TECENTRIQ- atezolizumab injection, solution
TECENTRIQ by
Drug Labeling and Warnings
TECENTRIQ by is a Prescription medication manufactured, distributed, or labeled by Genentech, Inc., Roche Diagnostics GmbH, F. Hoffmann-La Roche Ltd, F. Hoffmann-La Roche AG, Roche Singapore Technical Operation, Pte. Ltd. (RSTO). Drug facts, warnings, and ingredients follow.
Drug Details [pdf]
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use TECENTRIQ safely and effectively. See full prescribing information for TECENTRIQ.
TECENTRIQ® (atezolizumab) injection, for intravenous use
Initial U.S. Approval: 2016RECENT MAJOR CHANGES
Indications and Usage, Non-Small Cell Lung Cancer (1.2) 12/2019 Indications and Usage, Triple-Negative Breast Cancer (1.3) 3/2019 Indications and Usage, Small Cell Lung Cancer (1.4) 3/2019 Dosage and Administration (2.3) 12/2019 Dosage and Administration (2.1, 2.2, 2.4, 2.5, 2.7) 5/2019 Warnings and Precautions (5.1, 5.2, 5.3, 5.4) 3/2019 INDICATIONS AND USAGE
TECENTRIQ is a programmed death-ligand 1 (PD-L1) blocking antibody indicated:
Urothelial Carcinoma
- for the treatment of adult patients with locally advanced or metastatic urothelial carcinoma who:
- are not eligible for cisplatin-containing chemotherapy and whose tumors express PD-L1 (PD-L1 stained tumor-infiltrating immune cells [IC] covering ≥ 5% of the tumor area), as determined by an FDA-approved test, or
- are not eligible for any platinum-containing chemotherapy regardless of PD-L1 status, or
- have disease progression during or following any platinum-containing chemotherapy, or within 12 months of neoadjuvant or adjuvant chemotherapy. (1.1)
This indication is approved under accelerated approval based on tumor response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s). (1.1)
Non-Small Cell Lung Cancer (NSCLC)
- in combination with bevacizumab, paclitaxel, and carboplatin, for the first-line treatment of adult patients with metastatic non-squamous NSCLC with no EGFR or ALK genomic tumor aberrations. (1.2)
- in combination with paclitaxel protein-bound and carboplatin for the first-line treatment of adult patients with metastatic non-squamous NSCLC with no EGFR or ALK genomic tumor aberrations (1.2)
- for the treatment of adult patients with metastatic NSCLC who have disease progression during or following platinum-containing chemotherapy. Patients with EGFR or ALK genomic tumor aberrations should have disease progression on FDA-approved therapy for NSCLC harboring these aberrations prior to receiving TECENTRIQ. (1.2)
Triple-Negative Breast Cancer (TNBC)
- in combination with paclitaxel protein-bound for the treatment of adult patients with unresectable locally advanced or metastatic TNBC whose tumors express PD-L1 (PD-L1 stained tumor-infiltrating immune cells [IC] of any intensity covering ≥ 1% of the tumor area), as determined by an FDA approved test. This indication is approved under accelerated approval based on progression free survival. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s). (1.3)
Small Cell Lung Cancer (SCLC)
- in combination with carboplatin and etoposide, for the first-line treatment of adult patients with extensive-stage small cell lung cancer (ES-SCLC). (1.4)
DOSAGE AND ADMINISTRATION
Administer TECENTRIQ intravenously over 60 minutes. If the first infusion is tolerated, all subsequent infusions may be delivered over 30 minutes.
Urothelial Carcinoma (2.2)
- Administer TECENTRIQ as a single agent as 840 mg every 2 weeks, 1200 mg every 3 weeks, or 1680 mg every 4 weeks.
NSCLC (2.3)
- Administer TECENTRIQ as a single agent as 840 mg every 2 weeks, 1200 mg every 3 weeks, or 1680 mg every 4 weeks.
- When administering with chemotherapy with or without bevacizumab, administer TECENTRIQ 1200 mg every 3 weeks prior to chemotherapy and bevacizumab
- Following completion of 4-6 cycles of chemotherapy, and if bevacizumab is discontinued, administer TECENTRIQ 840 mg every 2 weeks, 1200 mg every 3 weeks, or 1680 mg every 4 weeks.
Metastatic Treatment of TNBC (2.4)
- Administer TECENTRIQ 840 mg, followed by 100 mg/m2 paclitaxel protein-bound. For each 28 day cycle, TECENTRIQ is administered on days 1 and 15, and paclitaxel protein-bound is administered on days 1, 8, and 15.
Small Cell Lung Cancer (2.5)
- When administering with carboplatin and etoposide, administer TECENTRIQ 1200 mg every 3 weeks prior to chemotherapy.
- Following completion of 4 cycles of carboplatin and etoposide, administer TECENTRIQ 840 mg every 2 weeks, 1200 mg every 3 weeks, or 1680 mg every 4 weeks.
DOSAGE FORMS AND STRENGTHS
Injection: 840 mg/14 mL (60 mg/mL) and 1200 mg/20 mL (60 mg/mL) solution in a single-dose vial (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
- Immune-Mediated Pneumonitis: Withhold or permanently discontinue based on severity of pneumonitis. (2.6, 5.1)
- Immune-Mediated Hepatitis: Monitor for changes in liver function. Withhold or permanently discontinue based on severity of transaminase or total bilirubin elevation. (2.6, 5.2)
- Immune-Mediated Colitis: Withhold or permanently discontinue based on severity of colitis. (2.6, 5.3)
-
Immune-Mediated Endocrinopathies (2.6, 5.4):
- Hypophysitis: Withhold based on severity of hypophysitis.
- Thyroid Disorders: Monitor for changes in thyroid function. Withhold based on severity of hyperthyroidism.
- Adrenal Insufficiency: Withhold based on severity of adrenal insufficiency.
- Type 1 Diabetes Mellitus: Withhold based on severity of hyperglycemia.
- Infections: Withhold for severe or life-threatening infection. (2.6, 5.6)
- Infusion-Related Reactions: Interrupt, slow the rate of infusion, or permanently discontinue based on severity of infusion reactions. (2.6, 5.7)
- Embryo-Fetal Toxicity: Can cause fetal harm. Advise females of reproductive potential of the potential risk to a fetus and use of effective contraception. (5.8, 8.1, 8.3)
ADVERSE REACTIONS
- Most common adverse reactions (reported in ≥ 20% of patients) with TECENTRIQ as a single-agent were fatigue/asthenia, nausea, cough, dyspnea, and decreased appetite. (6.1)
- Most common adverse reactions (reported in ≥ 20% of patients) with TECENTRIQ in combination with other antineoplastic drugs in patients with NSCLC and SCLC were fatigue/asthenia, nausea, alopecia, constipation, diarrhea, and decreased appetite (6.1)
- The most common adverse reactions (reported in ≥ 20% of patients) with TECENTRIQ in combination with paclitaxel protein-bound in patients with TNBC were alopecia, peripheral neuropathies, fatigue, nausea, diarrhea, anemia, constipation, cough, headache, neutropenia, vomiting, and decreased appetite. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Genentech at 1-888-835-2555 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 12/2019
- for the treatment of adult patients with locally advanced or metastatic urothelial carcinoma who:
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Urothelial Carcinoma
1.2 Non-Small Cell Lung Cancer
1.3 Locally Advanced or Metastatic Triple-Negative Breast Cancer
1.4 Small Cell Lung Cancer
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection for Treatment of Urothelial Carcinoma and Triple-Negative Breast Cancer
2.2 Recommended Dosage for Urothelial Carcinoma
2.3 Recommended Dosage for NSCLC
2.4 Recommended Dosage for Locally Advanced or Metastatic TNBC
2.5 Recommended Dosage for SCLC
2.6 Dosage Modifications for Adverse Reactions
2.7 Preparation and Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Immune-Mediated Pneumonitis
5.2 Immune-Mediated Hepatitis
5.3 Immune-Mediated Colitis
5.4 Immune-Mediated Endocrinopathies
5.5 Other Immune-Mediated Adverse Reactions
5.6 Infections
5.7 Infusion-Related Reactions
5.8 Embryo-Fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Immunogenicity
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
14.1 Urothelial Carcinoma
14.2 Non-Small Cell Lung Cancer
14.3 Locally Advanced or Metastatic Triple-Negative Breast Cancer
14.4 Small Cell Lung Cancer
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- * Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
1.1 Urothelial Carcinoma
TECENTRIQ is indicated for the treatment of adult patients with locally advanced or metastatic urothelial carcinoma who:
- are not eligible for cisplatin-containing chemotherapy and whose tumors express PD-L1 (PD-L1 stained tumor-infiltrating immune cells [IC] covering ≥ 5% of the tumor area), as determined by an FDA-approved test [see Dosage and Administration (2.1)], or
- are not eligible for any platinum-containing chemotherapy regardless of PD-L1 status, or
- have disease progression during or following any platinum-containing chemotherapy, or within 12 months of neoadjuvant or adjuvant chemotherapy
This indication is approved under accelerated approval based on tumor response rate and durability of response [see Clinical Studies (14.1)]. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
1.2 Non-Small Cell Lung Cancer
- TECENTRIQ, in combination with bevacizumab, paclitaxel, and carboplatin, is indicated for the first-line treatment of adult patients with metastatic non-squamous non-small cell lung cancer (NSCLC) with no EGFR or ALK genomic tumor aberrations.
- TECENTRIQ, in combination with paclitaxel protein-bound and carboplatin, is indicated for the first-line treatment of adult patients with metastatic non-squamous NSCLC with no EGFR or ALK genomic tumor aberrations.
- TECENTRIQ, as a single-agent, is indicated for the treatment of adult patients with metastatic NSCLC who have disease progression during or following platinum-containing chemotherapy. Patients with EGFR or ALK genomic tumor aberrations should have disease progression on FDA-approved therapy for NSCLC harboring these aberrations prior to receiving TECENTRIQ.
1.3 Locally Advanced or Metastatic Triple-Negative Breast Cancer
TECENTRIQ, in combination with paclitaxel protein-bound, is indicated for the treatment of adult patients with unresectable locally advanced or metastatic triple-negative breast cancer (TNBC) whose tumors express PD-L1 (PD-L1 stained tumor-infiltrating immune cells [IC] of any intensity covering ≥ 1% of the tumor area), as determined by an FDA-approved test [see Dosage and Administration (2.1)]. This indication is approved under accelerated approval based on progression free survival [see Clinical Studies (14.3)]. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
-
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection for Treatment of Urothelial Carcinoma and Triple-Negative Breast Cancer
Select cisplatin-ineligible patients with previously untreated locally advanced or metastatic urothelial carcinoma for treatment with TECENTRIQ based on the PD-L1 expression on tumor-infiltrating immune cells [see Clinical Studies (14.1)].
Select patients with locally advanced or metastatic triple-negative breast cancer for treatment with TECENTRIQ in combination with paclitaxel protein-bound based on the PD-L1 expression on tumor infiltrating immune cells [see Clinical Studies (14.3)].
Information on FDA-approved tests for the determination of PD-L1 expression in locally advanced or metastatic urothelial carcinoma or triple-negative breast cancer are available at: http://www.fda.gov/CompanionDiagnostics
2.2 Recommended Dosage for Urothelial Carcinoma
The recommended dosage of TECENTRIQ is:
- 840 mg every 2 weeks or
- 1200 mg every 3 weeks or
- 1680 mg every 4 weeks
administered intravenously over 60 minutes until disease progression or unacceptable toxicity. If the first infusion is tolerated, all subsequent infusions may be delivered over 30 minutes.
2.3 Recommended Dosage for NSCLC
Single Agent
The recommended dosage of TECENTRIQ is:
- 840 mg every 2 weeks or
- 1200 mg every 3 weeks or
- 1680 mg every 4 weeks
administered intravenously over 60 minutes until disease progression or unacceptable toxicity. If the first infusion is tolerated, all subsequent infusions may be delivered over 30 minutes.
TECENTRIQ with Platinum-based Chemotherapy
The recommended dosage of TECENTRIQ is 1200 mg intravenously every 3 weeks until disease progression or unacceptable toxicity.
Administer TECENTRIQ prior to chemotherapy and bevacizumab when given on the same day. Refer to the Prescribing Information for the chemotherapy agents or bevacizumab administered in combination with TECENTRIQ for recommended dosing information.
Following completion of 4-6 cycles of chemotherapy, and if bevacizumab is discontinued, the recommended dosage of TECENTRIQ is:
- 840 mg every 2 weeks or
- 1200 mg every 3 weeks or
- 1680 mg every 4 weeks
administered intravenously until disease progression or unacceptable toxicity.
Administer the initial infusion of TECENTRIQ over 60 minutes. If the first infusion is tolerated, all subsequent infusions may be delivered over 30 minutes.
2.4 Recommended Dosage for Locally Advanced or Metastatic TNBC
The recommended dosage of TECENTRIQ is 840 mg administered intravenously over 60 minutes, followed by 100 mg/m2 paclitaxel protein-bound.
For each 28 day cycle, TECENTRIQ is administered on days 1 and 15, and paclitaxel protein-bound is administered on days 1, 8, and 15 until disease progression or unacceptable toxicity.
TECENTRIQ and paclitaxel protein-bound may be discontinued for toxicity independently of each other.
If the first infusion is tolerated, all subsequent infusions may be delivered over 30 minutes. Refer to the Prescribing Information for paclitaxel protein-bound for recommended dosing information.
2.5 Recommended Dosage for SCLC
The recommended dosage of TECENTRIQ is 1200 mg intravenously every 3 weeks, when administered in combination with carboplatin and etoposide, until disease progression or unacceptable toxicity.
Administer TECENTRIQ prior to chemotherapy when given on the same day. Refer to the Prescribing Information for the chemotherapy agents administered in combination with TECENTRIQ for recommended dosing information.
Following completion of 4 cycles of carboplatin and etoposide, the recommended dosage of TECENTRIQ is:
- 840 mg every 2 weeks or
- 1200 mg every 3 weeks or
- 1680 mg every 4 weeks
administered intravenously until disease progression or unacceptable toxicity.
Administer the initial infusion of TECENTRIQ over 60 minutes. If the first infusion is tolerated, all subsequent infusions may be delivered over 30 minutes.
2.6 Dosage Modifications for Adverse Reactions
No dose reductions of TECENTRIQ are recommended. Recommendations for dosage modifications are provided in Table 1.
