ENTYVIO- vedolizumab injection, powder, lyophilized, for solution
Entyvio by
Drug Labeling and Warnings
Entyvio by is a Prescription medication manufactured, distributed, or labeled by Takeda Pharmaceuticals America, Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use ENTYVIO safely and effectively. See full prescribing information for ENTYVIO.
ENTYVIO (vedolizumab) for injection, for intravenous use
Initial U.S. Approval: 2014RECENT MAJOR CHANGES
INDICATIONS AND USAGE
DOSAGE AND ADMINISTRATION
- Recommended dosage in UC and CD: 300 mg infused intravenously over approximately 30 minutes at zero, two and six weeks, then every eight weeks thereafter. (2.3)
- Discontinue ENTYVIO in patients who do not show evidence of therapeutic benefit by Week 14. (2.3)
- Reconstitute ENTYVIO lyophilized powder with Sterile Water for Injection and dilute in 250 mL of sterile 0.9% Sodium Chloride Injection or sterile Lactated Ringer's Injection prior to administration. See Full Prescribing Information for complete reconstitution, dilution and storage instructions. (2.4)
- Bring patients up to date with all immunizations (according to current immunization guidelines) before initiating treatment with ENTYVIO. (2.2)
DOSAGE FORMS AND STRENGTHS
For injection: 300 mg vedolizumab in a single-dose vial. (3)
CONTRAINDICATIONS
Patients who have had a known serious or severe hypersensitivity reaction to ENTYVIO or any of its excipients. (4)
WARNINGS AND PRECAUTIONS
- Infusion-Related Reactions and Hypersensitivity Reactions: Discontinue ENTYVIO and initiate appropriate treatment if serious reactions occur. (5.1)
- Infections: Treatment with ENTYVIO is not recommended in patients with active, severe infections until the infections are controlled. Consider withholding ENTYVIO in patients who develop a severe infection while on treatment with ENTYVIO. (5.2)
- Progressive Multifocal Leukoencephalopathy (PML): Although unlikely, a risk of PML cannot be ruled out. Monitor patients for any new or worsening neurological signs or symptoms. (5.3)
ADVERSE REACTIONS
Most common adverse reactions (incidence ≥3% and ≥1% higher than placebo) are: nasopharyngitis, headache, arthralgia, nausea, pyrexia, upper respiratory tract infection, fatigue, cough, bronchitis, influenza, back pain, rash, pruritus, sinusitis, oropharyngeal pain, and pain in extremities. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Takeda Pharmaceuticals at 1-877-TAKEDA-7 (1-877-825-3327) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 3/2020
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Important Preparation and Administration Instructions
2.2 Prior to Administration of ENTYVIO
2.3 Dosage in Adults with Ulcerative Colitis or Crohn's Disease
2.4 Reconstitution and Dilution Instructions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Infusion-Related Reactions and Hypersensitivity Reactions
5.2 Infections
5.3 Progressive Multifocal Leukoencephalopathy
5.4 Liver Injury
5.5 Live and Oral Vaccines
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Immunogenicity
6.3 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Natalizumab
7.2 TNF Blockers
7.3 Live Vaccines
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Clinical Studies in Ulcerative Colitis
14.2 Clinical Studies in Crohn's Disease
15 REFERENCES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Important Preparation and Administration Instructions
- Administer ENTYVIO as an intravenous infusion over 30 minutes. Do not administer as an intravenous push or bolus.
- Reconstitute ENTYVIO lyophilized powder with Sterile Water for Injection and dilute in 250 mL of sterile 0.9% Sodium Chloride Injection or sterile Lactated Ringer's Injection prior to administration [see Dosage and Administration (2.4)].
- After the infusion is complete, flush with 30 mL of sterile 0.9% Sodium Chloride Injection or sterile Lactated Ringer's Injection.
- ENTYVIO should be administered by a healthcare professional prepared to manage hypersensitivity reactions including anaphylaxis, if they occur [see Warnings and Precautions (5.1)]. Appropriate monitoring and medical support measures should be available for immediate use. Observe patients during infusion and until the infusion is complete.
2.2 Prior to Administration of ENTYVIO
- Prior to initiating treatment with ENTYVIO, all patients should be brought up to date with all immunizations according to current immunization guidelines.
2.3 Dosage in Adults with Ulcerative Colitis or Crohn's Disease
The recommended dosage of ENTYVIO in adults with ulcerative colitis or Crohn's disease is 300 mg administered by intravenous infusion at zero, two and six weeks and then every eight weeks thereafter.
Discontinue therapy in patients who show no evidence of therapeutic benefit by Week 14.
2.4 Reconstitution and Dilution Instructions
Reconstitution Instructions
- Remove the flip-off cap from the single-dose vial and wipe with alcohol swab. Reconstitute ENTYVIO vial containing lyophilized powder with 4.8 mL of Sterile Water for injection at room temperature (20° to 25°C [68° to 77°F]), using a syringe with a 21- to 25- gauge needle.
- Insert the syringe needle into the vial through the center of the stopper and direct the stream of Sterile Water for Injection to the glass wall of the vial to avoid excessive foaming.
- Gently swirl the vial for at least 15 seconds to dissolve the lyophilized powder. Do not vigorously shake or invert.
- Allow the solution to sit for up to 20 minutes at room temperature to allow for reconstitution and for any foam to settle; the vial can be swirled and inspected for dissolution during this time. If not fully dissolved after 20 minutes, allow another 10 minutes for dissolution. Do not use the vial if the drug product is not dissolved within 30 minutes.
- Visually inspect the reconstituted ENTYVIO solution for particulate matter and discoloration prior to dilution. Solution should be clear or opalescent, colorless to light brownish yellow and free of visible particulates. Do not administer reconstituted solution showing uncharacteristic color or containing particulates.
- Once dissolved, gently invert vial three times.
- Immediately, withdraw 5 mL (300 mg) of reconstituted ENTYVIO solution using a syringe with a 21- to 25- gauge needle. Discard any remaining portion of the reconstituted solution in the vial.
Dilution Instructions
Add the 5 mL (300 mg) of reconstituted ENTYVIO solution to 250 mL of sterile 0.9% Sodium Chloride Injection or Lactated Ringer's Injection and gently mix the infusion bag. Do not add other medicinal products to the prepared infusion solution or intravenous infusion set. Once reconstituted and diluted, use the infusion solution as soon as possible.
Discard any unused portion of the infusion solution.
Storage
Specific storage conditions and timing for the reconstituted solution in vial and diluted solution in the infusion bag are outlined in Table 1.
Do not freeze the reconstituted solution in the vial or the diluted solution in the infusion bag.
Table 1. Storage Instructions Storage Condition Refrigeration
(2° to 8°C [36° to 46°F])Room temperature
(20° to 25°C [68° to 77°F])- * This time assumes the reconstituted solution is immediately diluted in the 0.9% Sodium Chloride Injection or Lactated Ringer's Injection and held in the infusion bag only. Any time that the reconstituted solution was held in vial should be subtracted from the time the solution may be held in the infusion bag.
