QFITLIA- fitusiran injection, solution
Qfitlia by
Drug Labeling and Warnings
Qfitlia by is a Prescription medication manufactured, distributed, or labeled by Genzyme Corporation, Vetter Pharma Fertigung GmbH & Co. KG (Langenargen Eisenbahnstrasse), Vetter Pharma Fertigung GmbH & Co. KG (Ravensburg Helmut-Vetter-Strasse), PPD Development Ireland Ltd., Sanofi-Aventis Deutschland GmbH, BioSpring GmbH, Vetter Pharma Fertigung GmbH & Co. KG (Ravensburg Schuetzenstrasse), Vetter Pharma Fertigung GmbH & Co. KG (Ravensburg Mooswiesen). Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use QFITLIA® safely and effectively. See full prescribing information for QFITLIA.
QFITLIA (fitusiran) injection, for subcutaneous use
Initial U.S. Approval: 2025WARNING: THROMBOTIC EVENTS and ACUTE AND RECURRENT GALLBLADDER DISEASE
See full prescribing information for complete boxed warning.
Thrombotic Events
- Serious thrombotic events have occurred in QFITLIA-treated patients with risk factors including persistent antithrombin (AT) activity less than 15%, QFITLIA 80 mg once monthly dosing, an indwelling venous catheter, and the post-operative setting when bleed management guidelines were not followed.
- Interrupt QFITLIA in patients with a thrombotic event and manage as clinically indicated. (5.1)
Acute and Recurrent Gallbladder Disease
- Gallbladder disease has occurred in QFITLIA-treated patients, some requiring cholecystectomy or developing complications (e.g., pancreatitis).
- Monitor for signs and symptoms of gallbladder disease. Consider interruption or discontinuation of QFITLIA if gallbladder disease occurs.
- Consider alternative treatment for hemophilia if history of symptomatic gallbladder disease. (5.2)
INDICATIONS AND USAGE
QFITLIA is an antithrombin-directed small interfering ribonucleic acid indicated for routine prophylaxis to prevent or reduce the frequency of bleeding episodes in adult and pediatric patients aged 12 years and older with hemophilia A or B with or without factor VIII or IX inhibitors. (1)
DOSAGE AND ADMINISTRATION
- For subcutaneous use only.
- Starting dose: 50 mg once every 2 months (2.1).
- Monitor AT activity using an FDA-cleared test.
- Maintain AT activity between 15–35% by adjusting the dose and/or frequency of administration (2.2).
See Full Prescribing Information for important preparation and administration instructions (2.4).
DOSAGE FORMS AND STRENGTHS
Injection:
- 50 mg/0.5 mL (100 mg/mL) in a single-dose prefilled pen
- 20 mg/0.2 mL (100 mg/mL) in a single-dose vial (3)
CONTRAINDICATIONS
- None (4)
WARNINGS AND PRECAUTIONS
- Hepatotoxicity: Obtain liver tests at baseline and then monthly for at least 6 months after initiating QFITLIA and after dose increases, and periodically thereafter. Liver test elevations may require QFITLIA interruption or discontinuation (5.3).
ADVERSE REACTIONS
Common adverse reactions (incidence >10%) are viral infection, nasopharyngitis, and bacterial infection (6.1).
To report SUSPECTED ADVERSE REACTIONS, contact Genzyme Corporation 1-800-745-4447 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
USE IN SPECIFIC POPULATIONS
Avoid use in patients with established hepatic impairment (Child-Pugh Class A, B and C) (8.6).
To report SUSPECTED ADVERSE REACTIONS, contact Genzyme Corporation 1-800-745-4447 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 9/2025
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: THROMBOTIC EVENTS AND ACUTE AND RECURRENT GALLBLADDER DISEASE
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
2.2 Dosage Modification
2.3 Bleed Management
2.4 Preparation and Administration Instructions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Thrombotic Events
5.2 Acute and Recurrent Gallbladder Disease
5.3 Hepatotoxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 Hypercoagulability with Concomitant Use of CFC or BPA
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.6 Immunogenicity
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage and Handling
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: THROMBOTIC EVENTS AND ACUTE AND RECURRENT GALLBLADDER DISEASE
Thrombotic Events
Serious thrombotic events have occurred in QFITLIA-treated patients with risk factors for thromboembolism including persistent antithrombin (AT) activity less than 15%, use of QFITLIA 80 mg once monthly, presence of indwelling venous catheters, and in the post-operative setting when bleed management guidelines were not followed.
Monitor AT activity using an FDA-cleared test and target AT activity 15–35% to reduce the risk of thrombosis. Monitor patients for signs and symptoms of thrombotic events. Interrupt QFITLIA in patients with a thrombotic event and manage as clinically indicated. (5.1)
Acute and Recurrent Gallbladder Disease
Acute and recurrent gallbladder disease, including cholelithiasis and cholecystitis have occurred in QFITLIA-treated patients, some of whom required cholecystectomy or had complications (e.g., pancreatitis) related to gallbladder disease. Monitor patients for signs and symptoms of acute and recurrent gallbladder disease.
Consider interruption or discontinuation of QFITLIA if gallbladder disease occurs. Consider alternative treatment for hemophilia in patients with a history of symptomatic gallbladder disease. (5.2)
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
For subcutaneous use only.
Use of QFITLIA is recommended under the supervision of a healthcare professional experienced in the treatment of hemophilia or bleeding disorders.
Measure AT activity prior to initiation of QFITLIA. Do not initiate QFITLIA dosing if AT activity is <60%.
Monitor AT activity using an FDA-cleared test. Information on FDA-cleared tests for AT activity is available at http://www.fda.gov/CompanionDiagnostics.
After QFITLIA is initiated, patients may continue their prior clotting factor concentrates (CFC) or bypassing agent (BPA) prophylaxis for the first 7 days of treatment. Discontinue CFC or BPA prophylaxis no later than 7 days after the initial dose of QFITLIA.
The starting dose of QFITLIA is 50 mg once subcutaneously every two months. Adjust the dose and/or dosing interval, if needed, to maintain AT activity between 15–35% [see Boxed Warning, Dosage and Administration (2.2) and Warnings and Precautions (5.1)].
2.2 Dosage Modification
Measure AT activity using an FDA-cleared test at Weeks 4 (Month 1), 12 (Month 3), 20 (Month 5) and 24 (Month 6) following the starting dose and after any dose modification.
- If any AT activity is <15%, a dose reduction is required. The lower dose should be initiated 3 months after the prior dose. AT measurements should be restarted after a dose reduction.
- If AT activity is >35% after 6 months, or if the patient has not achieved satisfactory bleed control, dose escalation should be considered. AT measurements should be restarted after a dose escalation.
Refer to Table 1 below for modified dosage based on AT activity levels.
Table 1: Dose Modification Based on Antithrombin Activity Levels Last Dosage Administered Antithrombin Activity Level Dose Modification 50 mg every 2 months Less than 15% 20 mg every 2 months 15% to 35% Continue current dosage Greater than 35% after 6 months 50 mg every month 20 mg every 2 months Less than 15% 10 mg every 2 months 15% to 35% Continue current dosage Greater than 35% after 6 months 20 mg every month 10 mg every 2 months Less than 15% Discontinue QFITLIA 15% to 35% Continue current dosage Greater than 35% after 6 months 10 mg every month Once the patient's target dose is identified based on AT activity 15–35%, measure AT activity annually. Additional AT measurements can be considered if bleeding control is not adequate.
After cessation of QFITLIA dosing, routine AT monitoring is not needed unless the patient is bleeding and treatment with CFC/BPA is required. Based on data from the clinical studies, a majority of patients have AT activity >60% by 6 months after the last QFITLIA dose, after which standard doses of CFC/BPA may be used.
2.3 Bleed Management
Breakthrough Bleed Management
If breakthrough bleeding requiring on-demand treatment with CFC or BPA occurs during the first 7 days after QFITLIA initiation, manage the bleed using the patient's prior dosing regimen of CFC or BPA.
If breakthrough bleeding occurs after 7 days from the first QFITLIA dose, bleeds should be managed with a reduced dose and frequency of CFC/BPA to minimize the risk of thrombotic events. Initially, the weight-based dose of a CFC/BPA should be reduced, and the dosing interval doubled compared to the standard dose. This reduced dosing is shown in Table 2. If adequate hemostatic control is not achieved, higher doses may be used based on clinical judgement. Combination use of antifibrinolytics with CFC or BPA has not been studied.
Table 2: Breakthrough Bleed Management While Treated with QFITLIA Factor VIII Factor IX
(SHL)Factor IX
(EHL)aPCC rFVIIa aPCC = activated prothrombin complex concentrate, EHL = extended half-life, rFVIIa = activated recombinant FVII, SHL = standard half-life. - * Use clinical judgement for situations requiring higher doses, more frequent administration, or multiple repeat doses.
