HESPAN- hetastarch in sodium chloride injection, solution
HESPAN by
Drug Labeling and Warnings
HESPAN by is a Prescription medication manufactured, distributed, or labeled by B. Braun Medical Inc.. Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use HESPAN® (6% HETASTARCH IN 0.9% SODIUM CHLORIDE INJECTION) safely and effectively. See full prescribing information for HESPAN® (6% HETASTARCH IN 0.9% SODIUM CHLORIDE INJECTION).
HESPAN® (6% HETASTARCH IN 0.9% SODIUM CHLORIDE INJECTION) IN EXCEL® CONTAINER, for intravenous use
Initial U.S. Approval: 1991WARNING: MORTALITY
RENAL REPLACEMENT THERAPYSee full prescribing information for complete boxed warning.
-
In critically ill adult patients, including patients with sepsis, use of hydroxyethyl starch (HES) products, including HESPAN®, increases risk of
- Mortality
- Renal replacement therapy
- Do not use HES products, including HESPAN®, in critically ill adult patients including patients with sepsis
INDICATIONS AND USAGE
DOSAGE AND ADMINISTRATION
For intravenous use only.
Recommended Dosage Dose Adults (2.1) 500 to 1000 mL Leukapheresis (2.2) 250 to 700 mL of HESPAN® (6% hetastarch in 0.9% sodium chloride injection) with citrate anticoagulant is added to the input line of the centrifugation apparatus. DOSAGE FORMS AND STRENGTHS
- 30 g hetastarch in 500 mL 0.9% sodium chloride injection. (3)
CONTRAINDICATIONS
- Do not use hydroxyethyl starch (HES) products, including HESPAN®, in critically ill adult patients, including patients with sepsis, due to increased risk of mortality and renal replacement therapy (RRT). (4)
- Do not use HES products, including HESPAN®, in patients with severe liver disease (4)
- Do not use HES products, including HESPAN®, in patients with known hypersensitivity to hydroxyethyl starch (4)
- Do not use HES products, including HESPAN®, in clinical conditions where volume overload is a potential problem. (4)
- Do not use HES products, including HESPAN®, in patients with pre-existing coagulation or bleeding disorders (4)
WARNINGS AND PRECAUTIONS
- Avoid use in patients with pre-existing renal dysfunction (5.1)
- Discontinue use of HESPAN® at the first sign of renal injury (5.1)
- Continue to monitor renal function in hospitalized patients for at least 90 days as use of RRT has been reported up to 90 days after administration of HES products, including HESPAN® (5.1)
- HESPAN® is not recommended for use as a cardiac bypass pump prime, while the patient is on cardiopulmonary bypass, or in the immediate period after the pump has been discontinued because of the risk of increasing coagulation abnormalities and bleeding in patients whose coagulation status is already impaired. Discontinue use of HESPAN® at first sign of coagulopathy (5.2)
-
Monitor liver function in patients receiving HES products, including HESPAN® (5.2)
ADVERSE REACTIONS
- The serious adverse events reported in clinical trials are Increased mortality and renal replacement therapy in critically ill patients (6.1)
- Most common adverse reactions are hypersensitivity, coagulopathy, hemodilution, circulatory overload and metabolic acidosis. (6.2)
To report SUSPECTED ADVERSE REACTIONS, contact B. Braun Medical Inc. at 1-800-227-2862 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
Revised: 3/2015
-
In critically ill adult patients, including patients with sepsis, use of hydroxyethyl starch (HES) products, including HESPAN®, increases risk of
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Adults
2.2 Leukapheresis
2.3 Direction for use for HESPAN®
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Renal Dysfunction
5.2 Coagulopathy
5.3 Hypersensitivity Reactions
5.4 Circulatory Overload
5.5 Liver Function Test
5.6 Drug/Laboratory Test Interactions
6 ADVERSE REACTIONS
6.1 Clinical Trial Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy Category C
8.3 Nursing Mothers
8.4 Pediatric Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
15 REFERENCES
16 HOW SUPPLIED/STORAGE AND HANDLING
- * Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: MORTALITY
RENAL REPLACEMENT THERAPY-
In critically ill adult patients, including patients with sepsis, use of hydroxyethyl starch (HES) products, including HESPAN®, increases risk of
- Mortality
- Renal replacement therapy
- Do not use HES products, including HESPAN®, in critically ill adult patients including patients with sepsis
-
In critically ill adult patients, including patients with sepsis, use of hydroxyethyl starch (HES) products, including HESPAN®, increases risk of
-
1 INDICATIONS AND USAGE
HESPAN® is indicated in the treatment of hypovolemia when plasma volume expansion is desired. It is not a substitute for blood or plasma.
