing regimen. (2.1)
DOSAGE FORMS AND STRENGTHS
- Oral solution: 1 mg/mL of terazosin (3)
CONTRAINDICATIONS
Hypersensitivity to terazosin hydrochloride or any other ingredient in TEZRULY. (4) (4)
WARNINGS AND PRECAUTIONS
- Syncope and “First-dose” Effect: Advise patients about the possibility of symptoms related to postural hypotension and to avoid situations where injury could result should syncope occur, especially when starting TEZRULY. (5.1)
- Orthostatic Hypotension: Dizziness, lightheadedness, and palpitations can occur with use of TEZRULY. Advise patients to take their first dose of TEZRULY at bedtime and to avoid driving or hazardous tasks for 12 hours after the first dose, after a dosage increase, and after interruption of therapy when treatment is resumed. (5.2)
- Risk of Hypotension with Concomitant Use of Other Antihypertensive Agents and Phospodiesterase Type 5 Inhibitors (PDE5-I): Concomitant administration of TEZRULY with antihypertensives or phosphodiesterase-5 (PDE-5) inhibitors can result in additive blood pressure lowering effects and symptomatic hypotension (5.3).
- Priapism: Advise patients about the possibility and seriousness of priapism. (5.4)
- Intraoperative Floppy Iris Syndrome: Advise patients considering cataract surgery to tell their ophthalmologist that they have taken terazosin as Intraoperative Floppy Iris Syndrome as been observed during cataract surgery in some patients. (5.5)
- Prostatic Cancer: Screen for the presence of prostatic cancer prior to treatment for BPH and at regular intervals afterwards. (5.6)
ADVERSE REACTIONS
- In treatment of BPH, the most common adverse reactions (≥1% of patients and at a higher incidence than placebo) were asthenia, flu syndrome, postural hypotension, nausea, somnolence, vertigo, dyspnea, nasal congestion/rhinitis, blurred vision/amblyopia and erectile dysfunction. (6.1)
- In treatment of hypertension, the most common adverse reactions where the incidence on terazosin was ≥ 5%, where the incidence on terazosin was at least 2% and greater than placebo or where the reaction was of particular interest were asthenia, back pain, pain in the extremities, headache, palpitations, postural hypotension, tachycardia, nausea, edema, peripheral edema, weight gain, depression, dizziness, libido, decreased, nervousness, paresthesia, somnolence, dyspnea, nasal congestion, sinusitis, blurred vision, and erectile dysfunction. (6.1)
(6)
To report SUSPECTED ADVERSE REACTIONS, contact ANI Pharmaceuticals, Inc. at 1-855-204-1431 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch. (6)
DRUG INTERACTIONS
- Co-administration of verapamil with terazosin increases the systemic exposure of terazosin and may lead to hypotension. Dosage reduction and re-titration of either agent may be necessary. (7)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 8/2024
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS & USAGE
1.1 Benign Prostatic Hyperplasia
1.2 Hypertension
2 DOSAGE & ADMINISTRATION
2.1 Recommended Dosage for Benign Prostatic Hyperplasia
2.2 Recommended Dosage for Hypertension
2.3 Administration Information
3 DOSAGE FORMS & STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Syncope and ‘‘First-dose’’ Effect
5.2 Orthostatic Hypotension
5.3 Risk of Hypotension with Concomitant Use of Other Antihypertensive Agents and Phosphodiesterase Type 5 Inhibitors (PDE5-I)
5.4 Priapism
5.5 Prostatic Cancer
5.6 Intraoperative Floppy Iris Syndrome
5.7 Laboratory Tests
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Post-marketing Experience
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis & Mutagenesis & Impairment Of Fertility
13.2 Animal Pharmacology & OR Toxicology
14 CLINICAL STUDIES
14.1 Benign Prostatic Hyperplasia
14.2 Hypertension
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS & USAGE
1.1 Benign Prostatic Hyperplasia
TEZRULY is indicated for the treatment of the signs and symptoms of benign prostatic hyperplasia (BPH) in adult males [see Clinical Studies (14.1)].
1.2 Hypertension
TEZRULY is indicated for the treatment of hypertension, to lower blood pressure in adults [see Clinical Studies (14.2)]. Lowering blood pressure reduces the risk of fatal and non-fatal cardiovascular events, primarily strokes and myocardial infarctions. These benefits have been seen in controlled trials of antihypertensive drugs from a wide variety of pharmacologic classes.
Control of high blood pressure should be part of comprehensive cardiovascular risk management, including, as appropriate, lipid control, diabetes management, antithrombotic therapy, smoking cessation, exercise, and limited sodium intake. Many patients will require more than one drug to achieve blood pressure goals. For specific advice on goals and management, see published guidelines, such as those of the National High Blood Pressure Education Program’s Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC).
Numerous antihypertensive drugs, from a variety of pharmacologic classes and with different mechanisms of action, have been shown in randomized controlled trials to reduce cardiovascular morbidity and mortality, and it can be concluded that it is blood pressure reduction, and not some other pharmacologic property of the drugs, that is largely responsible for those benefits. The largest and most consistent cardiovascular outcome benefit has been a reduction in the risk of stroke, but reductions in myocardial infarction and cardiovascular mortality also have been seen regularly.
Elevated systolic or diastolic pressure causes increased cardiovascular risk, and the absolute risk increase per mmHg is greater at higher blood pressures, so that even modest reductions of severe hypertension can provide substantial benefit. Relative risk reduction from blood pressure reduction is similar across populations with varying absolute risk, so the absolute benefit is greater in patients who are at higher risk independent of their hypertension (for example, patients with diabetes or hyperlipidemia), and such patients would be expected to benefit from more aggressive treatment to a lower blood pressure goal.
Some antihypertensive drugs have smaller blood pressure effects (as monotherapy) in black patients, and many antihypertensive drugs have additional approved indications and effects (e.g., on angina, heart failure, or diabetic kidney disease). These considerations may guide selection of therapy.
TEZRULY may be used alone or in combination with other antihypertensive agents.
-
2 DOSAGE & ADMINISTRATION
2.1 Recommended Dosage for Benign Prostatic Hyperplasia
Initial Dose: 1 mg orally once daily at bedtime. This dose should not be exceeded as an initial dose. Closely follow patients during initial administration in order to minimize the risk of severe hypotensive response, including syncope. For administration instructions, see Dosage and Administration (2.3).
