LARTRUVO- olaratumab injection, solution
LARTRUVO by
Drug Labeling and Warnings
LARTRUVO by is a Prescription medication manufactured, distributed, or labeled by Eli Lilly and Company. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use LARTRUVO safely and effectively. See full prescribing information for LARTRUVO.
LARTRUVO (olaratumab) injection, for intravenous use
Initial U.S. Approval: 2016INDICATIONS AND USAGE
LARTRUVO™ is a platelet-derived growth factor receptor alpha (PDGFR-α) blocking antibody indicated, in combination with doxorubicin, for the treatment of adult patients with soft tissue sarcoma (STS) with a histologic subtype for which an anthracycline-containing regimen is appropriate and which is not amenable to curative treatment with radiotherapy or surgery. (1)
This indication is approved under accelerated approval. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trial. (14)
DOSAGE AND ADMINISTRATION
- Administer LARTRUVO at 15 mg/kg as an intravenous infusion over 60 minutes on Days 1 and 8 of each 21-day cycle until disease progression or unacceptable toxicity. (2.1)
- For the first 8 cycles, LARTRUVO is administered with doxorubicin. (2.1)
- Premedicate with diphenhydramine and dexamethasone intravenously, prior to LARTRUVO on Day 1 of cycle 1. (2.2)
- For intravenous infusion only. Do not administer as an intravenous push or bolus. (2.4)
DOSAGE FORMS AND STRENGTHS
Injection: 500 mg/50 mL (10 mg/mL) or 190 mg/19 mL (10 mg/mL) solution in a single-dose vial (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
- Infusion-Related Reactions: Monitor for signs and symptoms during and following infusion. Discontinue LARTRUVO for Grade 3 or 4 infusion-related reactions. (2.2, 2.3, 5.1)
- Embryo-Fetal Toxicity: Can cause fetal harm. Advise females of potential risk to the fetus and to use effective contraception during treatment with LARTRUVO and for 3 months after the last dose. (5.2, 8.1, 8.3)
ADVERSE REACTIONS
The most common (≥20%) adverse reactions of LARTRUVO plus doxorubicin are nausea, fatigue, musculoskeletal pain, mucositis, alopecia, vomiting, diarrhea, decreased appetite, abdominal pain, neuropathy, and headache. (6.1)
The most common (≥20%) laboratory abnormalities were lymphopenia, neutropenia, thrombocytopenia, hyperglycemia, elevated aPTT, hypokalemia, and hypophosphatemia. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Eli Lilly and Company at 1-800-LillyRx (1-800-545-5979) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 8/2018
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
2.2 Premedication
2.3 Dosing Modifications
2.4 Preparation and Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Infusion-Related Reactions
5.2 Embryo-Fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Immunogenicity
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
LARTRUVO™ is indicated, in combination with doxorubicin, for the treatment of adult patients with soft tissue sarcoma (STS) with a histologic subtype for which an anthracycline-containing regimen is appropriate and which is not amenable to curative treatment with radiotherapy or surgery.
This indication is approved under accelerated approval [see Clinical Studies (14)]. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trial.
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
The recommended dose of LARTRUVO is 15 mg/kg administered as an intravenous infusion over 60 minutes on Days 1 and 8 of each 21-day cycle until disease progression or unacceptable toxicity. For the first 8 cycles, LARTRUVO is administered with doxorubicin, [see Clinical Studies (14)].
Refer to doxorubicin prescribing information for dosing, and dose modifications.
2.2 Premedication
- Premedicate with diphenhydramine (25 to 50 mg intravenously) and dexamethasone (10 to 20 mg intravenously) prior to LARTRUVO on Day 1 of cycle 1.
2.3 Dosing Modifications
Infusion-Related Reactions
- Permanently discontinue LARTRUVO for Grade 3 or 4 infusion-related reactions [see Dosage and Administration (2.2) and Warnings and Precautions (5.1)].
- Interrupt infusion of LARTRUVO for Grade 1 or 2 infusion-related reactions (IRR). After resolution, resume LARTRUVO infusion at 50% of the initial infusion rate. [see Warnings and Precautions (5.1)]
Neutropenia
- For neutropenic fever/infection or Grade 4 neutropenia lasting longer than 1 week, discontinue administration of LARTRUVO until the absolute neutrophil count is 1,000 /microliter or greater and then permanently reduce the dose to 12 mg/kg.