Use standard of care for adjunctive management of bleeding episodes and thrombotic events.Recommended dose 10 IU/kg
(maximum: 20 IU/kg)*20 IU/kg
(maximum: 30 IU/kg)*20 IU/kg
(maximum: 30 IU/kg)*30 U/kg
(maximum: 50 U/kg)*≤45 μg/kg Repeat dosing Should not repeat in <24 hours Should not repeat in <24 hours Should not repeat in <5–7 days Should not repeat in <24 hours Should not repeat in <2 hours Use of QFITLIA During Surgical Interventions
In clinical studies, patients with hemophilia A or B with or without inhibitors have undergone both major (N=60) and minor (N=71) surgical procedures without discontinuing QFITLIA prophylaxis. Utilize bleed management guidelines during the perioperative period for hemostatic management.
2.4 Preparation and Administration Instructions
QFITLIA is intended for use under the guidance of a healthcare provider. Provide proper training to patients and/or caregivers on the preparation and administration of QFITLIA prior to use, according to the Instructions for Use (IFU). A patient may self-inject QFITLIA or the patient's caregiver may administer QFITLIA. In pediatric patients 12 to 17 years of age, it is recommended that QFITLIA be administered by or under the supervision of an adult.
QFITLIA for subcutaneous administration is a clear, colorless to pale yellow solution. Do not use if the solution is discolored or cloudy, or if it contains visible flakes or particles. Do not use QFITLIA if it has been dropped or damaged.
If QFITLIA is stored at room temperature, it is ready for use. If QFITLIA is stored in the refrigerator, remove from the refrigerator and allow QFITLIA to reach room temperature, for at least 30 minutes.
Administer QFITLIA by subcutaneous injection in the thigh or abdomen region, except for the 2 inches (5 cm) around the navel. A caregiver can also inject QFITLIA in the outer area of the patient's upper arm. QFITLIA should not be injected into skin that is tender, damaged, bruised, or scarred. Do not inject into a vein.
To inject 10 mg or 20 mg of QFITLIA from the single-dose vial, it is recommended to use a sterile 1 mL Luer Lock syringe (polypropylene or polycarbonate), and a sterile 27 gauge ½ inch (13 mm) Luer Lock needle to withdraw and inject QFITLIA solution subcutaneously (see the Instructions for Use).
Discard unused product remaining in the prefilled pen or vial.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Thrombotic Events
Serious thrombotic events have been reported in QFITLIA-treated patients. Thrombotic events were reported in 2.6% of patients receiving the 80 mg once monthly dose (2.3 events per 100 person-years), including a fatal event of cerebral venous sinus thrombosis. The 80 mg once monthly dose is not approved or recommended for use. Thrombotic events were reported in 1.4% of patients receiving QFITLIA prophylaxis using the antithrombin-based dose regimen (AT-DR) that targeted AT activity 15-35% (0.8 events per 100 person-years). Participants with established thrombophilia or a history of thrombosis were generally excluded from studies with QFITLIA.
The risk of thrombosis is greater in patients with persistent AT activity <15%, with comorbidities that predispose to thrombosis, when bleed management guidelines are not followed in the post-operative setting, when there is an indwelling venous catheter, and with use of the 80 mg once monthly (non-AT-based) dose. Treatment of breakthrough bleeding episodes with CFC or BPA at a dose greater or more frequent than recommended may also increase thrombotic risk [see Dosage and Administration (2.3)]. The decision to utilize higher dosing regimens of CFC or BPA in the setting of inadequate hemostasis requires an assessment of the benefits and risks and close clinical monitoring.
Monitor AT activity using an FDA-cleared test and target AT activity 15–35% to reduce the risk of thrombosis [see Dosage and Administration (2.1, 2.2) and Warnings and Precautions (5.1)]. Monitor patients for signs and symptoms of thrombotic events. Interrupt QFITLIA prophylaxis in patients with a thrombotic event and manage as clinically indicated.
Inform patients treated with QFITLIA to monitor for and report signs and symptoms of thrombotic events. Consider the benefits and risks of resuming QFITLIA prophylaxis following resolution of the thrombotic event.
5.2 Acute and Recurrent Gallbladder Disease
Treatment with QFITLIA is associated with an increased occurrence of acute and recurrent gallbladder disease including cholelithiasis and cholecystitis. QFITLIA at a fixed dose (including 80 mg once monthly) is not approved or recommended for use. In the 270 patients in the QFITLIA clinical studies who received the fixed dose (non-AT-based dose) once monthly regimen, 17% experienced gallbladder events and 4% (11 patients) underwent cholecystectomy.
In 286 patients who received the AT-DR, 3.8% experienced gallbladder events and 0.3% (1 patient) underwent cholecystectomy.
All but one of the patients who underwent cholecystectomy resumed QFITLIA after surgery. One patient who started on fixed dosing experienced cholangitis and pancreatitis caused by gallstone disease more than a year after cholecystectomy while receiving AT-DR.
Patients diagnosed with acute or recurrent gallbladder disease most commonly presented with epigastric pain, generalized abdominal pain, indigestion, nausea and/or vomiting. If gallbladder disease is suspected, appropriate imaging and clinical follow-up are indicated.
Consider alternative treatment for hemophilia in patients with a history of symptomatic gallbladder disease. Consider interruption or discontinuation of QFITLIA if gallbladder disease occurs.
5.3 Hepatotoxicity
In the two randomized studies testing QFITLIA 80 mg once monthly, serum alanine transaminase (ALT) and aspartate transaminase (AST) elevations above 3 times the upper limit of normal (ULN) occurred in 32% of patients with hemophilia with inhibitors and 18% of patients with hemophilia without inhibitors compared to no events of AST or ALT elevation greater than 3× ULN in the control groups. There was one case of moderate hepatic injury (ALT elevation >300 U/L and total serum bilirubin >3 mg/dL) attributable to QFITLIA use. This patient had elevation of liver tests after a single 80 mg dose that continued to rise with repeated dosing of QFITLIA 80 mg once monthly. This patient's liver tests recovered with drug discontinuation. QFITLIA 80 mg once monthly is not approved or recommended for use.
On the AT-DR, 3.4% of patients treated with QFITLIA had at least one ALT value greater than 3× ULN with a median onset of 89 days after initial dosing (range 15 to 768 days).
Avoid use of QFITLIA in patients with hepatic impairment (Child-Pugh Class A, B and C).
Obtain baseline liver tests including AST, ALT, and total bilirubin prior to initiating QFITLIA, monthly for at least the first 6 months of QFITLIA use, and monthly for at least 6 months after a dose increase, and periodically thereafter as clinically indicated.
If new or worsening liver test abnormalities occur, perform appropriate diagnostic evaluations, initiate medical management as appropriate and monitor laboratory parameters until they return to baseline. If ALT or AST elevations greater than 5× ULN occur, interrupt QFITLIA treatment. Consider the benefits and risks of resuming QFITLIA prophylaxis following resolution of transaminase elevations. If you decide to restart QFITLIA, wait until liver tests have returned to baseline. If QFITLIA is restarted and ALT or AST elevations greater than 5× ULN reoccur or the patient experiences jaundice (total bilirubin ≥2.5 mg/dL) thought to be from hepatotoxicity with other causes of liver test elevation ruled out, permanently discontinue QFITLIA.
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Thrombotic Events [see Boxed Warning and Warnings and Precautions (5.1)]
- Acute and Recurrent Gallbladder Disease [see Boxed Warning and Warnings and Precautions (5.2)]
- Hepatotoxicity [see Warnings and Precautions (5.3)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The pooled safety population described in the WARNINGS AND PRECAUTIONS reflect exposure to QFITLIA as fixed doses and AT-DR (N=335).
The safety of the QFITLIA AT-DR was assessed in 286 adult and pediatric male patients with hemophilia A or B with or without inhibitors [see Clinical Studies (14)].
Among patients who received the AT-DR, 93% were exposed for 6 months or longer and 83% were exposed for 12 months or longer. The median duration of exposure across the studies was 674 days (with a maximum of 896 days).
Serious adverse reactions occurred in 4/286 (1.4%) patients who received the AT-DR, two of whom had serious adverse reactions of cholecystitis.
Permanent discontinuation of QFITLIA due to an adverse reaction occurred in 4/286 (1.4%) patients receiving the AT-DR and included liver injury, post-operative deep vein thrombosis, cerebral infarction and pruritus.
Dosage interruptions of QFITLIA due to an adverse reaction occurred in 2/286 (0.7%) patients receiving the AT-DR and included increased serum transaminases.
The most common adverse reactions (≥10%) reported in patients treated with the AT-DR were viral infection, nasopharyngitis, and bacterial infection.
Table 3: Adverse Reactions Reported in ≥5% of Patients from Pooled Clinical Studies with QFITLIA AT-DR Adverse Reaction Number of Patients
(%)
N=286- * Includes similar terms.
- † Hepatic injury includes: alanine aminotransferase increased, aspartate aminotransferase increased, liver injury.
- ‡ injection site reactions: includes injection site bruising, injection site erythema, injection site pain, injection site hematoma, injection site atrophy, injection site hemorrhage, injection site discomfort, injection site swelling, injection site discoloration, injection site pruritus, injection site induration, injection site nodule, injection site mass, injection site vesicles, injection site deformation, injection site rash, injection site joint pain and application site erythema.