The adjunctive use of HESPAN® in leukapheresis has also been shown to be safe and efficacious in improving the harvesting and increasing the yield of granulocytes by centrifugal means.
-
2 DOSAGE AND ADMINISTRATION
Dosage for Acute Use in Plasma Volume Expansion
HESPAN® is administered by intravenous infusion only. Total dosage and rate of infusion depend upon the amount of blood or plasma lost and the resultant hemoconcentration.
2.1 Adults
The amount usually administered is 500 to 1000 mL. Doses of more than 1500 mL per day for the typical 70 kg patient (approximately 20 mL per kg of body weight) are usually not required. Higher doses have been reported in postoperative and trauma patients where severe blood loss has occurred [see Warnings and Precautions (5)].
2.2 Leukapheresis
250 to 700 mL of HESPAN® (6% hetastarch in 0.9% sodium chloride injection) with citrate anticoagulant is administered by aseptic addition to the input line of the centrifugation apparatus at a ratio of 1:8 to 1:13 to venous whole blood. The HESPAN® and citrate should be thoroughly mixed to assure effective anticoagulation of blood as it flows through the leukapheresis machine.
2.3 Direction for use for HESPAN®
- Do not use plastic container in series connection. If administration is controlled by a pumping device, care must be taken to discontinue pumping action before the container runs dry or air embolism may result. If administration is not controlled by a pumping device, refrain from applying excessive pressure (>300mmHg) causing distortion to the container such as wringing or twisting. Such handling could result in breakage of the container.
- Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration whenever solution and container permit. Use only if solution is clear and container and seals are intact.
- Intended for intravenous administration using sterile equipment. It is recommended that intravenous administration apparatus be replaced at least once every 24 hours.
- Withdraw or expel all air from the bag through the medication port prior to infusion if administration is by pressure infusion.
-
For single use only. Discard unused portion.
CAUTION: Before administering to the patient, review these directions:
Visual Inspection
- Do not remove the plastic infusion container from its overwrap until immediately before use.
- Inspect each container. Read the label. Ensure solution is the one ordered and is within the expiration date.
- Invert container and carefully inspect the solution in good light for cloudiness, haze, or particulate matter.
- Any container which is suspect should not be used.
To Open
- Tear overwrap down at notch and remove solution container.
- Check for minute leaks by squeezing solution container firmly.
- If any leaks are found, discard solution as sterility may be impaired.
Preparation for Administration
- Remove plastic protector from sterile set port at bottom of container.
- Attach administration set. Refer to complete directions accompanying set.
When stored at room temperature, HESPAN® admixtures of 500-560 mL with citrate concentrations up to 2.5% were compatible for 24 hours. The safety and compatibility of additives other than citrate have not been established.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
- Do not use hydroxyethyl starch (HES) products, including HESPAN®, in critically ill adult patients, including patients with sepsis, due to increased risk of mortality and renal replacement therapy (RRT).
- Do not use HES products, including HESPAN®, in patients with severe liver disease
- Do not use HES products, including HESPAN®, in patients with known hypersensitivity to hydroxyethyl starch
- Do not use HES products, including HESPAN®, in clinical conditions where volume overload is a potential problem (such as, congestive heart failure or renal disease with anuria or oliguria not related to hypovolemia).
