These highlights do not include all the information needed to use APREMILAST TABLETS safely and effectively. See full prescribing information for APREMILAST TABLETS. APREMILAST tablets, for oral use Initial U.S. Approval: 2014
APREMILAST by
Drug Labeling and Warnings
APREMILAST by is a Prescription medication manufactured, distributed, or labeled by Amneal Pharmaceuticals NY LLC, Amneal Pharmaceuticals Private Limited. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
APREMILAST- apremilast
APREMILAST- apremilast tablet, film coated
Amneal Pharmaceuticals NY LLC
----------
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use APREMILAST TABLETS safely and effectively. See full prescribing information for APREMILAST TABLETS.
APREMILAST tablets, for oral use Initial U.S. Approval: 2014 INDICATIONS AND USAGEApremilast, an inhibitor of phosphodiesterase 4 (PDE4), is indicated for the treatment of:
DOSAGE AND ADMINISTRATIONTo reduce risk of gastrointestinal symptoms, titrate to recommended dosage as follows:
DOSAGE FORMS AND STRENGTHSTablets: 10 mg, 20 mg, 30 mg. (3) CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
ADVERSE REACTIONS
To report SUSPECTED ADVERSE REACTIONS, contact Amneal Pharmaceuticals LLC at 1-877-835-5472 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch. (6) USE IN SPECIFIC POPULATIONSSevere Renal Impairment: Increased systemic exposure of apremilast has been observed. For adults reduce dosage to 30 mg once daily. (2.2, 8.6) Pediatric use information is approved for Amgen Inc.'s Otezla (apremilast) Tablets. However, due to Amgen Inc.'s marketing exclusivity rights, this drug product is not labeled with that information. See 17 for PATIENT COUNSELING INFORMATION. Revised: 2/2025 |
FULL PRESCRIBING INFORMATION
1 INDICATIONS AND USAGE
1.2 Plaque Psoriasis
Apremilast tablets are indicated for the treatment of:
- Adult patients with plaque psoriasis who are candidates for phototherapy or systemic therapy.
Pediatric use information is approved for Amgen Inc.'s Otezla (apremilast) Tablets. However, due to Amgen Inc.'s marketing exclusivity rights, this drug product is not labeled with that information.
2 DOSAGE AND ADMINISTRATION
2.1 Dosage in Plaque Psoriasis
Adult Patients with Plague Psoriasis.
The recommended initial dosage titration of apremilast tablets from Day 1 to Day 5 is shown in Table 1. Following the 5-day titration, the recommended maintenance dosage is 30 mg twice daily taken orally starting on Day 6. This titration is intended to reduce the gastrointestinal symptoms associated with initial therapy.
Table 1: Dosage Titration Schedule for Adult Patients with Plaque Psoriasis
|
Day 1 |
Day 2 |
Day 3 |
Day 4 |
Day 5 |
Day 6 & thereafter |
|||||
|
AM |
AM |
PM |
AM |
PM |
AM |
PM |
AM |
PM |
AM |
PM |
|
10 mg |
10 mg |
10 mg |
10 mg |
20 mg |
20 mg |
20 mg |
20 mg |
30 mg |
30 mg |
30 mg |
Pediatric use information is approved for Amgen Inc.'s Otezla (apremilast) Tablets. However, due to Amgen Inc.'s marketing exclusivity rights, this drug product is not labeled with that information.
2.2 Dosage Adjustment in Patients with Severe Renal Impairment
Adult Patients with Plaque Psoriasis
For initial dosage titration in adult patients with severe renal impairment (creatinine clearance [CLcr] of less than 30 mL per minute estimated by the Cockcroft–Gault equation), titrate apremilast using only the AM schedule listed in Table 1 and skip the PM doses. Reduce apremilast maintenance dosage to 30 mg once daily in this group [see Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)].
Pediatric use information is approved for Amgen Inc.'s Otezla (apremilast) Tablets. However, due to Amgen Inc.'s marketing exclusivity rights, this drug product is not labeled with that information.
3 DOSAGE FORMS AND STRENGTHS
Apremilast tablets are available as oval shaped, film coated tablets in the following dosage strengths:
- Apremilast Tablets, 10 mg are supplied as pink colored, oval shaped, film-coated tablets, debossed with “C12” on one side and plain on other side.
- Apremilast Tablets, 20 mg are supplied as brown colored, oval shaped, film-coated tablet, debossed with "C13” on one side and plain on other side.
- Apremilast Tablets, 30 mg are supplied as beige colored, oval shaped, film-coated tablets, debossed with “C14" on one side and plain on other side.
4 CONTRAINDICATIONS
Apremilast tablets are contraindicated in patients with a known hypersensitivity to apremilast or to any of the excipients in the formulation [see Adverse Reactions (6.1)].
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity
Hypersensitivity reactions, including cases of angioedema and anaphylaxis, have been reported during post marketing surveillance. Avoid the use of apremilast in patients with known hypersensitivity to apremilast or to any of the excipients in the formulation. If signs or symptoms of serious hypersensitivity reactions develop during treatment, discontinue apremilast and institute appropriate therapy.