Subsequent Doses: The dose should be increased in a stepwise fashion from 2 mg to 10 mg orally once daily to achieve the desired improvement of symptoms and/or flow rates. Doses of 10 mg once daily are generally required for a clinical response. Therefore, treatment with 10 mg for a minimum of 4 weeks to 6 weeks may be required to assess whether a beneficial response has been achieved. Although some patients responded to 20 mg per daily, data are insufficient to draw definitive conclusions about this dose. There are insufficient data to support the use of doses higher than 20 mg daily. If TEZRULY is discontinued for more than a few days, therapy should be restarted using the initial dosing regimen. For administration instructions, see Dosage and Administration (2.3).
2.2 Recommended Dosage for Hypertension
- Initial Dose: 1 mg orally once daily at bedtime. Do not exceed the initial dosing regimen to minimize the potential for severe hypotensive effects, including syncope. For administration instructions, see Dosage and Administration (2.3).
- Subsequent Doses: Slowly increase the dose to achieve the desired blood pressure response. The usual recommended dose range is 1 mg to 5 mg orally once daily; however, some patients may benefit from doses as high as 20 mg per day. Doses over 20 mg do not appear to provide further blood pressure effect and doses over 40 mg have not been studied. Blood pressure should be monitored at the end of the dosing interval to ensure control throughout the dosing interval. It may also be helpful to measure blood pressure 2 hours to 3 hours after dosing to see if the maximum and minimum responses are similar, and to evaluate symptoms such as dizziness or palpitations which can result from excessive hypotensive response. If response is substantially diminished at 24 hours, consider increasing the dose or use a twice daily dosing regimen. In clinical trials, except for the initial dose, the dose was given in the morning. If TEZRULY is discontinued for more than a few days, therapy should be restarted using the initial dosing regimen. For administration instructions, see Dosage and Administration (2.3).
TEZRULY may be used alone or in combination with other antihypertensive agents. Adjust the dose of TEZRULY and the dose frequency (every 12 hours or 24 hours) based on the patient’s individual blood pressure response.
2.3 Administration Information
Take TEZRULY orally with or without food.
A calibrated measuring device, such as an oral syringe or oral dosing cup, is recommended to measure and deliver the prescribed dose accurately. A household measuring cup, teaspoon, or tablespoon is not an adequate measuring device.
- 3 DOSAGE FORMS & STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Syncope and ‘‘First-dose’’ Effect
TEZRULY, like other alpha-1-adrenoceptors antagonists, can cause marked lowering of blood pressure, especially postural hypotension, and syncope in association with the first dose or first few days of therapy [see Warnings and Precautions (5.2)]. A similar effect can be anticipated if therapy is interrupted for several days and then restarted. Syncope has also been reported with other alpha-1-adrenoceptors antagonists in association with rapid dosage increases or the introduction of another antihypertensive drug. Syncope is believed to be due to an excessive postural hypotensive effect, although occasionally the syncopal episode has been preceded by a bout of severe supraventricular tachycardia with heart rates of 120-160 beats per minute. Additionally, the possibility of the contribution of hemodilution to the symptoms of postural hypotension should be considered.
To decrease the likelihood of syncope or excessive hypotension, initiate treatment with a 1 mg dose of terazosin, given at bedtime. Higher doses (e.g., 2 to 10 mg) are not indicated as initial therapy. Then slowly increase the dose [see Dosage and Administration (2.1 and 2.2)], and add additional antihypertensive agents with caution. Advise patients to avoid situations, such as driving or hazardous tasks, where injury could result should syncope occur during initiation of therapy.
In early investigational studies, where increasing single doses up to 7.5 mg were given at 3-day intervals, tolerance to the first dose phenomenon did not necessarily develop and the ‘‘first-dose” effect could be observed at all doses. Syncopal episodes occurred in 3 of the 14 subjects given terazosin at doses of 2.5, 5 and 7.5 mg, which are higher than the recommended initial dose; in addition, severe orthostatic hypotension (blood pressure falling to 50/0 mmHg) was seen in two others and dizziness, tachycardia, and lightheadedness occurred in most subjects. These adverse effects all occurred within 90 minutes of dosing.
In three placebo-controlled BPH studies 1, 2, and 3 [see Clinical Pharmacology (12)], the incidence of postural hypotension in the terazosin treated patients was 5.1%, 5.2%, and 3.7% respectively.
In multiple dose clinical trials involving nearly 2000 hypertensive patients treated with terazosin, syncope was reported in about 1% of patients. Syncope was not necessarily associated only with the first dose.
If syncope occurs, place the patient in a recumbent position and treat supportively as necessary. There is evidence that the orthostatic effect of terazosin is greater, even in chronic use, shortly after dosing. The risk of the events is greatest during the initial seven days of treatment but continues at all time intervals.
5.2 Orthostatic Hypotension
While syncope is the most severe orthostatic effect of terazosin, other symptoms of lowered blood pressure [see Warnings and Precautions (5.1)], such as dizziness, lightheadedness and palpitations were more common and occurred in some 28% of patients in clinical trials of hypertension. In BPH clinical trials, 21% of the patients experienced one or more of the following: dizziness, hypotension, postural hypotension, syncope, and vertigo. Patients with occupations in which such events represent potential problems should be treated with particular caution. Advise patients to take their first dose of TEZRULY at bedtime and to avoid driving or hazardous tasks for 12 hours after the first dose, after a dosage increase, and after interruption of therapy when treatment is resumed.
5.3 Risk of Hypotension with Concomitant Use of Other Antihypertensive Agents and Phosphodiesterase Type 5 Inhibitors (PDE5-I)
Concomitant use of TEZRULY with other anti-hypertensive agents, especially the calcium channel blocker verapamil, can increase the risk of hypotension. To reduce the risk of hypotension, dosage reduction and re-titration of either agent may be necessary [see Warnings and Precautions (5.1 and 5.2)]. Hypotension has been reported when terazosin has been used with phosphodiesterase-5 (PDE-5) inhibitors. Therefore, PDE-5 inhibitor therapy should be initiated at the lowest dose in patients taking TEZRULY.
5.4 Priapism
Rarely (probably less than once in every several thousand patients) terazosin and other alpha-1-selective adrenoceptor antagonist have been associated with priapism (painful penile erection, sustained for hours and unrelieved by sexual intercourse or masturbation). Because this condition can lead to permanent impotence if not promptly treated, advise patients about the seriousness of the condition and the need to seek immediate medical attention at an emergency room.
5.5 Prostatic Cancer
Carcinoma of the prostate and BPH present with many of the same symptoms and frequently co-exist. Therefore, examine patients thought to have BPH prior to starting TEZRULY therapy to rule out the presence of carcinoma of the prostate.