Table 1: Recommended Dosage Modifications for Adverse Reactions Adverse Reaction Severity of Adverse Reaction* Dosage Modifications - * National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) version 4.0
Pneumonitis [see Warnings and Precautions (5.1)] Grade 2 Withhold dose until Grade 1 or resolved and corticosteroid dose is less than or equal to prednisone 10 mg per day (or equivalent) Grade 3 or 4 Permanently discontinue Hepatitis [see Warnings and Precautions (5.2)] AST or ALT more than 3 and up to 8 times the upper limit of normal or total bilirubin more than 1.5 and up to 3 times the upper limit of normal Withhold dose until Grade 1 or resolved and corticosteroid dose is less than or equal to prednisone 10 mg per day (or equivalent) AST or ALT more than 8 times the upper limit of normal or total bilirubin more than 3 times the upper limit of normal Permanently discontinue Colitis or diarrhea [see Warnings and Precautions (5.3)] Grade 2 or 3 Withhold dose until Grade 1 or resolved and corticosteroid dose is less than or equal to prednisone 10 mg per day (or equivalent) Grade 4 Permanently discontinue Endocrinopathies (including but not limited to hypophysitis, adrenal insufficiency, hyperthyroidism, and type 1 diabetes mellitus) [see Warnings and Precautions (5.4)] Grade 2, 3, or 4 Withhold dose until Grade 1 or resolved and clinically stable on hormone replacement therapy. Other immune-mediated adverse reactions involving a major organ [see Warnings and Precautions (5.5)] Grade 3 Withhold dose until Grade 1 or resolved and corticosteroid dose is less than or equal to prednisone 10 mg per day (or equivalent) Grade 4 Permanently discontinue Infections [see Warnings and Precautions (5.6)] Grade 3 or 4 Withhold dose until Grade 1 or resolved Infusion-Related Reactions [see Warnings and Precautions (5.7)] Grade 1 or 2 Interrupt or slow the rate of infusion Grade 3 or 4 Permanently discontinue Persistent Grade 2 or 3 adverse reaction (excluding endocrinopathies) Grade 2 or 3 adverse reaction that does not recover to Grade 0 or 1 within 12 weeks after last TECENTRIQ dose Permanently discontinue Inability to taper corticosteroid Inability to reduce to less than or equal to prednisone 10 mg per day (or equivalent) within 12 weeks after last TECENTRIQ dose Permanently discontinue Recurrent Grade 3 or 4 adverse reaction Recurrent Grade 3 or 4 (severe or life-threatening) adverse reaction Permanently discontinue 2.7 Preparation and Administration
Preparation
Visually inspect drug product for particulate matter and discoloration prior to administration, whenever solution and container permit. Discard the vial if the solution is cloudy, discolored, or visible particles are observed. Do not shake the vial.
Prepare the solution for infusion as follows:
- Select the appropriate vial(s) based on the prescribed dose.
- Withdraw the required volume of TECENTRIQ from the vial(s).
- Dilute into a 250 mL a polyvinyl chloride (PVC), polyethylene (PE), or polyolefin (PO) infusion bag containing 0.9% Sodium Chloride Injection, USP.
- Dilute with only 0.9% Sodium Chloride Injection, USP.
- Mix diluted solution by gentle inversion. Do not shake.
- Discard used or empty vials of TECENTRIQ.
Storage of Infusion Solution
This product does not contain a preservative.
Administer immediately once prepared. If diluted TECENTRIQ infusion solution is not used immediately, store solution either:
- At room temperature for no more than 6 hours from the time of preparation. This includes room temperature storage of the infusion in the infusion bag and time for administration of the infusion, or
- Under refrigeration at 2°C to 8°C (36°F to 46°F) for no more than 24 hours from time of preparation.
Do not freeze.
Do not shake.
Administration
Administer the initial infusion over 60 minutes through an intravenous line with or without a sterile, non-pyrogenic, low-protein binding in-line filter (pore size of 0.2–0.22 micron). If the first infusion is tolerated, all subsequent infusions may be delivered over 30 minutes.
Do not coadminister other drugs through the same intravenous line.
Do not administer as an intravenous push or bolus.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Immune-Mediated Pneumonitis
TECENTRIQ can cause immune-mediated pneumonitis or interstitial lung disease, defined as requiring use of systemic corticosteroids, including fatal cases. Monitor patients for signs and symptoms of pneumonitis. Evaluate patients with suspected pneumonitis with radiographic imaging. Administer corticosteroids, prednisone 1–2 mg/kg/day or equivalents, followed by a taper for Grade 2 or higher pneumonitis. Withhold or permanently discontinue TECENTRIQ based on the severity [see Dosage and Administration (2.6)].
In clinical studies enrolling 2616 patients with various cancers who received TECENTRIQ as a single-agent [see Adverse Reactions (6.1)], pneumonitis occurred in 2.5% of patients, including Grade 3 (0.6%), Grade 4 (0.1%), and Grade 5 (< 0.1%) immune-mediated pneumonitis. The median time to onset of pneumonitis was 3.6 months (3 days to 20.5 months) and median duration of pneumonitis was 1.4 months (1 day to 15.1 months). Pneumonitis resolved in 67% of patients. Pneumonitis led to discontinuation of TECENTRIQ in 0.4% of the 2616 patients. Systemic corticosteroids were required in 1.5% of patients, including 0.8% who received high-dose corticosteroids (prednisone ≥ 40 mg per day or equivalent) for a median duration of 4 days (1 day to 45 days) followed by a corticosteroid taper.
In clinical studies enrolling 2421 patients with NSCLC and SCLC who received TECENTRIQ in combination with platinum-based chemotherapy [see Adverse Reactions (6.1)], immune-mediated pneumonitis occurred in 5.5% of patients, including Grades 3-4 in 1.4% of patients. Systemic corticosteroids were required in 4.2% of patients, including 3.1% who received high-dose corticosteroids (prednisone ≥ 40 mg per day or equivalent) for a median duration of 5 days (1 day to 98 days) followed by a corticosteroid taper.
5.2 Immune-Mediated Hepatitis
TECENTRIQ can cause liver test abnormalities and immune-mediated hepatitis, defined as requiring use of systemic corticosteroids. Fatal cases have been reported. Monitor patients for signs and symptoms of hepatitis, during and after discontinuation of TECENTRIQ, including clinical chemistry monitoring. Administer corticosteroids, prednisone 1–2 mg/kg/day or equivalents, followed by a taper for Grade 2 or higher elevations of ALT, AST and/or total bilirubin. Interrupt or permanently discontinue TECENTRIQ based on the severity [see Dosage and Administration (2.6)].
In clinical studies enrolling 2616 patients with various cancers who received TECENTRIQ as a single-agent [see Adverse Reactions (6.1)], hepatitis occurred in 9% of patients, including Grade 3 (2.3%), Grade 4 (0.6%), and Grade 5 (< 0.1%). The median time to onset of hepatitis was 1.4 months (1 day to 25.8 months) and median duration was 24 days (1 day to 13 months). Hepatitis resolved in 71% of patients. Hepatitis led to discontinuation of TECENTRIQ in 0.4% of 2616 patients. Systemic corticosteroids were required in 2% of the patients, with 1.3% requiring high-dose corticosteroids (prednisone ≥ 40 mg per day or equivalent) for a median duration of 3 days (1 day to 35 days) followed by a corticosteroid taper.
In clinical studies enrolling 2421 patients with NSCLC and SCLC who received TECENTRIQ in combination with platinum-based chemotherapy [see Adverse Reactions (6.1)], immune-mediated hepatitis occurred in 14% of patients, including Grades 3-4 in 4.1% of patients. Systemic corticosteroids were required in 4.8% of patients, including 3.4% who received high-dose corticosteroids (prednisone ≥ 40 mg per day or equivalent) for a median duration of 6 days (1 day to 144 days) followed by a corticosteroid taper.
5.3 Immune-Mediated Colitis
TECENTRIQ can cause immune-mediated colitis or diarrhea, defined as requiring use of systemic corticosteroids. Monitor patients for signs and symptoms of diarrhea or colitis. Withhold treatment with TECENTRIQ for Grade 2 or 3 diarrhea or colitis. If symptoms persist for longer than 5 days or recur, administer corticosteroids, prednisone 1–2 mg/kg/day or equivalents, followed by a taper for Grade 2 diarrhea or colitis. Interrupt or permanently discontinue TECENTRIQ based on the severity [see Dosage and Administration (2.6) and Adverse Reactions (6.1)].
In clinical studies enrolling 2616 patients with various cancers who received TECENTRIQ as a single-agent [see Adverse Reactions (6.1)], diarrhea or colitis occurred in 20% of patients, including Grade 3 (1.4%) events. The median time to onset of diarrhea or colitis was 1.5 months (1 day to 41 months). Diarrhea and colitis resolved in 85% of the patients. Diarrhea or colitis led to discontinuation of TECENTRIQ in 0.2% of 2616 patients. Systemic corticosteroids were required in 1.1% of patients and high-dose corticosteroids (prednisone ≥ 40 mg per day or equivalent) was required in 0.4% patients with a median duration of 3 days (1 day to 11 days) followed by a corticosteroid taper.
In clinical studies enrolling 2421 patients with NSCLC and SCLC who received TECENTRIQ in combination with platinum-based chemotherapy [see Adverse Reactions (6.1)], diarrhea or colitis occurred in 29% of patients, including Grade 3-4 in 4.3% of patients. Systemic corticosteroids were required in 4.7% of patients, including 2.9% who received high-dose corticosteroids (prednisone ≥ 40 mg per day or equivalent) for a median duration of 4 days (1 day to 170 days) followed by a corticosteroid taper.
5.4 Immune-Mediated Endocrinopathies
TECENTRIQ can cause immune-mediated endocrinopathies, including thyroid disorders, adrenal insufficiency, and type 1 diabetes mellitus, including diabetic ketoacidosis, and hypophysitis/hypopituitarism.
Thyroid Disorders: Monitor thyroid function prior to and periodically during treatment with TECENTRIQ. Initiate hormone replacement therapy or medical management of hyperthyroidism as clinically indicated. Continue TECENTRIQ for hypothyroidism and interrupt for hyperthyroidism based on the severity [see Dosage and Administration (2.6)].
In clinical studies enrolling 2616 patients who received TECENTRIQ as a single-agent [see Adverse Reactions (6.1)], hypothyroidism occurred in 4.6% of patients, and 3.8% of patients required the use of hormone replacement therapy. Hyperthyroidism occurred in 1.6% of patients. One patient experienced acute thyroiditis.
In clinical studies enrolling 2421 patients with NSCLC and SCLC who received TECENTRIQ in combination with platinum-based chemotherapy [see Adverse Reactions (6.1)], hypothyroidism occurred in 11% of patients, including Grades 3-4 in 0.3% of patients; 8.2% of the 2421 patients required the use of hormone replacement therapy. The frequency and severity of hyperthyroidism and thyroiditis were similar whether TECENTRIQ was given as a single-agent in patients with various cancers or in combination with other antineoplastic drugs in NSCLC and SCLC.
Adrenal Insufficiency: Monitor patients for clinical signs and symptoms of adrenal insufficiency. For Grade 2 or higher adrenal insufficiency, initiate prednisone 1to 2 mg/kg/day or equivalents, followed by a taper and hormone replacement as clinically indicated. Interrupt TECENTRIQ based on the severity [see Dosage and Administration (2.6)].
In clinical studies enrolling 2616 patients who received TECENTRIQ as a single-agent, adrenal insufficiency occurred in 0.4% of patients, including Grade 3 (< 0.1%) adrenal insufficiency. Median time to onset was 5.7 months (3 days to 19 months). There was insufficient information to adequately characterize the median duration of adrenal insufficiency. Adrenal insufficiency resolved in 27% of patients. Systemic corticosteroids were required in 0.3% of 2616 patients, including 0.1% who required high-dose corticosteroids (prednisone ≥ 40 mg per day or equivalent). The frequency and severity of adrenal insufficiency were similar whether TECENTRIQ was given as a single-agent in patients with various cancers or in combination with other antineoplastic drugs in NSCLC and SCLC.
Type 1 Diabetes Mellitus: Monitor patients for hyperglycemia or other signs and symptoms of diabetes. Initiate treatment with insulin as clinically indicated. Interrupt TECENTRIQ based on the severity [see Dosage and Administration (2.6)].
In clinical studies enrolling 2616 patients who received TECENTRIQ as a single-agent, type 1 diabetes mellitus occurred in < 0.1% of patients. Insulin was required in one patient. The frequency and severity of diabetes mellitus were similar whether TECENTRIQ was given as a single-agent in patients with various cancers or in combination with other antineoplastic drugs in NSCLC and SCLC.
Hypophysitis: For Grade 2 or higher hypophysitis, initiate prednisone 1–2 mg/kg/day or equivalents, followed by a taper and hormone replacement therapy as clinically indicated. Interrupt TECENTRIQ based on the severity [see Dosage and Administration (2.6)].
In clinical studies enrolling 2616 patients who received TECENTRIQ as a single-agent, Grade 2 hypophysitis occurred in < 0.1% of patients. The frequency and severity of hypophysitis were similar whether TECENTRIQ was given as a single-agent in patients with various cancers or in combination with other antineoplastic drugs in NSCLC and SCLC.
5.5 Other Immune-Mediated Adverse Reactions
TECENTRIQ can cause severe and fatal immune-mediated adverse reactions. These immune-mediated reactions may involve any organ system. While immune-mediated reactions usually manifest during treatment with TECENTRIQ, immune-mediated adverse reactions can also manifest after discontinuation of TECENTRIQ.
For suspected Grade 2 immune-mediated adverse reactions, exclude other causes and initiate corticosteroids as clinically indicated. For severe (Grades 3 or 4) adverse reactions, administer corticosteroids, prednisone 1 to 2 mg/kg/day or equivalents, followed by a taper. Interrupt or permanently discontinue TECENTRIQ, based on the severity of the reaction [see Dosage and Administration (2.6)].
If uveitis occurs in combination with other immune-mediated adverse reactions, evaluate for Vogt-Koyanagi-Harada syndrome, which has been observed with other products in this class and may require treatment with systemic steroids to reduce the risk of permanent vision loss.
The following clinically significant, immune-mediated adverse reactions occurred at an incidence of < 1% in 2616 patients who received TECENTRIQ as a single-agent and in 2421 patients who received TECENTRIQ in combination with platinum-based chemotherapy or were reported in other products in this class [see Adverse Reactions (6.1)]:
Cardiac: myocarditis
Dermatologic: bullous dermatitis, pemphigoid, erythema multiforme, Stevens Johnson Syndrome (SJS)/toxic epidermal necrolysis (TEN).
Gastrointestinal: pancreatitis, including increases in serum amylase or lipase levels
General: systemic inflammatory response syndrome, histiocytic necrotizing lymphadenitis
Hematological: autoimmune hemolytic anemia, immune thrombocytopenic purpura.
Musculoskeletal: myositis, rhabdomyolysis.
Neurological: Guillain-Barre syndrome, myasthenia syndrome/myasthenia gravis, demyelination, immune-related meningoencephalitis, aseptic meningitis, encephalitis, facial and abducens nerve paresis, polymyalgia rheumatica, autoimmune neuropathy, and Vogt-Koyanagi-Harada syndrome.
Ophthalmological: uveitis, iritis.
Renal: nephrotic syndrome, nephritis.
Vascular: vasculitis
5.6 Infections
TECENTRIQ can cause severe infections including fatal cases. Monitor patients for signs and symptoms of infection. For Grade 3 or higher infections, withhold TECENTRIQ and resume once clinically stable [see Dosage and Administration (2.6) and Adverse Reactions (6.1)].