Viral infection* 29 Nasopharyngitis* 26 Bacterial infection* 11 Hepatic Injury† 8 Arthralgia* 8 Prothrombin fragment 1.2 increased 7 Injection site reaction‡ 6 Headache 5 Cough* 5 -
7 DRUG INTERACTIONS
7.1 Hypercoagulability with Concomitant Use of CFC or BPA
QFITLIA prophylaxis leads to increased thrombin generation with additive increase in peak thrombin when used concomitantly with CFC or BPA [see Boxed Warning, Dosage and Administration (2.3) and Warnings and Precautions (5.1)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no available data on QFITLIA use in pregnant women to evaluate for a drug-associated risk of major birth defects, miscarriage, or other adverse maternal or fetal outcomes. Reproduction studies in pregnant animals have not been conducted with fitusiran. It is not known whether QFITLIA can cause fetal harm when administered to a pregnant woman or can affect reproduction capacity. QFITLIA should be used during pregnancy only if the potential benefit justifies the potential risks, including those to the fetus.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2–4% and 15–20%, respectively.
8.2 Lactation
Risk Summary
There are no data on the presence of fitusiran or its metabolite in human milk, the effects on the breastfed child, or the effects on milk production. It is not known whether QFITLIA is safe for use during breastfeeding. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for QFITLIA and any potential adverse effects on the breastfed infant from QFITLIA or from the underlying maternal condition.
8.3 Females and Males of Reproductive Potential
Contraception
Females
Use of QFITLIA in women using hormonal contraceptives may increase the risk of thrombotic events. Estrogen based hormonal contraceptives are an established risk factor for thrombosis in women with inherited AT deficiency. Advise patients using hormonal contraceptives to use an alternative non-hormonal contraception prior to starting treatment with and while receiving QFITLIA.
8.4 Pediatric Use
The safety and effectiveness of QFITLIA for the treatment of hemophilia A or B with or without factor VIII or IX inhibitors have been established in pediatric patients aged 12 years and older. Use of QFITLIA in pediatric patients with hemophilia A and B is supported by evidence from adequate and well-controlled studies in adult and pediatric patients [see Adverse Reactions (6.1) and Clinical Studies (14)]. A total of 60 pediatric patients ages 12 to 17 were treated with QFITLIA in the clinical studies.
The safety and effectiveness of QFITLIA have not been established in pediatric patients below 12 years of age.
8.5 Geriatric Use
There were 3 patients with hemophilia 65 years of age and older in the clinical studies on QFITLIA. Clinical studies of QFITLIA did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects.
8.6 Hepatic Impairment
Serum transaminase elevations have been observed in the clinical studies [see Warnings and Precautions (5.3)]. Avoid use of QFITLIA in patients with established hepatic impairment (Child-Pugh Class A, B and C).
-
11 DESCRIPTION
QFITLIA injection contains fitusiran, an antithrombin-directed double-stranded small interfering ribonucleic acid (siRNA), which is covalently linked to a ligand containing a triantennary N-acetylgalactosamine (GalNAc) moiety. The molecular formula of fitusiran sodium is C520H636F21N175Na43O309P43S6 and the molecular weight is 17,193 Da. Fitusiran drug substance is a white to pale yellow powder and freely soluble in water and phosphate buffered saline. The pH of a 1% aqueous solution of fitusiran drug substance in 50 mM KCl is 5.4 (4.4–7.3). It has the following structural formula:
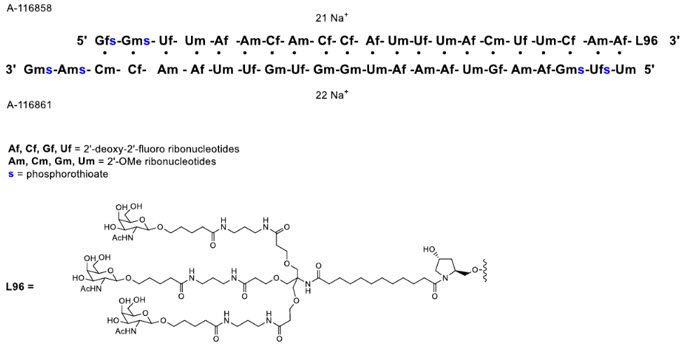
QFITLIA is supplied as a sterile, preservative-free, clear, colorless to pale yellow solution for subcutaneous administration.
Each 50 mg single-dose prefilled pen delivers 50 mg fitusiran (equivalent to 53.0 mg fitusiran sodium) in 0.5 mL, and also contains dibasic sodium phosphate (0.585 mg), monobasic sodium phosphate (0.044 mg), sodium chloride (2.455 mg), and Water for Injection, USP. Phosphoric acid (concentrated) and sodium hydroxide may be added to adjust to pH 7.0.
Each 20 mg single-dose vial delivers 20 mg fitusiran (equivalent to 21.2 mg fitusiran sodium) in 0.2 mL, and also contains dibasic sodium phosphate (0.234 mg), monobasic sodium phosphate (0.018 mg), sodium chloride (0.982 mg), and Water for Injection, USP. Phosphoric acid (concentrated) and sodium hydroxide may be added to adjust to pH 7.0.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
QFITLIA is a double-stranded siRNA that causes degradation of AT messenger RNA (mRNA) through RNA interference, reducing plasma AT levels.
12.2 Pharmacodynamics
In clinical studies with QFITLIA in hemophilia patients, the primary pharmacodynamic (PD) measure was plasma AT activity. Lower AT activity levels were associated with lower annualized bleeding rates (ABR); however, persistent AT activity <15% is a risk factor for thrombotic events.
In Study LTE15174, the mean (SD) AT activity on the AT-DR was 24% (4.64).
Simulations to estimate time for AT activity to reach steady state found that regardless of the dose regimen, more than 99% of participants reached steady state AT levels (defined as <10% variability among consecutive measures) within 23 weeks following initiation of QFITLIA dosing or following dose escalation or de-escalation. The duration of persistent AT activity <60% (non-negligible impact of QFITLIA on coagulation) following QFITLIA treatment discontinuation was estimated to be approximately 6 months, after which standard doses of CFC/BPA may be administered. In patients with AT activity <15%, the time required prior to initiation of a lower dose was estimated to be 12 weeks to maximize the AT activity recovery to the target range of 15% to 35%. Extent of AT reduction with QFITLIA did not differ between patients with hemophilia A or B, with or without inhibitors.
12.3 Pharmacokinetics
The pharmacokinetic (PK) properties of fitusiran were evaluated following administration of QFITLIA in patients with hemophilia A or B, with or without inhibitors as summarized in Table 4. QFITLIA PD is driven by liver PK rather than plasma PK. The QFITLIA dosing strategy is based on maintaining plasma AT activity levels between 15–35% with 10, 20 or 50 mg dosing every month or every two months.
Table 4: Pharmacokinetic Parameters of QFITLIA PK Parameters* QFITLIA 20 mg QFITLIA 50 mg AUC = area under the plasma concentration-time curve extrapolated to infinity; CL/F = apparent clearance; Cmax = maximum plasma concentration; CV = coefficient of variation; PK = pharmacokinetic(s); SD = standard deviation; t1/2 = terminal elimination half-life; tmax = time to maximum concentration; Vss/F = apparent volume of distribution - * Unless otherwise indicated, the PK parameters in this table are from Study LTE15174. All PK parameters are estimated using non-compartmental analysis and are presented as Mean (SD) [CV%], except for tmax which is presented as median (range)
- † Study TDR14767 (ALN-AT3SC-001)
- ‡ QFITLIA distributes primarily to the liver after subcutaneous injection.
- § In vitro data
General Information Steady State Exposure: Cmax (ng/mL) 34.4 (10.1) [29%] 84.1 (58.7) [70%] AUC (ng∙h/mL) 491 (83.8) [17%] 1290 (377) [29%] Dose Proportionality QFITLIA exhibited a dose proportional increase in plasma exposure after SC administration. Accumulation† No accumulation of QFITLIA was observed following multiple once monthly dosing. Absorption tmax (h) 2.88 (0.5 – 4.33) 3.78 (0.42 – 11.9) Distribution‡ Vss/F 431 (135) [31%] 570 (713) [125%] Protein Binding§ 96.6% at clinically relevant exposure (0.5 microgram/ml) Elimination t1/2 (h) 5.57 (2.36) [42%] 7.98 (4.74) [59%] CL/F 41.9 (8.39) [20%] 50.8 (71.1) [140%] Metabolism Primary Pathway QFITLIA is metabolized by endo- and exo-nucleases to oligonucleotides of varying lengths, which are further metabolized to oligonucleotides of even shorter lengths.
QFITLIA is not a substrate for CYP450 or transporters.Main Metabolite AS(N-1)3' Excretion Primary Pathway Not evaluated 14.6% (mean) of the administered 50 mg dose of QFITLIA is recovered unchanged in 0–24 hour urine Specific Populations
QFITLIA has only been studied in male patients. The pharmacokinetics of fitusiran is not influenced by race. QFITLIA has not been studied in children <12 years old.