5.6 Intraoperative Floppy Iris Syndrome
Intraoperative floppy iris syndrome (IFIS) has been observed during cataract surgery in some patients on/or previously treated with alpha-1-adrenoceptor antagonists. This variant of small pupil syndrome is characterized by the combination of a flaccid iris that billows in response to intraoperative irrigation currents, progressive intraoperative miosis despite preoperative dilation with standard mydriatic drugs, and potential prolapse of the iris toward the phacoemulsification incisions. The patient’s ophthalmologist should be prepared for possible implementation of intraoperative preventive measures and modifications to surgical techniques during phacoemulsification surgery to reduce overall complication rates. There does not appear to be a benefit of stopping alpha-1-adrenoceptor antagonists therapy prior to cataract surgery.
5.7 Laboratory Tests
Small but statistically significant decreases in hematocrit, hemoglobin, white blood cells, total protein and albumin were observed in controlled clinical trials. These laboratory findings suggested the possibility of hemodilution. Treatment with terazosin for up to 24 months had no significant effect on prostate specific antigen (PSA) levels.
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Syncope and ‘‘First-dose’’ Effect [see Warnings and Precautions (5.1)]
- Orthostatic Hypotension [see Warnings and Precautions (5.2)]
- Priapism [see Warnings and Precautions (5.3)]
- Intraoperative Floppy Iris Syndrome [see Warnings and Precautions (5.5)]
6.1 Clinical Trials Experience
Because clinical studies are conducted under widely varying conditions, adverse reactions rates observed in the clinical studies of a drug cannot be directly compared to rates in the clinical studies of another drug and may not reflect the rates observed in practice.
Benign Prostatic Hyperplasia
The incidence rates presented below are based on combined data from six placebo-controlled trials involving once-daily administration of terazosin at doses ranging from 1 to 20 mg. Table 1 summarizes those adverse events reported for patients in these trials when the incidence rate in the terazosin group was at least 1% and was greater than that for the placebo group, or where the reaction is of clinical interest.
Asthenia, postural hypotension, dizziness, somnolence, nasal congestion/rhinitis, and impotence were the only events that were significantly (p ≤ 0.05) more common in patients receiving terazosin than in patients receiving placebo. The incidence of urinary tract infection was significantly lower in the patients receiving terazosin than in patients receiving placebo. An analysis of the incidence rate of hypotensive adverse reactions [see Warnings and Precautions (5.1, 5.2)] adjusted for the length of drug treatment has shown that the risk of the reactions is greatest during the initial seven days of treatment but continues at all time intervals.
Table 1. Adverse Reactions Reported During Placebo-Controlled Studies of Terazosin in Patients with Benign Prostatic Hyperplasia
Body System
Terazosin
(N=636)
%
Placebo
(N=360)
%
BODY AS A WHOLE
†Asthenia
7*
3
Flu Syndrome
2
2
Headache
5
6
CARDIOVASCULAR SYSTEM
Hypotension
1
1
Palpitations
1
1
Postural Hypotension
4*
1
Syncope
1
0
DIGESTIVE SYSTEM
Nausea
2
1
METABOLIC AND NUTRITIONAL DISORDERS
Peripheral Edema
1
0
Weight Gain
1
0
NERVOUS SYSTEM
Dizziness
9*
4
Somnolence
4
2
Vertigo
1
0
RESPIRATORY SYSTEM
Dyspnea
2
1
Nasal Congestion/Rhinitis
2*
0
UROGENITAL SYSTEM
Erectile Dysfunction
2*
1
Urinary Tract Infection
1
4*
† Includes weakness, tiredness, lassitude and fatigue.
* p ≤ 0.05, comparison between groups.
Additional adverse reactions have been reported, but these are, in general, not distinguishable from symptoms that might have occurred in the absence of exposure to terazosin. The safety profile of patients treated in the long-term open-label study was similar to that observed in the controlled studies.
Adverse reactions were usually transient and mild or moderate in intensity, but sometimes were serious enough to interrupt treatment. In the placebo-controlled clinical trials, the rates of premature termination due to adverse reactions were not statistically different between the placebo and terazosin groups. Adverse reactions that were bothersome, reported as the reason for discontinuation of therapy by at least 0.5% of the terazosin group and reported more often than in the placebo group are shown in Table 2.
Table 2. Discontinuation Rates During Placebo-Controlled Studies of Terazosin in Patients with Benign Prostatic Hyperplasia
Body System
Terazosin
(N=636)
%
Placebo
(N=360)
%
BODY AS A WHOLE
Fever
0.5%
0.0%
Headache
1.1%
0.8%
CARDIOVASCULAR SYSTEM
Postural Hypotension
0.5%
0.0%
Syncope
0.5%
0.0%
DIGESTIVE SYSTEM
Nausea
0.5%
0.3%
NERVOUS SYSTEM
Dizziness
2.0%
1.1%
Vertigo
0.5%
0.0%
RESPIRATORY SYSTEM
Dyspnea
0.5%
0.3%
SPECIAL SENSES
Blurred Vision/Amblyopia
0.6%
0.0%
UROGENITAL SYSTEM
Urinary Tract Infection
0.6%
0.0%
Hypertension
The prevalence rates presented below are based on combined data from fourteen placebo-controlled studies involving once-daily administration of terazosin, as monotherapy or in combination with other antihypertensive agents, at doses ranging from 1 to 40 mg. Table 3 summarizes those adverse reactions reported for patients in these trials where the prevalence rate in the terazosin group was at least 5%, where the prevalence rate for the terazosin group was at least 2% and was greater than the prevalence rate for the placebo group, or where the reaction is of particular interest. Asthenia, blurred vision, dizziness, nasal congestion, nausea, peripheral edema, palpitations and somnolence were the only symptoms that were significantly (p < 0.05) more common in patients receiving terazosin than in patients receiving placebo. Similar adverse reaction rates were observed in placebo-controlled monotherapy trials.
Table 3. Adverse Reactions Reported During Placebo-Controlled Studies of Terazosin in Patients with Hypertension
Body System
Terazosin
(N=859)
%
Placebo
(N=506)
%
BODY AS A WHOLE
†Asthenia
11*
4
Back Pain
2
1
CARDIOVASCULAR SYSTEM
Palpitations
4*
1
Postural Hypotension
1
0
Tachycardia
2
1
DIGESTIVE SYSTEM
Nausea
4*
1
METABOLIC AND NUTRITIONAL DISORDERS
Peripheral Edema
6*
2
Weight Gain
1
0
MUSCULOSKELETAL SYSTEM
Pain-Extremities
4
3
NERVOUS SYSTEM
Dizziness
19*
8
Libido Decreased
1
0
Paresthesia
3
1
Somnolence
5*
3
RESPIRATORY SYSTEM
Dyspnea
3
2
Nasal Congestion
6*
3
Sinusitis
3
1
SPECIAL SENSES
Blurred Vision
2*
0
UROGENITAL SYSTEM
Erectile Dysfunction
1
1
† Includes weakness, tiredness, lassitude and fatigue.