2.4 Preparation and Administration
Preparation
- Inspect vial contents for particulate matter and discoloration prior to dilution [see Description (11)]. Discard the vial if particulate matter or discolorations are identified.
- Withdraw calculated dose and further dilute with 0.9% Sodium Chloride Injection, USP to a final volume of 250 mL for intravenous infusion. Do not use dextrose-containing or other solutions.
- Gently invert but do not shake.
- DO NOT FREEZE the diluted solution.
- Store the diluted solution for up to 24 hours under refrigeration at 2°C to 8°C (36°F to 46°F) and for up to an additional 8 hours at room temperature (below 25°C [77°F]). Storage times include the duration of infusion. If refrigerated, allow the diluted solution to come to room temperature prior to administration.
- Discard vial with any unused portion of LARTRUVO.
Administration
- Do not administer LARTRUVO as an intravenous push or bolus. Do not co-infuse with electrolytes or other medications through the same intravenous line.
- Visually inspect the diluted solution for particulate matter and discoloration prior to administration. If particulate matter or discolorations are identified, discard the solution.
- Administer diluted solution as an intravenous infusion over 60 minutes. Flush the line with 0.9% Sodium Chloride Injection, USP at end of infusion.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Infusion-Related Reactions
Infusion-related reactions (IRR) occurred in 70 (14%) of 485 patients who received at least one dose of LARTRUVO across clinical trials. For 68 of these 70 patients (97%), the first occurrence of IRR was in the first or second cycle. Grade ≥3 IRR occurred in 11 (2.3%) of 485 patients, with one (0.2%) fatality. Symptoms of IRR included flushing, shortness of breath, bronchospasm, or fever/chills, and in severe cases symptoms manifested as severe hypotension, anaphylactic shock, or cardiac arrest.
Infusion-related reactions required permanent discontinuation in 2.3% of patients and interruption of infusion in 10% of patients. All 59 patients with Grade 1 or 2 IRR resumed LARTRUVO; 12 (20%) of these patients had a Grade 1 or 2 IRR with rechallenge. The incidence of IRR in the overall safety database (N = 485) was similar (18% versus 12%) between those who did (56%) and those who did not (44%) receive premedication.
Monitor patients during and following LARTRUVO infusion for signs and symptoms of IRR in a setting with available resuscitation equipment. Immediately and permanently discontinue LARTRUVO for Grade 3 or 4 IRR [see Dosage and Administration (2.2, 2.3) and Adverse Reactions (6.1)].
5.2 Embryo-Fetal Toxicity
Based on animal data and its mechanism of action, LARTRUVO can cause fetal harm when administered to a pregnant woman. Animal knockout models link disruption of platelet-derived growth factor receptor alpha (PDGFR-α) signaling to adverse effects on embryo-fetal development. Administration of an anti-murine PDGFR-α antibody to pregnant mice during organogenesis caused malformations and skeletal variations. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with LARTRUVO and for 3 months after the last dose [see Use in Specific Populations (8.1, 8.3)].
-
6 ADVERSE REACTIONS
The following adverse drug reactions are described elsewhere in the labeling:
- Infusion-Related Reactions [see Warnings and Precautions (5.1)].
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data in the Warnings and Precautions section reflect exposure to LARTRUVO in 485 patients from three randomized, open-label, active-controlled clinical trials, which enrolled 256 patients with various tumors who received LARTRUVO in combination with chemotherapy (191 patients) or LARTRUVO as a single agent (65 patients); four open-label single-arm trials which enrolled 96 patients with various tumors who received LARTRUVO as a single agent at doses of 10 to 20 mg/kg; and two trials, including Trial 1, which enrolled 133 patients with soft tissue sarcoma who received LARTRUVO at doses of 15 to 20 mg/kg in combination with doxorubicin (103 patients) or LARTRUVO as a single agent (30 patients). Among the 485 patients, 25% were exposed to LARTRUVO for ≥6 months and 6% were exposed for ≥12 months.