No clinical study to evaluate the effect of hepatic impairment or renal impairment on the pharmacokinetics of QFITLIA was conducted. In the pivotal studies, 12 patients had mild renal impairment (eGFR ≥60 to <90 mL/min/1.73 m2) and 1 patient had moderate renal impairment (eGFR 30 mL/min/1.73 m2 to <60 mL/min/1.73 m2). Population PK analysis indicated no impact of renal impairment on the exposure (Cmax and AUC) of QFITLIA in patients with mild renal impairment.
Drug Interaction Studies
Clinical Studies
No clinical drug-drug interaction studies have been conducted to evaluate the potential of QFITLIA to interact with other co-administered drugs which are either substrates, inhibitors or inducers of CYP isozymes, or are substrates or inhibitors of drug transporters.
In Vitro Studies
Results from in vitro studies evaluating the potential of co-administered drugs to increase the PK exposure of QFITLIA, and the potential of QFITLIA to increase the PK exposure of co-administered drugs, indicate that CYP- or transporter-mediated interactions between QFITLIA and co-administered drugs are unlikely at clinically relevant concentrations.
12.6 Immunogenicity
The observed incidence of anti-drug antibodies (ADA) is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of ADA in the studies described below with the incidence of ADA in other studies, including those of QFITLIA or of other fitusiran products.
During QFITLIA treatment (up to 250 weeks) in four studies, 10 out of 290 adults with hemophilia (3.4%) developed ADAs. All patients had low ADA titers, and the majority of patients had transient ADAs. There was no identified clinically significant effect of ADAs on PK, PD, safety, or effectiveness of QFITLIA.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
In a 2-year carcinogenicity study in Sprague Dawley rats, fitusiran was not carcinogenic up to the highest dose tested following subcutaneous administration at doses of 0.03, 0.1 or 0.3 mg/kg/week in males and 0.1, 0.3, or 1 mg/kg/week in females. All doses in males and females were below the maximum recommended human dose (MRHD) of 50 mg per month based on body surface area (BSA) comparisons due to the sensitivity of wild type animals to the pharmacological effects of fitusiran. Fitusiran was not carcinogenic in Tg-rasH2 mice following subcutaneous administration at doses of 0.05, 0.15, or 0.5 mg/kg/week in males and 0.025, 0.08, or 0.25 mg/kg/week in females for 26 weeks.
Fitusiran was not mutagenic or clastogenic in a battery of genetic toxicity studies (i.e., in vitro bacterial reverse mutation assay, in vitro chromosomal aberration assay in human peripheral blood lymphocytes; in vivo bone marrow micronucleus test in rats).
In a chronic repeat dose toxicology study in Sprague Dawley rats, fitusiran-treated males received subcutaneous doses of 0.25, 0.5, or 1 mg/kg/week for 16 weeks then were cohabitated with fitusiran-naïve females. No adverse effects on male fertility endpoints were observed. All doses used in males were below the MRHD based on BSA comparisons due to the sensitivity of wild type animals to the pharmacological effects of fitusiran.
-
14 CLINICAL STUDIES
The efficacy and safety of QFITLIA in adult and pediatric patients aged 12 years and older with hemophilia A or B with or without inhibitors were established in two clinical studies:
- Hemophilia A or B with Inhibitory Antibodies: ATLAS-INH (NCT03417102)
- Hemophilia A or B without Inhibitory Antibodies: ATLAS-A/B (NCT03417245)
Patients in the above parent studies rolled over into the long-term extension study ATLAS-OLE (NCT03754790).
The clinical studies ATLAS-INH and ATLAS-A/B tested an 80 mg monthly fixed dose of QFITLIA. Because of thrombotic events with this dose, the QFITLIA AT-DR targeting AT activity of 15–35% was implemented in ATLAS-OLE. The AT-DR was initiated when studies ATLAS-INH and ATLAS-A/B were nearly completed, therefore, the efficacy of QFITLIA AT-DR treatment was assessed by comparing the QFITLIA AT-DR treatment data from the long-term extension study ATLAS-OLE to the control data from studies ATLAS-INH and ATLAS-A/B. The efficacy analyses were conducted according to the intent to treat (ITT) principle preserving the randomization in the parent studies.
ATLAS-INH
ATLAS-INH was a randomized, multicenter, open-label clinical study in 57 adult and pediatric males (aged ≥12 years) with hemophilia A or B with inhibitory antibodies to factor VIII (FVIII) or factor IX (FIX), who previously received on-demand (episodic) treatment with BPAs for bleeding. Eligible patients were randomized in a 2:1 ratio to receive QFITLIA prophylaxis at a fixed dose 80 mg SC monthly (N=38) or BPA on-demand for treatment of breakthrough bleeding episodes (N=19) for 9 months. The 80 mg dose of QFITLIA is not approved because of an increased risk of serious thrombotic events, gallbladder events (including the need for cholecystectomy) and hepatotoxicity [see Boxed Warning and Warnings and Precautions (5.1, 5.2, 5.3)].
Of the 57 enrolled patients all had inhibitors; 45 patients had Hemophilia A and 12 had Hemophilia B. All patients in the study were male. The mean age of patients was 28.4 years, and 10 (17.5%) patients were 12–17 years of age. A total of 68.4% of patients were Asian, 28.1% were White, 1.8% were other, and 1.8% were multiple races; 5.3% patients identified as Hispanic or Latino and 94.7% identified as not Hispanic or Latino. Generally, the demographic and patient characteristics at baseline were comparable between the patients with hemophilia A and B.
ATLAS A/B
ATLAS A/B was a randomized, multicenter, open-label clinical study in 120 adult and pediatric males (aged ≥12 years) with hemophilia A or B without inhibitory antibodies to FVIII or FIX, who previously received on-demand (episodic) treatment with CFC for bleeding. Eligible patients were randomized in a 2:1 ratio to receive QFITLIA prophylaxis at a fixed dose of 80 mg SC monthly (N=80) or CFCs on-demand to treat breakthrough bleeding episodes (N=40) for 9 months. The 80 mg dose of QFITLIA is not approved because of an increased risk of serious thrombotic events, gallbladder events (including the need for cholecystectomy) and hepatotoxicity [see Boxed Warning and Warnings and Precautions (5.1, 5.2, 5.3)].
Of the 120 enrolled patients none had inhibitors; 93 patients had Hemophilia A and 27 had Hemophilia B. All patients in the study were male. The mean age of patients was 33.8 years, and 14 (11.7%) patients were 12–17 years of age. A total of 59.2% of patients were Asian, 37.5% were White, 1.7% were Black or African American, and 1.7% were multiple races; 3.3% of patients identified as Hispanic or Latino, 90.8% identified as not Hispanic or Latino, and 5.8% had ethnicity information unreported. Generally, the demographic and patient characteristics at baseline were comparable between the patients with hemophilia A and B.
ATLAS-OLE
A total of 227 patients rolled over from two clinical studies (ATLAS-INH and ATLAS-A/B) and ATLAS-PPX, a crossover study in patients previously on CFC or BPA prophylaxis, and were treated with QFITLIA in ATLAS-OLE. This multicenter open-label extension study evaluated the long-term safety and efficacy of QFITLIA in adult and pediatric males aged ≥12 years with hemophilia A or B, with or without inhibitory antibodies to FVIII or FIX. Eligible patients initially received QFITLIA 80 mg SC once monthly. The study was amended to evaluate the efficacy and safety of the AT-DR. A total of 213 patients were subsequently transitioned to AT-DR targeting AT activity of 15–35%.
In the AT-DR, the QFITLIA starting dose was 50 mg every two months, and dosing was individually adjusted based on AT activity level using the INNOVANCE Antithrombin assay. The dose could be increased to 50 mg every month or 80 mg every month or decreased to 20 mg every two months or 20 mg every month. QFITLIA was discontinued if AT activity was <15% at the lowest dose. No patients required escalation to 80 mg every month to achieve the target AT range. The dose required to maintain AT activity 15–35% in patients who initiated dosing on 50 mg every two months was: 50 mg every two months (35.8% of patients), 50 mg every month (15.7% of patients), 20 mg every two months (30.9% of patients), or 20 mg every month (2.9% of patients). A total of 14.7% of patients discontinued QFITLIA due to more than one AT activity <15%.
Patients with known co-existing coagulation disorders other than hemophilia A or B, increased risk of thrombosis as assessed by history of arterial or venous thromboembolism, significant valvular disease or atrial fibrillation, or co-existing thrombophilic disorder (e.g., Factor V Leiden mutation), AT activity <60% at screening, platelet count ≤100,000/μL, eGFR ≤45 mL/min (using the MDRD), or clinically significant liver disease were not eligible for enrollment.
The efficacy of QFITLIA AT-DR in ATLAS-OLE was evaluated for a duration of 7 months (primary efficacy period) following a 6-month dose adjustment period. The median observed annualized bleeding rate (IQR) for treated bleeds was 3.7 (0.0; 7.5) overall, 1.9 (0.0; 5.6) in inhibitor patients and 3.8 (0.0; 11.2) in non-inhibitor patients.
QFITLIA Prophylaxis Compared to On-Demand BPA or CFC
The efficacy results of QFITLIA prophylaxis using AT-DR in ATLAS-OLE compared to on-demand BPA or CFC control data from studies ATLAS-INH and ATLAS-A/B with respect to rate of treated bleeds, treated spontaneous bleeds, and treated joint bleeds are shown in Table 5 (patients with inhibitors) and Table 6 (patients without inhibitors) below.