* Statistically significant at p=0.05 level.
The following adverse reactions associated with the use of terazosin were identified in clinical studies (≥1%) or during post-approval use of terazosin. Because some of these reactions were reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure:
Body as a Whole: edema, facial edema;
Cardiovascular System: chest pain, arrhythmia, vasodilation;
Digestive System: abdominal pain, constipation, diarrhea, dry mouth, dyspepsia, flatulence, vomiting;
Metabolic/Nutritional Disorders: gout;
Musculoskeletal System: arthralgia, arthritis, joint disorder, myalgia, neck pain, shoulder pain;
Nervous System: anxiety, insomnia, nervousness, depression;
Respiratory System: bronchitis, cold symptoms, epistaxis, increased cough, pharyngitis;
Skin and Appendages: pruritus, rash, sweating;
Special Senses: abnormal vision, conjunctivitis, tinnitus;
Urogenital System: urinary frequency, urinary incontinence primarily reported in postmenopausal women;
Adverse reactions were usually mild or moderate in intensity but sometimes were serious enough to interrupt treatment. The adverse reactions that were most bothersome, reported as reason for discontinuation therapy by at least 0.5% of the terazosin group and being reported more often than in the placebo group are shown in Table 4.
Table 4. Discontinuations During Placebo-Controlled Studies of Terazosin in Patients with Hypertension
Body System
Terazosin
(N=859)
%
Placebo
(N=506)
%
BODY AS A WHOLE
Asthenia
1.6%
0.0%
Headache
1.3%
1.0%
CARDIOVASCULAR SYSTEM
Palpitations
1.4%
0.2%
Postural Hypotension
0.5%
0.0%
Syncope
0.5%
0.2%
Tachycardia
0.6%
0.0%
DIGESTIVE SYSTEM
Nausea
0.8%
0.0%
METABOLIC AND NUTRITIONAL DISORDERS
Peripheral Edema
0.6%
0.0%
NERVOUS SYSTEM
Dizziness
3.1%
0.4%
Paresthesia
0.8%
0.2%
Somnolence
0.6%
0.2%
RESPIRATORY SYSTEM
Dyspnea
0.9%
0.6%
Nasal Congestion
0.6%
0.0%
SPECIAL SENSES
Blurred Vision
0.6%
0.0%
6.2 Post-marketing Experience
The following adverse reactions have been identified during postapproval use of terazosin. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Blood and Lymphatic System: thrombocytopenia
Cardiovascular System: atrial fibrillation
Skin and Appndages: allergic reactions, including anaphylaxis
Urogenital System: priapism
During cataract surgery, a variant of small pupil syndrome known as intraoperative floppy iris syndrome (IFIS) has been reported in association with alpha-1 antagonist therapy [see Warnings and Precautions (5.5)].
-
7 DRUG INTERACTIONS
Co-administration of verapamil with terazosin increases the systemic exposure of terazosin [see Clinical Pharmacology (12.3)], which may increase the risk of hypotenstion. To reduce the risk of hypotension, dosage reduction and re-titration of either agent may be necessary [see Warnings and Precautions (5.3)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
The limited available data on terazosin use in pregnant women are insufficient to inform a drug-associated risk of major birth defects, miscarriage, or other adverse maternal or fetal outcomes.
In animal reproduction studies, no adverse developmental effects were observed when terazosin was orally administered to pregnant rats and rabbits during the period of organogenesis at doses of up to 230 and 60 times, respectively, the maximum recommended human dose on a body surface area (mg/m2) basis. In the rat and rabbit oral doses at 230 and 60 times, respectively, the maximum recommended human dose on a body surface area basis, reduced fetal survival was observed [see Data].
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Data
Animal Data
Studies in rats and rabbits at oral doses up to 230 and 60 times, respectively, the maximum recommended human dose on a body surface area (mg/m2) basis, have revealed no evidence of adverse developmental effects. In rats, fetal resorptions were observed at 480 mg/kg/day, approximately 230 times the maximum recommended human dose on a body surface area basis. In rabbits,. increased fetal resorptions, decreased fetal weight and an increased number of supernumerary ribs were observed at 60 mg/kg/day, 60 times the maximum recommended human dose on a body surface area basis. These findings (in both species) were most likely secondary to maternal toxicity.
In a peri- and post-natal development study in rats, significantly more pups died at 120 mg/kg/day (60 times the maximum recommended human dose on a body surface area basis) than in the control group during the three-week postpartum period.
8.2 Lactation
Risk Summary
There are no available data on the presence of terazosin in human milk, effects on the breastfed infant, or the effects on milk production.
8.3 Females and Males of Reproductive Potential
Infertility
Males
Possible effects on male fertility could be observed based on findings in rats at exposures that were at least 15 times higher than at the maximum recommended human dose on a body surface area basis. The clinical relevance of this finding is unknown [see Nonclincal Toxicology (13.1)].
-
10 OVERDOSAGE
There is limited experience regarding overdosage with terazosin. Following a single oral dose of 300 mg (15 times the maximum recommended human dose of 20 mg), signs and symptoms of ovedosage included hypothermia, bradycardia and hypotension.
Management of overdose leading to hypotension should concentrate on support of the cardiovascular system. Restoration of blood pressure and normalization of heart rate may be accomplished by keeping the patient in the supine position. If this measure is inadequate, shock should first be treated with volume expanders. If necessary, vasopressors should then be used and renal function should be monitored and supported as needed. Close medical supervision and monitoring should continue until the patient recovers. Dialysis is unlikely to be effective as terazosin is highly protein bound.
Consult the National Capital Poison Center (1-800-222-1222 or www.poison.org) for up-to-date guidance and advice regarding a TEZRULY overdosage.
-
11 DESCRIPTION
TEZRULY (terazosin), an alpha-1-selective adrenoceptor antagonist, is a quinazoline derivative represented by the following chemical name and structural formula:
1-(4-amino-6,7-dimethoxy-2-quinazolinyl)-4-(tetrahydro-2-furoyl)piperazine monohydrochloride dihydrate
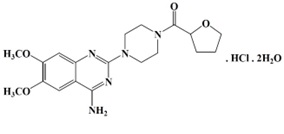
Terazosin hydrochloride is racemic and is a white to pale yellow crystalline substance, sparingly soluble in water. The molecular formula is C19H25N5O4. HCl. 2H2O and the molecular weight is 459.92.