5.2 Diarrhea, Nausea, and Vomiting
There have been reports of severe diarrhea, nausea, and vomiting associated with the use of apremilast. Most events occurred within the first few weeks of treatment. In some cases, patients were hospitalized. Patients 65 years of age or older and patients taking medications that can lead to volume depletion or hypotension may be at a higher risk of complications from severe diarrhea, nausea, or vomiting. Monitor patients who are more susceptible to complications of diarrhea or vomiting. Patients who reduced dosage or discontinued apremilast generally improved quickly. Consider apremilast dose reduction or suspension if patients develop severe diarrhea, nausea, or vomiting.
5.3 Depression
Treatment with apremilast is associated with an increase incidence of depression. Before using apremilast in patients with a history of depression and/or suicidal thoughts or behavior, carefully weigh the risks and benefits of treatment with apremilast. Advise patients, their caregivers, and families of the need to be alert for the emergence or worsening of depression, suicidal thoughts or other mood changes, and if such changes occur to contact their healthcare provider. Carefully evaluate the risks and benefits of continuing treatment with apremilast if such events occur.
Plaque Psoriasis: During the 16-week placebo-controlled period of the 3 controlled clinical trials in adult subjects with moderate to severe plaque psoriasis, 1.3% (12/920) of subjects treated with apremilast reported depression compared to 0.4% (2/506) treated with placebo. During the clinical trials, 0.1% (1/1,308) of subjects treated with apremilast discontinued treatment due to depression compared with none in placebo-treated subjects (0/506). Depression was reported as serious in 0.1% (1/1,308) of subjects exposed to apremilast, compared to none in placebo-treated subjects (0/506). Instances of suicidal behavior have been observed in 0.1% (1/1,308) of subjects while receiving apremilast, compared to 0.2% (1/506) in placebo-treated subjects. In the clinical trials, one subject treated with apremilast attempted suicide while one who received placebo committed suicide.
During the 16-week placebo-controlled period of the clinical trial in adults with mild to moderate plaque psoriasis, the incidence of subjects reporting depression was similar to what was observed in the adult moderate to severe plaque psoriasis trials.
5.4 Weight Decrease
Weight loss may occur in adult or pediatric patients treated with apremilast.
Regularly monitor the weight of patients treated with apremilast. If unexplained or clinically significant weight loss occurs, evaluate weight loss and consider discontinuation of apremilast [see Adverse Reactions (6.1)].
Weight Loss in Adult Patients
During the placebo-controlled period of the trials in adults with moderate to severe plaque psoriasis, weight decrease between 5% to 10% of body weight occurred in 12% (96/784) of subjects treated with apremilast compared to 5% (19/382) treated with placebo. Weight decrease of ≥ 10% of body weight occurred in 2% (16/784) of subjects treated with apremilast 30 mg twice daily compared to 1% (3/382) subjects treated with placebo.
During the placebo-controlled period of the clinical trial in adults with mild to moderate plaque psoriasis, weight decrease was similar to what was observed in the trials of adults with moderate to severe plaque psoriasis.
Pediatric use information is approved for Amgen Inc.'s Otezla (apremilast) Tablets. However, due to Amgen Inc.'s marketing exclusivity rights, this drug product is not labeled with that information.
5.5 Drug Interactions
Co-administration of strong cytochrome P450 enzyme inducer, rifampin, resulted in a reduction of systemic exposure of apremilast, which may result in a loss of efficacy of apremilast. Therefore, the use of cytochrome P450 enzyme inducers (e.g., rifampin, phenobarbital, carbamazepine, phenytoin) with apremilast is not recommended [see Drug Interactions (7.1) and Clinical Pharmacology (12.3)].
6 ADVERSE REACTIONS
The following adverse reactions are described elsewhere in the labeling:
- Hypersensitivity [see Warnings and Precautions (5.1)]
- Diarrhea, Nausea, and Vomiting [see Warnings and Precautions (5.2)]
- Depression [see Warnings and Precautions (5.3)]
- Weight Decrease [see Warnings and Precautions (5.4)]
- Drug Interactions [see Warnings and Precautions (5.5)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
Moderate to Severe Plague Psoriasis Clinical Trials
Adverse Reactions from Clinical Trials in Adults
The safety of apremilast was assessed in 1,426 subjects in 3 randomized, double-blind, placebo-controlled trials in adult subjects with moderate to severe plaque psoriasis who were candidates for phototherapy or systemic therapy. Subjects were randomized to receive apremilast 30 mg twice daily or placebo twice daily. Titration was used over the first 5 days [see Dosage and Administration (2.1)]. Subjects ranged in age from 18 to 83 years, with an overall median age of 46 years.
Diarrhea, nausea, and upper respiratory tract infection were the most commonly reported adverse reactions (see Table 4). The most common adverse reactions leading to discontinuation for subjects taking apremilast were nausea (1.6%), diarrhea (1.0%), and headache (0.8%). The proportion of subjects with plaque psoriasis who discontinued treatment due to any adverse reaction was 6.1% for subjects treated with apremilast 30 mg twice daily and 4.1% for placebo-treated subjects.