- † This period may include up to 12 hours at room temperature (20° to 25°C [68° to 77°F]).
Reconstituted Solution
(in Sterile Water for Injection inside vial)8 hours Use immediately after reconstitution Diluted Solution
(in 0.9% Sodium Chloride Injection)24 hours*,† 12 hours* Diluted Solution
(in Lactated Ringer's Injection)6 hours* Use immediately after dilution The combined storage time of reconstituted ENTYVIO solution in the vial and the diluted solution in the infusion bag with 0.9% Sodium Chloride Injection is a total of 12 hours at room temperature (20° to 25°C [68° to 77°F]) or 24 hours refrigerated (2° to 8°C [36° to 46°F]). This combined storage time may include up to eight hours of the reconstituted solution in the vial at 2° to 8°C.
The combined storage time of reconstituted ENTYVIO solution in the vial and the diluted solution in the infusion bag with Lactated Ringer's Injection is a total of six hours refrigerated (2° to 8°C [36° to 46°F]).
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
ENTYVIO is contraindicated in patients who have had a known serious or severe hypersensitivity reaction to ENTYVIO or any of its excipients (such as dyspnea, bronchospasm, urticaria, flushing, rash and increased heart rate) [see Warnings and Precautions (5.1)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Infusion-Related Reactions and Hypersensitivity Reactions
Infusion-related reactions and hypersensitivity reactions have been reported, including anaphylaxis, dyspnea, bronchospasm, urticaria, flushing, rash, and increased blood pressure and heart rate [see Adverse Reactions (6.1, 6.3)]. These reactions may occur with the first or subsequent infusions of ENTYVIO and may vary in their time of onset from during infusion or up to several hours post-infusion.
If anaphylaxis or other serious infusion-related or hypersensitivity reactions occur, discontinue administration of ENTYVIO immediately and initiate appropriate treatment.
5.2 Infections
Patients treated with ENTYVIO are at increased risk for developing infections [see Adverse Reactions (6.1)]. The most commonly reported infections in clinical trials occurring at a rate greater on ENTYVIO than placebo involved the upper respiratory and nasal mucosa (e.g., nasopharyngitis, upper respiratory tract infection). Serious infections have also been reported in patients treated with ENTYVIO, including anal abscess, sepsis (some fatal), tuberculosis, salmonella sepsis, Listeria meningitis, giardiasis and cytomegaloviral colitis.
ENTYVIO is not recommended in patients with active, severe infections until the infections are controlled. Consider withholding treatment in patients who develop a severe infection while on treatment with ENTYVIO. Exercise caution when considering the use of ENTYVIO in patients with a history of recurring severe infections. Consider screening for tuberculosis (TB) according to the local practice. For progressive multifocal leukoencephalopathy (PML), see Warnings and Precautions (5.3).
5.3 Progressive Multifocal Leukoencephalopathy
PML, a rare and often fatal opportunistic infection of the central nervous system (CNS), has been reported with systemic immunosuppressants, including another integrin receptor antagonist. PML is caused by the John Cunningham (JC) virus and typically only occurs in patients who are immunocompromised. One case of PML in an ENTYVIO-treated patient with multiple contributory factors has been reported in the postmarketing setting (e.g., human immunodeficiency virus [HIV] infection with a CD4 count of 300 cells/mm3 and prior and concomitant immunosuppression). Although unlikely, a risk of PML cannot be ruled out.
Monitor patients on ENTYVIO for any new onset, or worsening, of neurological signs and symptoms. Typical signs and symptoms associated with PML are diverse, progress over days to weeks, and include progressive weakness on one side of the body or clumsiness of limbs, disturbance of vision, and changes in thinking, memory, and orientation leading to confusion and personality changes. The progression of deficits usually leads to death or severe disability over weeks or months. If PML is suspected, withhold dosing with ENTYVIO and refer to a neurologist; if confirmed, discontinue dosing permanently.
5.4 Liver Injury
There have been reports of elevations of transaminase and/or bilirubin in patients receiving ENTYVIO. In general, the combination of transaminase elevations and elevated bilirubin without evidence of obstruction is generally recognized as an important predictor of severe liver injury that may lead to death or the need for a liver transplant in some patients. ENTYVIO should be discontinued in patients with jaundice or other evidence of significant liver injury [see Adverse Reactions (6.1)].
5.5 Live and Oral Vaccines
Prior to initiating treatment with ENTYVIO, all patients should be brought up to date with all immunizations according to current immunization guidelines [see Dosage and Administration (2.2)]. Patients receiving ENTYVIO may receive non-live vaccines (e.g., influenza vaccine injection) and may receive live vaccines if the benefits outweigh the risks. There are no data on the secondary transmission of infection by live vaccines in patients receiving ENTYVIO [see Adverse Reactions (6.1)].
-
6 ADVERSE REACTIONS
The following topics are also discussed in detail in the Warnings and Precautions section:
- Infusion-Related Reactions and Hypersensitivity Reactions [see Warnings and Precautions (5.1)]
- Infections [see Warnings and Precautions (5.2)]
- Progressive Multifocal Leukoencephalopathy [see Warnings and Precautions (5.3)]
- Liver Injury [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data described below reflect exposure to ENTYVIO in 3,326 patients and healthy volunteers in clinical trials, including 1,396 exposed for greater than one year, and 835 exposed for greater than two years.
The safety data described in Table 2 are derived from four controlled Phase 3 trials (UC Trials I and II, and CD Trials I and III); data from patients receiving open-label ENTYVIO treatment at Weeks 0 and 2 (prior to entry into UC Trial II and CD Trial III) and from Weeks 6 to 52 (non-responders at Week 6 of UC Trial I and CD Trial I) are included [see Clinical Studies (14.1, 14.2)].
In these trials, 1,434 patients received ENTYVIO 300 mg for up to 52 weeks, and 297 patients received placebo for up to 52 weeks. Of these, 769 patients had ulcerative colitis and 962 patients had Crohn's disease. Patients were exposed for a mean duration of 259 days (UC Trials I and II) and 247 days (CD Trials I and III).
Adverse reactions were reported in 52% of patients treated with ENTYVIO and 45% of patients treated with placebo (UC Trials I and II: 49% with ENTYVIO and 37% with placebo; CD Trials I and III: 55% with ENTYVIO and 47% with placebo). Serious adverse reactions were reported in 7% of patients treated with ENTYVIO compared to 4% of patients treated with placebo (UC Trials I and II: 8% with ENTYVIO and 7% with placebo; CD Trials I and III: 12% with ENTYVIO and 9%, with placebo).
The most common adverse reactions (reported by ≥3% of patients treated with ENTYVIO in the UC Trials I and II and CD Trials I and III combined group and ≥1% higher than in combined placebo group) were nasopharyngitis, headache, arthralgia, nausea, pyrexia, upper respiratory tract infection, fatigue, cough, bronchitis, influenza, back pain, rash, pruritus, sinusitis, oropharyngeal pain and pain in extremities (Table 2).