TEZRULY (terazosin) oral solution is supplied in one dosage strength containing 1 mg/mL of terazosin (equivalent to 1.1 mg/mL of terazosin hydrochloride) and the following inactive ingredients: anhydrous citric acid, artificial cherry flavor, glycerin, methylparaben, propylparaben, purified water, sodium citrate dihydrate and sucralose.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Benign Prostatic Hyperplasia
The symptoms associated with benign prostatic hyperplasia (BPH) are related to bladder outlet obstruction, which is comprised of two underlying components: static and dynamic. The static component is related to an increase in prostate size caused, in part by a proliferation of smooth muscle cells in the prostatic stroma. However, the severity of BPH symptoms and the degree of urethral obstruction do not correlate well with the size of the prostate. The dynamic component is a function of an increase in smooth muscle tone in the prostate and bladder neck. The degree of tone in this area is mediated by the alpha-1-adrenoceptors, which is reent in high density in the prostate stroma, prostatic capsule, prostatic urethra, and bladder neck. Blockade of the alpha-1 receptor decreases urethral resistance and may relieve the obstruction of BPH symptoms and improve urine flow
Hypertension
The mechanism of action of terazosin is selective blockade of the alpha-1 subtype of adrenergic receptors. The antihypertensive effect of terazosin results from a decrease in systemic vascular resistance. In animals, terazosin causes a decrease in blood pressure by decreasing total peripheral vascular resistance. The vasodilatory hypotensive action of terazosin appears to be produced mainly by antagonism of alpha-1 adrenoceptors. Terazosin decreases blood pressure within 15 minutes following oral administration.
12.2 Pharmacodynamics
Benign Prostatic Hyperplasia (BPH)
Administration of terazosin 10 mg to patients with BPH resulted in significant improvement in both symptoms and peak urinary flow rate compared to placebo [see Clinical Studies (14.1)].
Terazosin administration to normotensive men with BPH did not result in a clinically significant blood pressure lowering effect [see Clinical Studies (14.1)].
Hypertension
Terazosin’s antihypertensive effects usually persist throughout the dosing interval (usually 24 hours), with the supine systolic and diastolic responses 5 to 10 mmHg and 3.5 to 8 mmHg greater, respectively than placebo.
Limited data show that the peak blood pressure response measured 2 to 3 hours post-dose is greater than about twice the trough (24 hour) response during chronic administration [see Clinical Studies (14.2)].
12.3 Pharmacokinetics
Absorption
Following a single oral dose administration of TEZRULY in healthy subjects under fasted conditions, the median (range) time to reach peak plasma terazosin levels was 0.67 (0.50 - 1.75) hours. Mean (standard deviation) terazosin Cmax and AUC0-∞ were 27.09 (7.85) ng/mL and 285.62 (74.77) h*ng/mL, respectively.
Effect of Food
No clinically significant differences in TERZULY pharmacokinetics were observed following administration of a high-fat, high calorie meal [995.5 Kcalories; 38.31 g protein, 80.75 g carbohydrate, and 57.7 g fat].
Distribution
Terazosin is 90 to 94% bound to plasma proteins and binding is constant over the clinically observed concentration range.
Elimination
Terazosin has a half-life of approximately 13 hours.
Metabolism
Terazosin has been shown to undergo minimal hepatic first-pass metabolism and nearly all of the circulating dose is in the form of parent drug.
Elimination
Approximately 10% of an orally administered dose is excreted as parent drug in the urine and approximately 20% is excreted in the feces. The remainder is eliminated as metabolites. Overall, approximately 40% of the administered dose is excreted in the urine and approximately 60% in the feces.
Specific Populations
Geriatric Patients
In a study that evaluated the effect of age on terazosin pharmacokinetics, the mean plasma half-lives were 14.0 and 11.4 hours for the age group ≥ 70 years and the age group of 20-39 years, respectively. After oral administration, the plasma clearance decreased by 31.7% in patients ≥ 70 years of age compared to patients 20-39 years of age.
Patients with Renal Impairment
Impaired renal function had no significant effect on the elimination of terazosin. Dosage adjustment to compensate for the drug removal during hemodialysis (approximately 10%) does not appear to be necessary.
Drug Interactions
In a study (n=24) where terazosin and verapamil were co-administered, terazosin’s mean AUC0-24 increased 11% after the first verapamil dose. After 3 weeks of verapamil treatment, terazosin’s mean AUC0-24 increased by 24% with associated increases in Cmax (25%) and Cmin (32%) means and decrease in mean Tmax from 1.3 hours to 0.8 hours. Statistically significant differences were not found in the verapamil level with and without terazosin.
In a study (n=6) where terazosin and captopril were administered concomitantly, plasma disposition of captopril was not influenced by concomitant administration of terazosin and terazosin maximum plasma concentrations increased linearly with dose at steady-state after administration of terazosin plus captopril.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis & Mutagenesis & Impairment Of Fertility
Carcinogenesis
Terazosin, administered in the feed to rats at doses of 8, 40, and 250 mg/kg/day (48, 240, and 1500 mg/m2/day), for two years, was associated with a statistically significant increase in benign adrenal medullary tumors of male rats exposed to the 250 mg/kg dose. This dose is 120 times the maximum recommended human dose of 20 mg (12 mg/m2) on a body surface area basis. Female rats were unaffected. Terazosin was not oncogenic in mice when administered in feed for 2 years at a maximum tolerated dose of 32 mg/kg/day (96 mg/m2/day; 8 times the maximum recommended human dose on a body surface area basis). The absence of mutagenicity in a battery of tests, of tumorigenicity of any cell type in the mouse carcinogenicity assay, of increased total tumor incidence in either species, and of proliferative adrenal lesions in female rats, suggests a male rat species-specific event. Numerous other diverse pharmaceutical and chemical compounds have also been associated with benign adrenal medullary tumors in male rats without supporting evidence for carcinogenicity in man.
Mutagenesis
Terazosin was devoid of mutagenic potential when evaluated in vivo and in vitro (the Ames test, in vivo cytogenetics, the dominant lethal test in mice, in vivo Chinese hamster chromosome aberration test and V79 forward mutation assay).