In clinical studies enrolling 2616 patients with various cancers who received TECENTRIQ as a single-agent [see Adverse Reactions (6.1)], infections occurred in 42% of patients, including Grade 3 (8.7%), Grade 4 (1.5%), and Grade 5 (1%). In patients with urothelial carcinoma, the most common Grade 3 or higher infection was urinary tract infections, occurring in 6.5% of patients. In patients with NSCLC, the most common Grade 3 or higher infection was pneumonia, occurring in 3.8% of patients. The frequency and severity of infections were similar whether TECENTRIQ was given as a single-agent in patients with various cancers or in combination with other antineoplastic drugs in NSCLC and SCLC.
5.7 Infusion-Related Reactions
TECENTRIQ can cause severe or life-threatening infusion-related reactions. Monitor for signs and symptoms of infusion-related reactions. Interrupt, slow the rate of, or permanently discontinue TECENTRIQ based on the severity [see Dosage and Administration (2.6)]. For Grade 1 or 2 infusion-related reactions, consider using pre-medications with subsequent doses.
In clinical studies enrolling 2616 patients with various cancers who received TECENTRIQ as a single-agent [see Adverse Reactions (6.1)], infusion-related reactions occurred in 1.3% of patients, including Grade 3 (0.2%). The frequency and severity of infusion-related reactions were similar whether TECENTRIQ was given as a single-agent in patients with various cancers, in combination with other antineoplastic drugs in NSCLC and SCLC, and across the recommended dose range (840 mg Q2W to 1680 mg Q4W).
5.8 Embryo-Fetal Toxicity
Based on its mechanism of action, TECENTRIQ can cause fetal harm when administered to a pregnant woman. There are no available data on the use of TECENTRIQ in pregnant women. Animal studies have demonstrated that inhibition of the PD-L1/PD-1 pathway can lead to increased risk of immune-related rejection of the developing fetus resulting in fetal death.
Verify pregnancy status of females of reproductive potential prior to initiating TECENTRIQ. Advise females of reproductive potential of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with TECENTRIQ and for at least 5 months after the last dose [see Use in Specific Populations (8.1, 8.3)].
-
6 ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail in other sections of the label:
- Immune-Mediated Pneumonitis [see Warnings and Precautions (5.1)]
- Immune-Mediated Hepatitis [see Warnings and Precautions (5.2)]
- Immune-Mediated Colitis [see Warnings and Precautions (5.3)]
- Immune-Mediated Endocrinopathies [see Warnings and Precautions (5.4)]
- Other Immune-Mediated Adverse Reactions [see Warnings and Precautions (5.5)]
- Infections [see Warnings and Precautions (5.6)]
- Infusion-Related Reactions [see Warnings and Precautions (5.7)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data described in WARNINGS AND PRECAUTIONS reflect exposure to TECENTRIQ as a single-agent in 2616 patients in two randomized, active-controlled studies (POPLAR, OAK) and four open-label, single arm studies (PCD4989g, IMvigor210, BIRCH, FIR) which enrolled 524 patients with metastatic urothelial carcinoma, 1636 patients with metastatic NSCLC, and 456 patients with other tumor types. TECENTRIQ was administered at a dose of 1200 mg intravenously every 3 weeks in all studies except PCD4989g. Among the 2616 patients who received a single-agent TECENTRIQ, 36% were exposed for longer than 6 months and 20% were exposed for longer than 12 months. Using the dataset described for patients who received TECENTRIQ as a single-agent, the most common adverse reactions in ≥ 20% of patients were fatigue/asthenia (48%), decreased appetite (25%), nausea (24%), cough (22%), and dyspnea (22%).
In addition, the data reflect exposure to TECENTRIQ in combination with other antineoplastic drugs in 2421 patients with NSCLC (N = 2223) or SCLC (N = 198) enrolled in five randomized, active-controlled trials, including IMpower150, IMpower130 and IMpower133. Among the 2421 patients, 53% were exposed to TECENTRIQ for longer than 6 months and 29% were exposed to TECENTRIQ for longer than 12 months. Among the 2421 patients with NSCLC and SCLC who received TECENTRIQ in combination with other antineoplastic drugs, the most common adverse reactions in ≥20% of patients were fatigue/asthenia (49%), nausea (38%), alopecia (35%), constipation (29%), diarrhea (28%) and decreased appetite (27%).
Urothelial Carcinoma
Cisplatin-Ineligible Patients with Locally Advanced or Metastatic Urothelial Carcinoma
The safety of TECENTRIQ was evaluated in IMvigor 210 (Cohort 1), a multicenter, open-label, single-arm trial that included 119 patients with locally advanced or metastatic urothelial carcinoma who were ineligible for cisplatin-containing chemotherapy and were either previously untreated or had disease progression at least 12 months after neoadjuvant or adjuvant chemotherapy [see Clinical Studies (14.1)]. Patients received TECENTRIQ 1200 mg intravenously every 3 weeks until either unacceptable toxicity or disease progression. The median duration of exposure was 15 weeks (0 to 87 weeks).
Five patients (4.2%) who were treated with TECENTRIQ experienced one of the following events which led to death: sepsis, cardiac arrest, myocardial infarction, respiratory failure, or respiratory distress. One additional patient (0.8%) was experiencing herpetic meningoencephalitis and disease progression at the time of death.
Serious adverse reactions occurred in 37% of patients. The most frequent serious adverse reactions (≥ 2%) were diarrhea, intestinal obstruction, sepsis, acute kidney injury, and renal failure.
TECENTRIQ was discontinued for adverse reactions in 4.2% of patients. The adverse reactions leading to discontinuation were diarrhea/colitis (1.7%), fatigue (0.8%), hypersensitivity (0.8%), and dyspnea (0.8%).
Adverse reactions leading to interruption occurred in 35% of patients; the most common (≥ 1%) were intestinal obstruction, fatigue, diarrhea, urinary tract infection, infusion- related reaction, cough, abdominal pain, peripheral edema, pyrexia, respiratory tract infection, upper respiratory tract infection, creatinine increase, decreased appetite, hyponatremia, back pain, pruritus, and venous thromboembolism.
Tables 2 and 3 summarize the adverse reactions and Grades 3–4 selected laboratory abnormalities, respectively, in patients who received TECENTRIQ in IMvigor210 (Cohort 1).
Table 2: Adverse Reactions in ≥ 10% of Patients with Urothelial Carcinoma in IMvigor210 (Cohort 1) Adverse Reaction TECENTRIQ
N = 119All Grades
(%)Grades 3–4
(%)- * Includes fatigue, asthenia, lethargy, and malaise
- † Includes edema peripheral, scrotal edema, lymphedema, and edema
- ‡ Includes diarrhea, colitis, frequent bowel movements, autoimmune colitis
- § Includes abdominal pain, upper abdominal pain, lower abdominal pain, and flank pain
- ¶ Includes decreased appetite and early satiety
- # Includes rash, dermatitis, dermatitis acneiform, rash maculo-papular, rash erythematous, rash pruritic, rash macular, and rash papular
- Þ Includes urinary tract infection, urinary tract infection bacterial, cystitis, and urosepsis
- ß Includes cough and productive cough
- à Includes dyspnea and exertional dyspnea
General Fatigue* 52 8 Peripheral edema† 17 2 Pyrexia 14 0.8 Gastrointestinal Diarrhea‡ 24 5 Nausea 22 2 Vomiting 16 0.8 Constipation 15 2 Abdominal pain§ 15 0.8 Metabolism and Nutrition Decreased appetite¶ 24 3 Musculoskeletal and Connective Tissue Back/Neck pain 18 3 Arthralgia 13 0 Skin and Subcutaneous Tissue Pruritus 18 0.8 Rash# 17 0.8 Infections Urinary tract infectionÞ 17 5 Respiratory, Thoracic, and Mediastinal Coughß 14 0 Dyspneaà 12 0 Table 3: Grades 3–4 Laboratory Abnormalities in ≥ 1% of Patients with Urothelial Carcinoma in IMvigor210 (Cohort 1) Laboratory Abnormality Grades 3–4
(%)Chemistry Hyponatremia 15 Hyperglycemia 10 Increased Alkaline Phosphatase 7 Increased Creatinine 5 Hypophosphatemia 4 Increased ALT 4 Increased AST 4 Hyperkalemia 3 Hypermagnesemia 3 Hyperbilirubinemia 3 Hematology Lymphopenia 9 Anemia 7 Previously Treated Locally Advanced or Metastatic Urothelial Carcinoma
The safety of TECENTRIQ was evaluated in IMvigor210 (Cohort 2), a multicenter, open-label, single-arm trial that included 310 patients with locally advanced or metastatic urothelial carcinoma who had disease progression during or following at least one platinum-containing chemotherapy regimen or who had disease progression within 12 months of treatment with a platinum-containing neoadjuvant or adjuvant chemotherapy regimen [see Clinical Studies (14.1)]. Patients received TECENTRIQ 1200 mg intravenously every 3 weeks until unacceptable toxicity or either radiographic or clinical progression. The median duration of exposure was 12.3 weeks (0.1 to 46 weeks).
Three patients (1%) who were treated with TECENTRIQ experienced one of the following events which led to death: sepsis, pneumonitis, or intestinal obstruction.
TECENTRIQ was discontinued for adverse reactions in 3.2% of patients. Sepsis led to discontinuation in 0.6% of patients.
Serious adverse reactions occurred in 45% of patients. The most frequent serious adverse reactions (> 2%) were urinary tract infection, hematuria, acute kidney injury, intestinal obstruction, pyrexia, venous thromboembolism, urinary obstruction, pneumonia, dyspnea, abdominal pain, sepsis, and confusional state.
Adverse reactions leading to interruption occurred in 27% of patients; the most common (> 1%) were liver enzyme increase, urinary tract infection, diarrhea, fatigue, confusional state, urinary obstruction, pyrexia, dyspnea, venous thromboembolism, and pneumonitis.
Tables 4 and 5 summarize the adverse reactions and Grades 3–4 selected laboratory abnormalities, respectively, in patients who received TECENTRIQ in IMvigor210 (Cohort 2).
Table 4: Adverse Reactions in ≥ 10% of Patients with Urothelial Carcinoma in IMvigor210 (Cohort 2) Adverse Reaction TECENTRIQ
N = 310All Grades
(%)Grades 3–4
(%)General Fatigue 52 6 Pyrexia 21 1 Peripheral edema 18 1 Metabolism and Nutrition Decreased appetite 26 1 Gastrointestinal Nausea 25 2 Constipation 21 0.3 Diarrhea 18 1 Abdominal pain 17 4 Vomiting 17 1 Infections Urinary tract infection 22 9 Respiratory, Thoracic, and Mediastinal Dyspnea 16 4 Cough 14 0.3 Musculoskeletal and Connective Tissue Back/Neck pain 15 2 Arthralgia 14 1 Skin and Subcutaneous Tissue Rash 15 0.3 Pruritus 13 0.3 Renal and Urinary Hematuria 14 3 Table 5: Grades 3–4 Laboratory Abnormalities in ≥ 1% of Patients with Urothelial Carcinoma in IMvigor210 (Cohort 2) Laboratory Abnormality Grades 3–4
(%)Chemistry Hyponatremia 10 Hyperglycemia 5 Increased Alkaline Phosphatase 4 Increased Creatinine 3 Increased ALT 2 Increased AST 2 Hypoalbuminemia 1 Hematology Lymphopenia 10 Anemia 8 Non-small Cell Lung Cancer (NSCLC)
IMpower150
The safety of TECENTRIQ with bevacizumab, paclitaxel and carboplatin was evaluated in IMpower150, a multicenter, international, randomized, open-label trial in which 393 chemotherapy-naïve patients with metastatic non-squamous NSCLC received TECENTRIQ 1200 mg with bevacizumab 15 mg/kg, paclitaxel 175 mg/m2 or 200 mg/m2, and carboplatin AUC 6 mg/mL/min intravenously every 3 weeks for a maximum of 4 or 6 cycles, followed by TECENTRIQ 1200 mg with bevacizumab 15 mg/kg intravenously every 3 weeks until disease progression or unacceptable toxicity [see Clinical Studies (14.2)]. The median duration of exposure to TECENTRIQ was 8.3 months in patients receiving TECENTRIQ with bevacizumab, paclitaxel, and carboplatin.
Fatal adverse reactions occurred in 6% of patients receiving TECENTRIQ; these included hemoptysis, febrile neutropenia, pulmonary embolism, pulmonary hemorrhage, death, cardiac arrest, cerebrovascular accident, pneumonia, aspiration pneumonia, chronic obstructive pulmonary disease, intracranial hemorrhage, intestinal angina, intestinal ischemia, intestinal obstruction and aortic dissection.
Serious adverse reactions occurred in 44%. The most frequent serious adverse reactions (>2%) were febrile neutropenia, pneumonia, diarrhea, and hemoptysis.
TECENTRIQ was discontinued due to adverse reactions in 15% of patients; the most common adverse reaction leading to discontinuation was pneumonitis (1.8%).
Adverse reactions leading to interruption of TECENTRIQ occurred in 48%; the most common (>1%) were neutropenia, thrombocytopenia, fatigue/asthenia, diarrhea, hypothyroidism, anemia, pneumonia, pyrexia, hyperthyroidism, febrile neutropenia, increased ALT, dyspnea, dehydration and proteinuria.
Tables 6 and 7 summarize adverse reactions and laboratory abnormalities in patients receiving TECENTRIQ with bevacizumab, paclitaxel, and carboplatin in IMpower150. Study IMpower150 was not designed to demonstrate a statistically significant reduction in adverse reaction rates for TECENTRIQ, as compared to the control arm, for any specified adverse reaction or laboratory abnormality listed in Tables 6 and 7.
Table 6: Adverse Reactions Occurring in ≥15% of Patients with NSCLC Receiving TECENTRIQ in IMpower150 Adverse Reaction TECENTRIQ with Bevacizumab, Paclitaxel, and Carboplatin
N = 393Bevacizumab, Paclitaxel and Carboplatin
N = 394All Grades*
(%)Grades 3–4*
(%)All Grades*
(%)Grades 3–4*
(%)- * Graded per NCI CTCAE v4.0
- † Includes neuropathy peripheral, peripheral sensory neuropathy, hypoesthesia, paraesthesia, dysesthesia, polyneuropathy.
- ‡ Includes rash, rash maculo-papular, drug eruption, eczema, eczema asteatotic, dermatitis, contact dermatitis, rash erythematous, rash macular, pruritic rash, seborrheic dermatitis, dermatitis psoriasiform.
- § Includes pain in extremity, musculoskeletal chest pain, musculoskeletal discomfort, neck pain, backpain, myalgia, and bone pain.
- ¶ Includes diarrhea, gastroenteritis, colitis, enterocolitis.
- # Data based on Preferred Terms since laboratory data for proteinuria were not systematically collected.