Table 5: Annualized Bleed Rate for Treated Bleeds with QFITLIA Prophylaxis versus On-Demand BPA in Patients ≥12 Years of Age with Inhibitors Endpoint QFITLIA AT-DR
(N=38)On-Demand BPA
(N=19)ABR = annualized bleed rate; AT-DR = AT-Dosing Regimen; BPA = bypassing agents; CI = confidence interval. - * Based on negative binomial regression model.
All Treated Bleeds ABR (95% CI)* 5.1 (2.8, 9.5) 19.1 (11.8, 31.0) % reduction
p-value73%
p=0.0006Treated Spontaneous Bleeds ABR (95% CI)* 3.1 (1.8, 5.4) 17.1 (9.9, 29.6) % reduction
p-value82%
<0.0001Treated Joint Bleeds ABR (95% CI)* 4.0 (2.5, 6.2) 14.4 (9.0, 23.1) % reduction
p-value73%
p=0.0001Table 6: Annualized Bleed Rate for Treated Bleeds with QFITLIA Prophylaxis versus On-Demand CFC in Patients ≥12 Years of Age without Inhibitors Endpoint QFITLIA AT-DR
(N=80)On-Demand CFC
(N=40)ABR = annualized bleed rate; AT-DR = AT-Dosing Regimen; CFC = clotting factor concentrate; CI = confidence interval. - * Based on negative binomial regression model.
All Treated Bleeds ABR (95% CI)* 9.0 (5.6, 14.5) 31.4 (20.5, 48.2) % reduction
p-value71%
p<0.0001Treated Spontaneous Bleeds ABR (95% CI)* 5.4 (3.7, 8.0) 21.0 (14.0, 31.6) % reduction
p-value74%
p<0.0001Treated Joint Bleeds ABR (95% CI)* 6.2 (4.2, 9.2) 21.6 (14.6, 31.9) % reduction
p-value71%
p<0.0001 -
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
QFITLIA (fitusiran) is a clear, colorless to pale yellow solution supplied in a single-dose prefilled pen or a single-dose vial. Each prefilled pen is designed to deliver 50 mg of QFITLIA in 0.5 mL (NDC: 58468-0348-1). Each vial is designed to deliver 20 mg of QFITLIA in 0.2 mL (NDC: 58468-0347-1).
QFITLIA is available in cartons containing 1 prefilled pen or 1 vial.
16.2 Storage and Handling
50 mg Prefilled Pen
- Store refrigerated at 2°C to 8°C (36°F to 46°F) in the original carton to protect from light.
- QFITLIA may be stored at room temperature between 15°C to 30°C (59°F to 86°F) for a single period of up to 3 months within the expiration date printed on the label. Discard no later than 3 months after removal from the refrigerator or at the expiration date, whichever comes first. After storage at room temperature, do not return the product to the refrigerator.
20 mg Vial
- Store QFITLIA either in the refrigerator at 2°C to 8°C (36°F to 46°F) or at room temperature between 15°C to 30°C (59°F to 86°F), in the original carton to protect from light. After storage at room temperature, do not return the product to the refrigerator.
Do not shake QFITLIA at any time. Do not heat QFITLIA. Do not freeze. Do not put into direct sunlight.
-
17 PATIENT COUNSELING INFORMATION
- Advise the patient to read the FDA-approved patient labeling (Medication Guide and Instructions for Use).
Use of BPAs or CFCs
Advise the patient and/or caregiver to discontinue prophylactic use of BPA or CFC no later than 7 days after starting QFITLIA to reduce the risk of thrombotic events. Discuss the appropriate dosing and frequency of BPA or CFC for breakthrough bleed management with the patient and/or caregiver prior to starting QFITLIA prophylaxis [see Dosage and Administration (2.3)].
Thrombotic Events
Advise the patient and/or caregiver of the risk of thrombotic events while receiving QFITLIA. Inform the patient and/or caregiver of the need for periodic measurements of AT activity that may result in changes to the QFITLIA dose and/or frequency of administration to reduce the risk for thrombosis. Educate patients on the signs and symptoms of thrombotic events and to seek immediate medical attention if new symptoms of thrombotic events occur [see Warnings and Precautions (5.1)].
Acute and Recurrent Gallbladder Disease
Inform the patient and/or caregiver of the risk of acute and recurrent gallbladder disease while receiving QFITLIA. Educate patients on the signs and symptoms of gallbladder disease and to seek medical attention if new symptoms of gallbladder disease occur [see Warnings and Precautions (5.2)].
Hepatotoxicity
Inform the patient and/or caregiver of the risk of hepatotoxicity and that blood tests to monitor for this risk will be obtained before starting QFITLIA and periodically during treatment. Inform patients to seek medical attention if symptoms of hepatotoxicity occur [see Warnings and Precautions (5.3)].
Administration Instructions
Provide training to the patient and/or caregiver on proper subcutaneous injection technique, including aseptic technique, and the preparation and administration of QFITLIA prior to use [see Dosage and Administration (2.4)].
- SPL UNCLASSIFIED SECTION
-
MEDICATION GUIDE
This Medication Guide has been approved by the U.S. Food and Drug Administration. Issued: September 2025 MEDICATION GUIDE
QFITLIA® (kew fit lee ah)
(fitusiran)
injection, for subcutaneous useWhat is the most important information I should know about QFITLIA?
QFITLIA helps your blood form clots. Do not stop using QFITLIA without talking to your healthcare provider. If you miss doses or stop using QFITLIA, you may no longer be protected against bleeding.
Use of a clotting factor concentrate (CFC) or bypassing agent (BPA) to help protect against bleeding must be stopped within 7 days after your first dose of QFITLIA.
Your healthcare provider may prescribe on demand CFC (factor VIII or factor IX products) or BPA if you bleed during treatment with QFITLIA. Carefully follow your healthcare provider's instructions regarding when to use on-demand treatment with CFC or BPA, including the prescribed dose and timing of the CFC or BPA.
QFITLIA can cause serious side effects, including: - Abnormal blood clotting (thrombotic events). Serious blood clots have happened in people treated with QFITLIA. QFITLIA can cause blood clots to form in blood vessels in your arms, legs, lungs, heart, brain, eyes, or head. Your risk of blood clots is greater if your antithrombin (AT) blood level is persistently less than 15% or if you have certain other conditions. Get medical help right away if you get any of these signs or symptoms of blood clots during or after treatment with QFITLIA:
- swelling, pain or redness in arms or legs
- coughing up blood
- shortness of breath
- severe chest pain or tightness of the chest
- fast heart rate
- feeling faint or passing out
- severe or persistent headache
- difficulty speaking or understanding language
- feel confused
- numbness or weakness in your face, arms or legs
- sudden loss or changes in your vision, eye pain or swelling
- Gallbladder disease. QFITLIA can cause gallstones (cholelithiasis) and inflammation of your gallbladder (cholecystitis), which might require surgery to remove your gallbladder. Tell your healthcare provider right away if you develop stomach (abdomen) pain, indigestion, nausea or vomiting. Your healthcare provider may temporarily or permanently stop your treatment with QFITLIA if you develop any of these symptoms.
- Liver problems. QFITLIA can cause an increase in your blood liver enzymes. Your healthcare provider will do blood tests to check your liver function before and during treatment with QFITLIA.
- See "What are the possible side effects of QFITLIA?" for more information about side effects.
What is QFITLIA?
QFITLIA is a prescription medicine used for routine prophylaxis to prevent or reduce the frequency of bleeding episodes in adults and children 12 years and older with hemophilia A or B with or without factor VIII or IX inhibitors.
It is not known if QFITLIA is safe and effective in children younger than 12 years of age.Before receiving QFITLIA, tell your healthcare provider about all of your medical conditions, including if you: - have liver problems
- have a history of gallbladder disease
- are pregnant or plan to become pregnant. It is not known if QFITLIA may harm your unborn baby.
Females who are able to become pregnant: Hormonal birth control (contraception) may increase your risk of blood clots if used during treatment with QFITLIA. If you use hormonal birth control (contraception), talk to your healthcare provider about effective forms of non-hormonal birth control (contraception) options you can use before starting and during treatment with QFITLIA. - are breastfeeding or plan to breastfeed. It is not known if QFITLIA passes into your breast milk.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. Keep a list of them to show your healthcare provider and pharmacist when you get a new medicine. How should I use QFITLIA?
See the detailed "Instructions for Use" that comes with your QFITLIA for information on how to prepare and inject a dose of QFITLIA, and how to properly throw away (dispose of) the used prefilled pen, vial, needle and syringe.- Use QFITLIA exactly as prescribed by your healthcare provider.
- Your healthcare provider may change your dose or how often you inject QFITLIA based on your AT blood level to help reduce your chance of abnormal blood clots. Your healthcare provider will check your AT blood level:
- before starting QFITLIA and at month 1, 3, 5 and 6 during QFITLIA treatment.
- after any change in your dose of QFITLIA.
- at yearly routine visits after your healthcare provider determines your target dose.