Impairment of Fertility
The effect of terazosin on fertility was assessed in a standard fertility/reproductive performance study in which male and female rats were administered oral doses of 8, 30 and 120 mg/kg/day. Four of 20 male rats given 30 mg/kg (180 mg/m2; 15 times the maximum recommended human dose on a body surface area basis) and five of 19 male rats given 120 mg/kg (720 mg/m2; 60 times the maximum recommended human dose on a body surface area basis) failed to sire a litter. Testicular weights and morphology were unaffected by treatment. However, vaginal smears at 30 and 120 mg/kg/day appeared to contain less sperm than smears from control matings and good correlation was reported between sperm count and subsequent pregnancy.
13.2 Animal Pharmacology & OR Toxicology
Oral administration of terazosin for one or two years elicited a statistically significant increase in the incidence of testicular atrophy in rats exposed to 40 and 250 mg/kg/day (20 and 120 times the maximum recommended human dose on a body surface area basis), but not in rats exposed to 8 mg/kg/day (4 times the maximum recommended human dose on a body surface area basis). Testicular atrophy was also observed in dogs dosed with 300 mg/kg/day (> 490 times the maximum recommended human dose on a body surface area basis) for three months but not after one year when dosed with 20 mg/kg/day (30 times the maximum recommended human dose on a body surface area basis). This finding has also been seen with prazosin, another alpha-1 adrenergic antagonist.
The disposition of terazosin in animals is qualitatively similar to that in man. In animals, terazosin causes a decrease in blood pressure by decreasing total peripheral vascular resistance. The vasodilatory hypotensive action of terazosin appears to be produced mainly by antagonism of alpha-1-adrenoceptors.
-
14 CLINICAL STUDIES
14.1 Benign Prostatic Hyperplasia
Terazosin has been studied in 1222 men with symptomatic BPH. In three placebo-controlled studies, symptom evaluation and uroflowmetric measurements were performed approximately 24 hours following dosing. Symptoms were quantified using the Boyarsky Index which evaluated both obstructive (hesitancy, intermittency, terminal dribbling, impairment of size and force of stream, sensation of incomplete bladder emptying) and irritative (nocturia, daytime frequency, urgency, dysuria) symptoms by rating each of the 9 symptoms from 0-3, for a total score of 27 points. Results from these studies indicated that terazosin statistically significantly improved symptoms and peak urine flow rates over placebo (Table 6).
Table 5. Symptom and Uroflowmetry Scores 24 Hours Following Terazosin Dosing in Three Placebo-Controlled Studies in BPH
Symptom Score
(Range 0-27)
Peak Flow Rate
(mL/sec)
Mean
Mean
Mean
Mean
N
Baseline
Change
(%)
N
Baseline
Change
(%)
Study 1
(10 mg)a
Titration to fixed dose (12 wk)
Placebo
55
9.7
-2.3
(24)
54
10.1
+1.0
(10)
Terazosin
54
10.1
-4.5
(45)*
52
8.8
+3.0
(34)*
Study 2
(2, 5, 10, 20 mg)b
Titration to response (24 wk)
Placebo
89
12.5
-3.8
(30)
88
8.8
+1.4
(16)
Terazosin
85
12.2
-5.3
(43)*
84
8.4
+2.9
(35)*
Study 3
(1, 2, 5, 10 mg)c
Titration to response (24 wk)
Placebo
74
10.4
-1.1
(11)
74
8.8
+1.2
(14)
Terazosin
73
10.9
-4.6
(42)*
73
8.6
+2.6
(30)*
a Highest dose 10 mg shown.
b 23% of patients on 10 mg, 41% of patients on 20 mg.
c 67% of patients on 10 mg.
* Significantly (p ≤ 0.05) more improvement than placebo.
In all three studies, both symptom scores and peak urine flow rates showed statistically significant improvement from baseline in patients treated with terazosin from week 2 (or the first clinic visit) and throughout the study duration.
Analysis of the effect of terazosin on individual urinary symptoms demonstrated that terazosin significantly improved the symptoms of hesitancy, intermittency, impairment in size and force of urinary stream, sensation of incomplete emptying, terminal dribbling, daytime frequency and nocturia, compared to placebo.
Global assessments of overall urinary function and symptoms were also performed by investigators who were blinded to patient treatment assignment. In Studies 1 and 3, patients treated with terazosin had a significantly (p ≤ 0.001) greater overall improvement compared to placebo treated patients.
In a short-term study (Study 1), patients were randomized to either 2, 5 or 10 mg of terazosin or placebo. Patients randomized to the 10 mg group achieved a statistically significant response in both symptoms and peak flow rate compared to placebo (Figure 1).
Figure 1
Study 1
Mean Change in Total Symptom
Score from Baseline+
Mean Increase in Peak Flow
Rate (mL/sec) from Baseline+
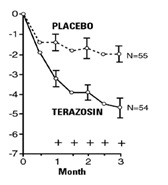
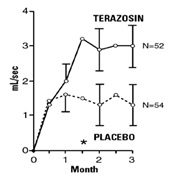
+ for baseline values see above table; * p ≤ 0.05, compared to placebo group
In a long-term, open-label, non-placebo controlled clinical trial, 181 men were followed for 2 years and 58 of these men were followed for 30 months. The effect of terazosin on urinary symptom scores and peak flow rates was maintained throughout the study duration (Figures 2 and 3):
Figure 2
Mean Change in Total Symptom Score from Baseline
Long-Term, Open-Label, Non-Placebo Controlled Study (N=494)
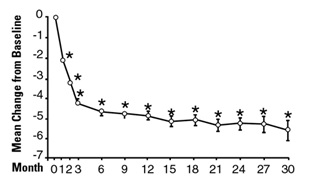
*p ≤ 0.05 vs. baseline; mean baseline = 10.7
Figure 3
Mean Change in Peak Flow Rate from Baseline
Long-Term, Open-Label, Non-Placebo Controlled Study (N=494)
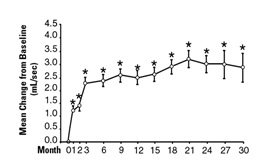
* p ≤ 0.05 vs. baseline; mean baseline = 9.9
In this long-term trial, both symptom scores and peak urinary flow rates showed statistically significant improvement suggesting a relaxation of smooth muscle cells.
Although antagonism of alpha-1-adrenoceptors also lowers blood pressure in hypertensive patients with increased peripheral vascular resistance, terazosin treatment of normotensive men with BPH did not result in a clinically significant blood pressure lowering effect (Table 6).