Nervous System Neuropathy† 56 3 47 3 Headache 16 0.8 13 0 General Fatigue/Asthenia 50 6 46 6 Pyrexia 19 0.3 9 0.5 Skin and Subcutaneous Tissue Alopecia 48 0 46 0 Rash‡ 23 2 10 0.3 Musculoskeletal and Connective Tissue Myalgia/Pain§ 42 3 34 2 Arthralgia 26 1 22 1 Gastrointestinal Nausea 39 4 32 2 Diarrhea¶ 33 6 25 0.5 Constipation 30 0.3 23 0.3 Vomiting 19 2 18 1 Metabolism and Nutrition Decreased appetite 29 4 21 0.8 Vascular Hypertension 25 9 22 8 Respiratory Cough 20 0.8 19 0.3 Epistaxis 17 1 22 0.3 Renal Proteinuria# 16 3 15 3 Table 7: Laboratory Abnormalities Worsening from Baseline Occurring in ≥20% of Patients with NSCLC Receiving TECENTRIQ in IMpower150 Laboratory Abnormality TECENTRIQ with Bevacizumab, Paclitaxel, and Carboplatin Bevacizumab, Paclitaxel and Carboplatin All Grades
(%)Grades 3–4
(%)All Grades
(%)Grades 3–4
(%)Each test incidence is based on the number of patients who had both baseline and at least one on-study laboratory measurement available: TECENTRIQ with bevacizumab, paclitaxel, and carboplatin range: 337-380); bevacizumab, paclitaxel, and carboplatin (range: 337-382). Graded per NCI CTCAE v4.0 - * NA = Not applicable. NCI CTCAE does not provide a Grades 3-4 definition for these laboratory abnormalities
Hematology Anemia 83 10 83 9 Neutropenia 52 31 45 26 Lymphopenia 48 17 38 13 Chemistry Hyperglycemia 61 0 60 0 Increased BUN 52 NA* 44 NA* Hypomagnesemia 42 2 36 1 Hypoalbuminemia 40 3 31 2 Increased AST 40 4 28 0.8 Hyponatremia 38 10 36 9 Increased Alkaline Phosphatase 37 2 32 1 Increased ALT 37 6 28 0.5 Increased TSH 30 NA* 20 NA* Hyperkalemia 28 3 25 2 Increased Creatinine 28 1 19 2 Hypocalcemia 26 3 21 3 Hypophosphatemia 25 4 18 4 Hypokalemia 23 7 14 4 Hyperphosphatemia 25 NA* 19 NA* IMpower130
The safety of TECENTRIQ with paclitaxel protein-bound and carboplatin was evaluated in IMpower130, a multicenter, international, randomized, open-label trial in which 473 chemotherapy-naïve patients with metastatic non-squamous NSCLC received TECENTRIQ 1200 mg and carboplatin AUC 6 mg/mL/min intravenously on Day 1 and paclitaxel protein-bound 100 mg/m2 intravenously on Day 1, 8, and 15 of each 21-day cycle for a maximum of 4 or 6 cycles, followed by TECENTRIQ 1200 mg intravenously every 3 weeks until disease progression or unacceptability toxicity [see Clinical Studies (14.2)]. Among patients receiving TECENTRIQ, 55% were exposed for 6 months or longer and 3.5% were exposed for greater than one year.
Fatal adverse reactions occurred in 5.3% of patients receiving TECENTRIQ; these included including pneumonia (1.1%), pulmonary embolism (0.8%), myocardial infarction (0.6%), cardiac arrest (0.4%) and pneumonitis (0.4%) and sepsis, septic shock, staphylococcal sepsis, aspiration, respiratory distress, cardiorespiratory arrest, ventricular tachycardia, death (not otherwise specified), and hepatic cirrhosis (0.2% each).
Serious adverse reactions occurred in 51% of patients receiving TECENTRIQ. The most frequent serious adverse reactions (≥2%) were pneumonia (6%), diarrhea (3%), lung infection (3.0%), pulmonary embolism (3%), chronic obstructive pulmonary disease exacerbation (2.5%), dyspnea (2.3%), and febrile neutropenia (1.9%).
TECENTRIQ was discontinued due to adverse reactions in 13% of patients; the most common adverse reactions leading to discontinuation were pneumonia (0.8%), pulmonary embolism (0.8%), fatigue (0.6%), dyspnea (0.6%), pneumonitis (0.6%), neutropenia (0.4%), nausea (0.4%), renal failure (0.4%), cardiac arrest (0.4%), and septic shock (0.4%).
Adverse reactions leading to interruption of TECENTRIQ occurred in 62% of patients; the most common (>1%) were neutropenia, thrombocytopenia, anemia, diarrhea, fatigue/asthenia, pneumonia, dyspnea, pneumonitis, pyrexia, nausea, acute kidney injury, vomiting, pulmonary embolism, arthralgia, infusion-related reaction, abdominal pain, chronic obstructive pulmonary disease exacerbation, dehydration, and hypokalemia.
Tables 8 and 9 summarize adverse reactions and laboratory abnormalities in patients receiving TECENTRIQ with paclitaxel protein-bound and carboplatin in IMpower130.
Table 8: Adverse Reactions Occurring in ≥20% of Patients with NSCLC Receiving TECENTRIQ in IMpower130 Adverse Reaction TECENTRIQ with Paclitaxel Protein-Bound and Carboplatin
(n=473)Paclitaxel Protein-Bound and Carboplatin
(n=232)All Grades
(%)Grades 3–4
(%)All Grades
(%)Grades 3–4
(%)Graded per NCI CTCAE v4.0 - * Includes diarrhea, colitis, and gastroenteritis
- † Includes back pain, pain in extremity, myalgia, musculoskeletal chest pain, bone pain, neck pain and musculoskeletal discomfort
- ‡ Includes neuropathy peripheral, peripheral sensory neuropathy, hypoesthesia, paresthesia, dysesthesia, polyneuropathy
- § Includes dyspnea, dyspnea exertional and wheezing
- ¶ Includes rash, rash maculo-papular, eczema, rash pruritic, rash erythematous, dermatitis, dermatitis contact, drug eruption, seborrheic dermatitis and rash macular.
General Fatigue/Asthenia 61 11 60 8 Gastrointestinal Nausea 50 3.4 46 2.2 Diarrhea * 43 6 32 6 Constipation 36 1.1 31 0 Vomiting 27 2.7 19 2.2 Musculoskeletal and Connective Tissue Myalgia/Pain † 38 3 22 0.4 Nervous System Neuropathy ‡ 33 2.5 28 2.2 Respiratory, Thoracic and Mediastinal Dyspnea § 32 4.9 25 1.3 Cough 27 0.6 17 0 Skin and Subcutaneous Tissue Alopecia 32 0 27 0 Rash ¶ 20 0.6 11 0.9 Metabolism and Nutrition Decreased appetite 30 2.1 26 2.2 Table 9: Laboratory Abnormalities Worsening from Baseline Occurring in ≥20% of Patients Receiving TECENTRIQ in IMpower130 Laboratory Abnormality TECENTRIQ with Paclitaxel Protein-Bound and Carboplatin
(n=473)Paclitaxel Protein-Bound and Carboplatin
(n=232)All Grades (%) Grades 3–4 (%) All Grades (%) Grades 3–4 (%) Each test incidence is based on the number of patients who had both baseline and at least one on-study laboratory measurement available: TECENTRIQ with paclitaxel protein bound and carboplatin (range: 423 - 467); paclitaxel protein bound and carboplatin (range: 218- 229). Graded per NCI CTCAE v4.0. - * NA = Not applicable. NCI CTCAE does not provide a Grades 3-4 definition for these laboratory abnormalities
Hematology Anemia 92 33 87 25 Neutropenia 75 50 67 39 Thrombocytopenia 73 19 59 13 Lymphopenia 71 23 61 16 Chemistry Hyperglycemia 75 8 66 8 Hypomagnesemia 50 3.4 42 3.2 Hyponatremia 37 9 28 7 Hypoalbuminemia 35 1.3 31 0 Increased ALT 31 2.8 24 3.9 Hypocalcemia 31 2.6 27 1.8 Hypophosphatemia 29 6 20 3.2 Increased AST 28 2.2 24 1.8 Increased TSH 26 NA* 5 NA* Hypokalemia 26 6 24 4.4 Increased Alkaline Phosphatase 25 2.6 22 1.3 Increased Blood Creatinine 23 2.8 16 0.4 Hyperphosphatemia 21 NA* 13 NA* Previously Treated Metastatic NSCLC
The safety of TECENTRIQ was evaluated in OAK, a multicenter, international, randomized, open-label trial in patients with metastatic NSCLC who progressed during or following a platinum-containing regimen, regardless of PD-L1 expression [see Clinical Studies (14.2)]. A total of 609 patients received TECENTRIQ 1200 mg intravenously every 3 weeks until unacceptable toxicity, radiographic progression, or clinical progression or docetaxel (n=578) 75 mg/m2 intravenously every 3 weeks until unacceptable toxicity or disease progression. The study excluded patients with active or prior autoimmune disease or with medical conditions that required systemic corticosteroids. The median duration of exposure was 3.4 months (0 to 26 months) in TECENTRIQ-treated patients and 2.1 months (0 to 23 months) in docetaxel-treated patients.
The study population characteristics were: median age of 63 years (25 to 85 years), 46% age 65 years or older, 62% male, 71% White, 20% Asian, 68% former smoker, 16% current smoker, and 63% had ECOG performance status of 1.
Fatal adverse reactions occurred in 1.6% of patients; these included pneumonia, sepsis, septic shock, dyspnea, pulmonary hemorrhage, sudden death, myocardial ischemia or renal failure.
Serious adverse reactions occurred in 33.5% of patients. The most frequent serious adverse reactions (>1%) were pneumonia, sepsis, dyspnea, pleural effusion, pulmonary embolism, pyrexia and respiratory tract infection.
TECENTRIQ was discontinued due to adverse reactions in 8% of patients. The most common adverse reactions leading to TECENTRIQ discontinuation were fatigue, infections and dyspnea. Adverse reactions leading to interruption of TECENTRIQ occurred in 25% of patients; the most common (>1%) were pneumonia, liver function test abnormality, dyspnea, fatigue, pyrexia, and back pain.
Tables 10 and 11 summarize adverse reactions and laboratory abnormalities, respectively, in OAK.
Table 10: Adverse Reactions Occurring in ≥10% of Patients with NSCLC Receiving TECENTRIQ in OAK Adverse Reaction* TECENTRIQ
N = 609Docetaxel
N = 578All Grades
(%)Grades 3-4
(%)All Grades
(%)Grades 3-4
(%)- * Graded per NCI CTCAE v4.0
- † Includes fatigue and asthenia
- ‡ Includes cough and exertional cough
- § Includes musculoskeletal pain, musculoskeletal stiffness, musculoskeletal chest pain, myalgia
- ¶ Includes rash, erythematous rash, generalized rash, maculopapular rash, papular rash, pruritic rash, pustular rash, pemphigoid
General Fatigue/Asthenia† 44 4 53 6 Pyrexia 18 <1 13 <1 Respiratory Cough‡ 26 <1 21 <1 Dyspnea 22 2.8 21 2.6 Metabolism and Nutrition Decreased appetite 23 <1 24 1.6 Musculoskeletal Myalgia/pain§ 20 1.3 20 <1 Arthralgia 12 0.5 10 0.2 Gastrointestinal Nausea 18 <1 23 <1 Constipation 18 <1 14 <1 Diarrhea 16 <1 24 2 Skin Rash¶ 12 <1 10 0 Table 11: Laboratory Abnormalities Worsening From Baseline Occurring in ≥20% of Patients with NSCLC Receiving TECENTRIQ in OAK Laboratory Abnormality TECENTRIQ Docetaxel All Grades
(%)Grades 3-4
(%)All Grades
(%)Grades 3-4
(%)Each test incidence is based on the number of patients who had both baseline and at least one on-study laboratory measurement available: TECENTRIQ (range: 546–585) and docetaxel (range: 532–560). Graded according to NCI CTCAE version 4.0 Hematology Anemia 67 3 82 7 Lymphocytopenia 49 14 60 21 Chemistry Hypoalbuminemia 48 4 50 3 Hyponatremia 42 7 31 6 Increased Alkaline Phosphatase 39 2 25 1 Increased AST 31 3 16 0.5 Increased ALT 27 3 14 0.5 Hypophosphatemia 27 5 23 4 Hypomagnesemia 26 1 21 1 Increased Creatinine 23 2 16 1 Metastatic Triple Negative Breast Cancer (TNBC)
The safety of TECENTRIQ in combination with paclitaxel protein-bound was evaluated in IMpassion130, a multicenter, international, randomized, double-blinded placebo-controlled trial in patients with locally advanced or metastatic TNBC who have not received prior chemotherapy for metastatic disease [see Clinical Studies (14.3)]. Patients received TECENTRIQ 840 mg (n=452) or placebo (n=438) intravenously followed by paclitaxel protein-bound (100 mg/m2) intravenously. For each 28 day cycle, TECENTRIQ was administered on days 1 and 15 and paclitaxel protein-bound was administered on days 1, 8, and 15 until disease progression or unacceptable toxicity. In the safety-evaluable population, the median duration of exposure to TECENTRIQ was 5.5 months (range: 0-32 months) and paclitaxel protein-bound was 5.1 months (range: 0-31.5 months) in the TECENTRIQ plus paclitaxel protein-bound arm. The median duration of exposure to placebo was 5.1 months (range: 0-25.1 months) and paclitaxel protein-bound was 5.0 months (range: 0-23.7 months) in the placebo plus paclitaxel protein-bound arm.
Fatal adverse reactions occurred in 1.3%) of patients in the TECENTRIQ and paclitaxel protein-bound arm; these included septic shock, mucosal inflammation, auto-immune hepatitis, aspiration, pneumonia, pulmonary embolism.
Serious adverse reactions occurred in 23% of patients. The most frequent serious adverse reactions were pneumonia (2%), urinary tract infection (1%), dyspnea (1%), and pyrexia (1%).
Adverse reactions leading to discontinuation of TECENTRIQ occurred in 6% (29/452) of patients in the TECENTRIQ and paclitaxel protein-bound arm. The most common adverse reaction leading to TECENTRIQ discontinuation was peripheral neuropathy (<1%).
Adverse reactions leading to interruption of TECENTRIQ occurred in 31% of patients; the most common (≥ 2%) were neutropenia, neutrophil count decreased, hyperthyroidism, and pyrexia.
Immune-related adverse reactions requiring systemic corticosteroid therapy occurred in 13% (59/452) of patients in the TECENTRIQ and paclitaxel protein-bound arm.
Tables 12 and 13 summarize adverse reactions and selected laboratory abnormalities worsening from baseline in the TECENTRIQ treated patients.