- It is recommended that QFITLIA be given by or under the supervision of an adult in children 12 to 17 years of age.
- QFITLIA is given as an injection under your skin (subcutaneous) by you or a caregiver.
- Your healthcare provider should show you or your caregiver how to prepare, measure, and inject your dose of QFITLIA before you inject yourself for the first time.
- Do not attempt to inject yourself or another person unless you have been taught how to do so by a healthcare provider.
- After your first dose of QFITLIA, your healthcare provider will let you know when to take your next dose and how much QFITLIA to inject.
- If you miss a dose of QFITLIA on your scheduled day, you should give the dose as soon as possible. After that, resume your dosing schedule either 1 time every month or 1 time every two months from the last dose, as instructed by your healthcare provider.
What are the possible side effects of QFITLIA? The most common side effects of QFITLIA include: - viral infection
- common cold symptoms
- bacterial infection
Call your doctor for medical advice about side effects. You may report side effects to the FDA at 1-800-FDA-1088.How should I store QFITLIA? -
50 mg Prefilled Pen
- Store QFITLIA prefilled pen in the refrigerator between 36°F to 46°F (2°C to 8°C) in the original carton to protect from light.
- QFITLIA prefilled pen may be stored at room temperature between 59°F to 86°F (15°C to 30°C) for a single period of up to 3 months within the expiration date printed on the label.
- Throw away (dispose of) the QFITLIA prefilled pen after 3 months at room temperature or at the expiration date, whichever comes first.
- Do not return QFITLIA prefilled pen to the refrigerator after storing at room temperature.
-
20 mg Vial
- Store QFITLIA vial in the refrigerator between 36°F to 46°F (2°C to 8°C) or at room temperature between 59°F to 86°F (15°C to 30°C) in the original carton to protect from light.
- Do not return QFITLIA vial to the refrigerator after storing at room temperature.
- Do not shake the QFITLIA vial or prefilled pen at any time.
- Do not heat the QFITLIA vial or prefilled pen.
- Do not freeze the QFITLIA vial or prefilled pen.
- Do not put the QFITLIA vial or prefilled pen into direct sunlight.
- Throw away (dispose of) any unused QFITLIA.
Keep QFITLIA and all medicines out of the reach of children.General information about the safe and effective use of QFITLIA.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use QFITLIA for a condition for which it was not prescribed. Do not give QFITLIA to other people even if they have the same symptoms you have. It may harm them. You can ask your healthcare provider or pharmacist for information about QFITLIA that is written for health professionals.What are the ingredients in QFITLIA?
Active ingredient: fitusiran
Inactive ingredients: dibasic sodium phosphate, monobasic sodium phosphate, sodium chloride, and Water for Injection, USP. Phosphoric acid (concentrated) and sodium hydroxide may be added to adjust pH to 7.
Manufactured by: Genzyme Corporation, Cambridge, MA 02141. A SANOFI COMPANY.
For patent information: https://www.sanofi.us/en/products-and-resources/patents
©2025 Genzyme Corporation. All rights reserved.
For more information, go to www.QFITLIA.com or call 1-800-745-4447. -
INSTRUCTIONS FOR USE
QFITLIA® (kew fit lee ah)
(fitusiran)
injection, for subcutaneous use
Single-Dose Prefilled Pen (50 mg/0.5 mL)This Instructions for Use contains information on how to inject QFITLIA.
Read this Instructions for Use before using the QFITLIA Prefilled Pen. Do not inject yourself or someone else until you have been shown how to inject QFITLIA Prefilled Pen. Your healthcare provider can show you or your caregiver how to prepare and inject a dose of QFITLIA before you try to do it yourself for the first time. Keep this Instructions for Use. Call your healthcare provider if you have any questions.
QFITLIA Prefilled Pen is only for use in adults and children 12 years of age and older. It is recommended that QFITLIA Prefilled Pen be given by adults or under adult supervision to children 12 to 17 years of age.
This QFITLIA Prefilled Pen is a single-dose device. It contains 50 mg of QFITLIA for injection under the skin (subcutaneous injection).
The parts of the QFITLIA Prefilled Pen are shown below:
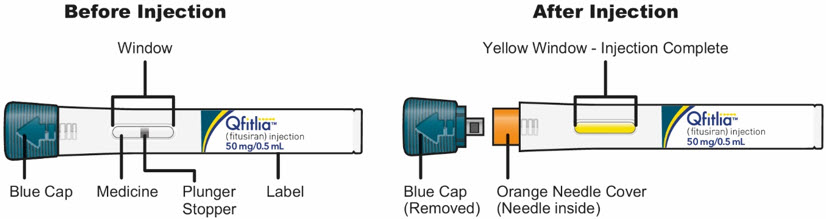
Important information to know before injecting QFITLIA
- Read all of the instructions carefully before using the QFITLIA Prefilled Pen.
- Ask your healthcare provider how often you will need to inject the medicine.
- Throw away (dispose of) the used QFITLIA Prefilled Pen right away after use.
- Do not re-use a QFITLIA Prefilled Pen.
- QFITLIA Prefilled Pen is for subcutaneous use only. Do not inject QFITLIA Prefilled Pen into a vein.
Storing QFITLIA Prefilled Pen
- Store QFITLIA Prefilled Pen in the refrigerator between 36°F to 46°F (2°C to 8°C) in the original carton to protect from light.
- QFITLIA Prefilled Pen may be stored at room temperature between 59°F to 86°F (15°C to 30°C) for a single period of up to 3 months within the expiration date printed on the label.
- Do not return QFITLIA Prefilled Pen to the refrigerator after storing at room temperature.
- Do not shake the QFITLIA Prefilled Pen at any time.
- Do not heat the QFITLIA Prefilled Pen.
- Do not freeze the QFITLIA Prefilled Pen.
- Do not put the QFITLIA Prefilled Pen into direct sunlight.
- Keep QFITLIA Prefilled Pen and all medicines out of the reach of children.
A. Get ready to inject
A1) Gather supplies
Find a clean, flat work surface. Make sure you have the following supplies:

A2) Check the QFITLIA Prefilled Pen
-
Note: The plunger stopper in the middle of the Window is normal.

- Do not use the QFITLIA Prefilled Pen if it has been dropped or damaged.
- Do not use the QFITLIA Prefilled Pen if the Blue Cap is missing or not securely attached.
-
Do not use the QFITLIA Prefilled Pen if the Window is yellow before use. A yellow Window means the QFITLIA Prefilled Pen has been used.
A3) Look at the label

- Check the label to be sure that you have the correct prescribed medicine and dose.

- Check the expiration date.
- Do not use the QFITLIA Prefilled Pen if the expiration date has passed.
A4) Check the medicine
- Look at the medicine through the Window, it should be clear and colorless to pale yellow.
- Note: You may see air bubbles, this is normal.
- Do not use the QFITLIA Prefilled Pen if the liquid is discolored or cloudy, or if it contains visible flakes or particles.
A5) If QFITLIA Prefilled Pen was stored in the refrigerator, warm up for 30 minutes.

- Lay the QFITLIA Prefilled Pen on a clean, flat surface and let it warm up at room temperature between 59°F to 86°F (15°C to 30°C) for at least 30 minutes.
- Do not heat the QFITLIA Prefilled Pen or put into direct sunlight.
B. Choose and prepare your injection site
B1) Wash your hands well with soap and water
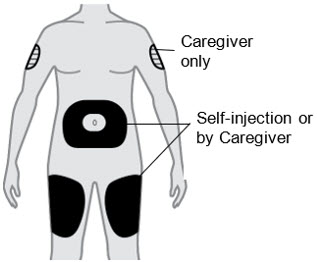
B2) Choose an injection site
- Thigh
- Stomach except for the 2 inches (5 cm) around your belly button (navel).
- A caregiver can also inject in the outer area of your upper arm.
- Do not inject into skin that is tender, damaged, has bruises or scars, or into areas with visible veins.
- For subcutaneous use only. Do not inject into a vein.
- Do not inject through clothes.
B3) Prepare the injection site
- Clean the injection site with an alcohol wipe.
- Let the skin dry before injecting.
- Do not touch the injection site again or blow on it before the injection.
C. Give the injection
C1) Remove Blue Cap
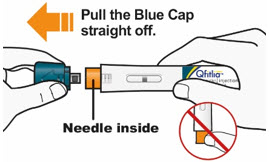
- Remove the Blue Cap by pulling it straight off as shown. Do not twist the Blue Cap off.
- Do not remove the Blue Cap until you are ready to inject.
- Do not touch the Orange Needle Cover with your fingers. The Needle is inside.
- Do not put the Blue Cap back on the QFITLIA Prefilled Pen after you have removed it.
- Note: You may see a droplet on the needle tip. This is normal.
C2) Place QFITLIA Prefilled Pen on injection site
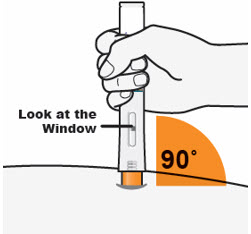
- Hold the QFITLIA Prefilled Pen as shown so you can see the Window.
- Place the Orange Needle Cover on the skin at a 90-degree angle.