Table 6. Mean Changes in Blood Pressure from Baseline to Final Visit in all Double-Blind, Placebo-Controlled Studies
Normotensive Patients
DBP ≤ 90 mm Hg
Hypertensive Patients
DBP > 90 mm Hg
Group
N
Mean Change
N
Mean Change
SBP
Placebo
293
-0.1
45
-5.8
(mm Hg)
Terazosin
519
-3.3*
65
-14.4*
DBP
Placebo
293
+0.4
45
-7.1
(mm Hg)
Terazosin
519
-2.2*
65
-15.1*
* p ≤ 0.05 vs. placebo
14.2 Hypertension
Patients in clinical trials of terazosin were administered once daily (the great majority) and twice daily regimens with total doses usually in the range of 5 to 20 mg/day, and had mild (about 77%, diastolic pressure 95 to 105 mmHg) or moderate (23%, diastolic pressure 105 to 115 mmHg) hypertension. Because terazosin, like all alpha-1 adrenergic antagonists can cause unusually large falls in blood pressure after the first dose or first few doses, the initial dose was 1 mg in virtually all trials, with subsequent titration to a specified fixed dose or titration to some specified blood pressure end point (usually a supine diastolic pressure of 90 mmHg).
Blood pressure responses were measured at the end of the dosing interval (usually 24 hours) and effects were shown to persist throughout the interval, with the usual supine responses 5 to 10 mmHg systolic and 3.5 to 8 mmHg diastolic greater than placebo. The responses in the standing position tended to be somewhat larger, by 1 to 3 mmHg, although this was not true in all studies. The magnitude of the blood pressure responses was similar to prazosin and less than hydrochlorothiazide (in a single study of hypertensive patients). In measurements 24 hours after dosing, heart rate was unchanged.
Limited measurements of peak response (2-3 hours after dosing) during chronic terazosin administration indicate that it is greater than about twice the trough (24 hour) response, suggesting some attenuation of response at 24 hours, presumably due to a fall in blood terazosin concentrations at the end of the dose interval. This explanation is not established with certainty, however, and is not consistent with the similarity of blood pressure response to once daily and twice daily dosing and with the absence of an observed dose-response relationship over a range of 5-20 mg, i.e., if blood concentrations had fallen to the point of providing less than full effect at 24 hours, a shorter dosing interval or larger dose should have led to increased response.
Blood pressure should be measured at the end of the dose interval; if response is not satisfactory, patients may be tried on a larger dose or twice daily dosing regimen. The latter should also be considered if blood pressure-related side effects, such as dizziness, palpitations, or orthostatic complaints are seen within a few hours after dosing.
The greater blood pressure effect associated with peak plasma concentrations (first few hours after dosing) appears somewhat more position-dependent (greater in the erect position) than the effect of terazosin at 24 hours and in the erect position there is also a 6-10 beat per minute increase in heart rate in the first few hours after dosing.
During the first 3 hours after dosing, 12.5% of patients had a systolic pressure fall of 30 mmHg or more from supine to standing, or standing systolic pressure below 90 mmHg with a fall of at least 20 mmHg, compared to 4% of a placebo group.
There was a tendency for patients to gain weight during terazosin therapy. In placebo-controlled monotherapy trials, male and female patients receiving terazosin gained a mean of 1.7 and 2.2 pounds respectively, compared to losses of 0.2 and 1.2 pounds respectively in the placebo group. Both differences were statistically significant.
During controlled clinical trials, patients receiving terazosin monotherapy had a small but statistically significant decrease (a 3% fall) compared to placebo in total cholesterol and the combined low-density and very-low-density lipoprotein fractions. No significant changes were observed in high-density lipoprotein fraction and triglycerides compared to placebo.
Analysis of clinical laboratory data following administration of terazosin suggested the possibility of hemodilution based on decreases in hematocrit, hemoglobin, white blood cells, total protein and albumin. Decreases in hematocrit and total protein have been observed with alpha-1-adrenoceptors antagonism and are attributed to hemodilution.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
TEZRULY 1 mg/mL oral solution is a clear, cherry flavored solution, free from visible particulate matter available in bottles of 150 mL with child resistant closure, NDC: 70954-592-10.
Storage and Handling
Store at 20°C to 25°C (68°F to 77°F); excursions permitted to 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature].
Keep the container tightly closed.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Administration Instructions
Inform patients that a calibrated measuring device, such as an oral syringe or oral dosing cup, should be obtained from the pharmacy to measure and deliver the prescribed dose accurately. A household measuring cup, teaspoon, or tablespoon is not an adequate measuring device.
Syncope and ‘‘First-dose’’ Effect and Orthostatic Hypotension
Inform patients about the possibility of syncopal and orthostatic symptoms, especially at the initiation of therapy, and to avoid driving or hazardous tasks for 12 hours after the first dose, after a dosage increase and after more than a few days of interruption of therapy when treatment is resumed. They should be cautioned to avoid situations where injury could result should syncope occur during initiation of terazosin therapy, including driving, operating machinery and performing hazardous tasks. They should also be advised of the need to sit or lie down when symptoms of lowered blood pressure occur, although these symptoms are not always orthostatic, and to be careful when rising from a sitting or lying position. If dizziness, lightheadedness, or palpitations are bothersome, advise patients to report these symptoms to their healthcare provider, so that dose adjustment can be considered. Inform patients that drowsiness or somnolence can occur with terazosin, requiring caution in people who must drive or operate heavy machinery [see Warnings and Precautions (5.1, 5.2)].
Risk of Hypotension when Tezruly is Taken Concomitantly with Other Antihypertensive Agents and/or Phosphodiesterase Type 5 Inhibitors (PDE5-I)
Advise patients that dosage reduction of their other antihypertensive agents and/or PDE5-I may be necessary to avoid the possibility of developing significant hypotension.
Priapism
Advise the patient about the possibility of priapism as a result of treatment with TEZRULY and other similar medications. Patients should be informed that this reaction is extremely rare, but if not brought to immediate medical attention, can lead to permanent erectile dysfunction (impotence) [see Warnings and Precautions (5.3)].
Screening for Prostate Cancer
Advise patients that prostate cancer and BPH frequently present with many of the same symptoms and may co-exist; therefore, inform patients that they should have screening for the presence of prostate cancer prior to treatment with TEZRULY and at regular intervals afterwards [see Warnings and Precautions (5.4)].
Intraoperative Floppy Iris Syndrome
Advise the patient when considering cataract surgery to tell their ophthalmologist that they have taken TEZRULY [see Warnings and Precautions (5.5)].
Trademarks are the property of their respective owners.
Distributed by:
ANI Pharmaceuticals, Inc.
Baudette, MN 56623
Issued: 07/2024
LB4752-01
TEZRULYTM is a pending trademark of ANI Pharmaceuticals, Inc.