Table 12: Adverse Reactions Occurring in ≥10% of Patients with TNBC in IMpassion130 Adverse Reaction* TECENTRIQ with Paclitaxel Protein-Bound
(n=452)Placebo with Paclitaxel Protein-Bound
(n=438)All Grades
(%)Grades 3–4
(%)All Grades
(%)Grades 3–4
(%)- * Graded per NCI CTCAE v4.0
- † Includes peripheral neuropathy, peripheral sensory neuropathy, paresthesia, and polyneuropathy
Skin and Subcutaneous Tissue Alopecia 56 <1 58 <1 Rash 17 <1 16 <1 Pruritus 14 0 10 0 Nervous System Peripheral neuropathies† 47 9 44 5 Headache 23 <1 22 <1 Dysgeusia 14 0 14 0 Dizziness 14 0 11 0 General Fatigue 47 4 45 3.4 Pyrexia 19 <1 11 0 Peripheral Edema 15 <1 16 1.4 Asthenia 12 <1 11 <1 Gastrointestinal Nausea 46 1.1 38 1.8 Diarrhea 33 1.3 34 2.1 Constipation 25 <1 25 <1 Vomiting 20 <1 17 1.1 Abdominal pain 10 <1 12 <1 Respiratory, Thoracic, and Mediastinal Cough 25 0 19 0 Dyspnea 16 <1 15 <1 Metabolism and Nutrition Decreased Appetite 20 <1 18 <1 Musculoskeletal and Connective Tissue Arthralgia 18 <1 16 <1 Back pain 15 1.3 13 <1 Myalgia 14 <1 15 <1 Pain in extremity 11 <1 10 <1 Endocrine Hypothyroidism 14 0 3.4 0 Infections Urinary tract infection 12 <1 11 <1 Upper respiratory tract infection 11 1.1 9 0 Nasopharyngitis 11 0 8 0 Table 13: Laboratory Abnormalities Worsening from Baseline Occurring in ≥20% of Patients with TNBC in IMpassion130 Laboratory Abnormality TECENTRIQ with Paclitaxel Protein-Bound
(n=452)Placebo in combination with Paclitaxel Protein-Bound
(n=438)All Grades*
(%)†Grades 3–4
(%)All Grades*
(%)†Grades 3–4
(%)- * Graded per NCI CTCAE v4.0, except for increased creatinine which only includes patients with creatinine increase based on upper limit of normal definition for grade 1 events (NCI CTCAE v5.0).
- † Based on the number of patients with available baseline and at least one on-treatment laboratory test.
Hematology Decreased Hemoglobin 79 3.8 73 3 Decreased Leukocytes 76 14 71 9 Decreased Neutrophils 58 13 54 13 Decreased Lymphocytes 54 13 47 8 Increased Prothrombin INR 25 <1 25 <1 Chemistry Increased ALT 43 6 34 2.7 Increased AST 42 4.9 34 3.4 Decreased Calcium 28 1.1 26 <1 Decreased Sodium 27 4.2 25 2.7 Decreased Albumin 27 <1 25 <1 Increased Alkaline Phosphatase 25 3.3 22 2.7 Decreased Phosphate 22 3.6 19 3.7 Increased Creatinine 21 <1 16 <1 Small Cell Lung Cancer (SCLC)
The safety of TECENTRIQ with carboplatin and etoposide was evaluated in IMpower133, a randomized, multicenter, double-blind, placebo-controlled trial in which 198 patients with ES-SCLC received TECENTRIQ 1200 mg and carboplatin AUC 5 mg/mL/min on Day 1 and etoposide 100 mg/m2 intravenously on Days 1, 2 and 3 of each 21-day cycle for a maximum of 4 cycles, followed by TECENTRIQ 1200 mg every 3 weeks until disease progression or unacceptable toxicity [see Clinical Studies (14.4)]. Among 198 patients receiving TECENTRIQ, 32% were exposed for 6 months or longer and 12% were exposed for 12 months or longer.
Fatal adverse reactions occurred in 2% of patients receiving TECENTRIQ. These included pneumonia, respiratory failure, neutropenia, and death (1 patient each).
Serious adverse reactions occurred in 37% of patients receiving TECENTRIQ. Serious adverse reactions in >2% were pneumonia (4.5%), neutropenia (3.5%), febrile neutropenia (2.5%), and thrombocytopenia (2.5%).
TECENTRIQ was discontinued due to adverse reactions in 11% of patients. The most frequent adverse reaction requiring permanent discontinuation in >2% of patients was infusion-related reactions (2.5%).
Adverse reactions leading to interruption of TECENTRIQ occurred in 59% of patients; the most common (>1%) were neutropenia (22%), anemia (9%), leukopenia (7%), thrombocytopenia (5%), fatigue (4.0%), infusion-related reaction (3.5%), pneumonia (2.0%), febrile neutropenia (1.5%), increased ALT (1.5%), and nausea (1.5%).
Tables 14 and 15 summarize adverse reactions and laboratory abnormalities, respectively, in patients who received TECENTRIQ with carboplatin and etoposide in IMpower133.
Table 14: Adverse Reactions Occurring in ≥20% of Patients with SCLC Receiving TECENTRIQ in IMpower133 Adverse Reaction TECENTRIQ with Carboplatin and Etoposide
N = 198Placebo with Carboplatin and Etoposide
N = 196All Grades*
(%)Grades 3–4*
(%)All Grades*
(%)Grades 3–4*
(%)- * Graded per NCI CTCAE v4.0
General Fatigue/asthenia 39 5 33 3 Gastrointestinal Nausea 38 1 33 1 Constipation 26 1 30 1 Vomiting 20 2 17 3 Skin and Subcutaneous Tissue Alopecia 37 0 35 0 Metabolism and Nutrition Decreased appetite 27 1 18 0 Table 15: Laboratory Abnormalities Worsening from Baseline Occurring in ≥20% of Patients with SCLC Receiving TECENTRIQ in IMpower133 Laboratory Abnormality TECENTRIQ with Carboplatin and Etoposide Placebo with Carboplatin and Etoposide All Grades
(%)Grades 3–4
(%)All Grades
(%)Grades 3–4
(%)Each test incidence is based on the number of patients who had both baseline and at least one on-study laboratory measurement available: TECENTRIQ (range: 181-193); Placebo (range: 181-196). Graded per NCI CTCAE v4.0 - * NA= Not applicable. NCI CTCAE v4.0 does not include these laboratories.
Hematology Anemia 94 17 93 19 Neutropenia 73 45 76 48 Thrombocytopenia 58 20 53 17 Lymphopenia 46 14 38 11 Chemistry Hyperglycemia 67 10 65 8 Increased Alkaline Phosphatase 38 1 35 2 Hyponatremia 34 15 33 11 Hypoalbuminemia 32 1 30 0 Decreased TSH3 28 NA* 15 NA* Hypomagnesemia 31 5 35 6 Hypocalcemia 26 3 28 5 Increased ALT 26 3 31 1 Increased AST 22 1 21 2 Increased Blood Creatinine 22 4 15 1 Hyperphosphatemia3 21 NA* 23 NA* Increased TSH3 21 NA* 7 NA* 6.2 Immunogenicity
As with all therapeutic proteins, there is a potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to atezolizumab in the studies described above with the incidence of antibodies in other studies or to other products may be misleading.
Among 565 patients with NSCLC in OAK, 30% tested positive for treatment-emergent anti-drug antibodies (ADA) at one or more post-dose time points. The median onset time to ADA formation was 3 weeks. The ability of these binding ADA to neutralize atezolizumab is unknown. Patients who tested positive for treatment-emergent ADA also had decreased systemic atezolizumab exposure [see Clinical Pharmacology (12.3)]. Exploratory analyses showed that the subset of patients who were ADA positive by week 4 (21%; 118/560) appeared to have less efficacy (effect on overall survival) as compared to patients who tested negative for treatment-emergent ADA by week 4 [see Clinical Studies (14.2)]. The presence of ADA did not have a clinically significant effect on the incidence or severity of adverse reactions.
Among 275 patients with urothelial carcinoma in IMvigor210 (Cohort 2), 42% tested positive for treatment-emergent ADA at one or more post-dose time points. Among 111 patients in IMvigor210 (Cohort 1), 48% tested positive for treatment-emergent ADA at one or more post-dose time points. Patients who tested positive for treatment-emergent ADA also had decreased systemic atezolizumab exposures. The presence of ADA did not have a clinically significant effect on the incidence or severity of adverse reactions.
Among 364 ADA-evaluable patients with NSCLC who received TECENTRIQ with bevacizumab, paclitaxel and carboplatin in IMpower150, 36% (n=132) tested positive for treatment-emergent ADA at one or more post-dose time points and 83% of these 132 patients tested ADA positive prior to receiving the second dose of atezolizumab. The ability of these binding ADA to neutralize atezolizumab is unknown. Patients who tested positive for treatment-emergent ADA had lower systemic atezolizumab exposure as compared to patients who were ADA negative [see Clinical Pharmacology (12.3)]. The presence of ADA did not increase the incidence or severity of adverse reactions [see Clinical Studies (14.2)].
Among 434 patients with TNBC in IMpassion130, 13% tested positive for treatment-emergent ADA at one or more post-dose time points. Among 178 patients in PD-L1 positive subgroup with TNBC in IMpassion130, 12% tested positive for treatment-emergent ADA at one or more post-dose time points. Patients who tested positive for treatment-emergent ADA had decreased systemic atezolizumab exposure [see Clinical Pharmacology (12.3)]. There are insufficient numbers of patients in the PD-L1 positive subgroup with ADA to determine whether ADA alters the efficacy of atezolizumab. The presence of ADA did not have a clinically significant effect on the incidence or severity of adverse reactions.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on its mechanism of action [see Clinical Pharmacology (12.1)], TECENTRIQ can cause fetal harm when administered to a pregnant woman. There are no available data on the use of TECENTRIQ in pregnant women.
Animal studies have demonstrated that inhibition of the PD-L1/PD-1 pathway can lead to increased risk of immune-related rejection of the developing fetus resulting in fetal death (see Data). Advise females of reproductive potential of the potential risk to a fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
Animal reproduction studies have not been conducted with TECENTRIQ to evaluate its effect on reproduction and fetal development. A literature-based assessment of the effects on reproduction demonstrated that a central function of the PD-L1/PD-1 pathway is to preserve pregnancy by maintaining maternal immune tolerance to a fetus. Blockage of PD-L1 signaling has been shown in murine models of pregnancy to disrupt tolerance to a fetus and to result in an increase in fetal loss; therefore, potential risks of administering TECENTRIQ during pregnancy include increased rates of abortion or stillbirth. As reported in the literature, there were no malformations related to the blockade of PD-L1/PD-1 signaling in the offspring of these animals; however, immune-mediated disorders occurred in PD-1 and PD-L1 knockout mice. Based on its mechanism of action, fetal exposure to atezolizumab may increase the risk of developing immune-mediated disorders or altering the normal immune response.
8.2 Lactation
Risk Summary
There is no information regarding the presence of atezolizumab in human milk, the effects on the breastfed infant, or the effects on milk production. As human IgG is excreted in human milk, the potential for absorption and harm to the infant is unknown. Because of the potential for serious adverse reactions in breastfed infants from TECENTRIQ, advise women not to breastfeed during treatment and for at least 5 months after the last dose.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Verify pregnancy status in females of reproductive potential prior to initiating TECENTRIQ [see Use in Specific Populations (8.1)].
Contraception
Females
Based on its mechanism of action, TECENTRIQ can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)]. Advise females of reproductive potential to use effective contraception during treatment with TECENTRIQ and for at least 5 months following the last dose.
Infertility
Females
Based on animal studies, TECENTRIQ may impair fertility in females of reproductive potential while receiving treatment [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of TECENTRIQ have not been established in pediatric patients.
8.5 Geriatric Use
Of 2481 patients with urothelial carcinoma, lung cancer, and triple-negative breast cancer who were treated with TECENTRIQ in clinical studies, 45% were 65 years and over and 11% were 75 years and over. No overall differences in safety or effectiveness were observed between patients aged 65 years or older and younger patients.
-
11 DESCRIPTION
Atezolizumab is a programmed cell death ligand 1 (PD-L1) blocking antibody. Atezolizumab is an Fc-engineered, humanized, non-glycosylated IgG1 kappa immunoglobulin that has a calculated molecular mass of 145 kDa.
TECENTRIQ (atezolizumab) injection for intravenous use is a sterile, preservative-free, colorless to slightly yellow solution in single-dose vials. Each 20 mL vial contains 1200 mg of atezolizumab and is formulated in glacial acetic acid (16.5 mg), L-histidine (62 mg), polysorbate 20 (8 mg), and sucrose (821.6 mg), with a pH of 5.8. Each 14 mL vial contains 840 mg of atezolizumab and is formulated in glacial acetic acid (11.5 mg), L-histidine (43.4 mg), polysorbate 20 (5.6 mg), and sucrose (575.1 mg) with a pH of 5.8.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
PD-L1 may be expressed on tumor cells and/or tumor infiltrating immune cells and can contribute to the inhibition of the anti-tumor immune response in the tumor microenvironment. Binding of PD-L1 to the PD-1 and B7.1 receptors found on T cells and antigen presenting cells suppresses cytotoxic T-cell activity, T-cell proliferation and cytokine production.
Atezolizumab is a monoclonal antibody that binds to PD-L1 and blocks its interactions with both PD-1 and B7.1 receptors. This releases the PD-L1/PD-1 mediated inhibition of the immune response, including activation of the anti-tumor immune response without inducing antibody-dependent cellular cytotoxicity. In syngeneic mouse tumor models, blocking PD-L1 activity resulted in decreased tumor growth.
12.3 Pharmacokinetics
Patients' exposure to atezolizumab increased dose proportionally over the dose range of 1 mg/kg to 20 mg/kg, including a dose of 1200 mg administered every 3 weeks. The clearance (CV%) was 0.20 L/day (29%), the volume of distribution at steady state was 6.9 L, and the terminal half-life was 27 days. Steady state was achieved after 6 to 9 weeks following multiple doses. The systemic accumulation ratio for every 2 weeks administration and every 3 weeks administration was 3.3- and 1.9- fold, respectively. Atezolizumab clearance was found to decrease over time, with a mean maximal reduction (CV%) from baseline value of approximately 17% (41%); however, the decrease in clearance was not considered clinically relevant.
Specific Populations
Age (21 to 89 years), body weight, sex, albumin levels, tumor burden, region or race, mild or moderate renal impairment [estimated glomerular filtration rate (eGFR) 30 to 89 mL/min/1.73 m2], mild hepatic impairment (bilirubin ≤ ULN and AST > ULN or bilirubin > 1 to 1.5 × ULN and any AST), level of PD-L1 expression, or performance status had no clinically significant effect on the systemic exposure of atezolizumab. In OAK, IMpower150 (TECENTRIQ, bevacizumab, paclitaxel, carboplatin arm only), and IMpassion130 (TECENTRIQ and paclitaxel protein-bound) atezolizumab clearance in patients who tested positive for treatment-emergent anti-drug antibodies (ADA) was 25%, 18%, and 22% higher, respectively, as compared to clearance in patients who tested negative for treatment-emergent ADA.
The effect of severe renal impairment or moderate or severe hepatic impairment on the pharmacokinetics of atezolizumab is unknown.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No studies have been performed to test the potential of atezolizumab for carcinogenicity or genotoxicity.
Animal fertility studies have not been conducted with atezolizumab; however, an assessment of the male and female reproductive organs was included in a 26-week, repeat-dose toxicity study in cynomolgus monkeys. Weekly administration of atezolizumab to female monkeys at the highest dose tested caused an irregular menstrual cycle pattern and a lack of newly formed corpora lutea in the ovaries. This effect occurred at an estimated AUC approximately 6 times the AUC in patients receiving the recommended dose and was reversible. There was no effect on the male monkey reproductive organs.
13.2 Animal Toxicology and/or Pharmacology
In animal models, inhibition of PD-L1/PD-1 signaling increased the severity of some infections and enhanced inflammatory responses. M. tuberculosis-infected PD-1 knockout mice exhibit markedly decreased survival compared with wild-type controls, which correlated with increased bacterial proliferation and inflammatory responses in these animals. PD-L1 and PD-1 knockout mice and mice receiving PD-L1 blocking antibody have also shown decreased survival following infection with lymphocytic choriomeningitis virus.