C3) Press down firmly → Watch Window turn fully yellow
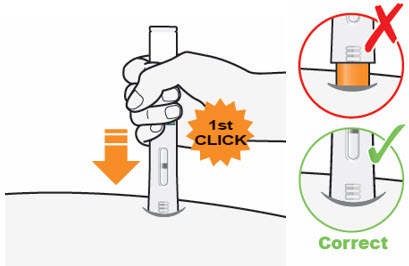
Press down and hold the QFITLIA Prefilled Pen firmly against the skin until you cannot see the Orange Needle Cover.
- There will be a "click" when the injection starts, and
- The Window will start to turn yellow.
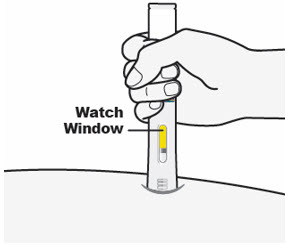
Keep pressing the QFITLIA Prefilled Pen against the skin and watch the Window.
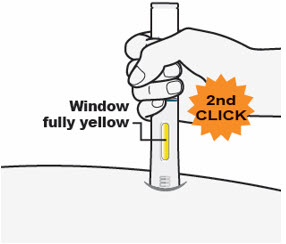
- The Window will turn completely yellow, and
- You may hear a 2nd "click." The injection is complete.
C4) Remove
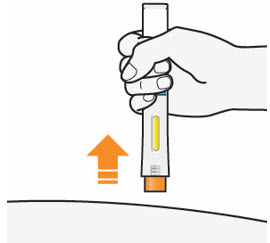
- After you have completed your injection pull straight up to remove QFITLIA Prefilled Pen from the skin. The Orange Needle Cover will cover the needle.
- If you see any blood at the site, lightly dab a cotton ball or gauze pad.
- Do not rub the skin after the injection.
- If the Window does not turn completely yellow, or if it looks like you may not have received a full dose, call your healthcare provider right away.
- Do not give a second dose without speaking to your healthcare provider.
D. Throw away (dispose of) used QFITLIA Prefilled Pen
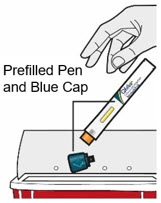
- Do not put the Blue Cap back on.
Keep your sharps disposal container out of the reach of children.
How to dispose of (throw away) QFITLIA Prefilled Pen
- Put your used QFITLIA Prefilled Pen and Blue Cap in an FDA-cleared sharps disposal container right away after use.
- Do not dispose of (throw away) QFITLIA Prefilled Pen and Blue Cap in your household trash.
- If you do not have an FDA-cleared sharps disposal container, you may use a household container that is:
- made of a heavy-duty plastic,
- can be closed with a tight-fitting, puncture-resistant lid, without sharps being able to come out,
- upright and stable during use,
- leak-resistant, and
- properly labeled to warn of hazardous waste inside the container.
- When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used QFITLIA Prefilled Pens.
- For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA's website at: http://www.fda.gov/safesharpsdisposal
- Do not dispose of your used sharps disposal container in your household trash unless your community guidelines permit this.
- Do not recycle your used sharps disposal container.
Manufactured by:
Genzyme Corporation
Cambridge, MA 02141
A SANOFI COMPANYQFITLIA is a registered trademark of GENZYME CORPORATION
© 2025 GENZYME CORPORATION. All rights reserved.For patent information: https://www.sanofi.us/en/products-and-resources/patents
For more information go to www.QFITLIA.com or call 1-800-745-4447.
This Instructions for Use has been approved by the US Food and Drug Administration.
Issued: September 2025 -
INSTRUCTIONS FOR USE
QFITLIA® (kew fit lee ah)
(fitusiran)
injection, for subcutaneous use
Single-Dose Vial (20 mg/0.2 mL)This Instructions for Use contains information on how to inject QFITLIA from a vial.
Read this Instructions for Use before using QFITLIA. Do not inject yourself or someone else until you have been shown how to inject QFITLIA from a vial. Your healthcare provider can show you or your caregiver how to prepare and inject a dose of QFITLIA from a vial before you do it yourself for the first time. Keep this Instructions for Use. Call your healthcare provider if you have any questions.
QFITLIA is only for use in adults and children 12 years of age and older. In children 12 to 17 years of age it is recommended that QFITLIA be given by or under the supervision of an adult.
This QFITLIA vial is for single-use only. It contains 20 mg of QFITLIA for injection under the skin (subcutaneous injection).
Important information to know before injecting QFITLIA
- Read all of the instructions carefully before using QFITLIA.
- Ask your healthcare provider how often you will need to inject the medicine.
- Check your prescription label to make sure you draw up the correct medicine dose prescribed by your healthcare provider.
- Throw away (dispose of) the used vial, syringe and needle right away after use.
- Only use the vial one time. After you inject your dose, throw away (dispose of) any unused QFITLIA left in the vial. Do not save any unused QFITLIA in the vial for later use.
- QFITLIA is for subcutaneous use only. Do not inject QFITLIA into a vein.
Storing QFITLIA vial
- Store QFITLIA vial either in the refrigerator between 36°F to 46°F (2°C to 8°C) or at room temperature between 59°F to 86°F (15°C to 30°C) in the original carton to protect from light.
- Do not return QFITLIA vial to the refrigerator after storing at room temperature.
- Do not shake the QFITLIA vial at any time.
- Do not heat the QFITLIA vial.
- Do not freeze the QFITLIA vial.
- Do not put the QFITLIA vial into direct sunlight.
- Keep QFITLIA vial and all medicines out of the reach of children.
A. Get ready to inject
A1) Gather the supplies.
Find a clean, flat work surface. Make sure you have the following supplies:
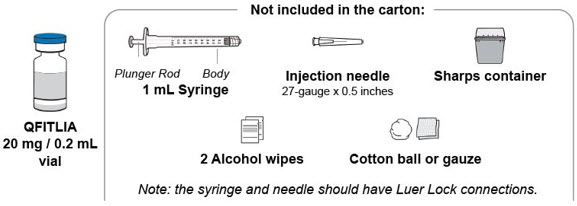
A2) Check the vial.
- Do not use the QFITLIA vial if it has been dropped or damaged.
- Check the label on the vial to be sure that you have the correct prescribed medicine and dose.
- Check expiration date (EXP:).
- Do not use QFITLIA if the expiration date on the vial has passed.
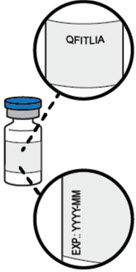
A3) Check the medicine.
- Look at the medicine in the vial to make sure it is clear and colorless to pale yellow.
- Do not use QFITLIA if the liquid in the vial is discolored or cloudy, or if it contains visible flakes or particles.
A4) If the QFITLIA vial was stored in the refrigerator, warm up for 30 minutes.
- Set the QFITLIA vial on a clean, flat surface and let it warm up at room temperature between 59°F to 86°F (15°C to 30°C) for at least 30 minutes.
- Do not heat the QFITLIA vial or put in direct sunlight.
A5) Wash your hands well with soap and water.
A6) Remove the protective cap from the QFITLIA vial.
Do not remove the stopper.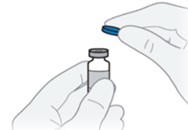
A7) Wipe the rubber stopper of the vial with an alcohol wipe and allow it to dry.
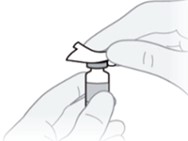
B. Choose and prepare the injection site
B1) Choose the injection site.
- Thigh
- Stomach except for the 2 inches (5 cm) around your belly button (navel).
- A caregiver can also inject in the outer area of your upper arm.
- Do not inject into skin that is tender, damaged, has bruises or scars, or into areas with visible veins.
- Do not inject into a vein. For subcutaneous use only.
- Do not inject through clothes.
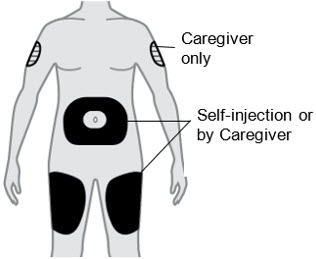
B2) Prepare the injection site.
- Clean the injection site with an alcohol wipe.
- Let the skin dry before injecting.
- Do not touch the injection site again or blow on it before the injection.
C. Prepare the injection
Contact your healthcare provider if you have any questions about preparing your dose.C1) Attach the needle to the syringe and remove the needle cap.
- Attach the needle to the syringe.
- Hold the syringe body and pull the needle cap straight off the syringe.
- Do not touch the needle.
- Do not recap the needle.
C2) Withdraw the solution.
Withdraw slightly more than your prescribed dose into the syringe.
- Push the needle through the center of the rubber vial stopper.
- Leave the needle in the vial and turn both the syringe and vial upside down (vial on top).
- Hold the syringe and vial firmly in one hand. Make sure the tip of the needle is in the QFITLIA solution.
- With your other hand, pull back on the plunger rod to withdraw slightly more than your prescribed dose into the syringe.
- Withdraw slightly more than 0.2 mL for a 20 mg dose.
- Withdraw slightly more than 0.1 mL for a 10 mg dose.