The data described below reflect exposure to LARTRUVO in 64 patients with metastatic soft tissue sarcoma enrolled in Trial 1, a multicenter, randomized (1:1), open-label, active-controlled trial comparing LARTRUVO plus doxorubicin with doxorubicin as a single agent. LARTRUVO was administered at 15 mg/kg as an intravenous infusion on Days 1 and 8 of each 21-day cycle until disease progression or unacceptable toxicity [see Clinical Studies (14)]. All patients received doxorubicin 75 mg/m2 as an intravenous infusion on Day 1 of each 21-day cycle for a maximum of eight cycles and received dexrazoxane, prior to doxorubicin in cycles 5 to 8. In Trial 1, no patients had received a prior anthracycline-containing regimen. The trial excluded patients with an ECOG performance status >2; left ventricular ejection fraction <50%; or unstable angina pectoris, angioplasty, cardiac stenting, or myocardial infarction within 6 months.
Baseline demographics and disease characteristics were: median age 58 years (range 22 to 86); 45% male; 87% White, 8% Black, 3% Asian, 2% Other; 57% ECOG PS 0, 39% ECOG PS 1, and 5% ECOG PS 2. The median duration of exposure to LARTRUVO was 6 months (range: 21 days to 29.4 months) with 36 (56%) patients receiving LARTRUVO for ≥6 months and 10 (16%) patients receiving LARTRUVO for ≥12 months. The median cumulative doxorubicin dose was 488 mg/m2 in the LARTRUVO plus doxorubicin arm and 300 mg/m2 in the doxorubicin arm.
In Trial 1, adverse reactions resulting in permanent discontinuation of LARTRUVO occurred in 8% (5/64) of patients. The most common adverse reaction leading to LARTRUVO discontinuation was infusion-related reaction (3%). Dose reductions of LARTRUVO for adverse reactions occurred in 25% (16/64) of patients; the most common adverse reaction leading to dose reduction was Grade 3 or 4 neutropenia (20%). Dose delays of LARTRUVO for adverse reactions occurred in 52% (33/64) of patients; the most common adverse reactions resulting in dose delays were neutropenia (33%), thrombocytopenia (8%), and anemia (5%).
Table 1 summarizes adverse reactions that occurred in at least 10% of patients receiving LARTRUVO in the randomized portion of the study. The most common adverse reactions reported in at least 20% of patients receiving LARTRUVO plus doxorubicin were nausea, fatigue, musculoskeletal pain, mucositis, alopecia, vomiting, diarrhea, decreased appetite, abdominal pain, neuropathy, and headache.
Table 1: Adverse Reactions Occurring in ≥10% (All Grades) of Patients in the LARTRUVO plus Doxorubicin Arm and at a Higher Incidence than in the Doxorubicin Arm (Between Arm Difference of ≥5% for All Grades or ≥2% for Grades 3 and 4) (Trial 1) a Abdominal pain includes: abdominal pain, lower abdominal pain, and upper abdominal pain.
b Fatigue includes: asthenia and fatigue.
c Musculoskeletal pain includes: arthralgia, back pain, bone pain, flank pain, groin pain, musculoskeletal chest pain, musculoskeletal pain, myalgia, muscle spasms, neck pain, and pain in extremity.
Adverse Reactions LARTRUVO plus Doxorubicin
N=64Doxorubicin
N=65All Grades
(%)Grade 3-4
(%)All Grades
(%)Grade 3-4
(%)Gastrointestinal Disorders Nausea 73 2 52 3 Mucositis 53 3 35 5 Vomiting 45 0 19 0 Diarrhea 34 3 23 0 Abdominal Paina 23 3 14 0 General Disorders and Administrative Site Conditions Fatigueb 69 9 69 3 Infusion-Related Reactions 13 3 3 0 Musculoskeletal and Connective Tissue Disorders Musculoskeletal Painc 64 8 25 2 Skin and Subcutaneous Tissue Disorders Alopecia 52 0 40 0 Metabolic and Nutritional Disorders Decreased Appetite 31 2 20 0 Nervous System Disorders Neuropathy 22 0 11 0 Headache 20 0 9 0 Psychiatric Disorder Anxiety 11 0 3 0 Eye Disorder Dry Eyes 11 0 3 0 In Trial 1, the most common laboratory abnormalities (≥20%) were lymphopenia, neutropenia, thrombocytopenia, hyperglycemia, elevated aPTT, hypokalemia, and hypophosphatemia as shown in Table 2.