Table 4: Adverse Reactions Reported in ≥ 1% of Adult Subjects with Moderate to Severe Plaque Psoriasis on Apremilast and With Greater Frequency Than in Subjects on Placebo up to Day 112 (Week 16)
|
Adverse Reactions |
Placebo (N=506) n (%) |
Apremilast 30 mg BID b (N=920) n (%) |
|
Diarrhea |
32 (6) |
160 (17) |
|
Nausea |
35 (7) |
155 (17) |
|
Upper respiratory tract infection |
31 (6) |
84 (9) |
|
Tension headache |
21 (4) |
75 (8) |
|
Headache |
19 (4) |
55 (6) |
|
Abdominal pain a |
11 (2) |
39 (4) |
|
Vomiting |
8 (2) |
35 (4) |
|
Fatigue |
9 (2) |
29 (3) |
|
Dyspepsia |
6 (1) |
29 (3) |
|
Decreased appetite |
5 (1) |
26 (3) |
|
Insomnia |
4 (1) |
21 (2) |
|
Back pain |
4 (1) |
20 (2) |
|
Migraine |
5 (1) |
19 (2) |
|
Frequent bowel movements |
1 (0) |
17 (2) |
|
Depression |
2 (0) |
12 (1) |
|
Bronchitis |
2 (0) |
12 (1) |
|
Tooth abscess |
0 (0) |
10 (1) |
|
Folliculitis |
0 (0) |
9 (1) |
|
Sinus headache |
0 (0) |
9 (1) |
|
a Two subjects treated with apremilast experienced serious adverse reaction of abdominal pain. b BID = twice daily. |
||
Severe worsening of psoriasis (rebound) occurred in 0.3% (4/1,184) subjects following discontinuation of treatment with apremilast.
Apremilast was evaluated in a Phase 3, multicenter, randomized, placebo-controlled trial (PSOR-3) in adults with moderate to severe plaque psoriasis of the scalp [see Clinical Studies (14.2)]. A total of 302 subjects were randomized to receive apremilast 30 mg twice daily or placebo twice daily. The most commonly reported adverse reactions that occurred at a higher rate in the apremilast group than in the placebo group were: diarrhea (31% vs. 11%), nausea (22% vs. 6%), headache (12% vs. 5%), and vomiting (6% vs. 2%). The proportion of subjects who discontinued treatment because of any adverse reaction during the 16-week placebo-controlled period of the trial was 6% for subjects who received apremilast 30 mg twice daily and 3% for subjects who received placebo. Gastrointestinal adverse reactions that led to discontinuation of treatment were diarrhea (3% vs. 0%), nausea (1.5% vs. 1%), and vomiting (1.5% vs. 0 %) in the apremilast group compared to placebo.
Mild to Moderate Plaque Psoriasis Clinical Trial in Adults
Apremilast was evaluated in a Phase 3, multicenter, randomized, placebo-controlled trial (PSOR-4) in adult subjects with mild to moderate plaque psoriasis [see Clinical Studies (14.4)]. A total of 595 subjects were randomized to receive apremilast 30 mg twice daily (297 subjects) or placebo twice daily (298 subjects) during the placebo- controlled phase of the trial. The trial also included an open label extension phase during which all subjects received apremilast 30 mg twice daily. Overall, the safety profile observed in the apremilast group during the placebo- controlled phase was consistent with the safety profile previously established in adult subjects with moderate to severe plaque psoriasis.
Other adverse reactions reported in subjects on apremilast in plaque psoriasis
- Gastrointestinal Disorders: Gastroesophageal reflux disease
- Immune System Disorders: Hypersensitivity
- Investigations: Weight decrease
- Metabolism and Nutrition Disorders: Decreased appetite*
- Nervous System Disorders: Migraine
- Respiratory, Thoracic, and Mediastinal Disorders: Cough
- Skin and Subcutaneous Tissue Disorders: Rash
*1 subject treated with apremilast 30 mg twice daily experienced a serious adverse reaction.
Pediatric use information is approved for Amgen Inc.'s Otezla (apremilast) Tablets. However, due to Amgen Inc.'s marketing exclusivity rights, this drug product is not labeled with that information.
7 DRUG INTERACTIONS
7.1 Strong CYP450 Inducers
Apremilast exposure is decreased when apremilast is co-administered with strong CYP450 inducers (such as rifampin) and may result in loss of efficacy [see Warnings and Precautions (5.5) and Clinical Pharmacology (12.3)].
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to apremilast during pregnancy. Information about the registry can be obtained by calling 1-877-311-8972.
Risk Summary
Available pharmacovigilance data with apremilast use in pregnant women have not established a drug-associated risk of major birth defects, miscarriage or adverse maternal or fetal outcomes, but these data are extremely limited. Based on findings from animal reproduction studies, apremilast may increase the risk for fetal loss. In animal embryo-fetal development studies, the administration of apremilast to pregnant cynomolgus monkeys during organogenesis resulted in dose-related increases in abortion/embryo-fetal death at dose exposures 2.1-times the maximum recommended human therapeutic dose (MRHD) and no adverse effect at an exposure of 1.4-times the MRHD. When administered to pregnant mice, during organogenesis there were no apremilast-induced malformations up to exposures 4.0-times the MRHD (see Data). Advise pregnant women of the potential risk of fetal loss. Consider pregnancy planning and prevention for females of reproductive potential.