-
14 CLINICAL STUDIES
14.1 Urothelial Carcinoma
Cisplatin-Ineligible Patients with Locally Advanced or Metastatic Urothelial Carcinoma
The efficacy of TECENTRIQ was investigated in IMvigor210 (Cohort 1) (NCT02951767), a multicenter, open-label, single-arm trial that included 119 patients with locally advanced or metastatic urothelial carcinoma who were ineligible for cisplatin-containing chemotherapy and were either previously untreated or had disease progression at least 12 months after neoadjuvant or adjuvant chemotherapy. Patients were considered cisplatin-ineligible if they met any one of the following criteria at study entry: impaired renal function [creatinine clearance (CLcr) of 30 to 59 mL/min], Eastern Cooperative Oncology Group (ECOG) performance status (PS) of 2, hearing loss of ≥ 25 decibels (dB) at two contiguous frequencies, or Grades 2-4 peripheral neuropathy. This study excluded patients who had: a history of autoimmune disease; active or corticosteroid-dependent brain metastases; administration of a live, attenuated vaccine within 28 days prior to enrollment; or administration of systemic immunostimulatory agents within 6 weeks or systemic immunosuppressive medications within 2 weeks prior to enrollment. Patients received TECENTRIQ 1200 mg as an intravenous infusion every 3 weeks until unacceptable toxicity or disease progression. Tumor response assessments were conducted every 9 weeks for the first 54 weeks and every 12 weeks thereafter. Major efficacy outcome measures included confirmed overall response rate (ORR) as assessed by independent review facility (IRF) using Response Evaluation Criteria in Solid Tumors (RECIST v1.1), duration of response (DoR) and overall survival (OS).
In this study, the median age was 73 years, 81% were male, and 91% were White. Thirty-five percent of patients had non-bladder urothelial carcinoma and 66% had visceral metastases. Eighty percent of patients had an ECOG PS of 0 or 1. Reasons for ineligibility for cisplatin-containing chemotherapy were: 70% had impaired renal function, 20% had an ECOG PS of 2, 14% had a hearing loss of ≥ 25dB, and 6% had Grades 2-4 peripheral neuropathy at baseline. Twenty percent of patients had disease progression following prior platinum-containing neoadjuvant or adjuvant chemotherapy.
Tumor specimens were evaluated prospectively using the VENTANA PD-L1 (SP142) Assay at a central laboratory, and the results were used to define subgroups for pre-specified analyses. Of the 119 patients, 27% were classified as having PD-L1 expression of ≥ 5% (defined as PD-L1 stained tumor-infiltrating immune cells [IC] covering ≥ 5% of the tumor area). The remaining 73% of patients were classified as having PD-L1 expression of < 5% (PD-L1 stained tumor-infiltrating IC covering < 5% of the tumor area).
Among the 32 patients with PD-L1 expression of ≥ 5%, median age was 67 years, 81% were male, 19% female, and 88% were White. Twenty-eight percent of patients had non-bladder urothelial carcinoma and 56% had visceral metastases. Seventy-two percent of patients had an ECOG PS of 0 or 1. Reasons for ineligibility for cisplatin-containing chemotherapy were: 66% had impaired renal function, 28% had an ECOG PS of 2, 16% had a hearing loss ≥ 25 dB, and 9% had Grades 2-4 peripheral neuropathy at baseline. Thirty-one percent of patients had disease progression following prior platinum-containing neoadjuvant or adjuvant chemotherapy.
Confirmed ORR in all patients and the two PD-L1 subgroups are summarized in Table 16. The median follow-up time for this study was 14.4 months. In 24 patients with disease progression following neoadjuvant or adjuvant therapy, the ORR was 33% (95% CI: 16%, 55%).
Table 16: Efficacy Results in IMvigor210 (Cohort 1) All Patients PD-L1 Expression Subgroups N = 119 PD-L1
Expression of < 5% in ICs*
N = 87PD-L1
Expression of ≥ 5% in ICs*
N = 32NR = Not reached - * PD-L1 expression in tumor-infiltrating immune cells (ICs)
- † Denotes a censored value
Number of IRF-assessed Confirmed Responders 28 19 9 ORR % (95% CI) 23.5% (16.2, 32.2) 21.8% (13.7, 32) 28.1% (13.8, 46.8) Complete Response (CR) (%) 6.7% 6.9% 6.3% Partial Response (PR) (%) 16.8% 14.9% 21.9% Median DoR, months NR NR NR (range) (3.7, 16.6†) (3.7, 16.6†) (8.1, 15.6†) IMvigor130 (NCT02807636) is an ongoing multicenter, randomized study in previously untreated patients with metastatic urothelial carcinoma who are eligible for platinum-containing chemotherapy. The study contains three arms: TECENTRIQ monotherapy, TECENTRIQ with platinum-based chemotherapy (i.e., cisplatin or carboplatin with gemcitabine), and platinum-based chemotherapy alone (comparator). Both cisplatin-eligible and cisplatin-ineligible patients are included in the study. Tumor specimens were evaluated prospectively using the VENTANA PD-L1 (SP142) Assay at a central laboratory. The independent Data Monitoring Committee (iDMC) for the study conducted a review of early data and found that patients classified as having PD-L1 expression of <5% when treated with TECENTRIQ monotherapy had decreased survival compared to those who received platinum-based chemotherapy. The iDMC recommended closure of the monotherapy arm to further accrual of patients with low PD-L1 expression, however, no other changes were recommended for the study, including any change of therapy for patients who had already been randomized to and were receiving treatment in the monotherapy arm.
Previously Treated Locally Advanced or Metastatic Urothelial Carcinoma
The efficacy of TECENTRIQ was investigated in IMvigor210 (Cohort 2) (NCT02108652), a multicenter, open-label, single-arm trial that included 310 patients with locally advanced or metastatic urothelial carcinoma who had disease progression during or following a platinum-containing chemotherapy regimen or who had disease progression within 12 months of treatment with a platinum-containing neoadjuvant or adjuvant chemotherapy regimen. This study excluded patients who had: a history of autoimmune disease, active or corticosteroid-dependent brain metastases, administration of a live, attenuated vaccine within 28 days prior to enrollment, or administration of systemic immunostimulatory agents within 6 weeks or systemic immunosuppressive medications within 2 weeks prior to enrollment. Patients received TECENTRIQ 1200 mg intravenously every 3 weeks until unacceptable toxicity or either radiographic or clinical progression. Tumor response assessments were conducted every 9 weeks for the first 54 weeks and every 12 weeks thereafter. Major efficacy outcome measures included confirmed ORR as assessed by IRF using RECIST v1.1 and DoR.
In this study, the median age was 66 years, 78% were male, 91% of patients were White. Twenty-six percent had non-bladder urothelial carcinoma and 78% of patients had visceral metastases. Sixty-two percent of patients had an ECOG PS of 1 and 35% of patients had a baseline CLcr < 60 mL/min. Nineteen percent of patients had disease progression following prior platinum-containing neoadjuvant or adjuvant chemotherapy. Forty-one percent of patients had received 2 or more prior systemic regimens in the metastatic setting. Seventy-three percent of patients received prior cisplatin, 26% had prior carboplatin, and 1% were treated with other platinum-based regimens.
Tumor specimens were evaluated prospectively using the VENTANA PD-L1 (SP142) Assay at a central laboratory and the results were used to define subgroups for pre-specified analyses. Of the 310 patients, 32% were classified as having PD-L1 expression of ≥ 5%. The remaining 68% of patients were classified as having PD-L1 expression of < 5%.
Confirmed ORR and median DOR in all patients and the two PD-L1 subgroups are summarized in Table 17. The median follow-up time for this study was 32.9 months. In 59 patients with disease progression following neoadjuvant or adjuvant therapy, the ORR was 22.0% (95% CI: 12.3%, 34.7%).
Table 17: Efficacy Results in IMvigor210 (Cohort 2) All Patients PD-L1 Expression Subgroups N = 310 PD-L1
Expression of < 5% in IC*
N = 210PD-L1
Expression of ≥ 5% in IC*
N = 100- * PD-L1 expression in tumor-infiltrating immune cells (IC)
- † Denotes a censored value
Number of IRF-assessed Confirmed Responders 46 20 26 ORR % (95% CI) 14.8% (11.2, 19.3) 9.5% (5.9, 14.3) 26% (17.7, 35.7) Complete Response (CR) (%) 5.5% 2.4% 12.0% Partial Response (PR) (%) 9.4% 7.1% 14.0% Median DOR, months 27.7 20.9 29.7 (range) (2.1†, 33.4†) (2.1†, 33.4†) (4.2, 31.2†) 14.2 Non-Small Cell Lung Cancer
Metastatic Chemotherapy-Naive Non-Squamous NSCLC
IMpower150
The efficacy of TECENTRIQ with bevacizumab, paclitaxel, and carboplatin was evaluated in IMpower150 (NCT02366143), a multicenter, international, randomized (1:1:1), open-label trial in patients with metastatic non-squamous NSCLC. Patients with stage IV non-squamous NSCLC who had received no prior chemotherapy for metastatic disease, but could have received prior EGFR or ALK kinase inhibitor if appropriate, regardless of PD-L1 or T-effector gene (tGE) status and ECOG performance status 0 or 1 were eligible. The trial excluded patients with a history of autoimmune disease, administration of a live attenuated vaccine within 28 days prior to randomization, active or untreated CNS metastases, administration of systemic immunostimulatory agents within 4 weeks or systemic immunosuppressive medications within 2 weeks prior to randomization, or clear tumor infiltration into the thoracic great vessels or clear cavitation of pulmonary lesions as seen on imaging. Randomization was stratified by sex, presence of liver metastases, and PD-L1 expression status on tumor cells (TC) and tumor-infiltrating immune cells (IC) as follows: TC3 and any IC vs. TC0/1/2 and IC2/3 vs. TC0/1/2 and IC0/1. Patients were randomized to one of the following three treatment arms.
- Arm A: TECENTRIQ 1200 mg, paclitaxel 175 mg/m2 or 200 mg/m2 and carboplatin AUC 6 mg/mL/min on Day 1 of each 21-day cycle for a maximum of 4 or 6 cycles
- Arm B: TECENTRIQ 1200 mg, bevacizumab 15 mg/kg, paclitaxel 175 mg/m2 or 200 mg/m2, and carboplatin AUC 6 mg/mL/min on Day 1 of each 21-day cycle for a maximum of 4 or 6 cycles
- Arm C: bevacizumab 15 mg/kg, paclitaxel 175 mg/m2 or 200 mg/m2, and carboplatin AUC 6 mg/mL/min on Day 1 of each 21-day cycle for a maximum of 4 or 6 cycles
Patients who had not experienced disease progression following the completion or cessation of platinum-based chemotherapy, received:
- Arm A: TECENTRIQ 1200 mg intravenously on Day 1 of each 21-day cycle until disease progression or unacceptable toxicity
- Arm B: TECENTRIQ 1200 mg and bevacizumab 15 mg/kg intravenously on Day 1 of each 21-day cycle until disease progression or unacceptable toxicity
- Arm C: bevacizumab 15 mg/kg intravenously on Day 1 of each 21-day cycle until disease progression or unacceptable toxicity
Tumor assessments were conducted every 6 weeks for the first 48 weeks following Cycle 1, Day 1 and then every 9 weeks thereafter. Tumor specimens were evaluated prior to randomization for PD-L1 tumor expression using the VENTANA PD-L1 (SP142) assay at a central laboratory. Tumor tissue was collected at baseline for expression of tGE signature and evaluation was performed using a clinical trial assay in a central laboratory prior to the analysis of efficacy outcome measures.
Major efficacy outcome measures for comparison of Arms B and C were progression free survival (PFS) by RECIST v1.1 in the tGE-WT (patients with high expression of T-effector gene signature [tGE], excluding those with EGFR- and ALK-positive NSCLC [WT]) and in the ITT-WT subpopulations and overall survival (OS) in the ITT-WT subpopulation. Additional efficacy outcome measures for comparison of Arms B and C or Arms A and C were PFS and OS in the ITT population, OS in the tGE-WT subpopulation, and ORR/DoR in the tGE-WT and ITT-WT subpopulations.
A total of 1202 patients were enrolled across the three arms of whom 1045 were in the ITT-WT subpopulation and 447 were in the tGE-WT subpopulation. The demographic information is limited to the 800 patients enrolled in Arms B and C where efficacy has been demonstrated. The median age was 63 years (range: 31 to 90), and 60% of patients were male. The majority of patients were White (82%), 13% of patients were Asian, 10% were Hispanic, and 2% of patients were Black. Clinical sites in Asia (enrolling 13% of the study population) received paclitaxel at a dose of 175 mg/m2 while the remaining 87% received paclitaxel at a dose of 200 mg/m2. Approximately 14% of patients had liver metastases at baseline, and most patients were current or previous smokers (80%). Baseline ECOG performance status was 0 (43%) or 1 (57%). PD-L1 was TC3 and any IC in 12%, TC0/1/2 and IC2/3 in 13%, and TC0/1/2 and IC0/1 in 75%. The demographics for the 696 patients in the ITT-WT subpopulation were similar to the ITT population except for the absence of patients with EGFR- or ALK-positive NSCLC.
The trial demonstrated a statistically significant improvement in PFS between Arms B and C in both the tGE-WT and ITT-WT subpopulations, but did not demonstrate a significant difference for either subpopulation between Arms A and C based on the final PFS analyses. In the interim analysis of OS, a statistically significant improvement was observed for Arm B compared to Arm C, but not for Arm A compared to Arm C. Efficacy results for the ITT-WT subpopulation are presented in Table 18 and Figure 1.
Table 18: Efficacy Results in ITT-WT Population in IMpower150 Arm C: Bevacizumab, Paclitaxel and Carboplatin Arm B: TECENTRIQ with Bevacizumab, Paclitaxel, and Carboplatin Arm A: TECENTRIQ with Paclitaxel, and Carboplatin N = 337 N = 359 N = 349 CI=confidence interval - * Based on OS interim analysis .
- † Stratified by sex, presence of liver metastases, and PD-L1 expression status on TC and IC
- ‡ Based on the stratified log-rank test compared to Arm C
- § Compared to the allocated α=0.0174 (two sided) for this interim analysis.
- ¶ Compared to the allocated α=0.0128 (two sided) for this interim analysis.
- # As determined by independent review facility (IRF) per RECIST v1.1 (Response Evaluation Criteria in Solid Tumors v1.1)
- Þ Compared to the allocated α=0.006 (two sided) for the final PFS analysis.