Table 2: Laboratory Abnormalities Worsening from Baseline in >10% (All Grades) of Patients in the LARTRUVO plus Doxorubicin Arm and Occurring at a Higher Incidence than in the Doxorubicin Arm (Between Arm Difference ≥5% for All Grades or ≥2% for Grades 3 and 4) (Trial 1) a The incidence is based on the number of patients who had both baseline and at least one on-study laboratory measurement: LARTRUVO plus doxorubicin arm (range 60 to 63 patients) and doxorubicin arm (range 39 to 62 patients).
b aPTT = activated partial thromboplastin time
Laboratory Abnormality LARTRUVO plus Doxorubicina Doxorubicina All Grades
(%)Grades 3-4
(%)All Grades
(%)Grades 3-4
(%)Chemistry Hyperglycemia 52 2 28 3 Increased aPTTb 33 5 13 0 Hypokalemia 21 8 15 3 Hypophosphatemia 21 5 7 3 Increased Alkaline Phosphatase 16 0 7 0 Hypomagnesemia 16 0 8 0 Hematology Lymphopenia 77 44 73 37 Neutropenia 65 48 63 38 Thrombocytopenia 63 6 44 11 6.2 Immunogenicity
As with all therapeutic proteins, there is the potential for immunogenicity. In clinical trials, 13/370 (3.5%) of evaluable LARTRUVO-treated patients tested positive for treatment-emergent anti-olaratumab antibodies by an enzyme-linked immunosorbent assay (ELISA). Neutralizing antibodies were detected in all patients who tested positive for treatment-emergent anti-olaratumab antibodies. The effects of anti-olaratumab antibodies on efficacy, safety, and exposure could not be assessed due to the limited number of patients with treatment-emergent anti-olaratumab antibodies.
The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of incidence of antibodies to LARTRUVO with the incidences of antibodies to other products may be misleading.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on animal data and its mechanism of action, LARTRUVO can cause fetal harm [see Clinical Pharmacology (12.1)]. There are no available data on LARTRUVO use in pregnant women. No animal studies using olaratumab have been conducted to evaluate its effect on female reproduction and embryo-fetal development. Animal knockout models link disruption of platelet-derived growth factor receptor alpha (PDGFR-α) signaling to adverse effects on embryo-fetal development. Administration of an anti-murine PDGFR-α antibody to pregnant mice during organogenesis at exposures less than the exposure at the maximum recommended human dose caused malformations and skeletal variations [see Data].
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Data
Animal Data
No animal studies have been conducted using olaratumab to evaluate the effect of blocking PDGFR-α signaling on reproduction and embryo-fetal development. In PDGFR-α knockout mice, disruption of PDGFR-α signaling resulted in embryo-fetal lethality and teratogenicity, including cleft face and spina bifida. Intravenous administration of an anti-murine PDGFR-α antibody once every 3 days to pregnant mice during organogenesis at 50 and 150 mg/kg resulted in increased malformations (abnormal eyelid development) and skeletal variations (additional ossification sites in the frontal/parietal skull). Increased post-implantation loss occurred at a dose of 5 mg/kg. The effects on fetal development in mice administered this antibody occurred at exposures less than the AUC exposure at the maximum recommended human dose of 15 mg/kg LARTRUVO.
8.2 Lactation
Risk Summary
There are no data on the presence of olaratumab in human milk, or its effects on the breastfed infant or on milk production. Because of the potential risk for serious adverse reactions in breastfeeding infants from olaratumab, advise women not to breastfeed during treatment with LARTRUVO and for 3 months following the last dose.
8.3 Females and Males of Reproductive Potential
Contraception
Females
Based on its mechanism of action, LARTRUVO can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)]. Advise females of reproductive potential to use effective contraception during treatment with LARTRUVO and for 3 months after the last dose.
Infertility
Males
Based on animal models, LARTRUVO may impair male fertility [see Nonclinical Toxicology (13.1)].
-
11 DESCRIPTION
Olaratumab is a recombinant human IgG1 monoclonal blocking antibody that binds specifically to human platelet-derived growth factor receptor alpha (PDGFR-α). LARTRUVO has an approximate molecular weight of 154 kDa. LARTRUVO is produced in genetically engineered mammalian NS0 cells.