The background risk of major birth defects and miscarriage for the indicated populations is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
In an embryo-fetal developmental study, pregnant cynomolgus monkeys were administered apremilast at doses of 20, 50, 200, or 1,000 mg/kg/day during the period of organogenesis (gestation Days 20 through 50). There was a dose-related increase in spontaneous abortions, with most abortions occurring during Weeks 3 to 4 of dosing in the first trimester, at doses approximately 2.1-times the MRHD and greater (on an area under the curve [AUC] basis at doses ≥ 50 mg/kg/day). No abortifacient effects were observed at a dose approximately 1.4-times the MRHD (on an AUC basis at a dose of 20 mg/kg/day). Although, there was no evidence for a teratogenic effect at doses of 20 mg/kg/day and greater when examined at day 100, aborted fetuses were not examined.
In an embryo-fetal development study in mice, apremilast was administered at doses of 250, 500, or 750 mg/kg/day to dams during organogenesis (gestation Day 6 through 15). In a combined fertility and embryo-fetal development study in mice, apremilast was administered at doses of 10, 20, 40, or 80 mg/kg/day starting 15 days before cohabitation and continuing through gestation Day 15. No teratogenic findings attributed to apremilast were observed in either study; however, there was an increase in post-implantation loss at doses corresponding to a systemic exposure of 2.3-times the MRHD and greater (≥ 20 mg/kg/day). At doses of ≥ 20 mg/kg/day skeletal variations included incomplete ossification sites of tarsals, skull, sternebra, and vertebrae. No effects were observed at a dose approximately 1.3-times the MRHD (10 mg/kg/day).
Apremilast distributed across the placenta into the fetal compartment in mice and monkeys.
In a pre and postnatal study in mice, apremilast was administered to pregnant female mice at doses of 10, 80, or 300 mg/kg/day from Day 6 of gestation through Day 20 of lactation, with weaning on Day 21. Dystocia, reduced viability, and reduced birth weights occurred at doses corresponding to ≥ 4.0-times the MRHD (on an AUC basis at doses ≥ 80 mg/kg/day). No adverse effects occurred at a dose 1.3-times the MRHD (10 mg/kg/day). There was no evidence for functional impairment of physical development, behavior, learning ability, immune competence, or fertility in the offspring at doses up to 7.5-times the MRHD (on an AUC basis at a dose of 300 mg/kg/day).
8.2 Lactation
Risk Summary
There are no data on the presence of apremilast in human milk, the effects on the breastfed infant, or the effects on milk production. However, apremilast was detected in the milk of lactating mice. When a drug is present in animal milk, it is likely that the drug will be present in human milk. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for apremilast and any potential adverse effects on the breastfed infant from apremilast or from the underlying maternal condition.
Data
In mice, following a single oral administration of 10 mg/kg to dams on postpartum day 13, apremilast concentrations in milk were approximately 1.5-times that of simultaneously collected blood samples.
8.4 Pediatric Use
Plaque Psoriasis
The safety and effectiveness of apremilast have not been established in pediatric patients below the age of 6 years or weighing less than 20 kg with moderate to severe plaque psoriasis.
Pediatric use information is approved for Amgen Inc.'s Otezla (apremilast) Tablets. However, due to Amgen Inc.'s marketing exclusivity rights, this drug product is not labeled with that information.
8.5 Geriatric Use
Of the 1,257 subjects who enrolled in two placebo-controlled plaque psoriasis trials (PSOR-1 and PSOR-2), a total of 108 plaque psoriasis patients were 65 years of age and older, including 9 patients who were 75 years of age and older. No overall differences were observed in the safety or effectiveness in geriatric patients ≥ 65 years of age and younger adult patients < 65 years of age in the clinical trials.
Because patients 65 years of age or older may be at a higher risk of complications such as volume depletion or hypotension from severe diarrhea, nausea, or vomiting, monitor geriatric patients closely for such complications [see Warning and Precautions (5.2)].
8.6 Renal Impairment
Apremilast pharmacokinetics were characterized in adult subjects with mild, moderate, and severe renal impairment as defined by a creatinine clearance of 60 to 89, 30 to 59, and less than 30 mL per minute, respectively, by the Cockcroft–Gault equation. No dosage adjustment is needed in patients with mild or moderate renal impairment. In adult patients with severe renal impairment, reduce the maintenance dosage of apremilast to 30 mg once daily. [see Dosage and Administration (2.2) and Clinical Pharmacology (12.3)].
Pediatric use information is approved for Amgen Inc.'s Otezla (apremilast) Tablets. However, due to Amgen Inc.'s marketing exclusivity rights, this drug product is not labeled with that information.
11 DESCRIPTION
The active ingredient in apremilast tablets is apremilast. Apremilast is a phosphodiesterase 4 (PDE4) inhibitor. Apremilast is known chemically as N-[2-[(1S)-1-(3-ethoxy-4-methoxyphenyl)-2-(methylsulfonyl)ethyl]-2,3-dihydro-1,3-dioxo-1H-isoindol-4-yl]acetamide. Its molecular formula is C22H24N2O7S and the molecular weight is 460.50 g/mol.