Table 2. Adverse Reactions in ≥3% of ENTYVIO-Treated Patients and ≥1% Higher than in Placebo (UC Trials I and II* and CD Trials I and III*) Adverse Reaction ENTYVIO†
(N=1434)Placebo‡
(N=297)- * Data from patients receiving open-label ENTYVIO treatment at Weeks 0 and 2 (prior to entry into UC Trial II and CD Trial III) and from Weeks 6 to 52 (non-responders at Week 6 of UC Trial I and CD Trial I) are included.
- † Patients who received ENTYVIO for up to 52 weeks.
- ‡ Patients who received placebo for up to 52 weeks.
Nasopharyngitis 13% 7% Headache 12% 11% Arthralgia 12% 10% Nausea 9% 8% Pyrexia 9% 7% Upper respiratory tract infection 7% 6% Fatigue 6% 3% Cough 5% 3% Bronchitis 4% 3% Influenza 4% 2% Back pain 4% 3% Rash 3% 2% Pruritus 3% 1% Sinusitis 3% 1% Oropharyngeal pain 3% 1% Pain in extremities 3% 1% Safety data for patients (n=279) in UC Trials I and II and CD Trials I and III who received ENTYVIO at Weeks 0 and 2 and were then randomized to placebo at Week 6 for up to 52 weeks, and for patients (n=416) in CD Trial II, a 10 week Crohn's disease trial, are similar to those listed in Table 2.
Infusion-Related Reactions and Hypersensitivity Reactions
Serious infusion-related reactions and hypersensitivity reactions including anaphylaxis have been reported following ENTYVIO administration in clinical trials [see Warnings and Precautions (5.1)]. In UC Trials I and II and Crohn's Trials I and III, one case of anaphylaxis [one out of 1,434 patients treated with ENTYVIO (0.07%)] was reported by a Crohn's disease patient during the second infusion (symptoms reported were dyspnea, bronchospasm, urticaria, flushing, rash and increased blood pressure and heart rate) and was managed with discontinuation of infusion and treatment with antihistamine and intravenous hydrocortisone.
In UC Trials I and II and CD Trials I and III, 4% of patients treated with ENTYVIO and 3% of patients treated with placebo experienced an infusion-related reaction (IRR). The most frequently observed IRR in the patients treated with ENTYVIO (reported more than twice) were nausea, headache, pruritus, dizziness, fatigue, infusion-related reaction, pyrexia, urticaria and vomiting (each of these adverse reactions occurred in <1% in all patients treated with ENTYVIO) and no individual adverse reaction reported occurred at a rate above 1%. These reactions generally occurred within the first two hours after the infusion and resolved with no treatment or following antihistamine and/or IV hydrocortisone treatment. Less than 1% of patients treated with ENTYVIO had IRRs assessed by the investigator as severe, and IRRs requiring discontinuation of study treatment occurred in <1%.
In clinical trials, for patients with mild IRRs or hypersensitivity reactions, physicians were allowed to pretreat with standard medical treatment (e.g., antihistamine, hydrocortisone and/or acetaminophen) prior to next infusion.
Infections
In UC Trials I and II and CD Trials I and III, the rate of infections was 0.85 per patient-year in the patients treated with ENTYVIO and 0.7 per patient-year in the patients treated with placebo [see Warnings and Precautions (5.2)]. The infections consisted primarily of nasopharyngitis, upper respiratory tract infection, sinusitis, and urinary tract infection. Two percent of patients discontinued ENTYVIO due to infections.
In UC Trials I and II and CD Trials I and III, the rate of serious infections was 0.07 per patient-year in patients treated with ENTYVIO and 0.06 per patient-year in patients treated with placebo. Serious infections were more common in Crohn's disease patients than ulcerative colitis patients, and anal abscesses were the most frequently reported serious adverse reaction in Crohn's disease patients. Over 48 months, there was no increase in the rate of serious infections.
In controlled- and open-label long-term extension trials in adults treated with ENTYVIO, serious infections have been reported, including anal abscess, sepsis (some fatal), tuberculosis, salmonella sepsis, Listeria meningitis, giardiasis and cytomegaloviral colitis.
In UC Trials I and II and CD Trials I and III, sepsis, including bacterial sepsis and septic shock, was reported in four of 1,434 (0.3%) patients treated with ENTYVIO and in two of 297 patients treated with placebo (0.7%). During these trials, two Crohn's disease patients treated with ENTYVIO died due to reported sepsis or septic shock; both patients had significant comorbidities and a complicated hospital course that contributed to the deaths. In an open-label, long-term extension trial, additional cases of sepsis (some fatal), including bacterial sepsis and septic shock, were reported. The rate of sepsis in patients with ulcerative colitis or Crohn's disease receiving ENTYVIO was two per 1,000 patient-years.
In clinical trials, all patients were screened for tuberculosis. One case of latent, pulmonary tuberculosis was diagnosed during the controlled trials with ENTYVIO. Additional cases of pulmonary tuberculosis were diagnosed during the open-label trial. All of these observed cases occurred outside the United States, and none of the patients had extrapulmonary manifestations.
Liver Injury
There have been reports of elevations of transaminase and/or bilirubin in patients receiving ENTYVIO [see Warnings and Precautions (5.4)]. In UC Trials I and II and CD Trials I and III, three patients reported serious adverse reactions of hepatitis, manifested as elevated transaminases with or without elevated bilirubin and symptoms consistent with hepatitis (e.g., malaise, nausea, vomiting, abdominal pain, anorexia). These adverse reactions occurred following two to five ENTYVIO doses; however, based on case report information it is unclear if the reactions indicated drug-induced or autoimmune etiology. All patients recovered following discontinuation of therapy with some requiring corticosteroid treatment. In controlled trials, the incidence of ALT and AST elevations ≥3× ULN was <2% in patients treated with ENTYVIO and in patients treated with placebo. In the open-label trial, one additional case of serious hepatitis was observed.
Malignancies
In UC Trials I and II and CD Trials I and III, malignancies (excluding dysplasia and basal cell carcinoma) were reported in six of 1,434 (0.4%) patients treated with ENTYVIO, including colon cancer (n=2), transitional cell carcinoma (n=1), breast cancer (n=1), carcinoid tumor of the appendix (n=1) and squamous cell carcinoma (n=1). Malignancy was reported in one of 297 (0.3%) patients treated with placebo (squamous cell carcinoma).
Malignancies (excluding dysplasia and basal cell carcinoma) observed during the ongoing open-label long-term extension trial included B-cell lymphoma, breast cancer, colon cancer, malignant hepatic neoplasm, malignant lung neoplasm, malignant melanoma, lung cancer of primary neuroendocrine carcinoma, renal cancer and squamous cell carcinoma. Overall, the number of malignancies in the clinical trials was small; however, long-term exposure was limited.
Live and Oral Vaccines
There are no data on the secondary transmission of infection by live vaccines in patients receiving ENTYVIO.