- Do not use HES products, including HESPAN®, in patients with pre-existing coagulation or bleeding disorders
-
5 WARNINGS AND PRECAUTIONS
5.1 Renal Dysfunction
- Avoid use in patients with pre-existing renal dysfunction
- Discontinue use of HESPAN® at the first sign of renal injury
- Continue to monitor renal function in hospitalized patients for at least 90 days as use of RRT has been reported up to 90 days after administration of HES products, including HESPAN®
5.2 Coagulopathy
- HESPAN® is not recommended for use as a cardiac bypass pump prime, while the patient is on cardiopulmonary bypass, or in the immediate period after the pump has been discontinued because of the risk of increasing coagulation abnormalities and bleeding in patients whose coagulation status is already impaired. Discontinue use of HESPAN® at first sign of coagulopathy1-2
HESPAN® has not been adequately evaluated to establish its safety in uses over extended periods other than leukapheresis. HESPAN® has been associated with coagulation abnormalities in conjunction with an acquired, reversible von Willebrand’s-like syndrome and/or Factor Vlll deficiency when used over a period of days. Replacement therapy should be considered if a severe Factor Vlll deficiency is identified. If a coagulopathy develops, it may take several days to resolve. Certain conditions may affect the safe use of HESPAN® on a chronic basis. For example, in patients with subarachnoid hemorrhage where HESPAN® is used repeatedly over a period of days for the prevention of cerebral vasospasm, significant clinical bleeding may occur. Intracranial bleeding resulting in death has been reported.3
Slight declines in platelet counts and hemoglobin levels have been observed in donors undergoing repeated leukapheresis procedures using HESPAN® due to the volume expanding effects of hetastarch and to the collection of platelets and erythrocytes. Hemoglobin levels usually return to normal within 24 hours. Hemodilution by HESPAN® may also result in 24 hour declines of total protein, albumin, calcium, and fibrinogen levels. Regular and frequent clinical evaluation and complete blood counts (CBC) are necessary for proper monitoring of HESPAN® use during leukapheresis. If the frequency of leukapheresis is to exceed the guidelines for whole blood donation, you may wish to consider the following additional tests: total leukocyte and platelet counts, leukocyte differential count, hemoglobin and hematocrit, prothrombin time (PT), and partial thromboplastin time (PTT).
5.3 Hypersensitivity Reactions
Life threatening anaphylactic/anaphylactoid reactions including death have been rarely reported with HESPAN®. Patients may develop hypersensitivity reaction to corn starch from which this product is made. If a hypersensitivity reaction occurs, administration of the drug should be discontinued immediately and the appropriate treatment and supportive measures should be undertaken until symptoms have resolved.
5.4 Circulatory Overload
HESPAN® has not been adequately evaluated to establish its safety in situations other than treatment of hypovolemia in elective surgery.
Large volumes of HESPAN® may transiently alter the coagulation mechanism due to hemodilution and a direct inhibitory action on Factor Vlll. Administration of volumes of HESPAN® that are greater than 25% of the blood volume in less than 24 hours may cause significant hemodilution reflected by lower hematocrit and plasma protein values. Administration of packed red cells, platelets, or fresh frozen plasma should be considered if clinically indicated.
When using HESPAN® for plasma volume expansion, caution should be taken to avoid excessive hemodilution and circulatory overload especially in those patients at risk for developing congestive heart failure and pulmonary edema. HESPAN® is primarily excreted via the kidneys so caution should be exercised in patients who have impaired renal function. Although the risk of circulatory overload is largely dependent on the clinical circumstances, use of doses higher than 20 mL/kg/24h will increase the risk significantly. Increased risk of coagulation abnormalities and bleeding is also associated with higher doses. Monitor patients' vital signs and hemoglobin, hematocrit, platelet count, prothrombin time and partial thromboplastin time.