The chemical structure is:
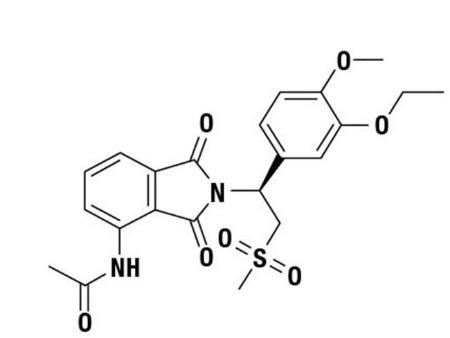
Apremilast is a white to pale yellow powder. It is soluble in acetone and practically insoluble in water.
Apremilast tablets are supplied in 10 mg, 20 mg, and 30 mg strengths for oral administration. Each tablet contains apremilast as the active ingredient and the following inactive ingredients: croscarmellose sodium, ferrosoferric oxide (30 mg), iron oxide red, iron oxide yellow (20 mg and 30 mg), lactose monohydrate, magnesium stearate, polyethylene glycol, polyvinyl alcohol, talc and titanium dioxide.
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Apremilast is an oral small molecule inhibitor of phosphodiesterase 4 (PDE4) specific for cyclic adenosine monophosphate (cAMP). PDE4 inhibition results in increased intracellular cAMP levels. The specific mechanism(s) by which apremilast exerts its therapeutic action is not well defined.
12.3 Pharmacokinetics
Absorption
Apremilast when taken orally is absorbed with an absolute bioavailability of ~73%, with peak plasma concentrations (Cmax) occurring at a median time (tmax) of ~2.5 hours. Co-administration with food does not alter the extent of absorption of apremilast.
Distribution
Human plasma protein binding of apremilast is approximately 68%. Mean apparent volume of distribution (Vd) is 87 L.
Metabolism
Following oral administration in humans, apremilast is a major circulating component (45%) followed by inactive metabolite M12 (39%), a glucuronide conjugate of O-demethylated apremilast. It is extensively metabolized in humans with up to 23 metabolites identified in plasma, urine and feces. Apremilast is metabolized by both cytochrome (CYP) oxidative metabolism with subsequent glucuronidation and non-CYP mediated hydrolysis. In vitro, CYP metabolism of apremilast is primarily mediated by CYP3A4, with minor contributions from CYP1A2 and CYP2A6.
Elimination
The plasma clearance of apremilast is about 10 L/hr in healthy subjects, with a terminal elimination half-life of approximately 6 to 9 hours. Following oral administration of radiolabeled apremilast, about 58% and 39% of the radioactivity is recovered in urine and feces, respectively, with about 3% and 7% of the radioactive dose recovered as apremilast in urine and feces, respectively.
Specific Populations
Patients with Hepatic Impairment: The pharmacokinetics of apremilast is not affected by moderate or severe hepatic impairment.
Patients with Renal Impairment: The pharmacokinetics of apremilast is not affected by mild or moderate renal impairment. In 8 adult subjects with severe renal impairment administered a single dose of 30 mg apremilast, the AUC and Cmax of apremilast increased by approximately 88% and 42%, respectively [see Dosage and Administration (2.2) and Use in Specific Populations (8.6)].
Geriatric Patients: A single oral dose of 30-mg apremilast was studied in young adults and elderly healthy subjects. The apremilast exposure in elderly subjects (65 to 85 years of age) was about 13% higher in AUC and about 6% higher in Cmax than in young subjects (18 to 55 years of age) [see Use in Specific Populations (8.5)].
Male and Female Patients: In pharmacokinetic trials in healthy volunteers, the extent of exposure in females was about 31% higher and Cmax was about 8% higher than that in male subjects.
Racial or Ethnic Groups: The pharmacokinetics of apremilast in Chinese and Japanese healthy male subjects is comparable to that in White healthy male subjects. In addition, apremilast exposure is similar among White subjects (including Hispanic or Latino and not Hispanic or Latino subjects) and Black or African American subjects.
Drug Interactions
In vitro data: Apremilast is not an inhibitor of CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, or CYP3A4 and not an inducer of CYP1A2, CYP2B6, CYP2C9, CYP2C19, or CYP3A4. Apremilast is a substrate, but not an inhibitor of P-glycoprotein (P-gp) and is not a substrate or an inhibitor of organic anion transporter (OAT)1 and OAT3, organic cation transporter (OCT)2, organic anion transporting polypeptide (OATP)1B1 and OATP1B3, or breast cancer resistance protein (BCRP).
Drug interaction trials were performed with apremilast and CYP3A4 substrates (oral contraceptive containing ethinyl estradiol and norgestimate), CYP3A and P-gp inhibitor (ketoconazole), CYP450 inducer (rifampin) and frequently co-administered drug in this patient population (methotrexate).
No significant pharmacokinetic interactions were observed when 30-mg oral apremilast was administered with either oral contraceptive, ketoconazole, or methotrexate. Co-administration of the CYP450 inducer rifampin (600 mg once daily for 15 days) with a single oral dose of 30-mg apremilast resulted in reduction of apremilast AUC and Cmax by 72% and 43%, respectively [see Warnings and Precautions(5.5) and Drug Interactions (7.1)].