LARTRUVO is a sterile, preservative-free, clear to slightly opalescent, and colorless to slightly yellow solution. LARTRUVO injection is supplied in single-dose vials for intravenous use following dilution. Each vial contains 500 mg LARTRUVO in 50 mL (10 mg/mL) or 190 mg LARTRUVO in 19 mL (10 mg/mL). Each mL contains 10 mg olaratumab, glycine (7.5 mg), L-histidine (0.3 mg), L-histidine monohydrochloride (1.7 mg), mannitol (13.7 mg), polysorbate 20 (0.2 mg), sodium chloride (2.9 mg), and water for injection, USP, pH 5.2 to 5.8.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Olaratumab is a human IgG1 antibody that binds platelet-derived growth factor receptor alpha (PDGFR-α). PDGFR-α is a receptor tyrosine kinase expressed on cells of mesenchymal origin. Signaling through this receptor plays a role in cell growth, chemotaxis, and mesenchymal stem cell differentiation. The receptor has also been detected on some tumor and stromal cells, including sarcomas, where signaling can contribute to cancer cell proliferation, metastasis, and maintenance of the tumor microenvironment. The interaction between olaratumab and PDGFR-α prevents binding of the receptor by the PDGF-AA and -BB ligands as well as PDGF-AA, -BB, and -CC-induced receptor activation and downstream PDGFR-α pathway signaling. Olaratumab exhibits in vitro and in vivo anti-tumor activity against selected sarcoma cell lines and disrupted the PDGFR-α signaling pathway in in vivo tumor implant models.
12.2 Pharmacodynamics
Olaratumab exposure-response relationships and the time course of the pharmacodynamics response are unknown.
12.3 Pharmacokinetics
Elimination
The mean clearance (CV%) for olaratumab was 0.56 L/day (33%). The estimated elimination half-life was approximately 11 days (range 6 to 24 days).
Specific Populations
Age (22 to 85 years), sex (47% females), race (86% Whites), mild to moderate renal impairment [calculated creatinine clearance (CLcr) 30-89 mL/min as estimated by the Cockcroft-Gault formula (C-G)], and mild [total bilirubin within upper limit of normal (ULN) and AST greater than ULN or total bilirubin greater than 1.0 and up to 1.5 times ULN and any AST] to moderate (total bilirubin greater than 1.5 and up to 3.0 times ULN and any AST) hepatic impairment had no clinically important effect on the pharmacokinetics of olaratumab. The pharmacokinetics of olaratumab in patients with severe renal impairment (CLcr 15-29 mL/min as estimated by C-G) or with severe hepatic impairment (total bilirubin greater than 3.0 times ULN and any AST) are unknown. Body weight (range 37 to 151 kg) correlates with clearance and volume of distribution of olaratumab.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No studies have been performed to assess the potential of olaratumab for carcinogenicity or genotoxicity.
Fertility studies have not been performed with olaratumab; however, in animal knockout models, loss of PDGFR-α pathway signaling resulted in progressive reduction in testicular size, Leydig cell loss, and spermatogenic arrest.
-
14 CLINICAL STUDIES
The efficacy of LARTRUVO was demonstrated in Trial 1, an open-label, randomized, active-controlled study. Eligible patients were required to have soft tissue sarcoma not amenable to curative treatment with surgery or radiotherapy, a histologic type of sarcoma for which an anthracycline-containing regimen was appropriate but had not been administered, ECOG PS of 0-2, and tumor specimen available for assessment of PDGFR-α expression by an investigational use assay. Patients were randomized (1:1) to receive LARTRUVO in combination with doxorubicin or doxorubicin as a single agent. PDGFR-α expression (positive versus negative), number of previous lines of treatment (0 versus 1 or more), histological tumor type (leiomyosarcoma versus synovial sarcoma versus all others), and ECOG PS (0 or 1 versus 2) were used to allocate patients in the randomization. LARTRUVO was administered at 15 mg/kg as an intravenous infusion on Days 1 and 8 of each 21-day cycle until disease progression or unacceptable toxicity. All patients received doxorubicin 75 mg/m2 as an intravenous infusion on Day 1 of each 21-day cycle for a maximum of eight cycles and were permitted to receive dexrazoxane prior to doxorubicin in Cycles 5 to 8. Patients randomized to receive doxorubicin as a single agent were offered LARTRUVO at the time of disease progression. The efficacy outcome measures were overall survival (OS), and progression-free survival (PFS) and objective response rate (ORR) as assessed by investigator and by independent review according to RECIST v1.1.