5.5 Liver Function Test
- Monitor liver function in patients receiving HES products, including HESPAN®
5.6 Drug/Laboratory Test Interactions
Bilirubin Levels
Indirect bilirubin levels of 8.3 mg/L (normal 0.0-7.0 mg/L) have been reported in 2 out of 20 normal subjects who received multiple infusions of HESPAN® (6% hetastarch in 0.9% sodium chloride injection). Total bilirubin was within normal limits at all times; indirect bilirubin returned to normal by 96 hours following the final infusion. The significance, if any, of these elevations is not known; however, caution should be observed before administering HESPAN® to patients with a history of liver disease.
Serum Amylase Levels
Elevated serum amylase levels may be observed temporarily following administration of HESPAN® although no association with pancreatitis has been demonstrated. Serum amylase levels cannot be used to assess or to evaluate for pancreatitis for 3-5 days after administration of HESPAN®. Elevated serum amylase levels persist for longer periods of time in patients with renal impairment. Hetastarch has not been shown to increase serum lipase.
-
6 ADVERSE REACTIONS
The serious adverse events reported in clinical trials are increased mortality and renal replacement therapy renal in critically ill patients.
Most common adverse reactions are hypersensitivity, coagulopathy, hemodilution, circulatory overload and metabolic acidosis.
6.1 Clinical Trial Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Three randomized controlled trials (RCTs) followed critically ill adult patients treated with different HES products for 90 days.
One trial (N=804) in severe sepsis patients using HES product (not approved in the U.S.) reported increased mortality (relative risk, 1.17; 95% CI, 1.01 to 1.36; p=0.03) and RRT (relative risk, 1.35; 95% CI, 1.01 to 1.80; p=0.04) in the HES treatment arm.4
Another trial (N=196) using different HES in severe sepsis patients reported no difference in mortality (relative risk,1.20; 95% CI, 0.83 to 1.74; p=0.33) and a trend for RRT (relative risk, 1.83; 95% CI, 0.93 to 3.59; p=0.06) in HES patients.5
A third trial (N=7000) using different HES in a heterogeneous patient population consisting of critically ill adult patients admitted to the ICU reported no difference in mortality (relative risk, 1.06; 95% CI, 0.96 to 1.18; p=0.26) but increased use of RRT (relative risk, 1.21; 95% CI, 1.00 to 1.45; p=0.04) in HES patients.6
6.2 Postmarketing Experience
Because adverse reactions are reported voluntarily post-approval from a population of uncertain size, it is not always possible to reliably estimate the frequency of these reactions or establish a causal relationship to product exposure.
The following adverse reactions have been identified and reported during the post-approval use of HES products:
Hypersensitivity reactions
including death, life-threatening anaphylactic/anaphylactoid reactions, cardiac arrest, ventricular fibrillation, severe hypotension, non-cardiac pulmonary edema, laryngeal edema, bronchospasm, angioedema, wheezing, restlessness, tachypnea, stridor, fever, chest pain, bradycardia, tachycardia, shortness of breath, chills, urticaria, pruritus, facial and periorbital edema, coughing, sneezing, flushing, erythema multiforme, and rash [see Warnings and Precautions (5.3)].
Cardiovascular reactions
including circulatory overload, congestive heart failure, and pulmonary edema [see Warnings and Precautions (5.4)].
Hematologic reactions
including intracranial bleeding, bleeding and/or anemia due to hemodilution [see Warnings and Precautions (5.4)] and/or Factor Vlll deficiency, acquired von Willebrand’s-like syndrome, and coagulopathy including rare cases of disseminated intravascular coagulopathy and hemolysis.
Other reactions
including vomiting, peripheral edema of the lower extremities, submaxillary and parotid glandular enlargement, mild influenza-like symptoms, headaches, and muscle pains. Hydroxyethyl starch-associated pruritus has been reported in some patients with deposits of hydroxyethyl starch in peripheral nerves.