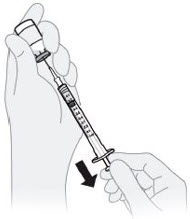
C3) Check for air bubbles.
- Keep the needle in the vial and make sure both syringe and vial are upside down (vial on top).
- If air bubbles are in the syringe, hold the syringe straight up and tap the side of the syringe until the bubbles float to the top.
- Push the plunger rod up to push the bubbles out of the syringe.
C4) Adjust plunger to your prescribed dose.
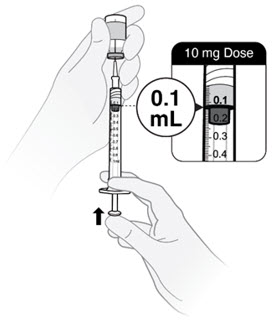
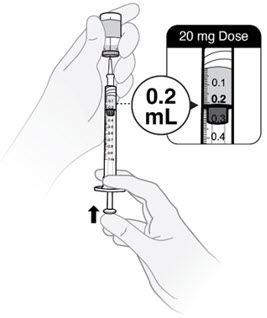
- Push the plunger rod to push solution back into the vial until you have the prescribed dose in the syringe.
- Push plunger rod to 0.2 mL for a 20 mg dose.
- Push plunger rod to 0.1 mL for a 10 mg dose.
- If the medicine in the syringe is less than your prescribed dose, repeat steps C2, C3 and C4 until you have your correct dose.
- Remove the needle from the vial. Do not let the needle touch anything.
D. Inject QFITLIA
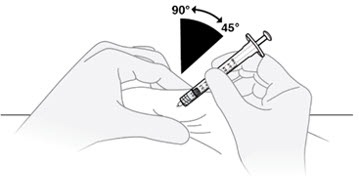
D1) Pinch a fold of skin and insert the needle.
Gently pinch a fold of skin at the cleaned injection site. Insert the needle into the skin fold at a 45° to 90° angle.
D2) Inject QFITLIA.
Relax the skin pinch. Slowly push the plunger rod all the way to the bottom of the syringe body and inject all of the QFITLIA solution.
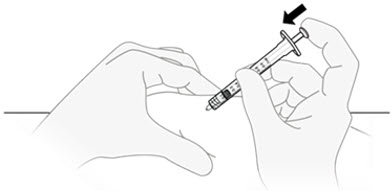
D3) Gently pull out the needle and syringe from the injection site.
- Do not recap the used needle to avoid a needle-stick injury.
- Do not rub the skin after the injection.
- If you see any blood at the site, lightly dab it with a cotton ball or gauze pad. If your injection site becomes red or sore, call your healthcare provider right away.
E. Throw away (dispose of) used vial, needle and syringe
E1) Throw away (dispose of) your used vial, needle and syringe in an FDA-cleared sharps disposal container right away after use.
Do not re-use the vial, needle, or syringe.
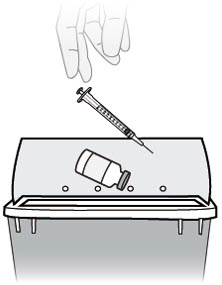
Keep your sharps disposal container out of the reach of children.
If you do not have an FDA-cleared sharps disposal container, you may use a household container that is:
- made of a heavy-duty plastic,
- can be closed with a tightfitting, puncture-resistant lid, without sharps being able to come out,
- upright and stable during use,
- leak-resistant, and
- properly labeled to warn of hazardous waste inside the container.
When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used QFITLIA needles and syringes.
For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA's website at: http://www.fda.gov/safesharpsdisposal
- Do not dispose of your used sharps disposal container in your household trash unless your community guidelines permit this.
- Do not recycle your used sharps disposal container.
Manufactured by:
Genzyme Corporation
Cambridge, MA 02141
A SANOFI COMPANYQFITLIA is a registered trademark of GENZYME CORPORATION
© 2025 GENZYME CORPORATION. All rights reserved.For patent information: https://www.sanofi.us/en/products-and-resources/patents
For more information go to www.QFITLIA.com or call 1-800-745-4447.
This Instructions for Use has been approved by the US Food and Drug Administration.
Issued: September 2025 -
PRINCIPAL DISPLAY PANEL - 0.5 mL Syringe Carton
NDC: 58468-0348-1
Rx onlyQfitlia™
(fitusiran) injection
50 mg/0.5 mLFor Subcutaneous Use Only
Dispense the enclosed Medication Guide to each patient.
One 0.5 mL Single-Dose Prefilled Pen
sanofi
-
PRINCIPAL DISPLAY PANEL - 0.2 mL Vial Carton
NDC: 58468-0347-1
Rx onlyQfitlia™
(fitusiran) injection
20 mg/0.2 mLFor Subcutaneous Use Only
Dispense the enclosed Medication
Guide to each patient.One 0.2 mL Single-Dose Vial.
Discard Unused Portion.sanofi
-
INGREDIENTS AND APPEARANCE
QFITLIA
fitusiran injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 58468-0348 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength FITUSIRAN (UNII: SV9W47ZLE1) (FITUSIRAN - UNII:SV9W47ZLE1) FITUSIRAN 50 mg in 0.5 mL Inactive Ingredients Ingredient Name Strength SODIUM PHOSPHATE, DIBASIC, HEPTAHYDRATE (UNII: 70WT22SF4B) 0.585 mg in 0.5 mL SODIUM PHOSPHATE, MONOBASIC, MONOHYDRATE (UNII: 593YOG76RN) 0.044 mg in 0.5 mL SODIUM CHLORIDE (UNII: 451W47IQ8X) 2.455 mg in 0.5 mL WATER (UNII: 059QF0KO0R) PHOSPHORIC ACID (UNII: E4GA8884NN) SODIUM HYDROXIDE (UNII: 55X04QC32I) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 58468-0348-1 1 in 1 CARTON 03/28/2025 1 0.5 mL in 1 SYRINGE; Type 2: Prefilled Drug Delivery Device/System (syringe, patch, etc.) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA219019 03/28/2025 QFITLIA
fitusiran injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 58468-0347 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength FITUSIRAN (UNII: SV9W47ZLE1) (FITUSIRAN - UNII:SV9W47ZLE1) FITUSIRAN 20 mg in 0.2 mL Inactive Ingredients Ingredient Name Strength SODIUM PHOSPHATE, DIBASIC, HEPTAHYDRATE (UNII: 70WT22SF4B) 0.234 mg in 0.2 mL SODIUM PHOSPHATE, MONOBASIC, MONOHYDRATE (UNII: 593YOG76RN) 0.018 mg in 0.2 mL SODIUM CHLORIDE (UNII: 451W47IQ8X) 0.982 mg in 0.2 mL WATER (UNII: 059QF0KO0R) PHOSPHORIC ACID (UNII: E4GA8884NN) SODIUM HYDROXIDE (UNII: 55X04QC32I) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 58468-0347-1 1 in 1 CARTON 03/28/2025 1 0.2 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA219019 03/28/2025 Labeler - Genzyme Corporation (025322157) Establishment Name Address ID/FEI Business Operations Vetter Pharma Fertigung GmbH & Co. KG (Langenargen Eisenbahnstrasse) 344217323 ANALYSIS(58468-0347, 58468-0348) , MANUFACTURE(58468-0347) Establishment Name Address ID/FEI Business Operations Vetter Pharma Fertigung GmbH & Co. KG (Ravensburg Helmut-Vetter-Strasse) 341629292 ANALYSIS(58468-0347, 58468-0348) Establishment Name Address ID/FEI Business Operations PPD Development Ireland Ltd. 985036175 ANALYSIS(58468-0347, 58468-0348) Establishment Name Address ID/FEI Business Operations Sanofi-Aventis Deutschland GmbH 313218430 ANALYSIS(58468-0348) , PACK(58468-0348) , LABEL(58468-0348) Establishment Name Address ID/FEI Business Operations BioSpring GmbH 537401556 ANALYSIS(58468-0347, 58468-0348) , API MANUFACTURE(58468-0347, 58468-0348) Establishment Name Address ID/FEI Business Operations Genzyme Corporation 050424395 ANALYSIS(58468-0347, 58468-0348) , PACK(58468-0347, 58468-0348) , LABEL(58468-0347, 58468-0348) Establishment Name Address ID/FEI Business Operations Vetter Pharma Fertigung GmbH & Co. KG (Ravensburg Schuetzenstrasse) 316126754 ANALYSIS(58468-0348) , MANUFACTURE(58468-0348) Establishment Name Address ID/FEI Business Operations Vetter Pharma Fertigung GmbH & Co. KG (Ravensburg Mooswiesen) 312670654 ANALYSIS(58468-0348)
Trademark Results [Qfitlia]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 QFITLIA 98054571 not registered Live/Pending |
Genzyme Corporation 2023-06-22 |
 QFITLIA 97546562 not registered Live/Pending |
Genzyme Corporation 2022-08-12 |
 QFITLIA 90836577 not registered Live/Pending |
Genzyme Corporation 2021-07-19 |
 QFITLIA 87813309 not registered Live/Pending |
GENZYME CORPORATION 2018-02-27 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.