A total of 133 patients were randomized, 66 patients to the LARTRUVO plus doxorubicin arm and 67 patients to the doxorubicin arm. Baseline demographics and disease characteristics were: median age of 58 years (range 22 to 86); 44% men; 86% White, 8% Black, 3% Asian, and 2% Other; 56% ECOG PS 0 and 39% ECOG PS 1; 65% no prior chemotherapy (excluding adjuvant and neoadjuvant therapy); 38% leiomyosarcoma, 1.5% synovial sarcoma, and 61% other histologies [17% liposarcoma (8% dedifferentiated, 4% myxoid, 3% well-differentiated, 1.5% pleomorphic, 1% liposarcoma not otherwise specified (NOS)), 11% undifferentiated pleomorphic sarcoma, 5% angiosarcoma, 5% undifferentiated sarcoma NOS, 3% extraskeletal myxoid chondrosarcoma, 2% malignant peripheral nerve sheath tumor, 2% myxofibrosarcoma, 2% malignant solitary fibrous tumor, 2% endometrial stromal sarcoma, 1.5% chondrosarcoma, 1.5% epithelioid sarcoma, 1.5% fibrosarcoma, 1.5% low-grade fibromyxoid sarcoma, and 5% other histologies with one patient each]. All patients had metastatic disease and were enrolled at U.S. sites. Among patients randomized to doxorubicin, 30 (45%) patients received LARTRUVO as a single agent at the time of disease progression.
Trial 1 demonstrated a significant improvement in overall survival. The efficacy results are summarized in Table 3 and Figure 1.
Table 3: Efficacy Results in Trial 1 Abbreviations: CI = confidence interval, CR = complete response, PR = partial response
a Unstratified Cox model.
b Based on independent review.
LARTRUVO + Doxorubicin
N=66Doxorubicin
N=67Overall Survival Number of deaths (%) 39 (59%) 52 (78%) Median, months (95% CI) 26.5 (20.9, 31.7) 14.7 (9.2, 17.1) Hazard Ratio (95% CI)a 0.52 (0.34, 0.79) p-value p<0.05 Progression-Free Survivalb Number of events (%) 37 (56%) 34 (51%) Median, months (95% CI) 8.2 (5.5, 9.8) 4.4 (3.1, 7.4) Hazard Ratio (95% CI)a 0.74 (0.46, 1.19) Objective Response Rate (CR + PR)b (95% CI) 18.2% (9.8, 29.6) 7.5% (2.5, 16.6) CR, n (%) 3 (4.5%) 1 (1.5%) PR, n (%) 9 (13.6%) 4 (6%) -
16 HOW SUPPLIED/STORAGE AND HANDLING
LARTRUVO is supplied in single-dose vials as a sterile, preservative-free, clear to slightly opalescent and colorless to slightly yellow solution.
- NDC: 0002-7190-01
190 mg/19 mL (10 mg/mL) single-dose vial, individually packaged in a carton - NDC: 0002-8926-01
500 mg/50 mL (10 mg/mL) single-dose vial, individually packaged in a carton
Store vials in a refrigerator at 2°C to 8°C (36°F to 46°F) until time of use. Keep the vial in the outer carton to protect from light. DO NOT FREEZE OR SHAKE the vial.
- NDC: 0002-7190-01
-
17 PATIENT COUNSELING INFORMATION
Infusion-Related Reactions
Advise patients to report signs and symptoms of infusion reactions [see Dosage and Administration (2.2) and Warnings and Precautions (5.1)].
Embryo-Fetal Toxicity
Advise pregnant women of the potential risk to the fetus. Advise females of reproductive potential of the potential risk to the fetus, to use effective contraception during treatment with LARTRUVO and for 3 months after the last dose, and to inform their healthcare provider of a known or suspected pregnancy [see Use in Specific Populations (8.1, 8.3)].