- 7 DRUG INTERACTIONS
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy Category C
Hetastarch has been shown to have an embryocidal effect on New Zealand rabbits when given intravenously over the entire organogenesis period in a daily dose 1/2 times the maximum recommended therapeutic human dose (1500 mL) and on BD rats when given intraperitoneally, from the 16th to the 21st day of pregnancy, in a daily dose 2.3 times the maximum recommended therapeutic human dose. When hetastarch was administered to New Zealand rabbits, BD rats, and swiss mice with intravenous daily doses of 2 times, 1/3 times, and 1 times the maximum recommended therapeutic human dose respectively over several days during the period of gestation, no evidence of teratogenicity was evident.
There are no adequate and well-controlled studies in pregnant women. HESPAN® should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
-
11 DESCRIPTION
HESPAN® (6% hetastarch in 0.9% sodium chloride injection) is a sterile, nonpyrogenic solution for intravenous administration.
Each 100 mL contains:
Hetastarch.................................................. 6 g
Sodium Chloride, USP............................... 0.9 g
Water for Injection, USP............................. qs
pH adjusted with Sodium Hydroxide, NF if necessary
Concentration of Electrolytes (mEq/L): Sodium 154, Chloride 154
pH: approximately 5.9 with negligible buffering capacity
Calc. Osmolarity: approximately 309 mOsM
Hetastarch is a synthetic colloid derived from a waxy starch composed almost entirely of amylopectin. Hydroxyethyl ether groups are introduced into the glucose units of the starch, and the resultant material is hydrolyzed to yield a product with a molecular weight suitable for use as a plasma volume expander and erythrocyte sedimenting agent. The molar substitution is approximately 0.75 which means hetastarch has an average of approximately 75 hydroxyethyl groups for every 100 glucose units. The weight average molecular weight is approximately 600,000 with a range of 450,000 to 800,000 and with at least 80% of the polymers falling within the range of 20,000 to 2,600,000. Hydroxyethyl groups are attached by ether linkage primarily at C-2 of the glucose unit and to a lesser extent at C-3 and C-6. The polymer resembles glycogen, and the polymerized D-glucose units are joined primarily by α-1,4 linkages with occasional α-1,6 branching linkages.
The chemical name for hetastarch is hydroxyethyl starch.
The structural formula is as follows:
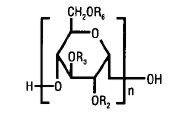
Amylopectin derivative in which R2 and R3 are H or CH2CH2OH and R6 is H, CH2CH2OH, or a branching point in the starch polymer connected through an α-1,6 link to additional D-glucopyranosyl units.
HESPAN® is a clear, pale yellow to amber solution. Exposure to prolonged adverse storage conditions may result in a change to a turbid deep brown or the formation of a crystalline precipitate. Do not use the solution if these conditions are evident.
Not made with natural rubber latex, PVC or DEHP.
The plastic container is made from a multi-layered film specifically developed for parenteral drugs. It contains no plasticizers and exhibits virtually no leachables. The solution contact layer is a rubberized copolymer of ethylene and propylene. The container is nontoxic and biologically inert. The container-solution unit is a closed system and is not dependent upon entry of external air during administration. The container is overwrapped to provide protection from the physical environment and to provide an additional moisture barrier when necessary.
The closure system has two ports; the one for the administration set has a tamper evident plastic protector.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
The plasma volume expansion produced by HESPAN® approximates that of 5% Albumin (Human). Intravenous infusion of HESPAN® results in expansion of plasma volume.
12.2 Pharmacodynamics
HESPAN® results in expansion of plasma volume that decreases over the succeeding 24 to 36 hours. The degree of plasma volume expansion and improvement in hemodynamic state depend upon the patient’s intravascular status.
12.3 Pharmacokinetics
Hetastarch molecules below 50,000 molecular weight are rapidly eliminated by renal excretion. A single dose of approximately 500 mL of HESPAN® (approximately 30 g) results in elimination in the urine of approximately 33% of the dose within 24 hours. This is a variable process but generally results in an intravascular hetastarch concentration of less than 10% of the total dose injected by two weeks. A study of the biliary excretion of HESPAN® in 10 healthy males accounted for less than 1% of the dose over a 14 day period. The hydroxyethyl group is not cleaved by the body but remains intact and attached to glucose units when excreted. Significant quantities of glucose are not produced as hydroxyethylation prevents complete metabolism of the smaller polymers.