Pediatric use information is approved for Amgen Inc.'s Otezla (apremilast) Tablets. However, due to Amgen Inc.'s marketing exclusivity rights, this drug product is not labeled with that information.
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term studies were conducted in mice and rats with apremilast to evaluate its carcinogenic potential. No evidence of apremilast-induced tumors was observed in mice at oral doses up to 8.8-times the Maximum Recommended Human Dose (MRHD) on an AUC basis (1,000 mg/kg/day) or in rats at oral doses up to approximately 0.08- and 1.1-times the MRHD, (20 mg/kg/day in males and 3 mg/kg/day in females, respectively).
Apremilast tested negative in the Ames assay, in vitro chromosome aberration assay of human peripheral blood lymphocytes, and the in vivo mouse micronucleus assay.
In a fertility study of male mice, apremilast at oral doses up to approximately 3-times the MRHD based on AUC (up to 50 mg/kg/day) produced no effects on male fertility. In a fertility study of female mice, apremilast was administered at oral doses of 10, 20, 40, or 80 mg/kg/day. At doses ≥ 1.8-times the MRHD (≥ 20 mg/kg/day), estrous cycles were prolonged, due to lengthening of diestrus which resulted in a longer interval until mating. Mice that became pregnant at doses of 20 mg/kg/day and greater also had increased incidences of early post-implantation losses. There was no effect of apremilast approximately 1.0-times the MRHD (10 mg/kg/day).
14 CLINICAL STUDIES
14.2 Adult Moderate to Severe Plaque Psoriasis
Two multicenter, randomized, double-blind, placebo-controlled trials (PSOR-1 [NCT01194219] and PSOR-2 [NCT01232283]) enrolled a total of 1,257 subjects 18 years of age and older with moderate to severe plaque psoriasis [body surface area (BSA) involvement of ≥ 10%, static Physician Global Assessment (sPGA) of ≥ 3 (moderate or severe disease), Psoriasis Area and Severity Index (PASI) score ≥ 12, candidates for phototherapy or systemic therapy]. Subjects were allowed to use low potency topical corticosteroids on the face, axilla and groin. Subjects with plaque psoriasis of the scalp were allowed to use coal tar shampoo and/or salicylic acid scalp preparations on scalp lesions.
Trial PSOR-1 enrolled 844 subjects and trial PSOR-2 enrolled 413 subjects. In both trials, subjects were randomized 2:1 to apremilast 30 mg twice daily (BID) or placebo for 16 weeks. Both trials assessed the proportion of subjects who achieved PASI-75 at Week 16 and the proportion of subjects who achieved an sPGA score of clear (0) or almost clear (1) at Week 16. Across both trials, subjects ranged in age from 18 to 83 years, with an overall median age of 46 years. The mean baseline BSA involvement was 25.2% (median 21.0%), the mean baseline PASI score was 19.1 (median 16.8), and the proportion of subjects with an sPGA score of 3 (moderate) and 4 (severe) at baseline were 70.0% and 29.8%, respectively. Approximately 30% of all subjects had received prior phototherapy and 54% had received prior conventional systemic and/or biologic therapy for the treatment of psoriasis with 37% receiving prior conventional systemic therapy and 30% receiving prior biologic therapy. Approximately one-third of subjects had not received prior phototherapy, conventional systemic nor biologic therapy.
Clinical Response in Adult Subjects with Moderate to Severe Plaque Psoriasis
The proportion of subjects who achieved PASI-75 responses, and an sPGA score of clear (0) or almost clear (1), are presented in Table 8.
Table 8: Clinical Response at Week 16 in Adult with Moderate to Severe Plaque Psoriasis in Trials PSOR-1 and PSOR-2
|
Trial PSOR-1 |
Trial PSOR-2 |
|||
|
Placebo |
Apremilast 30 mg BID |
Placebo |
Apremilast 30 mg BIDd |
|
|
Na |
N=282 |
N=562 |
N=137 |
N=274 |
|
PASIb -75, n (%) |
15 (5.3) |
186 (33.1) |
8 (5.8) |
79 (28.8) |
|
sPGAc of Clear or Almost Clear, n (%) |
11 (3.9) |
122 (21.7) |
6 (4.4) |
56 (20.4) |
|
a N is number of randomized and treated subjects. b PASI=Psoriasis Area and Severity Index. c sPGA=Static Physician Global Assessment. d BID = twice daily. |
||||
The median time to loss of PASI-75 response among the subjects re-randomized to placebo at Week 32 during the Randomized Treatment Withdrawal Phase was 5.1 weeks.
Plaque Psoriasis Involving the Scalp Area
A randomized, double-blind, placebo-controlled trial (PSOR-3 [NCT03123471]) was conducted in 303 adult subjects with moderate to severe plaque psoriasis of the scalp. Enrolled subjects had a Scalp Physician Global Assessment (ScPGA) score of ≥ 3, Scalp Surface Area (SSA) involvement of ≥ 20%, an inadequate response or intolerance to at least one topical therapy for plaque psoriasis of the scalp, and moderate to severe plaque psoriasis (BSA involvement of ≥ 10%, sPGA of ≥ 3 [moderate or severe disease], and PASI score ≥ 12).