Lactation
Advise patients not to breastfeed during treatment with LARTRUVO and for 3 months after the last dose [see Use in Specific Populations (8.2)].
-
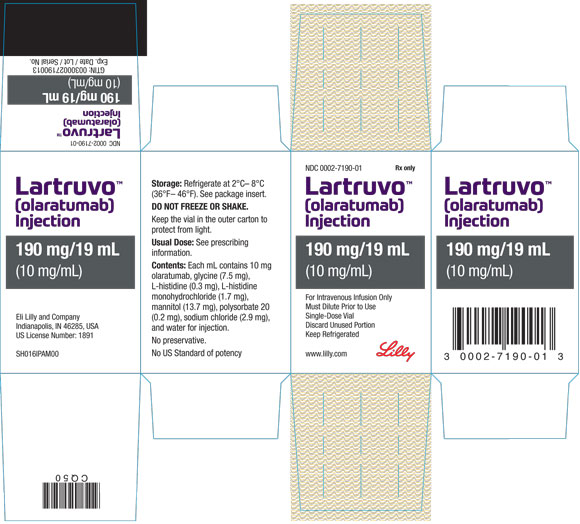
PRINCIPAL DISPLAY PANEL
PACKAGE CARTON – LARTRUVO 500 mg/50 mL single-use vial
NDC: 0002-8926-01
Rx only
LARTRUVOTM
(olaratumab)
Injection
500 mg/50 mL
(10 mg/mL)
For Intravenous Infusion Only
Must Dilute Prior to Use
Single-Dose Vial
Discard Unused Portion
Keep Refrigerated
www.lilly.com
Lilly
-
PRINCIPAL DISPLAY PANEL
PACKAGE CARTON – LARTRUVO 190 mg/19 mL single-use vial
NDC: 0002-7190-01
Rx only
LARTRUVOTM
(olaratumab)
Injection
190 mg/19 mL
(10 mg/mL)
For Intravenous Infusion Only
Must Dilute Prior to Use
Single-Dose Vial
Discard Unused Portion
Keep Refrigerated
www.lilly.com
Lilly
-
INGREDIENTS AND APPEARANCE
LARTRUVO
olaratumab injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0002-8926 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength olaratumab (UNII: TT6HN20MVF) (olaratumab - UNII:TT6HN20MVF) olaratumab 10 mg in 1 mL Inactive Ingredients Ingredient Name Strength histidine (UNII: 4QD397987E) 0.3 mg in 1 mL histidine monohydrochloride (UNII: 1D5Q932XM6) 1.7 mg in 1 mL glycine (UNII: TE7660XO1C) 7.5 mg in 1 mL sodium chloride (UNII: 451W47IQ8X) 2.9 mg in 1 mL mannitol (UNII: 3OWL53L36A) 13.7 mg in 1 mL polysorbate 20 (UNII: 7T1F30V5YH) 0.2 mg in 1 mL water (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0002-8926-01 1 in 1 CARTON 10/19/2016 11/27/2020 1 50 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761038 10/19/2016 LARTRUVO
olaratumab injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0002-7190 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength olaratumab (UNII: TT6HN20MVF) (olaratumab - UNII:TT6HN20MVF) olaratumab 10 mg in 1 mL Inactive Ingredients Ingredient Name Strength histidine (UNII: 4QD397987E) 0.3 mg in 1 mL histidine monohydrochloride (UNII: 1D5Q932XM6) 1.7 mg in 1 mL glycine (UNII: TE7660XO1C) 7.5 mg in 1 mL sodium chloride (UNII: 451W47IQ8X) 2.9 mg in 1 mL mannitol (UNII: 3OWL53L36A) 13.7 mg in 1 mL polysorbate 20 (UNII: 7T1F30V5YH) 0.2 mg in 1 mL water (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0002-7190-01 1 in 1 CARTON 02/14/2017 11/27/2020 1 19 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761038 10/19/2016 Labeler - Eli Lilly and Company (006421325)


Trademark Results [LARTRUVO]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 LARTRUVO 86842906 5536174 Live/Registered |
Eli Lilly and Company 2015-12-08 |
 LARTRUVO 86430008 5453832 Live/Registered |
Eli Lilly and Company 2014-10-21 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.