The addition of hetastarch to whole blood increases the erythrocyte sedimentation rate. Therefore, HESPAN® is used to improve the efficiency of granulocyte collection by centrifugal means.
- 13 NONCLINICAL TOXICOLOGY
-
14 CLINICAL STUDIES
Surgical Patients Comparative Studies
In randomized, controlled, comparative studies of HESPAN® (6% hetastarch in 0.9% sodium chloride injection) (n=92) and Albumin (n=85) in surgical patients, no patient in either treatment group had a bleeding complication and no significant difference was found in the amount of blood loss between the treatment groups.7-10
Pediatric Postoperative Volume Expander Study
In one small double-blind study, 47 infants, children, and adolescents (ages 1 year to 15.5 years) scheduled for repair of congenital heart disease with moderate hypothermia were randomized to receive either HESPAN® or Albumin as a postoperative volume expander during the first 24 hours after surgery. Thirty-eight children required colloid replacement therapy, of which 20 children received HESPAN®. No differences were found in the coagulation parameters or in the amount of replacement fluids required in the children receiving 20 mL/kg or less of either colloid replacement therapy. In children who received greater than 20 mL/kg of HESPAN®, an increase in prothrombin time was demonstrated (p=0.006).11 There were no neonates included in this study [see Use in Specific Populations (8.4)].
Adult Critically Ill Studies
Three randomized controlled trials (RCTs) followed critically ill adult patients treated with different HES products for 90 days.
One trial (N=804) in severe sepsis patients using HES product (not approved in the U.S.) reported increased mortality (relative risk, 1.17; 95% CI, 1.01 to 1.36; p=0.03) and RRT (relative risk, 1.35; 95% CI, 1.01 to 1.80; p=0.04) in the HES treatment arm.4
Another trial (N=196) using different HES in severe sepsis patients reported no difference in mortality (relative risk,1.20; 95% CI, 0.83 to 1.74; p=0.33) and a trend for RRT (relative risk, 1.83; 95% CI, 0.93 to 3.59; p=0.06) in HES patients.5
A third trial (N=7000) using different HES in a heterogeneous patient population consisting of critically ill adult patients admitted to the ICU reported no difference in mortality (relative risk, 1.06; 95% CI, 0.96 to 1.18; p=0.26) but increased use of RRT (relative risk, 1.21; 95% CI, 1.00 to 1.45; p=0.04) in HES patients.6
-
15 REFERENCES
- Knutson JE., et al., Does Intraoperative Hetastarch Administration Increase Blood Loss and Transfusion Requirements After Cardiac Surgery? Anesthesia Analg., 2000;90:801-7.
- Cope JT., et al., Intraoperative Hetastarch Infusion Impairs Hemostasis After Cardiac Operations. The Annals of Thoracic Surgery, 1997;63:78-83.
- Damon L., Intracranial Bleeding During Treatment with Hydroxyethyl Starch. New England Journal of Medicine, 1987;317(15):964-965.
- Perner A, et al., Hydroxyethyl starch 130/0.42 versus Ringer"s acetate in severe sepsis patients. The New England Journal of Medicine, 2012 July 12;367(2):124-34.
- Guidet B, et al., Assessment of hemodynamic efficacy and safety of 6% hydroxyethyl starch 130/0.4 vs 0.9% NaCl fluid replacement in patients with severe sepsis: The CRYSTMAS Study. Critical Care, 2012 May 24;16(3):R94.
- Myburgh JA, et al., Hydroxyethyl starch or saline for fluid resuscitation in intensive care. The New England Journal of Medicine, 2012 November 15;367(20):1901-11.
- Diehl J., et al., Clinical Comparison of Hetastarch and Albumin in Postoperative Cardiac Patients. The Annals of Thoracic Surgery, 1982;34(6):674-679.