Overall Survival* Deaths (%) 197 (59%) 179 (50%) 179 (51%) Median, months 14.7 19.2 19.4 (95% CI) (13.3, 16.9) (17.0, 23.8) (15.7, 21.3) Hazard ratio† (95% CI) --- 0.78 (0.64, 0.96) 0.84 (0.72, 1.08) p-value‡ --- 0.016§ 0.204¶ Progression-Free Survival# Number of events (%) 247 (73%) 247 (69%) 245 (70%) Median, months 7.0 8.5 6.7 (95% CI) (6.3, 7.9) (7.3, 9.7) (5.6, 6.9) Hazard ratio† (95% CI) --- 0.71 (0.59, 0.85) 0.94 (0.79, 1.13) p-value‡ --- 0.0002Þ 0.5219 Objective Response Rate# Number of responders (%) 142 (42%) 196 (55%) 150 (43%) (95% CI) (37, 48) (49, 60) (38, 48) Complete response 3 (1%) 14 (4%) 9 (3%) Partial response 139 (41%) 182 (51%) 141 (40%) Duration of Response# n = 142 n = 196 n = 150 Median (months) 6.5 10.8 9.5 (95% CI) (5.6, 7.6) (8.4, 13.9) (7.0, 13.0) Figure 1: Kaplan-Meier Curves for Overall Survival in ITT-WT Population in IMpower150
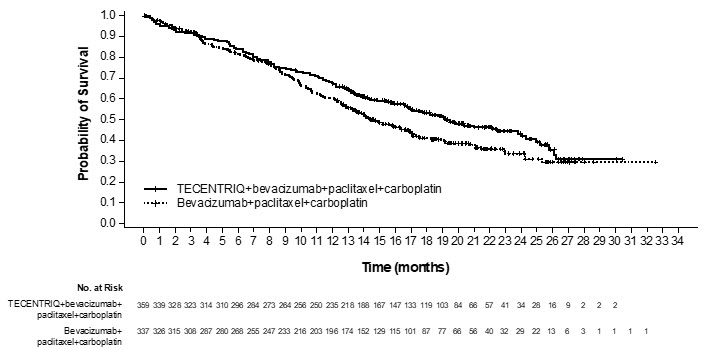
Exploratory analyses showed that the subset of patients in the four drug regimen arm who were ADA positive by week 4 (30%) appeared to have similar efficacy (effect on overall survival) as compared to patients who tested negative for treatment-emergent ADA by week 4 (70%) [see Adverse Reactions (6.2), Clinical Pharmacology (12.3)]. In an exploratory analysis, propensity score matching was conducted to compare ADA positive patients in the TECENTRIQ, bevacizumab, paclitaxel, and carboplatin arm with a matched population in the bevacizumab, paclitaxel, and carboplatin arm. Similarly ADA negative patients in the TECENTRIQ, bevacizumab, paclitaxel, and carboplatin arm were compared with a matched population in the bevacizumab, paclitaxel, and carboplatin arm. Propensity score matching factors were: baseline sum of longest tumor size (BSLD), baseline ECOG, baseline albumin, baseline LDH, sex, tobacco history, metastatic site, TC level, and IC level. The hazard ratio comparing the ADA-positive subgroup with its matched control was 0.69 (95% CI: 0.44, 1.07). The hazard ratio comparing the ADA-negative subgroup with its matched control was 0.64 (95% CI: 0.46, 0.90).
IMpower130
The efficacy of TECENTRIQ with paclitaxel protein-bound and carboplatin was evaluated in IMpower130 (NCT02367781), a multicenter, randomized (2:1), open-label trial in patients with stage IV non-squamous NSCLC. Patients with Stage IV non-squamous NSCLC who had received no prior chemotherapy for metastatic disease, but could have received prior EGFR or ALK kinase inhibitor, if appropriate, were eligible. The trial excluded patients with history of autoimmune disease, administration of live attenuated vaccine within 28 days prior to randomization, administration of immunostimulatory agents within 4 weeks or systemic immunosuppressive medications within 2 weeks prior to randomization, and active or untreated CNS metastases. Randomization was stratified by sex, presence of liver metastases, and PD-L1 tumor expression according to the VENTANA PD-L1 (SP142) assay as follows: TC3 and any IC vs. TC0/1/2 and IC2/3 vs. TC0/1/2 and IC0/1. Patients were randomized to one of the following treatment regimens:
- TECENTRIQ 1200 mg on Day 1, paclitaxel protein-bound 100 mg/m2 on Days 1, 8, and 15, and carboplatin AUC 6 mg/mL/min on Day 1 of each 21-day cycle for a maximum of 4 or 6 cycles followed by TECENTRIQ 1200 mg once every 3 weeks until disease progression or unacceptable toxicity, or
- Paclitaxel protein-bound 100 mg/m2 on Days 1, 8 and 15 and carboplatin AUC 6 mg/mL/min on Day 1 of each 21-day cycle for a maximum of 4 or 6 cycles followed by best supportive care or pemetrexed.
Tumor assessments were conducted every 6 weeks for the first 48 weeks, then every 9 weeks thereafter. Major efficacy outcome measures were PFS by RECIST v1.1 and OS in the subpopulation of patients evaluated for and documented to have no EGFR or ALK genomic tumor aberrations (ITT-WT).
A total of 724 patients were enrolled; of these, 681 (94%) were in the ITT-WT population. The median age was 64 years (range: 18 to 86) and 59% were male. The majority of patients were white (90%), 2% of patients were Asian, 5% were Hispanic, and 4% were Black. Baseline ECOG performance status was 0 (41%) or 1 (58%). Most patients were current or previous smokers (90%). PD-L1 tumor expression was TC0/1/2 and IC0/1 in 73%; TC3 and any IC in 14%; and TC0/1/2 and IC2/3 in 13%.
Efficacy results for the ITT-WT population are presented in Table 19 and Figure 2.
Table 19: Efficacy Results from IMpower130 TECENTRIQ with Paclitaxel Protein-Bound and Carboplatin Paclitaxel Protein-Bound and Carboplatin CI=confidence interval - * Based on OS interim analysis
- † Stratified by sex and PD-L1 tumor expression on tumor cells (TC) and tumor infiltrating cells (IC)
- ‡ Based on the stratified log-rank test
- § Compared to the allocated α=0.0428 (two sided) for this interim analysis
- ¶ As determined by independent review facility (IRF) per RECIST v1.1 (Response Evaluation Criteria in Solid Tumors v1.1)
- # Compared to the allocated α=0.006 (two sided) for the final PFS analysis
- Þ Confirmed response
Overall Survival* n=453 n=228 Deaths (%) 228 (50%) 131 (57%) Median, months 18.6 13.9 (95% CI) (15.7, 21.1) (12.0, 18.7) Hazard ratio† (95% CI) 0.80 (0.64, 0.99) p-value‡ 0.0384§ Progression-Free Survival¶ n=453 n=228 Number of events (%) 330 (73%) 177 (78%) Median, months 7.2 6.5 (95% CI) (6.7, 8.3) (5.6, 7.4) Hazard ratio† (95% CI) 0.75 (0.63, 0.91) p-value‡ 0.0024# Overall Response Rate¶,Þ n=453 n=228 Number of responders (%) 207 (46%) 74 (32%) (95% CI) (41, 50) (26, 39) Complete response 22 (5%) 2 (1%) Partial response 185 (41%) 72 (32%) Duration of Response¶,Þ n=207 n=74 Median (months) 10.8 7.8 (95% CI) (9.0, 14.4) (6.8, 10.9) Figure 2: Kaplan-Meier Curves for Overall Survival in IMpower130 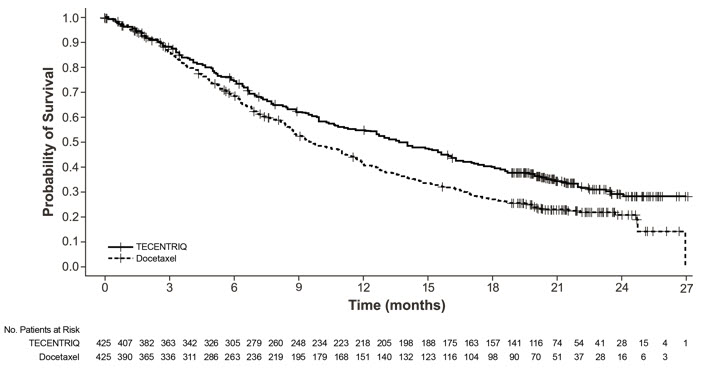
Previously Treated Metastatic NSCLC
The efficacy of TECENTRIQ was evaluated in a multicenter, international, randomized (1:1), open-label study (OAK; NCT02008227) conducted in patients with locally advanced or metastatic NSCLC whose disease progressed during or following a platinum-containing regimen. Patients with a history of autoimmune disease, symptomatic or corticosteroid-dependent brain metastases, or requiring systemic immunosuppression within 2 weeks prior to enrollment were ineligible. Randomization was stratified by PD-L1 expression tumor-infiltrating immune cells (IC), the number of prior chemotherapy regimens (1 vs. 2), and histology (squamous vs. non-squamous).
Patients were randomized to receive TECENTRIQ 1200 mg intravenously every 3 weeks until unacceptable toxicity, radiographic progression, or clinical progression or docetaxel 75 mg/m2 intravenously every 3 weeks until unacceptable toxicity or disease progression. Tumor assessments were conducted every 6 weeks for the first 36 weeks and every 9 weeks thereafter. Major efficacy outcome measure was overall survival (OS) in the first 850 randomized patients and OS in the subgroup of patients with PD-L1-expressing tumors (defined as ≥ 1% PD-L1 expression on tumor cells [TC] or immune cells [IC]). Additional efficacy outcome measures were OS in all randomized patients (n = 1225), OS in subgroups based on PD-L1 expression, overall response rate (ORR), and progression free survival as assessed by the investigator per RECIST v.1.1.
Among the first 850 randomized patients, the median age was 64 years (33 to 85 years) and 47% were ≥ 65 years old; 61% were male; 70% were White and 21% were Asian; 15% were current smokers and 67% were former smokers; and 37% had baseline ECOG PS of 0 and 63% had a baseline ECOG PS of 1. Nearly all (94%) had metastatic disease, 74% had non-squamous histology, 75% had received only one prior platinum-based chemotherapy regimen, and 55% of patients had PD-L1-expressing tumors.
Efficacy results are presented in Table 20 and Figure 3.
Table 20: Efficacy Results in OAK TECENTRIQ Docetaxel CI=confidence interval; NE=not estimable - * Stratified by PD-L1 expression in tumor infiltrating immune cells, the number of prior chemotherapy regimens, and histology
- † Based on the stratified log-rank test
- ‡ Compared to the pre-specified allocated α of 0.03 for this analysis
- § Per RECIST v1.1 (Response Evaluation Criteria in Solid Tumors v1.1)
- ¶ Compared to the allocated α of 0.0177 for this interim analysis based on 86% information using O'Brien-Fleming boundary
Overall Survival in first 850 patients Number of patients N=425 N=425 Deaths (%) 271 (64%) 298 (70%) Median, months 13.8 9.6 (95% CI) (11.8, 15.7) (8.6, 11.2) Hazard ratio* (95% CI) 0.74 (0.63, 0.87) p-value† 0.0004‡ Progression-Free Survival Number of Patients N=425 N=425 Events (%) 380 (89%) 375 (88%) Progression (%) 332 (78%) 290 (68%) Deaths (%) 48 (11%) 85 (20%) Median, months 2.8 4.0 (95% CI) (2.6, 3.0) (3.3, 4.2) Hazard ratio* (95% CI) 0.95 (0.82, 1.10) Overall Response Rate § Number of Patients N=425 N=425 ORR, n (%) 58 (14%) 57 (13%) (95% CI) (11%, 17%) (10%, 17%) Complete response 6 (1%) 1 (0.2%) Partial response 52 (12%) 56 (13%) Duration of Response‡ N=58 N=57 Median (months) 16.3 6.2 (95% CI) (10.0, NE) (4.9, 7.6) Overall Survival in all 1225 patients Number of patients N=613 N=612 Deaths (%) 384 (63%) 409 (67%) Median, months 13.3 9.8 (95% CI) (11.3, 14.9) (8.9, 11.3) Hazard ratio* (95% CI) 0.79 (0.69, 0.91) p-value† 0.0013¶ Figure 3: Kaplan-Meier Curves of Overall Survival in the First 850 Patients Randomized in OAK 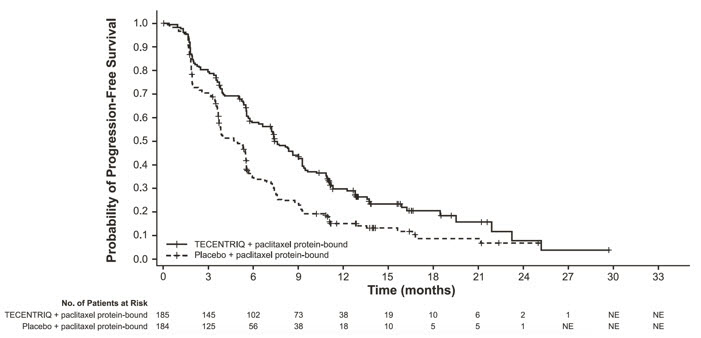
Tumor specimens were evaluated prospectively using the VENTANA PD-L1 (SP142) Assay at a central laboratory and the results were used to define the PD-L1 expression subgroups for pre-specified analyses. Of the 850 patients, 16% were classified as having high PD-L1 expression, defined as having PD-L1 expression on ≥ 50% of TC or ≥ 10% of IC. In an exploratory efficacy subgroup analysis of OS based on PD-L1 expression, the hazard ratio was 0.41 (95% CI: 0.27, 0.64) in the high PD-L1 expression subgroup and 0.82 (95% CI: 0.68, 0.98) in patients who did not have high PD-L1 expression.
Exploratory analyses showed that the subset of patients who were ADA positive by week 4 (21%) appeared to have less efficacy (effect on overall survival) as compared to patients who tested negative for treatment-emergent ADA by week 4 (79%) [see Adverse Reactions (6.2), Clinical Pharmacology (12.3)]. ADA positive patients by week 4 appeared to have similar OS compared to docetaxel-treated patients. In an exploratory analysis, propensity score matching was conducted to compare ADA positive patients in the atezolizumab arm with a matched population in the docetaxel arm and ADA negative patients in the atezolizumab arm with a matched population in the docetaxel arm. Propensity score matching factors were: baseline sum of longest tumor size (BSLD), baseline ECOG, histology (squamous vs. non-squamous), baseline albumin, baseline LDH, gender, tobacco history, metastases status (advanced or local), metastatic site, TC level, and IC level. The hazard ratio comparing the ADA positive subgroup with its matched control was 0.89 (95% CI: 0.61, 1.3). The hazard ratio comparing the ADA negative subgroup with its matched control was 0.68 (95% CI: 0.55, 0.83).
14.3 Locally Advanced or Metastatic Triple-Negative Breast Cancer
The efficacy of TECENTRIQ in combination with paclitaxel protein-bound was investigated in IMpassion130 (NCT02425891), a multicenter, international, double-blinded, placebo-controlled, randomized (1:1) trial that included 902 unresectable locally advanced or metastatic triple-negative breast cancer patients that had not received prior chemotherapy for metastatic disease. The trial excluded patients with a history of autoimmune disease, administration of a live attenuated vaccine within 4 weeks prior to randomization, administration of systemic immunostimulatory agents within 4 weeks or systemic immunosuppressive medications within 2 weeks prior to randomization; or untreated or corticosteroid-dependent brain metastases.