Subjects were randomized 2:1 to receive either apremilast 30 mg twice daily (n =201) or placebo twice daily (n = 102) for 16 weeks. The primary endpoint was the proportion of subjects who achieved an ScPGA response at Week 16 (defined as ScPGA score of clear [0] or almost clear [1] with at least a 2-point reduction from baseline at Week 16). Secondary endpoints included the proportion of subjects with Whole Body Itch Numeric Rating Scale (NRS) response (defined as ≥ 4-point reduction from baseline) and the proportion of subjects with a Scalp Itch NRS response (defined as ≥ 4-point reduction from baseline).
Subjects had a mean age of 46.9 years, 61.7% were men and 75.6 % were white. At baseline, 76.9% of subjects had moderate plaque psoriasis of the scalp (ScPGA of 3), 23.1% had severe plaque psoriasis of the scalp (ScPGA of 4), 71.6% of subjects were biologic naïve, and 58.8% had failed 1 or 2 topical treatments. At baseline, the mean Whole- Body Itch NRS score was 7.2 and the mean Scalp Itch NRS score was 6.7 with the scales ranging from 0 to 10. The mean baseline SSA involvement was 60.6% and the mean baseline BSA involvement was 19.8%.
The proportion of subjects who achieved an ScPGA response, Whole Body Itch NRS response, and Scalp Itch NRS response at Week 16 are presented in Table 9.
Figure 1 displays the proportion of subjects achieving Whole Body Itch NRS response at each visit, while Figure 2 displays the proportion of subjects achieving Scalp Itch NRS response at each visit.
Table 9: Efficacy Results at Week 16 in Adults with Plaque Psoriasis of the Scalp in Trial PSOR-3
|
Trial PSOR-3 |
|||
|
Placebo |
Apremilast 30 mg twice daily |
Treatment Differencea,b (95% CIc) |
|
|
Number of subjects randomized |
N=102 |
N=201 | |
|
ScPGA responsed |
13.7% |
43.3% |
29.6% (19.5%, 39.7%) |
|
Number of subjects with baseline Whole Body Itch NRS Score ≥4 |
N=94 |
N=185 | |
|
Whole Body Itch NRS response |
22.5% |
45.5% |
23.0% (11.5%, 34.6%) |
|
Number of subjects with baseline Scalp Itch NRS Score ≥4 |
N=90 |
N=175 | |
|
Scalp Itch NRS response |
21.1% |
47.1% |
26.2% (13.9%, 38.5%) |
|
a Apremilast – Placebo. b Adjusted difference in proportions is the weighted average of the treatment differences across baseline ScPGA scores with the Cochran-Mantel-Haenszel weights. c CI = confidence interval. d ScPGA score of clear [0] or almost clear [1] with at least a 2-point reduction from baseline. |
|||
Figure 1: Proportion (± SE) of Subjects Achieving Whole Body Itch NRS Response through Week 16

Figure 2: Proportion (± SE) of Subjects Achieving Scalp Itch NRS Response through Week 16

Pediatric use information is approved for Amgen Inc.'s Otezla (apremilast) Tablets. However, due to Amgen Inc.'s marketing exclusivity rights, this drug product is not labeled with that information.
14.4 Adult Mild to Moderate Plaque Psoriasis
A multicenter, randomized, double-blind, placebo-controlled trial (PSOR-4 [NCT03721172]) was conducted in 595 adult subjects with mild to moderate plaque psoriasis (BSA involvement of 2-15%, sPGA score of 2-3 [mild or moderate disease], and PASI score of 2-15). Enrolled subjects had an inadequate response or were intolerant to at least one topical therapy and had not received prior biologic therapy. Subjects were allowed to use unmedicated emollients for lesions on non-scalp areas of the body and non-medicated shampoos for lesions on the scalp.
Subjects were randomized 1:1 to receive either apremilast 30 mg twice daily (n = 297) or placebo twice daily (n = 298) for 16 weeks. At Week 16, the placebo group was switched to receive apremilast and the apremilast group remained on drug through Week 32. The primary endpoint was the proportion of subjects who achieved an sPGA response (defined as an sPGA score of clear [0] or almost clear [1] with at least a 2-point reduction from baseline) at Week 16. Subjects with mild disease (sPGA = 2 at baseline) were required to be clear (sPGA = 0) to achieve an sPGA response. Other evaluated endpoints include the proportion of subjects with a Whole-Body Itch NRS response (defined as a ≥ 4-point reduction from baseline) at Week 16 among subjects with a baseline Whole Body Itch NRS ≥ 4 and the proportion of subjects with an ScPGA response (defined as an ScPGA score of clear [0] or almost clear [1] with at least a 2-point reduction from baseline) at Week 16 among subjects with a baseline ScPGA score ≥ 2.
Subjects ranged in age from 18 to 85 years, with an overall median age of 50 years. The mean baseline BSA involvement was 6.4%, the mean baseline PASI score was 6.5, and the proportions of subjects with an sPGA score of 2 (mild) and 3 (moderate) at baseline were 30.6% and 69.4%, respectively.
Clinical Response in Subjects with Mild to Moderate Plaque Psoriasis
The proportions of subjects who achieved an sPGA response, Whole Body Itch NRS response, and an ScPGA response at Week 16 are presented in Table 12.