In a placebo-controlled study of healthy volunteers, 61 subjects were given a single ENTYVIO 750 mg dose (2.5 times the recommended dose), and 62 subjects received placebo followed by intramuscular vaccination with Hepatitis B surface antigen and oral cholera vaccine. After intramuscular vaccination with three doses of recombinant Hepatitis B surface antigen, those treated with ENTYVIO did not have lower rates of protective immunity to Hepatitis B virus. However, those exposed to ENTYVIO did have lower seroconversion rates and anti-cholera titers relative to placebo after receiving the two doses of a killed, oral cholera vaccine. The impact on other oral vaccines and on nasal vaccines in patients is unknown.
6.2 Immunogenicity
As with all therapeutic proteins, there is potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors, including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to vedolizumab in the studies described below with the incidence of antibodies in other studies or to other products may be misleading.
In UC Trials I and II and CD Trials I and III, in patients who received ENTYVIO, the frequency of antibodies detected in patients was 13% at 24 weeks after the last dose of study drug (greater than five half-lives after last dose). During treatment, 56 of 1,434 (4%) of patients treated with ENTYVIO had detectable anti-vedolizumab antibody at any time during the 52 weeks of continuous treatment. Nine of 56 patients were persistently positive (at two or more study visits) for anti-vedolizumab antibody and 33 of 56 patients developed neutralizing antibodies to vedolizumab. Among eight of these nine subjects with persistently positive anti-vedolizumab antibody and available vedolizumab concentration data, six had undetectable and two had reduced vedolizumab concentrations [see Clinical Pharmacology (12.3)]. None of the nine subjects with persistently positive anti-vedolizumab antibody achieved clinical remission at Weeks 6 or 52 in the controlled trials.
6.3 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of ENTYVIO. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Immune system disorders: Anaphylaxis [see Warnings and Precautions (5.1)]
-
7 DRUG INTERACTIONS
7.1 Natalizumab
Because of the potential for increased risk of PML and other infections, avoid the concomitant use of ENTYVIO with natalizumab.
7.2 TNF Blockers
Because of the potential for increased risk of infections, avoid the concomitant use of ENTYVIO with TNF blockers.
7.3 Live Vaccines
Live vaccines may be administered concurrently with ENTYVIO only if the benefits outweigh the risks [see Warnings and Precautions (5.5)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to ENTYVIO during pregnancy. Information about the registry can be obtained by calling 1-877-TAKEDA7 (1-877-825-3327).
Risk Summary
Available pharmacovigilance data, data from the ongoing pregnancy registry, and data from published case reports and cohort studies in pregnant women have not identified an ENTYVIO associated risk of major birth defects, miscarriage or adverse maternal or fetal outcomes. There are risks to the mother and the fetus associated with inflammatory bowel disease in pregnancy (see Clinical Considerations). No fetal harm was observed in animal reproduction studies with intravenous administration of vedolizumab to rabbits and monkeys at dose levels 20 times the recommended human dosage (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated populations is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and miscarriage is 15 to 20%, respectively.
Clinical Considerations
Disease-associated maternal and embryo/fetal risk
Published data suggest that the risk of adverse pregnancy outcomes in women with inflammatory bowel disease (IBD) is associated with increased disease activity. Adverse pregnancy outcomes include preterm delivery (before 37 weeks of gestation), low birth weight (less than 2,500 g) infants, and small for gestational age at birth.
Fetal/Neonatal adverse reactions
ENTYVIO administered during pregnancy could affect immune responses in the in utero exposed newborn and infant. The clinical significance of low levels of ENTYVIO in utero-exposed infants is unknown. The safety of administering live or live-attenuated vaccines in exposed infants is unknown.
Data
Animal Data
A reproduction study has been performed in pregnant rabbits at single intravenous doses up to 100 mg/kg administered on gestation Day 7 (about 20 times the recommended human dosage) and has revealed no evidence of impaired fertility or harm to the fetus due to vedolizumab. A pre- and post-natal development study in monkeys showed no evidence of any adverse effect on pre- and post-natal development at intravenous doses up to 100 mg/kg (about 20 times the recommended human dosage).
8.2 Lactation
Risk Summary
Available published literature suggests the presence of vedolizumab in human milk. The effects of local gastrointestinal exposure and expected low systemic exposure to vedolizumab on the breastfed infant are unknown. There are no data on the effects of vedolizumab on the breastfed infant, or the effects on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for ENTYVIO and any potential adverse effects on the breastfed infant from ENTYVIO or from the underlying maternal condition.
8.4 Pediatric Use
Safety and effectiveness of ENTYVIO in pediatric patients have not been established.
8.5 Geriatric Use
Clinical trials of ENTYVIO did not include sufficient numbers of subjects aged 65 and over (46 Crohn's and ulcerative colitis patients aged 65 and over were treated with ENTYVIO during controlled Phase 3 trials) to determine whether they respond differently from younger subjects. However, no overall differences in safety or effectiveness were observed between these patients and younger patients, and other reported clinical experience has not identified differences in responses between the elderly and younger patients.
-
11 DESCRIPTION
Vedolizumab, an integrin receptor antagonist, is a humanized IgG1 monoclonal antibody produced in Chinese hamster ovary cells that binds to the human α4β7 integrin. ENTYVIO has an approximate molecular weight of 147 kilodaltons.
ENTYVIO (vedolizumab) for injection is supplied as a sterile, white to off-white, preservative-free, lyophilized cake for intravenous infusion. After reconstitution with 4.8 mL Sterile Water for Injection, USP, the final concentration is 60 mg/mL with a deliverable volume of 5 mL (300 mg) and the resulting pH is approximately 6.3.
Each single-dose vial contains 300 mg vedolizumab, L-arginine hydrochloride (131.7 mg), L-histidine (23 mg), L-histidine monohydrochloride (21.4 mg), polysorbate 80 (3 mg), and sucrose (500 mg).
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Vedolizumab is a humanized monoclonal antibody that specifically binds to the α4β7 integrin and blocks the interaction of α4β7 integrin with mucosal addressin cell adhesion molecule-1 (MAdCAM-1) and inhibits the migration of memory T-lymphocytes across the endothelium into inflamed gastrointestinal parenchymal tissue. Vedolizumab does not bind to or inhibit function of the α4β1 and αEβ7 integrins and does not antagonize the interaction of α4 integrins with vascular cell adhesion molecule-1 (VCAM-1).
The α4β7 integrin is expressed on the surface of a discrete subset of memory T-lymphocytes that preferentially migrate into the gastrointestinal tract. MAdCAM-1 is mainly expressed on gut endothelial cells and plays a critical role in the homing of T-lymphocytes to gut lymph tissue. The interaction of the α4β7 integrin with MAdCAM-1 has been implicated as an important contributor to the chronic inflammation that is a hallmark of ulcerative colitis and Crohn's disease.
12.2 Pharmacodynamics
In clinical trials with ENTYVIO at doses ranging from 0.2 to 10 mg/kg (which includes doses outside of the recommended dose), saturation of α4β7 receptors on subsets of circulating lymphocytes involved in gut-immune surveillance was observed.