- Gold M., et al., Comparison of Hetastarch to Albumin for Perioperative Bleeding in Patients Undergoing Abdominal Aortic Aneurysm Surgery. Annals of Surgery, 1990;211(4):482-485.
- Kirklin J., et al., Hydroxyethyl Starch versus Albumin for Colloid Infusion Following Cardiopulmonary Bypass in Patients Undergoing Myocardial Revascularization. The Annals of Thoracic Surgery, 1984;37(1):40-46.
- Moggio RA., et al., Hemodynamic Comparison of Albumin and Hydroxyethyl Starch in Postoperative Cardiac Surgery Patients. Critical Care Medicine, 1983;11(12):943-945.
- Brutocao D., et al., Comparison of Hetastarch with Albumin for Postoperative Volume Expansion in Children After Cardiopulmonary Bypass. Journal of Cardiothoracic and Vascular Anesthesia, 1996;10(3):348-351.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
HESPAN® (6% hetastarch in 0.9% sodium chloride injection) is supplied sterile and nonpyrogenic in 500 mL EXCEL® Containers packaged 12 per case.
NDC REF Volume
0264-1965-10 L6511 500 mLExposure of pharmaceutical products to heat should be minimized. Avoid excessive heat. Protect from freezing.
Store at room temperature (25°C); however, brief exposure up to 40°C does not adversely affect the product.
Storage in automated dispensing machines: Brief exposure up to 2 weeks to ultraviolet or fluorescent light does not adversely affect the product labeling legibility; prolonged exposure can cause fading of the red label. Rotate stock frequently.
- SPL UNCLASSIFIED SECTION
-
PRINCIPAL DISPLAY PANEL - 500 mL
6% hetastarch in
0.9% sodium chloride injectionHESPAN®
NDC: 0264-1965-10
500 mL
EXCEL® CONTAINERFor intravenous use only.
Each 100 mL contains: Hetastarch 6 g
Sodium Chloride USP 0.9 g in Water for Injection, USP
pH adjusted with Sodium Hydroxide, NFElectrolytes (mEq/L): Sodium 154 Chloride 154
If administration is by pressure infusion, all air should be withdrawn
or expelled from the bag through the medication port prior to infusion.Sterile. Single dose container. Discard unused solution.
Recommended Storage: Store at room temperature, 25°C (77°F). Avoid
excessive heat. Protect from freezing. Usual Dosage: See package insert for
complete information.Do not remove overwrap until ready for use. If leaks are found, discard solution as
sterility may be impaired. DO NOT INTRODUCE ADDITIVES OTHER THAN CITRATE INTO BAG.REF L6511
Not made with natural rubber latex, PVC or DEHP.
Rx only

B. Braun Medical Inc.
Bethlehem, PA 18018-3524 USA
1-800-227-2862
www.bbraun.comY94-003-305 LD-152-7
EXP LOT

- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
HESPAN
hetastarch in sodium chloride injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 0264-1965 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength HETASTARCH (UNII: 875Y4127EA) (HETASTARCH - UNII:875Y4127EA) HETASTARCH 6 g in 100 mL SODIUM CHLORIDE (UNII: 451W47IQ8X) (SODIUM CATION - UNII:LYR4M0NH37, CHLORIDE ION - UNII:Q32ZN48698) SODIUM CHLORIDE 0.9 g in 100 mL Inactive Ingredients Ingredient Name Strength WATER (UNII: 059QF0KO0R) SODIUM HYDROXIDE (UNII: 55X04QC32I) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 0264-1965-10 12 in 1 CASE 1 500 mL in 1 CONTAINER; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA BN890105 04/04/1991 Labeler - B. Braun Medical Inc. (002397347) Establishment Name Address ID/FEI Business Operations B. Braun Medical Inc. 037425308 LABEL(0264-1965) , MANUFACTURE(0264-1965) , PACK(0264-1965)

Trademark Results [HESPAN]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 HESPAN 72378335 0933806 Live/Registered |
AMERICAN HOSPITAL SUPPLY CORPORATION 1970-12-10 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.