Table 12: Efficacy Results at Week 16 in Adults with Mild to Moderate Plaque Psoriasis in Trial PSOR-4
|
Trial PSOR-4 |
|||
|
Placebo |
Apremilast 30 mg twice daily |
Treatment Differencea,b (95% CIc) |
|
|
Number of Subjects Randomized |
N = 298 |
N = 297 | |
|
sPGA Responsed |
4.1% |
21.6% |
17.5% (12.2%, 22.8%) |
|
Number of Subjects with Baseline Whole Body Itch NRS Score ≥ 4 |
N = 249 |
N = 253 | |
|
Whole Body Itch NRS Responsee |
18.6% |
43.2% |
24.7% (16.5%, 32.8%) |
|
Number of Subjects with Baseline ScPGA Score ≥ 2 |
N = 199 |
N = 212 | |
|
a Apremilast – Placebo. b Adjusted difference in proportions is the weighted average of the treatment differences across baseline sPGA scores with the Cochran-Mantel-Haenszel weights. c CI = confidence interval. d sPGA score of clear [0] or almost clear [1] with at least a 2-point reduction from baseline. e Whole Body Itch NRS score reduction of ≥ 4-points from baseline. f ScPGA score of clear [0] or almost clear [1] with at least a 2-point reduction from baseline. |
|||
16 HOW SUPPLIED/STORAGE AND HANDLING
Apremilast Tablets, 10 mg are supplied as pink colored, oval shaped, film-coated tablets, debossed with “C12” on one side and plain on other side.
Apremilast Tablets, 20 mg are supplied as brown colored, oval shaped, film-coated tablet, debossed with "C13” on one side and plain on other side.
Apremilast Tablets, 30 mg are supplied as beige colored, oval shaped, film-coated tablets, debossed with “C14" on one side and plain on other side.
Tablets are supplied with the following strengths and package configurations:
|
Package Configuration |
Tablet Strength |
NDC number |
|
Bottles of 60 |
30 mg |
NDC: 60219-1410-6 |
|
Two-week starter pack |
13-tablet blister titration pack containing: (4) 10-mg, (4) 20-mg, and (5) 30-mg tablets with an additional (14) 30-mg tablets | NDC: 60219-1744-7 |
Storage and Handling
Store at 20° to 25°C (68° to 77°F) [see USP Controlled Room Temperature].
17 PATIENT COUNSELING INFORMATION
-
Administration Instructions
Instruct patients to take apremilast only as prescribed [see Dosage and Administration (2.1 and 2.2)]. Advise patients to take apremilast with or without food. Instruct patients to swallow tablets whole and not to crush, split, or chew prior to swallowing [see Dosage and Administration (2.3)].
-
Hypersensitivity
Inform patients that hypersensitivity reactions can occur following administration of apremilast. Instruct patients to contact their healthcare provider if they experience symptoms of an allergic reaction [see Warnings and Precautions (5.1)].
-
Diarrhea, Nausea, and Vomiting
Advise patients of the potential complications of severe diarrhea, nausea, or vomiting and instruct them to contact their healthcare provider if they experience these adverse reactions, especially if the patient is 65 years of age or older [see Warnings and Precautions (5.2)].
-
Depression
Inform patients that treatment with apremilast is associated with an increased incidence of depression. Advise patients, their caregivers, and families of the need to be alert for the emergence or worsening of depression, suicidal thoughts or other mood changes, and if such changes occur to contact their healthcare provider [see Warnings and Precautions (5.3)].
-
Weight Decrease
Inform patients that treatment with apremilast is associated with potential weight loss. Instruct patients or caregivers to have their weight monitored regularly and, if unexplained or clinically significant weight loss occurs, to contact their healthcare provider for evaluation of the weight loss [see Warnings and Precautions (5.4)].
- Pregnancy
Advise pregnant patients and patients of reproductive potential of the potential risk to a fetus. Advise patients to inform their prescriber of a known or suspected pregnancy [see Use in Specific Populations (8.1)].
Pediatric use information is approved for Amgen Inc.'s Otezla (apremilast) Tablets. However, due to Amgen Inc.'s marketing exclusivity rights, this drug product is not labeled with that information.
Manufactured by:
Amneal Pharmaceuticals Pvt. Ltd.
Oral Solid Dosage Unit
Ahmedabad 382213, INDIA
Distributed by:
Amneal Pharmaceuticals LLC
Bridgewater, NJ 08807
Rev. 02-2025-01
PRINCIPAL DISPLAY PANEL
NDC: 60219-1744-7Apremilast Tablets, Two week starter pack
Rx Only
Blister of 27 Tablets
Amneal Pharmaceuticals LLC

NDC: 60219-1410-6
Apremilast Tablets, 30 mg
Rx Only
60 Tablets
Amneal Pharmaceuticals LLC

| APREMILAST
apremilast kit |
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
| APREMILAST
apremilast tablet, film coated |
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
| Labeler - Amneal Pharmaceuticals NY LLC (123797875) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Amneal Pharmaceuticals Private Limited | 650762060 | analysis(60219-1744, 60219-1410) , label(60219-1744, 60219-1410) , manufacture(60219-1744, 60219-1410) , pack(60219-1744, 60219-1410) | |