In clinical trials with ENTYVIO at doses ranging from 0.2 to 10 mg/kg and 180 to 750 mg (which include doses outside of the recommended dose) in healthy subjects and in patients with ulcerative colitis or Crohn's disease, vedolizumab did not elevate neutrophils, basophils, eosinophils, B-helper and cytotoxic T-lymphocytes, total memory helper T-lymphocytes, monocytes or natural killer cells.
A reduction in gastrointestinal inflammation was observed in rectal biopsy specimens from Phase 2 ulcerative colitis patients exposed to ENTYVIO for four or six weeks compared to placebo control as assessed by histopathology.
In a study of 14 healthy subjects, ENTYVIO did not affect the CD4+ lymphocyte cell counts, CD8+ lymphocyte cell counts, or the CD4+:CD8+ ratios in the CSF [see Clinical Pharmacology (12.3)].
12.3 Pharmacokinetics
Similar pharmacokinetics were observed in ulcerative colitis and Crohn's disease patients administered 300 mg ENTYVIO as a 30 minute intravenous infusion on Weeks 0 and 2, followed by 300 mg ENTYVIO every eight weeks starting from Week 6 (Table 3).
Table 3. Mean ± SD Vedolizumab Concentrations in Patients* with Ulcerative Colitis and Crohn's Disease Patient Population Weeks 0 to 6 Weeks 6 to 52
ENTYVIO Every 8 WeeksTrough Serum Concentration at Week 6 (mcg/mL) Trough Serum Concentration at Week 46† (mcg/mL) - * Data from patients in UC Trials I and II and CD Trials I and III with pharmacokinetic data available; data from patients with anti-vedolizumab antibody were excluded.
- † Steady-state trough serum concentration.
Ulcerative Colitis 26.3 ± 12.9
(N=210)11.2 ± 7.2
(N=77)Crohn's Disease 27.4 ± 19.2
(N=198)13.0 ± 9.1
(N=72)The presence of persistent anti-vedolizumab antibody was observed to substantially reduce serum concentrations of vedolizumab, either to undetectable or negligible levels at Weeks 6 and 52 (n=8).
Vedolizumab clearance depends on both linear and nonlinear pathways; the nonlinear clearance decreases with increasing concentrations. Population pharmacokinetic analyses indicated that the linear clearance was approximately 0.157 L/day, the serum half-life was approximately 25 days at 300 mg dosage, and the distribution volume was approximately 5 L.
Vedolizumab was not detected in the cerebrospinal fluid (CSF) of 14 healthy subjects at five weeks after a single intravenous administration of 450 mg ENTYVIO (1.5 times the recommended dosage).
Special Populations
Population pharmacokinetic analysis showed that the severity of disease state, body weight, prior treatment with TNF blocker therapy, age (18 to 78 years), serum albumin, coadministered immunomodulators (including azathioprine, 6-mercaptopurine, methotrexate), and coadministered aminosalicylates did not have a clinically meaningful effect on the pharmacokinetics of ENTYVIO.
Pharmacokinetics of vedolizumab in patients with renal or hepatic insufficiency have not been studied.
- 13 NONCLINICAL TOXICOLOGY
-
14 CLINICAL STUDIES
14.1 Clinical Studies in Ulcerative Colitis
The safety and efficacy of ENTYVIO were evaluated in two randomized, double-blind, placebo-controlled trials (UC Trials I and II) in adult patients with moderately to severely active ulcerative colitis (UC) defined as Mayo score of six to 12 with endoscopy subscore of two or three. The Mayo score ranges from zero to 12 and has four subscales that are each scored from zero (normal) to three (most severe): stool frequency, rectal bleeding, findings on endoscopy, and physician global assessment. An endoscopy subscore of two is defined by marked erythema, lack of vascular pattern, friability, and erosions; an endoscopy subscore of three is defined by spontaneous bleeding and ulceration.
Enrolled patients in the United States (US) had over the previous five-year period an inadequate response or intolerance to immunomodulator therapy (i.e., azathioprine or 6-mercaptopurine) and/or an inadequate response, loss of response, or intolerance to a TNF blocker. Outside the US, prior treatment with corticosteroids was sufficient for entry if over the previous five-year period the patients were corticosteroid dependent (i.e., unable to successfully taper corticosteroids without a return of the symptoms of UC) or had an inadequate response or intolerance to corticosteroids.
Patients that had received natalizumab ever in the past, and patients that had received a TNF blocker in the past 60 days were excluded from enrollment. Concomitant use of natalizumab or a TNF blocker was not allowed.
UC Trial I
In UC Trial I, 374 patients were randomized in a double-blind fashion (3:2) to receive ENTYVIO 300 mg or placebo by intravenous infusion at Week 0 and Week 2. Efficacy assessments were at Week 6. Concomitant stable dosages of aminosalicylates, corticosteroids (prednisone dosage ≤30 mg/day or equivalent), and immunomodulators (azathioprine or 6-mercaptopurine) were permitted through Week 6.
At baseline, patients received corticosteroids (54%), immunomodulators (azathioprine or 6-mercaptopurine) (30%), and/or aminosalicylates (74%). Thirty-nine percent of patients had an inadequate response, loss of response, or intolerance to a TNF blocker therapy. Eighteen percent of patients had an inadequate response, inability to taper or intolerance to prior corticosteroid treatment only (i.e., had not received prior immunomodulators or TNF blockers). The median baseline Mayo score was nine in the ENTYVIO group and eight in the placebo group.
In UC Trial I, a greater percentage of patients treated with ENTYVIO compared to patients treated with placebo achieved clinical response at Week 6 (defined in Table 4). A greater percentage of patients treated with ENTYVIO compared to patients treated with placebo also achieved clinical remission at Week 6 (defined in Table 4). In addition, a greater percentage of patients treated with ENTYVIO had improvement of endoscopic appearance of the mucosa at Week 6 (defined in Table 4).
Table 4. Proportion of Patients Meeting Efficacy Endpoints at Week 6 (UC Trial I) Endpoint Placebo
N=149ENTYVIO
N=225p-value Treatment Difference and 95% CI - * Clinical response: reduction in complete Mayo score of ≥3 points and ≥30% from baseline with an accompanying decrease in rectal bleeding subscore of ≥1 point or absolute rectal bleeding subscore of ≤1 point.
- † Clinical remission: complete Mayo score of ≤2 points and no individual subscore >1 point.
- ‡ Improvement of endoscopic appearance of the mucosa: Mayo endoscopy subscore of 0 (normal or inactive disease) or 1 (erythema, decreased vascular pattern, mild friability).
Clinical response* at Week 6 26% 47% <0.001 22%
(12%, 32%)Clinical remission† at Week 6 5% 17% 0.001 12%
(5%, 18%)Improvement of endoscopic appearance of the mucosa‡ at Week 6 25% 41% 0.001 16%
(6%, 26%)UC Trial II
In order to be randomized to treatment in UC Trial II, patients had to have received ENTYVIO and be in clinical response at Week 6. Patients could have come from either UC Trial I or from a group who received ENTYVIO open-label.