Randomization was stratified by presence of liver metastases, prior taxane treatment, and by PD-L1 expression status in tumor infiltrating immune cells (IC) (PD-L1 stained tumor-infiltrating immune cells [IC] <1% of tumor area vs. ≥ 1% of the tumor area) by the VENTANA PD-L1 (SP142) Assay. Of the 902 patients in the intent to treat population (ITT), 41% (369 patients) were classified as PD-L1 expression ≥ 1%. Patients were randomized to receive TECENTRIQ 840 mg or placebo intravenously on Days 1 and 15 of every 28-day cycle with paclitaxel protein-bound 100 mg/m2 intravenously on Days 1, 8 and 15 of every 28-day cycle. Patients received treatment until radiographic disease progression per RECIST v1.1, or unacceptable toxicity. Tumor assessments were performed every 8 weeks (± 1 week) for the first 12 months after Cycle 1, day 1 and every 12 weeks (± 1 week) thereafter. Major efficacy outcomes were investigator-assessed progression free survival (PFS) in the ITT and PD-L1 expressing patient population per RECIST v1.1 and overall survival (OS) in the ITT population.
In IMpassion130, the median age was 55 years (range: 20-86). Overall, most patients were women (99.6%) and the majority of patients were white (68%), Asian (18%), Black or African American (7%), and American Indian or Alaskan Native (4.4%). The demographic and baseline disease characteristics of the study population were well balanced between the treatment arms. Baseline ECOG performance status was 0 (58%) or 1 (41%). Overall, 41% of enrolled patients had PD-L1 expression ≥ 1%, 27% had liver metastases and 7% brain metastases at baseline. Approximately half the patients had received a taxane (51%) or anthracycline (54%) in the (neo)adjuvant setting. Patient demographics and baseline tumor disease in the PD-L1 expressing population were generally representative of the broader study population.
Tumor specimens (archival or fresh) were evaluated prospectively using the VENTANA PD-L1 (SP142) Assay at a central laboratory and the results were used as a stratification factor for randomization and to define the PD-L1 expression subgroups for pre-specified analyses.
Overall survival data were immature with 43% deaths in the ITT population. The efficacy results of IMpassion130 for the patient population with PD-L1 expression ≥ 1% are presented in Table 21 and Figure 4.
Table 21: Efficacy Results from IMpassion130 in Patients with PD-L1 Expression ≥ 1% PD-L1 Expression ≥ 1%* TECENTRIQ in combination with paclitaxel protein-bound Placebo in combination with paclitaxel protein-bound PFS=Progression-Free Survival; CI=Confidence Interval; ORR=Objective Response Rate; DOR=Duration of Response; NE=Not Estimable - * PD-L1 expression in tumor-infiltrating immune cells (IC)
- † As determined by investigator assessment
- ‡ per RECIST v1.1 (Response Evaluation Criteria in Solid Tumors v1.1)
- § Stratified by presence of liver metastases, and by prior taxane treatment
- ¶ patients with measurable disease at baseline
- # confirmed responses
Progression-Free Survival†,‡ (n=185) (n=184) Events (%) 136 (74) 151 (82) Median, months 7.4 (6.6, 9.2) 4.8 (3.8, 5.5) Stratified Hazard ratio (95% CI)§ 0.60 (0.48, 0.77) p-value <0.0001 Objective Response Rate†,‡,¶,# n=185 n=183 Number of responders (%) 98 (53) 60 (33) (95% CI) (45.5, 60.3) (26.0, 40.1) Complete response (%) 17 (9) 1 (<1) Partial response (%) 81 (44) 59 (32) Duration of Response†,‡,# n=98 n=60 Median (months) 9.2 6.2 (95% CI) (7.5, 11.9) (5.5, 8.8) Figure 4: Kaplan-Meier Plot of Progression-Free-Survival in IMpassion130 in Patients with PD-L1 Expression ≥1%
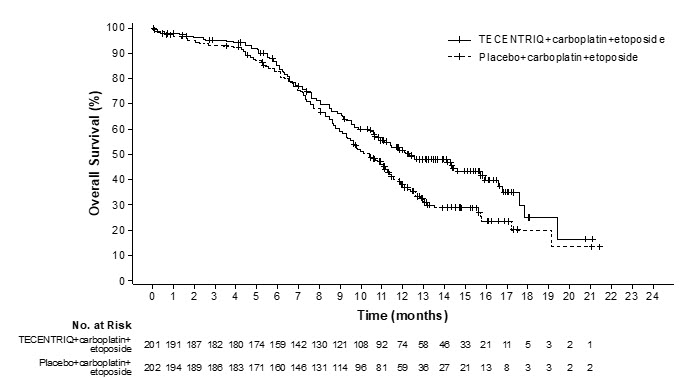
14.4 Small Cell Lung Cancer
The efficacy of TECENTRIQ with carboplatin and etoposide was investigated in IMpower133 (NCT02763579), a randomized (1:1), multicenter, double-blind, placebo-controlled trial in 403 patients with ES-SCLC. IMpower133 enrolled patients with ES-SCLC who had received no prior chemotherapy for extensive stage disease and ECOG performance status 0 or 1. The trial excluded patients with active or untreated CNS metastases, history of autoimmune disease, administration of a live, attenuated vaccine within 4 weeks prior to randomization, or administration of systemic immunosuppresive medications within 1 week prior to randomization. Randomization was stratified by sex, ECOG performance status, and presence of brain metastases. Patients were randomized to receive one of the following two treatment arms:
- TECENTRIQ 1200 mg and carboplatin AUC 5 mg/mL/min on Day 1 and etoposide 100 mg/m2 intravenously on Days 1, 2 and 3 of each 21-day cycle for a maximum of 4 cycles followed by TECENTRIQ 1200 mg once every 3 weeks until disease progression or unacceptable toxicity, or
- placebo and carboplatin AUC 5 mg/mL/min on Day 1 and etoposide 100 mg/m2 intravenously on Days 1, 2, and 3 of each 21-day cycle for a maximum of 4 cycles followed by placebo once every 3 weeks until disease progression or unacceptable toxicity.
Administration of TECENTRIQ was permitted beyond RECIST-defined disease progression. Tumor assessments were conducted every 6 weeks for the first 48 weeks following Cycle 1, Day 1 and then every 9 weeks thereafter. Patients treated beyond disease progression had tumor assessment conducted every 6 weeks until treatment discontinuation.
Major efficacy outcome measures were OS and PFS as assessed by investigator per RECIST v1.1 in the intent-to-treat population. Additional efficacy outcome measures included ORR and DoR as assessed by investigator per RECIST v1.1.
A total of 403 patients were randomized, including 201 to the TECENTRIQ arm and 202 to the chemotherapy alone arm. The median age was 64 years (range 26 to 90) and 65% were male. The majority of patients were White (80%); 17% were Asian, 4% were Hispanic and 1% were Black. Baseline ECOG performance status was 0 (35%) or 1 (65%); 9% of patients had a history of brain metastases, and 97% were current or previous smokers.
Efficacy results are presented in Table 22 and Figure 5.
Table 22: Efficacy Results from IMpower133 TECENTRIQ with Carboplatin and Etoposide Placebo with Carboplatin and Etoposide CI=confidence interval - * Stratified by sex and ECOG performance status
- † Based on the stratified log-rank test
- ‡ Compared to the allocated α of 0.0193 for this interim analysis based on 78% information using O'Brien-Fleming boundary
- § As determined by investigator assessment
- ¶ per RECIST v1.1 (Response Evaluation Criteria in Solid Tumors v1.1)
- # Compared to the allocated α of 0.05 for this analysis.
- Þ Confirmed response
Overall Survival N=201 N=202 Deaths (%) 104 (52%) 134 (66%) Median, months 12.3 10.3 (95% CI) (10.8, 15.9) (9.3, 11.3) Hazard ratio* (95% CI) 0.70 (0.54, 0.91) p-value†, ‡ 0.0069 Progression-Free Survival§,¶ N=201 N=202 Number of events (%) 171 (85%) 189 (94%) Median, months 5.2 4.3 (95% CI) (4.4, 5.6) (4.2, 4.5) Hazard ratio* (95% CI) 0.77 (0.62, 0.96) p-value†, # 0.0170 Objective Response Rate§,¶,Þ N=201 N=202 Number of responders (%) 121 (60%) 130 (64%) (95% CI) (53, 67) (57, 71) Complete response 5 (2%) 2 (1%) Partial response 116 (58%) 128 (63%) Duration of Response§,¶,Þ N=121 N=130 Median (months) 4.2 3.9 (95% CI) (4.1, 4.5) (3.1, 4.2) Figure 5: Kaplan-Meier Plot of Overall Survival in IMpower133

-
16 HOW SUPPLIED/STORAGE AND HANDLING
TECENTRIQ injection is a sterile, preservative-free, and colorless to slightly yellow solution for intravenous infusion supplied as a carton containing one 840 mg/14 mL single-dose vial (NDC: 50242-918-01) or 1200 mg/20 mL single-dose vial (NDC: 50242-917-01).
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Immune-Mediated Adverse Reactions
Inform patients of the risk of immune-mediated adverse reactions that may require corticosteroid treatment and interruption or discontinuation of TECENTRIQ, including:
- Pneumonitis: Advise patients to contact their healthcare provider immediately for any new or worsening cough, chest pain, or shortness of breath [see Warnings and Precautions (5.1)].
- Hepatitis: Advise patients to contact their healthcare provider immediately for jaundice, severe nausea or vomiting, pain on the right side of abdomen, lethargy, or easy bruising or bleeding [see Warnings and Precautions (5.2)].
- Colitis: Advise patients to contact their healthcare provider immediately for diarrhea, blood or mucus in stools, or severe abdominal pain [see Warnings and Precautions (5.3)].
- Endocrinopathies: Advise patients to contact their healthcare provider immediately for signs or symptoms of hypophysitis, hyperthyroidism, hypothyroidism, adrenal insufficiency, or type 1 diabetes mellitus, including diabetic ketoacidosis [see Warnings and Precautions (5.4)].
- Other Immune-Mediated Adverse Reactions: Advise patients to contact their healthcare provider immediately for signs or symptoms of other potential immune-mediated adverse reactions [see Warnings and Precautions (5.5)].
Infections
Advise patients to contact their healthcare provider immediately for signs or symptoms of infection [see Warnings and Precautions (5.6)].
Infusion-Related Reactions
Advise patients to contact their healthcare provider immediately for signs or symptoms of infusion-related reactions [see Warnings and Precautions (5.7)].
Embryo-Fetal Toxicity
Advise females of reproductive potential that TECENTRIQ can cause harm to a fetus and to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.8), Use in Specific Populations (8.1, 8.3)].
Advise females of reproductive potential to use effective contraception during treatment and for at least 5 months after the last dose of TECENTRIQ [see Use in Specific Populations (8.3)].
Lactation
Advise female patients not to breastfeed while taking TECENTRIQ and for at least 5 months after the last dose [see Use in Specific Populations (8.2)].
- SPL UNCLASSIFIED SECTION
-
MEDICATION GUIDE
-
SPL UNCLASSIFIED SECTION
Representative sample of labeling (see the HOW SUPPLIED section for complete listing):
-
PRINCIPAL DISPLAY PANEL - 20 mL Vial Carton
NDC: 50242-917-01
Tecentriq®
(atezolizumab)
Injection1200 mg/20 mL
(60 mg/mL)For Intravenous Infusion After Dilution
Single-Dose Vial
Discard Unused Portion
No preservative.Attention Pharmacist: Dispense the
accompanying Medication Guide
to each patient.1 vial
Rx only
Genentech
10199144

-
PRINCIPAL DISPLAY PANEL - 14 mL Vial Carton
NDC: 50242-918-01
Tecentriq®
(atezolizumab)
Injection840 mg/14 mL
(60 mg/mL)For Intravenous Infusion After Dilution
Single-Dose Vial
Discard Unused Portion
No preservative.Attention Pharmacist: Dispense the
accompanying Medication Guide
to each patient.1 vial
Rx only
Genentech10198487

-
INGREDIENTS AND APPEARANCE
TECENTRIQ
atezolizumab injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 50242-917 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength atezolizumab (UNII: 52CMI0WC3Y) (atezolizumab - UNII:52CMI0WC3Y) atezolizumab 1200 mg in 20 mL Inactive Ingredients Ingredient Name Strength histidine (UNII: 4QD397987E) 62 mg in 20 mL acetic acid (UNII: Q40Q9N063P) 16.5 mg in 20 mL sucrose (UNII: C151H8M554) 821.6 mg in 20 mL polysorbate 20 (UNII: 7T1F30V5YH) 8 mg in 20 mL water (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 50242-917-01 1 in 1 CARTON 05/18/2016 1 20 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product 2 NDC: 50242-917-86 1 in 1 CARTON 08/11/2016 2 20 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761034 05/18/2016 TECENTRIQ
atezolizumab injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC: 50242-918 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength atezolizumab (UNII: 52CMI0WC3Y) (atezolizumab - UNII:52CMI0WC3Y) atezolizumab 840 mg in 14 mL Inactive Ingredients Ingredient Name Strength histidine (UNII: 4QD397987E) 43.4 mg in 14 mL acetic acid (UNII: Q40Q9N063P) 11.5 mg in 14 mL sucrose (UNII: C151H8M554) 575.1 mg in 14 mL polysorbate 20 (UNII: 7T1F30V5YH) 5.6 mg in 14 mL water (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC: 50242-918-01 1 in 1 CARTON 03/08/2019 1 14 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product 2 NDC: 50242-918-86 1 in 1 CARTON 03/08/2019 2 14 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761034 03/08/2019 Labeler - Genentech, Inc. (080129000) Establishment Name Address ID/FEI Business Operations Roche Diagnostics GmbH 315028860 MANUFACTURE(50242-917) , ANALYSIS(50242-917, 50242-918) Establishment Name Address ID/FEI Business Operations F. Hoffmann-La Roche Ltd 485244961 ANALYSIS(50242-917, 50242-918) , LABEL(50242-917, 50242-918) , MANUFACTURE(50242-917, 50242-918) , PACK(50242-917, 50242-918) Establishment Name Address ID/FEI Business Operations Genentech, Inc. 833220176 LABEL(50242-917, 50242-918) , PACK(50242-917, 50242-918) Establishment Name Address ID/FEI Business Operations F. Hoffmann-La Roche Ltd 482242971 API MANUFACTURE(50242-917, 50242-918) , ANALYSIS(50242-917, 50242-918) Establishment Name Address ID/FEI Business Operations Genentech, Inc. 080129000 ANALYSIS(50242-917, 50242-918) Establishment Name Address ID/FEI Business Operations Genentech, Inc. 146373191 ANALYSIS(50242-917, 50242-918) Establishment Name Address ID/FEI Business Operations Roche Singapore Technical Operation, Pte. Ltd. (RSTO) 937189173 ANALYSIS(50242-917, 50242-918) Establishment Name Address ID/FEI Business Operations Roche Diagnostics GmbH 323105205 ANALYSIS(50242-917, 50242-918)
Trademark Results [TECENTRIQ]
Mark Image Registration | Serial | Company Trademark Application Date |
|---|---|
 TECENTRIQ 86441478 5027722 Live/Registered |
GENENTECH, INC. 2014-10-31 |
© 2026 FDA.report
This site is not affiliated with or endorsed by the FDA.