-
PATIENT INFORMATION
PATIENT INFORMATION
TEZRULY (tez-ROO-lee)(terazosin)
oral solution
What is TEZRULY?
TEZRULY is a prescription medicine that contains terazosin hydrochloride and is called an “alpha-blocker”. TEZRULY is used to treat:
- the symptoms of benign prostatic hyperplasia (BPH)
- high blood pressure (hypertension)
Do not take TEZRULY if you:
are allergic to terazosin or any of the ingredients in TEZRULY. See the end of this Patient Information leaflet for a complete list of ingredients in TEZRULY.Before taking TEZRULY, tell your healthcare provider about all of your medical conditions, including if you: - have had low blood pressure, especially after taking other medicine. Signs of low blood pressure include fainting, dizziness, and lightheadedness.
- have any planned eye surgery.
- have prostate cancer or a history of prostate cancer. Your healthcare provider may have you checked for prostate cancer before you start taking and while you take TEZRULY.
- are pregnant or plan to become pregnant. It is not known if TEZRULY will harm your unborn baby.
- are breastfeeding or plan to breastfeed. It is not known if TEZRULY passes into your breastmilk. Talk to your healthcare provider about the best way to feed your baby if you take TEZRULY.
Especially tell your healthcare provider if you take:
- other medicine for high blood pressure, particularly the calcium channel blocker verapamil. The use of TEZRULY with verapamil can lead to a drop in blood pressure or to fainting.
- medicines to treat erectile dysfunction (ED) called phosphodiesterase type 5 (PDE-5) inhibitors. The use of TEZRULY with PDE-5 inhibitors can lead to a drop in blood pressure or to fainting.
How should I take TEZRULY? - Take TEZRULY by mouth with or without food.
- The starting dose of TEZRULY is 1 mg (1 mL) at bedtime. Do not take more than 1 mg as your starting dose. This is to reduce the chance of a sudden drop in blood pressure which can cause fainting, dizziness, and lightheadedness. Your healthcare provider will then increase your dose of TEZRULY as needed.
- Your healthcare provider will tell you how much TEZRULY to take and when to take it.
- Your healthcare provider may need to change your dose of TEZRULY until it is the right dose for you.
- Take TEZRULY using a marked (calibrated) measuring device, such as an oral syringe or oral dosing cup. Ask your pharmacist for a measuring device and instructions on how to measure and take the correct dose of medicine. A household measuring cup, teaspoon, or tablespoon is not an adequate measuring device.
What should I avoid while taking TEZRULY?
Do not drive or perform any hazardous task until at least 12 hours after you have taken TEZRULY if you are taking:
- your first dose of TEZRULY
- TEZRULY for the first time after your healthcare provider has increased your dose of TEZRULY
- TEZRULY for the first time after any breaks (interruptions) in your treatment with TEZRULY
What are the possible side effects of TEZRULY?
TEZRULY may cause serious side effects, including:
- A sudden drop in blood pressure, especially when you first start treatment or when there is an increase in your dose of TEZRULY, is common but can also be serious. This may cause you to have a feeling that your heart is pounding or racing (palpitations), faint, have a spinning feeling (vertigo), or to feel dizzy or lightheaded. If you have a spinning feeling, dizziness or lightheadedness, you should sit or lie down and be careful when rising from a sitting or lying position. Your risk of having this problem may be increased if you take TEZRULY with certain other medicines that lower blood pressure including verapamil and PDE-5 inhibitors. Your healthcare provider may monitor your blood pressure while you take TEZRULY. See “What should I avoid while taking TEZRULY?”
- A painful erection that will not go away. TEZRULY can cause a painful erection (priapism), which cannot be relieved by having sex or masturbation. If this happens, get medical help right away. If priapism is not treated, you may not be able to get an erection in the future
- Eye problems during cataract surgery. A condition called Intraoperative Floppy Iris Syndrome (IFIS) can happen during cataract surgery if you take or have taken alpha-blockers such as TEZRULY. If you need to have cataract surgery, be sure to tell your ophthalmologist (eye doctor) if you take or have taken TEZRULY.
The most common side effects of TEZRULY include:
- weakness
- dizziness
- puffy feet or hands
- lack of energy
- nasal congestion
- drowsiness
These are not all the possible side effects of TEZRULY.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
General information about the safe and effective use of TEZRULY.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use TEZRULY for a condition for which it was not prescribed. Do not give TEZRULY to other people, even if they have the same symptoms you have. It may harm them.
You can ask your healthcare provider or pharmacist for information that is written for health professionals.What are the ingredients in TEZRULY?
Active ingredient: terazosin hydrochloride
Inactive ingredients: artificial cherry flavor, citric acid anhydrous, glycerin, methylparaben, propylparaben, purified water, sodium citrate dihydrate and sucralose.Distributed by:
ANI Pharmaceuticals, Inc.
Baudette, MN 56623
TEZRULYTM is a pending trademark of ANI Pharmaceuticals, Inc.
For more information, call 1-855-204-1431.
This Patient Information has been approved by the U.S. Food and Drug Administration.
Issued: 07/2024
LB4752-01
- PACKAGE LABEL.PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
TEZRULY
terazosin solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 70954-592 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength TERAZOSIN HYDROCHLORIDE (UNII: D32S14F082) (TERAZOSIN - UNII:8L5014XET7) TERAZOSIN 1 mg in 1 mL Inactive Ingredients Ingredient Name Strength ALCOHOL (UNII: 3K9958V90M) ANHYDROUS CITRIC ACID (UNII: XF417D3PSL) GLYCERIN (UNII: PDC6A3C0OX) METHYLPARABEN (UNII: A2I8C7HI9T) PROPYLPARABEN (UNII: Z8IX2SC1OH) WATER (UNII: 059QF0KO0R) TRISODIUM CITRATE DIHYDRATE (UNII: B22547B95K) SUCRALOSE (UNII: 96K6UQ3ZD4) Product Characteristics Color Score Shape Size Flavor CHERRY Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 70954-592-10 150 mL in 1 BOTTLE; Type 0: Not a Combination Product 04/18/2025 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA218139 04/18/2025 Labeler - ANI Pharmaceuticals, Inc. (145588013) Establishment Name Address ID/FEI Business Operations Novitium Pharma LLC 080301870 ANALYSIS(70954-592) , LABEL(70954-592) , MANUFACTURE(70954-592) , PACK(70954-592)

Trademark Results [TEZRULY]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 TEZRULY 97808542 not registered Live/Pending |
ANI Pharmaceuticals, Inc. 2023-02-23 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.