In UC Trial II, 373 patients were randomized in a double-blind fashion (1:1:1) to one of the following regimens beginning at Week 6: ENTYVIO 300 mg every eight weeks, ENTYVIO 300 mg every four weeks or placebo every four weeks. Efficacy assessments were at Week 52. Concomitant aminosalicylates and corticosteroids were permitted through Week 52. Concomitant immunomodulators (azathioprine or 6-mercaptopurine) were permitted outside the US but were not permitted beyond Week 6 in the US.
At Week 6, patients were receiving corticosteroids (61%), immunomodulators (azathioprine or 6-mercaptopurine) (32%) and aminosalicylates (75%). Thirty-two percent of patients had an inadequate response, loss of response or intolerance to a TNF blocker therapy. At Week 6, the median Mayo score was eight in the ENTYVIO every eight week group, the ENTYVIO every four week group, and the placebo group. Patients who had achieved clinical response at Week 6 and were receiving corticosteroids were required to begin a corticosteroid-tapering regimen at Week 6.
In UC Trial II, a greater percentage of patients in groups treated with ENTYVIO as compared to placebo achieved clinical remission at Week 52, and maintained clinical response (clinical response at both Weeks 6 and 52) (Table 5). In addition, a greater percentage of patients in groups treated with ENTYVIO as compared to placebo were in clinical remission at both Weeks 6 and 52, and had improvement of endoscopic appearance of the mucosa at Week 52 (Table 5). In the subgroup of patients who achieved clinical response at Week 6 and were receiving corticosteroid medication at baseline, a greater proportion of patients in groups treated with ENTYVIO as compared to placebo discontinued corticosteroids and were in clinical remission at Week 52 (Table 5).
The ENTYVIO every four week dosing regimen did not demonstrate additional clinical benefit over the every eight dosing week regimen. The every four week dosing regimen is not the recommended dosing regimen [see Dosage and Administration (2.3)].
Table 5. Proportion of Patients Meeting Efficacy Endpoints at Week 52* (UC Trial II) Endpoint Placebo†
N=126ENTYVIO Every 8 Weeks
N=122p-value Treatment Difference and 95% CI - * Patients must have achieved clinical response at Week 6 to continue into UC Trial II. This group includes patients that were not in clinical remission at Week 6.
- † The placebo group includes those patients who received ENTYVIO at Week 0 and Week 2 and were randomized to receive placebo from Week 6 through Week 52.
- ‡ Improvement of endoscopic appearance of the mucosa: Mayo endoscopy subscore of 0 (normal or inactive disease) or 1 (erythema, decreased vascular pattern, mild friability) at Week 52.
- § Corticosteroid-free clinical remission: Assessed in the subgroup of patients who were receiving corticosteroids at baseline and who were in clinical response at Week 6 (n=72 for placebo and n=70 for ENTYVIO every eight weeks). Corticosteroid-free clinical remission was defined as the proportion of patients in this subgroup that discontinued corticosteroids by Week 52 and were in clinical remission at Week 52.
Clinical remission at Week 52 16% 42% <0.001 26%
(15%, 37%)Clinical response at both Weeks 6 and 52 24% 57% <0.001 33%
(21%, 45%)Improvement of endoscopic appearance of the mucosa‡ at Week 52 20% 52% <0.001 32%
(20%, 44%)Clinical remission at both Weeks 6 and 52 9% 21% 0.008 12%
(3%, 21%)Corticosteroid-free clinical remission§ 14%§ 31%§ 0.012 18%
(4%, 31%)14.2 Clinical Studies in Crohn's Disease
The safety and efficacy of ENTYVIO were evaluated in three randomized, double-blind, placebo-controlled clinical trials (CD Trials I, II, and III) in adult patients with moderately to severely active Crohn's disease (CD) (Crohn's Disease Activity Index [CDAI] score of 220 to 450).1
Enrolled patients in the United States (US) had over the previous five-year period an inadequate response or intolerance to immunomodulator therapy (i.e., azathioprine, 6-mercaptopurine, or methotrexate) and/or an inadequate response, loss of response, or intolerance to one or more TNF blockers. Outside the US, prior treatment with corticosteroids was sufficient for entry if over the previous five-year period the patients were corticosteroid dependent (i.e., unable to successfully taper corticosteroids without a return of the symptoms of CD) or had an inadequate response or intolerance to corticosteroids.
Patients that had received natalizumab ever in the past, and patients that had received a TNF blocker in the past 30 to 60 days were excluded from enrollment. Concomitant use of natalizumab or a TNF blocker was not allowed.
CD Trial I
In CD Trial I, 368 patients were randomized in a double-blind fashion (3:2) to receive ENTYVIO 300 mg or placebo by intravenous infusion at Week 0 and Week 2. Efficacy assessments were at Week 6. Concomitant stable dosages of aminosalicylates, corticosteroids (prednisone dosage ≤30 mg/day or equivalent), and immunomodulators (azathioprine, 6-mercaptopurine or methotrexate) were permitted through Week 6.
At baseline, patients were receiving corticosteroids (49%), immunomodulators (azathioprine, 6-mercaptopurine, or methotrexate) (35%), and/or aminosalicylates (46%). Forty-eight percent of the patients had an inadequate response, loss of response, or intolerance to a TNF blocker therapy. Seventeen percent of patients had inadequate response, inability to taper, or intolerance to prior corticosteroid treatment only (i.e., had not received prior immunomodulators or TNF blockers). The median baseline CDAI score was 324 in the ENTYVIO group and 319 in the placebo group.
In CD Trial I, a statistically significantly higher percentage of patients treated with ENTYVIO achieved clinical remission (defined as CDAI ≤150) as compared to placebo at Week 6 (Table 6). The difference in the percentage of patients who demonstrated clinical response (defined as a ≥100-point decrease in CDAI score from baseline), was however, not statistically significant at Week 6.
CD Trial II
Compared to CD Trial I, CD Trial II enrolled a higher number of patients who had over the previous five-year period had an inadequate response, loss of response, or intolerance to one or more TNF blockers (76%); this was the primary analysis population. In CD Trial II, 416 patients were randomized in a double-blind fashion (1:1) to receive either ENTYVIO 300 mg or placebo at Weeks 0, 2 and 6. Efficacy assessments were at Weeks 6 and 10. Concomitant aminosalicylates, corticosteroids, and immunomodulators (azathioprine, 6-mercaptopurine, or methotrexate) were permitted through Week 10.
At baseline, patients were receiving corticosteroids (54%), immunomodulators (azathioprine, 6-mercaptopurine, or methotrexate) (34%), and aminosalicylates (31%). The median baseline CDAI score was 317 in the ENTYVIO group and 301 in the placebo group.
For the primary endpoint (clinical remission at Week 6), treatment with ENTYVIO did not result in statistically significant improvement over placebo (Table 6). Secondary endpoints including assessments at Week 10 were not tested because the primary endpoint was not statistically significant.
Table 6. Proportion of Patients in Clinical Remission at Week 6 (CD Trials I and II) Placebo ENTYVIO p-value Treatment Difference and 95% CI - * Clinical Remission: CDAI ≤150
- † Adjusted p-value for multiple comparisons of two primary endpoints
- ‡ The primary analysis population for CD Trial II was patients that had an inadequate response, loss of response, or intolerance to one or more TNF blockers (76% of the overall population)
- § NS: Not significant (Secondary endpoints including assessments at Week 10 were not tested because the CD Trial II primary endpoint was not statistically significant)
CD Trial I:
Clinical Remission* at Week 67%
(10/148)15%
(32/220)0.041† 8%
(1%, 14%)CD Trial II‡:
Clinical Remission* at Week 612%
(19/157)15%
(24/158)NS§ 3%
(-5%, 11%)CD Trial III
In order to be randomized to treatment in CD Trial III, patients had to have received ENTYVIO and be in clinical response (defined as a ≥70-point decrease in CDAI score from baseline) at Week 6. Patients could have come from either CD Trial I or from a group who received ENTYVIO open-label.
In CD Trial III, 461 patients were randomized in a double-blind fashion (1:1:1) to one of the following regimens beginning at Week 6: ENTYVIO 300 mg every eight weeks, ENTYVIO 300 mg every four weeks or placebo every four weeks. Efficacy assessments were at Week 52. Concomitant aminosalicylates and corticosteroids were permitted through Week 52. Concomitant immunomodulators (azathioprine, 6-mercaptopurine, or methotrexate) were permitted outside the US but were not permitted beyond Week 6 in the US.
At Week 6, patients were receiving corticosteroids (59%), immunomodulators (azathioprine, 6-mercaptopurine, or methotrexate) (31%), and aminosalicylates (41%). Fifty-one percent of patients had an inadequate response, loss of response, or intolerance to a TNF blocker therapy. At Week 6, the median CDAI score was 322 in the ENTYVIO every eight week group, 316 in the ENTYVIO every four week group, and 315 in the placebo group. Patients who had achieved clinical response (≥70 decrease in CDAI score from baseline) at Week 6 and were receiving corticosteroids were required to begin a corticosteroid-tapering regimen at Week 6.
In CD Trial III, a greater percentage of patients in groups treated with ENTYVIO as compared to placebo were in clinical remission (defined as CDAI score ≤150) at Week 52. A greater percentage of patients in groups treated with ENTYVIO as compared to placebo had a clinical response (defined as ≥100 decrease in CDAI score from baseline) at Week 52 (Table 7). In the subgroup of patients who were receiving corticosteroids at baseline and who were in clinical response at Week 6 (defined as ≥70 decrease in CDAI score from baseline), a greater proportion of patients in groups treated with ENTYVIO as compared to placebo discontinued corticosteroids by Week 52 and were in clinical remission at Week 52 (Table 7).
The ENTYVIO every four week dosing regimen did not demonstrate additional clinical benefit over the every eight dosing week regimen. The every four week dosing regimen is not the recommended dosing regimen [see Dosage and Administration (2.3)].
Table 7. Proportion of Patients Meeting Efficacy Endpoints at Week 52* (CD Trial III) Placebo†
N=153ENTYVIO Every 8 Weeks
N=154p-value Treatment Difference and 95% CI - * This group includes patients that were not in clinical remission at Week 6. Patients must have achieved clinical response (defined as ≥70 decrease in CDAI from baseline) at Week 6 to continue into CD Trial III.
- † The placebo group includes those patients who received ENTYVIO at Week 0 and Week 2, and were randomized to receive placebo from Week 6 through Week 52
- ‡ Clinical remission: CDAI ≤150
- § Clinical response: ≥100 decrease in CDAI from baseline
- ¶ Corticosteroid-free clinical remission: Assessed in the subgroup of patients who were receiving corticosteroids at baseline and who were in clinical response (defined as ≥70 decrease in CDAI from baseline) at Week 6 (n=82 for placebo and n=82 for ENTYVIO every eight weeks). Corticosteroid-free clinical remission was defined as the proportion of patients in this subgroup that discontinued corticosteroids by Week 52 and were in clinical remission at Week 52.
Clinical remission‡ at Week 52 22% 39% 0.001 17%
(7%, 28%)Clinical response§ at Week 52 30% 44% 0.013 13%
(3%, 24%)Corticosteroid-free clinical remission¶ 16%¶ 32%¶ 0.015 16%
(3%, 29%) - 15 REFERENCES
-
16 HOW SUPPLIED/STORAGE AND HANDLING
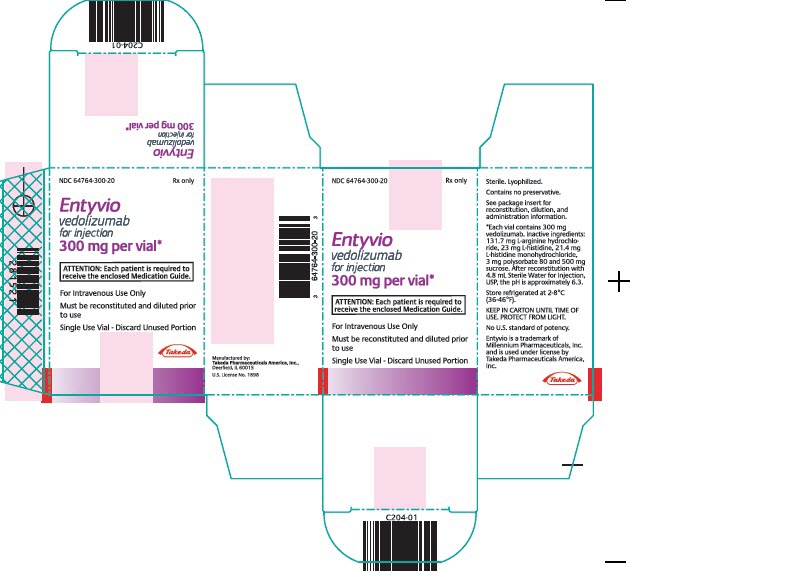
ENTYVIO (vedolizumab) for injection is supplied in sterile 20 mL single-dose glass vials, containing 300 mg of vedolizumab as a white to off-white lyophilized cake.
NDC: 64764-300-20 300 mg single-dose vial in individual carton -
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Infusion-Related and Hypersensitivity Reactions
Instruct patients to report immediately if they experience symptoms consistent with a hypersensitivity reaction during or following an infusion of ENTYVIO [see Warnings and Precautions (5.1)].
Infections
Inform patients that they may be more likely to develop infections when taking ENTYVIO. Instruct patients to tell their healthcare provider if they develop any signs or symptoms of an infection [see Warnings and Precautions (5.2)].
Progressive Multifocal Leukoencephalopathy
Inform patients that progressive multifocal leukoencephalopathy (PML) has occurred in patients who received some integrin receptor antagonist and systemic immunosuppressant products. Instruct patients to report if they experience any new onset or worsening of neurological signs and symptoms immediately, as these could be indicative of PML [see Warnings and Precautions (5.3)].
Liver Injury
Inform patients that elevated transaminase levels with or without elevated bilirubin has occurred in patients who received ENTYVIO. Instruct patients to report promptly any symptoms that may indicate liver injury, including fatigue, anorexia, right upper abdominal discomfort, dark urine or jaundice [see Warnings and Precautions (5.4)].
Pregnancy
Inform patients that there is a pregnancy registry to monitor pregnancy outcomes of women who are pregnant or become pregnant while exposed to ENTYVIO [see Use in Specific Populations (8.1)].
- SPL UNCLASSIFIED SECTION
-
MEDICATION GUIDE
This Medication Guide has been approved by the U.S. Food and Drug Administration VMB245 R4 Revised: March 2020 MEDICATION GUIDE
ENTYVIO (en ti' vee oh)
(vedolizumab)
for injection, for intravenous useWhat is the most important information I should know about ENTYVIO?
ENTYVIO may cause serious side effects, including:- Infusion-related and serious allergic reactions. These reactions can happen while you are receiving ENTYVIO or several hours after treatment. You may need treatment if you have an allergic reaction. Tell your healthcare provider or get medical help right away if you get any of these symptoms during or after an infusion of ENTYVIO: rash, itching, swelling of your lips, tongue throat or face, shortness of breath or trouble breathing, wheezing, dizziness, feeling hot, or palpitations (feel like your heart is racing).
- Infections. ENTYVIO may increase your risk of getting a serious infection. Before receiving ENTYVIO and during treatment with ENTYVIO, tell your healthcare provider if you think you have an infection or have symptoms of an infection such as fever, chills, muscle aches, cough, shortness of breath, runny nose, sore throat, red or painful skin or sores on your body, tiredness, or pain during urination.
- Progressive Multifocal Leukoencephalopathy (PML). People with weakened immune systems can get progressive multifocal leukoencephalopathy (PML) (a rare, serious brain infection caused by a virus). Although unlikely while receiving ENTYVIO, a risk of PML cannot be ruled out. PML can result in death or severe disability. There is no known treatment, prevention, or cure for PML. Tell your healthcare provider right away if you have any of the following symptoms: confusion or problems thinking, loss of balance, change in the way you walk or talk, decreased strength or weakness on one side of the body, blurred vision, or loss of vision.
- Liver Problems. Liver problems can happen in people who receive ENTYVIO. Tell your healthcare provider right away if you have any of the following symptoms: tiredness, loss of appetite, pain on the right side of your stomach (abdomen), dark urine, or yellowing of the skin and eyes (jaundice).
What is ENTYVIO?
ENTYVIO is a prescription medicine used in adults for the treatment of:- moderately to severely active ulcerative colitis.
- moderately to severely active Crohn's disease.
Who should not receive ENTYVIO?
Do not receive ENTYVIO if you have had an allergic reaction to ENTYVIO or any of the ingredients in ENTYVIO. See the end of this Medication Guide for a complete list of ingredients in ENTYVIO.Before receiving ENTYVIO, tell your healthcare provider about all of your medical conditions, including if you: - have an infection, think you may have an infection or have infections that keep coming back (see "What is the most important information I should know about ENTYVIO?").
- have liver problems.
- have tuberculosis (TB) or have been in close contact with someone with TB.
- have recently received or are scheduled to receive a vaccine. Talk to your healthcare provider about bringing your vaccines up-to-date before starting treatment with ENTYVIO.
- are pregnant or plan to become pregnant. It is not known if ENTYVIO will harm your unborn baby. Tell your healthcare provider right away if you become pregnant while receiving ENTYVIO.
- Pregnancy Registry: There is a pregnancy registry for women who use ENTYVIO during pregnancy. The purpose of this registry is to collect information about the health of you and your baby. Talk with your healthcare provider about how you can take part in this registry or you may contact the registry at 1-877-825-3327 to enroll.
- are breastfeeding or plan to breastfeed. ENTYVIO can pass into your breast milk. Talk to your healthcare provider about the best way to feed your baby if you take ENTYVIO.
How will I receive ENTYVIO? - ENTYVIO is given through a needle placed in a vein (intravenous infusion) in your arm.
- ENTYVIO is given to you over a period of about 30 minutes.
- Your healthcare provider will monitor you during and after the ENTYVIO infusion for side effects to see if you have a reaction to the treatment.
What are the possible side effects of ENTYVIO?
ENTYVIO may cause serious side effects, see "What is the most important information I should know about ENTYVIO?".
The most common side effects of ENTYVIO include: common cold, headache, joint pain, nausea, fever, infections of the nose and throat, tiredness, cough, bronchitis, flu, back pain, rash, itching, sinus infection, throat pain, and pain in extremities.
These are not all of the possible side effects of ENTYVIO.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.General information about ENTYVIO
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. You can ask your pharmacist or healthcare provider for information about ENTYVIO that is written for health professionals.What are the ingredients in ENTYVIO?
Active ingredient: vedolizumab
Inactive ingredients: L-arginine hydrochloride, L-histidine, L-histidine monohydrochloride, polysorbate 80 and sucrose
Manufactured by: Takeda Pharmaceuticals America, Inc. Deerfield, IL 60015
U.S. License No. 1898
ENTYVIO is a trademark of Millennium Pharmaceuticals Inc. and is used under license by Takeda Pharmaceuticals America, Inc.
All other trademark names are the property of their respective owners.
©2014 – 2020 Takeda Pharmaceuticals America, Inc.
For more information, go to www.ENTYVIO.com or call 1-877-TAKEDA-7 (1-877-825-3327). - PRINCIPAL DISPLAY PANEL – 300 MG NDC: 64764-300-20Rx only Entyvio vedolizumabfor injection300 mg per vial*ATTENTION: Each patient is required to receive the enclosed Medication Guide.For Intravenous Use OnlyMust be reconstituted and diluted prior to useSingle Use Vial – Discard Unused Portion
-
INGREDIENTS AND APPEARANCE
ENTYVIO
vedolizumab injection, powder, lyophilized, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 64764-300 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength VEDOLIZUMAB (UNII: 9RV78Q2002) (VEDOLIZUMAB - UNII:9RV78Q2002) VEDOLIZUMAB 300 mg in 5 mL Inactive Ingredients Ingredient Name Strength HISTIDINE (UNII: 4QD397987E) 23 mg in 5 mL HISTIDINE MONOHYDROCHLORIDE (UNII: 1D5Q932XM6) 21.4 mg in 5 mL ARGININE HYDROCHLORIDE (UNII: F7LTH1E20Y) 131.7 mg in 5 mL SUCROSE (UNII: C151H8M554) 500 mg in 5 mL POLYSORBATE 80 (UNII: 6OZP39ZG8H) 3 mg in 5 mL Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 64764-300-20 1 in 1 CARTON 05/20/2014 1 5 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125476 05/20/2014 Labeler - Takeda Pharmaceuticals America, Inc. (830134016)
Trademark Results [Entyvio]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 ENTYVIO 85438158 4580498 Live/Registered |
Millennium Pharmaceuticals, Inc. 2011-10-03 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.